1
Energia słoneczna (paliwa)
Reakcję fotochemiczną, prowadzącą do powstania paliwa można zapisać następująco:
C
F
B
A
h
kat
+
→
←
+
ν
,
(1)
A+B – reagenty;
F – paliwo (najczęściej wodór);
C – inne produkty reakcji (najczęściej tlen).
Warunki, jakie muszą być spełnione przy generowaniu paliwa z użyciem energii słonecznej:
•
Reakcja musi być endotermiczna;
•
Po absorpcji światła przez fotosensybilizator muszą następować reakcje termiczne
bądź fotochemiczne prowadzące do zajścia reakcji (1), a jednocześnie odtwarzające
fotosensybilizator i substancje pośredniczące w reakcji;
•
Reakcja odwrotna, która prowadzi do nieodwracalnej degradacji reagentów musi być
wyeliminowana;
•
Reakcja odwrotna musi być bardzo powolna w warunkach atmosferycznych, a szybka
w warunkach katalitycznych;
•
Reakcja (1) musi następować pod wpływem światła w jego szerokim zakresie
spektralnym;
•
Wydajność kwantowa produkcji paliwa F musi być jak najwyższa;
•
Paliwo powinno być łatwe do magazynowania i transportu;
•
Reagenty powinny być tanie i nietoksyczne.
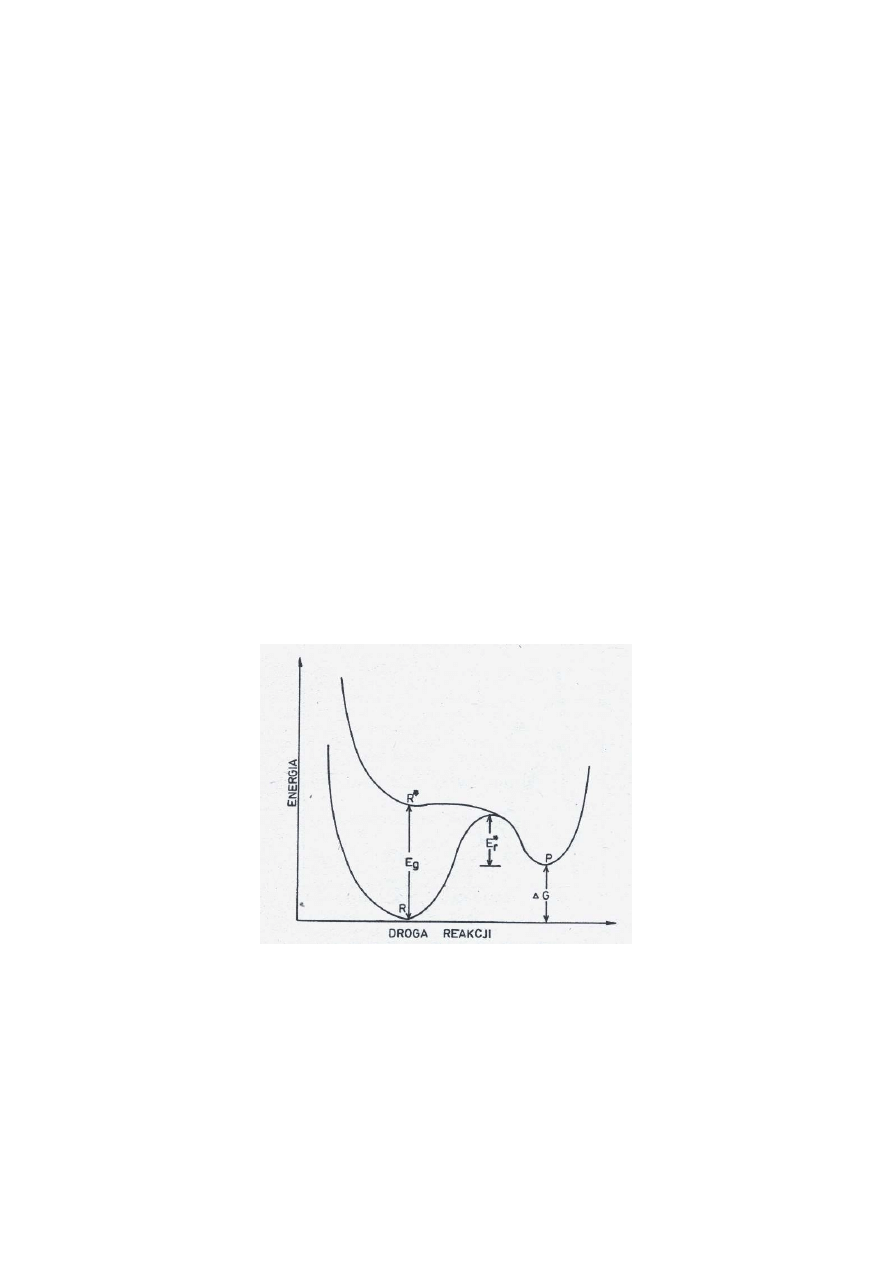
Generowanie paliwa przy użyciu energii słonecznej przebiega w jednym lub kilku etapach
endotermicznych.
•
Fotony o energii niższej niż szerokość pasma zabronionego E
g
nie mogą być
zaabsorbowane i użyte w procesie fotochemicznym;
•
Fotony o energii wyższej od E
g
są absorbowane, a wzbudzony reagent, R* z wyższych
poziomów wibracyjnych bardzo szybko przechodzi na poziom E
g
oddając nadmiarową
energię do otoczenia w postaci ciepła;
•
Przejście od R* do P musi być egzotermiczne w celu stworzenia bariery
energetycznej(E*
r
) dla reakcji odwrotnej do (1).
2
Rozważania termodynamiczne i kinetyczne pozwalają oszacować, że maksymalna wartość
wydajności procesu magazynowania energii słonecznej w procesie fotochemicznym wynosi
12 – 13%. Jest to duża wartość w porównaniu z wydajnością fotosyntezy, przy idealnych
warunkach wynoszącą 6% dla wodorostów i 3,2% dla roślin uprawnych. Większym
problemem jest warunek, aby fotosensybilizator pracował z bardzo dużą liczbą cykli (nie
wchodził w reakcje uboczne prowadzące do jego dezaktywacji). Dla chlorofilu liczba cykli
wynosi 10
5
, a dla fotosensybilizatorów 50 – 100.
Schemat procesów prowadzących do separacji ładunku:
*
+
+
→
S
S
h
ν
'
0
*
ν
h
S
S
k
+
→
+
+
[
]
+
+
→
←
+
+
⋅⋅
⋅
+
−
2
*
2
*
MV
S
MV
S
d
k
d
k
[
]
•
+
+
+
+
+
→
⋅⋅
⋅
MV
S
MV
S
et
k
2
2
*
+
+
•
+
+
+
→
+
2
2
MV
S
MV
S
bc
k
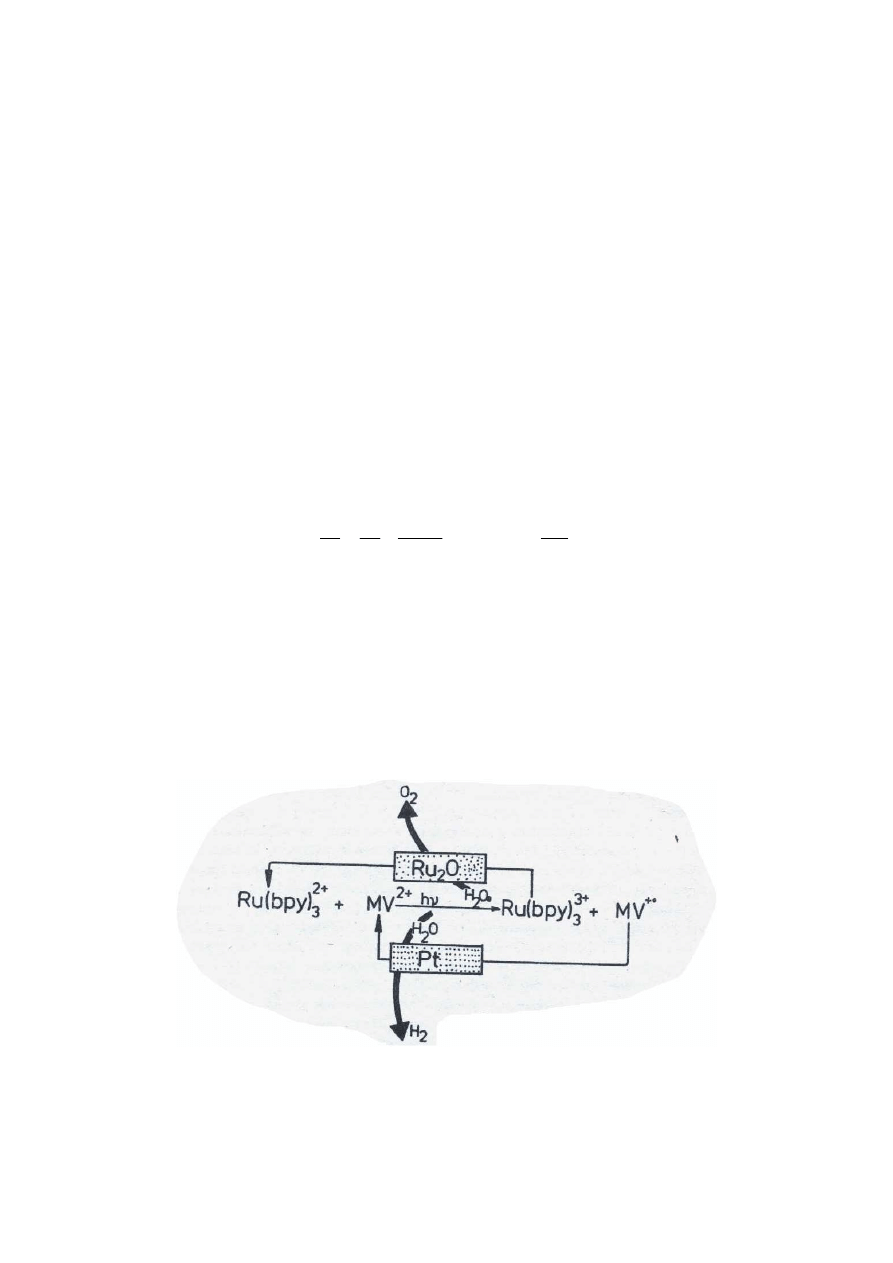
S – fotosensybilizator;
MV – metylowiologen – akceptor elektronów
et
d
d
q
k
K
k
k
1
1
1
+
=
d
d
d
k
k
K
−
=
.
Jako fotouczulacze można stosować:
•
Porfiryny
•
Chloryny
•
Związki kompleksowe rutenu
•
Ftalocyjaniny
Związek ofiarny – związek ulegający nieodwracalnym zmianom kosztem którego powstaje
paliwo, np. EDTA.
Układ do cyklicznego, fotochemicznego rozkładu wody na tlen i wodór:
Nie udało się znaleźć efektywnej metody chemicznej konwersji energii słonecznej na paliwo.
Wynika to z małej liczby cykli. Znane są natomiast metody fizyczne polegające na
generowaniu prądu za pomocą światła.
1
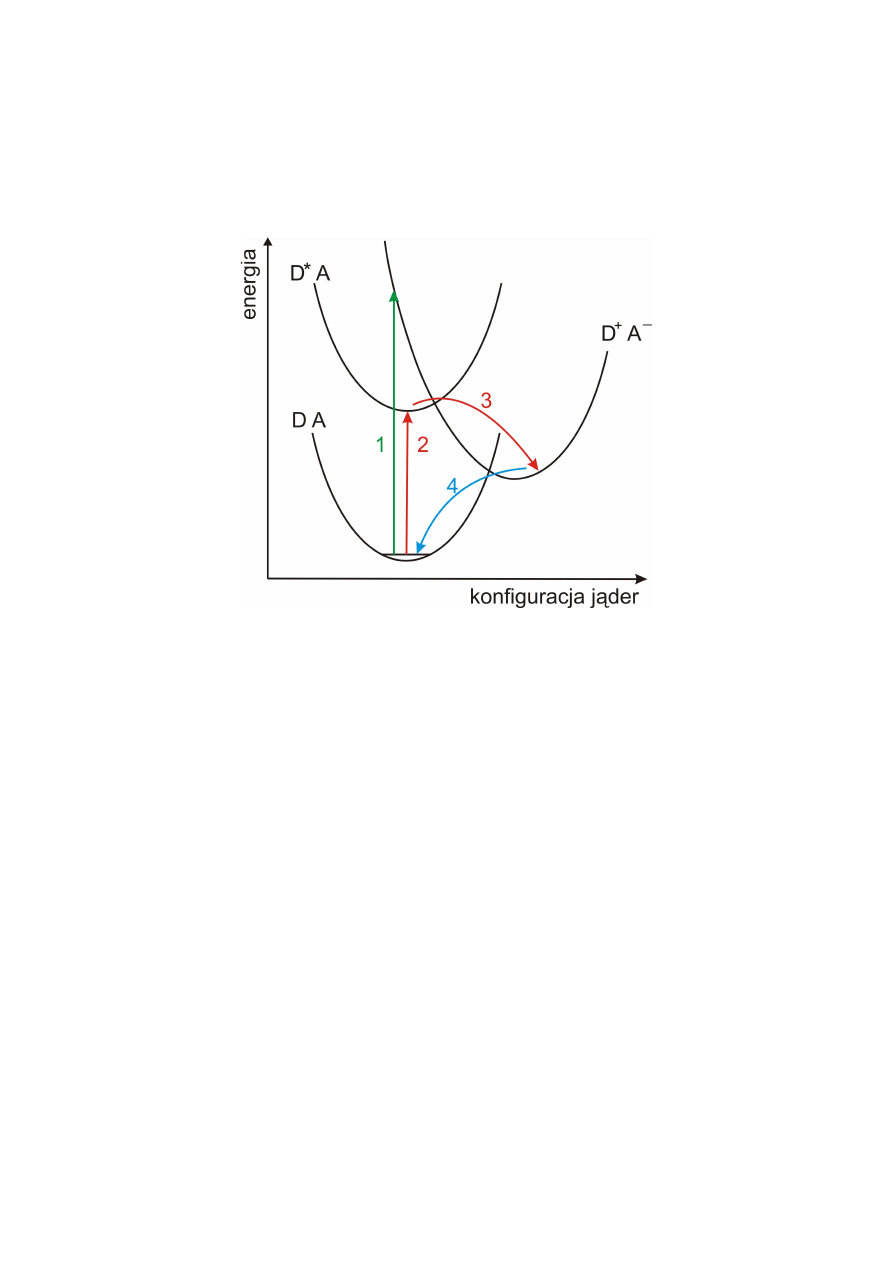
Teoria Markusa
Przeniesienie elektronu pomiędzy donorem (D) a akceptorem (A) jest elementarnym
procesem redox, leżącym u podstaw ogromnej liczby reakcji utleniania-redukcji.
Przeniesienie elektronu może być indukowane w różny sposób:
1 – proces optyczny
2, 3 – proces fotoindukowany
3, 4 – proces termiczny
Proces optyczny występuje rzadko (głównie dla związków kompleksowych metali).
Polega na bezpośrednim (nie przechodzącym przez stadium cząsteczki wzbudzonej)
przeniesieniu elektronu w wyniku pochłonięcia fotonu światła przez układ.
Najważniejszymi procesami przeniesienia elektronu są procesy fotoindukowane. Biegną one
przez stan wzbudzony.
Każdy proces przeniesienia elektronu jest procesem izoenergetycznym.
Jeżeli nie odseparuje się D
+
A
-
, nastąpi rekombinacja. Sumarycznie proces będzie polegał na
zamianie światła na ciepło. Aby temu przeciwdziałać, dodaje się związki ofiarne, które
reagują z D
+
lub A
-
.
W teorii Markusa rozważa cię wielowymiarowe powierzchnie energii potencjalnej
opisujące stan energetyczny reagentów i produktów wraz z otaczającymi je cząsteczkami
rozpuszczalnika. Powierzchnie te są funkcjami wielu współrzędnych oscylacyjnych
reagentów oraz współrzędnych orientacyjnych cząsteczek rozpuszczalnika. W teorii stanu
przejściowego wprowadza się tzw. Współrzędną reakcji dzięki czemu powierzchnie energii
potencjalnej mogą być zredukowane do jednowymiarowego profilu. Również Marcus
wprowadził współrzędną reakcji, definiując ją jako różnicę energii potencjalnej U
P
-U
S
.
Wzdłuż
tej
współrzędnej
Marcus
obliczał
entalpię
swobodną
G
S
układu
substraty+rozpuszczalnik oraz entalpię G
P
układu produkty+rozpuszczalnik.
Rozpuszczalnik był traktowany jako ośrodek dielektryczny, którego cząsteczki podlegają
polaryzacji elektronowej i orientacyjnej. Pierwsza z nich pozostaje w każdej chwili w
2
równowadze z rozkładem ładunku reagujących cząsteczek, a druga stara się nadążyć za
zmianami tego rozkładu poprzez powolne ruchy dipoli rozpuszczalnika.
Marcus przyjął tu przybliżenie liniowej odpowiedzi, zgodnie z którym jakakolwiek zmiana
ładunku reagentów powoduje proporcjonalną do niej zmianę polaryzacji orientacyjnej
ośrodka. Po zastosowaniu tego przybliżenia entalpie swobodne G
S
i G
P
stały się funkcjami
kwadratowymi współrzędnej reakcji.
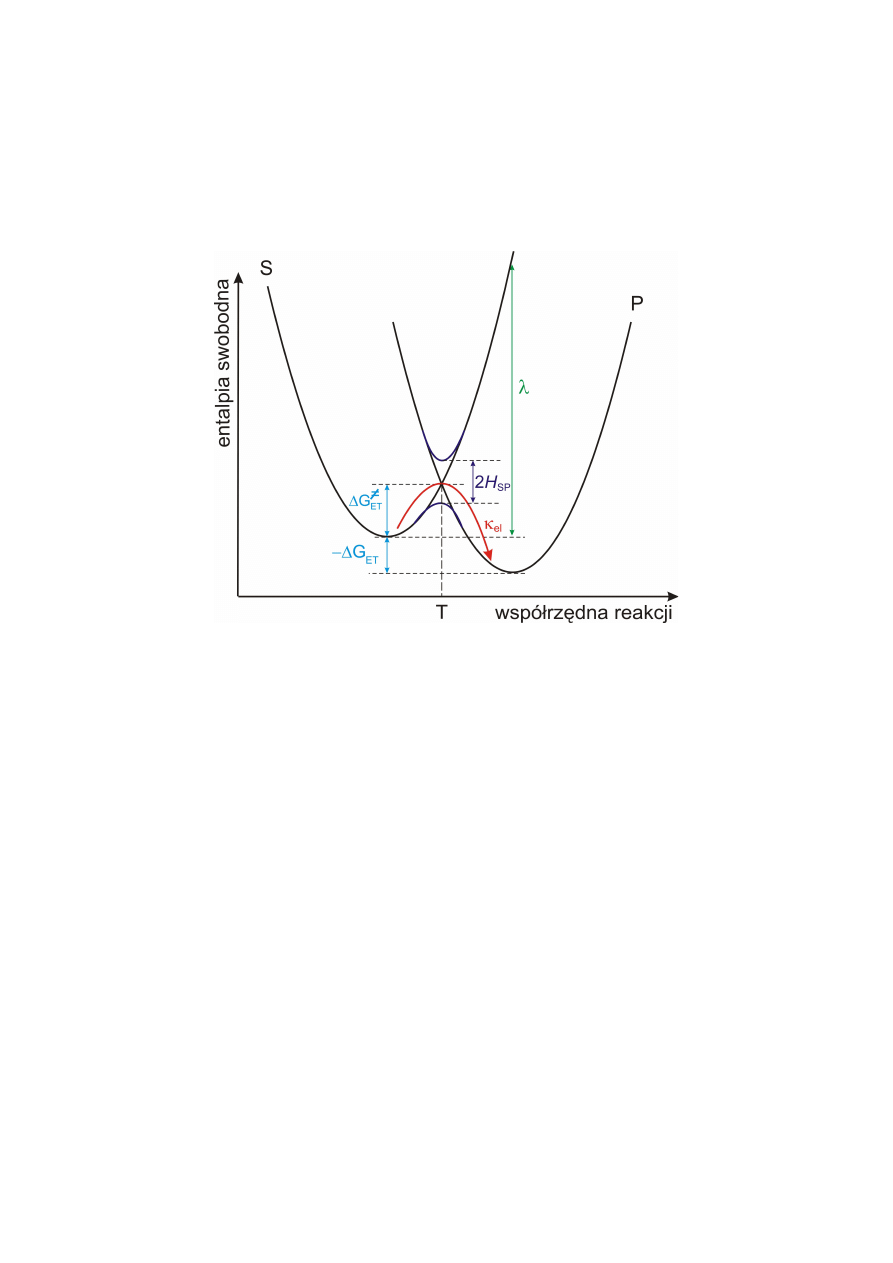
Entalpia swobodna układu substraty+rozpuszczalnik S i produkty+rozpuszczalnik P jako
funkcja współrzędnej reakcji przeniesienia elektronu.
ET
G
∆
- standardowa entalpia reakcji ET;
≠
∆
ET
G
- bariera aktywacji procesu ET;
SP
H
- efekt macierzowy sprzężenia stanów elektronowych substratu i produktu;
λ
- energia reorganizacji.
Stan przejściowy reprezentuje punkt T. Wartość entalpii swobodnej w tym punkcie w
odniesieniu do entalpii swobodnej stanu równowagowego substratów określa entalpię
aktywacji procesu ET
(
)
≠
∆
ET
G
.
Zgodnie z klasyczną teorią stanu przejściowego stała szybkości pierwszego rzędu dla reakcji
przeniesienia elektronu
ET
k
wynosi:
(
)
T
k
G
k
B
ET
n
el
ET
/
exp
≠
∆
−
=
ν
κ
n
ν
- częstotliwość przejść pomiędzy powierzchniami entalpii swobodnej substratów i
produktów;
el
κ
- elektronowy współczynnik przejścia.
Na drodze prostych rozważań geometrycznych można wyznaczyć entalpię aktywacji procesu
ET.
3
(
)
λ
λ
4
2
ET
ET
G
G
∆
+
=
∆
≠
Po podstawieniu:
(
)
∆
+
−
=
T
k
G
k
B
ET
n
el
ET
λ
λ
ν
κ
4
exp
2
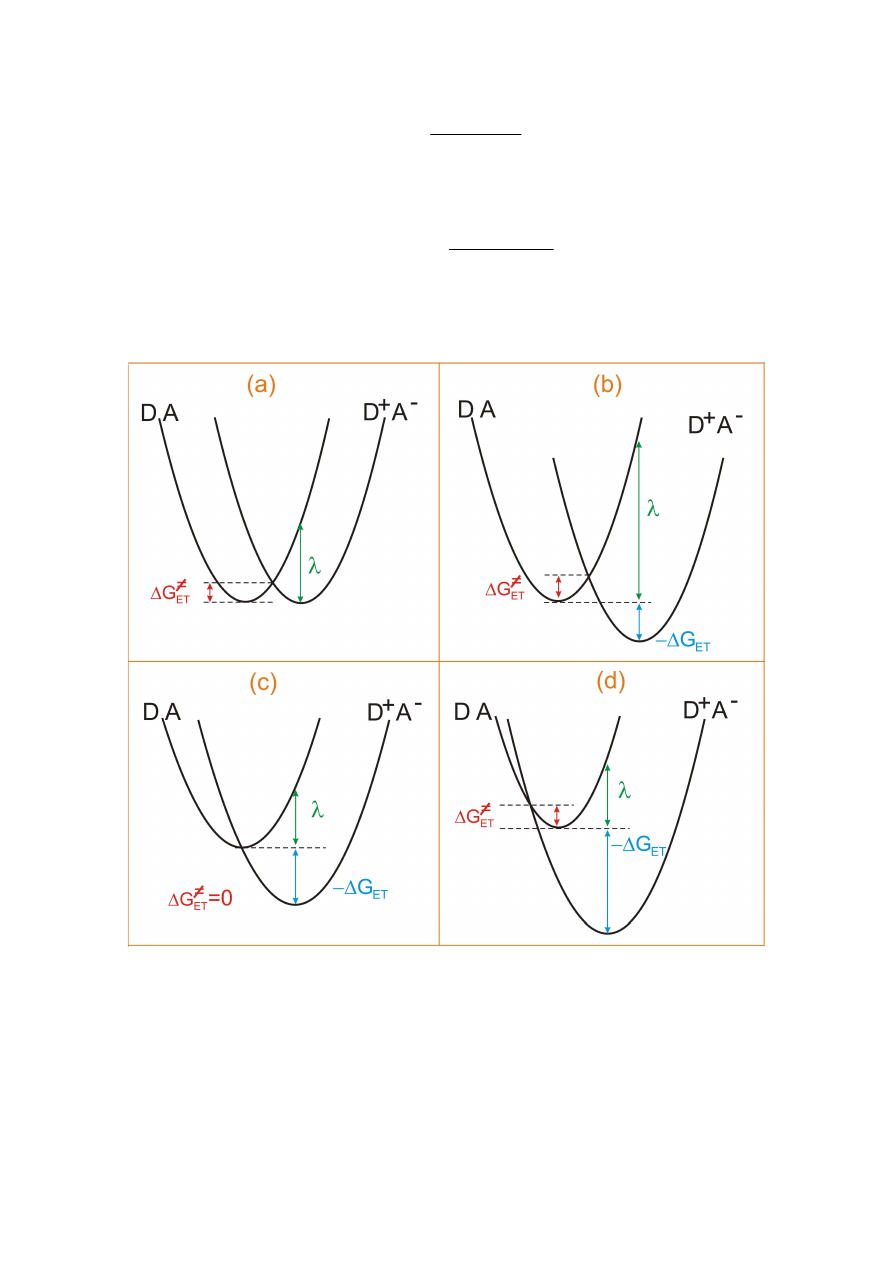
Możliwe są 4 charakterystyczne układy krzywych entalpii swobodnych substratów DA i
produktów D
+
A
-
:
a)
0
=
∆
ET
G
;
b)
λ
≤
∆
−
≤
ET
G
0
, normalny obszar Markusa,
ET
k
rośnie wraz ze wzrostem
ET
G
∆
−
;
c)
λ
=
∆
−
ET
G
, maksymalna wartość
ET
k
;
d)
λ
>
∆
−
ET
G
, odwrotny obszar Markusa,
ET
k
maleje ze wzrostem
ET
G
∆
−
.
Kształt parabol substratów i produktów jest jednakowy. Istotnym parametrem jest energia
reorganizacji
λ
, zdefiniowana jako różnica entalpii swobodnej substratów w ich

4
równowagowej konfiguracji i entalpii swobodnej substratów w konfiguracji odpowiadającej
równowagowej konfiguracji produktów reakcji.
Rysunek a) odpowiada przypadkowi, gdy entalpia swobodna reakcji
ET
G
∆
(siła napędowa)
jest równa zero. Przykładem takiego procesu jest reakcja samowymiany:
(
)
(
)
(
)
(
)
+
+
+
+
+
⇔
+
2
6
2
3
6
2
3
6
2
2
6
2
O
H
Fe
O
H
Fe
O
H
Fe
O
H
Fe
.
Rysunek b) przedstawia sytuację gdy
λ
≤
∆
−
≤
ET
G
0
. Dla takiego przedziału wartości
ET
G
∆
stała szybkości reakcji przeniesienia elektronu rośnie wraz ze wzrostem siły napędowej
reakcji.
Dla
λ
=
∆
−
ET
G
energia aktywacji
0
=
∆
≠
ET
G
;
max
=
ET
k
.
Dalszy wzrost
ET
G
∆
−
powoduje wzrost entalpii aktywacji, a w konsekwencji obniżenie
stałej szybkości reakcji przeniesienia elektronu.
Istnienie odwrotnego obszaru Markusa zostało udowodnione eksperymentalne.
Energia reorganizacji składa się z energii reorganizacji wewnętrznej
i
λ
i energii reorganizacji
zewnętrznej
s
λ
. Energia
i
λ
wynika z różnic w strukturze pomiędzy równowagowymi
konfiguracjami substratów i produktów.
(
)
∑
−
=
i
eq
P
eq
S
i
r
r
f
2
1
2
1
λ
i
f
- zredukowana stała siłowa oscylacji i-tego wiązania;
eq
S
r
i
eq
P
r
- równowagowe długości wiązań substratu i produktu.
i
λ
jest zwykle niewielka i wynosi 0,1 – 0,3eV (w niektórych związkach kompleksowych
1,6eV).
Energia reorganizacji zewnętrznej jest nazywana energią reorganizacji rozpuszczalnika,
ponieważ wynika z różnic pomiędzy polaryzacją cząsteczek rozpuszczalnika wokół
substratów i produktów reakcji ET.
Jeżeli rozpuszczalnik jest traktowany jako ciągły ciągły ośrodek dielektryczny.
−
+
−
=
r
d
a
e
s
s
2
1
1
1
1
2
0
2
ε
ε
λ
0
ε
- optyczna przenikalność elektryczna, równa w przybliżeniu kwadratowi współczynnika
załamania światła w danym rozpuszczalniku;
s
ε
- statyczna przenikalność elektryczna rozpuszczalnika;
d
i
a
- promienie donora i akceptora;
r
- odległość donor – akceptor.
1
Wypełnienia
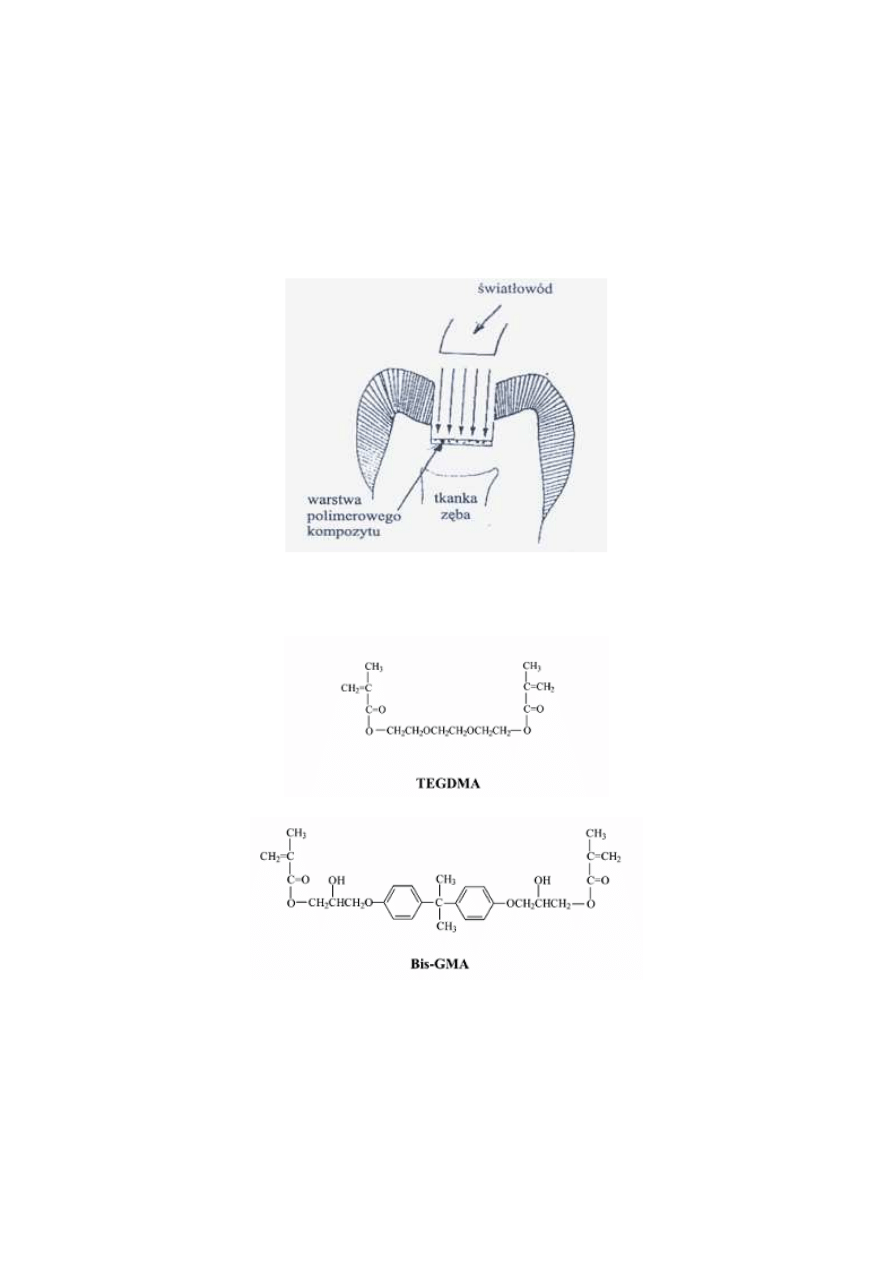
Fotopolimeryzacja stała się podstawową metodą stosowaną w nowoczesnej
stomatologii do utwardzania in vivo polimerowych wypełnień.
Po oczyszczeniu zęba z zalegającej próchnicy i dokładnym wysuszeniu jego
powierzchni dentysta wprowadza warstwę fotoczułego monomerowego kompozytu, który
utwardza za pomocą światła (fotopolimeryzacja).
Polimerowe materiały kompozytowe składają się z mieszaniny:
•
Monomerów jedno i wielofunkcyjnych:
2
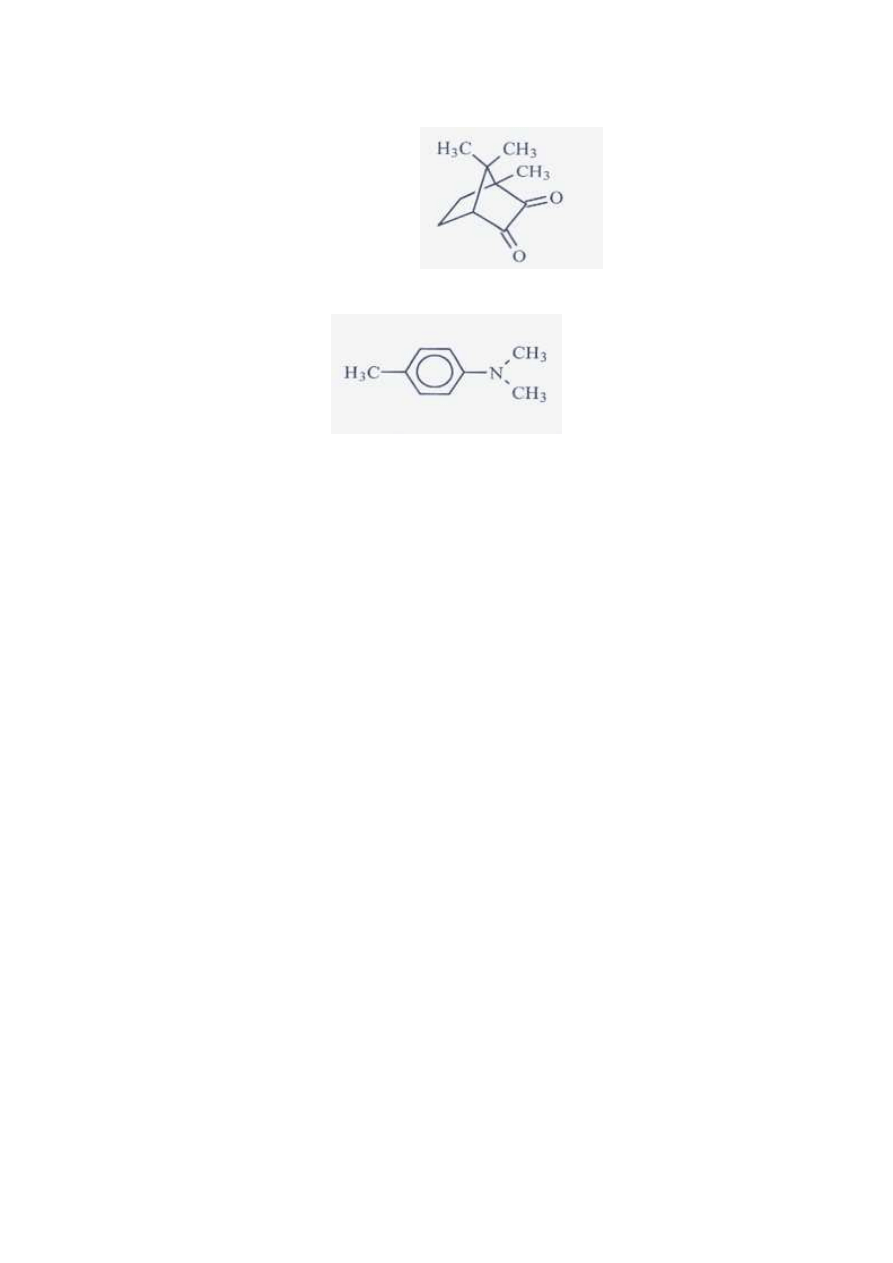
•
Fotoinicjatora – kamforchinon – sam w obecności monomeru fotoinicjuje reakcję
polimeryzacji stosunkowo powoli.
W celu przyśpieszenia
polimeryzacji dodaje się koinicjatory (aminy aromatyczne), np.:
•
Wypełniaczy wzmacniających (kwarcu, szkła krzemianowego, zmielonego polimeru).
Dodatek ten służy poprawie adhezji.
•
Dodatków
takich,
jak
inhibitory
(zapobiegających
polimeryzacji
podczas
przechowywania), fotostabilizatorów (zapobiegających zmianie koloru wypełnienia),
związków pozwalających dobrać kolor wypełnienia do naturalnego koloru uzębienia
pacjenta.
Fotopolimeryzację prowadzi się w jamie ustnej pacjenta. Dlatego nie można stosować
promieniowania UV, które mogłoby powodować oparzenia i raka. Stosuje się światło
niebieskie. (dla kamforchinonu 470 nm).
Jednym z podstawowych wymagań wypełnień dentystycznych jest odpowiednia
wytrzymałość mechaniczna (na ścieranie, ściskanie, wypłukiwanie). Aby ją zwiększyć,
stosuje się różnego rodzaju wypełniacze (do 70%).
Ze względu na obecność wypełniaczy, fotopolimeryzację trzeba prowadzić w cienkich
warstwach (do 1mm). Ze względu na komfort pacjenta czas fotopolimeryzacji warstwy
powinien wynosić 20 – 40 sekund. Aby zapewnić odpowiednią jakość i trwałość wypełnienia,
sposób naświetlania musi być odpowiedni (np. światło musi obejmować całą powierzchnię
wypełnienia).
Poważnym mankamentem fotoutwardzalnych wypełnień jest efekt skurczu
polimeryzacyjnego dochodzący nawet do kilkunastu %. Powoduje to tworzenie się szczeliny
pomiędzy wypełnieniem a tkanką zęba, co może być przyczyną wtórnej próchnicy. Ponadto
skurcz powoduje powstanie naprężeń mechanicznych w wypełnieniu, co znacznie obniża jego
wytrzymałość mechaniczną. Stosowanie wypełniaczy zmniejsza skurcz o 1 – 3%, ale
obecność dużej ilości wypełniacza ogranicza głębokość penetracji światła, co może prowadzić
do niecałkowitego przereagowania monomeru. Wydostające się z biegiem czasu z
wypełnienia monomery mogą powodować alergie.
Stosowane jako koinicjator aminy aromatyczne są kancerogenne.
Istnieje technika oświetlania wypełnienia przez ząb. Wówczas proces polimeryzacji
rozpoczyna się od strony zęba co ogranicza problem skurczu.
3
Inicjatory:
Kamforchinon:
Wyszukiwarka
Podobne podstrony:
Mat wys czys II
mat wys czys znowu
mat wys czys WOLSZCZAK odt
Wyklad2 mat
Mat 10 Ceramika
Mat dla stud 2
Wyklad7 mat
mat skale pomiarowe
logika mat
Magn mat
7Komunikacja org mat
mat bud 006 (Kopiowanie) (Kopiowanie)
Materialy do seminarium inz mat 09 10 czesc III
mat bud 102 (Kopiowanie) (Kopiowanie)
mat 2013 k11
Mat 3
więcej podobnych podstron