
str. 1
GENETYKA
Wykład I 2.11.07r.
Gen – jednostka dziedziczenia, która w sensie klasycznym zajmuje swoistą pozycję (locus)w genomie lub w
chromosomie; jednostka, która wpływa w jeden lub więcej sposobów na fenotyp organizmu, która może
mutować do różnych form allelicznych i ulegać rekombinacji z innymi jednostkami tego typu. Wyróżnia
się obecnie 3 klasy genów:
1) strukturalne, przepisywane na mRNA, które ulegają translacji na sekwencję aminokwasową łańcuchów
polipeptydowych
2) przepisywane na rRNA
3) na tRNA
Fenotyp – obserwowane, strukturalne lub funkcjonalne cechy organizmu (morfologia, fizjologia, zachowanie),
będące rezultatem złożonej sieci powiązań między różnymi genami oraz pomiędzy genami a
środowiskiem.
Genotyp – całość informacji genetycznej organizmu, w odróżnieniu od całości cech fizycznych (fenotyp).
Cechy mendlowskie – monogenowe, np. polidaktylia (dodatkowy palec); wieloczynnikowe, poddawane
działaniu środowiska np. kolor włosów.
Fenokopia – cecha warunkowana środowiskowo a sprawiająca wrażenie dziedzicznej.
Wnioski z eksperymentów (pięciornik – Potentilla glandulosa, szczepy szczurów) :
dany genotyp wykształca różne fenotypy w różnych środowiskach,
różnice fenotypowe pomiędzy dwoma genotypami zmieniają się wraz ze środowiskiem. Genotyp, który
jest najlepiej przystosowany (fitness) w jednym środowisku, nie musi być najlepszy w innym.
!
Genotyp nie określa jednoznacznie fenotypu. Raczej genotyp determinuje zakres fenotypów jakie może
wykształcić. Jest to NORMA REAKCJI genotypu.
Determinizm genetyczny – „jesteśmy naszymi genami”; cecha dziedziczna jest niezmienna a jej pojawienie jest
nieuchronne; idea, która może być niebezpieczna;
Np., mutacje w genie BRCA1 powodują ok. 5% wszystkich przypadków nowotworu piersi.
Prawdopodobieństwo wystąpienia choroby u kobiet:
- z Żydowskich rodzin wschodnioeuropejskich – 86%
- z innych grup etnicznych – 45%
Typ dziki – normalny, osobnik zdrowy, występujący licznie w danym środowisku;
Ryzyko tła – typ dziki bez wpływu szkodliwego środowiska, występuje 1.0 możliwości ryzyka zachorowania;
Wariant – ze szkodliwą mutacją;
Gregor Mendel:
- Badania odmian grochu od 1856r.
- publikacja wyników w 1866r.
Rok 1900 De Vries – Holandia
Correns – Niemcy
Tschermak – Austria
Podstawowe prawa dziedziczności, bo stosują się do wszystkich organizmów diploidalnych:
I.
segregacji (czystości gamet)
II.
niezależnego dziedziczenia
Linia czysta – linia homozygotyczna, w której obrębie nie występuje rozszczepienie cech.
Uczelnia bli
żej Ciebie

str. 2
Homozygota – 2 allele identyczne
Heterozygota – 2 allele inne
P
1
:
RRYY × rryy
F
1
:
RrYy
F
1
:
RrYy
× RrYy
1/16 homozygota recesywna
F
2
:
9:3:3:1
MEJOZA:
1. wczesna profaza I – nici chromatyny stają się coraz grubsze, tworzą się chromosomy
2. późna profaza I – pojawia się wrzeciono podziałowe, widoczne chromosomy, fragmentacja błony,
kondensacja chromosomów
3. metafaza I – chromosomy homologiczne układają się równikowo (równoleżnikowo)
4. anafaza I – migracja chromosomów
5. telofaza I – częściowo odbudowuje się błona, podział komórek
II MEJOZA = MITOZA
1. profaza II – wrzeciono podziałowe, fragmentacja błony jądrowej
2. metafaza II – chromosomy układają się równoleżnikowo
3. anafaza II – migracja do biegunów
4. telofaza II – odbudowanie błony komórkowej, zanik wrzeciona
↓
4 komórki potomne nieidentyczne
Mejoza:
I – podział redukcyjny komórki diploidalnej → powstają 2 komórki haploidalne
II – podział mitotyczny → powstają 4 komórki haploidalne (w sumie)
Mitoza
- 1 podział
- 2 komórki potomne w cyklu podziałowym
- komórki potomne genetycznie identyczne
- liczba chromosomów jest diploidalna (2n)
- w komórkach somatycznych
- przez cały okres życia
- rozmnażanie bezpłciowe, do wzrostu i naprawy
(regeneracji)
Mejoza
- 2 podziały
- 4 komórki potomne
- komórki potomne genetycznie różne
- n
- w komórkach linii zarodkowej
- u ludzi po osiągnięciu dojrzałości płciowej
- rozmnażanie płciowe
Cykl komórkowy:
1. Interfaza:
G
1
– odbudowywane struktury komórkowe; może przejść w fazę G
0
S – replikacja
G
2
– komórka przygotowuje się do podziału
Niezależna segregacja
Z 2 alleli do gamet trafia tylko jeden, dzieje się to losowo, więc nie wiadomo który
I Prawo Mendla – segregacja genów (czystości gamet)
II Prawo Mendla – niezależnego dziedziczenia
- różne cechy dzielą się niezależnie gdy znajdują się na różnych chromosomach
Oddziaływania alleliczne
Dominacja – efekt funkcjonalny bądź fizjologiczny
Rośliny wysokie – ekspresja genu umożliwiającego produkcję gibereliny (hormonu wpływającego na
wydłużanie łodygi)
Nasiona gładkie – ekspresja genu kodującego białko łączące cukry proste w rozgałęzione polisacharydy
(skrobia), efekt to wiązanie wody

str. 3
Dominacja niezupełna
fenotyp heterozygoty jest pośredni (brak dominacji)
np. w rodzinnej hipercholesterolemii (FH), brak receptorów na komórkach wątroby, które wiążą
cholesterol z krwi
jednak w chorobie Tay-Sachsa (degeneracja systemu nerwowego) dominacja niezupełna wyraża się
tylko na poziomie molekularnym; połowa normalnej ilości enzymu wystarczy by być zdrowym
Kodominacja
dziedziczenie grup krwi
determinanty antygenowe: A – N-acetylogalaktozoamina
B – D-glukoza
produkty genów ABO to glikozylotransferazy
aa
AA
fenotyp
fenotyp
Aa
Aa
Aa
Dominacja niekompletna
d.kompletna
naddominacja
- np. anemia sierpowata
- odnosi się do wart. Przystosowawczej
- zamiast glutaminy jest valina
-
heterozygota
fenotypowo
jest
(korzystniejsza) faworyzowana
Wykład II 9.10.07r.
KODOMINACJA – obydwa allele są równocenne, heterozygota ma fenotyp różny od fenotypów aa, AA, ale nie
pośredni, np. grupa krwi AB
Czynniki zmieniające fenotypowe cechy mendlowskie:
Zjawisko
- allele letalne
- allele wielokrotne
- dominacja niekompletna
- kodominacja
- epistaza
- penetracja
- ekspresywność
- plejotropia
- fenokopia
- różnorodność genetyczna
Efekt fenotypowy
klasa fenotypowa ginie
wiele wariantów fenotypu
fenotyp heterozygoty pośredni
fenotyp heterozygoty różny, ale nie pośredni
gen maskuje efekt innego
genotyp nie zawsze kształtuje typowy fenotyp
różna intensywność fenotypu
wiele symptomów fenotypu
symptomy środowiskowe przypominają genetyczne
różne genotypy a ten sam fenotyp
Przykład
poronienie
ABO
FH (cholesterolemia)
ABO
Bombay
polidaktylia
polidaktylia
fenyloketonuria
infekcje
upośledzenie słuchu
Geny letalne – warunkują nieprawidłowości rozwojowe i zakłócenia procesów fizjologicznych; śmiertelność
ponad 90%
Semiletalne – śmiertelność 50-90%
Subwitalne – śmiertelność 10-50%
Y/+
×
Y/+
Y/Y
Y/+
Y/+
+/+
Ginie
żółta
żółta
szara
25%
25%
25%
25%
Allele wielokrotne
Np. Allele warunkujące grupy krwi
allele
n
Rodzaje genotypów
Rodzaje homozygot
n
Rodzaje heterozygot

str. 4
Dawca
Biorca
A
B
AB
0
A
I
A
I
A
(AA)
I
A
i (A0)
-
agl.
-
agl.
B
I
B
I
B
(BB)
I
B
i (B0)
agl.
-
-
agl.
AB
I
A
I
B
(AB)
agl.
agl.
-
-
0
i
0
i
0
(00)
-
-
-
-
Układ grupowy Rh
Antygeny Rh są kodowane przez geny:
RHD antygen D
RHCE antygen C/c E/e (zlokalizowane na jednym polipeptydzie)
Osoby RhD
+
stanowią 85% populacji kaukaskiej (białej)
RHD
RHCE
Osoby RhD
-
stanowią 15% populacji kaukaskiej
-
RHCE
U Rh
-
Japończyków i Afrykańczyków gen RHD obecny, ale nieaktywny (zmieniony).
Choroba hemolityczna noworodków:
- niezgodność Rh
Jeżeli matka jest Rh(-) a płód Rh(+), komórki płodu mogą stymulować syntezę matczynych przeciwciał
anty-Rh (D); częstość niezgodności – 10% ciąż, ale tylko 2-5% potomstwa ma anemię hemolityczną.
- niezgodność ABO
matka A
ojciec B, AB
B
A, AB
O
A, B, AB
Przeciwciała anty-A i anty-B → immunoglobuliny M (IgM), nie przekraczają bariery łożyska
Przeciwciała anty-D → IgG łatwo przechodzą przez łożysko
Epistaza – fenotyp Bombay
Jeden gen wpływa na ekspresję innego
antygen H (część cukru)
Allel H → enzym transferaza +
Glikoproteina (na powierzchni komórki) = glikoproteina H
antygen H
allel h → enzym nieaktywny
glikoproteina
0 nie zmienione
glikoproteina H A Antygen A
genotyp hh → grupa 0
B Antygen B
Penetracja i ekspresywność
Np. polidaktylia
penetracja – ekspresja genotypu w kategorii „wszystko albo nic” (1 dodatkowy palec)
ekspresywność – zmienna intensywność cechy fenotypowej (dodatkowy palec może być na więcej niż 1
kończynie)
Penetracja i ekspresywność dominującego genu lobe u D. melanogaster
Penetracja wynosi 75%
Plejotropia
defekt któregoś genu powoduje zablokowanie powstania produktu
jeden gen wywiera szereg efektów fenotypowych
str. 5
Gen A
Gen B
Gen C
Enzym A
Enzym B
Enzym C
Substrat
Produkt A
Produkt B
Produkt C
Plejotropia rzekoma – złożony fenotyp nie wynika z podstaw genetycznych, jest to cecha pozorna np. szurpata
kura.
Efekty plejotropowe:
- alkaptonuria – ciemnienie moczu, paznokci, skóry
- fenyloketonuria
białka pokarmowe
hydroksylaza fenyloalaniny
tyrozynaza
fenyloalanina
tyrozyna
melaniny
jodo- i dwujodotyrozyna
oksydaza
parahydroksyfenylo-
hipotetyczny enzym
kw. fenylopirogronowy
pirogronianowa
sprzęgający jod
tyroksyna
kw. homogentyzynowy
trójjodotyronina
oksydaza alkaptonu
kw. homogentyzynowego
CO
2
, H
2
O
fenyloketonuria
albinizm
fenyloalanina
tyrozyna
melaniny
tyrozynaza=tyrozynemia
hipotyreoza=kretynizm
kw. homogentyzynowy
tyroksyna, trójjodotyronina
alkaptonuria
CO
2
, H
2
O
Formy współdziałania genów nieallelicznych:
1) epistaza
2) komplementarność
3) oddziaływania modyfikatorowe (+/-)
4) oddziaływania poligonowe
Test komplementacji (czy cechy recesywne są alleliczne)
P
a
1
a
1
×
a
2
a
2 ;
aaBB
×
AAbb
F
1
a
1
a
2
AaBb
Fenotyp
mutant
typ dziki → wtedy cecha recesywna nie jest alleliczna

str. 6
Wniosek
1 locus
2 loci
Odstępstwa od klasycznego stosunku rozszczepień w F
2
(9:3:3:1)
Przewidywane stosunki fenotypowe w F
2
(3:1)
n
n - l. genów
(3:1)
2
= (3:1)(3:1) = 9:3:3:1
P w pełni heterozygotyczni: liczba rodzajów gamet – 2
n
liczba klas genotypowych – 3
n
liczba klas fenotypowych – 2
n
AB
Ab
aB
ab
AB
AABB
1
AABb
2
AaBB
3
AaBb
4
Ab
AABb
5
AAbb
6
AaBb
7
Aabb
8
aB
AaBB
9
AaBb
10
aaBB
11
aaBb
12
ab
AaBb
13
Aabb
14
aaBb
15
Aabb
16
9 : 3 : 4
Gen pary A, jeżeli jest homozygotycznie recesywny, jest epistatyczny do genu pary B.
Przykład – umaszczenie sierści myszy
A: kolor dominuje nad albinos
B: agouti dominuje nad czarnym
9/16 agouti A_B_ (1,2,3,4,5,7,9,10,13)
3/16 czarny A_bb (6,8,14)
4/16 albinos aa__ (11,12,15,16)
Interakcja: homozygotyczny albinos jest epistatyczny do agouti i czarnego.
9 : 7
Którykolwiek z genów, homozygotycznie recesywny jest epistatyczny do pozostałego genu.
Przykład – kolor kwiatów słodkiego groszku
A: fiolet dominuje nad białym
B: kolor dominuje nad brakiem koloru (biel)
9/16 fiolet A_B_ (1,2,3,4,5,7,9,10,13)
7/16 biały aa__ (6,8,11,12,14)
__bb (15,16)
Interakcja: efektem homozygoty recesywnej w genie A bądź B jest kolor biały.
12 : 3 : 1
Dominujący gen A epistatyczny do genu B.
Przykład – kolor owocu dyni
A: biel dominuje nad kolorem
B: żółty dominuje nad zielonym
12/16 biały A___ (1-10,13,14)
3/16 żółty aaB_ (11,12,15)
1/16 zielony aabb (16)
Interakcja: dominujący biały maskuje efekt żółci lub zieleni.
15 : 1
Którykolwiek gen dominujący, epistatyczny do pozostałego genu.
Przykład – kształt torebek nasiennych tasznika pospolitego
A: trójkątny dominuje
B: trójkątny dominuje
15/16 trójkątny (1-15)

str. 7
1/16 owal aabb (16)
Interakcja: allel dominujący w parze A lub B maskuje efekt owalu.
13 : 3
Dominujący gen A epistatyczny do genu B, który jeżeli homozygotycznie recesywny, epistatyczny do genu A.
Przykład – kolor piór u drobiu
A: inhibicja dominuje nad kolorem
B: kolor dominuje nad bielą
13/16 biel A___ (1-10, 13,14,16)
__bb
3/16 kolor aaB_ (11,12,15)
Interakcja: dominujący inhibitor maskuje kolor nawet gdyby mógł on się ujawnić, gen koloru gdy
homozygotycznie recesywny, uniemożliwia ujawnienie się koloru nawet gdyby brak inhibitora.
9 : 6 : 1
Współdziałanie między dwoma dominantami w celu wytworzenia nowego fenotypu.
Przykład – kształt owocu dyni
A: kształt kulisty dominuje nad podłużnym
B: --------//---------
9/16 dysk A_B_ (1-5, 7,9,10,13)
6/16 kula A_bb (6,8,11,12,14,15)
aaB_
1/16 podłużny aabb (16)
Interakcja: allele dominujące genów A i B razem tworzą kształt dyskoidalny.
Gen lub genotyp
epistatyczny
Gen hipostatyczny
Nazwa epistazy
Rozszczepienie w F
2
aa
A
A
bb
aa
bb
A
B
B oraz b
B oraz b
B oraz b
A oraz a
B oraz b
A oraz a
B oraz b
A oraz a
recesywna
dominacyjna
dominacyjno –
recesywna
podwójnie
recesywna
podwójnie
dominacyjna
9:3:4
12:3:1
13:3
9:7
15:1
Kompleksowy przykład współdziałania genów: umaszczenie ssaków
warstwa komórek przezroczystych
rdzeń, komórki brylaste
kora, komórki wrzecionowate
Melaniny: 1) eumelaniny – brązowe lub czarne, pochodne DOPA-chromu
2) feomelaniny – żółte lub czerwone, pochodne cysteinylo-DOPA-chinonu
Melaniny są wytwarzane pod kontrolą genetyczną w melanocytach.
Geny specyficznie aktywowane w melanocytach:
a) locus E (extension) – koduje białko błonowe melanocytów będące receptorem MSH (hormon
melanotropowy; produkt przysadki)
b) locus C (albino) – koduje enzym tyrozynazę, katalizujący wytwarzanie melanin; ekspresja pod kontrolą
MSH; wysoki poziom tyrozynazy → eumelaniny; niski → feomelaniny. Mutacje: np. u królików seria
alleli wielokrotnych c (albinizm) – enzym nieaktywny, brak wytwarzania pigmentu; c
h
(himalajski) –
str. 8
stanowi o zmianie w glikozylacji, co wywołuje efekt termowrażliwości, końce ciała są ciemniejsze; c
ch
(szynszyl) – stanowi o uwrażliwieniu tyrozynazy na inaktywację proteolityczną co osłabia katalizę;
c) locus B (brown) – ma strukturę podobną do locus C i koduje białko o cechach tyrozynazy; allel
zmutowany (mutacja zmiany sensu: Cys→Tyr) powoduje wytwarzanie brązowej eumelaniny;
d) locus D (dilute) – koduje strukturalne białko dendrytów melanocytu, allel zmutowany determinuje
słabszy rozwój dendrytów, taki melanocyt zaopatruje komórki włosa mniejszą ilość cząsteczek
pigmentu, co daje rozjaśnienie barwy
Gen specyficznie aktywowany w komórkach mieszka włosowego w sąsiedztwie melanocytów:
a) locus A (agouti) – koduje białko wydzielane na zewnątrz, będące antagonistą MSH co do receptora
MSH błony melanocytów. Allel A
+
podlega przerywanej ekspresji, co owocuje zmianami poziomu
tyrozynazy w melanocytach, które wytwarzają zatem na przemian eu- i feomelaniny, co daje strefowość
barwy pojedynczego włosa
Inne geny:
a) locus P (plebald) – gen środkowego ubarwienia i locus Vitiligo – gen nabytego bielactwa kontrolują
liczbę i rozmieszczenie melanocytów w skórze właściwej
geny kontrolujące wędrówkę melanocytów
geny kodujące inne niż tyrozynaza enzymy katalizujące syntezę pigmentów itd.
locus
allele
fenotyp
A
A
+
A
w
a
A
Y
a
t
agouti
agouti z białym brzuchem
non-agouti (jednolite)
żółty, zarodek zamiera
czarny podpalony
B
B
b
agouti z A
+
, czarny z aa, cynamonowy z A
+
bb
brązowy z aabb
C
C
c
c
h
barwny
bezbarwny (cc=albinos)
himalajski
D
D (D1)
d (D2)
pełna ekspresja barwy
barwa „rozcieńczona”
S
S
s
nie plamisty
plamisty
Wykład III 16.10.07r.
Eksperymenty Johannsena z fasolą
Cechy monogenowe są wyrażone fenotypowo bardzo wyraźnie, cech takich jest niewiele.
Dziedziczenie poligenowe
Np. dziedziczenie barwy ziaren pszenicy
białe
– aabbcc
jasno-czerwone
– Aabbcc
czerwone
– AABBcc
ciemno – czerwone
– AABBCC
białe × jasno czerwone
białe × czerwone
białe × ciemno czerwone
F
1
: pośrednie
jasno czerwone
pośrednie
F
2
: białe, pośrednie, j. czerwone
1 : 2 : 1
5 klas
1 : 4 : 6 : 4 : 1
7 klas
1 : 6 : 15 : 20 : 15 : 6 : 1
Zmienność barwy oka – model dwugenowy tłumaczy istnienie pięciu kolorów oka u ludzi;
Zmienność koloru skóry – model trójgenowy.
Transgresja (+/-)

str. 9
Przekraczanie zakresu zmienności form rodzicielskich przez potomstwo.
Np. (+) Aabbcc × aabbCc
F
1
: AabbCc
- wtedy ciemniejszy niż rodzice
(-) Aabbcc × aabbCc
F
1
: aabbcc
- wtedy jaśniejszy niż rodzice
Wariancja ogólna (cechy)
V
PH
= V
E
+ V
GE
+ V
GA
+ V
GD
+ V
GI
V
PH
= V
E
+ V
G
V
E
– wariancja środowiskowa (składnik niegenetyczny)
V
GE
– interakcja genotyp - środowisko
V
GA
– genetyczny składnik addytywny
V
GD
– genetyczny składnik dominujący
V
GI
– interakcja między genami (epistaza)
Odziedziczalność ogólna
Odziedziczalność w wąskim sensie
(addytywny komponent wariancji genetycznej, V
GA
= V
A
przechodzącej bez zmian na potomstwo)
Mierzenie odziedziczalności (kontrolowana hodowla)
Linie wsobne homozygotyczne P
1
× P
2
= F
1
jednorodne genetycznie V
PH
= V
E
W F
2
pełny zakres zmienności genetycznej
Przykład:
Przeciętna wariancja wewnątrz każdej z dwóch odmian rodzicielskich i potomstwa F
1
jest np. 8,76
V
E
= 8,76
W F
2
całkowita wariancja fenotypowa V
PH
= 40,96
ponieważ V
PH
= V
G
+ V
E
V
G
= V
PH
– V
E
= 40,96 – 8,76 = 32,20
Odziedziczalność tej cechy
Metoda bliźniąt
V
I
– wariancja fenotypowa u bliźniąt jednojajowych
V
F
– wariancja fenotypowa u bliźniąt dwujajowych
V
I
= V
E
bo bliźnięta identyczne
V
F
= 1/2V
G
+ V
E
V
F
– V
I
= 1/2V
G
+ V
E
– V
E
= 1/2V
G
Odziedziczalność między proporcją wariancji fenotypowej pomiędzy osobnikami, która zależy od wariancji
genetycznej.
str. 10
Współczynnik korelacji – proporcje genów wspólnych dla dwóch osobników w określonym stopniu
spokrewnionych.
Odpowiedź na selekcję jako funkcja odziedziczalności.
Determinacja płci – dziedziczenie cech sprzężonych z płcią
Poziom chromosomowy
autosomy i chromosomy płci
Henking 1891; ciałko X
płeć homo – i hetero gametyczna
♀
♂
komórka jajowa plemniki
XX
X0
X
X + -
Np. wiele Ortoptera i Hemiptera
XX
XY
X
X + Y
ZW ZZ
Z + W
Z
Motyle, niektóre ryby, ptaki, gady
X
Y
X
Y
Bryophyta
Homogametyczna – 1 rodzaj gamet
Heterogametyczna – 2 rodzaje gamet
Poziom genowy
Asparagus officinalis – dwupienna, rośliny męskie i żeńskie w równych ilościach, sygnał jednopienności
- w kwiatach słupkowych zawiązki pręcików
- w kwiatach pręcikowych zawiązki słupków (wyjątek nasiona)
Serie 198 wyjątkowych nasion dała 155 męskich i 43 żeńskich (3 : 1)
Hipoteza: para alleli, męski dominujący
P:
Aa × Aa
Wyjątkowe nasiona ¼ AA ; 2/4 Aa ; ¼ aa
3 męskie : 1 żeńskie
Falsyfikacja hipotezy: krzyżówka – rośliny męskie z wyjątkowych nasion × żeńskie
W F
1
otrzymano 2/3 męskich i 1/3 żeńskich
Interpretacja zgodna z hipotezą
1/3 AA × aa →1∕3 Aa (męskie)
2∕3 Aa × aa → 1∕3 Aa (męskie) i 1∕3 aa(żeńskie)
Bonellia viridis (Echiuroidae)
~90% larw niezdeterminowanych płciowo
Płeć męska na skutek: - zetknięcia się larwy z ryjkiem samicy
- obecności w wodzie homogenatu ryjków samic
- dodatku jonów K i Cu
- ubytku jonów Mg i SO
4
W innych przypadkach: płeć żeńska
Wniosek: płeć męska pod wpływem nieswoistego zahamowania ekspresji genów determinujących płeć żeńską.
Habrobracon (Hymenoptera)
Seria alleli wielokrotnych: xa, xb, xc, xd, …
np. xa, xaxa- samiec
xaxd, xbxd – samica
Wniosek: dopełniające się działanie różnych alleli szeregu x → płeć żeńska
Gynandroidy
Samica xaxb; kom. jajowa: xa
xb
Wyjątkowo dwujądrowa kom. jajowa: xaxb po I podziale → xa
xb
xa – męski, xb – męski, xaxb – żeński
D. melanogaster – płeć jako kontinuum
X/A
przykłady
fenotyp ogólny
str. 11
>1
1
>0,5<1
0,5
<0,5
XXXA
XXXXAAA
XXAA
XXXAAAA
XXAAA
XYAA
X0AA
XYAAA
XYAAAA
metasamica
samica
interseks
samiec
samiec sterylny
metasamiec
U ssaków płeć determinowana jest przez obecność chromosomu Y!
Nondysjunkcja u ludzi i myszy: heterozygoty X0/2A z jajnikami żeńskimi
XXY/2A z jądrami męskimi
Dziedziczenie(Sxr) sex reversed u myszy, jeśli dołączy się do X, to odwraca płeć, staje się osobnikiem męskim,
ale jest bezpłodny.
Chromosom Y, ma rejony odpowiedzialne za:
- SRY – płeć,
- tworzenie emalii zębów
krótkie ramię
- rozwój kości
- spermatogenezę
długie ramię
a) Na większości chromosomu Y nie zachodzi rekombinacja, jednak posiada dwa rejony PAR1 i PAR2, które
rekombinują z X zwane są rejonami pseudoautosomalnymi. PAR1 na krótkim ramieniu, PAR2 na długim (na
ich zakończeniach).
b) Granica głównego regionu pseudoautosomalnego przebiega przez gen kodujący proteinę układu grupowego
krwi XG (na chromosomie X)
c) Gen SRY koduje białko, tzw. „czynnik transkrypcyjny”, który reguluje ekspresję innych genów.
d) Chromosom Y jest wysoce repetytywny.
e) 5% chromosomu Y stanowią regiony pseudoautosomalne
95% to region specyficzny dla ♂ tzw. MSY (koduje 27 białek), z czego
→ 10-15% stanowią sekwencje w 99% identyczne z odpowiednikami na X
→ 20% sekwencje zdegenerowane o pewnym podobieństwie do X
→ pozostałe sekwencje stanowią liczne PALINDROMY, np. „Madam, I’m Adam”
f) hemizygota – osobnik męski XY, bo ma geny zlokalizowane na jednym chromosomie (X)
Ewolucja chromosomów płci ssaków rozpoczęła się najprawdopodobniej od pary autosomów.
Wykład IV 30.10.07r.
Człowiek – poziomy płciowości
hormony
induktory w kom. niezróżnicowane
gonady
przewody rozrodcze
embrionalnych gonady
i zew. genitalia
XX
część korowa
jajniki
żeńskie
XY
część rdzeniowa
jądra
męskie
płeć genetyczna
płeć gonadowa
płeć genitalna
II-rzędowe cechy
wychowanie
str. 12
Płciowe
w rodzinie
żeńskie
kobieta
męskie
mężczyzna
płeć somatyczna
płeć socjo-psychologiczna
Do 5 tygodnia ciąży nie wiemy jaka jest płeć (rozwijają się jednocześnie przewody Wolfa♂ i Millera♀). Jeżeli
obecny jest Y, to zanika przewód Millera w 6 tygodniu i rozwija się płeć ♂, jeżeli nie ma Y, to zanika przewód
Wolfa i rozwija się płeć ♀.
SRY (aktywny)
jądra
hormon antymillerowski
niszczy przewody Millera,które mogłyby się rozwinąć w ♀
komórki śródmiąższowe
produkują testosteron, gdy
produkcja zostaje zablokowana,
testosteron
DHT
to jest osobnikiem pseudoherma-
dihydrotestosteron
frodytycznym (II-rzędowe cechy
wrodzona hyperplazja
płciowe mogą być żeńskie)
błona
nadnerczy (nie produkuje
plazmatyczna
DHT)
(przechodzi)
receptory
na chromosomie X są
receptory, z którymi
łączy się testosteron
zew. struktury męskie
Syndrom niewrażliwości na androgeny (uszkodzenie genu kodującego receptory), struktury wewnętrzne męskie
nie są wytwarzane (pęcherzyki, przewody, itd.)
Spermatogeneza
spermatangium (2n) → mitoza → pierwotny spermatocyt (2n) → mejoza I → wtórne spermatocyty (n) (dwa) →
mejozaII → 4 spermatydy (n) → plemniki (n)
Oogeneza
oogonium (2n) → mitoza → pierwotny oocyt (2n) → mejoza I →1) ciałko biegunowe (może zaniknąć) →
2) wtórny oocyt (n), większa komórka →
mejoza II → a) komórka jajowa (n) → dojrzałe jajo (n)
b) ciałko biegunowe II rzędu (n) ginie
Zapłodnione jajo daje początek komórkom macierzystym, a z nich rozwijają się inne komórki, np. szlak kom.
skóry, kom. nerwowe, łojowe; drugi szlak tworzy tkanki łączne (krew, tk. łączną, tk. kostną).
Kompensacja dawki
- Np. kotka szyldkretowa
- inaktywację chromosomu X kontroluje gen XIST (jednego w organizmie kobiety)
- transkrypcja dużej ilości RNA, który pokrywa chromosom, modyfikując DNA i związane z nim białka
- 15% genów pozostaje aktywnych (rejony PAR); (pseudoautosomalny rejon)
- 75% genów jest permanentnie wyłączonych
- 10% genów ulega ekspresji w niektórych inaktywowanych X
Ciałko Baara – wyłączony chromosom
Piętno genomowe (imprinting)
1% genów ludzkich; zjawisko epigenetyczne (zew.) – wiązanie grup metylowych
str. 13
Zaburzenia piętnowania → ok. 30 dolegliwości
Syndrom Prader – Willie’go:
wyłącznie ekspresja regionu chromosomu 15, matczynego
małe dłonie i stopy, nie osiąga dojrzałości płciowej
Syndrom Angelmana:
ekspresja genu(-ów) ojcowskiego na chromosomie 15; brak genu matczynego
opóźnienie umysłowe, słaba koordynacja mięśni, konwulsje
Przyczyna niekompletnej penetracji?
W pewnym momencie następuje usunięcie piętna i w gametach pojawia się od nowa – zachowanie w
kolejnych pokoleniach.
sprzężone z płcią
zależne od płci
geny związane z płcią
ograniczone przez płeć
niekompletnie sprzężone z płcią
geny na heterochromosomie Y – holandryczne
geny zależne od płci
np. człowiek
Wniosek: dominacja w parze alleli zależy od płci.
Geny ograniczone przez płeć
np. kury domowe
♀
♂
HH
Hh
hh
upierzenie kurze
upierzenie kurze
upierzenie kurze
upierzenie kurze
upierzenie kurze
upierzenie kogucie
Usunięcie gonad → upierzenie kogucie po najbliższym pierzeniu
Gen H → upierzenie kurze w obecności hormonów ♀ lub ♂
upierzenie kogucie przy braku tych hormonów
gen h → upierzenie kogucie przy braku hormonu ♀
upierzenie kurze przy obecności hormonu ♀
Wniosek: ekspresja fenotypowa zdeterminowana obecnością lub brakiem jednego z hormonów płciowych.
Geny sprzężone z płcią
- gen na chromosomie X
- 3 typy komórek siatkówki (stożkowych) zawierają różne fotopigmenty, złożone z części będącej pochodną wit.
A i części białkowej – opsyny
Mechanizm powstawania ślepoty na barwy (daltonizm):
Na chromosomach znajdują się geny czerwonej i zielonej opsyny, niesymetryczne ułożenie
chromosomów podczas crossing-over prowadzi do tego, iż na jednym z chromosomów występują dwa geny
czerwonej i jeden zielonej opsyny, a na drugim chromosomie tylko gen zielonej opsyny.
deuterotopia – ślepota na barwę zieloną (daltonizm); (średnie długości fal)
protanopia – ślepota na barwę czerwoną (długie fale)
♀
♂
BB
Bb
bb
łysa
nie łysa
nie łysa
łysy
łysy
nie łysy

str. 14
Cechy mitochondrialnego DNA:
brak crossing-over
brak naprawy DNA
dziedziczenie matczyne (tylko kom. jajowa ma wystarczającą ilość cytoplazmy)
wiele kopii w mitochondrium i komórce
narażenie na działanie wolnych rodników
37 genów
brak histonów i intronów
tempo mutacji wielokrotnie większe niż w DNA jądrowym
heteroplazmia – zmienna ekspresywność
Upośledzenia DNA mitochondrialnego prowadzą przede wszystkim do różnych miopatii, upośledzenia widzenia
i słyszenia.
Zdarza się, że nie wszystkie kopie DNA mitochondrialnego znajdującego się w jednej komórce są identyczne
(heteroplazmia – np. gdy mamy dużo DNA w wyniku którego powstaje jakieś białko to jest większa
ekspresywność).
Egipska dynastia Ptolemeuszy – kojarzenie w pokrewieństwie.
Małżeństwo kuzynów a ryzyko ekspresji alleli recesywnych
A) dziedziczenie autosomalne dominujące
cecha przejawia się w każdym pokoleniu
dotyczy obojga płci i przenoszona przez obie płcie
np. przodozgryz żuchwowy
B) dziedziczenie autosomalne recesywne
osoby dotknięte dolegliwością mają zwykle zdrowych rodziców
rodzice osób chorych są zwykle nosicielami
np. albinizm, alkaptonuria, fenyloketonuria
C) dziedziczenie cechy recesywnej sprzężonej z płcią (X)
dotyczy głównie mężczyzn
mężczyźni dotknięci dolegliwością mają zwykle zdrowych rodziców
brak transmisji ♂ → ♂
np. hemofilia, daltonizm, dystrofia mięśni Duchenne’a
D) dziedziczenie cechy dominującej sprzężonej z płcią (X)
dotyczy obu płci, ale raczej kobiet niż mężczyzn
wszystkie córki mężczyzny dotkniętego dolegliwością także cierpią na tę dolegliwość ale synowie nie
np. krzywica hipofosfatemiczna
E) dziedziczenie cechy sprzężonej z chromosomem Y
dotyczy tylko mężczyzn
przekazywana przez mężczyznę wszystkim synom
np. owłosienie uszu
Wykład V 6.11.07r.
Jak się utrwalają nowe mutacje mitochondrialne?
Zapłodniony oocyt – 100000 cząsteczek DNA
Efekt szyjki butelki – po zapłodnieniu – z wielkiej liczby cząsteczek DNA zostaje niewiele (losowo są gubione).
Komórki rozwijającego się embrionu – 100 – 500 kopii.
Segregacja genotypów mitochondrialnych (haplotypów) w matczynej linii zarodkowej
Komplementacja: rodzice a wadą słuchu (cecha autosomalna recesywna) mają normalnie słyszące potomstwo.
Sprzężenie genów
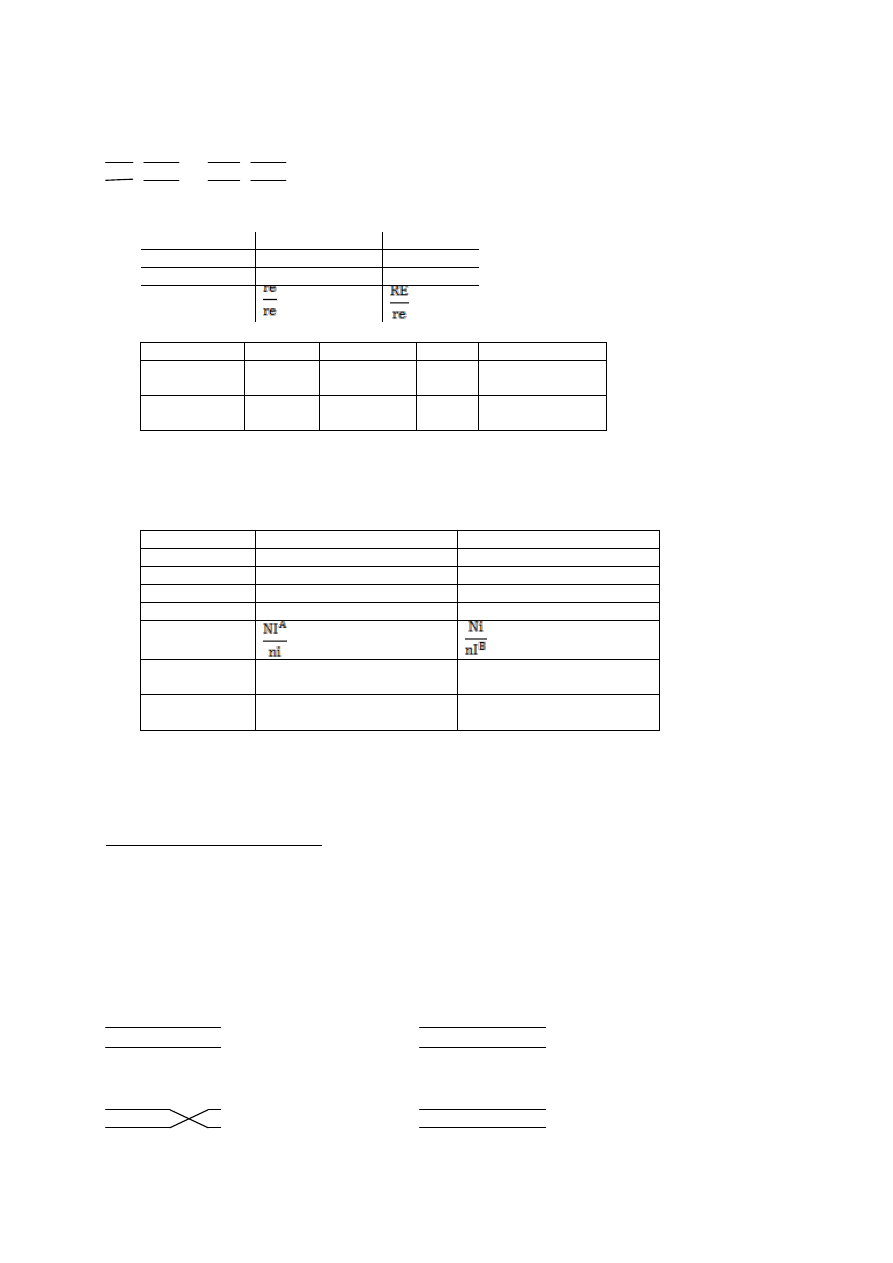
str. 15
Oczekiwane rezultaty krzyżówki dihybrydowej (heterozygot) 9 : 3 : 3 : 1 czy 3 : 1?
Crossing-over wpływa na sprzężenia.
Sprzężenie absolutne – nigdy nie zachodzi między genami zjawisko crossing-over (tylko gdy geny są bardzo
blisko siebie).
Konfiguracja alleli jest istotna..
● P L ● P l
● p l ● p L
Cis
trans
rodzice 1
rodzice 2
fenotyp
Rh
-
, brak anemii
Rh
+
, anemia
genotyp
rree
RrEe
konfiguracja
alleli
gamety
plemniki
częstotliwość oocyty
rodzicielskie
re
48%
48%
RE
re
Rh
+
, anemia
Rh
-
, brak anemii
rekombinanty
re
2%
2%
Re
rE
Rh
+
, brak anemii
Rh
-
, anemia
Jakie jest prawdopodobieństwo, że dziecko rodziców cierpiących na syndrom braku paznokci i bolesnego
artretyzmu będzie zdrowe i z grupą krwi 0? (geny zlokalizowane na chromosomie 9 w odległości 10
jednostek mapowych); N – syndrom +
P (dziadkowie)
♀ syndrom +; A / ♂ zdrowy; 0 ♀ syndrom +; 0 / ♂ zdrowy; B
NI
A
/ ni cis
Ni / nI
B
trans
ojciec
matka
fenotyp
syndrom +; A
syndrom +; B
genotyp
NnI
A
_
NnI
B
_
konfiguracja
alleli
rodzicielskie
NI
A
45% plemniki
ni 45% plemniki
Ni oocyty
nI
B
oocyty
rekombinanty
Ni 5%
nI
A
5%
NI
B
ni
Dziecko zdrowe / grupa 0 jedynie gdy plemnik ni (częstość 45%) a oocyt ni (częstość 5%).
Prawdopodobieństwo tego zdarzenia → 2,25%
0,45 × 0,05 = 0,0225 → 2,25%
Cechy mejotycznego crossing-over:
1) rekombinacja wzajemna
2) zachodzi między 2 spośród 4 chromatyd
3) złożony c – o, może dotyczyć 2 – 4 chromatyd, jeśli podwójny, to:
regresywny – te same dwie chromatydy
progresywny – jedna z chromatyd dwa razy, dwie inne po razie
dygresywny – dwie różne pary chromatyd
Zjawisko crossing-over w odniesieniu do trzech genów
A
B
C
brak c-o
A
B
C
a
b
c
a
b
c
A
B
c
pojedynczy c-o
A
B
c

str. 16
a
b
C
a
b
C
A
b
c
pojedynczy c-o
A
b
c
a
B
C
a
B
C
A
b
C
podwójny c-o
A
b
C
w wyniku c-o gen środkowy
zmienił swoje miejsce
a
B
c
a
B
c
Krzyżówka trzypunktowa jako metoda mapowania chromosomów.
Jednostka mapowa (map unit) – 1% rekombinantów
Jednostka Morgana – 100% rekombinantów
1 m.u. = 1 cM
♀
Y W M
×
y w m
♂
y w m
Y
rodzicielskie
Y W M
6972
y w m
pojedyncze c-o
Y W m
3454
y w M
pojedyncze c-o
y W M
60
Y w m
Podwójne c-o
y W m
9
Y w M
_______________
10495
w i m
→ 33% = 33 cM
y i w
→ 0,7% = 0,7 cM
y i m
→ 33,5% = 33,5 cM
Częstość rekombinacji może wyrażać szacunek prawdopodobieństwa rekombinacji między niezbyt oddalonymi
genami.
Na tej podstawie można określić, czy przypadki rekombinacji wewnątrz chromosomu są wzajemnie niezależne.
Interferencja
I = 1 – K
K - współczynnik koincydencji
m. n. > 45 → I = 0 (zero)
E = 0,330 × 0,007 = 0,00231
O = 9/10495 = 0,00086
I = 1 – 0,00086/0,00231 = 1 – 0,374 = 0,626
Interferencja ujemna (negatywna)
y/o
pan18
trp
+
pan
+
(pan18 + pan22) -
niezmutowany
y/o
+
pan22
trp
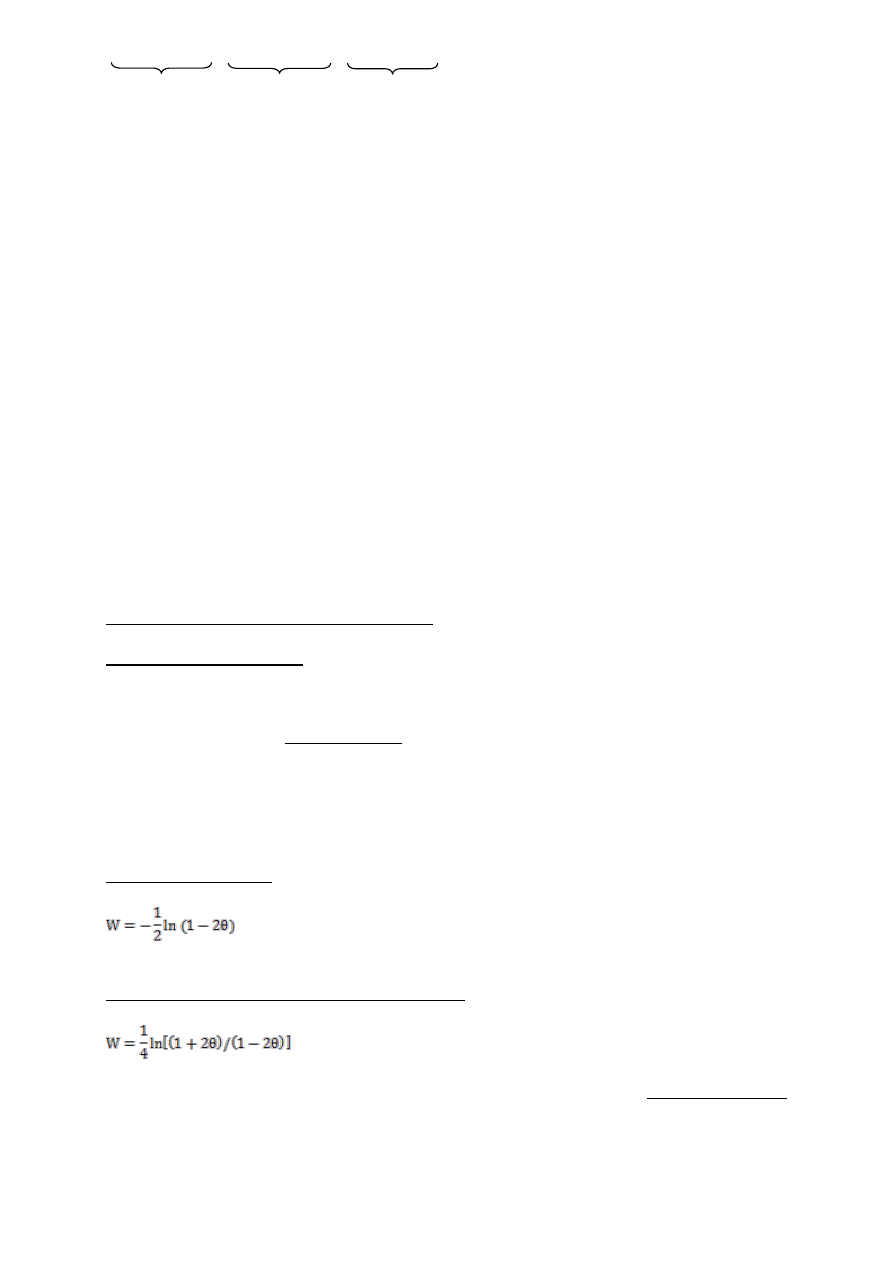
str. 17
region I
II
III
A
potrójne c-o w regionie I, II, III → y/o
+
pan18 pan22 trp
+
y/o pan
+
trp
B
podwójne c-o w regionie I i II → y/o
+
pan18 pan22 trp
y/o pan
+
trp
+
C
podwójne c-o w regionie II i III → y/o
+
pan
+
trp
y/o pan18 pan22 trp
+
Oczekiwane
Obserwowane
A
0,3%
71%
B
3,3%
10%
C
7,4%
13%
U Neurospora pozycja askospor w worku umożliwia precyzyjną analizę segregacji i rekombinacji genów.
Preredukcja – segregacja alleli w I podziale mejotycznym.
Postredukcja – w II podziale mejotycznym (w przypadku gdy c-o zachodzi między centromerem a pierwszym
genem).
Worki normalne i abberacyjne ( zawartość alleli dominujących i recesywnych nie są sobie równe, np. 6
dominujących i 2 recesywne) jako rezultat krzyżówki szczepu dzikiego i zmutowanego.
Ogólny model rekombinacji Holliday’a
Tworzenie heterodupleksów DNA.
2 sposoby rozwiązania połączonej struktury: z rekombinacją markerów oskrzydlających lub bez.
W wyniku cięcia wschód – zachód, pojedynczy c-o sprawia wrażenie podwójnego w wyniku dezintegracji
heterodupleksów (środkowy segment wymieniony).
Symetryczne fragmenty heterodupleksów DNA wewnątrz heterozygotycznego rejonu są warunkiem wstępnym
KONWERSJI GENÓW.
Interferencja ujemna jest rezultatem konwersji genów.
Mapowanie genetyczne u ludzi
Cel: określenie, jak często dwa loci są rozdzielane przez mejotyczną rekombinację.
LOCI SYNTENICZNE – zlokalizowane na tym samym chromosomie.
Proporcje rekombinantów w potomstwie stanowi frakcję rekombinacji –θ (theta)
Frakcja rekombinacji określa dystans genetyczny, który nie jest tożsamy z dystansem fizycznym.
Frakcja rekombinacji nie przekracza wartości 0,5 , jakkolwiek duży byłby dystans fizyczny.
Zatem, dla szeregu loci, wartości te nie sumują się wprost, wzdłuż mapy genetycznej.
Jeżeli seria loci A, B, C, D, ……. jest rozmieszczona w odstępach 5 cM na mapie, locus M może być w
odległości 60 cM od locus A, ale frakcja rekombinacji pomiędzy A i M nie będzie 60%.
Funkcje mapowe (Haldone’a i Kosambi’ego) określają relacje między frakcją rekombinacji a dystansem
genetycznym.
Frakcja mapowa Haldone’a
W – odległość mapowa; θ – frakcja rekombinacji
Funkcja mapowa z poprawką na interferencję Kosamb’iego
Wykład VI 13.11.07r.

str. 18
Poziomy precyzji map genetycznych:
- cytogenetyczna – 5000 kb (5 000 000 zasad)
- fizyczna – 10 kb
- genetyczna (sprzężeniowa) – 100 kb
- sekwencyjna – ostateczna mapa genetyczna
Relacje między dystansem genetycznym a fizycznym w genomie nie jest stała.
U człowieka, w mejozie u osobnika ♂ zachodzi przeciętnie 49 crossing-over na komórkę. Ponieważ każdy c-o
daje 50% rekombinantów, całkowita długość męskiej mapy genetycznej wyniosłaby 2450 cM. Aktualna
wersja Location Database podaje 2851 cM.
Chiazmy są częstsze w mejozie żeńskiej, a całkowita długość żeńskiej mapy genetycznej wg. Location Database
wynosi 4296 cM (z wyłączeniem chromosomu X).
Zatem, w ponad 3000 Mb genomie autosomalnym, 1 męski cM ~ 1,05 Mb, natomiast 1 żeński cM ~ 0,70 Mb
Średnio dla obu płci 1 cM = 0,88 Mb
Największa dysproporcja występuje w regionie pseudoautosomalnym, na końcu krótkich ramion chromosomów
X i Y.
U mężczyzn zachodzi obligatoryjny crossing-over w tym 2,6 Mb regionie, tak że jego długość oblicza się na 50
cM. Czyli dla tego regionu, u mężczyzn 1 Mb = 19 cM, natomiast u kobiet 1 Mb = 2,7 cM.
Główny region pseudoautosomalny charakteryzuje wysoka częstość rekombinacji i istotna różnica częstości
między płciami.
Wzajemne relacje mapy fizycznej i genetycznej chromosomu 19 człowieka, oparte na 180 markerach
zmapowanych obu technikami. Zwraca uwagę nierówne natężenie rekombinacji wzdłuż chromosomu
(wzrasta ku telomerom) i u płci (mapa ♀ jest o ok. 10% dłuższa niż ♂).
U kobiet częściej zachodzi rekombinacja w pobliżu centromeru niż u mężczyzn.
W rodzinie segregują allele A
1
i A
2
z locus A oraz B
1
i B
2
z locus B. Osoby w pokoleniu III, które otrzymały od
swojego ojca allele A
1
B
1
lub A
2
B
2
powstały z niezrekombinowanej spermy, osoby które otrzymały od
ojca A
1
B
2
lub A
2
B
1
są rekombinantami. Rodowód nie pozwala na zaklasyfikowanie żadnej z osób z
pokolenie I i II jako rekombinanta lub nie rekombinanta, ani na zidentyfikowanie rekombinantów
wynikających z oogenezy u II
2
.
A) ♂A
1
A
1
× ♀A
2
A
2
B) ♂ A
1
A
2
× A
1
A
2
♀
C) ♂ A
1
A
2
× A
1
A
2
♀
D) ♂ A
1
A
2
× A
3
A
4
♀
A
1
A
2
A
1
A
2
A
1
A
1
A
1
A
4
Mejozy informujące i nieinformujące o sprzężeniu badanego genu z daną kopią genu markerowego. Założenie:
ojciec ma cechę dominującą, którą dziedziczy wraz z markerową kopią allela A
1
:
A – mejoza nieinformatywna: markerowy allel u homozygotycznego ojca jest nierozpoznawalny
B – mejoza nieinformatywna: dziecko mogło odziedziczyć A
1
od ojca a A
2
od matki, bądź odwrotnie
C, D – mejozy informatywne: dziecko odziedziczyło A
1
od ojca.
Obliczanie wartości lod
Zakładając, że loci są sprzężone, a frakcja rekombinacji wynosi θ, prawdopodobieństwo mejozy
nierekombinacyjnej wynosi (1 – θ), a rekombinacyjnej θ.
Jeżeli loci są niesprzężone, prawdopodobieństwo mejozy rekombinacyjnej czy nierekombinacyjnej
wynosi ½.
Współczynnik lod (logarytm szans faworyzujących sprzężenie)
Z = log
10
(Pr/Pi)
Pr – określone na podstawie analizy rodowodów prawdopodobieństwo, że dany gen wykazuje określoną wartość
rekombinacji (r) z genem markerowym
Pi – prawdopodobieństwo rekombinacji przy założeniu, że oba elementy segregują niezależnie.
Wartość +3 jest uważana za dowód sprzężenia.
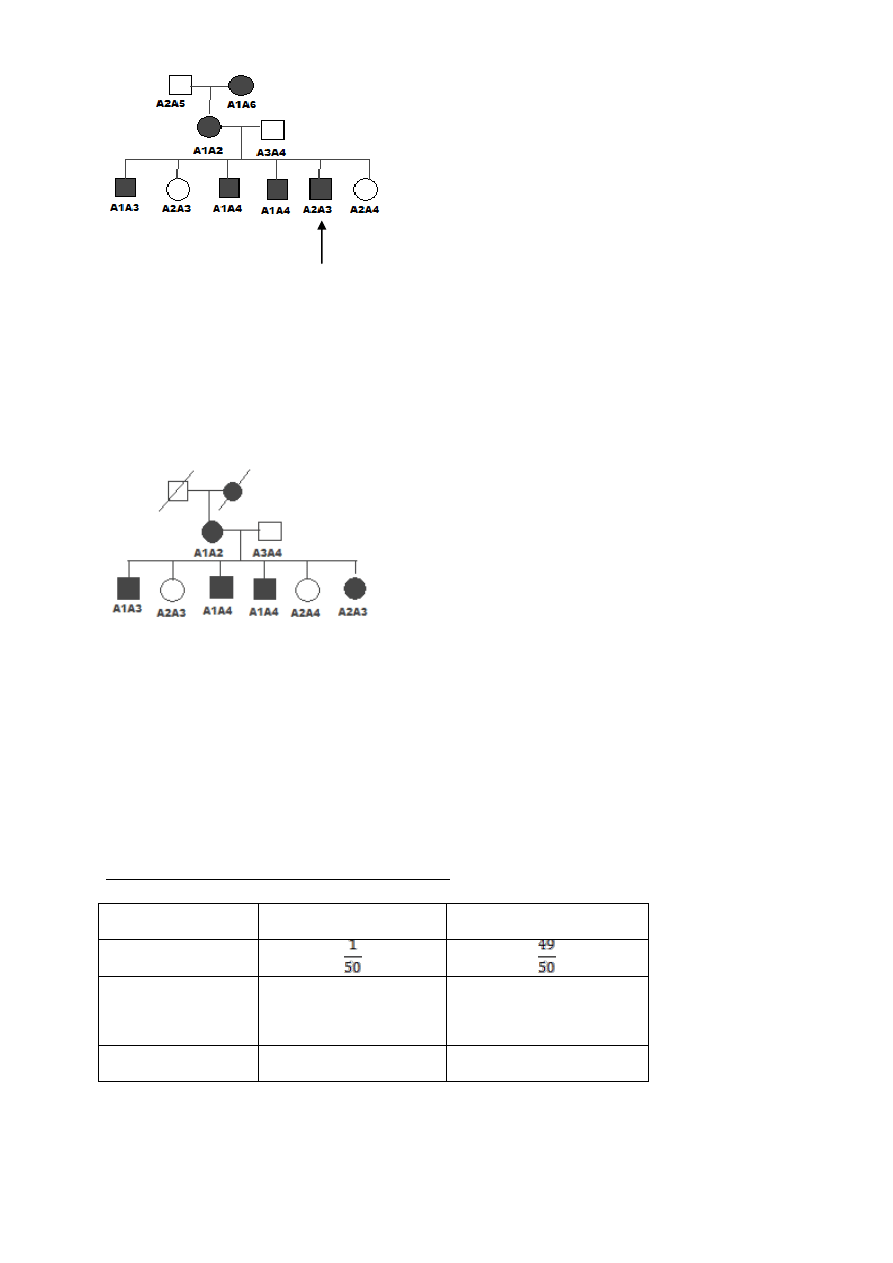
str. 19
rekombinant
Rodzina A
Jest 5 nierekombinantów i 1 rekombinant. Ogólne prawdopodobieństwo, zakładając sprzężenie wynosi
(1 – θ)
5
× θ. Prawdopodobieństwo zakładające brak sprzężenia wynosi (1/2)
6
. Stosunek prawdopodobieństw
(1 – θ)
5
× θ / (1/2)
6
.
Wartość lod, Z jest logarytmem stosunku prawdopodobieństw
θ
0
0,1
0,2
0,3
0,4
0,5
Z
-∞
0,577
0,623
0,509
0,299
0(dziedziczenie niezależne)
sprzężenie
absolutne
Rodzina B
Jeden układ alleli nieznany.
Jeżeli odziedziczyła A
1
wywołujący chorobę, jest 5 nierekombinantów i 1 rekombinant.
Jeżeli odziedziczyła A
2
wywołujący chorobę, jest 5 rekombinantów i 1 nierekombinant.
Ogólne prawdopodobieństwo jest:
½ [ (1 – θ )
5
× θ / ( ½ )
6
] + ½ [ (1 – θ) × θ
5
/ ( ½ )
6
]
To pozwala uwzględnić każdy możliwy układ alleli z jednakowym prawdopodobieństwem
θ
0
0,1
0,2
0,3
0,4
0,5
Z
-∞
0,276
0,323
0,222
0,076
0
Najbardziej prawdopodobna frakcja rekombinacji jest ta, której wartość lod jest najwyższa.
Obliczanie progu sprzężenia(+3; -2) wg formuły Bayes’a
Prawdopodobieństwo sprzężenia dwóch loci (a priori) szacuje się na 1/50.
Hipoteza
loci są sprzężone
(frakcja rekombinacji = θ)
loci są niesprzężone
(frakcja rekombinacji = 0,5)
Prawdopodobieństwo a
priori
Prawdopodobieństwo
warunkowe: 1000 : 1
szans sprzężenia lod
Z(θ) = 3,0
1000
1
Łączne
prawdopododobieństwo
20
~ 1

str. 20
Ponieważ jest niskie a priori, prawdopodobieństwo sprzężenia dwóch przypadkowo wybranych loci, wymagany
jest dowód dający 1000 : 1 szans na korzyść sprzężenia, by ostatecznie dać 20 : 1 szans na korzyść
sprzężenia.
To odpowiada konwencjonalnemu progowi istotności statystycznej p = 0,05.
Wartości – 2 = brak sprzężenia
Rozwój ludzkich markerów genetycznych
Typ markera
Liczba loci
Grupy krwi (1910 – 1960)
~ 20
Warianty elektroforetyczne
ruchliwości protein
surowicy (1960 – 1975)
~ 30
Typy HLA (1970 - )
1
(haplotyp)
DNA RFLPs (1975 - )
>10
5
(potencjalnie)
Minisatelity (1985 - )
>10
4
(potencjalnie)
Mikrosatelity (1989 - )
>10
5
(potencjalnie)
DNA SNPs (1998 - )
>10
6
(potencjalnie)
RFLP – polimorfizm długości fragmentów restrykcyjnych
Mikrosatelitarny DNA – powtórzenie sekwencji 2,3,4 nukleotydów (np. (TTA)
8
); jest markerem genetycznym
Mapowanie fizyczne
1) Niskiej rozdzielczości
Najmniejsza jednostka mapowa jaka może być wyróżniona wynosi 1 – kilka megazasad DNA
panele hybryd komórek somatycznych
hybrydy monochromosomalne generowane przez fuzję mikrokomórek
mapowanie subchromosomalne
mapowanie STS (sequence tegget site) w panelach komórek hybrydowych zawierających określone
fragmenty ludzkiego genomu. CMGT (chromosome – mediated gene transfer; hybrydy radiacyjne)
2) Wysokiej rozdzielczości
Setki kb do pojedynczego nukleotydu
chromosome walking
Wykorzystanie chromosomów politenicznych z gruczołów śliniankowych D. melanogaster
powstają na skutek ENDOMITOZ – replikacja DNA bez podziału jądra (1024 chromatydy)
pojedyncza nić – chromonema
pętelki na nici – chromonemy
Fragmenty chromosomów politenicznych wskazujące na cytologiczny zasięg pewnych deficjencji (i duplikacji).
Tworzenie linii komórek hybrydowych człowiek – gryzoń.
W odpowiedniej pożywce umieszcza się komórki człowieka i gryzonia, dodaje się wirusa Sendai
(ułatwiają kontakt między dwoma komórkami), w odpowiednich warunkach dochodzi do fuzji, powstaje
dikarion, potem synkarion (jednojądrowa). Potem przenosi się te komórki do medium selektywnego, gdzie
dochodzi do oddzielenia komórek hybrydowych od niehybrydowych.
Połączenie jąder (synkarion) – pozostaje komplet chromosomów gryzonia, chromosomy ludzkie ulegają
zgubieniu, mogą pozostać pojedyncze chromosomy i wtedy można je badać.

str. 21
CMGT – transfer genów za pośrednictwem chromosomu
- komórki myszy pozbawione HPRT (fosforybozylotransferaza hipoksantynowa)
- chromosom chomika HPRT+
- HAT – hipoksantyna, aminopteryna, tymidyna (pożywka selekcyjna) komórek hybrydowych
kinaza tymidynowa TK
Tymidyna
TMP
Hipoksantyna
HPRT
IMP
GMP
Guanina
aminopteryna
inne prekursory
TMP, IMP, GMP – jednofosforany nukleotydów
Mapowanie genów wewnątrz chromosomów z wykorzystaniem anomalii chromosomowych.
Delecje fragmentów chromosomu zawierającego gen kodujący kwaśną fosfatazę 1 czerwonokrwinkową.
Translokacja między ludzkimi chromosomami X i 14:
Ekspresja genów zlokalizowanych na X w linii kom. hybrydowych zawierających ludzki 14/X
HPRT; PGK (kinaza fosfoglicerynianowa)
G6PDH (dehydrogenaza glukozo – 6 – fosforanowa)
Względna aktywność enzymu w kom. może dostarczyć informacji pozwalającej mapować duplikowane geny.
GOT – transaminaza glutaminianowo – szczawiooctowa
Wykład VII 20.11.07r.
Subchromosomowa lokalizacja genu przez mapowanie w panelu komórek hybrydowych zawiarających
chromosomy translokacyjne i delecyjne.
Wykorzystanie panelu hybryd radiacyjnych w mapowaniu fizycznym.
letalna dawka
fuzja z TK
-
kom.
promieniowania
chomika
normalne ludzkie
kom., w których
komórki hybrydowe (TK
+
)
fibroblasty
chromosomy uległy
fragmentacji
wybierane 100 – 200 hybryd,
każda zawiera 25 – 30%
STS jaka ma być zmapowana
kawałków genomu człowieka
PCR amplifikacja
w każdej hybrydzie
hybrydyzacja ze
znanymi mapowanymi
STSs
porównanie
baza danych
centralnego serwera
lokalizacja
Hybrydyzacja klonów
Seria klonów DNA z nakładającymi się insertami (contig) powstała przez częściowe trawienie genomowego
DNA enzymami restykcyjnymi podczas konstrukcji „biblioteki” DNA.

str. 22
Dwie drogi sekwencjonowania genomu ludzkiego:
1) Pocięcie całego genomu na fragmenty i sekwencjonowanie
2) Kawałek o znanej sekwencji
Identyfikacja funkcji genu i jego lokalizacji.
Profile ekspresji genu i proteomika.
Zastosowanie informacji o genomie ludzkim:
1) w diagnostyce i medycynie zapobiegawczej
2) przemysł farmaceutyczny, rolnictwo
3) badania ewolucyjne
4) genetyka populacji (można śledzić historię gatunku)
5) identyfikacja funkcji genu – działanie terapeutyczne
6) jeśli znamy sekwencję genomu, można powiedzieć o jego organizacji
Chromosomy
Chromatyna:
30% białka histonowe (rdzeń – tworzą 4 białka; nić DNA jest na nią nawinięta = NUKLEOSOM)
30% białka wiążące DNA
30% DNA
10% RNA
Włókno chromatynowe – ściśle upakowane nukleosomy, spiralnie ułożone
Włókno owinięte jest na białkach.
Euchromatyna - barwi się jasno; geny kodujące białka się tu znajdują; struktura luźniejsza
Heterochromatyna – barwi się ciemno; zawiera wiele powtórzeń DNA, pozbawiona aktywności genowej;
struktura skondensowana.
konstytutywna – pozbawiona aktywności genowej bezwzględnie!
fakultatywna – np. ciałko Baara; aktywność genów jest wyłączona.
Niezbędne elementy chromosomu:
CENTROMER, białka z nim związane tworzą KINETOCHOR, strukturą łączącą się z włóknami
wrzeciona kariokinetycznego
ORIGIN REPLIKACJI
TELOMERY – chronią końce chromosomu przed degradacją, z każdym podziałem komórki zostają o
nie skrócone;
Chromosomy:
a) TELOCENTRYCZNE – nie mają krótkich ramion(p); nie ma ich u ludzi
b) AKROCENTRYCZNE – mają bardzo krótkie ramiona p
c) SUBMETACENTRYCZNY – ramię p jest krótsze od q
d) METACENTRYCZNY – ramiona p i q są sobie równe
Nomenklatura:
regiony: p1, p2, p3, etc. (na zewnątrz od centromeru)
regiony: q1, q2, q3, etc.
regiony podzielone na prążki, np. p1 1, p1 2, p1 3, etc.
prążki dzielą się na podprążki, np. p1 1.1, p1 1.2, etc.
podprążki dzielą się na dalsze podprążki, np. p1 1.2 1, p1 1.2 2
centromer = „cen”
telomer = „ter”
fragment proksymalny (najbliższy centromerowi)
fragment dystalny (najdalszy od centromeru, najbliższy telomerowi)
str. 23
Barwienie chromosomów:
prążki G – wynik barwienia barwnikiem Giemsa; barwią się na ciemno; barwią te regiony, które są
bogate w pary A – T
prążki Q – wynik barwienia barwnikiem Quinacrine; barwią te regiony, które są bogate w pary A – T;
barwnik fluorescencyjny
prążki R – barwią odwrotne regiony niż G i Q, czyli bogate w pary G – C
prążki T – barwią rejony telomerowe; są kategorią prążków R (są bogate w G – C)
prążki C – związane z centromerami (barwią konstytutywną heterochromatynę głównie w rejonach
centromerów)
4p11.21
chromosom region
podprążek
ramię prążek podprążek
Kariotyp – zestaw chromosomów komórki, organizmu…
W postaci diagramu skonstruowanego na podstawie wielkości i cech fizycznych chromosomów (wzór
prążkowy chromosomów ludzkich
zdjęcie).
X i Y są w różnej grupie chromosomów (wg wielkości i wzoru prążkowego).
Nienormalności chromosomowe:
strukturalne
DUPLIKACJA odcinka chromosomu – powtórzenie
np. „syndrom inv dup (15)” – kolejność jest zamieniona na chromosomie 15; słabe napięcie
mięśniowe, mały rozmiar dziecka, opóźnienie umysłowe, skolioza, itp.
tandemowa
tandemowa inwersyjna
przemieszczona
Duplikacja i delecja genu (chromosom 16)
- osoba z trzema genami
-globiny jest zdrowym nosicielem
- z dwoma kopiami – cierpi na łagodną anemię
- z jedną kopią – dotkliwa anemia;
-thalassemia
-thalassemia – mutacja w genie
-globiny (niedobór ładunków
)
Względny nadmiar łańcuchów
- redukcja czerwonych krwinek
Uwolnione jony Fe niszczą serce, wątrobę, gruczoły dokrewne
Izochromosomy – mają identyczne ramiona
normalnie

str. 24
duplikacja i delecja(bo nie ma w ogóle części, która powinna się znaleźć)
ABC C B A
DELECJA
np. 5p
-
syndrom „cri – du – chat” (płaczącego kota); opóźnienie umysłowe i rozwojowe;
gen telomerazy (odwrotna transkryptaza) – może zostać utracony, co powoduje skrócenie
życia osobnika bo gen odpowiada za podziały komórkowe;
INWERSJA PARACENTRYCZNA – nie obejmuje centromeru, dotyczy ramienia,
dwa pęknięcia, odcinek został wstawiony w to samo miejsce tylko odwrotnie;
DELECJA INTERSTYCJALNA – wypadł środkowy (wew.) odcinek chromosomu,
dwa pęknięcia;
DELECJA TERMINALNA – wypada końcowy fragment chromosomu, 1 pęknięcie;
INWERSJA PERICENTRYCZNA – odcinek uległ odwróceniu w obrębie
centromeru, pęknięcie na dwóch ramionach;
CHROMOSOM PIERŚCIENIOWY – gdy końcowe odcinki zostają zgubione na obu
ramionach i to ulegnie połączeniu (ta pozostała część chromosomu).
Inwersja paracentryczna
Tworzenie pętli w trakcie koniugacji ratuje przed inwersją, jednak może dojść do c-o i wtedy
powstają gamety: 1) normalne
2) dicentryczna (ginie)
3) z inwersją
4) acentryczne (ginie)
Powstają nieprawidłowe gamety.
Inwersja pericentryczna
Podobne konsekwencje – powstaje połowa gamet nieprawidłowych.
TRANSLOKACJA – przeniesienie fragmentu na niehomologiczny chromosom
WZAJEMNA – gdy dwie pary niehomologicznych chromosomów się wymieniają;
można to zobaczyć dzięki metodzie FISH – fluorescence in situ hybridization;
INTRACHROMOSOMALNA – w obrębie jednego chromosomu mają miejsce trzy
pęknięcia, geny zawarte pomiędzy dwoma pęknięciami zostają przeniesione w
miejsce trzeciego pęknięcia;
INTERCHROMOSOMALNA – w wyniku dwóch pęknięć, geny zawarte między
pęknięciami zostają przeniesione na niehomologiczny chromosom.
Wykład VIII 27.11.07r.
Powstawanie translokacji
- powstanie co najmniej dwóch pęknięć
Fragmenty centryczne
z centromerami
Fragmenty acentryczne
bez centromeru
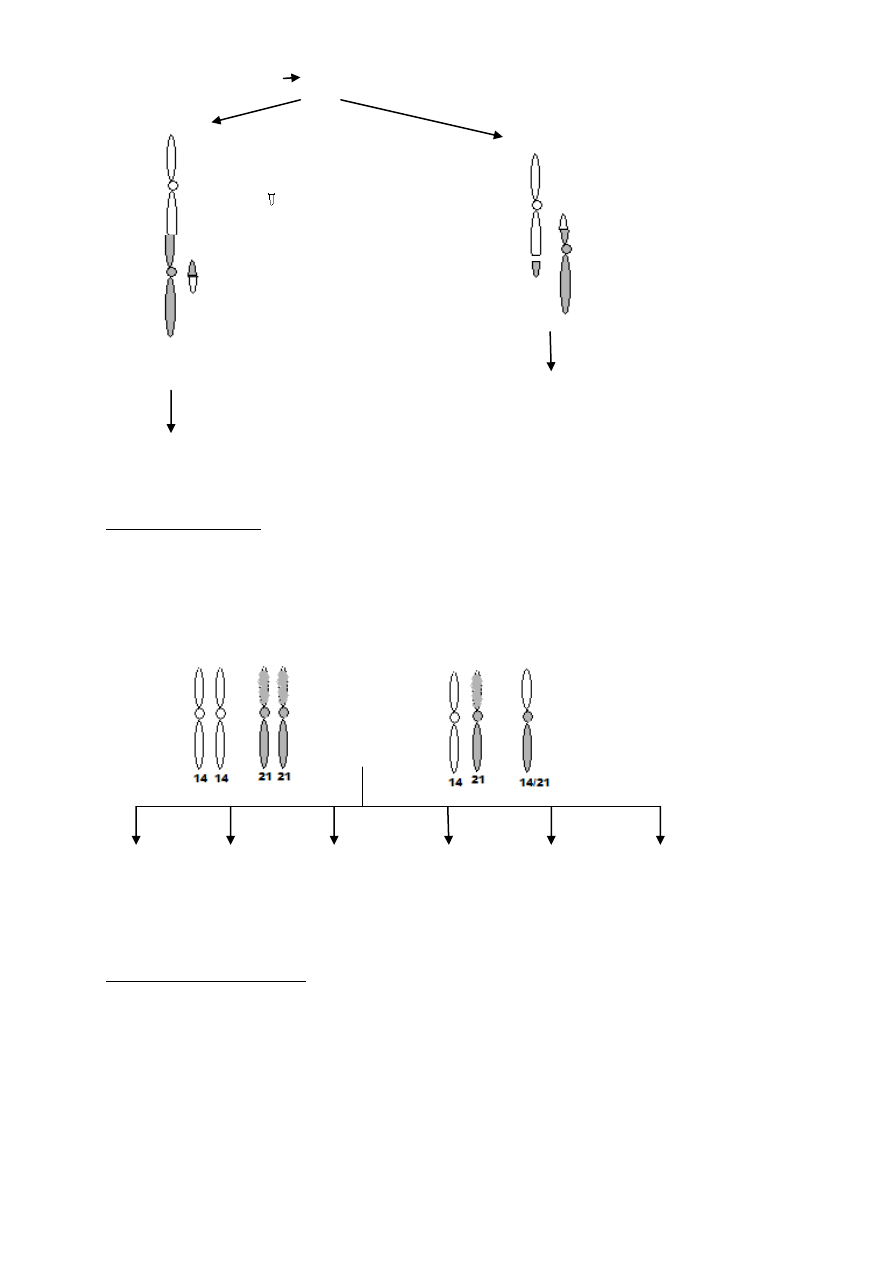
str. 25
p ter
p ter
q ter
p ter
q ter
p ter
chromosom acentryczny (2)
q ter
q ter
chromosom dicentryczny (1)
stabilna translokacja wzajemna
sytuacja niestabilna
(2) ginie (nie wchodzi do mitozy)
(1) rozerwany
Translokacja Robertson’a
- z reguły dotyczy chromosomów akrocentrycznych (1 ramię b. krótkie)
- pęknięcie tuż obok lub w samej strukturze centromeru
- następuje zmniejszenie liczby chromosomów komórki o jeden
- jeżeli nieistotne fragmenty ulegną utraceniu, to nie ma większych konsekwencji dla danego osobnika
- jeżeli osobnik jest nosicielem translokacji to konsekwencje ujawniają się w produkcji gamet
×
14, 21; 14, 21
14, 21; 14/21
14, 21; 21, 14/21
14, 21; 14, 14/21
14, 21; 14
14, 21; 21
kariotyp
normalny
nosiciel
translokacji
nadmiar
chromosomu
21
(translokacyjny
syndrom Downa)
nadmiar
chromosomu 14
(spontaniczne
poronienie)
brak
chromosomu 21
(spontaniczne
poronienie)
brak
chromosomu 14
(spontaniczne
poronienie)
Nienormalności chromosomowe:
1) Strukturalne
delecja terminalna – ubytek końcowego fragmentu, np. 46, XY, del(4) (p16.3)
delecja interstycjalna – ubytek wewnętrzny, np. 46, XX, del(5) (q13 q33)
inwersja – np. 46, XY, inv(11) (p11 p15)
duplikacja – np. 46, XX, dup(1) (q22 q25)
insercja – np. 46, XX, ins(2) (p13 p21 p31)
pierścień (ring) – np. 46, XY, r(7) (p22 q36)

str. 26
dodatkowy niezidentyfikowany chromosom (marker) – np. 47, XX + mar
translokacja wzajemna – np. 46, XX, t(2; 6) (q35; p21.3)
translokacja Robertsona – np. 45, XY, der(14; 21) (q10; q10) ; der – zrównoważony nosiciel; q10 –
oznacza centromer
translokacja syndrom Downa – np. 46, XX, der(14; 21) (q10; q10) + 21
2) liczbowe
triploid – np. 69, XXX; 69,XXY; 69, XYY – zwiększenie całego garnituru chromosomów
trisomik – np. 47, XX + 21
monosomik – np. 45, X
mozaika – np. 47, XXX / 46, XX
Powstawanie poliploidów:
1) TRIPLOIDY:
- 66% - jednoczesne zapłodnienie komórki jajowej przez dwa plemniki
- 24% - zapłodnienie komórki jajowej przez diploidalny plemnik
- 10% - zapłodnienie diploidalnej komórki jajowej przez plemnik
2) TETRAPLOIDY:
- duplikacja DNA bez podziału komórki
endomitoza
3) ANEUPLOIDY (dodatkowy lub brakujący chromosom):
- rezultat nondysjunkcji w pierwszym podziale mejotycznym
- wtórny spermatocyt ma dwa chromosomy, a drugi wtórny nic
- mejoza II – połowa plemników bez danego chromosomu 2 pary, połowa z dodatkowym
chromosomem
- połączenie z euploidalnym oocytem – 50% trisomia, 50% monosomia
- nondysjunkcja w II podziale mejotycznym
- ¼ gamet ma dwa chromosomy homologiczne z danej pary
¼ gamet brak chromosomu
½ gamet normalna
- nondysjunkcja w II podziale powoduje, że kom. zawiera 2 chromatydy jednego chromosomu
- połączenie z euploidalnym oocytem
50% - euploidy
25% - monosomie
25% - trisomie
Konsekwencje liczbowych abberacji chromosomowych
1) Poliploidy
- triploidy (69, XXX; 69, XXY; 69, XYY) – 1-3% poczęć; prawie nigdy nie dochodzi do narodzin, nie
są w stanie przetrwać
2) Aneuploidy
- Autosomy:
Nullisomia – letalne w stanie preimplantacji; brak obu chromosomów z danej pary
Monosomia – letalne w stadium embrionalnym
Trisomia – zwykle letalne w stadium embrionalnym lub płodowym
- trisomia 13 – syndrom Patau
- trisomia 18 – syndrom Edwardsa, mogą przeżyć do urodzenia
- trisomia 21 – syndrom Downa, mogą przeżyć do lat 40 i dłużej
Chromosomy płci:
1) XXX; XXY; XYY – względnie małe problemy, normalna długość życia
2) 45, X – syndrom Turnera – 99% spontaniczne poronienia, przeżywający są bezpłodni, o normalnej
inteligencji (syndrom Klinefeltera)
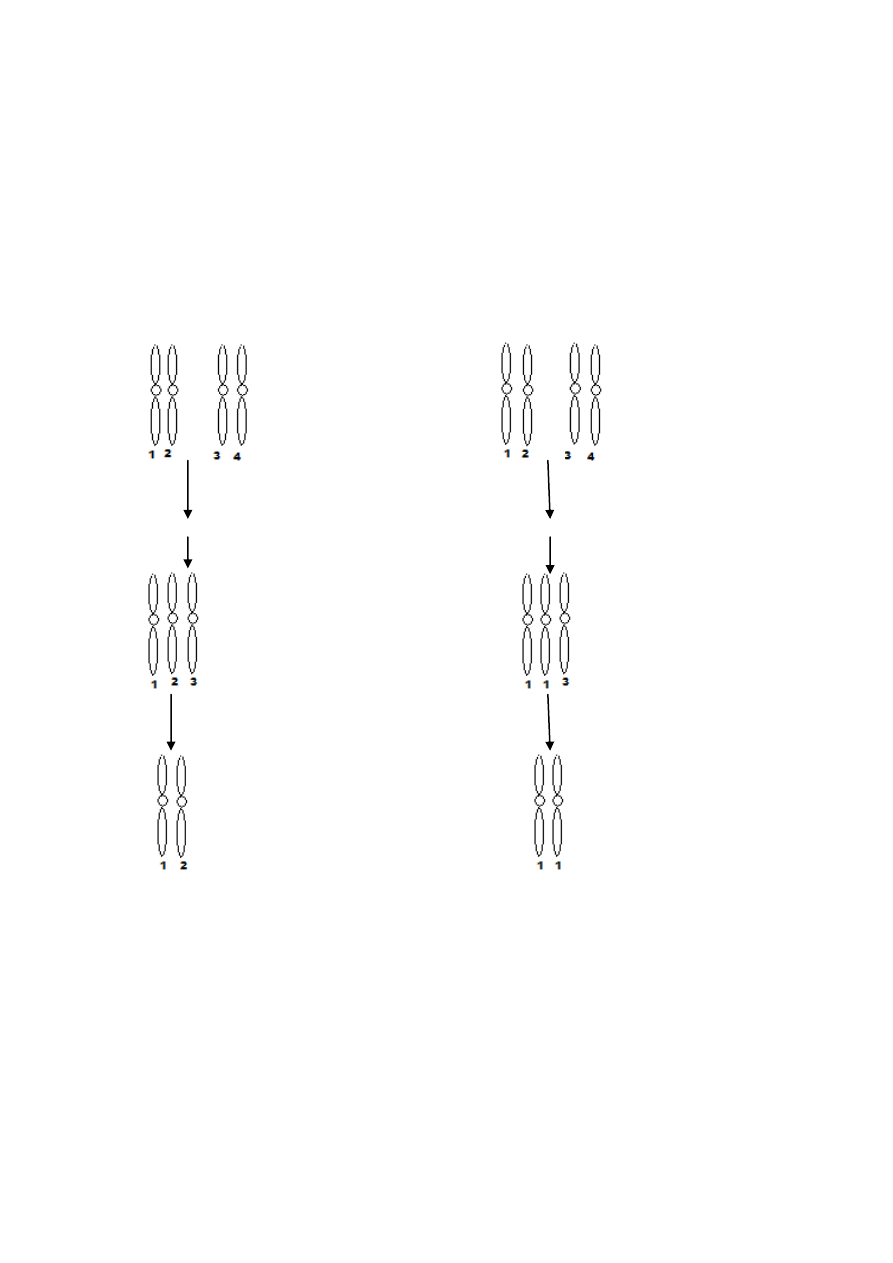
str. 27
Najczęstsze dodatkowe autosomy: 13, 18, 21 (mało genów na nich)
urodzenia
(1 rok)
13 (Patau)
18 (Edwards)
21 (Down)
1/12500 – 21700
1/6000 – 10000
1/800 – 826
5%
5%
85%
Trisomia 18 (syndrom Edwardsa)
- zaburzenia fizyczne i umysłowe
- defekty serca, przemieszczenie wątroby
- opóźnienie wzrostu
Podwójny układ jednego z rodziców – mechanizm powstawania (izo)disomii jednorodzicielskiej
P:
P:
I podział mejotyczny
II podział mejotyczny
nondysjunkcja
nondysjunkcja
zygota trisomiczna
tak naprawdę są to 2
chromatydy jednego
chromosomu
utrata jednego homologa
Disomia jednorodzicielska
izodisomia jednorodzicielska
Symptomy chroby, gdy konsekwencją jest stan homozygotycznie recesywny lub gdy disomia dotyczy genu
piętnowanego.
MUTACJE GENOWE
Mutacje: ● somatyczne – konsekwencje miejscowe, np. w tkankach)
● w komórkach linii rozrodczej (dotyczy całego organizmu)
Mutacje spontaniczne – najczęściej podczas replikacji DNA (złe podstawienie nukleotydu)
Mutacje punktowe:
1) tranzycje - A ↔ G (puryny); C ↔ T (pirymidyny)

str. 28
2) transwersje – A ↔ T lub G ↔ C
Tranzycje zachodzą znacznie częściej – mniejsze konsekwencje dla DNA).
Mutacje odcinkowe – dotyczą większej niż 1 liczby nukleotydów
Typy mutacji:
1) zapis normalny;
2) zmiana sensu – nukleotyd podstawiony innym (zmiana aminokwasów w strukturze białka)
zmiana synonimowa – inny nukleotyd, ale nadal kodowany jest ten sam aminokwas;
zmiana antysynonimowa – podstawiony inny nukleotyd, w wyniku czego kodowane jest inne
białko;
3) nonsens – zmiana nukleotydu, powstaje kodon STOP! (krótsze białko);
4) przesunięcie ramki odczytu – poprzez wstawienie nowego nukleotydu;
5) delecja – usunięcie nukleotydu (najkorzystniej gdy wypadnie cała trójka nukleotydów);
6) insercja – dodanie (najkorzystniej gdy wstawiona zostanie cała trójka)
7) duplikacja – powtórzenie fragmentu (najkorzystniej j/w)
Mutacje poszerzające zasięg (ekspansja powtórzeń trójnukleotydowych) – poszerza się z pokolenia na pokolenie.
pok. 1TEN KOT TAM BYŁ ALE …
pok. 2 TEN KOT TAM BYŁ BYŁ ALE …
pok. 3 TEN KOT TAM BYŁ BYŁ BYŁ ALE …
Mechanizm nieprawidłowości spowodowanych ekspansją powtórzeń trójnukleotydowych:
czynniki transkrypcyjne, oddziałują w sposób zróżnicowany z sekwencjami zawierającymi powtórzenia
i bez powtórzeń;
powtórzenie CTG i CGG nadają podwójnej większą elastyczność;
DNA z wieloma powtórzeniami CTG jest ściślej zwinięty wokół białek histonowych tworząc
nukleosomy w porównaniu z innymi sekwencjami DNA, wpływając na dostępność genów w procesie
transkrypcji;
DNA z wieloma powtórzeniami może składać się tworząc rozmaite struktury w czasie replikacji lub
transkrypcji: pętle; potrójne i poczwórne helisy; struktury umożliwiające „poślizg nici”, zwiększając
szanse mutacji.
Dystrofia miotoniczna – typ dystrofii mięśniowej
Antycypacja genetyczna – nasilenie objawów, z pokolenia na pokolenie pojawiają się one coraz szybciej
(wcześniej).
wiek
fenotyp
liczba powtórzeń CTG
(norma 5 – 37)
późny
dojrzały
łagodne osłabienie mięśni
przedramion
50 – 80
średni
dojrzały
umiarkowane osłabienie
mięśni kończyn
80 – 700
dzieciństwo
silne osłabienie mięśni,
trudności w oddychaniu,
wczesna śmierć
>700
Analiza DNA – elektroforeza (im dłuższy DNA tym wolniej migruje).
Mechanizm:
Niekiedy mutacja udaremnia ekspresję genu na etapie przed translacją
dystrofia miotoniczna typu I – ekspansja trójnukleotydów dotyczy początkowego, nie ulegającego
translacji regionu genu;
dystrofia miotoniczna typu II – (CCTG)
n
gen na chromosomie 3, dotyczy intronu.
Wniosek: cząsteczka mRNA jest zbyt duża by wydostać się z jądra komórki.
typ I: nadmiar materiału w początkowym odcinku

str. 29
typ II: w intronie, który nie jest wycinany
Także: zespół łamliwego X
norma (CGG) × 6 – 50 w genie (FMR1 – fragile X mental retardation – opóźnienie umysłowe)
łagodne symptomy 50 – 200 kopii
nasilone objawy 200 – 2000 kopii
FMR1 – koduje białko, które z nadmiernymi powtórzeniami może reagować z mRNA, które koduje białka
reagujące z neuronami (?)
Choroba Huntingtona:
konsekwencje mutacji zależą od jej pozycji w genie
mutacje miejsc składania (splite sites) – typ zmiany sensu
na granicy intronów i egzonów są sekwencje dające znak do cięcia
mutacje w tym miejscu powodują niewystąpienie cięcia!
Intron+ (mukowiscydoza)
EgzonA
Intron1
EgzonB
Egzon – (BRCA 1) – gen mający wpływ na rozwój raka piersi
EgzonA EgzonB / ← brak egzonu C
Mukowiscydoza (zwłóknienie torbielowate trzustki, cystic fibrosis):
gen autosomalny recesywny
częstość w Europie 1 : 2500
mutacje (ok. 500) w genie CFTR (cystic fibrosis transmembrane conductance regulator)
defekt przewodnictwa przez błonę komórkową (kanały chlorkowe i sodowe)
najczęściej trójnukleotydowe delecje ∆ F508 – białko się rozpada
missense – 40%
H
2
O
przesunięcie ramki odczytu – 30%
Cl
-
nonsens – 20%
gęstniejący śluz
nieprawidłości składania – 10% - białko jest za duże, nie może się wbudować w błonę.
Zaburzenia precyzyjnej struktury kolagenu:
gen
1 kolagenu koduje 2 łańcuchy polipeptydowe
gen
2 kolagenu koduje trzeci polipeptyd
wiele powtórzeń sekwencji gly – pro
mutacje w genach kolagenu są szczególnie niszczące, ponieważ białko ma bardzo precyzyjną
konformację, łatwo naruszaną przez niewielkie zmiany
kolagen – 60% białek kości i chrząstek
50 – 90% suchej masy skóry, wiązadeł, ścięgien, zębiny
prokolagen
kolagen
odcięcie nieregularnych końców
mutacja blokująca skracanie łańcuchów prokolagenu skutkuje rozciągliwą skórą (syndrom Ehlers –
Daulosa typu I)
Wykład IX 4.12.07r.
Jedna z przyczyn choroby Alzheimera (wczesne objawy):
gen na chromosomie 14 (PS 1) koduje białko – presenilinę 1, działające jako receptor zakotwiczony w
błonie aparatu Golgiego;
białko monitoruje wykorzystanie przez komórkę amyloidowego białka beta;
mutacje w genie preseniliny powodują zaburzenia produkcji, struktury i funkcjonowania amyloidu;
zmienione białko tnie prekursorowe białka amyloidu na fragmenty o niewłaściwej długości, które łączą
się i akumulują na zewnątrz komórki.
Cl
-

str. 30
ORGANIZACJA GENOMU LUDZKIEGO:
1) genom jądrowy
zawiera 3300 Mb
wchodzi ok. 80000 genów
ok. 75% genomu to pozagenowy DNA:
w 60% składa się z sekwencji występujących w pojedynczej lub niskiej liczbie kopii
40% to sekwencje umiarkowanie bądź wysoce repetytywne (powtarzalne)
powtórzone tandemowo
powtórzenia rozproszone w genomie
ok. 25% to geny i sekwencje związane z genami (pojedyncze lub umiarkowanie repetytywne):
90% to niekodujący DNA:
pseudogeny
fragmenty genów
introny i sekwencje nie ulegające translacji
10% to kodujący DNA (ok. 2,5% całego DNA)
2) genom mitochondrialny
składa się z 16,6 kb
wchodzi 37 genów:
2 geny rRNA
22 geny tRNA
13 genów kodujących polipeptydy
GENOM MITOCHONDRIALNY:
1) komórki ludzkie zawierają tysiące kopii DNA;
2) jakkolwiek pojedyncza cząsteczka mtDNA zawiera 1/8000 tej ilości DNA jaka jest umieszczona w
przeciętnym chromosomie, to cały mtDNA stanowi 0,5% DNA komórki somatycznej;
3) ludzki genom mitochondrialny jest wysoce efektywny pod względem informacji genetycznej – 93%
sekwencji DNA to sekwencje kodujące;
4) ograniczona autonomia genomu mitochondrialnego:
składniki systemu oksydatywnej fosforylacji – kodowane przez genom mitochondrialny (13
podjednostek) przez genom jądrowy (>80 podjednostek);
składniki aparatu syntezy białek – 24 podjednostki, a przez genom jądrowy ~ 80.
5) geny kodujące białka: ND1, ND2, CO1, CO2, ATPaza 6, CO3, ND3, ND4L, ND4, ND5, CYB;
6) pętla D – zawiera origin replikacji; jedyny fragment niekodujący;
GENOM JĄDROWY:
1) jest rozmieszczony na 24 różnych typach dupleksów DNA (chromosomy), wyróżnionych na podstawie
wielkości, położenia centromeru i wzoru prążkowego;
2) wykazują one znaczną zmienność regionalną pod względem składu zasad i gęstości genów
zawartość GC: 42%
C = G = 0,21 → CpG (0,21)
2
= 0,0441
Obserwowana częstość, to 1/5 tej wartości
→ niedostatek dinukleotydów CpG (5’ – 3’)
5 – metylocytozyna
dezaminacja
CpG → TpG
3) wyspy CpG – mają oczekiwaną częstość CpG (0,0441);
związana jest z nimi aktywność transkrypcyjna;
związane są z 56% genów ludzkich;

str. 31
wysp jest ok. 45000 → 80000 genów; chromosom 4, X, Y mają mało genów, chromosomy 19,
20, 22 jest dużo genów
Telomery:
głównym składnikiem jest DNA złożony z 6 – nukleotydowych powtórzeń TTAGGG (50%
udziału par GC); długość 10 – 15 kb;
subtelomer składa się z 8000 do 300000 zasad; jest zaraz za telomerem;
synteza powtórzeń telomerowych: telomeraza – odwrotna transkryptaza zawierająca RNA
(RNA, to matryca AAUCCC)
telomeraza zapobiega degradacji końców chromosomów z każdym podziałem (u kom.
płciowych, kom. krwi, nowotworowe); odnawiają się powtórzenia telomerowe;
w kom. somatycznych z każdym podziałem tracone są powtórzenia telomerowe, po utracie
telomerów komórki przestają się dzielić.
Heterochromatyna:
zawiera repetytywny DNA
ciemne prążki G charakteryzuje niższa zawartość GC niż prążki jasne
Zagęszczenie genów w genomie ludzkim:
3000 genów / chromosom;
zagęszczenie wysokie w rejonach subtelomerowych;
na niektórych chromosomach jest wiele genów: np. chromosom 19 i 22;
na innych mało: chromosom 4 i 18;
ciemne prążki G ubogie w geny, jasne obfite;
zawartość DNA chromosomów ludzkich: chromosom 1 → 263 Mb DNA
10 → 144 Mb DNA
16 → 98 Mb DNA
X → 164 Mb DNA
Y → 59 Mb DNA
4) genom jądrowy zawiera ok. 65000 – 80000 genów, ale tylko 3% genomu to sekwencje kodujące;
5) ok. 5% (3000 – 4000) genów jądrowych koduje cząsteczki RNA
Rodziny genów kodujących RNA często mają licznych członków rodziny;
geny rybosomalnego RNA (rRNA) – 28S, 18S i 5,8S; cytoplazmatyczny rRNA kodowany
jest przez pojedynczą jednostkę transkrypcyjną, tandemowo powtórzoną ok. 250 razy tworząc
5 grup sprzężeniowych (chusters), liczących po ok. 50 powtórzeń rozmieszczonych na
krótkich ramionach chromosomów 13, 14, 15, 21, 22; 5S cytoplazmatyczny rRNA jest
kodowany przez kilkaset kopii (przynajmniej 3 grupy sprzężeniowe na długim ramieniu
chromosomu 1.
geny transferowego RNA (tRNA) – bardzo duże i rozproszone; rodzina genów obejmująca
ponad 40 podrodzin, każda z szeregiem genów kodujących różne rodzaje cytoplazmatycznego
tRNA. Także pseudogeny (defektywne kopie genów).
geny małego jądrowego RNA (snRNA) – kilkaset rodzajów; niektóre tworzą
rybonukleoproteiny; odgrywają istotną rolę w reakcjach cięcia i modyfikacji zasad podczas
dojrzewania (splicing, maturation) rybosomalnego RNA.
Geny funkcjonalnie identyczne:
niedawno duplikowane, np.
-globiny
niektóre geny na różnych chromosomach kodują identyczne polipeptydy, np. podrodziny
genów histonowych: H1, histon łącznikowy; H2A, H2B, H3 i H4, histony rdzenia; najwięcej
jest ich na 6, 1.
Geny funkcjonalnie podobne:
blisko spokrewnione, ale nie identyczne co do sekwencji

str. 32
rzadko są sprzężone w ludzkim genomie, częściej są rozproszone na różnych chromosomach
geny, które kodują wyraźnie spokrewnione produkty a są ulokowane na różnych
chromosomach, są generalnie mniej spokrewnione, np. geny
i
-globiny.
Geny funkcjonalnie związane:
kodują produkty, które mogą różnić się strukturą ale są związane funkcjonalnie, np.
podjednostki tego samego białka, komponenty tego samego szlaku rozwojowego,
metabolicznego.
6) geny ludzkie są istotnie zmienne pod wzgl. wielkości i zawartości egzonów
geny krótsze niż 10 kb – zawartość egzonów ok. 100%, 33%, 46%
geny krótsze niż 100 kb – zawartość egzonów kilkadziesiąt do 3%
geny dłuższe niż 100 kb – zawartość egzonów kilka procent
Im dłuższy gen – tym zawartość egzonów mniejsza.
7) geny ludzkie wykazują ogromną zmienność wielkości i organizacji wewnętrznej
olbrzymie rozmiary niektórych genów oznaczają, że transkrypcja może być czasochłonna, np.
gen dystrofiny – 16 godzin;
odwrotna korelacja między wielkością genu i proporcją długości, która ulega ekspresji na
poziomie RNA.
8) znane są rzadkie przykłady genów nakładających się i genów w genach;
9) przeciętna gęstość genów w genomie ludzkim wynosi:
1 gen / 40 – 45 kb; jeżeli 1 gen, to przeciętnie 10 – 15 kb; to geny powinny być oddzielone
odcinkiem 30 kb niegenowego DNA
E. coli – 1 gen / 1 kb
S. cerevisiae – 1 gen / 2 kb
C. elegans – 1 gen / 5 kb
Dwa geny w jednym, np. PSA – antygen właściwy dla prostaty (5 egzonów i 4 introny); PSA – LM – białko
związane z PSA, kodowane przez egzon pierwszy PSA i czwarty intron; są to białka o funkcjach
antagonistycznych.
Produkt jednego genu jest ostatecznie cięty na dwa białka:
DPP – fosfoproteina zębiny niedobór któregoś → dentinogenesis imperfecta
DSP – sialoproteina zębiny
Prekursor – sialofosfoproteina DSPP → jest cięty na te dwa wyżej.
Geny w intronach – intron genu neurofibrominy zawiera trzy geny na przeciwległej nici DNA.
Nerwiakowłókniakowatość typu 1 (NF 1) – dziedziczenie autosomalne dominujące.
Rodziny wielogenowe i repetytywny kodujący DNA.
Ludzki genom składa się z trzech (szeroko rozumianych) składników:
DNA wyst. w pojedynczej kopii – 60%
umiarkowanie repetytywny – 30%
wysoce repetytywny – 10%
Rodziny genów ludzkich są zmienne pod względem pokrewieństwa różnych członków i co do stopnia, w jakim
szczególnie konserwowane sekwencje subgenowe definiują daną rodzinę:
klasyczne rodziny genów (rRNA; histony);
rodziny genów kodujące produkty z dużymi konserwatywnymi domenami (czynnikami transkrypcji TF;
geny Homeoboxu – 30 genów HOX); homodomena ~ 60 aminokwasów
rodziny genów kodujące produkty z bardzo krótkimi konserwatywnymi motywami aminokwasowymi
(DEAD box – produkty funkcjonują jako helikazy DNA); Asp – Glu – Ala – Asp

str. 33
nadrodziny genów – członkowie nadrodziny Ig są białkami powierzchniowymi
Rodziny genów mogą występować jako geny ściśle sprzężone, o specyficznej, subchromosomowej lokalizacji
lub jako geny szeroko rozproszone.
Uważa się, że geny w pojedynczej grupie powstały w wyniku tandemowych duplikacji. Wyraźne są różne
warianty organizacji grupy:
tandemowy układ genów (rRNA);
bliskie sprzężenie (nawet wspólny mechanizm regulacyjny; np. geny
i
-globiny)
złożona grupa genów (fizyczny związek między genami nie tak bliski, pomiędzy nimi mogą być
zawarte geny niespokrewnione co do sekwencji i funkcji – kompleks HLA na chromosomie 6 p21.3
Duplikacja genu może prowadzić do nabycia nowej funkcji lub powstania pseudogenu.
Rodziny genów zorganizowane w grupy wielokrotne:
o wysokiej homologii sekwencji (rRNA, chromosomy 13, 14, 15, 21, 22; rodzina genów receptorów
węchowych kodujących zróżnicowany zestaw receptorów pozwalających rozróżnić tysiące zapachów
~1000 genów)
o niskiej homologii sekwencji; rodzina genów globiny obejmuje geny o trzech lokalizacjach – grupa
-
globiny na chromosomie 16p;
-globiny na 11p, a mioglobiny na 22q; geny z jednej grupy są bliżej
spokrewnione niż geny z różnych grup.
Rozproszone rodziny genów:
przypuszczalnie pochodzące od dwóch genomów
będące rezultatem duplikacji starożytnego genomu
będące rezultatem retrotranspozycji – większość kopii niefunkcjonalnych
Opracowane pseudogeny powstają przez odwrotną transkrypcję z RNA.
Pseudogeny – niekompletne kopie genów i fragmenty genów są często spotykane w rodzinach wielogenowych:
nieopracowane – konwencjonalne pseudogeny (nie ulegają ekspresji);
nieopracowane ale ulegające ekspresji pseudogeny
opracowane pseudogeny
opracowane i ulegające ekspresji pseudogeny
niekompletne geny i fragmenty genów; 5’ albo 3’ fragment lub niewielki segment genu –
prawdopodobnie rezultat niesymetrycznego c-o lub niesymetrycznej wymiany chromatyd siostrzanych.
Wykład X 11.12.07r.
-globina oprócz genów ekspresywnych, zawiera 3 pseudogeny i pseudogen ulegający ekspresji.
Podobnie jak rodziny wielogenowe, niekodujący repetytywny DNA wykazuje dwa główne typy ogranizacji:
1) powtórzenia tandemowe (satelitarny, mini- , mikrosatelitarny DNA)
2) rozproszone powtórzone sekwencje
Główne klasy tandemowe powtórzonego ludzkiego DNA:
● satelitarny DNA
(bloki od 100 kb –
kilka Mb długości)
5 – 171 bp
szczególnie w centromerach
● minisatelitarny DNA
(bloki 0,1 – 20 kb)
6 – 64 bp
w lub blisko telomerów
wszystkich chromosomów
● telomerowy
6 bp (TTAGGG)
wszystkie telomery
● mikrosatelitarny DNA
1 – 4 bp
rozproszone

str. 34
(bloki krótsze niż 150 bp)
Rodziny wysoce powtórzonego rozproszonego DNA zawierają niewielki procent aktywnie przenoszących się
elementów:
- SINE – krótkie rozproszone elementy jądrowe
- LINE – długie rozproszone elementy jądrowe
Klasy sekwencji DNA ssaków, które przechodzą transpozycję za pośrednictwem RNA:
endogenne retrowirusy – przypominają retrowirusy, ale nie mogą infekować nowych komórek;
ograniczone są tylko do danego genomu. Zawierają sekwencje mające Long terminal repeats (LTRs) i
inne elementy funkcjonalnych retrowirusów;
retrotranspozony – nie mają LTRs; kodują odwrotną transkryptazę, co umożliwia niezależną
transpozycję, np. LINE-1;
retropseudogeny – wykorzystują odwrotną transkryptazę innych elementów; SINEs, powtórzenia Alu.
Jeżeli jakaś sekwencja przenosi się w obrębie DNA – transpozycja.
Jeżeli transpozycja za pomocą RNA – retrotranspozycja.
Dimery Alu – powtórzenia ok. 130pz, wstawka 32pz; występują w 1 mln kopii u naczelnych.
Element LINE-1 – koduje odwrotną transkryptazę.
STRUKTURA GENETYCZNA POPULACJI
Obliczanie frekwencji alleli:
Genotyp
F / F
F / S
S / S
Suma
l. osobników
l. alleli F
l. alleli S
l. alleli F+S
4
8
0
8
7
7
7
14
5
0
10
10
16
15
17
32
Frekwencja allelu F = 15 / 32 = 0,469
Frekwencja allelu S = 17 / 32 = 0,531
Mikroewolucja – zmiany frekwencji alleli w populacjach.
Makroewolucja – takie nagromadzenie zmian mikroewolucyjnych, które zapobiega posiadaniu płodnego
potomstwa przez dwa płodne organizmy płci przeciwnych.
Obliczanie średniej heterozygotyczności (w czterech loci):
l. osobników
locus
heterozygot
suma
heterozygotyczność
1
2
3
4
25
42
9
0
100
100
100
100
25/100 = 0,25
42/100 = 0,42
9/100 = 0,09
0/100 = 0
Przeciętnie: 0,19
str. 35
Polimorfizm – obecność w populacji allelu o częstości większej niż 1% (5%); allel rzadszy musi mieć wartość
większą niż 5%.
Prawo Hardy’ego – Weinberg’a
Założenia przyjęte w modelu określającym frekwencje genotypów:
rozpatrywany organizm jest diploidalny
reprodukcja jest płciowa
pokolenia nie nakładają się
kojarzenie jest losowe
wielkość populacji jest bardzo duża (nieograniczona)
migracja jest nieistotna
mutacje można pominąć
naturalna selekcja nie dotyczy rozpatrywanych genów
Jeżeli w locus są dwa allele, frekwencja allelu A wynosi p, frekwencja allelu a wynosi q.
p + q = 1
p = A
q = a
Frekwencja trzech możliwych genotypów wynoszą
p = A pp = AA
pq = Aa
(p + q)
2
= p
2
+ 2pq + q
2
q = a
pq = Aa
qq = aa
Prawo sformułowane w 1908r. (prawo równowagi):
frekwencje alleli nie zmieniają się z pokolenia na pokolenie;
frekwencje genotypów w populacji będącej w stanie równowagi genetycznej wyraża powyższy wzór
(rozwinięty kwadrat dwumianu), one także nie ulegają zmianom;
populacja osiąga stan równowagi (i zrównoważone frekwencje genotypów) po jednym pokoleniu
kojarzeń losowych, jeśli frekwencje alleli są równe u obu płci.
p = frekwencja allelu D – normalna długość palca = 0,7
q = frekwencja allelu d – krótki środkowy palec = 0,3
F
1
genotyp
DD
normalne palce
Dd
normalne palce
dd
krótki palec
frekwencja
genotypu
p
2
= (0,7)
2
= 0,49
2pq = 0,42
q
2
= 0,09
frekwencja
gamet
0,49
D
0,21
D
0,21
d
0,09
d
0,7
0,3
F
2
DD
Dd
dd
0,49
0,42
0,09
Równowaga Hardy’ego – Weinberg’a
Prawo H – W dla trzech alleli w locus
p
2
+ 2pq + q
2
+ 2pr + 2qr + r
2
= 1
Grupy krwi ABO
Frekwencje w pewnej populacji:
A
1
A
2
A
3
p
A
1
A
1
A
1
p
2
A
1
A
2
pq
A
1
A
3
pr
q
A
2
A
1
A
2
pq
A
2
A
2
q
2
A
2
A
3
qr
r
A
3
A
1
A
3
pr
A
2
A
3
qr
A
3
A
3
r
2
A (I
A
I
A
, I
A
i)
= 0,45

str. 36
Jakie są frekwencje alleli?
frekwencja genotypu ii = r
2
zatem r =
= 0,60
łączna frekwencja grup B + 0 jest (q + r)
2
= 0,13 + 0,36 = 0,49
q + r =
= 0,70
q = 0,70 – 0,60 = 0,1
p = 1 – q – r = 1 – 0,7 = 0,3
Profile DNA – wykorzystywane w kryminalistyce, w badaniach genetycznych zgodności, np. DNA
podejrzanego z dowodami.
Czynniki wpływające na zmiany częstości alleli w populacjach:
1) Nielosowe kojarzenie
- kryteria doboru partnera (80% podobni do nas; 1/3 z sąsiedztwa);
- chiński imigrant Arnold w RPA: 7 żon → 356 potomków – u 70 poważna wada uzębienia;
- chromosom Y Dzyngis Chana – ma 16 mln mężczyzn między Płn. Chinami a Afganistanem 1/200;
- Kaszubi – rzadkie uszkodzenie genu 30/100;
2) Migracje
- zmienność klinalna – częstość danego allelu wraz z odległością wzrasta lub maleje;
- deficjencja kinazy galaktozowej w Europie (proces fosforylacji). Cecha autosomalna recesywna
powodująca zaćmę u noworodków;
- w pewnym rejonie Bułgarii częstość tej deficjencji wynosi 1 : 1600 – 2500 i maleje wraz z odległością
Przypuśćmy, że osobnik z otaczających populacji migruje z pewną częstością do populacji lokalnej i
krzyżują się z rezydentami. Jeśli proporcja migrantów jest m , to w następnym pokoleniu 1 – m genów
pochodzi od rezydentów a m od migrantów. Jeśli w zewnętrznej populacji allel A
1
ma przeciętną
frekwencję P , a w populacji lokalnej p
0
, to w następnej generacji jego frekwencja będzie:
P
1
= (1 – m)p
0
+ mP = p
0
– m(p
0
– P)
mP
zmiana frekwencji allelu ∆p = p
1
– p
0
∆p = p
0
– m(p
0
– P) – p
0
= - m(p
0
– P)
różnica w częstościach allelu między populacją lokalną a zew. po pierwszym pokoleniu będzie:
p
1
– P = p
0
– m(p
0
– P) – P = m
0
– mp
0
– P + mP = (1 – m)p
0
– (1 – m)P = (1 – m)(p
0
– P)
po drugim pokoleniu:
p
2
– P = (1 – m)
2
(p
0
– P)
po t pokoleń migracji:
p
t
– P = (1 – m)
t
(p
0
– P)
Ten wzór pozwala obliczyć efekt t pokoleń migracji o proporcji m jeśli znane są początkowe
frekwencje allelu (p
0
i P).
p
t
= (1 – m)
t
(p
o
– P) + P
np. w USA, w populacji ludzi reprezentujących typ kaukaski, frekwencja allelu R
0
z locus Rh wynosi P
= 0,028. Wśród afrykańskich populacji, z których pochodzą przodkowie Afroamerykanów, frekwencja
allelu R
0
= 0,630. Afrykańscy przodkowie Afroamerykanów przybyli do USA ok. 300 lat lub 10
pokoleń temu. Zatem t = 10. Obecnie frekwencja R
0
w tej populacji jest p
t
= 0,446.
(1 – m)
t
=
1 – m =
m = 0,036
Zatem przepływ genów z populacji typu kaukaskiego (białej) do populacji afroamerykańskiej zachodzi
w tempie 3,6% na pokolenie. 10 pokoleń przepływu genów pozostawiło 0,694 wszystkich genów w
B (I
B
I
B
, I
B
i)
= 0,13
AB ( I
A
I
B
)
= 0,06
0 ( i i )
= 0,36
p
0

str. 37
populacji Afroamerykanów wywodzących się od afrykańskich przodków, a 0,306 ich genów wywodzi
się od przodków kaukaskich.
Wykład XI 18.12.07r.
3) DRYF GENETYCZNY
Przypadkowy dryf genetyczny odnosi się do zmian w frekwencjach alleli na skutek odchyleń w
losowej segregacji alleli do gamet i losowym łączeniu się gamet w zygoty.
Im mniejsza liczba krzyżujących się osobników w populacji, tym większe zmiany frekwencji alleli
na skutek dryfu.
W populacjach frekwencje allelu w danym pokoleniu określa prawdopodobieństwo wystąpienia
tego allelu w następnym pokoleniu.
Jeśli frekwencja allelu w pokoleniu zmienia się z np. 0,5 do 0,6 , to w następnym pokoleniu jego
frekwencja będzie 0,6.
Zmiany w frekwencji alleli kumulują się w kolejnych pokoleniach. Ostatecznie allel może zostać
zgubiony lub utrwalony tzn. jego frekwencja osiąga wartość 0 lub 1, wtedy proces dryfu ustaje.
EFEKT ZAŁOŻYCIELA
40 rodzin w Płn. Finlandii XVII w – obecnie populacja 18000; ryzyko schizofrenii 3,2% - 3 ×
wyższe niż w reszcie kraju;
Qubec – tylko 4 allele BRCA 1; na świecie 500;
Dunker – populacja przybyła z Niemiec do Pensylwanii 1720 – 30
Wielkość
populacji
(N)
Gamety
Wariancja
(pq/2N)
odchylenie
standardowe
(V
/2N)
Oczekiwany zakres
p w 95% populacji
1) p = q = 0,5
5
50
500
10
100
1000
0,025
0,0025
0,00025
0,16
0,05
0,016
0,18 - 0,82
0,40 - 0,60
0,468 - 0,532
2) p=0,3; q=0,7
5
50
500
10
100
1000
0,021
0,0021
0,00021
0,145
0,046
0,0145
0,01 - 0,59
0,208 - 0,392
0,271 - 0,329
Liczebność ma bardzo duży wpływ na wielkość dryfu genetycznego.
EFEKT SZYJKI BUTELKI
Im mniej liczna populacja tym większy efekt.
Związany z jakimiś katastrofami zmniejszając liczebność populacji, w której losowo jest
przenoszona część genów. Z czasem może zostać zwiększona ilość genów.
4) MUTACJE
Przyjmijmy, że są dwa allele A
1
i A
2
w locus. A
1
mutuje w A
2
z częstością u / gametę / pokolenie
→ frekwencja A
1
w danym czasie jest p
0
. W następnym pokoleniu frakcja u wszystkich alleli A
1
zmutuje do A
2
, zatem:
p
1
= p
0
– up
0
= p
0
(1 – u)
w następnym pokoleniu:
p
2
= p
1
– up
1
= p
1
(1 – u)
p
2
= p
1
(1 – u) = p
0
(1 – u)(1 – u) = p
0
(1 – u)
2
po t pokoleń, frekwencja A
1
będzie:
p
t
= p
0
(1 – u)
t
tempo zmian jest wolne: 10
-5
gameta/pokolenie
1000 pokoleń – zmiana frekwencji A
1
z 1,00 na 0,99
2000 pokoleń – zmiana frekwencji A
1
z 0,50 na 0,49

str. 38
10000 pokoleń – zmiana frekwencji A
1
z 0,10 na 0,09
Im mniejsza frekwencja allelu tym więcej czasu zajmuje osiągnięcie danego poziomu zmian.
A
2
mutuje do A
1
z częstością v, wyjściowe frekwencje alleli są: p
0
i q
0
, dla A
1
: p
1
= p
0
– up
0
+ vq
0
zmiana w frekwencji A
1
: ∆p
∆p = p
1
– p
0
∆p = (p
0
– up
0
+ vq
0
) – p
0
= vq
0
– up
0
równowaga istnieje, gdy ∆p = 0
up = vq
up = v(1 – p)
up + vp = v
ponieważ p + q = 1, to
Przykład:
Przypuśćmy, że u = 10
-5
a v = 10
-6
wtedy będzie równowaga
5) DOBÓR NATURALNY
negatywny lub pozytywny;
wrażliwość na gorzki smak cenna w środowisku zawierającym toksyczne rośliny;
jeżeli infekcja zabija przed osiągnięciem wieku reprodukcyjnego, to ostatecznie usuwa z populacji
allele uwrażliwiającą na nią; eliminowane najgroźniejsze bakterie (-) i zwiększony udział alleli
warunkujących oporność w kolejnych pokoleniach (+) zanim odkryto antybiotyki (gruźlica
ewoluowała od poważnej, ostrej infekcji systemowej (1880) do rzadkiej chronicznej infekcji płuc)
anemia sierpowata i malaria (odporność na nią), co prowadzi do:
zrównoważonego polimorfizmu (utrzymywanie szkodliwych alleli w heterozygotach);
deficjencja G6PD a malaria (glukozo-6-fosfodehydrogenaza) – cecha sprzężona z X
recesywna, ryzyko anemii sp. warunkach. Nadreprezentacja ♀ G g i ♂ g w środowisku
malarii: pasożyt infekuje krwinki ale nie może się rozmnażać; także zróżnicowany
polimorfizm;
fenyloketonuria i infekcje grzybów pleśniowych – heterozygotyczność pod względem
genu PKU chroni płód – wysoki poziom fenyloalaniny inaktywuje ochratoksynę A
(Aspergilus, Penicillium); PKU częsta w Irlandii i Szkocji
mukowiscydoza (CF) a tyfus i cholera – Salmonella infekuje kom. jelita tylko jeżeli
obecne są funkcjonalne kanały chlorkowe (CFTR); u heterozygot część kanałów jest
uszkodzona;
Mukowiscydoza – zwłóknienie torbielowate trzustki
- gen autosomalny recesywny;
- częstość w Europie 1 : 2500;
- mutacje (ok. 500) w genie CFTR;
- defekt przewodnictwa przez błonę kom. (kanały chlorkowe i sodowe)
Bakterie cholery produkują toksyny otwierające kanały w kom. jelita. U nosicieli CF jest to
niemożliwe; „wątpliwe dobrodziejstwo zrównoważonego polimorfizmu”
H
2
O
H
2
O →
gęstniejący śluz
Cl →
glikoproteina P a niewrażliwość na terapeutyki przeciw AIDS; proteina P – część
białkowa w błonie kom., część glikolityczna sterczy na zewnątrz; allele C i T
CC – nadprodukcja glikoproteiny – najszybciej pozbywają się szkodliwych
subst.(terapeutyków); TC – pośrednia ilość; TT – niewiele cząsteczek; glikoproteina na
powierzchni limfocytów T i kom. wyściełających jelito umożliwia intensywne usuwanie
szkodliwych substancji; genotyp CC chroni przed infekcjami bakteryjnymi i wirusowymi
Cl+

str. 39
szlaku żołądkowo – jelitowego (nieżyt). Jednocześnie obniża efektywność leczenia AIDS
intensyfikując wydalanie środków chemioterapeutycznych.
Zjawiska zmieniające częstości alleli w populacjach:
1) równowaga H-W → brak zmiany częstości w populacji
2) nielosowe kojarzenie → wzrost częstości jakiegoś genotypu
3) migracje
4) dryf genetyczny
5) mutacje
6) naturalna selekcja (dobór naturalny) – np. któryś nie reprodukuje
Genealogia genu (zapis pochodzenia):
np. zmienność frekwencji allelu CF - ∆F508 (wywołuje mukowiscydozę)
przebieg historyczny bierzemy pod uwagę oraz czas
migracje ze środkowego Wschodu do Europy (nedit lub nawet paledit – pochodząc od Basków, gdzie
frekwencja jest bardzo wysoka, migracja)
Genomika porównawcza
genom ludzki nie jest dużo większy niż u innych org. i większość sekwencji DNA jest wspólna. Na
poziomie wielkości genomu jesteśmy podobni do ryby (pufferfish) i brukselki. W istocie, różnice
wynikają z organizacji genomu i sekwencji DNA.
Wskaźnik hybrydyzacji DNA określa stopień pokrewieństwa.
Są miejsca wspólne dla wszystkich gatunków – konserwatywne, mają istotne znaczenie!
Modele zwierzęce
2/3 genów powodujących zaburzenia cech mendlowskich u ludzi ma swoje odpowiedniki u D.
melanogaster
Syndrom Waardenburga – jedna mutacja może wywołać podobne spektrum efektów u różnych
gatunków ( upośledzenie słuchu, niebieskie i duże oczy oraz pasek siwiejących włosów na czole).
Porównanie człowieka – szympans
98,7% podobieństwa na poziomie genomu (hybrydyzacja, lata 70-te); identyczność sekwencji białek
96,6% uwzględniając insercje/delecje
Różnice fenotypowe są uwarunkowane indywidualnymi cechami, często determinowanymi przez poj. geny:
1) owłosienie – szympansy/goryle, ekspresja genu keratyny, którego odpowiednik u ludzi został
wyciszony przez mutację nonsens i ma status pseudogenu;
2) zdolność mowy – pojedynczy gen, także obecny u szympansów ale w zmienionej formie;
3) hemoglobina – gen kontrolujący przejście od hemoglobiny embrionalnej do płodowej – prymitywne
naczelne nie mają hemoglobiny płodowej; u wyższych naczelnych przedłużony okres płodowy –
zwiększone wymiary mózgu;
4) także różnice nie w sekwencji genomu, a raczej w ekspresji genów; profil ekspresji genów w wątrobie
bardziej zbliżony niż w mózgu.
Struktura hemoglobiny:
faza embrionalna – 2 łańcuchy ε (epsilon) + 2 łańcuchy δ (zeta)
faza płodowa – 2 łańcuchy γ + 2 łańcuchy α
dorośli – 2 łańcuchy β + 2 łańcuchy α
Podobieństwa sekwencji aminokwasów odzwierciedlają pokrewieństwo ewolucyjne. Między człowiekiem a
szympansem nie ma różnic w cytochromie c.
Ewolucja genu

str. 40
Porównanie tego samego genu między gatunkami naczelnych ujawnia jak elementy genomu powstały w
wyniku mutacji, przemieszczenia i duplikacji.
Początek: chromosom 12 – występuje tam gen MCH ( prekursor neuropeptydu, funkcjonuje w mózgu);
nastąpiła retrotranspozycja i fragment DNA wstawiony został w ramię p na chromosomie 5. Pojawił się
intron między DNA, więcej intronów i mutacje (niekodujący fragment → w kodujący). Duplikacje
sekwencji na ramieniu q. powstały PMCHL 1 – ekspresja w mózgu, oraz PMCHL 2 – ekspresja w
jądrze.
Wykład XII 8.01.08r.
Czy genom ludzki uległ duplikacji?
W czasie 50 mln lat dywergencja od przodka kręgowców:
- podwójna duplikacja i utrata niektórych genów;
- pojedyncza duplikacja i dodatkowa duplikacja niektórych sekwencji.
Geny homeoboxu
(HOX, homeotyczne), odpowiadają za organizację części ciała.
Kontrolują kolejność w jakiej włączane są geny u embrionu i ta kaskada genów zapewnia prawidłowy
rozwój anatomicznych części ciała we właściwych miejscach.
Poszczególne geny ulegają ekspresji w takiej kolejności, w czasie lub pozycji anatomicznej, w jakiej są
zlokalizowane na chromosomie.
Mutanty: antennopedia u D. melanogaster → odnóża w miejsce czułków na głowie.
Organizacja klasterów genów HOX u ssaków i Amphioxus sugeruje możliwość pojedynczej lub podwójnej
duplikacji pierwotnego genomu.
U D. melamogaster 2 grypy : ultrabithorax i antennopedia;
U kręgowców w 4 grupach: A, B, C, D stanowiąc 39 genów.
geny paralogiczne – geny bądź fragmenty chromosomu, które wykazują bardzo wysoki stopień homologii
wewnątrz gatunku.
Mutacje w genach homeoboxu u ludzi:
forma białaczki – białe krwinki powstające z komórek pierwotnych wprowadzone na niewłaściwy szlak
rozwojowy zachowują zdolność szybkich podziałów powodując nowotwór;
syndrom DiGeorge – brak niektórych części ciała (grasica) i nieprawidłowy rozwój innych;
synpolidaktylia
Transfer genów jednego gatunku do komórek innego dowodzi wysokiej konserwatywności Homeoboxu.
mysi gen antennopodia → D. melanogaster odnóża na głowie
ludzki → zaburzenia rozwoju głowy u myszy
Genetyka zachowania
(badanie zmian funkcjonalnych ukl. nerwowego) determinizm genetyczny (dzieci sławnych rodziców);
Cechy behawioralne, to m.in.: - zdolności
- uczucia
- nastroje
- osobowość
- inteligencja
- reakcje na stres, zagrożenie
Zaburzenia o symptomach behawioralnych: - fobie
- niepokoje – stany lękowe
- demencje

str. 41
- psychozy
- zmiany nastroju
- uzależnienia
Geny wpływają na większość cech behawioralnych.
Większość zaburzeń behawioralnych zgodnych z klasycznym (złożonym) profilem dolegliwości:
raczej częste > 1/1000
poligenowe
wieloczynnikowe
Monogenowych niewiele – np. pląsawica Huntingtona
Nowe metody badania biologicznych podstaw cech behawioralnych:
analiza związków, korelacji markerów genetycznych (SNP) i poszczególnych symptomów;
analiza mutacji w „podejrzanych” genach, które są obecne wyłącznie u osobników przejawiających
daną cechę.
Geny kontrolują syntezę, poziom i dystrybucję neurotansmitterów.
Wiele genów wpływających za zachowanie koduje białka związane z neurotransmisją.
Transdukcja – zmiana formy.
Bodziec → receptor w błonie (ocenia, przekazuje i wzmacnia sygnał) → receptor przekazuje sygnał,
aktywuje regulator → regulator aktywuje enzym → utworzenie cAMP (wtórny posłaniec) z ATP →
odpowiedź komórki (tu następuje wzmocnienie)
Defekty
- NF 1 ( nerwiakowłókniakowatość) – komórka nie blokuje transmisji sygnału czynnika wzrostu do podziału
komórki; leczenie: blokada receptorów czynnika wzrostu;
Geny wpływające na dziedziczne składniki zaburzeń nastroju i chorób umysłowych (norma?) związane są z
neurotransmisją i transdukcją sygnału.
ADHD (attention deficit hyperactivity disorder)
-
leczenie skierowane na dopaminę i kontrolę jej funkcjonowania;
Autyzm (zaburzenie komunikowania)
- „skanowanie genomu” wskazało „podejrzane” geny na 14 różnych chromosomach.
Zaburzenia odżywiania:
- anoreksja 0,5%; najwyższe ryzyko śmierci 15 – 21%
- bulimia 2,5%
Odziedziczalność 0,5 – 0,8%
Geny kontrolują wagę ciała
Adipocyty (kom. tłuszczowe) – uwalniają leptynę (hormon białkowy) podczas jedzenia, z kwiobiegiem
przenoszona do mózgu (do jadra łukowatego, w podwzgórzu), gdzie wiązana jest przez receptory na
powierzchni neuronów, to stymuluje wydzielanie MSH (hormonu melanotropowego) wiązanego przez
receptor melanokortyny (MC4R), kompleks ten aktywuje supresory apetytu (tracimy apetyt) i blokowane
są stymulatory apetytu. Uwalniany jest enzym SCD-1 (jego wysoki poziom przyczynia się do
metabolizowania, spalany jest tłuszcz i uwalniana jest energia; przy małej jego ilości odkładany jest
tłuszcz).
Przy niskiej ilości leptyny uwalniany jest neuropeptyd Y, który stymuluje receptory apetytu.
Leptyna jest lekarstwem dla 15% otyłych (gdy otyłość wynika z wad syntezy leptyny).
Pozostałe osoby mają problem z receptorami leptyny.

str. 42
Leptyna – stymuluje kom. podwzgórza do obniżania apetytu i metabolizmu.
Neuropeptyd Y – podnosi apetyt.
Grelina – hormon peptydowy, sygnalizuje głód (pochodzi z żołądka) w krótkim czasie przesyła sygnał do
mózgu, stymulując neuropeptyd Y
PYY – w przypadku sytości daje sygnał do mózgu i spada apetyt
Sen (narkolepsja):
- w 1999 zidentyfikowano mutacje w genie kodującym receptor neuropeptydu (hipokretyny);
- mechanizm:
Receptor nie dociera do powierzchni pewnych komórek mózgu, które nie mogą otrzymać sygnału, by
trwać w stanie przebudzenia.
Receptor niefunkcjonalny.
- w 1998 odkryto białko (oreksynę) wiążące się z receptorem hipokretyny; myszy pozbawione genu oreksyny
nagle zapadają w sen podczas jedzenia.
- gen receptora hipokretyny/oreksyny jest zlokalizowany
u psów na chromosomie 12
u ludzi na chromosomie 6
- mózg ludzi cierpiących na narkolepsję wykazuje deficyt hipokretyny/oreksyny
- FASPS – 19:30 - 4:30;
gen period na końcach chromosomu 2q u ludzi 1 podstawienie nukleotydowe w tym genie;
cecha autosomalna dominująca;
ta mutacja uniemożliwia wiązanie grupy fosforanowej przez białko, co jest konieczne, by przekazać
sygnał synchronizujący cykl sen – czuwanie ze wschodem i zachodem Słońca;
gen period ma odpowiedniki u chomika i muszki owocowej, wywołujące zmiany cyklu sen – czuwanie
także inne przyczyny syndromu FASPS.
Inteligencja:
odnosi się do: zdolności rozumowania, uczenia się, zapamiętywania, kojarzenia, dedukcji i tworzenia;
1904, Alfred Binet, Sorbona – test IQ
Pytania werbalne, numeryczne i obrazkowe
Przeciętna – 100 pkt.; 2/3 ludzi 85 – 115; za łagodne opóźnienie umysłowe uważa się 50 – 70 pkt.
Niewielka rola środowiska → IQ dzieci adoptowanych jest zbliżony do ich rodziców biologicznych;
Genetyczne podłoże różnic w inteligencji:
niemal wszystkie syndromy wynikające z anomalii chromosomowych (Downa, łamliwego
chromosomu X) związane są z opóźnieniem umysłowym;
pojedyncze geny wpływające na inteligencję → białka kontrolujące neurotransmisję;
stwierdzono silną korelację pewnego profilu SNP genu kodującego cząst. zaangażowaną w
połączenie kom. nerwowych (N-CAM, neural cellular adhesion molecule) z wysokim IQ!
metody badań:
związek genu o znanej funkcji z inteligencją
skanowanie genomu w celu lokalizacji genów, których produkty białkowe wpływają na
połączenia nerwowe
fragment chromosomu 4 zawiera geny (3) związane z inteligencją co stwierdzono przez porównanie
147 markerów w grupie dzieci o wysokim IQ i grupie kontrolnej (przeciętnych).
Uzależnienie:
ośrodek uzależnień w mózgu zlokalizowano przez koncentrację badań receptorów powierzchniowych
komórek, które wiążą neurotransmitery;
zmiany w mózgu przyczyniające się do uzależnienia zachodzą w grupie funkcjonalnie podobnych
struktur tworzących układ limbiczny;
uwarunkowanie uzależnień na poziomie molekularnym, oddziaływań neuron – neuron… odpowiedź
behawioralna;
białka zaangażowane w uzależnienie, to te które:
str. 43
są elementem szlaków biosyntezy neurotransmiterów, jak np. enzymy;
tworzą transportery odbiorcze, które usuwają nadmiar neurotransmiterów z synapsy;
tworzą receptory na neuronach postsynaptycznych, które są aktywowane lub inaktywowane gdy
jest wiązany specyficzny neurotransmiter;
są częścią szlaku transdukcji sygnału w neuronie postsynaptycznym.
opium, kokaina, itd. wiążą się z receptorami neuronów ludzkich → mamy własne wersje tych
substancji;
ludzkie ekwiwalenty opioidów to: endorfiny i enkefaliny.
Zaburzenia nastroju:
depresja, depresja maniakalna;
przyczyna:
brak serotoniny (neurotransmiter), która wpływa na nastrój, emocje, apetyt i sen;
istotny jest także poziom norepinefryny;
anomalie występują w białkach transportujących neurotransmitery z synapsy do „pomp odbiorczych” w
neuronie presynaptycznym;
transportery nadaktywne lub w nadmiarze obniżają poziom neurotransmiterów;
leki antydepresyjne (SSRI) – zapobiegają odbieraniu serotoniny z synapsy przez neurony
presynaptyczne;
w efekcie zwiększona jest dostępność neurotransmitera i wyższa stymulacja neuronu
postsynaptycznego;
leczenie: blokowane są receptory wiążące na powrót serotoninę w neuronie presynaptycznym.
Wykład XIII 15.01.08r.
Genetyka nowotworów:
wg Hipokratesa „karkinos” – rak
nowotwór – jest zespołem zaburzeń, które powstają na skutek zmian w genach;
powstaje wówczas, gdy kom. wymyka się spod kontroli mechanizmów decydujących o tempie i
przebiegu jej cyklu komórkowego: wzrost, różnicowanie i śmierć;
białkowe czynniki wzrostu i cząsteczki związane z sygnałem – zew. kom.
czynniki transkrypcji – wewnątrz kom.
droga nieograniczonych podziałów
czynnik wzrostu
transdukcja
sygnału
komórka nowotworowa staje się niewrażliwa na sygnały
biochemiczne z otaczających komórek: podział, stop,
ekspresja zestawów genów specjalizujących komórkę.
HeLa – linia komórkowa, hodowana sztucznie na pożywce
tkankowej, pochodzi od Henrietty Lacks, która zmarła na
raka w 1951r., a jej komórki nadal się dzielą
(nieograniczone podziały).
Punkty kontrolne cyklu komórkowego:
punkt kontroli uszkodzeń DNA – może pojawić się w fazie S (replikacja) i gdy powstaną jakieś błędy,
zostaje wydłużona faza S aby mogły zostać naprawione;
w G
1
w G
2
Czynniki transkrypcyjne
Działanie telomerazy

str. 44
punkt apoptozy – pojawią się między G
2
a profazą; odpowiedni poziom surviviny warunkuje zajście
apoptozy (jej akumulacja), bądź przejście do mitozy;
punkt wrzeciona podziałowego – w anafazie; tu powstają pytania: czy powstało wrzeciono?; czy
chromosomy prawidłowo wiążą się z wrzecionem?; czy prawidłowo zostają ulokowane chromosomy?
Apoptoza:
gdy coś niedobrego dzieje się z komórką, zostają uruchomione kaspazy (niszczą wnętrze komórki) i
dzielona ona jest na fragmenty;
fagocyty trawią pozostałości.
Nowotwór raczej na poziomie komórkowym niż całego organizmu:
10% to zaburzenia monogenowe (w kom. generatywnych) – wtedy nowotwór całego organizmu;
podstawy dziedziczne;
reszta nowotworów powstaje w komórkach somatycznych
łagodny – ograniczony tylko do jednej tkanki; ograniczony zasięg;
złośliwy – gdy przerzuty na inne tkanki i narządy
system immunologiczny niszczy większość komórek nowotworowych po rozpoznaniu specyficznych
antygenów na ich powierzchni
Etapy karcinogenezy:
I. pre–inicjacja – ekspozycja organizmu na karcinogeny (powodują zmiany w DNA, genach - mutacje)
II. inicjacja – nagromadzenie tych mutacji
III. progresja – nowotwór zaczyna się rozwijać
Zaczyna się od pojedynczej mutacji do rozległych zmian w ekspresji genów (prowadzi do niekontrolowanych
podziałów).
Dzięki mikromacierzy DNA (związana z ekspresją genów) ujawniono ukryty typ białaczki (MLL):
ALL – ostra białaczka limfatyczna
MLL – białaczka linii mieszanej (mixed-lineage); wiek dziecięcy
AML – ostra białaczka szpikowa
W 2002 odkryto różnicę między MLL a ALL wyrażającą się ekspresją genów; 200 genów wykazywało
nadekspresję, a 1000 było o obniżonej ekspresji genów.
Nowotwory dziedziczne:
pierwsza mutacja w linii zarodkowej, która uwrażliwia na powstanie nowotworu;
druga mutacja w tkance.
Nowotwory spontaniczne – obydwie mutacje w jednej tkance.
Np. Siatkówczak sporadyczny i dziedziczny:
chromosom 13 (na ramieniu q) – tam gen odpowiedzialny
½ dzieci u których rozwija się siatkówczak (RB – retinoblastoma), dziedziczy podatność na nowotwór;
mutacja genu supresora jest delecją pozbawiającą funkcji;
normalnie, białko 928AA wiąże czynniki transkrypcji uniemożliwiając mitozę;
sporadyczny – atakuje najczęściej tylko jedno oko, a dziedziczny – oba;
sporadyczny – obydwa allele są pierwotnie normalne, w którymś momencie następuje zmiana jednego z
alleli na nieprawidłowy, w wyniku delecji drugiego allela powstaje siatkówczak;
dziedziczny – mutacja podczas tworzenia gamet, zygota już ma jeden uszkodzony, wystarczy tylko
jedna mutacja i oba allele będą uszkodzone.
Pacjent 1
Pacjent 2
Pacjent 3
Pacjent 4
S T
S T
S T
S T
→
→
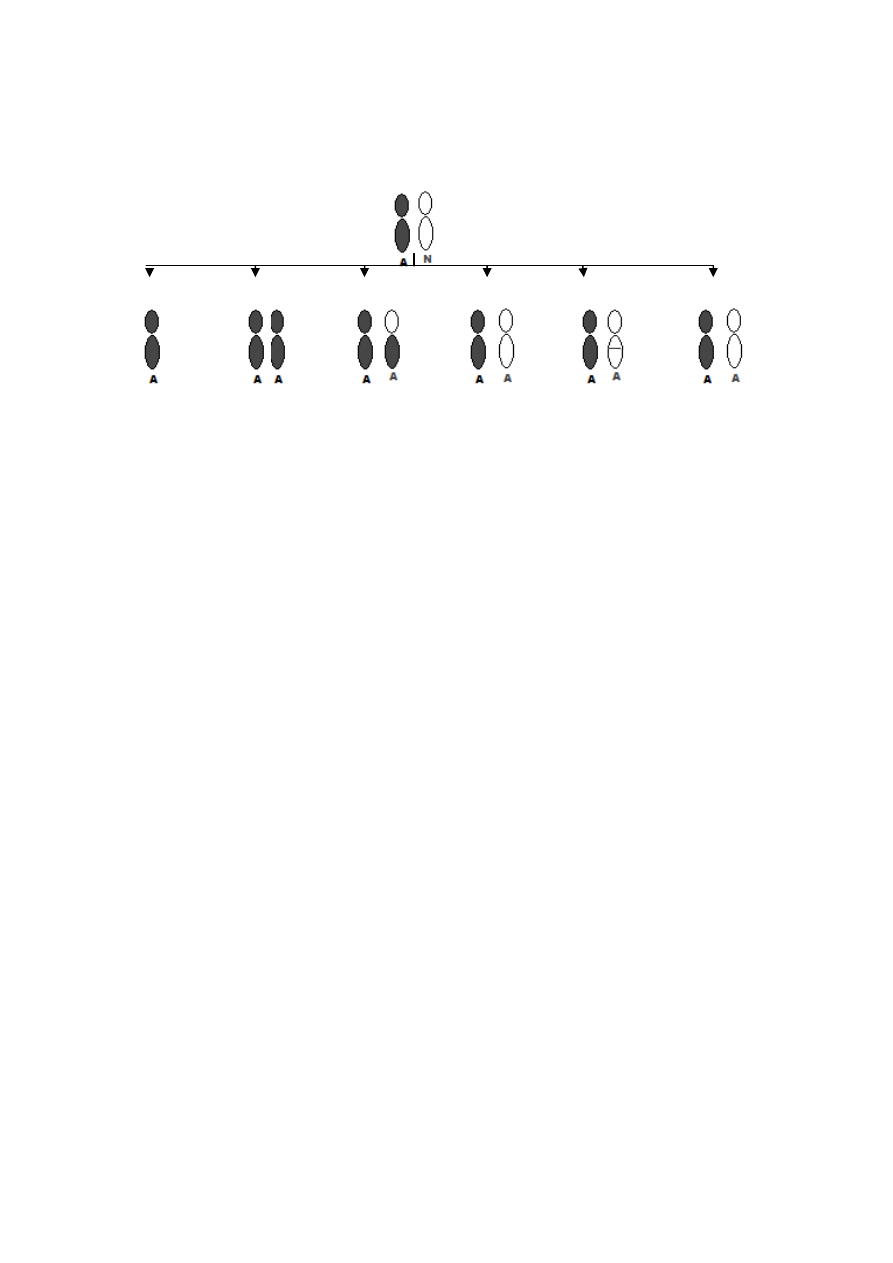
str. 45
Utrata heterozygotyczności allelu.
S – tkanka somatyczna; T – guz
obniżony poziom
Sześć możliwych par chromosomu 13, które mogą powstać w kom. siatkówczaka u osoby z predyspozycją do
rozwoju tego nowotworu.
Para chromosomów 13 w prawidłowej kom. osoby
z predyspozycją do rozwoju siatkówczaka
nondysjunkcja
(utrata punktowa)
nondysjunkcja i
reduplikacja
mitotyczna
rekombinacja
konwersja genu
delecje
mutacje
Charakterystyka komórek nowotworowych:
śliskie, oleiste, mniej przyległe;
utrata kontroli cyklu komórkowego;
dziedziczność – jeżeli komórka nowotworowa się dzieli, to każda z potomnych jest też nowotworowa;
zdolność transplantacji – kom. nowotworowa, gdy znajdzie się w innym organizmie tego samego
gatunku, też jest nowotworowa;
utrata zróżnicowania (specjalizacji);
utrata inhibicji kontaktu – normalne komórki w kontakcie z sobą uniemożliwiają podział, kom.
nowotworowe nakładają się jedne na drugie tworząc stosy;
zdolność angiogenezy – zdolność do tworzenia dodatkowych naczyń zaopatrujących w tlen i związki
odżywcze;
inwazyjność – atakują inne tkanki;
podniesione tempo mutacji;
zdolność rozprzestrzeniania (metastaza).
Angiogeneza – wzrost nowych naczyń krwionośnych; komórki wewnątrz guza nie mają dostępu do tlenu,
dlatego wydzielają naczyniowy, śródbłonkowy czynnik wzrostu, dzięki któremu możliwe jest tworzenie nowych
naczyń.
Typy genów powodujących nowotwory:
1) Onkogen – aktywnie promuje nowotwór. Normalna wersja, proto-onkogen kontroluje cykl komórkowy.
Onkogen aktywuje podział komórki w niewłaściwym czasie lub miejscu;
NABYCIE NOWEJ FUNKCJI
2) Mutacja genu supresora nowotworu – mutacja (delecja) znosi zwykłą supresję podziału komórki;
UTRATA FUNKCJI
3) Mutacja genu naprawy DNA – efekt pośredni. Uszkodzony gen pozwala na akumulację wielu mutacji,
niektóre w proto-onkogenach i genach supresorowych.
Od 1976 wykryto: > 100 onkogenów, > 30 genów supresorowych – których delecja lub inaktywacja powoduje
nowotwór.
Wyższa ekspresja genu w nowej lokalizacji (onkogeny):
1) Inwersja chromosomu 11 – nowotwór gruczołów przytarczycy
Proto-onkogen obok sekwencji DNA kontrolującej transkrypcję genu hormonu przytarczycy. Gdy
gruczoł syntetyzuje hormon – następuje ekspresja onkogenu. Komórka dzieli się tworząc guz
nowotworowy.
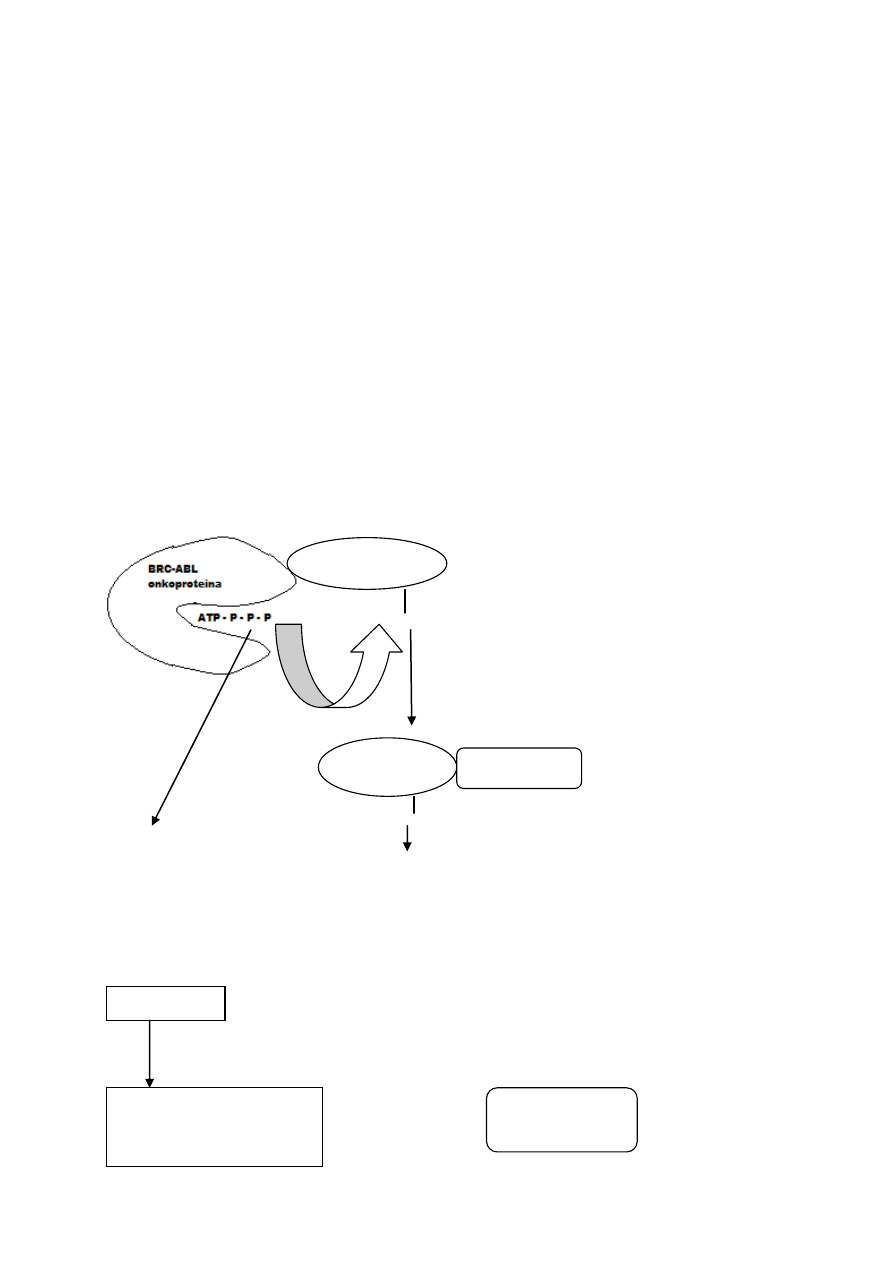
str. 46
+ substrat
substrat
efektor
onkogen
Białko powierzchniowe-
receptor czynnika wzrostu
(kinaza tyrozynowa)
Epidermalny
czynnik wzrostu
2) System immunologiczny przyczynia się do powstania nowotworu, gdy translokacja lub inwersja
umieszcza proto-onkogen obok genu przeciwciała.
3) Infekcje wirusowe – nowotwór wątroby w wyniku zapalenia wątroby; gdy proto-onkogen omyłkowo
aktywowany łącznie z genami przeciwciał.
Translokacja przyczyną nowotworu:
np. chłoniak Burkitta – infekcja wirusem Epstein – Barra; podczas tworzenia przeciwciał translokacja 8,
14 i wtedy oba geny znajdują się obok siebie (na jednym chromosomie), wobec czego podczas
aktywacji genu przeciwciał, aktywowany jest też onkogen.
Białka fuzyjne (połączone z dwóch innych) o nowych funkcjach:
np. chromosom Philadelphia (Ph’) wynikiem translokacji 9, 22; występuje u chorych na przewlekłą
białaczkę szpikową (CML); końcówka chromosomu 9 zostaje przeniesiona na chromosom 22, a
fragment 22 na koniec 9. Skrócony chromosom 22 – Ph’. Onkogen ABL przeniesiony 9 – 22, łączy się
z sekwencją BCR – forma enzymu kinazy tyrozynowej.
Białko połączone „fusion protein” a chroniczna białaczka szpikowa – takie białko ma przedłużoną aktywność!
Tyrozyna
P – tyrozyna
ATP – P – P
Chroniczna białaczka szpikowa
Leczenie: lek – Gleevec wchodzi w miejsce wiązania ATP i nie dopuszcza do ufosforylowania tyrozyny w
substracie, w wyniku czego nie może przyłączyć się efektor.
Zbyt mocny sygnał do podziału komórki; ok. 25% przypadków nowotworu piersi
koduje
przyłącza się
str. 47
normalnie jest ich 20000, a może
wzrosnąć do 2 mln kopii
transkrypcja genów stymulujących
podział komórki
Normalnie białko p53 zapobiega wielu nowotworom – koduje czynnik transkrypcyjny, który decyduje czy
komórka naprawia błędy replikacji DNA czy ginie na drodze apoptozy.
p53 – gdy jest uszkodzony gen , komórka dzieli się dalej, co jest powodem 50% nowotworów!
RB, p53, BRCA 1, BRCA 2 – to geny supresorowe, które wzajemnie reagują ze sobą w jądrze – oddziałują w
celu naprawy dwuniciowych uszkodzeń DNA (zmiany struktury chromosomów).
Środowisko inicjuje mutacje lub zmiany w ekspresji genu p53 prowadzące do nowotworu:
wirusy
toksyny
zmiany w p53
mutacje somatyczne
rak
promieniowanie
Seria zmian genetycznych transformuje normalne ostrocyty(?) wzmacniające kom. nerwowe w mózgu, w szybko
rozwijający się nowotwór.
utrata lub mutacja
utrata genów chrom. 9
aktywacja
utrata chromosomu 10,
obu alleli p53,
kodujących interferony
onkogenu
nieznana rola
supresorów raka
i supresory raka
podnosi sygnał wzrostu
Szereg genów przyczynia się do rozwoju nowotworu jelita:
uszkodzenie genu supresorowego (jego skrócenie), którego produkt białkowy chroni przed przerostem
nabłonka; normalne kom. żyją 3 dni;
uszkodzenia genów akumulują się w miarę upływu czasu
prawidłowy nabłonek
stadium wczesne
stadium pośrednie
gruczolaka
gruczolaka
gruczolaka
inaktywacja genu
aktywacja onkogenu
supresorowego APC
KRAS
inaktywacja genu
supresorowego DDC
przerzuty
rak jelita grubego
stadium późne
gruczolaka
inne mutacje
inaktywacja genu
supresorowego p53
Rys. Etapy progresji prawidłowego nabłonka jelita grubego do guza przerzutowego.
Warzywa krzyżowe (brukselka, brokuły) obniżają ryzyko nowotworu.
Wykład XIV 22.01.08r.
P
Wyszukiwarka
Podobne podstrony:
Filozofia skrypt do nauki id 47 Nieznany
GENETYKA WYKLADY PROPS id 18759 Nieznany
8 wyklad rynek pracy WIGE id 47 Nieznany (2)
LOGIKA wyklad 5 id 272234 Nieznany
ciagi liczbowe, wyklad id 11661 Nieznany
0 konspekt wykladu PETid 1826 Nieznany
AF wyklad1 id 52504 Nieznany (2)
Genetyka test koncowy styczen 2 Nieznany
Neurologia wyklady id 317505 Nieznany
genetyka wykład 3
MATERIALY DO WYKLADU CZ VIII i Nieznany
MATERIALY DO WYKLADU CZ V id 2 Nieznany
ZP wyklad1 id 592604 Nieznany
GOGN Wyklad 6 scalenie i podzia Nieznany
CHEMIA SA,,DOWA WYKLAD 7 id 11 Nieznany
or wyklad 1 id 339025 Nieznany
II Wyklad id 210139 Nieznany
cwiczenia wyklad 1 id 124781 Nieznany
więcej podobnych podstron