
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
ROZDZIAŁ 3
3.2.1 Interakcje leków między sobą
1. Interakcja – zjawisko polegające na wzajemnym oddziaływaniu podawanych jednocześnie leków, w
wyniku którego zmienia się końcowy wynik działania niektórych z nich. Jest to wpływ jednego leku
na końcowy wynik działania drugiego, jednocześnie zastosowanego.
2. Niepożądana interakcja leków – to terapeutycznie niepożądane działanie, które może wystąpić
podczas jednoczesnego zastosowania u chorego dwu lub więcej leków, polegające na osłabieniu lub
nasileniu działania, pojawieniu się objawów toksycznych lub jakościowo odmiennego od
spodziewanego działania farmakologicznego.
3. Kryteria klinicznego znaczenia interakcji leków:
- niebezpieczne następstwo interakcji – zwykle obserwowane w przypadku leków o wąskim
współczynniku terapeutycznym;
- występowanie interakcji u chorych potwierdzone dokumentacją;
- częstość występowania u chorych
4. Czynniki ryzyka zwiększające prawdobieństwo interakcji:
- polifarmakoterapia (powyżej 5 stosowanych leków ryzyko interakcji wzrasta ponadproporcjonalnie);
- stosowanie silnie działających leków o małym współczynniku terapeutycznym;
- stosowanie leków o nieliniowej farmakokinetyce (teofilina, fenytoina);
- współistnienie chorób, zwłaszcza nerek i wątroby;
- starszy lub bardzo młody wiek;
- inne, mniej istotne: powszechna dostępność leków OTC, leczenie się pacjentów u wielu lekarzy
jednocześnie, samoleczenie się chorych, reklama.
5. Zaburzenia jakich procesów farmakokinetycznych spowodują osłabienie działania
farmakologicznego leków?
- zaburzenia wchłaniania (pokarm, inne stosowane leki – np. alkalizujące, IPP, antybiotyki powod.
zespół złego wchłaniania, choroby przewodu pokarmowego i zaburzenia krążenia);
- zaburzenia dystrybucji (hiperproteinemia, zbyt wysoka zaw. tkanki tłuszczowej w przyp. leków
lipofilnych, zaburzenia krążenia m.in. w stanach zagrożenia życia);
- zaburzenia metabolizmu (indukcja enzymów lub ich inhibicja jeśli lek jest prolekiem);
- zaburzenia wydalania (osłabienie działania leku przez nasilenie jego wydalania w wyniku zmiany
odczynu moczu).
6. Zaburzenia jakich procesów farmakokinetycznych spowodują nasilenie działania
farmakologicznego leków?
- zaburzenia wchłaniania (pokarm i stosowane inne leki mogą zwiększać wchałanianie; np. leki
alkalizujące, IPP);
- zaburzenia dystrybucji (hipoproteinemia, wypieranie z połączeń z białkami, wiek – inna zaw. wody i
tkanki tłuszczowej u osób starszych);
- zaburzenia metabolizmu (inhibicja enzymów albo indukcja w przypadku proleków; choroby nerek
i/lub wątroby);
- zaburzenia wydalania (choroby nerek i/lub wątroby, zmiana odczynu moczu, a w konsekwencje
upośledzenie wydalania leku).
www.farmacja.e-lama.pl
1
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
7. Leki i grupy leków, których interakcje z innymi lekami powodują niebezpieczne klinicznie
następstwa.
- p/zakrzepowe pochodne kumaryny (doustne antykoagulanty);
- doustne leki p/cukrzycowe pochodne sulfonylomocznika;
- p/padaczkowe;
- p/arytmiczne i glikozydy naparstnicy;
- teofilina;
- statyny;
- inhibitory kanałów Ca i leki hipotensyjne;
- TLPD
- p/histaminowe II gen. - terfenadyna i astemizol -> Torsades de Pointes
- NLPZ
- antybiotyki
8. Przyczyny farmakodynamicznych interakcji leków.
Interakcje w fazie farmakodynamicznej polegają na zmianie czasu, siły i działania leku pod wpływem
działania farmakodynamicznego drugiego, jednocześnie zastosowanego leku. Mogą być receptorowe,
enzymatyczne (leki działają na ten sam receptor lub enzym) albo fizjologiczne = czynnościowe (leki
działając na różne receptory lub enzymy powodują takie same lub przeciwne skutki farmakologiczne).
- synergizm (może być addycyjny lub hiperaddycyjny);
- antagonizm
9. Przyczyny farmakokinetycznych interakcji leków na etapie wchłaniania.
- zmiana pH treści żołądkowo – jelitowej; alkalizacja sprzyja wchłanianiu: pochodnych kumaryny,
doustnych leków p/cukrzycowych, digoksyny, nifedypiny;
- absorpcja – węgiel aktywowany, cholestyramina – absorbują salicylany, tetracykliny, propranolol
- chelatowanie i tworzenie kompleksów – Ca, Fe, Mg, Al tworzą kompleksy z tetracyklinami;
cholestyramina kompleksuje digoksynę i warfarynę;
- zespół złego wchłaniania – po neomycynie i paromomycynie;
- zmiany motoryki przewodu pokarmowego – metoklopramid działa prokinetycznie; zmniejsza zatem
czas przebywania leków w przew. pok., przez co wchłanianie digoksyny, kumaryn i środków
antykoncepcyjnych zmniejszy się;
- hamowanie aktywności CYP3A4 w ścianie jelita przez składniki soku grejpfrutowego – spadek
efektu I przejścia, zwiększenie wchłaniania inhibitorów kanału Ca z grupy dihydropirydyny,
estradiolu, karbamazepiny, statyn, diazepamu;
- wpływ na aktywność P-glikoproteiny; ryfampicyna zwiększa aktywność P-gp, przez co osłabia
wchłanianie i zmniejsza stęż. we krwi m.in. digoksyny; cyklosporyna natomiast zmniejsza aktywność
P-gp.
10. Przyczyny farmakokinetycznych interakcji leków na etapie wiązania z białkami krwi.
- wypieranie leków o mniejszym powinowactwie do białek przez inne leki silniej wiążące się z
białkami; w rezultacie – wzrost stężenia wolnej frakcji tych pierwszych i nasilenie ich działania.
- wypieracze: NLPZ (asa, ibuprofen, diklefenak, fenylbutazon), sulfonamidy, klofibrat, chinydyna,
werapamil, amiodaron
- leki podatne na wypieranie: doustne antykoagulanty, doustne leki p/cukrzycowe, leki p/padaczkowe,
penicyliny, hydrokortyzon, metotreksat, digoksyna
www.farmacja.e-lama.pl
2

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
11. Przyczyny farmakokinetycznych interakcji leków na etapie metabolizmu.
Zmiana aktywności enzymów metabolizujących przez równocześnie stosowane leki.
Induktory enzymatyczne
Inhibitory enzymatyczne
Barbiturany (zwłaszcza fenobarbital),
karbamazepina, fenytoina, ryfampicyna,
gryzeofulwina, dziurawiec, WWA z dymu
tytoniowego, przewlekłe leczenie się alkoholem
Allopurynol, amiodaron, chinidyna,
chloramfenikol, cymetydyna, cyprofloksacyna,
diltiazem, disulfiram, enoksacyna, fenylbutazon,
flukonazol, fluoksetyna, izoniazyd, indynawir,
itrakonazol, ketokonazol, klarytromycyna,
metronidazol, omeprazol, rytonawir, werapamil,
alkohol w dawce jednorazowej, sok grejpfrutowy
12. Przyczyny farmakokinetycznych interakcji leków na etapie wydalania.
- zmiana pH moczu;
leki o charakterze kwaśnym (lepiej wydalane z zasadowym moczem): sulfonamidy, fenylbutazon,
barbiturany, salicylany;
leki o charakterze zasadowym (lepiej wydalane z kwaśnym moczem): kodeina, imipramina.
Alkalizacja moczu – po podaniu wodowęglanu lub mleczanu sodu. Zakwaszenie moczu – po podaniu
chlorku amonu.
- zmiany aktywnego transportu w cewkach nerkowych – leki mogą konkurować między sobą o
systemy przenośnikowe uczestniczące w aktywnym wydzielaniu z krwi do moczu:
:: dikumarol hamuje wydzielanie pochodnych sulfonylomocznika,
:: probenecyd hamuje wydzielanie penicyliny,
:: kwas aminosalicylowy ogranicza wydzielanie izoniazydu
- zmiany przepływu krwi przez nerki – przepływ ten zależny jest od prostaglandyn, których syntezę
hamują NLPZ i w ten mogą upośledzić wydalanie przez nerki leków (m.in. soli litu).
13. Interakcje leków z pochodnymi kumaryny powodujące osłabienie ich działania p/zakrzepowego.
- leki podnoszące pH soku żołądkowego zmniejszają wchłanianie pochodnych kumaryny;
- antybiotyki (neomycyna) powodując zespół złego wchłaniania, zmniejszają dostępność biologiczną
pochodnych kumaryn;
- cholestyramina kompleksuje warfarynę (spadek wchłaniania);
- leki prokinetycznie (metoklopramid) utrudniają wchłanianie pochodnych kumaryny;
- witamina K, metyloksantyny, estrogeny, glikokortykosteroidy antagonizują działanie kumaryn przez
zwiększanie w surowicy stężenia protrombiny i innych czynników krzepnięcia.
- induktory enzymatyczne pobudzają biotransformację kumaryn w wątrobie (zmniejszając ich stężenie
we krwi, a więc osłabiając działanie).
14. Interakcje leków z pochodnymi kumaryny powodujące nasilenie ich działania p/zakrzepowego.
- antybiotyki aminoglikozydowe (neomycyna) wyjaławiają przewód pokarmowy z bakterii
produkujących witaminę K. Brak naturalnego antagonisty nasila działanie pochodnych kumaryny.
- leki silnie wiążące się z białkami (fenylbutazon, indometacyna, celekoksyb, salicylany, fenytoina,
sulfonamidy, leki p/cukrzycowe i penicyliny) wypierają z połączeń z białkami pochodne kumaryny.
- NLPZ hamują syntezę protrombiny i tromboksanu (mają własne p/zakrzepowe potęgujące działanie
kumaryn).
- synergistycznie z kumarynami działają: sterydy anaboliczne (metka), tyroksyna i preparaty tarczycy,
chinidyna, heparyna, glukagon, klofibrat, niektóre cefalosporyny, klopidogrel, tiklopidyna
- inhibitory enzymatyczne
www.farmacja.e-lama.pl
3
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
15.
16. Interakcje leków p/cukrzycowych z innymi lekami.
Leki osłabiające działanie hipoglikemizujące pochodnych sulfonylomocznika:
a) glikokortykosteroidy, sympatykomimetyki (efedryna, pseudoefedryna), prep. tarczycy, glukagon,
doustne środki antykoncepcyjne (upośledzają tolerancję glukozy), fenytoina, leki moczopędne ->
antagonizm działania
b) ryfampicyna -> indukcja metabolizmu
Leki nasilające hipoglikemizujące działanie pochodnych sulfonylomocznika:
a) NLPZ: (1) same działają hipoglikemizująco w dużych dawkach, (2) wypierają leki p/cukrzycowe z
połączeń z białkami, (3) konkurują z lekami p/cukrzycowymi o systemy transportu w kanalikach
nerkowych
b) wypieracze – zwiększają wolną frakcję leków p/cukrzycowych we krwi;
c) inhibitory enzymatyczne – hamowanie biotransformacji poch. sulfonylomocznika
d) steroidy anaboliczne (metka) – synergistycznie działanie hipoglikemizujące;
e) beta-blokery – działają hipoglikemizująco na skutek antagonizmu do amin katecholowych w
działaniu na gospodarkę cukrową.
Doustne leki p/cukrzycowe hamują metabolizm etanolu (objawy reakcji disulfiramopodobnej).
17.
18. Interakcje NLPZ z innymi lekami.
FARMAKODYNAMICZNE:
- osłabiają hipotensyjne działanie leków obniżających ciśnienie krwi oraz działanie moczopędne
diuretyków, przez zahamowanie syntezy prostaglandyn rozszerzających naczynia (w tym naczynia
nerkowe),
- potęgują działanie pochodnych kumaryny, tiklopidyny, klopidogrelu przez hamowanie syntezy
protrombiny i tromboksanu,
- potęgują działanie leków p/cukrzycowych, ponieważ same działają hipoglikemizująco,
- nasilają nefrotoksyczność cyklosporyny i inhibitorów konwertazy angiotensynowej;
- alkohol zwiększa ryzyko uszkodzeń błony śluzowej przew. pokarmowego po NLPZ
FARMAKOKINETYCZNE:
- wypierają z połączeń z białkami wiele grup leków, nasilając ich działanie (poch. kumaryny,
p/cukrzycowe, metotreksat, digoksyna, fenytoina),
- zmniejszają wydalanie nerkowe innych leków (wpływ na transport aktywny oraz przesączanie
kłębkowe): p/cukrzycowe, metotreksat, digoksyna, aminoglikozydy, fenytoina, kortykosteroidy
19. Interakcje glikozydów nasercowych z innymi lekami.
- leki zwiększające stężenie Ca w osoczu (prep. wapnia) lub zmniejszające stężenie K (moczopędne –
furosemid, kwas etakrynowy, indapamid, glikokortykosteroidy, leki przeczyszczające, amfoterecyna
B) nasilają działanie glikozydów nasercowych i ich arytmogenność,
- prep. potasu oraz środki kompleksujące wapń (wersenian i cytrynian sodu) osłabiają działanie
glikozydów,
- epinefryna, norepinefryna, iMAO nasilają zaburzenia rytmu serca po glikozydach,
- węgiel, metoklopramid, cholestyramina, neomycyna, sulfasalazyna upośledzają wchłanianie
digoksyny z przewodu pokarmowego,
- induktory enzymatyczne osłabiają działanie glikozydów nasercowych,
- inhibitory enzymatyczne nasilają działanie glikozydów nasercowych,
- werapamil, diltiazem, chinidyna, amiodaron, propafenon – jako leki p/arytmiczne nasilają działanie
glikozydów,
- cyklosporyna hamuje aktywność P-glikoproteiny w jelitach i kanalikach nerkowych, przez co
www.farmacja.e-lama.pl
4

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
zwiększa wchłanianie digoksyny i zmniejsza jej wydalanie (wzrost stężenia we krwi -> nasilenie
działania),
- gentamycyna i cefradyna zmniejszają klirens nerkowy glikozydów (nasilenie działania).
20. Interakcje leków hipotensyjnych z innymi lekami.
A. Beta-blokery (LBA)
- inhibitory enzymatyczne (chinidyna, cymetydyna, amiodaron, propafenon, enkainid, flekainid,
SSRI) nasilają działanie hipotensyjne LBA, bradykardię i zaburzenia rytmu,
- inne leki hipotensyjne oraz p/arytmiczne (alfa-adrenolityki, werapamil, diltiazem, itd) – synergizm
farmakodynamiczny; -> hipotonia, zaburzenia przewodnictwa w sercu, nasilenie ujemnego działania
dromotropowego,
- NLPZ – osłabiają działanie hipotensyjne LBA przez hamowanie syntezy prostaglandyn,
- leki p/cukrzycowe – zwiększenie ryzyka hipoglikemii.
B. Leki moczopędne
- furosemid i kwas etakrynowy nasilają oto- i nefrotoksyczne działanie aminoglikozydów (synergizm
działań niepożądanych),
- NLPZ osłabiają działanie leków moczopędnych (bo zmniejszają przepływ krwi przez nerki),
- amilorid nasila nefrotoksyczność cyklosporyny,
- indapamid zwiększa ryzyko wystąpienia atypowego częstoskurczu komorowego po lekach
p/arytmicznych,
C. Blokery kanałów wapniowych
- inhibitory CYP3A4 (w tym sok grejpfrutowy) – hamowanie metabolizmu blokerów Ca, a w
rezultacie nasilenie hipotonii, ryzyka zab. rytmu serca, zawrotów, bolów głowy, obrzęków
obwodowych.
- induktory CYP3A4 (ryfampicyna, fenobarbital) – zmniejszają skuteczność terapeutyczną blokerów
kanału wapniowego.
- chinidyna i amiodaron hamują metabolizm werapamilu i diltiazemu (bradykardia, hipotonia, zab.
przewodnictwa przedsionkowo – komorowego, nasilenie objawów niewydolności serca),
- diltiazem, werapamil, amlodypina (jako inhibitory enz.) hamują metabolizm cyklosporyny (wzrost
jej nefrotoksyczności); statyn (rabdomioliza), p/padaczkowych (neurotoksyczność),
- synergizm z LBA i innymi hipotensyjnymi,
- przedłużenie działania zwiotczającego mięśnie szkieletowe tubokuraryny i atrakurium.
D. Sartany – antagoniści receptora angiotensyny II
- stosowane z lekami moczopędnymi oszczędzającymi potas mogą być przyczyną hiperkaliemi
(sartany przez blokadę receptora AT-1 zmniejszają wydzielanie aldosteronu; potas jest zatem
zatrzymywany w organiźmie, a nie wymieniany na sód).
21. Interakcje inhibitorów konwertazy angiotensynowej z innymi lekami.
- z diuretykami sodopędnymi (tiazydy i pętlowe) – nadmierna hipotonia i ostra niewydolność nerek;
- z diuretykami oszczędzającymi potas (amilorid, triamteren, spironolakton) – hiperkaliemia,
zagrażająca życiu(!),
- z diuretykami – zwiększenie stężenia mocznika we krwi,
- kaptopril w dawce 25 mg zmniejsza diuretyczne działanie furosemidu (konkurencyjne hamowanie
kanalikowego wydalania furosemidu);
- kaptopril w dawce 1 mg lub 12,5 mg podawanej przewlekle nasila moczopędne działanie
furosemidu,
- NLPZ osłabiają działanie hipotensyjne i efekty hemodynamiczne IKA (najsłabiej – ibuprofen),
- z lekami p/cukrzycowymi – wzrost hipoglikemii, ponieważ poprawiają wrażliwość tkanek
obwodowych na insulinę,
www.farmacja.e-lama.pl
5

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
- z cyklosporyną – ostra niewydolność nerek u chorych po przeszczepie nerek,
- IKA hamują nerkowe wydalanie węglanu litu, zwiększając jego toksyczność,
- z chlorpromazyną i klozapiną – hipotonia i omdlenia ortostatyczne.
22.
23. Interakcje azotanów z innymi lekami.
- z lekami hipotensyjnymi (blokery kanału Ca, LBA) – nadmierna hipotonia,
- z sildenafilem – gwałtowne rozszerzenie naczyń i spadek ciśnienia tętniczego krwi (nie wolno podać
nitratów w ciągu 24 godz. po zażyciu sildenafilu).
24. Interakcje leków hipolipemicznych z innymi lekami.
- inhibitory CYP3A4 – hamują biotransformację statyn (z wyjątkiem fluwastatyny) – miopatie,
rabdomioliza;
- fibraty, kwas nikotynowy – zmniejszają eliminację statyn (miopatie, rabdomioliza),
- fluwastatyna /inhibitor CYP2C9/ hamuje metabolizm warfaryny (zwiększa jej działanie),
- żywice jonowymienne – upośledzają wchłanianie wielu leków – patrz pyt. 9
- induktory enzymatyczne – osłabiają działanie statyn
25.
26. Interakcje aminoglikozydów z innymi lekami.
- ze środkami zwiotczającymi mięśnie szkieletowe – nasilenie działania zwiotczającego,
- z furosemidem i kwasem etakrynowym – nasilenie ototoksyczności i nefrotoksyczności,
- z niektórymi cefalosporynami, wankomycyną, amfoterecyną, cisplatyną, cyklosporyną – nasilenie
nefrotoksyczności,
- leki p/wymiotne (poch. fenotiazyny i dimenhydrynat) maskują objawy ototoksyczności
27. Interakcje tetracyklin z innymi lekami.
- z lekami zaw. jony Ca, Fe, Mg, Al, Bi, Zn, Cu, mlekiem i zasadami – tworzenie chelatów,
upośledzenie wchłaniania.
- induktory (fenytoina, barbiturany, karbamazepina) – nasilenie metabolizmu i osłabienie działania
doksycykliny,
- z fenotiazynami – nasilenie hepatotoksyczności,
- z warfaryną – nasilenie działania p/zakrzepowego, w wyniku doprowadzenia do dysbakteriozy
(zahamowanie prod. wit. K przez bakterie przew. pok.).
28. Interakcje makrolidów z innymi lekami.
- I i II generacja (erytromycyna, troleandomycyna, roksytromycyna, klarytromycyna) – inhibitory
enzymatyczne – nasilenie działania bardzo wielu leków!!!
- III generacja (azytromycyna, spiramycyna) – brak istotnych klinicznie interakcji.
29. Interakcje fluorochinolonów z innymi lekami.
- większość jest inhibitorami enzymatycznymi (cyprofloksacyna, pefloksacyna, enoksacyna, kwas
pipemidowy). Słabo – ofloksacyna; najsłabiej – lewofloksacyna, kwas nalidyksowy,
- leki zaw. jony Al, Mg, Ca, Zn, Fe – upośledzają wchłanianie fluorochinolonów z przewodu
pokarmowego.
30. Interakcje penicylin z innymi lekami.
- środki neutralizujące kwas solny i neomycyna – zmniejszają wchłanianie z przew. pok.,
- NLPZ i sulfonamidy – wypierają penicyliny z wiązania z białkami i przez to przyspieszają ich
eliminację (!),
- metotreksat działa silniej i toksyczniej, bo penicyliny konkurencyjnie hamują jego wydalanie w
kanalikach nerkowych,
www.farmacja.e-lama.pl
6

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
- probenecyd przedłuża działanie penicylin, bo hamuje ich wydalanie
- z warfaryną – nasilenie działania p/zakrzepowego (spadek synt. wit. K w wyniku dysbakteriozy po
szerokozakresowych penicylinach).
31. Interakcje cefalosporyn z innymi lekami.
- środki wpływające na wzrost pH w żołądku – zmniejszają wchłanianie cefalosporyn,
- z alkoholem dają reakcję disulfiramopodobną,
- z warfaryną – nasilenie działania p/zakrzepowego (spadek synt. wit. K w wyniku dysbakteriozy po
szerokozakresowych cefalosparynach),
- z furosemidem – nasilenie nefrotoksyczności
32. Interakcje leków p/gruźliczych z innymi lekami.
- ryfampicyna jest induktorem prawie wszystkich izoenzymów CYP. Pełna indukcja wystąpuje po
tygodniu stosowania i utrzymuje się dwa tygodnie od zakończenia terapii. Ryfampicyna znosi
skuteczność m.in. beznodiazepin, statyn, blokerów kanału Ca, osłabia działanie leków p/grzybiczych
(azoli), leków p/wirusowych, cyklosporyny, opiatów (w tym metadonu), doustnych środków
antykoncepcyjnych, pochodnych kumaryny, leków p/cukrzycowych, teofiliny, TLPD, neuroleptyków,
IKA, kortykosteroidów.
- ryfampicyna jest induktorem P-glikoproteiny (hamowanie wchłaniania z przew. pok. i/lub
zwiększanie wydalania z moczem niektórych leków).
- izoniazyd jest inhibitorem enzymatycznym (nasila działanie teofiliny, leków p/padaczkowych,
- środki neutralizujące kwas solny upośledzają wchłanianie izoniazydu.
33.
34. Interakcje leków p/grzybiczych z innymi lekami.
- amfoterecyna B zwiększa nefrotoksyczność aminoglikozydów i cyklosporyny,
- amfoterecyna B stosowana z diuretykami (furosemid) zwiększa hipokaliemię i nasila toksyczność
digoksyny,
- gryzeofulwina jest induktorem enzymatycznym – osłabia działanie innych leków,
- barbiturany osłabiają działanie gryzeofulwiny (indukują jej metabolizm),
- gryzeofulwina potęguje działanie alkoholu,
- azole są inhibitorami enzymatycznymi – nasilają działanie innych leków,
- induktory enzymatyczne (ryfampicyna, fenobarbital, fenytoina) osłabiają działanie azoli,
- leki podnoszące pH soku żołądkowego utrudniają wchłanianie azoli,
35.
36. Interakcje metronidazolu z innymi lekami.
- jest inhibitorem enz. (nasilenie działania warfaryny, fenytoiny, fluorouracylu, litu),
- powodują reakcję disulfiramopodobną,
- inne inhibitory (cymetydyna) nasilają jego działanie,
- induktory (barbiturany) osłabiają działanie metronidazolu.
37. Interakcje TLPD z innymi lekami.
- nasilają działanie doustnych antykoagulantów i leków p/cukrzycowych,
- działają synergistycznie z benzodiazepinami – nasilenie działania hamującego OUN oraz działania
cholinolitycznego,
- znoszą działanie hipotensyjne klonidyny i guanetydyny,
- z iMAO mogą powodować zespół serotoninergiczny,
- inhibitory (flukonazol, cymetydyna, werapamil, diltiazem, propafenon, chinidyna, rytonawir) hamują
metabolizm TLPD i nasilają ich działanie oraz toksyczność,
- pochodne fenotiazyny (tiorydazyna) hamują metabolizm TLPD i potęgują kardiotoksyczność i
działanie cholinolityczne,
www.farmacja.e-lama.pl
7
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
- SSRI – hamowanie metabolizmu TLPD, synergizm działania, zespół serotoninergiczny,
- doustne środki antykoncepcyjne – zwiększają wchłanianie i hamują metabolizm TLPD (nasilenie ich
działania i toksyczności),
- induktory enzymatyczne (barbiturany, ryfampicyna) zwiększają metabolizm TLPD i osłabiają ich
działanie.
38. Interakcje selektywnych inhibitorów zwrotnego wychwytu serotoniny z innymi lekami.
- są inhibitorami enzymatycznymi – nasilają działanie innych leków (teofiliny, neuroleptyków,
fenytoiny),
- z TLPD, iMAO, dziurawcem, sympatykomimetykami – zespół serotoninergiczny.
39.
40.
41.
42.
43. Interakcje metotreksatu z innymi lekami.
- cholestyramina i antybiotyki powodujące zespół złego wchłaniania (neomycyna, paromomycyna) –
utrudniają wchłanianie MTX,
- hydrokortyzon, prednizon, cefalotyna – zmniejszają transport MTX do komórek osłabiając jego
działanie p/nowotworowe,
- induktory enzymatyczne (ryfampicyna, gryzeofulwina, fenobarbital) zwiększają metabolizm MTX i
osłabiają jego działanie p/nowotworowe,
- asparaginaza działa antagonistycznie wobec MTX, znacznie osłabiając jego działanie,
- kanamycyna wybija jelitową florę biorącą udział w metabolizmie MTX, zwiększa zatem jego
wchłanianie i nasila działanie p/nowotworowe,
- wypieracze (NLPZ, sulfonamidy) zwiększają wolną frakcję MTX, nasilając jego działanie,
- salicylany, sulfonamidy, cefalotyna, omeprazol, probenecyd, penicyliny konkurencyjnie hamują
wydalanie MTX w kanalikach nerkowych,
- aminoglikozydy, cisplatyną, cyklosporyna, prokarbazyna upośledzają czynność nerek, a więc
zmniejszają wydalanie MTX (nasilenie jego działania i toksyczości),
- cisplatyna uszkadzając kanaliki nerkowe, zmniejsza wchłanianie zwrotne MTX i kwasu foliowego, a
przez to zmniejsza skuteczność ochrony folinianem wapnia,
- fenylbutazon nasila mielosupresyjne działanie MTX,
44. Interakcje teofiliny z innymi lekami.
- inhibitory enzymatyczne (fluorochinolony, cymetydyna, makrolidy, zileuton, doustne środki
antykoncepcyjne) nasilają działanie i toksyczność teofiliny powodując: nudności, wymioty, niepokój,
drżenia, tachykardię, drgawki, zaburzenia rytmu serca, a nawet śmierć,
- induktory enzymatyczne (ryfampicyna, fenytoina, barbiturany, dziurawiec) osłabiają skuteczność
teofiliny – wymagane zwiększenie dawki.
45. Interakcje cyklosporyny z innymi lekami.
- inhibitory (makrolidy I i II gen., leki p/grzybicze z grupy azoli, diltiazem, werapamil i sok
grejpfrutowy) hamują metabolizm cyklosporyny, nasilają jej działanie i toksyczność,
- induktory (fenobarbital, ryfampicyna, fenytoina, karbamazepina, dziurawiec) pobudzają metabolizm
cyklosporyny, zwiększają więc ryzyko odrzucenia przeszczepu,
- aminoglikozydy, NLPZ, amiloryd, inne leki nefrotoksyczne nasilają nefrotoksyczność cyklosporyny,
- cyklosporyna hamuje aktywność P-glikoproteiny, przez to zwiększa stężenie digoksyny i nasila jej
działanie,
- z IKA – ostra niewydolność nerek u chorych po przeszczepie nerek.
www.farmacja.e-lama.pl
8
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
3.2.4. Interakcje leków z żywnością
1. Interakcje leków z żywnością na etapie wchłaniania.
Ograniczenie wchłaniania:
a) przez adsporpcję leku;
- pektyny i błonnik adsorbują glikozydy nasercowe i TLPD,
- kwas cytrynowy asorbuje erytromycynę i benzylopenicylinę.
b) przez kompleksowanie;
- mleko, sery, jogurty (zaw. jony Ca) oraz jarzyny -zielona sałata, szpinak, buraki (zaw. jony Fe),
utrudniają wchłanianie tetracyklin i fluorochinolonów,
- kazeiniany i skrobia kukurydziana znacznie zmniejszają wchłanianie fenytoiny,
- duża ilość cukrów złożonych w pokarmie zmniejsza wchłanianie niektórych fluorochinolonów
(pefloksacyny) oraz makrolidów (azytromycyny).
c) przez wytrącanie leku;
- garbniki (kawa, herbata) wytrącają alkaloidy, pochodne fenotiazyny i preparaty żelaza.
d) przez zmianę pH;
- soki owocowe, mięso, ryby, sery zakwaszają treść żołądkowo – jelitową,
- dieta jarska alkalizuje treść żołądkowo – jelitową.
e) przez skrócenie czasu przebywania leku w przewodzie pokarmowym;
- produkty zawierające dużo błonnika (otręby, jabłka i ogórki ze skórą
Zwiększenie wchłaniania:
- tłuszcze przyspieszają wchłanianie leków o dużej lipofilności: gryzeofulwina, leki
p/pierwotniakowe, leki p/pasożytnicze, TLPD, teofilina, lipofilne beta-blokery (atenolol, metoprolol,
oksprenolol, propranolol),
- pokarm zwiększa dostępność biologiczną itrakonazolu,
- sok pomarańczowy zwiększa nawet 10-krotnie wchłanianie glinu z prep. alkalizujących (wzrost
ryzyka neurotoksyczności glinu).
2. Interakcje leków z pokarmem na etapie metabolizmu.
- flawonoidy soku grejpfrutowego hamują aktywnosć CYP3A4 w ścianie jelita, przez co ograniczają
efekt I przejścia i zwiększają stężenie we krwi następujących leków: blokerów kanałów Ca (poch.
dihydropirydyny i werapamilu), cyklosporyny, statyn, leków p/histaminowych (ebastyna, loratydyna),
a także: karbamazepiny, buspironu, diazepamu, midazolamu, triazolamu, metadonu, sildenafilu.
- dieta wysokobiałkowa i niskowęglowodanowa – nasila metabolizm leków (indukuje enzymy),
osłabienie działania teofiliny, tolbutamidu, fenacetyny.
- dieta ubogobiałkowa – zmniejsza aktywność enzymów wątrobowych, a więc zwiększa toksyczność,
- kwas foliowy (jarzyny liściaste i drożdże) nasila metabolizm difenylohydantoiny i sulfonamidów,
- produkty zaw. tyraminę stosowane z iMAO – przełom nadciśnieniowy,
- alkohol etylowy – zastosowany jednorazowo w dużej dawce hamuje aktywność enzymów
wątrobowych, a przewlekle nadużywany indukuje enzymy.
- obecność pokarmu w przew. pokarmowym osłabia efekt I przejścia: propranololu, metoprololu,
lidokainy, paracetamolu, TLPD, hydrochlorotiazydu.
3.
4. Interakcje leków z pokarmem na etapie wydalania.
- mięso, jaja, ryby, sery, chleb, makaron, rabarbar, owoce cytrusowe, żurawina – zakwaszają mocz, a
www.farmacja.e-lama.pl
9

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
więc zmniejszają wydalanie leków o odczynie kwaśnym (salicylany, sulfonamidy, barbiturany,
pochodne kumaryny, fenylbutazon, streptomycyna, ampicylina, atropina), zwiększają wydalanie
leków zasadowych (amfetamina, aminofenazon, chinidyna, imipramina, petydyna, prokaina, teofilina,
erytromycyna),
- mleko, jarzyny i większość owoców alkalizują mocz, powodując działanie przeciwne do w/w,
- zbyt niskie spożycie soli kuchennej może być zwłaszcza u osób starszych przyczyną zab. czynności
nerek, a w konsekwencji powodować zwiększenie resorpcji zwrotnej węglanu litu i nasilenie jego
toksyczności.
5. Synergizm składników żywności z lekami.
- kofeina (zaw. w kawie, herbacie), może nasilać działanie teofiliny, zwiększając ryzyko wystąpienia
działań toksycznych;
- kofeina jest także składnikiem złożonych prep. p/bólowych; ich stosowanie razem z kawą i herbatą
może spowodować objawy przedawkowania kofeiny (bezsenność, niepokój, zaburzenia rytmu serca,
tachykardię).
- saponiny zaw. w korzeniu lukrecji ze względu na swoją steroidową budową mogą wykazywać
działanie charakterystyczne dla aldosteronu – zatrzymanie sodu, kosztem wydalenia potasu
(hipokaliemię). Jest to szczególnie niebezpieczne podczas terapii glikozydami nasercowymi (wzrost
kardiotoksyczności) oraz diuretykami pętlowymi (jeszcze większa hipokaliemia -> osłabienie, bolesne
skurcze mięśni, zaburzenia przewodzenia i rytmu serca, a nawet śmierć),
- substytuty soli kuchennej (KCl) z diuretykami oszczędzającymi potas i/lub IKA powodują
hiperkaliemię (zaburzenia przewodzenia, rytmu i zatrzymanie akcji serca).
6. Sposoby unikania interakcji lek – pokarm.
- leki popijać czystą (...wodą);
- leki, których działanie może być zaburzone przed pokarm brać 1-2 godz. przed posiłkiem lub 2
godziny po;
- nie zażywać suplementów zawierających sole mineralne i witaminy oraz substytutów soli kuchennej
w tym samym czasie co leki;
- nie należy zażywać leków z alkoholem.
3.3. Zmiany działania leków uwarunkowane zaburzeniami kinetyki
w stanach patologicznych.
1. Patofarmakokinetyka – dziedzina farmakokinetyki klinicznej zajmująca się badaniem wpływu
stanów patologicznych na losy leków w organizmie. Badania patofarmakokinetyczne umożliwiają
prowadzenie tzw. farmakoterapii indywidualizowanej, pozwalającej na uniknięcie szkodliwych
następstw klinicznych schematycznej farmakoterapii nieuwzględniającej związanych z chorobą
odmienności osobniczych dotyczących wchłaniania, wiązania z białkami krwi, dystrybucji,
metabolizmu i wydalania leków.
2. Wpływ stanów patologicznych na wchłanianie leków.
- nadkwaśność lub niedokwaśność soku żołądkowego (zmiana pH wpływa na wchłanianie),
- nieprawidłowa motoryka przewodu pokarmowego może zakłócić wchłanianie leków.
=> zwolnione opróżnianie żołądka upośledza wchłanianie paracetamolu, lewodopy, chlorpromazyny,
ponieważ leki te, przebywając dłużej w przewodzie pokarmowym, w większym stopniu ulegną
efektowi I przejścia.
=> przyspieszenie pasażu jelitowego w stanach zapalnych żołądka oraz biegunkach może upośledzać
wchłanianie doustnych środków antykoncepcyjnych, digoksyny lub innych leków.
- ilościowe lub jakościowe zmiany powierzchni wchłaniania.
=> po resekcji żołądka utrudnione wchłanianie chinydyny, etambutolu,
www.farmacja.e-lama.pl
10
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
=> po resekcji jelita grubego – utrudnione wchłanianie sulfasalazyny,
=> w stanach zapalnych jelit – zmienione wchłanianie leków (większe lub mniejsze),
- unaczynienie przewodu pokarmowego:
=> we wstrząsie kardiogennym, zawale serca, zastoinowej niewydolności krążenia na skutek
zmniejszonego przepływu trzewnego, zmniejsza się wchłanianie m.in. chinidyny, prokainamidu,
digoksyny; leki te powinno się podawać wówczas tylko dożylnie!
- choroby wątroby:
=> zaburzenia wydzielania i/lub transportu żółci upośledzają wchłanianie leków i witamin
lipofilnych,
=> marskość wątroby, może przebiegać z opóźnieniem czasu opróżniania żołądka.
- choroby żoładka (brak czynnika wewnętrznego IF) – nie wchłania się witamina B-12,
- niewydolność nerek – przebiega zwykle z wymiotami, zapaleniem błony śluzowej żołądka lub jelit i
biegunkami, upośledzeniem wchłaniania wapnia (spowodowane zmniejszoną hydroksylacją
aktywnego metabolitu witaminy D3).
3. Wpływ chorób na dystrybucję leków i ich wiązanie z białkami.
- we wstrząsie kardiogennym zwiększa się dystrybucja leków do tkanek dobrze ukrwionych (mózg,
serce), a zmniejsza do tkanki tłuszczowej, mięśni.
- podobnie – we niewydolności serca, wiele leków osiąga znacznie wyższe stężenia we krwi
(chinidyna, lidokaina, meksyletyna, digoksyna, furosemid),
- w obrzękach zmienia się objętość kompartmentów organizmu, zmienia się zatem również
dystrybucja leków,
- zaburzenia równowagi kwasowo – zasadowej mogą powodować zakłócenie przenikania leków do
tkanek,
- w otyłości – wzrost objętości dystrybucji leków lipofilnych (fenytoina, diazepam, TLPD, prazosyna,
ifosfamid),
- w chorobach wątroby ze współistniejącą hipoalbuminemią i hiperbilirubinemią, upośledzona jest
zdolność białek osocza do wiązania takich leków, jak: tiopental, chinidyna, digoksyna, diazepam,
propranolol (mają mniejsze powinowactwo do albumin od bilirubiny),
- w chorobach nerek z powodu zmniejszenia stężenia białek w osoczu oraz zmian struktury tych
białek, zmniejsza się wiązanie wielu leków z albuminami. Leki mogą być również wypierane z wiązań
z białkami przez toksyny mocznicowe. W chorobach nerek upośledzone jest wiązanie z białkami
leków o odczynie kwaśnym (sulfonamidy, tyroksyna, fenytoina, klofibrat, kwas walproinowy,
salicylany, penicyliny, barbiturany, fenylbutazon, warfaryna, furosemid) oraz diazepamu.
Mniejszy stopień wiązania leku z białkami powoduje: nasilenie jego działania, wzrost ryzyka wyst.
działań niepożądanych, toksycznych. Jednocześnie czas działania ulega skróceniu (zwiększona jest
eliminacja). Należy zmniejszyć dawkę leku oraz skrócić przerwy między kolejnymi dawkami, przy
czym dobowa dawka powinna być zmniejszona.
4. Wpływ chorób wątroby na proces metabolizmu leków.
Choroby wątroby powodują następujące zaburzenia w metaboliźmie leków:
- upośledzenie zdolności komórek wątrobowych do wychwytywania (ekstrakcji) leków z krwi,
metabolizowania ich przez układy enzymatyczne i wydalania do żółci,
- zmiany przepływu wątrobowego,
- połączenia (przecieki) między krążeniem wrotnym a ogólnoustrojowym, zarówno wewnątrz, jak i
poza wątrobą,
- ilościowe i jakościowe zmiany białek osocza.
Grupa I [aminofenazon, paracetamol, kofeina, teofilina, fenobarbital, prokainamid]
- eliminacja zależy głównie od wydolności metabolicznej komórek wątroby
- nie zależy od przepływu wątrobowego
www.farmacja.e-lama.pl
11
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
- nie zależy od wiązania z białkami
w niewydolności wątroby zmniejszyć dawki tych leków
Grupa II [diazepam, fenytoina, tolbutamid, warfaryna, chlorpromazyna, ryfampicyna]
- eliminacja zależy od wydolności metabolicznej komórek wątroby
- zależy także od stopnia wiązania z białkami
- nie zależy od przepływu wątrobowego
w niewydolności wątroby zmniejszyć dawkę lub wydłużyć przedziały dawkowania na podstawie
oznaczenia klirensu wolnej frakcji tych leków
Grupa III [propranolol, lidokaina, petydyna, pentazocyna, morfina, nortryptylina]
- eliminacja zależy tylko od przepływu wątrobowego
- nie zależy od wydolności metabolicznej
- nie zależy od stopnia wiązania z białkami
w niektórych chorobach wątroby o ciężkim przebiegu zmniejsza się przepływ wątrobowy, przez co
znacznie zwiększa się dostępność biologiczna leków grupy III (nie ulegają efektowi I przejścia).
Należy znacznie zmniejszyć dawki.
Choroby wątroby w różny sposób wpływają na aktywność poszczególnych izoenzymów cytochromu
P450. Najbardziej wrażliwy jest CYP3A. Leczenie należy rozpoczynać od małych dawek.
Ale(!!) - gdy podany lek jest metabolizowany w wątrobie do postaci aktywnej (IKA) – należy u
chorych z niewydolnością wątroby zwiększyć dawki, aby uzyskać stężenie terapeutyczne.
Modyfikację dawkowania w chorobach wątroby oblicza się na podstawie:
- stężenia bilirubiny w surowicy
- stężenia albumin w surowicy
- czasu protrombinowego
- obecności (i stopnia) lub braku wodobrzusza
- obecności (i stopnia) lub braku encefalopatii
(metoda półilościowa Childa-Pugha)
Sposób modyfikacji dawkowania w niewydolności wątroby zależy od drogi podania leków:
:: gdy podanie doustne to wydłużamy przedziały dawkowania, ale nie zmniejszamy dawek;
:: gdy podanie pozajelitowe – zmniejszyć dawki i ewentualnie wydłużyć przedzał dawkowania.
5.
6.
7. Wpływ chorób nerek na metabolizm leków.
W nerkach zachodzi oksydacja, redukcja, hydroliza i sprzęganie niektórych leków. W chorobach nerek
upośledzeniu ulegają procesy redukcji, hydrolizy i acetylacji (rodzaj sprzęgania). Stwierdzono:
- zwolnienie redukcji kortyzolu
- zwolnienie acetylacji izoniazydu, kwasu aminosalicylowego
- zahamowanie hydrolizy insuliny, prokainy, cefalotyny
Może dojść do kumulacji leków, trzeba zatem zmniejszyć dawki i wydłużyć przedziały dawkowania.
8. Wpływ innych chorób na metabolizm leków.
- nadczynność tarczycy – wzrost intensywności metabolicznej (zmniejszenie stężenia np. digoksyny i
spadek jej okresu półtrwania);
- niedoczynność tarczycy – spadek aktywności enzymów;
- niewydolność krążenia - zmniejsza przepływ wątrobowy, a więc i metabolizm leków;
- gorączka – wzrost aktywności enzymów (wzrasta np szybkość biotransformacji digoksyny).
www.farmacja.e-lama.pl
12
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
9.
10. Na czym polegają zaburzenia wydalania leków w niewydolności nerek?
- zmniejsza się stopień przesączania kłębuszkowego;
- zaburzenie aktywnego wydzielania do kanalików;
- zaburzenia resorpcji zwrotnej z kanalików.
Postępowanie w niewydolności nerek:
- zmniejszyć dawki leków (wydalanych przez nerki),
- wydłużyć odstępy między kolejnymi podaniami,
- zmniejszyć dawki i wydłużyć odstępy.
Zmniejsza się zwykle dawki podtrzymujące, a dawkę początkową, służącą do osiągnięcia stężenia
terapeutycznego pozostawia zwykle bez zmian.
11. Leki wymagające modyfikacji dawkowania w niewydolności nerek.
Amfoterecyna B, aminoglikozydy, niektóre cefalosporyny, ko-trymoksazol, penicyliny (amoksycylina,
ampicylina, tykarcylina, karbenicylina), tetracykliny, salicylany, fenylbutazon, metadon, fenobarbital,
pochodne fenotiazyny, diuretyki (tiazydy, kwas etakrynowy, acetazolamid, spironolakton, triamteren),
IKA, nifedypina, digoksyna, cyklofosfamid, metotreksat, tolbutamid, chlorpropamid, klofibrat.
3.4. Farmakoterapia noworodków, niemowląt i dzieci
1. Wchłanianie leków u chorych w młodym wieku.
Noworodki i niemowlęta:
- mała powierzchnia przewodu pokarmowego,
- mniejsze wytwarzanie kwasu solnego i pepsynogenu,
- wolniejsze opróżnianie żołądka,
- słaba aktywność enzymów trzustkowych,
- słabe wydzielanie żółci,
- wolniejsza perystaltyka przewodu pokarmowego,
- znacznie zwiększona przepuszczalność błony śluzowej przewodu pokarmowego.
Zmniejszone wchłanianie
wolniejsze wchłanianie
zwiększone wchłanianie
fenobarbital, fenytoina,
paracetamol, kwas nalidyksowy
amoksycylina, flukloksacylina
penicylina G, ampicylina
- niska aktywność CYP3A4 w ścianie jelit = zwiększona biodostępność leków ulegających
metabolizmowi I przejścia w jelicie;
- niedojrzałość białek transportujących (P-glikoproteiny),
- różne ukrwienie mięśni – więc lepiej podawać leki dożylnie niż domięśniowo,
- lepsze przenikanie leków przez skórę (możliwość wystąpienia zatrucia).
2. Dystrybucja i wiązanie się leków z białkami u chorych w młodym wieku.
- większa zawartość wody w organizmie, zwłaszcza w płynie pozakomórkowym (leki hydrofilne
rozmieszczają się głównie w płynie pozakomórkowym, a we krwi osiągają niższe stężenia niż jest to u
dorosłych),
- mniejsza zawartość tkanki tłuszczowej (inne rozmieszczenie leków zależnych od współczynnika
podziału O/W),
- hipoproteinemia (zwłaszcza hipoalbuminemia), różnice jakościowe białek w porównaniu do osób
dorosłych oraz różnice ilościowe w zdolności wiązania leków,
- salicylany i sulfonamidy mogą spowodować wyparcie bilirubiny z wiązań z białkami (żółtaczka
www.farmacja.e-lama.pl
13

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
jąder podstawy mózgu),
- niska zawartość kwaśnej alfa1-glikoproteiny sprzyja toksyczności leków kationowych (u dorosłych
w znacznym stopniu wiązanych przez to białko): lidokaina, bupiwakaina, propranolol, werapamil,
chinidyna,
- zwiększona przepuszczalność bariery krew-mózg – większa wrażliwość na leki działające
ośrodkowo i etanol.
3. Metabolizm u chorych w młodym wieku.
- enzymy wątrobowe nie są jeszcze prawidłowo sprawne (upośledzona biotransformacja wielu leków,
silniejsze i dłuższe ich działania, toksyczność). Upośledzone sprzęganie chloramfenikolu z kwasem
glukuronowym => zespół szarego dziecka (zapaść krążenia, szarość powłok, wiotkie porażenie).
Również powstawanie glukuronianów: kwasu nalidyksowego, salicylanów, steroidów, sulfonamidów,
witaminy K jest u noworodków upośledzone,
- zmniejszona zdolność oksydacji (fenobarbital, mepiwakaina, lidokaina, diazepam), acetylacji,
sprzęgania z glicyną,
- sprzęganie z kwasem siarkowym i demetylacja przebiega tak, jak u dorosłych,
- aktywność enzymów można zwiększyć podając fenobarbital (indukcja), aby pobudzić czynność
odtruwającą wątroby i zmniejszyć stężenie bilirubiny u noworodka.
4.
5. Wydalanie przez nerki u chorych w młodym wieku.
- czynność nerek u noworodków stanowi 30-40 proc. czynności tych narządów u dorosłych.
Eliminacja leków jest zatem zmniejszona, a przedłuża się ich okres półtrwania w organizmie (dot.
leków wydalanych z moczem). Od 4 tygodnia życia czynność nerek osiąga sprawność zbliżoną do
osób dorosłych.
3.5. Farmakoterapia osób starszych
1. Podstawowe biologiczne, morfologiczne i czynnościowe różnice między organizmem starym a
dojrzałym.
- zmniejsza się wydolność układu krążenia, płuc, wątroby, nerek, pojemność wyrzutowa serca, a w
następstwie przepływ krwi przez tkanki; ze słabszą perfuzją tkanek wiąże się gorszy transport leków
do narządów do narządów docelowych,
- wzrasta zawartość tkanki tłuszczowej i płynu międzykomórkowego,
- zmniejsza się zawartość wody w całym organizmie i płynu wewnątrzkomórkowego,
- zmniejsza się masa mięśniowa,
- zmniejsza się liczba komórek w różnych narządach, zwłaszcza w OUN,
- słabną zdolności adaptacyjne i autoregulacyjne,
- słabnie ruchliwość, wydolność manualna, percepcja, wzrok, słuch.
2. Wchłanianie u osób starszych.
- alkalizacja soku żołądkowego,
- osłabienie wydzielania kwasu solnego,
- zwolniona perystaltyka i opóźnione opróżnianie przewodu pokarmowego,
- zmniejszony przepływ krwi przez przewód pokarmowy,
- zmniejszona powierzchnia wchłaniania,
- osłabiony transport aktywny
- wchłanianie leków przez transport bierny nie jest na tyle upośledzone, by mogło powodować
konsekwencje terapeutyczne,
- wchłanianie levodopy jest znacznie większe (bo w wieku starszym spada aktywność dopa-
dekarboksylazy w przewodzie pokarmowym),
www.farmacja.e-lama.pl
14

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
- wzrasta biodostępność i nasila się działanie leków podlegających metabolizmowi I przejścia
(propranolol, werapamil, nifedypina, molsydomina, labetalol, lidokaina, azotany),
- gorsze ukrwienie skóry, tkanki podskórnej i mięśniowej – upośledzenie wchłaniania w przypadku
podania przez skórę lub domięśniowego.
3. Dystrybucja i wiązanie z białkami u ludzi w podeszłym wieku.
- zwiększa się objętość dystrybucji leków lipofilnych (wzrost ilości tkanki tłuszczowej u osób
starych); dłuższy okres półtrwania, ryzyko kumulacji i wystąpienia objawów niepożądanych
werapamilu, diazepamu, lidokainy, haloperydolu, TLPD, barbituranów,
- spada objętość dystrybucji leków hydrofilnych i rozmieszczających się w beztłuszczowej masie ciała
(w tym mięśniach), natomiast rośnie ich stężenie we krwi i niebezpieczeństwo toksyczności:
aminoglikozydy, hydrofilne beta-blokery, teofilina, digoksyna.
- spada stężenie albumin we krwi – zwiększa się zatem wolna frakcja leków o charakterze kwaśnym
(cymetydyna, furosemid, fenylbutazon, salicylany, pochodne sulfonylomocznika, fenytoina);
- wzrost stężenia kwaśnej alfa1-glikoproteiny wiążącej leki zasadowe: propranolol, imipramina i inne
TLPD, lidokaina.
4. Metabolizm u ludzi w starszym wieku.
- zmniejszone: przepływ wątrobowy, masa wątroby, aktywność enzymów wątrobowych;
- w związku z powyższym: dłuższy czas półtrwania i mniejszy klirens leków metabolizowanych
głównie lub całkowicie w wątrobie poprzez reakcje I fazy (barbiturany, fenylbutazon, paracetamol,
teofilina, chinidyna, niektóre benzodiazepiny – diazepam, chlordiazepoksyd, flurazepam),
- okres półtrwania i klirens leków metabolizowanych tylko w procesach II fazy (sprzęgania) nie
ulegają istotnym zmianom (oksazepam, lorazepam, temazepam),
- zwiększenie biodostępności leków ulegających efektowi I przejścia w wątrobie (propranolol,
werapamil, lidokaina, nifedypina, labetalol, azotany).
5. Wydalanie u osób starszych.
- zmniejszone: masa nerek i ich czynność, perfuzja (wskutek spadku rzutu minutowego serca),
upośledzenie filtracji kłębuszkowej, wydalania kanalikowego, resorpcja zwrotna.
- w związku z tym spadek klirensu nerkowego i rośnie okres półtrwania leków wydalanych głównie z
moczem: aminoglikozydy, IKA, digoksyna, niektórych cefalosporyn, leków moczopędnych,
pochodnych sulfonylomocznika.
6. Wpływ starszego wieku na właściwości farmakodynamiczne leków.
Tkanki docelowe osób starszych wykazują zwiększoną wrażliwość na leki, z powodu osłabienia
mechanizmów utrzymujących homeostazę. Dlatego istnieje niebezpieczeństwo:
- hipotonii ortostatycznej, podczas stosowania leków hipotensyjnych;
- zaburzeń rytmu serca, czynności przewodu pokarmowego i OUN po glikozydach;
- hipokaliemii po lekach moczopędnych;
- nasilenie działania i toksyczności leków hamujących OUN (neuroleptyków, TLPD, benzodiazepin);
zmiany w OUN: -zmniejszenie masy mózgu, liczby neuronów, zaw. neuroprzekaźników; w
konsekwencji słabsze działanie leków pobudzających OUN, silniejsze leków hamujących OUN.
- większe ryzyko krwawień po antykoagulantach;
- większa hipoglikemia po lekach p/cukrzycowych;
- zmniejszona wrażliwość receptorów beta, a więc słabsze działanie beta-blokery i beta-mimetyków;
- zwiększona wrażliwość receptorów muskarynowych na cholinomimetyki, a zmniejszona na
cholinolityki.
7. Leki działające na układ krążenia stwarzające największe ryzyko powikłań u osób starszych.
a). glikozydy nasercowe – nie stosować; zaburzenia rytmu, brak łaknienia, zaburzenia widzenia,
zaburzenia psychiczne (depresja); w niewydolności serca lepiej tolerowane i skuteczniejsze u osób
www.farmacja.e-lama.pl
15

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
starszych są inhibitory konwertazy angiotensyny (IKA), ale małe dawki!
b). leki hipotensyjne – powodują podciśnienie ortostatyczne – w wyniku gwałtownego spadku
ciśnienia może dojść do niedokrwienia mózgu, serca, nerek;
c). beta-blokery – przeciwskazane u starszych, zwłaszcza propranolol – mogą nasilać lub ujawniać
niewydolność serca i niewydolność oddechową, powodować zaburzenia OUN (depresja); dobrze
tolerowane są bisoprolol, metoprolol i karwedilol, ale rozpocząć od małych dawek.
d). rezerpina, klonidyna, metyldopa – BEZWZGLĘDNIE PRZECIWSKAZANE – powodują objawy
głębokiej depresji.
e). alfa-adrenolityki (prazosyna) i pochodne hydralazyny – nie stosować – powodują gwałtowne
spadki ciśnienia, odruchową tachykardię, a więc wzrost zapotrzebowania serca na tlen.
f). blokery kanałów wapniowych – nie stosować krótko działających; długo działające (amlodypina,
felodypina) są bezpieczne.
g). IKA i sartany – można stosować, ale ostrożnie (zmniejszyć dawki).
h). leki moczopędne – powodują hipokaliemię, nadmierne odwodnienie, zaburzenia elektrolitów;
podawać w mniejszych dawkach, z preparatami potasu.
i). warfaryna – konieczne zmniejszenie dawki (bo hipoalbuminemia u osób starszych); dawkę
dostosować w oparciu o wskaźnik INR.
8.
9.
10. Grupy leków działających na OUN o zwiększonym ryzyku powikłań u osób starszych.
a). barbiturany – bezwzględnie przeciwskazane (mogą działać toksycznie na ośrodek
naczynioruchowy i oddechowy).
b). benzodiazepiny – mogą powodować zaburzenia świadomości, splątanie, pobudzenie paradoksalne,
zaburzenia koordynacji ruchowej prowadzące do urazów i złamań. Nie stosować długodziałających
BDA (diazepam, chlordiazepoksyd). Zmniejszyć o połowę dawkę nitrazepamu, estazolamu,
flurazepamu. Bezpieczne są krótko działające – oksazepam, temazepam albo zolpidem, zopiklon,
zaleplon.
c). TLPD – raczej nie stosować – powodują zaburzenia świadomości, podciśnienie ortostatyczne,
objawy cholinolityczne, bardzo niebezpieczne dla serca (wydłużenie QT, torsade de pointes),
majaczenie. Można stosować SSRI.
d). neuroleptyki poch. fenotiazyny – zaburzenia ruchowe (hiperkinetyczne i dyskinetyczne, objawy
pozapiramidowe), podciśnienie ortostatyczne, depresja, działanie cholinolityczne. Tiorydazyna,
haloperydol i droperidol ponadto – torsade de pointes. Bezpieczniejsze są atypowe neuroleptyki:
risperidon, klozapina, olanzapina.
e). leki p/padaczkowe – stosować o połowę zmniejszone dawki! U osób starszych ujawnia się
kardiotoksyczne działanie fenytoiny, karbamazepiny; ponadto: powikłania hematologiczne, zaburzenia
percepcji.
f). leki stosowane w chorobie Parkinsona – levodopa wywołuje pobudzenie, dezorientację,
podciśnienie ortostatyczne, psychozy. Selegilina – pobudzenie i dezorientacja. Cholinolityki –
nasilenie działania (zaparcia, zatrzymanie moczu, suchość w ustach).
11.
12. NLPZ-y w podeszłym wieku.
a). PRZECIWSKAZANE: fenylbutazon, indometacyna, piroksykam, diklofenak (ryzyko krawień z
przewodu pokarmowego, hepato-, mielo-, nefrotoksyczność i zatrzymanie płynów – wzrost ciśnienia).
b). DOPUSZCZALNE: naproksen, ibuprofen, ketoprofen, ewentualnie koksyby (jeśli nie ma
przeciwskazań ze strony serca).
13. Antybiotyki przeciwskazane u osób starszych.
a). aminoglikozydy – nie stosować bo mają przedłużony czas półtrwania – wzrost nefro- i
ototoksyczności;
www.farmacja.e-lama.pl
16

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
b). fluorochinolony – stosować tylko w wyjątkowych przypadkach pod ścisłym nadzorem. Potęgują
zaburzenia OUN (dezorientacja, osłabienie, drżenie, depresja), mogą nasilać uszkodzenie ścięgien i
przedłużać odstęp QT.
3.6. Farmakogenetyka
1. Farmakogenetyka – dział farmakologii klinicznej, zajmujący się wpływem genotypu i fenotypu
człowieka na działanie i losy leków w organizmie. Farmakogenetyka ma zapewnić pomoc w
podawaniu i dawkowaniu chorym takich leków, których stosowanie strwarza największe
prawdopodobieństwo odniesienia korzyści terapeutycznych i najmniejsze prawdopodobieństwo
wystąpienia reakcji niepożądanych.
2. Dziedzicznie uwarunkowane odmienności działania leków u człowieka mogą być spowodowane:
- zmienioną farmakokinetyką na wszystkich etapach,
- zmienionymi właściwościami farmakodynamicznymi leków – zmiana ich wpływu na organizm
wiąże się z innymi właściwościami receptorów, kanałów jonowych, enzymów, transporterów
neuroprzekaźników.
3. Niepożądane następstwa kliniczne genetycznych zmian kinetyki leków.
a). zaburzenia wchłaniania w wyniku zwiększonej aktywności P-glikoproteiny;
b). znaczne różnice osobnicze wartości stężenia stacjonarnego we krwi mimo podania tej samej dawki
w wyniku zaburzeń metabolizmu;
c). oporność wielolekowa (gen ABCB1).
4. Genetycznie uwarunkowane zaburzenia utleniania leków i ich kliniczne następstwa.
Za utlenianie leków odpowiadają izoenzymy CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6,
CYP2E1, CYP3A4, z których zwłaszcza CYP2C9, CYP2C19 i CYP2D6 wykazują polimodalność
(istnienie w populacji grup wyraźnie odmiennie reagujących na leki).
CYP2C9
CYP2C19
CYP2D6
- warfaryna, acenokumarol
- diklofenak, ibuprofen, naproksen,
piroksykam, celekoksyb
- tolbutamid, glipizyd
- losartan
- fenytoina
- fluwastatyna
- fluoksetyna
- amitryptylina
- tamoksyfen
- omeprazol, lansoprazol,
pantoprazol
- diazepam
- fenytoina
- cyklofosfamid
- progesteron
- propranolol
- debryzochina, sparteina
- metoprolol, karwedilol, tymolol
- flekainid, enkainid (prolek!),
propafenon, meksyletyna
- perheksylina
- TLPD
- SSRI
- neuroleptyki
- ondansetron, tropisetron
- dekstrometorfan, kodeina
(prolek!), tramadol (prolek!)
Fenotyp oksydacji, obok wpływu na skuteczność i działania niepożądane wyżej wymienionych leków,
może predysponować do wystąpienia niektórych chorób, takich jak układowy toczeń rumieniowaty,
choroba Parkinsona, niektóre nowotwory.
Współczynnik metaboliczny MR (Metabolic Ratio) to stosunek ilości wydalanej z moczem substancji
macierzystej do ilości wydalanych metabolitów.
www.farmacja.e-lama.pl
17
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
5. Genetycznie uwarunkowane zaburzenia acetylacji leków i ich kliniczne następstwa.
Za acetylację odpowiada enzym N-acetylotransferaza, kodowana przez geny: NAT1 i NAT2, z których
ten drugi wykazuje wyraźny polimorfizm genetyczny.
Acetylowane są: kofeina, dihydralazyna, izoniazyd, nitrazepam, prokainamid, niektóre sulfonamidy,
acebutolol.
Populacje: szybcy acetylatorzy (RA) i wolni acetylatorzy (SA). Fenotyp szybki (RA) jest dominujący
w stosunku do wolnego. Acetylatorzy wolni są homozygotami recesywnymi.
Podawanie dawek standardowych szybkim acetylatorom prowadzi zwykle do braku skuteczności
terapii – rzadziej obserwuje się remisję, częściej nawroty choroby i oporność na leki. U wolnych
acetylatorów skuteczność terapii jest większa, ale znacznie częściej występują toksyczne powikłania
polekowe, np. uszkodzenie wątroby i neuropatie po izoniazydzie, polekowy toczeń rumieniowaty po
hydralazynie.
Wolni acetylatorzy mają zwiększone ryzyko wystąpienia raka pęcherza moczowego, żołądka, wątroby,
krtani i płuc.
6. Genetycznie uwarunkowane stany patologiczne towarzyszące zmienionym reakcjom organizmu
na leki.
a). polimorfizm butyrylocholinoesterazy (u osób posiadających w surowicy atypowy enzym o niższym
powinowactwie do sukcynylocholiny, działanie zwiotczające mięśnie trwa znacznie dłużej);
b). polimorfizm metylotransferazy tiopuryny (u osób posiadających enzym o zmniejszonej aktywności
merkaptopuryna, tioguanina i azatiopryna osiągają znacznie wyższe stężenia we krwi, w komórkach
gromadzą się toksyczne metabolity, nasila się mielosupresja);
c). polimorfizm dehydrogenazy dihydropirymidyny (u osób posiadających enzym o zmniejszonej
aktywności fluorouracyl osiąga znacznie wyższe stężenia we krwi, nasila się mielosupresja i
neurotoksyczne działanie leku);
d). polimorfizm glukuronylotransferazy; osoby z niższą aktywnością UGT1A1 mają upośledzone
sprzęganie z kwasem glukuronowym (->hiperbilirubinemie). Jeśli wystąpi żółtaczka, nie wolno
stosować: chloramfenikolu, sulfonamidów, barbituranów, morfiny, kodeiny, paracetamolu (będą
konkurencyjnie hamować powstawanie glukuronianów bilirubiny); przeciwwskazane są też
wypieracze (salicylany). U słabych metabolizerów irynotekan (lek p/nowotworowy) ulega kumulacji,
powoduje ciężką biegunkę i małopłytkowość (działania toksyczne);
e). polimorfizm S-transferazy glutationu (metabolizm cyklofosfamidu);
f). polimorfizm syntazy tymidylowej – enzym jest hamowany przez fluorouracyl; polimorfizm może
być odpowiedzialny za oporność na ten lek;
g). polimorfizm dehydrogenazy glukozo-6-fosforanowej – ostra niedokrwistość hemolityczna u osób z
niedoborem tego enzymu po podaniu: prymachiny, akrydyny, kwasu acetylosalicylowego,
aminofenazonu, tolbutamidu, sulfonamidów, pochodnych sulfonylomocznika, pochodnych chinoliny,
nitrofurantoiny. Po 2-3 dniach od przyjęcia w/w leków pojawiają się silne bóle głowy, brzucha, bóle w
okolicy lędźwiowej, osłabienie, żółtaczka, rozwija się methemoglobinemia. Mogą wystąpić
niepohamowane wymioty, stan śpiączki, a nawet bezmocz i mocznica. Charakterystyczne czarne
zabarwienie moczu z powodu obecności w nim hemoglobiny i produktów jej rozpadu.
h). hipertermia złośliwa – u osób z nieprawidłowym receptorem rianodynowym (RYDR) zachodzi
niekontrolowany napływ wapnia do komórki i wystąpienie objawów hipertermii złośliwej po
zastosowaniu anestetyków ogólnych oraz środków zwiotczających.
Gwałtownie wzrasta ciepłota ciała (43-44 stopnie w czasie 1 stopień na 5 minut!), wzrasta napięcie
mięśni, przyspiesza się akcja serca, pojawia się duszność, sinica, kwasica metaboliczna i oddechowa,
wzrasta stężenie potasu, zmniejsza się stężenie wapnia we krwi, rozwija się rabdomioliza. Nieleczona
www.farmacja.e-lama.pl
18

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
hipertermia kończy się śmiercią w wyniku niewydolności krążenia i nerek.
Aktywne usuwanie z komórek jonów wapnia przez dążący do utrzymania homeostazy organizm
wymaga dostarczania dużych ilości energii. To wiąże się z wytwarzaniem dużych ilości ciepła i
zużyciem zasobów energetycznych komórki. Do krążenia przedostają się w wyniku rozpadu komórek:
wapń, fosfor, potas, fosfokinaza kreatyniny, mioglobina.
7. Leki a hipertermia złośliwa u osób predysponowanych.
Do najsilniejszych aktywatorów hipertermii złośliwej należą:
- anestetyki dozylne: ketamina
- anestetyki wziewne: halotan, izofluran, enfluran, eter dietylowy, metoksyfluran, desfluran.
- środki zwiotczające: suksametonium, deksametonium, tubokuraryna
- sympatykomimetyki
- leki nasercowe – glikozydy nasercowe
Względnie bezpieczne są:
- anestetyki dożylne: barbiturany, etomidat, benzodiazepiny
- leki p/bólowe: opioidy
- leki zwiotczające: pankuronium, atrakurium
- anestetyki wziewn: podtlenek azotu
- środki znieczulające miejscowo: pochodne amidowe i estrowe
Leczenie: DANTROLEN (zmniejsza śródkomórkowe stężenie wapnia).
3.7. Farmakoterapia w okresie ciąży
1. Kinetyka leków w ciąży.
a). wchłanianie
- wolniejsza motoryka przewodu pokarmowego (opóźnienie opróżniania żołądka).
- mniejsze wydzielanie kwaśnego soku żołądkowego, większa produkcja śluzu (w rezultacie wzrost
pH w żołądku).
- przedłużony czas przebywania leków w przew. pok., czyli spadek biodostępności leków ulegających
efektowi I przejścia w jelicie
- nudności i wymioty ciążowe – upośledzenie wchłaniania leków
b). dystrybucja
- więcej jest wody w organizmie; wzrasta objętość osocza i płynu pozakomórkowego, a więc leki
hydrofilne osiągają niższe stężenia
- hormony łożyskowe wypierają leki z wiązania z albuminami. Wzrost wolnej frakcji leku powoduje,
że większa jego ilość przenika do krążenia płodowego
c). metabolizm
- progesteron i estrogeny zwiększają aktywność enzymów I fazy metabolizmu, ale jednocześnie
zmniejszają aktywność procesów II fazy (zwłaszcza sprzęgania z kwasem glukuronowym i
siarkowym). Upośledzenie czynności odtruwającej wątroby stwarza niebezpieczeństwo kumulacji i
zwiększenia toksyczności wielu leków.
- hormony powodują też zastój żółci, co zmniejsza eliminację niektórych leków (ryfampicyna).
d). wydalanie
- w ciąży wzrasta przepływ nerkowy i przesączanie kłębuszkowe, co może zwiększyć stopień
nerkowego wydalania leków eliminowanych w postaci niezmienionej (digoksyna, aminoglikozydy,
sole litu).
www.farmacja.e-lama.pl
19
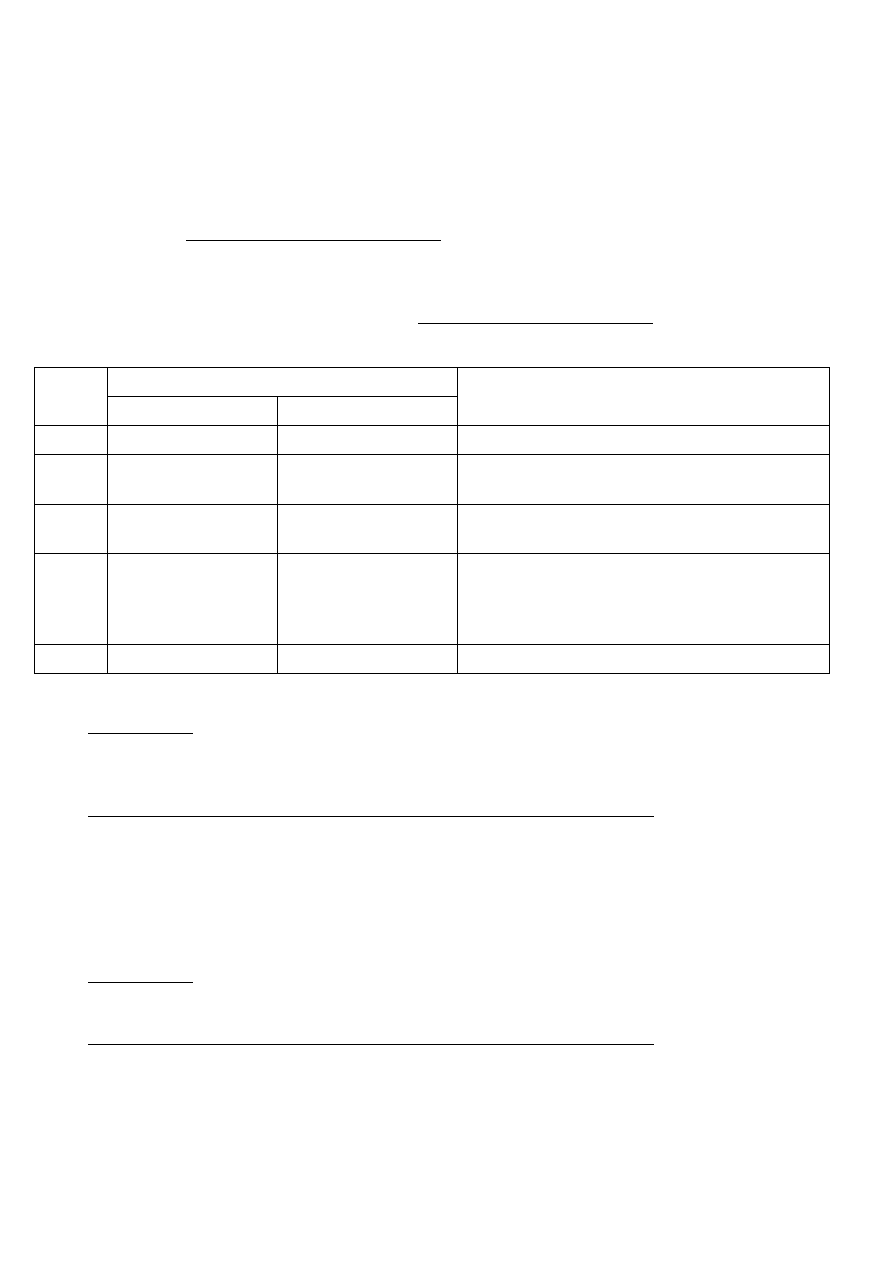
Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
2. Klasyfikacja leków w aspekcie ich szkodliwego działania na płód.
- A – badania na kobietach w ciąży nie wykazały szkodliwego działania na płód,
- B – badania na zwierzętach nie wykazały ryzyka dla płodu, ale nie przeprowadzono badań na babach
- C – badania na zwierzętach wykazały szkodliwość dla płodu, ale nie przeprowadzono badań na
kobietach. Leki te można stosować, gdy korzyść dla matki będzie większa niż ryzyko dla płodu.
- D – istnieje udokumentowane ryzyko dla płodu. Można zastosować te leki u ciężarnych tylko w
stanach zagrożenia życia matki, gdy leki z grup A, B i C okażą się nieskuteczne lub nie mogą być
zastosowane.
- X – udokumentowane działanie szkodliwe na płód u ludzi i ryzyko stosowania u kobiety ciężarnej
przewyższa wszelkie możliwe korzyści. Leki bezwzględnie przeciwwskazane u kobiet w ciąży i
kobiet w wieku reprodukcyjnym, które chcą i mogą zajść w ciążę.
Kat.
teratogenność potwierdzona w badaniach
na ludziach
na zwierzętach
Przykłady leków
A
-
B
brak badań
-
Makrolidy, metronidazol, poch. nitrofuranu,
penicyliny, cefalosporyny, ryfampicyna
C
brak badań
+
Izoniazyd, aminoglikozydy, chinolony,
desipramina
D
+
Aminoglikozydy, chloramfenikol, chinolony,
barbiturany, nortryptylina, diazepam, nitrazepam,
chlordiazepoksyd, oksazepam, warfaryna, leki
moczopędne, IKA, sartany
X
+
Estazolam, klobazam, temazepam
3. Stosowanie w ciąży leków przeciwbakteryjnych (antybiotyki, sulfonamidy, leki p/gruźlicze).
Nie stosować: aminoglikozydy, tetracykliny, chloramfenikol, karbenicylina, tykarcylina,
ureidopenicyliny, polimiksyny, wankomycyna, nefrotoksyczne cefalosporyny (cefalorydyna,
cefazolina), karbapenemy, trimetoprym, sulfonamidy, prawie wszystkie przeciwgruźlicze
Leki alternatywne, dopuszczone w przypadku uzasadnionej konieczności: penicyliny
(krystaliczna, prokainowa, benzatynowa, ampicylina), cefalosporyny pozbawione działania
nefrotoksycznego (cefaklor, cefuroksym, ceftriakson), makrolidy z wyjątkiem klarytromycyny,
pochodne nitrofuranu. Izoniazyd i ryfampicynę można stosować w uzasadnionych przypadkach po I
trymestrze ciąży.
4. Stosowanie leków przeciwpasożytniczych i przeciwgrzybiczych w ciąży?
Nie stosować: w I trymesterze ciąży nie stosować ŻADNYCH leków p/pasożytniczych. Z leków
przeciwgrzybiczych nie stosować: flukonazolu, flucytozyny, ketokonazolu, mikonazolu.
Leki alternatywne, dopuszczone w przypadku uzasadnionej konieczności: Po I trymestrze:
nystatyna, natamycyna, klotrimazol, terbinafina, amfoterycyna B. Chlorochina, metronidazol,
prazykwantel, pyrantel, mepakryna, niridazol – gdy korzyść dla matki przewyższa ryzyko dla płodu.
www.farmacja.e-lama.pl
20

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
5. NLPZ-ty i leki p/bólowe w ciąży.
Kwas acetylosalicylowy i inne NLPZ, p/bólowe i p/gorączkowe – szczególnie niebezpieczne w I, a
zwłaszcza III trymesterze ciąży; podobnie – morfinopodobne środki przeciwbólowe.
W przypadkach uzasadnionej konieczności wolno przez krótki okres stosować paracetamol.
6.
7. Leki psychotropowe w ciąży.
Nie stosować: barbiturany, benzodiazepiny (estazolam, klobazam, temazepam – kategoria X – apatia,
zaburzenie ssania, obniżenie ciśnienia tętniczego, zespół odstawienny, wady twarzoczaszki, układu
sercowo -naczyniowego i przew. pokarmowego), pochodne butyrofenonu, pochodne fenotiazyny,
prawie wszystkie leki przeciwdepresyjne, sole litu
Leki alternatywne, dopuszczone w przypadku uzasadnionej konieczności: zolpidem, neuroleptyki
(dopiero po I trymesterze, w najmniejszych dawkach, nie w formie depot), przeciwdepresyjne: z
wyboru: nortryptylina, dezipramina, fluoksetyna
8. Leki przeciwpadaczkowe w ciąży.
Nie stosować: walproiniany, fenytoina, karbamazepina (wszystkie kat. D), powodują wady wrodzone:
rozszczep podniebienia lub wargi, swoista wada cewy nerwowej – rozszczep kręgosłupa, niedorozwój
palców i paznokci, zespół hydantoinowy płodu (zahamowanie rozwoju wewnątrzmacicznego,
upośledzenie rozwoju umysłowego, małogłowie, zniekształcenie twarzy).
Leki alternatywne, dopuszczone w przypadku uzasadnionej konieczności: wigabatryna,
lamotrygina, gabapentyna, tiagabina
9. Leki działające na układ sercowo-naczyniowy i leki moczopędne w ciąży.
Nie stosować: beta-blokery niewybiórcze, IKA i sartany(!) - mogą wywołać ciężkie uszkodzenie
płodu i jego śmierć, wszystkie leki moczopędne – zmniejszają objętość krążącego osocza co może
spowodować niedokrwienie łożyska i zahamowanie rozwoju płodu, antagoniści aldosteronu i
triamteren mogą powodować obumarcie płodu i ostre zapalenie trzustki u matki; nie stosować też
p/arytmicznych: flekainidu, propafenonu i amiodaronu. Warfaryna, zwłaszcza stosowana w I
trymesterze, powoduje niedorozwój kości nosa, kości kończyn, nieprawidłowy rozwój chrząstek
stawowych, wady narządu słuchu i wzroku, niedorozwój umysłowy, zmniejszenie masy ciała
Leki alternatywne, dopuszczone w przypadku uzasadnionej konieczności: metoprolol, atenolol,
digoksyna, lidokaina, chinidyna, dizopyramid, metyldopa, hydralazyna, heparyna
10. Leki hormonalne i przeciwcukrzycowe w ciąży.
Nie stosować: kortykosteroidy (niewykształcenie się kory nadnerczy i związane z tym niebezpieczne
odwodnienie z towarzyszącą zapaścią po urodzeniu, debilizm u dziecka), estrogeny (niedorozwój
narządów rozrodczych u płodów), androgeny, I gen. pochodnych sulfonylomocznika (hipoglikemia
u noworodków).
Leki alternatywne, dopuszczone w przypadku uzasadnionej konieczności: preparaty tarczycy,
insulina, glibenklamid, gestageny
www.farmacja.e-lama.pl
21

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
3.9. Wpływ czynników środowiska oraz rytmu dobowego na działanie leków.
1. Wpływ czynników środowiska na działanie leków.
- induktory enzymów wątrobowych: policykliczne węglowodory z dymu tytoniowego, insektycydy,
herbicydy, olejki eteryczne, konserwanty żywności
- inhibitory enzymów wątrobowych: zatrucie ołowiem, tlenkiem węgla
2. Chronofarmakologia kliniczna – dziedzina zajmująca się zmianami działania leków u ludzi w
zależności od pory doby.
Chronofarmakokinetyka – zmiany losów leku w organizmie w zależności od rytmu dobowego;
Chronestezja – rytm wrażliwości receptora, przepuszczalności błon i wewnątrzkomórkowych
procesów metabolicznych;
Chronergia – rytm końcowego, zarówno pożądanego, jak i niepożądanego efektu działania leku w
organizmie, który zależy od farmakokinetyki i chronestezji;
Chronofarmakoterapia – optymalizacja leczenia farmakologicznego przez dawkowanie leków
dostosowane do biorytmu organizmu.
3. Wpływ rytmu dobowego na kinetykę leków.
- leki podane doustnie osiągają zwykle większą biodostępność w dzień niż w godzinach wieczornych
lub nocnych;
- największa aktywność metaboliczna wątroby przypada na godziny popołudniowe. Ma to szczególne
znaczenie w przypadku leków ulegających w dużym stopniu efektowi I przejścia, które zażyte
popołudniu osiągają niskie stężenia we krwi (np. propranolol);
- wydalanie z moczem również zależy od rytmu dobowego. Cisplatyna jest wydalana przez nerki we
wczesnych godzinach rannych w największym stężeniu; wtedy jest też znacznie bardziej
nefrotoksyczna. Powinna zatem być podawana wieczorem.
- leki uspokajające i nasenne działają znacznie silniej w nocy niż rano.
- preparaty naparstnicy silniej dziłają wieczorem (wtedy czynność serca jest zwolniona);
- heparyna podawana w ciągłym wlewie najsilniejsze działanie hamujące krzepnięcie wykazuje o 4, a
najsłabsze o 8 rano;
- dawki glikokortykosteroidów dostosowuje się do chronofizjologii osi podwzgórzowo- przysadkowo
– nadnerczowej podając 75% dawki o godz. 7-8, a pozostałą część o 14-15.
- objawy astmy nasilają się zwykle w nocy, dlatego teofilinę (SR), beta-adrenomimetyki należy
podawać wieczorem.
- największe wydzielanie kwasu solnego w żołądku ma miejsce w godzianach późno popołudniowych,
dlatego zaleca się podawania antagonistów H2 a także omeprazolu w późnych godzinach
popołudniowych.
3.10. Farmakoterapia monitorowana stężeniami leków w organizmie
1. Cel i istota terapii monitorowanej.
Celem terapii monitorowanej jest ustalenie takiego dawkowania leku, aby uzyskane stężenia mieściły
się w zakresie przedziału terapeutycznego, czyli w zakresie stężeń, w którym osiągnie się wysoki
stopień skuteczności przy jednoczesnym małym ryzyku wystąpienia objawów toksycznych.
Istotą terapii monitorowanej jest założenie zależności między działaniem farmakologicznym, a
stężeniem leku we krwi lub innym dostępnym do analizy materiale biologicznym. Wykazano bowiem
ścisłą zależność między stężeniem wielu leków we krwi a ich działaniem leczniczym i toksycznym;
znacznie natomiast mniejszy stopień zależności między dawką leku a wielkością reakcji
farmakologicznej.
www.farmacja.e-lama.pl
22

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
2. Leki, których stosowanie powinno być monitorowane ich stężeniami w organizmie.
- glikozydy nasercowe (digoksyna, digitoksyna)
- leki przeciwarytmiczne (amiodaron, dizopyramid, flekainid, lidokaina, prokainamid, propafenon,
propranolol)
- leki przeciwpadaczkowe (fenytoina, fenobarbital, karbamazepina, prymidon, etosuksimid, kwas
walproinowy)
- TLPD (amitryptylina, dezipramina, imipramina, nortryptylina)
- preparaty litu
- aminoglikozydy (gentamycyna, tobramycyna, netylmycyna, amikacyna, dibekacyna, streptomycyna,
kanamycyna)
- teofilina
- metotreksat
- cyklosporyna, takrolimus
3. Kryteria wyboru leków do monitorowania.
- mały współczynnik terapeutyczny
- niebezpieczne działania toksyczne i trudny do uchwycenia końcowy efekt kliniczny
- znaczna współzależność między stężeniem a działaniem
- długotrwała terapia
- zastosowanie w stanach zagrożenia życia
- znaczne różnice osobnicze w farmakokinetyce
- farmakokinetyka nieliniowa, gdy niewielkie zmiany dawki leku mogą powodować
nieproporcjonalnie duże zmiany jego stężenia i niebezpieczne objawy toksyczne
- duży współczynnik dystrybucji
4. Wskazania kliniczne do podjęcia terapii monitorowanej.
- brak spodziewanego wyniku leczniczego lub wystąpienie nieoczekiwanych objawów toksycznych
mimo zaplanowanego schematu dawkowania;
- brak możliwości odpowiedniej klinicznej lub laboratoryjnej kontroli skuteczności i siły
farmakologicznego działania leku, zwłaszcza wtedy, gdy lek jest stosowany długotrwale lub
zapobiegawczo;
- choroby, w których objawy związane z nieskutecznie leczoną chorobą są takie same jak toksyczne
efekty działania leku;
- różnice międzyosobnicze farmakokinetyki zależne przede wszytskim od wieku i genotypu;
- choroby wątroby lub nerek, choroby przewodu pokarmowego, zaburzenia ilości białek, gospodarki
wodno-elektrolitowej, kwasowo- zasadowej
- jednoczesne stosowanie innych leków, zwłaszcza gdy istnieje możliwość wystąpienia miedzy nimi
interakcji
- ochrona przed toksycznym działaniem niektórych leków celowo stosowanych w dużych dawkach dla
uzyskania lepszego wyniku leczniczego
- ocena wartości terapeutycznej nowych leków
5. Czynniki, które należy uwzględnić w interpretacji stężenia leku.
- stan kliniczny chorego
- czynniki zmieniające kinetykę (choroby współistniejące nerek, wątroby, przew. pok., tarczycy)
- zaburzenia wiązania leków z białkami
- reaktywność receptorów
- jednoczesne stosowanie innych leków (i interakcje)
- zmienność osobniczą uwarunkowaną genotypem, płcią, wiekiem
- niewłaściwe dawkowanie
- zła biodostępność zastosowanego preparatu
www.farmacja.e-lama.pl
23

Farmakoterapia – opracowanie pytań z podręcznika (rozdział 3)
- czynniki środowiska
- zła współpraca ze strony chorego
- zakłócenia analityczne
6. W jakim czasie należy pobierać próbki krwi w celu oznaczania stężenia leku?
- przy wielokrotnym dawkowaniu pomiar stężenia prowadzić po osiągnięciu stanu stacjonarnego, gdy
ustali się równowaga pomiędzy stężeniem leku w surowicy i w miejscu działania leku na receptor
- w przypadku leków o długim okresie półtrwania, próbkę pobiera się do oznaczenia, przed
osiągnieciem stanu równowagi
- próbki krwi pobierać w czasie, gdy lek osiąga stężenie maksymalne i/lub w czasie, gdy osiąga
stężenie minimalne, tzn tuż przed podaniem następnej dawki i po zakończeniem procesu dystrybucji
7.
8. Wskazania do oznaczania wolnej frakcji leku.
- stany chorobowe wątroby i nerek z hipoalbuminemią
- stosowanie kilku leków silnie wiążących się z białkami krwi (np. salicylany z fenytoiną)
- nieliniowość procesu wiązania z białkami niektórych leków (salicylany, prednizolon, fenylbutazon,
teofilina, dizopyramid). Wzrost stężenia całkowitego tych leków w surowicy prowadzi, z powodu
wysycenia miejsc wiążących, do nieproporcjonalnego zwiększenia ich frakcji wolnej.
- stany patologiczne (jak zawał serca), w których następuje zwiększenie alfa1-kwaśnej glikoproteiny, a
więc silniejsze wiązanie leków zasadowych (spadek stężenia ich wolnej frakcji).
www.farmacja.e-lama.pl
24
Wyszukiwarka
Podobne podstrony:
fizjo zwierząt krwionośny nerwowy pytania od marioli
interakcje żywności z lekami
Interakcje międzypopulacyjne
DYKTAFON Prawo miedzynarodowe 3 zjazd od Joasi, prawo międzynarodowe(3)
DYKTAFON Prawo miedzynarodowe 4 zjazd od Joasi, prawo międzynarodowe(3)
T7 Interakcje międzygatunkowe, pwr, W7 wydział inżynierii środowiska, Pwr OŚ Ochrona Środowiska, Sem
38. Koewolucja rozwijanie interakcji międzygatunkowych , biologia, licencjat eksperyment
MODELOWANIE DYNAMIKI INTERAKCJI MIĘDZYLUDZKICH
SEPLENIENIE MIĘDZYZĘBOWE, Logopedia, od mbeata231
Interakcje pomiędzy lekami a dodatkami do diety
DYKTAFON Prawo miedzynarodowe 2 zjazd od Joasi, prawo międzynarodowe(3)
Interakcje międzygatunkowe między populacjami
więcej podobnych podstron