
1
1. Definicja procesów jednostkowych.
Procesy jednostkowe:
* są to te czynności jednostkowe, z którymi związane jest zachodzenie reakcji chemicznych
* (ang. Unit Process) jest to wyodrębniony zespół przemian fizycznych i chemicznych materii,
charekterystych ze względu na zachodzacą reakcję chemiczna
2. Definicja operacji jednostkowych, podział z punktu widzenia istoty operacji.
Operacje jednostkowe:
* są to te czynności jednostkowe, które polegają na zachodzeniu przemian fizycznych
* (ang. unit operation) jest to wyodrębniony zespół fizycznych przemian materii (bez reakcji chemicznej),
charakterystyczny ze względu na ich skutek
* całokształt procesów fizycznych i fizykochemicznych zachodzących w wyodrębnionej części aparatury
chemicznej ciągu technologicznego (np. wieży absorpcyjnej, komorach pyłowych, wyparce)
* dzieli się je na:
- dyfuzyjne (wymiana masy)
- mechaniczne
- cieplne (wymiana ciepła)
- hydromechaniczne – flotacja
Zaprojektowanie operacji jednostkowych wymaga uwzględnienia bilansów – materiałowego i
energetycznego, stanu równowagi osiąganej przez układ oraz parametrów wpływajacych na przebieg
operacji jednostkowych, których odpowiednie zmiany mają doprowadzid do uzyskania stanu –
równowagi.
3.
Przykłady operacji jednostkowych i procesów jednostkowych
*oczyszczanie gazu przez absorpcję szkodliwych domieszek w wodzie możemy zaliczyd do operacji
jednostkowych, mimo że absorpcji mogą towarzyszyd reakcje chemiczne. Celem jest tu bowiem fizyczne
oddzielenie zanieczyszczeo a nie prowadzenie tych chemicznych reakcji i otrzymywanie produktów
* absorpcja dwutlenku węgla w solance nasyconej amoniakiem jest procesem jednostkowym. Jest to
jeden z ważnych procesów jednostkowych w metodzie Solvaya otrzymywania sody amoniakalnej. Celem
tej czynności jest otrzymywanie kwaśnego węglanu amonu NH
4
HCO
3
i to ten cel sprawia, że nie
zaliczamy jej do operacji jednostkowych, mimo, że udział zjawisk fizycznych jest tu znaczny.
PROCESY
− spalanie i utlenianie w wysokich temperaturach
− zgazowanie
− reakcje gazowe bez udziału kontaktu
− reakcje gazowe kontaktowe
− reakcje między gazami i cieczami
− zobojętnianie
− podwójna wymiana w roztworach
− podwójna wymiana między fazą stałą i ciekłą
− wymiana jonowa
− prażenie i wypalanie
− redukcja w wysokich temperaturach
− elektroliza
− procesy elektrotermiczne
OPERACJE
2) Przepływ płynów.

2
3) Sedymentacja
4) Filtracja
5) Rozdrabnianie ciał stałych
6) Mieszanie
7) Przewodzenie ciepła
8) Promieniowanie cieplne
9) Ogrzewanie i chłodzenie
10) Wrzenie i kondensacja
11) Przenikanie masy
12) Destylacja
13) Rektyfikacja
14) Absorpcja
15) Ekstrakcja
16)
Suszenie
1. Definicja i elementy składowe chemicznego procesu produkcyjnego
Jest to całokształt czynności technicznych związanych z utrzymaniem zakładu produkcyjnego w
ruchu.
Rozpoczyna się w momencie pobrania surowców z magazynu, a kooczy w momencie przekazania
do magazynu wyrobów gotowych. Jest procesem złożonym.
SKŁADOWE:
- proces technologiczny (czynności przetwórcze i obróbcze)
- transport wewnętrzny
- kontrola techniczna
- magazynowanie
- gospodarka energetyczna i cieplna
- gospodarka wodno-ściekowa
- gospodarka odpadami
2. Schemat ideowy procesu.
Schemat ideowy (blokowy) procesu jest to graficzne przedstawienie procesu technologicznego
polegajacego na zestawieniu poszczególnych procesów i operacji jednostkowych w kolejności ich
realizacji oraz wszystkich występujących strumieni masowych.
Magazynowani
e surowców
3
3. Rodzaje surowców i produktów procesu.
Podzial surowców:
PODSTAWOWE - to materiały, względnie substancje wyjściowe, które są przetwarzane w procesie
technologicznym na produkt finalny (wchodza w skład produktu finalnego)
POMOCNICZE – to materiały, które w prawdzie są konieczne do przeprowadzenia procesu
technologicznego, ale nie wchodza w skład produktu finalnego (po procesie są bardzo często
regenerowane)
Ze względu na ich pochodzenie oraz krotnośd przetwarzania dzieli się je na:
*naturalne – naturalnie występujące w przyrodzie (rudy skalne, sole organiczne)
* pierwotne – to materiały przypisane do danego procesu technologicznego jako materialy wyjściowe i
po raz pierwszy przetwarzane często są to surowce naturalne, mogą byd nimi surowce podstawowe i
pomocnicze
* wtórne – zazwyczaj przetwarzane produkty uboczne lub odpadowe, które są wykorzystywane w
procesie technologicznym jako substytut surowców pierwotnych (recykling)
PRODUKTY:
- materiały, względnie substancje wytwarzane w procesie technologicznym w wyniku przerobu
surowców
* PRODUKT GŁÓWNY ( koocowy, finalny) – jest zasadniczym celem organizacji określonego procesu
technologicznego. Powstaje zawsze w ostatniej fazie złożonego z kilku faz procesu technologicznego
* PRODUKTY POŚREDNIE ( półprodukty) – powstają w trwałej postaci w określonym stadium złożonego z
kilku faz przeróbki procesu technologicznego. Produkt ten może byd wydzielony w ramach tego samego
procesu technologicznego przetwarzany w produkt finalny lub wydzialny i kierowany do innego procesu
technologicznego w celu dalszej przeróbki
* PRODUKT UBOCZNY – wynik procesu technologicznego, ale nie stanowi zasadniczego celu procesu.
Najcześciej wykorzystywany do innych celów (surowiec wtórny lub może służyd do regeneracji surowców
pomocniczych)
*PRODUKTY ODPADOWE – nie mają żadnego zastosowania w procesie macierzystym, w którym powstały
(np. produkt uciążliwy dla środowiska i nie przydatny bez zastosowania dodatkowych zabiegów
przetwórczych) Po przeróbce może on byd wykorzystywany jako surowiec wtórny w innych procesach
technologicznych.
Klasyfikacja: procesy i operacje
procesy chemiczne reakcja
operacje fzyczne absorpcja, adsorpcja
Typy reakcji w procesach:

4
proste
złożone równoległe
złożone następcze
złożone mieszane
Reakcje pomiędzy składnikami
redukcja – utlenianie
kwasowo –zasadowe
Odwracalnośd (fizyczna i chemicza)
obszar kinetyczny
obszar dyfuzyjny
- dyfuzja zewnętrzna
- obszar pośredni – dyfuzja zewnętrzna i dyfuzja wewnętrzna
- dyfuzja wewnętrzna
1. Układ homo- i heterogeniczny procesu, wyjaśnid różnice, podad przykłady.
UKŁAD HOMOGENICZNY
- Składniki jednego rodzaju- gaz (G), ciecz (C), ciało stałe (S)
- Reakcja w układach homogenicznych zachodzi z reguły szybciej niż w układach heterogenicznych
- Układy homogeniczne są prostsze i łatwiejsze do sterowania
- Obserwuje się tendencję, aby składniki mieszaniny reakcyjnej przeprowadzid do jednego stanu, np.
składniki stałe rozpuszcza się lub topi, gazy absorbuje
- Procesy homogeniczne z reguły są kontrolowane przez kinetykę reakcji
UKŁAD HETEROGENICZNY
- układ zawierający składniki w formie dwóch lub więcej faz
- teoretycznie możliwe są następujące fazy
gaz -ciało stałe
gaz –ciecz
ciecz –ciecz
ciecz –ciało stałe
ciało stale –ciało stałe
- z reguły są kontrolowane przez dyfuzję
- w przemyśle mamy częściej doczynienia z procesami heterogenicznymi
* reakcje spalania cieczy i ciał stałych
* rozpuszczanie metali w kwasach i zasadach
2. Modele przepływu strumieni przez aparat, modele przepływu tłokowego i idealnego mieszania.
Dwa modele przepływu strumienia przez aparat
1. Model przepływu tłokowego
- wszystkie elementy strumienia przebywają przez jednakowy czas w aparacie (zwykle rurowym);
występuje wówczas pełne przemieszanie poprzeczne, nie ma natomiast przemieszania wzdłużnego
2. Model idealnego mieszania
5
- strumieo dopływający do aparatu miesza się nieskooczenie szybko z objętością układu znjadującego się
w aparacie z reguły z mieszadłem; skutkiem tego jest jednakowy rozkład stężenia w calej objętości
reaktora
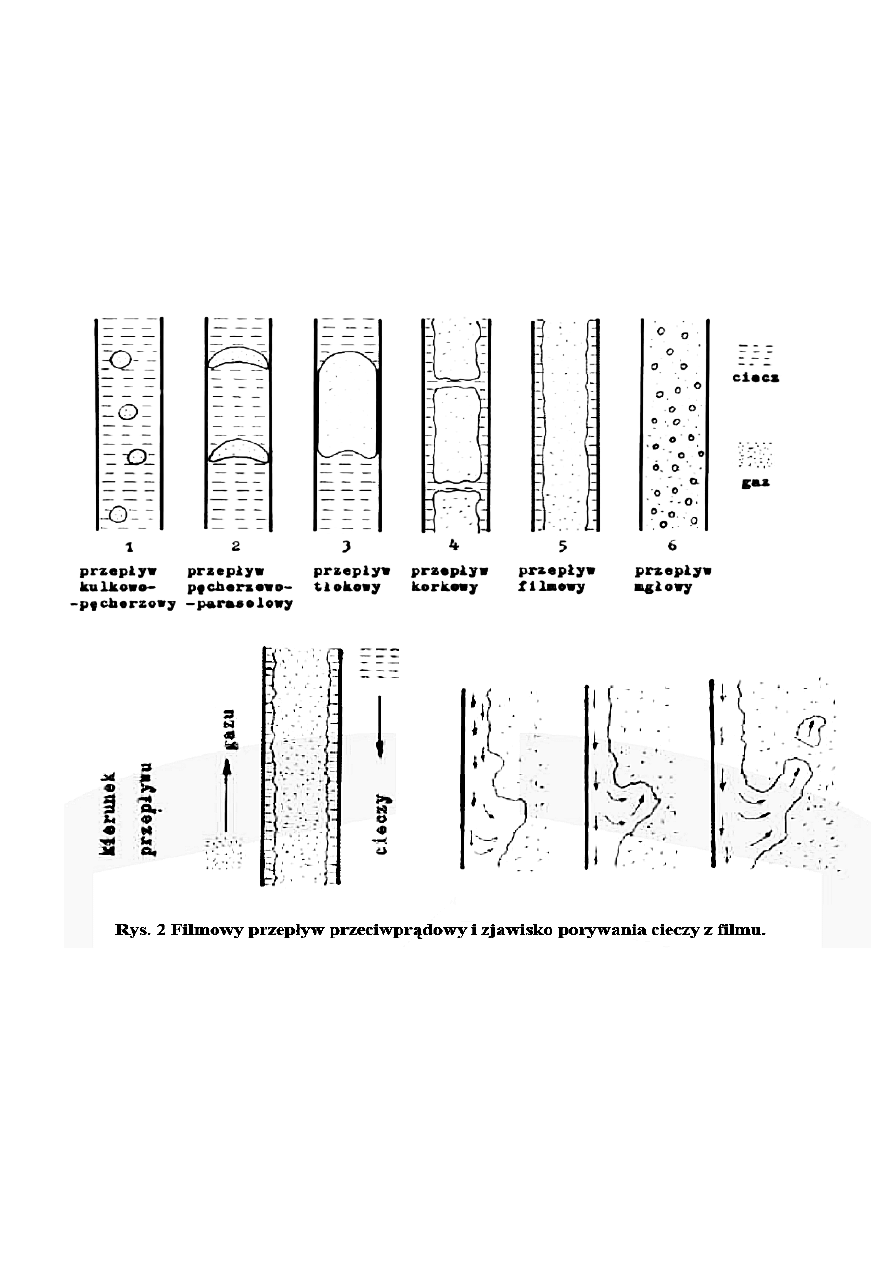
Przepływ gazów przez ciecz w reaktorze
- przepływ kulkowo –pęcherzykowy
- przepływ pęcherzykowo –prasaolowy
- przepływ tłokowy
- przepływ korkowy
- przepływ filmowy
- przepływ mgłowy
nmkkkk
3. Model przepływu gazów przez ciecz w reaktorze pionowym
4. Podział procesów z punktu widzenia efektu termicznego reakcji
- izotermicznie – nadmiar energii usuwamy, a niedomiar energii uzupełniamy
6
- adiabatyczne – całe ciepło reakcji jest oddawane lub odbierane przez mieszaninę reagentów i
produktów; zmiana temperatury mieszaniny reagentów jest funkcją stopnia przemiany
- polytermiczne tj. ze sterowaną temperaturą – energia strumienia jest uzupełniana lub usuwana ze
strumienia w zalezności od potrzeb
Przeważnie mamy do czynienia z systemem politermicznym czasami zbliżonym do izotermicznego lub
adiabatycznego.
11.Objaśnid definicje: wydajnośd produktu, stopieo konwersji.
Wydajnośd produktu – to stosunek masy uzyskiwanego produktu do maksymalnej ilości produktu.
X = G
e
/ G
max
Wydajnośd liczy się w stosunku do najważniejszego i najdroższego surowca:
np. SO
2
+ ½ O
2
= SO
3
składnikiem głównym jest SO
2
Stopieo konwersji – to stosunek ilości zużytego składnika głównego do jego ilości na początku procesu
X = (G
1
-G
2
) / G
1
12. Definicja siły napędowej reakcji
C i innych procesów, absorpcji, adsorpcji i kondensacji.
SIŁA NAPĘDOWA
Siła napędowa reakcji ΔC i innych procesów, absropcji, adsorpcji i kondensacji jest wyrażeniem
stanowiacym różnicę aktualnego stężenia C i stężenia równowagowego C*
ΔC=C-C*
Siła napędowa może byd zwiększona przez zwiększenie stężenia aktualnego, zmniejszenie stężenia
równowagowego lub zmianę obydwu.
Siła napędowa reakcji w układzie współprądowym i przeciwprądowym
WSPÓŁPRĄD
ΔC = C
i
– C
i
*
ΔC = C
f
– C
f
*
PRZECIWPRĄD
ΔC = C
i
–C
f
*

7
ΔC = C
f
–C
i
*
13.
Wymienid i omówid metody zwiększenia szybkości reakcji
A) Zwiększenie stężenia reagentów w surowcu zwiększa proporcjonalnie szybkośd reakcji.
W przypadku ciała stałego mówimy o wzbogaceniu mieszaniny reakcyjnej, w przypadku fazy ciekłej
mówimy o zwiększaniu stężenia.
Najcześciej stosowaną metodą zwiększania szybkości (intensyfikacji) procesu
B) Zwiększanie ciśnienia zwiększa szybkośc reakcji, szybkośd osiągnięcia równowagi reakcji odwracalnych
i zmienia położenie stanu równowagi. Zwiększenie ciśnienia ma istotny wpływ w układzie homogennym
lub heterogennym gaz–ciecz lub gaz-ciało stałe. W fazie ciekłej lub ciekłej i stałej wpływ ciśnienia jest
mniejszy.
Transport gazu do cieczy lub ciała stałego (ab- i adsorpcja, kondensacja) szybkośd reakcji podana jest
funkcją
r =dG/dτ = kFΔP lub r= kFP jeżeli reakcja jest daleka od stanu równowagi,
p – ciśnienie
F – powierzchnia wymiany
K -stała szybkości
Zwiększanie ciśnienia gazu w układzie homogennym gazowym lub heterogennym, w którym gaz bierze
udział w reakcji zwiększenia ciśnienia zmniejsza objętośd fazy gazowej i jest równoważna zwiększeniu
jego stężenia.

8
C) Sterowanie temperaturą procesu w celu zwiększenia siły napędowej.
- Siła napędowa procesu adsorpcji, absorpcji i kondencacji
ΔC= C- C*
C- stężenie składnika A w fazie gazowej
C* - stężenie składnika A nad cieczą w stanie równowagi nad cieczą
Przez obniżenie temperatury fazy ciekłej ciśnienie równowagowe parcjalne składników fazy gazowej
ponad nią tj. C* zostaje obniżone, a siła napędowa ΔC i całkowita szybkośd procesu przejścia gazu do fazy
ciekłej lub stałej „r” ulega zwiększeniu.
- Siła napędowa procesu desorpcji i odparowania
ΔC = C* - C
Przy stałym stężeniu gazu dla tych reakcji równowaga może byd przesunięta, a szybkośd desorpcji gazu z
cieczy zwiększona przez podniesienie temperatury cieczy, przed wejściem do aparatu (wymiennik ciepła)
lub w nim (spaliny, para wodna)
Powoduje to zwiększoną desorpcję lub odparowanie składnika z cieczy lub z adsorbenta.
D) Usuwanie produktów reakcji ze strefy reakcji zwiększa szybkośd reakcji przez zmniejszenie reakcji
odwrotnej r
2
r
c
= r
1
– r
2
lub zwiększenie siły napędowej ΔC= C – C* reakcji heterogenicznej przez zmniejszenie lub redukcję C*
nawet do zera.
Jeżeli reakcja zachodzi w fazie gazowej usunięcie produktu do fazy ciekłej, np. przez ab- lub adsorpcję
powoduje przesunięcia reakcji w prawo.
14. Wymienid i omówid metody zwiększenia stałej szybkości reakcji
r = k
r
(C
A
)
n
A) Podwyższenie temperatury reagującego układu
Podwyższenie temperatury powoduje ostre zwiększenie stałej szybkości reakcji i w mniejszym
stopniu dyfuzyjności.
Podwyższenie temperatury powoduje całkowite zwiększnie szybkości reakcji aż do pewnej
granicy, powyżej której rośnie szybkości reakcji odwrotnej lub reakcji ubocznych (k
2
, k
s1
, k
s2
–są duże).
Wpływ temperatury na stałą szybkości reakcji obrazuje prawo Arrheniusa
K=k
0
exp((-E/RT)
2,3log k
2
/k
1
= E/RT (1/T
1
– 1/T
2
)
Współczynnik β=k
T+t0
/k
T
Według zasady van’t Hoffa współczynnik β ma wartośd pomiędzy 2 a 4.
Obowizuje w zakresie temperatur 10-400ºC i dla energii aktywacyjnej 60-120kJ/mol
Bardziej realne jest to w warunkach procesu kinetycznego niż dyfuzyjnego, chociaż isnieje oczywisty
wpływ temperatury na dyfuzyjnośd.
B) Zastosowanie katalizatora
Zastosowanie katalizatora powoduje czasami gwałtowne zwiększenie stałej szybkości reakcji,
dzięki zmniejszeniu energii aktywacji. Obrazowo katalizator umożliwia zmianę jednego procesu z dużą
energią aktywacji na kilka z małymi energiami aktywacji.
A + B = AB
9
A + [kat] = [A*kat]
B + [A*kat] = AB + [kat]
Katalizatory heterogenne i homogenne
C) Intensyfikację regującego układu
Mieszanie zwiększa współczynnik wymiany masy lub stałą szybkości reakcji przez zmianę dyfuzji
cząsteczkowej dyfuzją konwekcyjną tj. przez zmniejszenie oporów dyfuzji które ograniczają możliwości
kontaktu reagentów.
Celowe jest podwyższanie szybkośdi mieszania aż do uzyskania obszaru kinetycznego procesu.
W układzie homogennym zwiększenie intensywności mieszania pomaga wyrównad stężęnia
reagentów i możliwości kontaktu.
Mieszanie zwiększa powierzchnię pomiędzy reagującymi fazami.
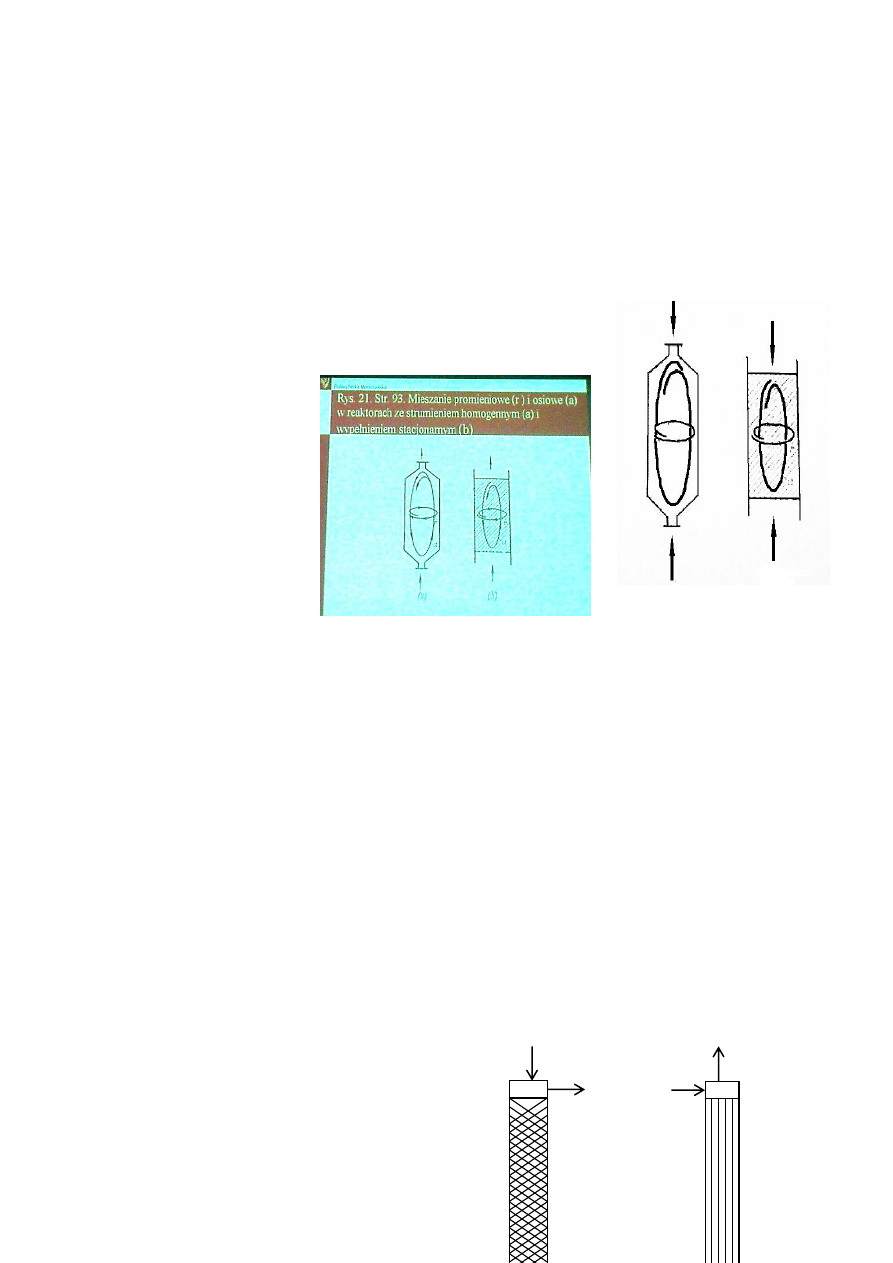
Mieszanie promieniowe (r) i osiowe (a) w
reaktorach ze strumieniem homogennym (a)
i wypełnieniem stacjonarnym (b).
Powierzchnia międzyfazowa jest zwiększana w układzie
heterogennym różnymi metodami w zależności od układu G-C, G-S, C-C, S-S
oraz wymaganych warunków procesu, ciśnienia, temperatury,
stężenia reagentów i obecności katalizatora.
Celem jest zawsze zwiększenie powierzchni najcięższego składnika, przeważnie fazy stałej w
układzie G-S, C-S i cieczy w układzie G-C
W układzie G-C główną metodą stosowaną do utworzenia interfazy jest:
1. Utworzenie cienkiego filmu na wypełnieniu kolumn (wież). Wymiana masy typu filmowego.
2. Rozpylanie fazy ciekłej, mechaniczne lub pneumatyczne w strumeniu gazu (wieże rozpyłowe).
Powierzchnia międzyfazowa to powierzchnia kropelek. Większa intensywnośd niż wież z wypełnieniem
ale mniejsza stabilnośd i rzadziej stosowana w przemyśle.
15. Zasada pracy reaktora dla reakcji heterogennych w układzie gaz – ciecz (przykładowy rysunek)
Reaktory wieżowe, typ filmowy
A – z wypełnieniem
B- z ruramy lub wypełnieniem
płytami równoległymi
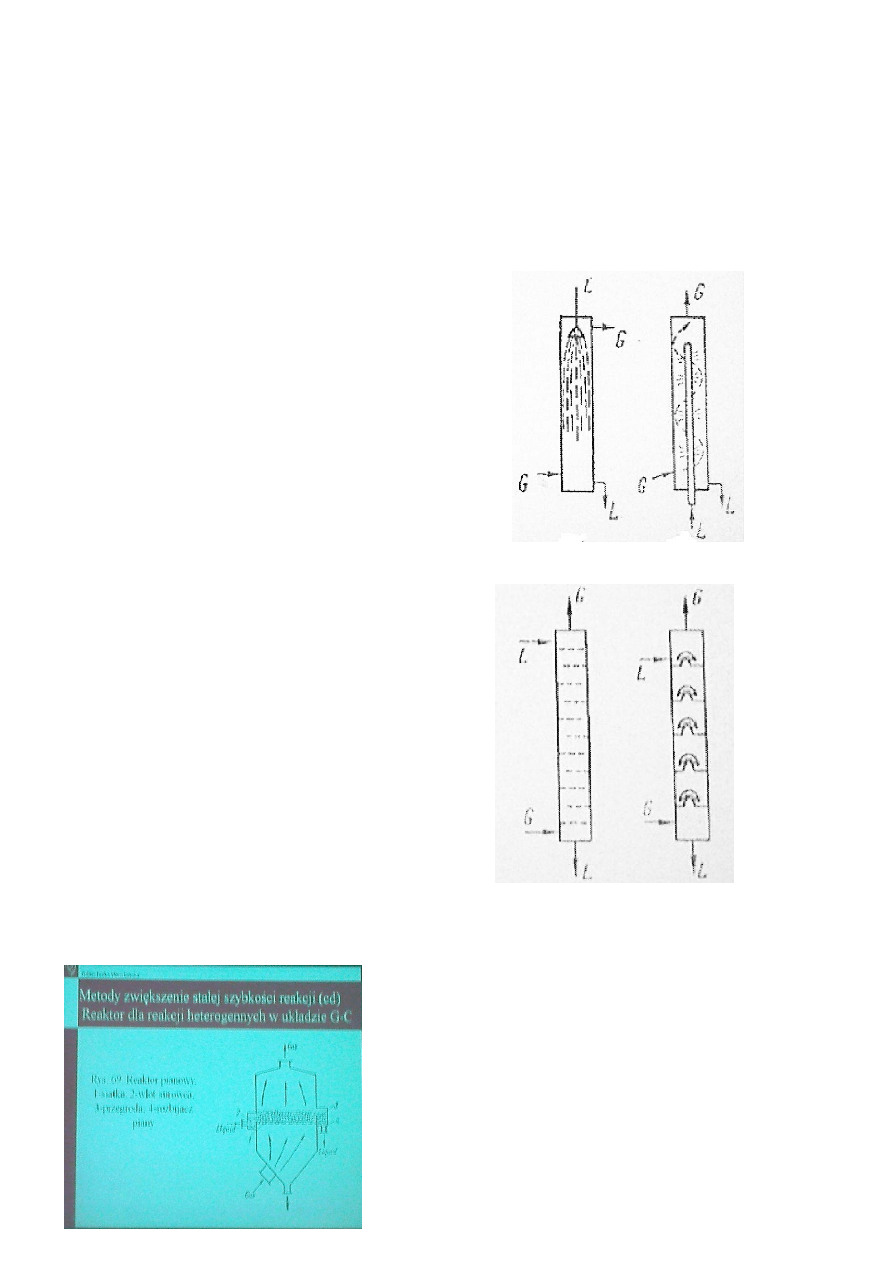
10
(absorpcja, desorpcja, prosta konstrukcja
niezawodne, łatwo sterowalne, małe
zużycie energii)
Reaktory rozpyłowy
A – z wieżą
B – ze skruberem cyklonowym
(absorpcja, desorpcja, polimeryzacja
prosta konstrukcja, mała stabilnośd pracy
łatwo sterowalne, małe zużycie energii)
Półka pęcherzykowa
A – z półkami sitowymi
B – z półkami kołpakowymi
(adsorpcja, desorpcja, prosta konstrukcja
niezawodne, łatwo sterowalne, duże
zużycie energii)
16. Zasada pracy reaktor dla reakcji heterogennych w układzie gaz – ciało stałe (przykładowy rysunek)
W układzie G-S i C-S rozwinięcie powierzchni uzyskuje się
głownie przez rozdrobnienie ciała stałego lub zastosowanie
ciała porowatego. Cztery metody zwiększenia powierzchni
wymiany:
1. Mieszanie dobrze zmielonego ciała stałego mieszadłami
mechanicznymi na tacach aparatu, przez które przepływa gaz
lub ciecz.
2. Mieszanie dobrze zmielonego ciała stałego ze strumieniem
cieczy lub gazu, aparaty komorowe w których drobno

11
zmielony materiał stały jest rozpylany przez dysze i opada w strumieniu gazu z którym reaguje.
Reaktory dla procesów w układzie
heterogennym (C-S) z ciałem stałym
na złożu filtrującym, przepływ tłokowy
adsorpcja, wymiana jonowa, ługowanie
5. Możliwe jest także zastosowanie
mieszania do układu C-C ( nie mieszajace się). Powstaje emulsja, lekka ciecz płynie do góry, cięższa do
dołu. Ciecz o większym napięciu powierzchniowym rozbija się w kuleczki. Możliwe jest mieszanie
mechaniczne lub pneumatyczne.
6. W ukaładzie S-S mieszanie mechaniczne lub pneumatyczne lub przy pomocy bębnów obrotowych. Są
one także stosowane chętnie w innch układach G-S, C-S i G-C.

12
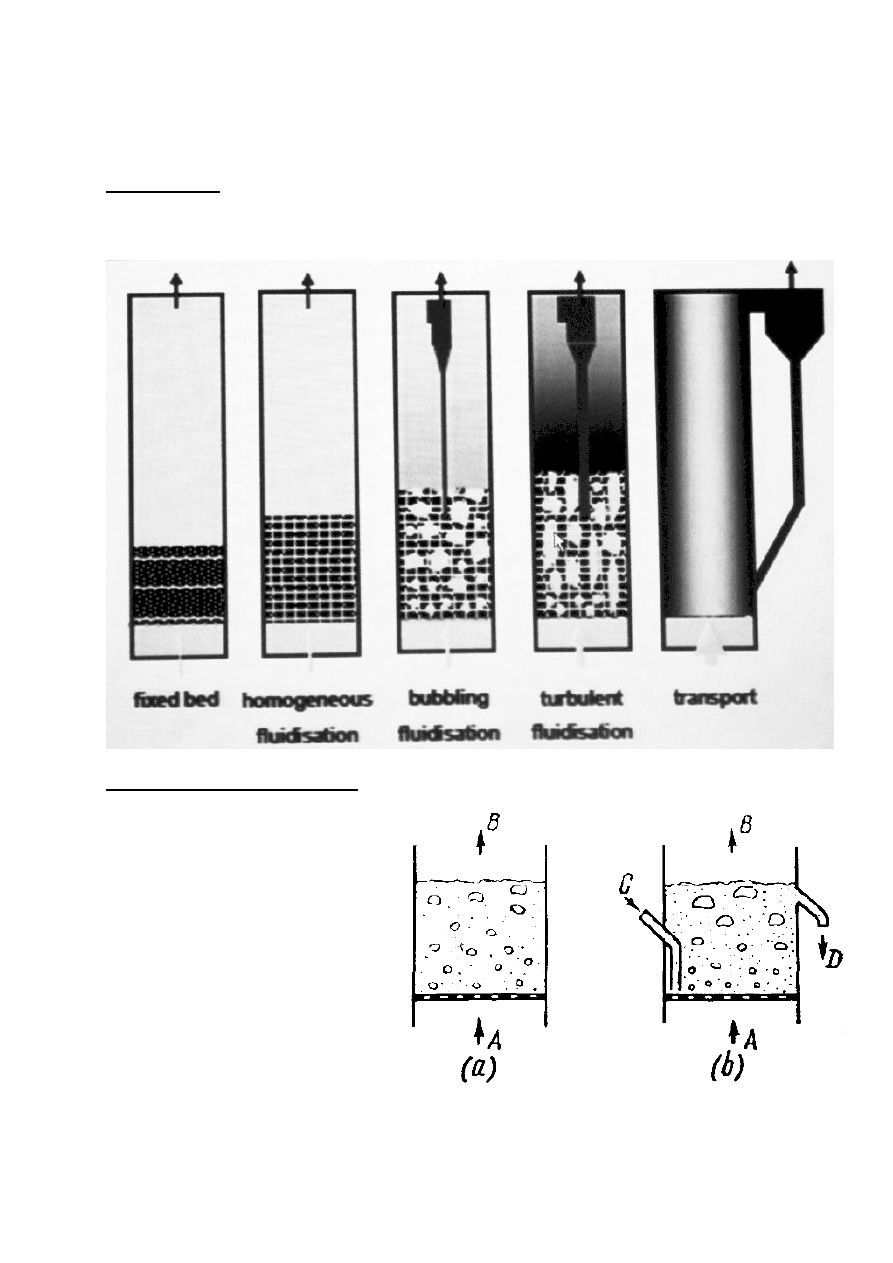
18. Zasada pracy reaktora ze złożem fluidalnym, przykładowy rysunek
Złoże fluidalne:
W czasie przepływu płynu (gaz, ciecz) stanowiacego tzw. fazę ciągłą przez warstwę
materiału stałego (złoze) tworzącą tzw, fazę rozproszoną można w zależności od prędkości
przepływu płynu przez złoże rozróżnid etapy:
Zasada działania złoża fluidalnego
Schemat złoża fluidalnego
a – bez regeneracji katalizatora
b – z regeneracja katalizatora
A –gazy reakcyjne
B – produkt reakcji
C – regenerowany katalizator
D – katalizator do regeneracji

19. Otrzymywanie gaz syntezowego z węgla jako surowca.
GAZ SYNTEZOWY
Gaz syntezowy – mieszanina tlenku węgla i wodoru o różnym stosunku molowym składników
Surowiec główny –materiały zawierające węgiel i wodór: węgiel kamienny, węgiel brunatny,
frakcje ropy naftowej, gaz ziemny, biomasa i inne
Czynniki gazyfikujące – tlen i para wodna
Podstawowe reakcje gazyfikacji węgla
Reakcje utleniania węgla
C + O
2
=> CO
2
ΔH= -395kJ
2C + O
2
=> 2CO ΔH= -219 kJ
Reakcje węgla z dwutlenkiem węgla i parą wodną.
C + CO
2
=> 2CO ΔH = 176 kJ
C + H
2
O => 2CO + H
2
ΔH= 133 kJ
20. Skład orientacyjny gazu syntezowego i kierunki jego zastosowania
Surowy gaz syntezowy: tlenek węgla i wodór (>80% obj. H
2
:CO = 0,6:0,7) i dwutlenek węgla (i
H
2
S i COS)
Zastosowanie:
Surowiec do syntez chemicznych
Gaz o małej wartości opałowej dla celów energetycznych
Gaz o właściwościach redukcyjnych, hutnictwo i przemysł maszynowy
Surowiec do wytwarzania syntetycznego gazu metanowego (metanizacja)
18. Procesy techniczne zgazowania węgla w kierunku wytwarzania gazu syntezowego
Zgazowanie prowadzi się w reaktorach zwanych dawniej generatorami obecnie
zgazowywaczami. Skłąd gazu syntezowego zależy od składu chemicznego węgla.
Koppers-Totzek
Przebieg procesu Koppers-Totzek
- Mielenie i suszenie węgla gazmi spalinowymi 90μm 2% wilgotność
- Transport pneumatyczny w strumieniu azotu
- Rozdział powietrza na tlen i azot. Zużycie tlenu 200 do 550 Nm
3
/tonę węgla
(duży udział w kosztach zgazowania)
- Zgazowanie, ciśnienie atmosferyczne, temp. 1500-1900ºC
- Chlodzenie dostrzykiem wody (wypłukiwanie popiołu)
- Elektrofiltry – usuwanie popiołu ze strumienia
gazu

Przykładowe dane charakteryzujące proces zgazowania węgla kamiennego i brunatnego
metodą
Koppersa-Totzka (wg materiałów informacyjnych tej firmy)
18. Reakcje konwersji metanu do gazu syntezowego.
Konwersja metanu z parą wodną
CH
4
+ H
2
O CO + 3H
2
ΔH = 206kJ
Rekacja silnie endotermiczna
Lub konwersja utleniajaca węglowodorów z parą wodną + tlenem
(półspalanie w reakcji z tlenem)
CH
4
+ ½ O
2
CO + 2H
2
ΔH= -35,7kJ
CH4 + H2O CO + 3H2 ΔH= 206 kJ
C + H2O CO + H2
Kompensacja energetyczna
Reakcje z parą wodna
CH4 + H2O CO + 3H2 ΔH= 206 kJ
(reakcja silnie endotermiczna)
19.
Oczyszczanie gazu ziemnego przed konwersją z parą wodną.
Reakcje hydrogenolizy związków siarki (5 do 30mg/m
3
) na katalizatorze CoMo/Al
2
O
3
CS
2
+ 4H
2
=> 2H
2
S
+ CH
4
RSH + H
2
=> H
2
S + RH
COS + 4H
2
=> H
2
S + CH
4
+ H
2
O
ZnO + H
2
S => ZnS + H
2
O
20. Sterowanie składem gazu syntezowego (otrzymanym z konwersji węgla i metanu z parą
wodną)
21. Zasada produkcji wodoru z gazu syntezowego
1. Oczyszczanie, usuwanie – związków siarki i ditlenku węgla (metanol-Rectisol),
metanoloaminy
2. Konwersja tlenku węgla, metanizacja
Cel: wytworzenie wodoru do produkcji amoniaku, procesów uwodnienia w rafineriach,
uwodornienie tłuszczy
18.
Wpływ parametrów procesu, stosunku para H
2
O: metan, ciśnienia i temperatury na
stopieo przereagowania metanu z parą wodną
Wpływ stosunku para H
2
O : metan na stopieo przereagowania metanu z parą wodną
Reakcja CH
4
+ H
2
O CO + 3H
2

Wpływ temepratury procesu na skład gazu
Równowagowy skład gazu przy konwersji metanu z parą wodną w funkcji temperatury
p = 3Mpa H
2
O : CH
4
= 4 : 1
19. Reakcje syntezy metanolu z gazu syntezowego, parametry katalizator
CO + 2H
2
CH
3
OH ΔH = -90,73kJ
CO
2
+ 2H
2
CH
3
OH + H
2
O ΔH = -49,53kJ
Reakcja egzotermiczna, stała równowagi silnie przesunięta w prawo w temperaturze 300ºC
ale zachodzi powoli
Zastosowanie katalizatora CuO-ZnO-Al2O3 pozwala na przsypieszenie reakcji w temperaturze
poniżej 300ºC, przy ciśnieniu rzędu powyżej 5 Mpa
Gazy inertne: azot, argon, metan do 15%obj, w gazie syntezowym
20. Selektywnośd reakcji syntezy metanolu, reakcje uboczne
CO + 3H
2
CH
4
+ H
2
O ΔH = -209kJ
2CO + 2H
2
CH
4
+ CO
2
ΔH = -252kJ
2CO CO
2
+ C
CO + H
2
CH
2
O ΔH = -8,4kJ
2CH
3
OH CH
3
OCH
3
+ H
2
O ΔH = -209kJ
2CH
3
OH + nCO + 2nH
2
CH-(CH
2
)
n
OH + nH
2
O
2CH
3
OH + H
2
CH
4
+ H
2
O
Duża selesktywnośc katalizatora umożliwia ograniczenie reakcji ubocznych tj. głównie
reakcji metanizacji CO i CO
2
eteryfikacji
Selektywnośd powyżej 95%
Skład gazu syntezowego wproadzany do procesu zbliżony do stechiometrycznego jeżeli
zachodzi korelacja
(H
2
+CO
2
) – (CO+CO
2
)= 2,05-2,15
Katalizator ZnO + Cr
2
O
3
stosunek CO/H
2
= 14 do 18
18. Reakcje elektrodowe w procesie elektrolizy roztworu chlorku sodu (katoda i anoda)
Reakcje elektrodowe w procesie elektrolizy roztworów NaCl
Elektroda
REakcje
Potencjał
φº (V)
Katoda
Na
+
+ e
-
Na
H
2
O + e
-
OH
-
+ ½ H
2
-2,714
-0,415
Anoda
H
2
O – 2e
-
2H
+
+ ½ O
2
2Cl
-
- e
-
Cl
2
+0,815
+1,359
19.
Napięcie i nadnapięcie rozkładowe elektrolitu.
Różnica potencjałów na eletrodach, przy którym zaczyna się rozkład elektrolitu
W warunkach rzeczywistych (przemysłowych) potrzebne napięcie rozkładowe jest
kilkadziesiat procent wyższe niż teoretyczne napięcie rozkładowe.

Wartosc nadnapięcia jest funkcją
* rodzaju materiału elektrod
* stanu ich powierzchni
* temperatury ( zmniejsza się z jej wzrostem)
* stężenia elektrolitu
* intensywności procesu
20. Wpływ stężenia (chlorku sodu) na potencjał rozkładowy elektrolitu
φ
sc
= φº - (RT/zF) lnC
φ
sc
- rzeczywisty potencjał rozładowania
φº - standardowy potencjał rozładowania
R – stała gazowa
T – temperatura
F – stała Faradaya (96 500 coulomb)
z – ładunek jonu
c – stężenie soli
Zwiększenie stężenia soli NaCl zmniejsza potencjal rozładowania chloru
21. Reakcje uboczne w procesie elektrolizy chlorku sodu
Reakcje uboczne
Cl
2
+ H
2
O HOCl + HCl
W przypadku kontaktu jonów OH
-
z anodą lub mechanicznego mieszania produktów anody i
katody są neutralizowane przez alkalia
HOCl + NaOH NaOCl + H
2
O
HCl + NaOH NaCl + H
2
O
Utlenianie: ClO
-
ClO
3
-
+ H
2
O (stad membrana azbestowa)
22. Schemat ideowy elektrolizera
Przepona separacyjna
Przestrzenie katodowe i anodowa
powinny byd oddzielone przeponą gdyż na
anodzie wydziela się chlor, który
wchodziłby z NaOH w niepożądaną
reakcję uboczną:
2NaOH + Cl
2
NaOCl + NaCl + H
2
O
Zasada pracy elektrolizera roztwór NaCl z
przeponą separacyjną (metoda
przeponowa)

23. Przebieg procesu elektrolizy chlorku sodu w zależności od rodzaju materiału katody
Procesy przemysłowe
Proces prowadzi się w urządzeniu zwanym elektrolizerem, wypełnionym elektrolitem w
którym za nużone są elektrody
Surowiec – wodny roztwór chlorku sodu (około 315g/l)
Elektrody:
Anoda – grafit (zawsze)
Katoda – metaliczna (żelaza- stała lub rtęd – ciekła)
Rodzaje katod:
Katoda żelazna – wydzielid się może sód lub wodór ale ze względu na znacznie
bardziej elektroujemny potencjał rozkładowy sodu wydziela się tylko wodór
Katoda rtęciowa – potencjał rozkładowy jonów sodu wynosi tu praktycznie ok. -0,9V
(rozpuszczalnośc sodu w rtęci i tworzenie amalgamatu). Nadnapięcie wodoru na rtęci jest
duże, wydziela się zatem sód.
24. Operacje jednostkowe w procesie elektrolizy chlorku sodu, przygotowanie surowca i
przeróbka produktów
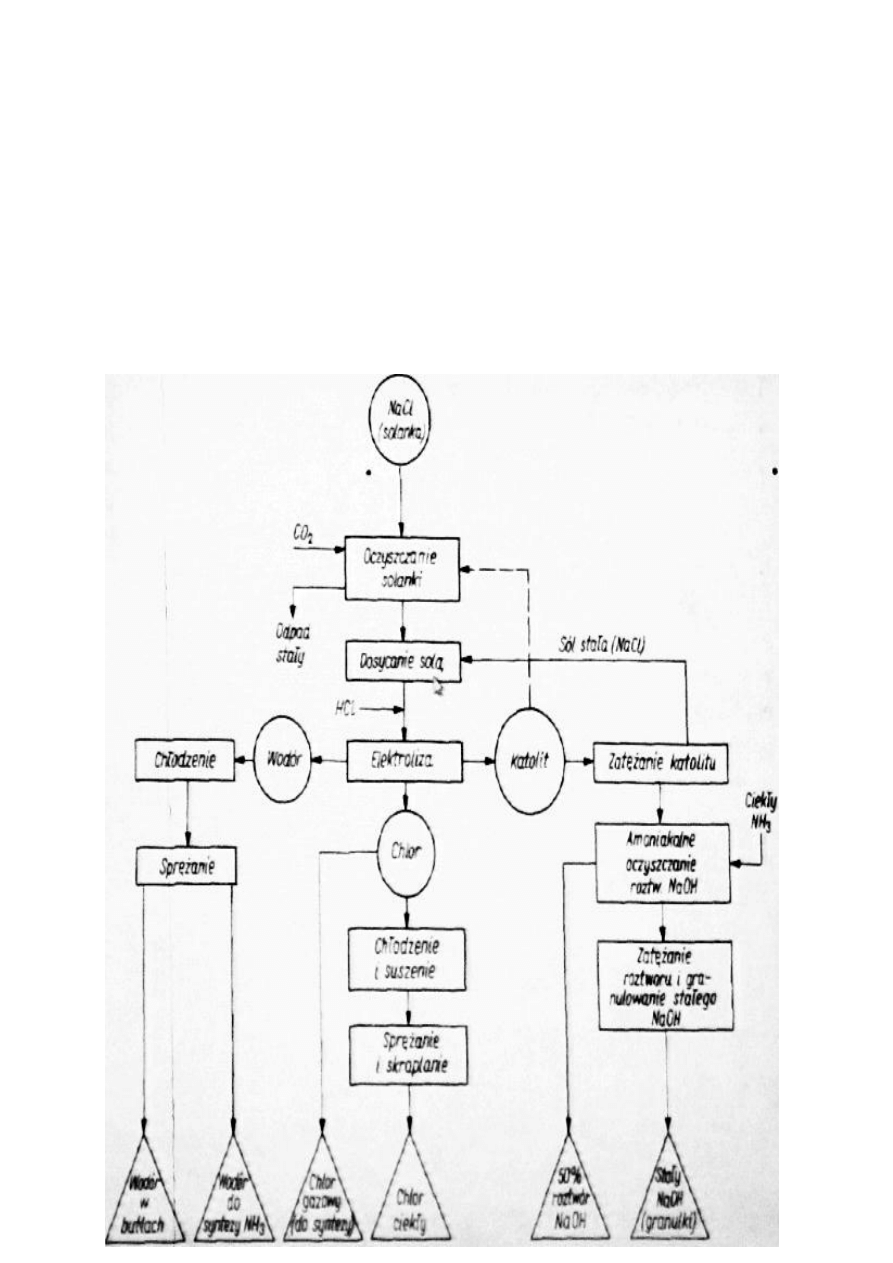
Schemat produkcji chloru
Korpus elektrolizera – betonowy
Płyty grafitowe – umocowane pionowo
Pomiedzy płytami grafitowymi klatki stalowe przymocowane do ścian elektrolizera.
Na siatkach płyty azbestowe.
Jony sodowe przenikaja do komory katodowej.
Wodór z przestrzeni katodowej.
Chlor z przestrzeni anodowej.
Modernizacja zamiarst grafitu – anody metalowe pokryte platyną
Parametry porcesu:
Temperatura – 94-97ºC
Przepływ prądu przez komorę – 25000-50000A
Napięcie – 2,4-3,75V
Proces z katodą rtęciową Anoda 2Cl
-
- 2e Cl
Katoda Na
+
+ e Na
Na + Hg
n
NaHg (amalgamat rtęciowy
NaHg
n
+ H
2
O NaOH + 1/2H
2
+ nHg
Napięcie - 4,6 -4,7V
Natęzenie - 150 000 – 300 000A
Aparatura:
Eletrolizer (reakcje elektrodowe)
Rozkład solanki od stężenia 15 do 260 g/dm
3
Zatężanie solanki i powrót do procesu
Urządzenie do dekompozycji amalgamatu
Zalety procesu: bardzo czysty NaOH
Wada procesu: wieksze zużycie energii

Osuszanie i upłynnianie chloru.
Osuszanie:
- Schładzanie
-Środki odwadniające, np. stężony kwas siarkowy
Upłynnianie:
- Sprężanie do ciśnienia 10-12atm.
- Schłodzenie do temperatury -50ºC
- Spreżenie do ciśnienia 3-6 atm. I schłodzenie do -5 ÷ -25ºC
Schemat produkcji chloru
- Solanka z kopalni 310-315 g/dm
3
, 55ºC
- Zanieczyszczenia – jony Mg
2+
, Ca
2+
, SO
4
2-
- Oczyszczanie przez zawrót katolitu i węglanem sodu (Nierozpuszczalne osady węglanów i
siarczanu Ca i Mg i wodorotlenki Fe)
- Filtracja
- Zakwaszanie (pH = 4-5)
- Konieczna separacja NaCl z katolitu
- Zatężanie roztworu powoduje zmniejszenie rozpuszczalności NaCl w roztworze NaOH,
odwirowanie
- z 99% r-ru NaOH ekstrahuje się NaCl i inne zanieczyszczenia ciekłym amoniakiem.
Zatężanie wodorotlenku sodu.
Separacja NaCl (wirówki): rozpuszczalnośd NaCl zmniejsza się ze stężeniem NaOH
25. Charakterystyka zjawiska przepływu płynu przez łoże stałe
Złoże fluidalne:
W czasie przepływu płynu (gaz, ciecz) stanowiacego tzw. fazę ciągłą przez warstwę
materiału stałego (złoze) tworzącą tzw, fazę rozproszoną można w zależności od prędkości
przepływu płynu przez złoże rozróżnid etapy:

26. Przykłady złóż fluidalnych, zalety złoża fluidalnego
* warstwa nieruchoma – przy niewielkich prędkościach, warstwa materialu sypkiego
pozostaje nieruchoma względem siebie i ścian aparatu, ze wzrostem predkości przeplywu
rośnie spadek cisniania na złożu
* warstwa fluidalna ( wrząca, pseudopłynna) – przy wzroście prędkości przepływu do
pewnej granicznej wartości zwanej dolną krytyczną prędkością fluidyzacji (wk1) złoże
ulega niewielkiej ekspansji
* transport pneumatyczny - gdy prękośc płynu wzrośnie do tzw. górnej krytycznej
prędkości fluidyzacji (wk2) cząstki tworzące złoże zostają porywane z aparatu.
38.Zalety złoża fluidalnego:
Fluidyzacja może zwiększyd efektywnośd procesu ponieważ:
- umożliwia uciąglenie procesu
- możliwe jest dodawanie i odbieranie ciała stałego i utrzymanie w ten sposób aktywności
katalizatora
- kontrolowanie zanieczyszczeo reaktora
- unikanie operacji zamykania instalacji na czyszczenie reaktora
- umożliwia kontrolowanie szybkości transportu ciepła
- w porównaniu ze zlożem stałym znacznie poprawione zostają współczynniki wymiany ciepła
- umożliwia zastosowanie drobnych cząstek i utrzymanie spadku cisnienia na racjonalnym
poziomie
- umożliwia ograniczenie oporów dyfuzji wewnętrznej kiedy szybkośc reakcji jest
wysoka lub kiedy wielkie molekuły biora udział w procesie
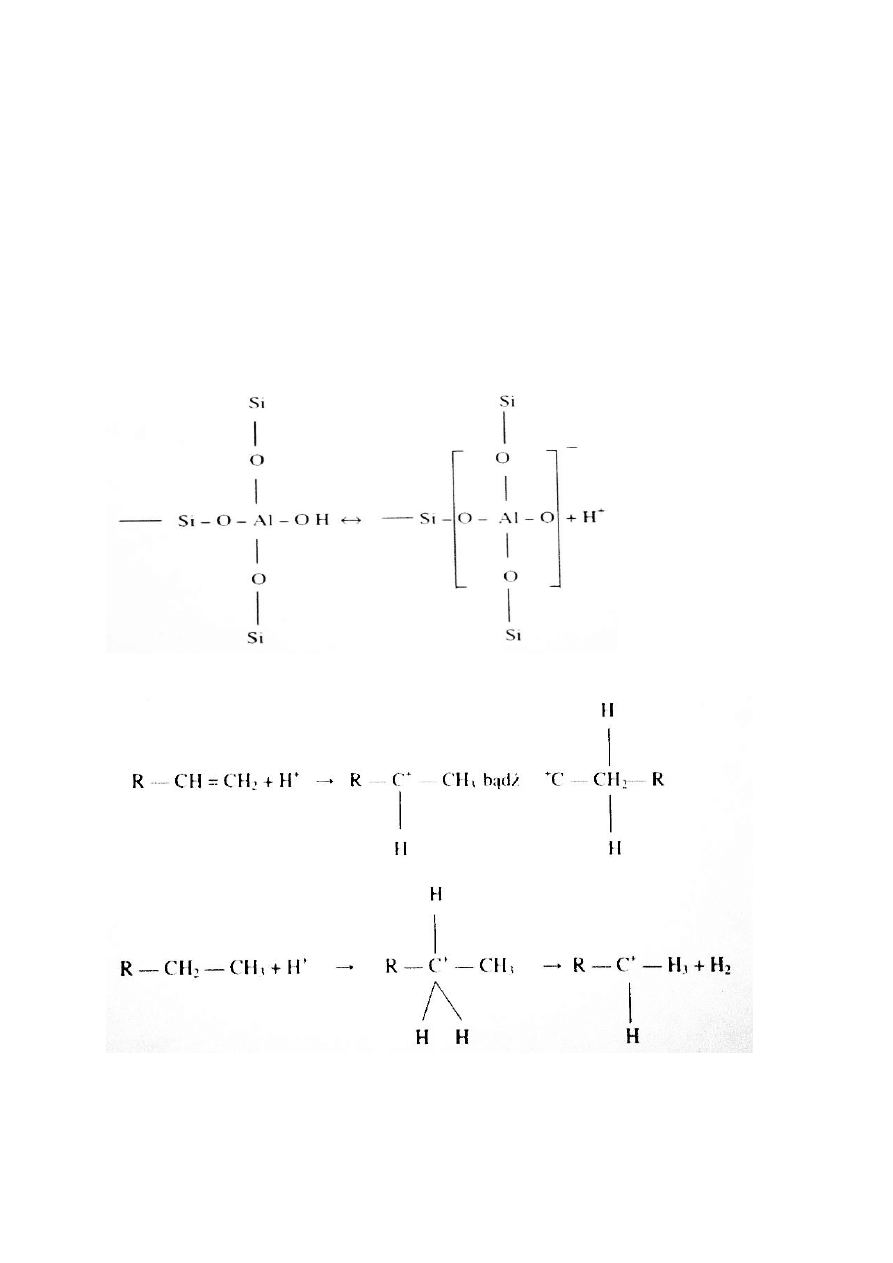
38.Cele procesu krakingu katalitycznego fluidalnego
* W fluidalnym krakingu katalitycznym zachodzi konwersja cieżkich frakcji naftowych do
benzyn, olejów napędowych.
* Cel procesu: zwiększenie ilości benzyn uzyskiwanych z ropy naftowej, przetworzenie
ciężkich frakcji ropy naftowej
* 40-50% ropy przetwarza się tą metodą na benzyny.
39. Kwasowośd katalizatorów krakingu
Wysoka kwasowośd glinokrzemianów wynika z faktu, że po oddaniu protonu ładunek ujemny
jest zdelokalizowany na tetraedrze [ AlO
4
]
Mechanizm procesu, tworzenie jonu karbeniowego:
40. Mechanizm krakingu katalitycznego, główne reakcje
Rekacja główna: pękanie, rozpad (crack) na mniejsze, gaz benzyny i frakcje olejowe
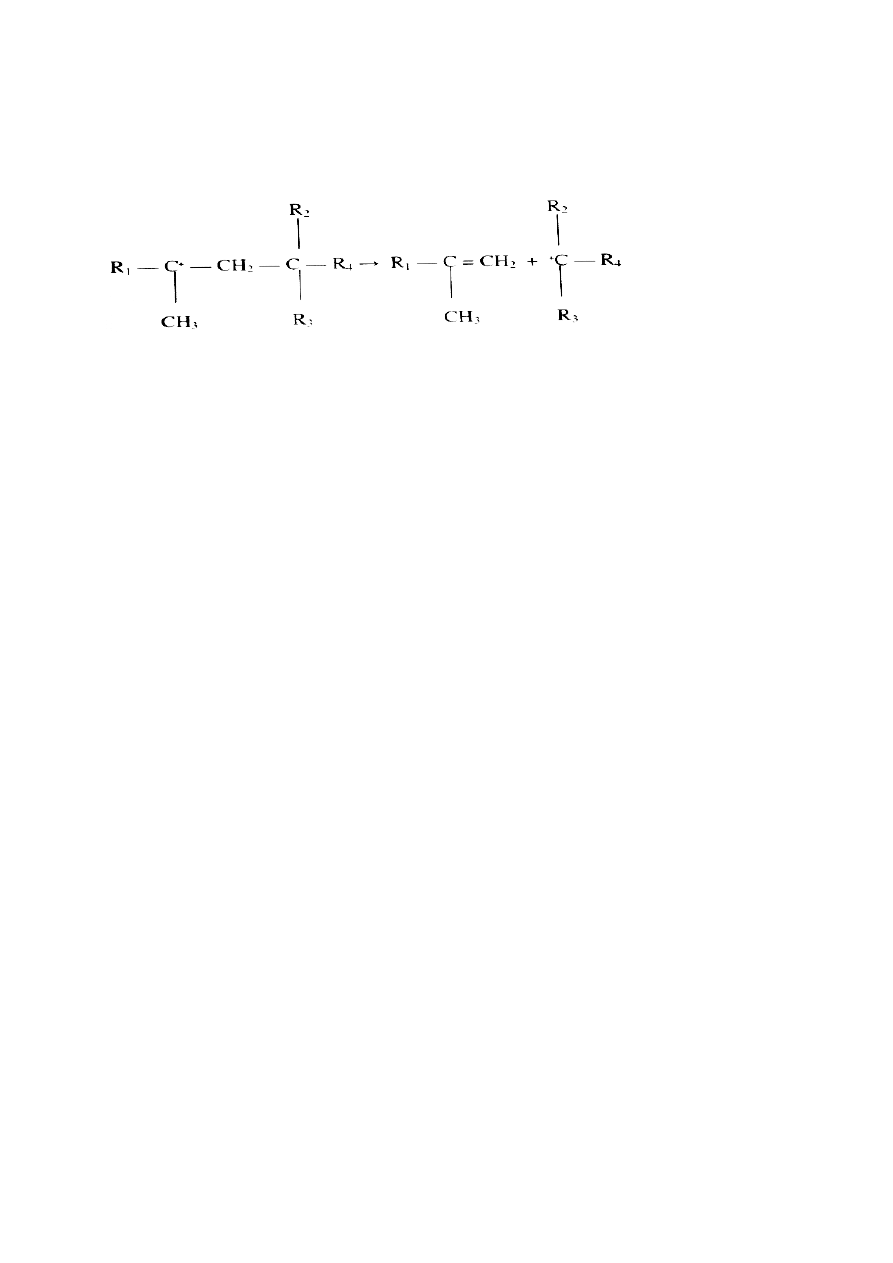
Rozerwanie wiązao z wytworzeniem olefin i parafin, np.:
C
16
H
34 (stały)
C
8
H
18 (ciekły)
+ C
8
H
16 (ciekły)
Główną reakcją jonu karbeniowego jest jego rozpad połączony z pękaniem wiązao C-C
W wyniku reakcji powstają mniejsze karbokationy oraz olefiny według następujących
prawideł:
* łaocuchy krótsze niż C
7
są krakowane w niewielkim stopniu lub wcale
* zrywanie wiązania zachodzi w pozycji β do umiejscowienia ładunku „+”
* powstają 1-alkeny
41. Podstawowe produkty procesu krakingu katalitycznego
* Benzyna z krakingu katalitycznego (w porównaniu z termicznym bez katalizatora) zawiera
znacznie mniej węglowodorów olefinowych (ponizej 10%) i więcej aromatycznych (5razy)
oraz znacznie więcej izoparafin.
* Benzyna z KK charakteryzuje duża liczbą oktanową (78-80)
* Frakcja gazowa zawiera duże ilości C
3
i C
4
, surowiec dla przemysłu petrochemicznego,
stosunkowo małe ilości metanu i etanu.
42.
Podstawowe operacje w procesie fluidalnego krakingu katalitycznego.
* Katalizator w reaktorze i regeneratorze w fazie fluidalnej
* Cyklony do oddzielania katalizatora od fazy gazowej
* Wężownice do odbioru ciepła spalania w regeneratorze
43. Opis procesu kraking katalityczny, przebieg procesu
1. Surowiec o temperaturze 350-500ºC podgrzewa się w wymiennikach 8 i piecu 7 do
temperatury pieca 400ºC, surowiec częściowo odparowuje.
2.
Do strumienia dodatkowo wprowadza się wodę i pozostałośd z kolumny
rektyfikacyjnej (6) i przez 11b zregenerowany katalizator krakingu o temp. 550-600ºC
w rurze transportowej podgrzewa surowiec do 440-450ºC.
3. Reakcja krakingu w fazie fluidalnej, gazowej.
4.
Produkty reakcji z parą wodną przez baterię cyklonów kierowane są do kolumny
rekatyfikacyjnej (6). Pył katalizatora z cyklonów zawracany jest do procesu.
5. Zakoksowany katalizator przez przeparnik 2 trafia do regeneratora 3, węglowodory do
reaktory
6.
Kontrolowane wypalanie koksu na katalizatorze, 570-590ºC, powietrze dmuchawą 5,
w złożu fluidalnym zostaje 0,2-0,3% koksu. Para wodna z wężownic trafia do procesu.
7.
Katalizator trafia do stojaka zasypowego 11 skad jest pobierany do procesu

REŻIM TECHNOLOGICZNY PROCESU
Pojęcie reżimu technologicznego – optymalne parametry procesu
- temperatura T
-
ciśnienie p
-
stosowany katalizator, rodzaj, ilość, aktywność
-
stężenie reagujących substratów
-
sposób i intensywność mieszania
- inne zmienne specyficzne
np. w procesach elektrochemicznych:
-
napięcie prądu
-
natężenie prądu
-
gęstość prądu
Znaczenie procesu technologicznego determinują konstrukcje (rodzaj) stosowanego
reaktora
* Parametry określane terminem reżimu technologicznego korespondują z terminem
maksymalnej wydajności aparatu.
* Parametry procesu są wzajemnie powiązane ze sobą
* Zmiana jednego parametru powoduje zmianę innego parametru
* Sposób i intensywność mieszania ma silny wpływ na konstrukcję rekatora, która w
dużym stopniu zależy od fazy składników mieszaniny reakcyjnej
Wyszukiwarka
Podobne podstrony:
ppj pytania ogol
ppj - wypisywanie liczb pierwszych w javie, Do nauki, Pytania, rozwiązania, prace
REAKCJA ALKILOWANIA IV-RZĘDOWYCH SOLI AMONIOWYCH, Uczelnia PWR Technologia Chemiczna, Semestr 5,
SPRAWOZDANIE ĆW 7 PPJ
PPJ Ćwiczenie 6
ppj pytania z egzaminow
ćw6 ppj sprawko
PPJ ćw 4
membrany sprawozdanie ppj membrany
skradzione ppj
SPRAWOZDANIE ĆW 5 PPJ
Zerówka PPJ
PPJ PYTANIA TERMIN 0(1), PWr W3, semestr 5, PPJ
ISO PPJ, Prace dyplomowe i magisterskie, praca dyplomowa, materiały z internetu, iso 9000
Procesy jednostkowe - pytania zbiorcze, PPJ egzamin
PPj opracowania(1)
Procesy jednostkowe - pytania zbiorcze (2), PWr W3, semestr 5, PPJ
SPRAWOZDANIE ĆW 6 PPJ
więcej podobnych podstron