Mechanizmy niewydolności krążenia-
rola układu neurohormonalnego serca
Zakład Nadciśnienia Tętniczego
Katedry Nefrologii i Nadciśnienia Tętniczego
Uniwersytetu Medycznego w Łodzi

Niewydolnośd serca
Zespół kliniczny spowodowany nieprawidłowością serca o
charakterystycznym okresie hemodynamicznym, któremu
towarzyszy upośledzenie funkcji nerek oraz odpowiedź układu
nerwowego i humoralnego.
wg Poole-Wilsona 1985
Niewydolnośd serca jest stanem, w którym serce jako pompa nie
zabezpiecza odpowiedniego do potrzeb ustroju dopływu krwi do tkanek i
narządów, mimo prawidłowego wypełnienia łożyska naczyniowego.
wg Steada D. 1948
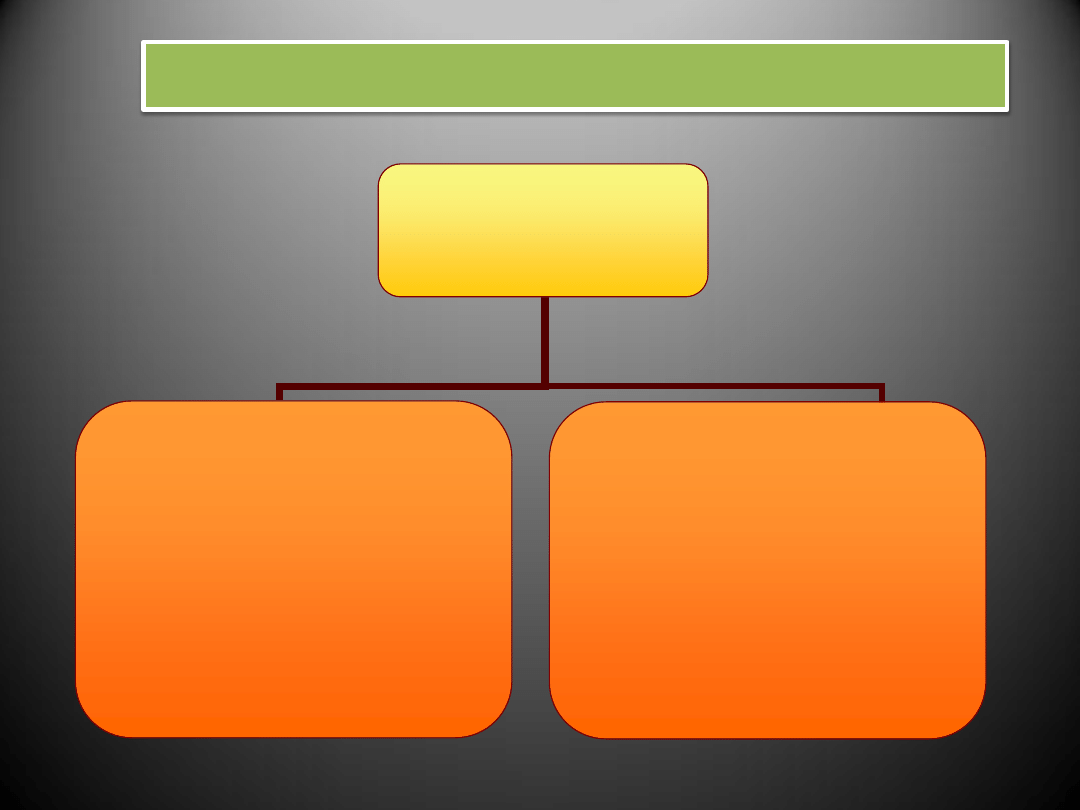
Niewydolnośd
serca
objętośd
wyrzutowa
pojemnośd
minutowa
Objętośd wyrzutowa
kurczliwośd mięśnia sercowego
obciążenie wstępne (wielkośd ciśnienia
późnorozkurczowego)
obciążenie następcze (wielkośd ciśnienia
skurczowego w komorach i aorcie lub tętnicy
płucnej)
częstośd i miarowośd rytmu serca
Skurczowa (spadek frakcji
wyrzutowej)
przewlekła
Rozkurczowa (zaburzenie
relaksacji komory)
ostra
Prawokomorowa
Lewokomorowa
Niewydolnośd serca
Niewydolnośd prawokomorowa
•
powikłanie niewydolności lewokomorowej
•
pierwotne przeciążenie prawej komory (nadciśnienie płucne, izolowana niedomykalnośd
zastawki trójdzielnej powodująca napływ dodatkowej krwi do prawej komory podczas
rozkurczu, zaciskające zapalenie osierdzia.
•
zawał prawej komory
Niewydolnośd lewokomorowa
•
stan po zawale mięśnia sercowego
•
nadciśnienie tętnicze,
•
wada mitralna, aortalna
•
choroba niedokrwienna serca
Ostra niewydolnośd serca
•
może byd wywołana nagle pojawiającym się upośledzeniem kurczliwości serca (np. zawał
serca), masywny zator tętnicy płucnej, jego nasilenia (np. nagły wzrost ciśnienia tętniczego)
bądź nałożenia się dodatkowych czynników (np. częstoskurcz) na już istniejące przeciążenie
hemodynamiczne serca.
•
objawami ciężkiej niewydolności serca są obrzęk płuc i wstrząs kardiogenny
Etiologia niewydolności serca
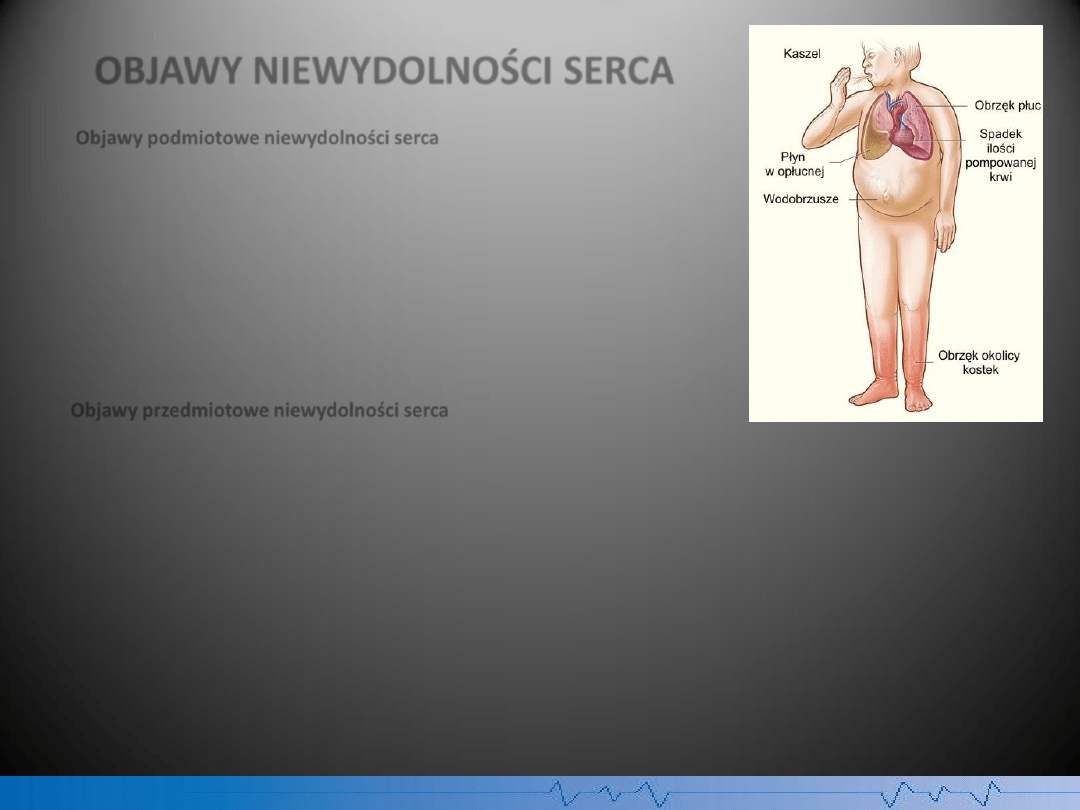
OBJAWY NIEWYDOLNOŚCI SERCA
Objawy podmiotowe niewydolności serca
* pogorszenie tolerancji wysiłku
* dusznośd, dychawica sercowa
* kaszel
* nocne oddawanie moczu, skąpomocz
* obrzęki
* ból brzucha
Objawy przedmiotowe niewydolności serca
* bladośd skóry, ochłodzenie skóry, sinica
* nadmierne wypełnienie żył szyjnych (związane z podwyższonym ciśnieniem żylnym), objaw wątrobowo-
szyjny (często nieobecne w ciężkiej niewydolności serca)
* powiększenie wątroby
* tachykardia (mało swoisty objaw)
* trzeci ton serca (nieswoisty objaw ciężkiej niewydolności serca)
* stwierdzane osłuchowo trzeszczenia nad polami płucnymi (objaw zastoju w płucach): objaw nieswoisty, o
małej wartości prognostycznej
* płyn w jamie opłucnej i jamie otrzewnej
* oddech Cheyne'a-Stokesa (oddech periodyczny)
Rys. Objawy niewydolności prawokomorowej serca.

Niewydolnośd serca
Niewydolnośd spowodowana uszkodzeniem funkcji mięśnia serca charakteryzuje się
zazwyczaj
zredukowanym przepływem krwi w tkankach.
Istotą niewydolności serca nie są
doraźne zaburzenia hemodynamiczne, lecz postępujące pogarszanie się sprawności serca.
Przeciążenie ciśnieniowe lewej komory prowadzi do
przerostu
i następowego
osłabienia
,
bądź ostrego wypadnięcia
czynności skurczowej
znacznego obszaru komory, co w
konsekwencji prowadzi do nadmiernego wydzielania katecholamin, podwyższonego
poziomu angiotensyny II i wazopresyny oraz wzrostu wydzielania mineralokortykoidów.
W wyniku tych procesów następuje uszkodzenie mięśnia sercowego, a zmiany w głównej mierze
dotyczą:
•
degeneracji kardiomiocytów
•
przebudowy zrębu łącznotkankowego (włóknienie)
•
zmniejszenia łożyska naczyniowego
•
wyczerpania mechanizmów adaptacyjnych mięśnia serca
•
spadku zasobów noradrenaliny w włóknach mięśniowych
•
spadku gęstości beta-adrenoreceptorów
•
obniżenia wrażliwości na działanie katecholamin
•
obnizeniu odpowiedzi inotropowej włókien mięśniowych
Efektem tego jest niezdolnośd do poprawy wydolności serca.
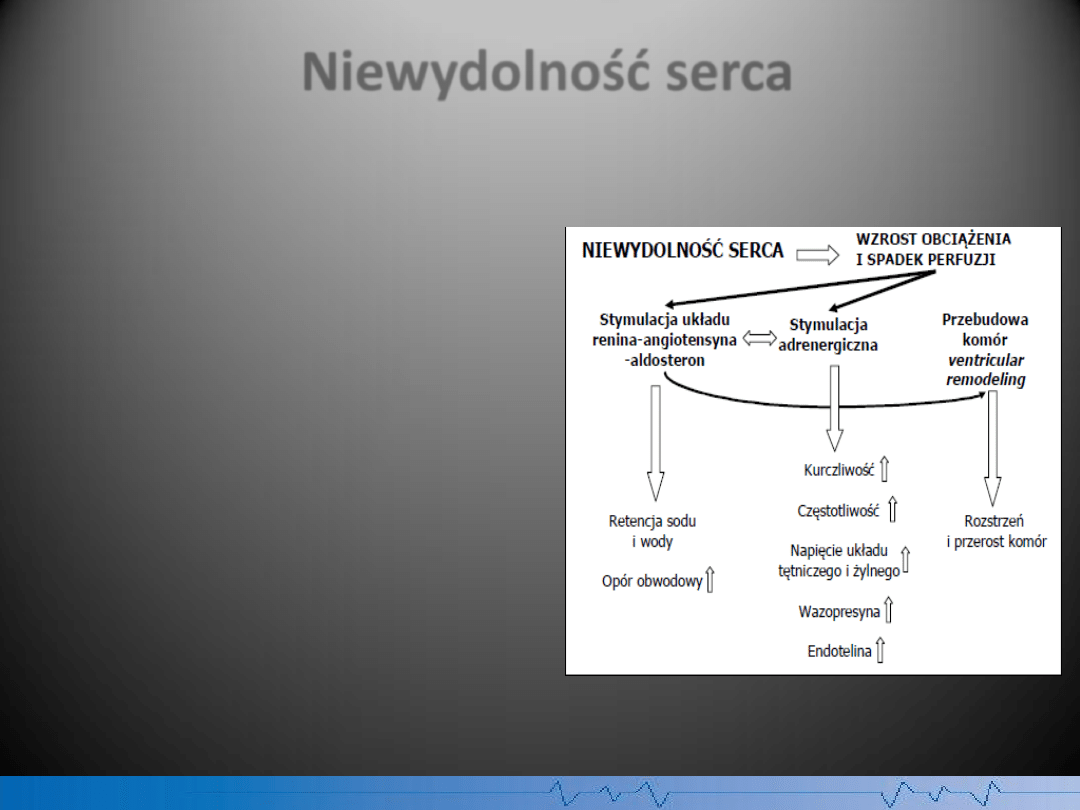
Niewydolnośd serca
Układy regulacyjne, których aktywacja
odgrywa istotną rolę w patogenezie
niewydolności serca, to
układ współczulny
i
układ renina-angiotensyna-aldosteron
(RAA). Efektem ich oddziaływania jest
szereg niekorzystnych zmian
hemodynamicznych w przebiegu
niewydolności serca (wzrost oporu
obwodowego, zwiększenie wolemii,
tachykardia, wzrost naprężenia ściany) oraz
liczne zaburzenia humoralne i
metaboliczne.
Trzecim bardzo istotnym narządem, którego
dysfunkcja odgrywa rolę w patogenezie
niewydolności serca jest śródbłonek.
Dysfunkcja śródbłonka
objawia się między
innymi przesunięciem równowagi pomiędzy
jego właściwościami wazorelaksacyjnymi,
zależnymi od tlenku azotu, a
wazokonstrykcyjnymi, zależnymi od
endoteliny.
Hipoteza zapalna niewydolności serca
•
Badania ostatnich lat wskazują, że przewlekłą niewydolnośd krążenia traktowad
można jako przewlekły proces zapalny, w którym istotną rolę odgrywają cytokiny
prozapalne.
•
Spośród wielu cytokin (interleukiny 1,2,3,4,6, TNF-α, interferon γ), wskazuje się na
kluczową rolę czynnika martwicy nowotworów (TNF-α) oraz interleukiny 1 i 6 (IL-1,
IL-6) w patogenezie niewydolności serca.
•
W badaniu SOLVD (Studies of Left Ventricular Dysfunction) stężenie TNF-α
korelowało z nasileniem objawów wg klasyfikacji NYHA (New York Heart
Association), a wysokie jego stężenie stanowiło niezależny czynnik prognozujący
zgon.
•
TNF-α wywiera biologiczne działania, które mogą odpowiadad za powstawanie
objawów niewydolności serca.
•
Do potencjalnych mechanizmów uszkodzenia i powstawania NS indukowanej przez
czynnik martwicy nowotworów należy m.in.: upośledzenie funkcji skurczowej lewej
komory, obrzęk płuc, indukowany procesu przebudowy, rozprężenie beta-receptora
z cyklazą adenylową, zaburzenie procesów energetycznych w mitochondriach,
apoptoza kardiomiocytów oraz komórek endotelium.
Hipoteza cytokinowa
•
Hipoteza cytokinowa niewydolności serca zakłada, że nadmierna ekspresja cytokin
zarówno w obrębie tkanek mięśnia sercowego, jak również w krążeniu ogólnym i
tkankach obwodowych powoduje powstawanie lub nasilenie już istniejących
zaburzeo i objawów niewydolności serca.
•
Istnieje kilka potencjalnych źródeł, mogących generowad cytokiny w przebiegu
niewydolności serca.
•
Najbardziej oczywista jest hipoteza sugerująca produkcję cytokin przez układ
immunologiczny w odpowiedzi na uszkodzenie tkanek serca.
•
Inna sugeruje możliwośd syntezy cytokin (np. IL-6) w niedokrwionych tkankach
obwodowych.
•
Kolejna koncepcja opiera się na założeniu aktywacji syntezy cytokin przez układ
odpornościowy w wyniku zwiększonego przenikania do krążenia endotoksyn z
przewodu pokarmowego.
•
Wreszcie ostatnia hipoteza wiąże zwiększone wydzielanie cytokin z aktywacją układu
adrenergicznego, która ma miejsce w niewydolności serca. Wiąże się to z obserwacją,
że podwyższone stężenia cAMP (cyklicznego adenozynomonofosforanu) powodują
zwiększoną ekspresję niektórych mediatorów zapalnych.
Dysfunkcja śródbłonka w przebiegu
niewydolności serca
•
Kluczowym zaburzeniem w patofizjologii niewydolności serca wydaje się ogrywad dysfunkcja
śródbłonka.
•
Do mechanizmów, za pomocą których cytokiny powodują dysfunkcję śródbłonka, należy: zaburzona
synteza tlenku azotu (NO), nasilenie stresu oksydacyjnego oraz uruchomienie procesu apoptozy.
•
Podstawowym objawem zaburzonej czynności śródbłonka jest upośledzenie jego właściwości
wazorelaksacyjnych, wyrażające się m.in. zaburzoną reakcją wazodilatacyjną na acetylocholinę, przy
zachowanej reakcji na podanie egzogennego donora NO.
•
Stres oksydacyjny u chorych z niewydolnością serca pojawia się jako wynik nadmiernej syntezy
wolnych rodników tlenowych lub w wyniku upośledzenia komórkowych mechanizmów
antyoksydacyjnych. Jest jednym z mechanizmów remodelingu serca w przebiegu niewydolności,
prowadzi także do apoptozy komórek miokardialnych oraz komórek endotelium, przyczyniając się do
postępu choroby.
•
Mięsieo sercowy podlega wzmożonemu stresowi oksydacyjnemu zarówno w wyniku niedokrwienia,
jak i w momencie reperfuzji.
•
Jedna z hipotez zakłada, że zaburzenie równowagi pomiędzy produkcją wolnych rodników a
mechanizmami antyoksydacyjnymi odpowiada za przejście od fazy przerostu do fazy dekompensacji
serca w procesie powstawania niewydolności serca.
•
Zarówno angiotensyna II, jak i niektóre cytokiny nasilają produkcję wolnych rodników tlenowych,
zwiększając tym samym stres oksydacyjny. Aktywacja układu RAA, będąca skutkiem upośledzenia
wydolności serca, prowadzi do zwiększonej produkcji wolnych rodników tlenowych.
•
W wyniku powstania stresu oksydacyjnego dochodzi do aktywacji genów zależnych od potencjału
oksydo-redukcyjnego. Częśd z tych genów odpowiada za produkcję prozapalnych cytokin (IL-6), co
prowadzi do nasilenia stresu oksydacyjnego tworząc w mechanizmie dodatniego sprzężenia
zwrotnego klasyczne "błędne koło" postępującej dysfunkcji narządu.

Układ renina-angiotensyna-aldosteron (RAAS)
Układ renina-angiotensyna-aldosteron bierze udział w patogenezie wielu chorób układu sercowo-
naczyniowego włączając:
•
zawał mięśnia sercowego
•
hipertrofie
•
kardiomiopatie
•
migotanie przedsionków
•
zastoinową niewydolnośd serca
•
miażdżyca
System RAAS działa jak układ wewnątrzwydzielniczy.
Renina jest syntetyzowana w postaci proreniny, przechowywana w ziarnistościach i uwalniana do krążenia
w nerkach, gdy pojawią się następujące bodźce:
•
obniżenie ciśnienia transmuralnego w tętniczkach doprowadzających
•
zmniejszenie zawartości sodu w plamce gęstej nefronu
•
pobudzenie receptorów beta-adrenergicznych aparatu przykłębuszkowego
•
obecnośd prostaglandyny E2, I2 oraz kininy
Krążąca renina katalizuje konwersję
angiotensynogenu
do
angiotensyny I
, przekształcanej dalej do
angiotensyny II
.
Układ renina-angiotensyna-aldosteron (RAAS) odgrywa istotną rolę w regulacji
systemu fibrynolitycznego
,
gdyż jak zostało udowodnione zarówno w badaniach in vitro jak i in vivo, angiotensyna II zwiększa
poziom PAI-1, który jest głównym fizjologicznym inhibitorem fibrynolizy
.
Produkcja zarówno ACE jak i
angiotensyny jest znacznie zwiększona miejscu, w którym naczynie jest uszkodzone. Składniki systemu
RAA występują lokalnie w wielu tkankach, gdzie pełnią
rolę prozapalną
i
sprzyjają zwłóknieniu
.
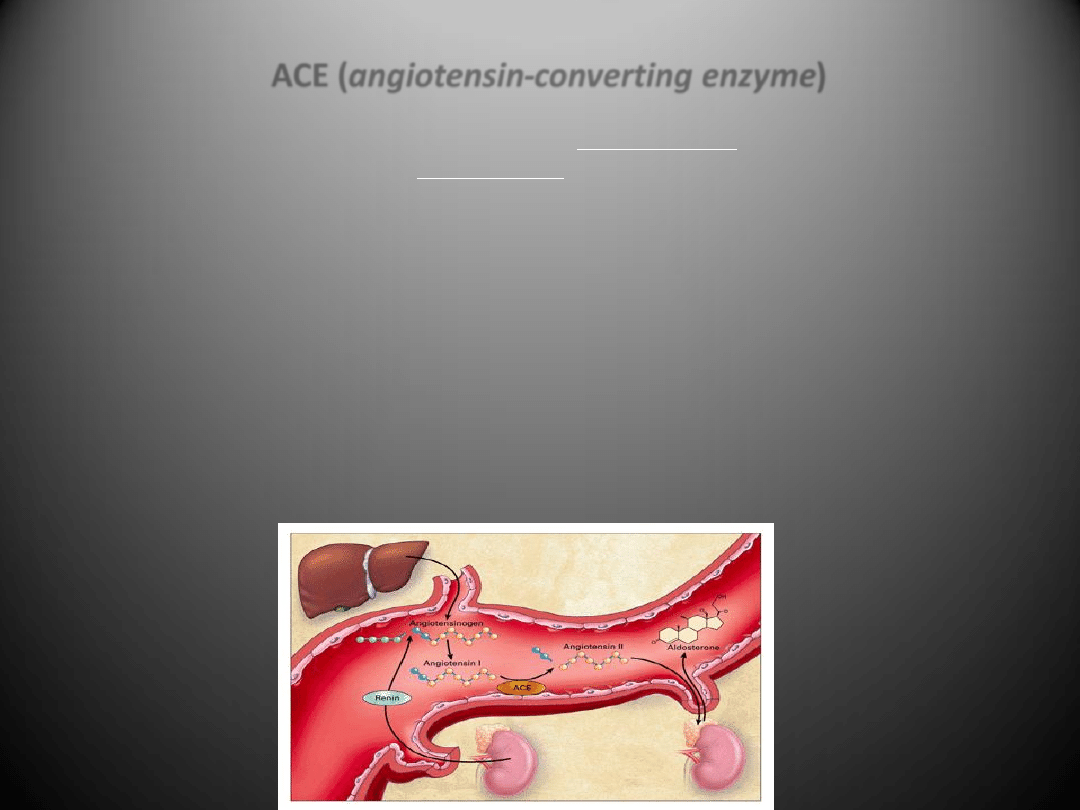
ACE (angiotensin-converting enzyme)
•
Enzym ACE jest odpowiedzialny za powstawanie z angiotensyny I aktywnego
wazokonstrykcyjnego peptydu - angiotensyny II. Odcina on od angiotensyny I C-koocowy
dipeptyd His-Leu
•
ACE może także działad na bradykininę odcinając z jej C-kooca dipeptyd Phe-Arg, co prowadzi do
jej inaktywacji
•
ACE jest głównie białkiem powierzchniowym, jego produkcja w zdrowym sercu jest niewielka i
ogranicza się głównie do śródbłonka naczyo, wzrasta natomiast znacznie w przeroście i
niewydolności serca
•
Może on występowad w tkankach w formie związanej z błoną komórkową lub w formie
rozpuszczalnej w płynach ustrojowych
•
ACE odgrywa znaczącą rolę w regulacji napięcia naczyo, utrzymywaniu homeostazy, zwiększa
aktywację i agregację płytek krwi, oraz wpływa na remodeling naczyniowy

Znaczenie ACE w chorobach układu krążenia
•
Niekorzystny wpływ tego enzymu na układ krążenia jest dwojaki. Poza generowaniem
angiotensyny II
, ACE
hydrolizuje bradykininę
do nieaktywnych związków znosząc jej korzystny
wpływ na śródbłonek naczyo
(bradykinina stymulująca produkcję i wydzielanie prostacykliny,
tlenku azotu i tkankowego aktywatora plazminogenu t-PA, ma działanie wazorelaksacyjne,
przeciwpłytkowe i fibrynolityczne)
•
Zwiększone stężenie ACE może prowadzid do zwiększonego wytwarzania angiotensyny II i w
konsekwencji do rozwoju miażdżycy. Zwiększoną ekspresję ACE odnotowano w makrofagach
kumulujących się wokół osłabionej pokrywy włóknistej blaszki. Przypuszcza się, że wyższa
aktywnośd tkankowa ACE może przyczyniad się do zmniejszenia stabilności blaszki miażdżycowej.
•
Zwiększoną ekspresję i aktywnośd ACE zaobserwowano także w obrębie „przerośniętej” komory
serca oraz w komórkach mięśni gładkich naczyo szczurów z uszkodzonym śródbłonkiem.
•
Udział ACE w patogenezie niewydolności serca potwierdzają liczne badania. Stosowanie
inhibitorów ACE (ACE-I)
przyczynia się do zmniejszenia częstości występowania incydentów
niedokrwiennych
oraz do poprawy funkcjonowania śródbłonka. Inhibitory ACE wpływają
pozytywnie na funkcjonowanie śródbłonka, prawdopodobnie przez to, że zmniejszają poziom
angiotensyny II, a przez to i endoteliny, obniżają stężenie reaktywnych form tlenu oraz zwiększają
stężenie rozszerzającej naczynia i stymulującej produkcję i uwalnianie NO bradykininy i
prostacykliny.
•
Inhibicja ACE przyczynia się do niezależnego od obciążenia zmniejszenia hipertrofii serca,
zapobiega także rozszerzeniu i remodelingowi komory po przebytym zawale mięśnia sercowego.

Budowa i powstawanie białka angiotensyny II
•
Angiotensyna jest hormonem peptydowym o budowie
(H-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-OH).
•
Aktywna angiotensyna II powstaje głównie na drodze odcięcia dipeptydu od angiotensyny I
przez enzym ACE. Peptyd ten może także powstawad w tkankach w sposób niezależny od ACE,
poprzez trawienie przez niespecyficzne karboksypeptydazy lub białka podobne do
chymotrypsyny np. chymazę.
•
Chymazowy szlak generowania angiotensyny II odnotowano w sercu, komórkach śródbłonka i
komórkach tucznych
Angiotensyna może byd generowana przez dwa systemy:
krążący we krwi układ RAA, który odpowiada za regulację krótkotrwałą (renina uwalniana do
krążenia z aparatu przykłębuszkowego pod wpływem obniżonej perfuzji kłębuszkowej
katalizuje transformację angiotensynogenu do angiotensyny I; system ten pełni funkcje
endokrynną)
tkankowy RAA biorący udział w długotrwałych zmianach (bierze udział w narządowym -
serce, naczynia krwionośne, nerki i tkankowym działaniu para-i autokrynnym)
•
W warunkach patologicznych (niewydolnośd serca, przerost lewej komory) działające lokalnie
(np. w sercu) czynniki systemu RAA wydają się mied większe znaczenie niż te krążące w osoczu.
•
Angiotensyna II może ulegad dalszemu przekształceniu w angiotensynę III (powstającą poprzez
odcięcie kwasu asparaginowego od oktapeptydu), a nawet w angiotensynę IV. Jednakże oba te
peptydy mają znacznie słabsze zdolności kurczenia naczyo niż AngII. Angiotensyna III stymuluje
syntezę aldosteronu i procesy zapalne (poprzez receptor AT2, natomiast angiotensyna IV
działając poprzez AT1 wywołuje skurcz naczyo, a poprzez AT4 wzmaga produkcję PAI-1.
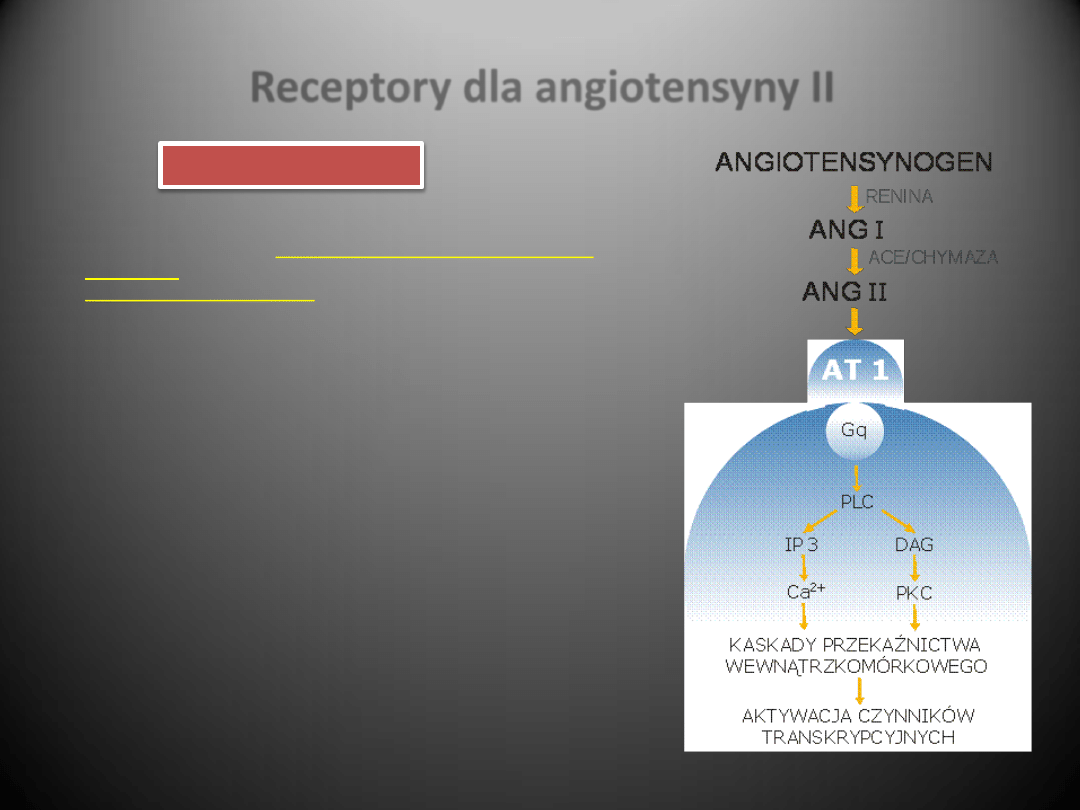
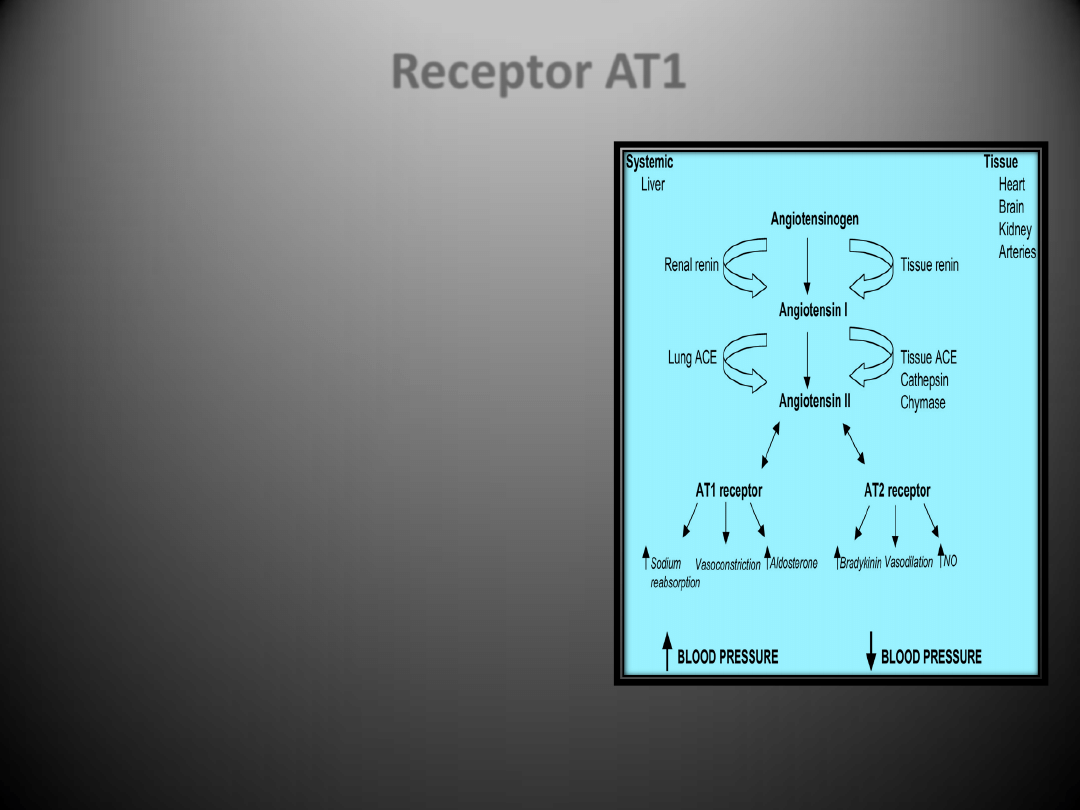
Receptory dla angiotensyny II
•
Receptor
AT1
ulega ekspresji w bardzo wielu tkankach i
narządach włączając
serce, nerki, wątrobę, płuca, mózg,
nadnercza
. Receptory te znajdują się także na powierzchni
monocytów/makrofagów
.
•
Wpływ na ekspresję receptora AT1 poza angiotensyną II,
której podwyższony poziom obniża jego aktywację, mają
czynniki wzrostu (PDGF, FGF, EGF – powodują obniżenie
ekspresji) oraz inne czynniki takie jak: glukokortykoidy,
aldosteron, forskolina, TNF-
, cytokiny, NO, insulina, LDL,
estrogeny, progesteron, sód, wolne rodniki tlenowe, IGF-1 i
izoprenalina.
•
Istnieją co najmniej cztery mechanizmy regulacji związane z
receptorem AT1.
•
Receptor AT1 sprzężony jest z białkiem Gq, jego aktywacja
prowadzi do stymulacji fosfolipazy C (PLC), a w dalszej
kolejności do powstania IP3 i IDG, mobilizacji Ca
2+
i aktywacji
białkowej kinazy C (PKC). IDG prawdopodobnie pośredniczy
w naczynioskurczowym działaniu AngII, a także w aktywacji
kinaz tyrozynowych/serynowych, które z kolei przyczyniają
się do stymulacji wzrostu indukowanego angiotensyną II
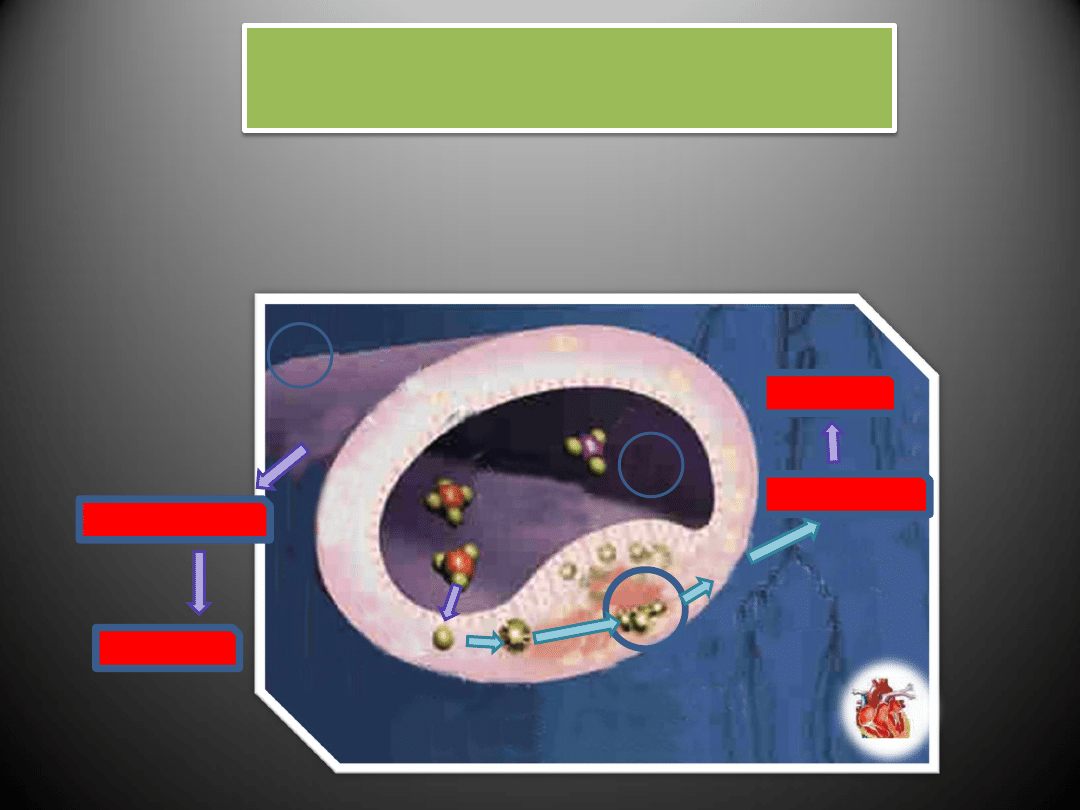
RENINA
ACE/CHYMAZA
Receptor AT1
Receptor AT1
Do efektów działania angiotensyny II, w której
pośredniczy AT1 zalicza się:
promowanie wzrostu komórkowego,
regulacja ekspresji substancji bioaktywnych
takich jak: hormony wazokonstrykcyjne, czynniki
wzrostu, cytokiny, aldosteron oraz składniki
macierzy zewnątrzkomórkowej
pośredniczenie w skurczu naczyo,
uwalnianianie katecholamin z zakooczeo
współczulnych włókien nerwowych
inicjowanie reakcji sprzężenia zwrotnego w
systemie RAS np. stymulowanie ekspresji
angiotensynogenu poprzez oddziaływanie na
fragment promotora zwany elementem
odpowiedzi ostrej fazy
hamowanie sekrecję reniny
Zwiększona produkcja angiotensyny II wraz z
podwyższonym stężeniem receptorów AT1 (dzięki
obecności w procesie miażdżycowym czynników, które
przyczyniają się do jego regulacji w górę) prowadzą do
większej wydajności systemu RAA, a przez to do
nasilonego niekorzystnego działania jego wszystkich
składników.
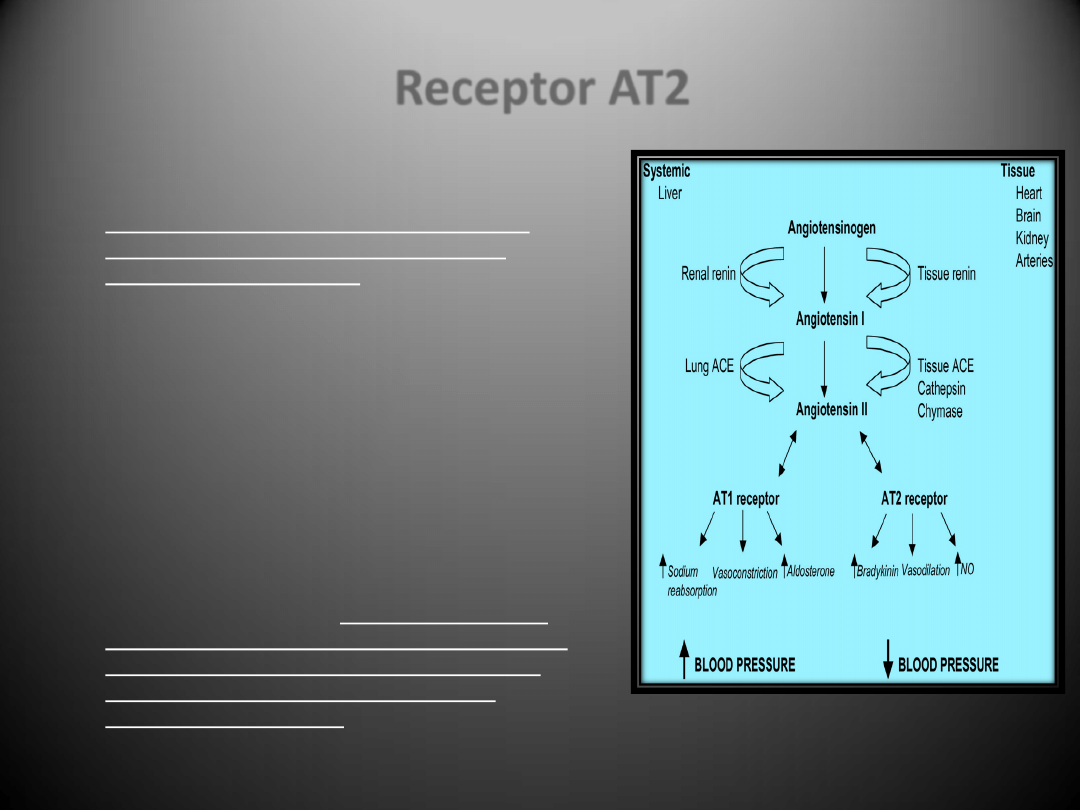
Receptor AT2
Receptor AT2 jest sprzężony z
fosfatazą
fosfotyrozynową
i występuje głównie w
przedsionkach serca, nabłonku naczyo,
macicy, jajnikach, rdzeniu nadnerczy,
trzustce oraz komorach.
Lokalizacja AT2 w śródmiąższowych
fibroblastach w sercu może sugerowad, że
może on także brad udział w rozwoju stanu
zapalnego i zwłóknienia.
Pomimo, iż zazwyczaj receptorowi AT2
przypisuje się pośredniczenie w działaniu
antyproliferacyjnym
i w procesie apoptozy,
może on również odgrywad rolę w stymulacji
wzrostu komórkowego.
Ekspresja receptorów dla angiotensyny II w
sercu jest odmienna: receptory AT2 są w
przewadze w zdrowym sercu, natomiast w
przypadku niewydolności receptory AT1
ulegają regulacji w dół, podczas gdy
ekspresja AT2 wzrasta

Funkcje angiotensyny II
Działanie:
–
syntezy i wydzielania aldosteronu
– skurcz naczyo
–
ADH (wazopresyna)
–
ACTH (kortykotropina)
–
resorbcji Na w kanalikach nerkowych
–
pragnienie
Aktywacja receptora AT1 zlokalizowanego na powierzchni komórek śródbłonka, mięśni gładkich
naczyo oraz monocytów wywołuje nie tylko szybkie uruchomienie szlaku fosfolipazy C i wapnia
wewnątrzkomórkowego, ale i pośredniczy w długofalowym efekcie AngII poprzez indukcje
zmian w transkrypcji genów.
Lokalnie syntetyzowana AngII wywołuje bezpośredni efekt inotropowy i poprawia funkcję
skurczową serca poprzez ułatwianie uwalniania noradrenaliny z zakooczeo nerwów
współczulnych, i o ile takie krótkotrwale działanie jest korzystne w stanie pozawałowym, to
przedłużona aktywacja może prowadzid do postępującego uszkodzenia mięśnia sercowego.
Angiotensyna II może stymulowad syntezę kolagenu typu I i III w ludzkich fibroblastach, a także
hamowad aktywnośd metaloproteinazy I

Rola angiotensyny II w dysfunkcji śródbłonka
AngII bierze udział w rozwoju nadciśnienia tętniczego
(ważnego czynnika ryzyka choroby wieocowej) poprzez
wywołanie skurczu naczyo (bezpośrednia stymulacja komórek
mięśni gładkich) i zwiększenie objętości
wewnątrznaczyniowej wody w organizmie na drodze
stymulacji kory nadnerczy do wydzielania aldosteronu.
Angiotensyna II zwiększa także wydzielanie endoteliny silnie
kurczącej naczynia krwionośne i wazopresyny (z przysadki
mózgowej) oraz zmniejsza biodostępnośd tlenku azotu
(NO), co w znaczący sposób wpływa na zależną od
śródbłonka wazodylatację naczyo.
Angiotensyna II stymulując oksydazę NADPH wytwarzającą
wolne rodniki tlenowe w komórkach mięśni gładkich naczyo i
śródbłonka przyczynia się do powstania stresu oksydacyjnego
oraz zmniejszenia biodostępności i aktywności NO. Efekt
przeciwny jest wywierany poprzez receptor AT2, który zwiększa
wytwarzanie bradykininy i stymuluje aktywnośd syntazy tlenku
azotu.

Rola Ang II w incydentach zakrzepowych
System RAA moduluje
równowagę fibrynolityczną
poprzez hamowanie układu
fibrynolitycznego i aktywowanie
procesów zakrzepowych na
drodze wpływania na kaskadę
koagulacji i aktywnośd płytek.
Zwiększa ekspresję czynnika
tkankowego (TF), który odgrywa
istotną rolę w inicjacji kaskady
krzepnięcia (jest ważnym
kofaktorem czynnika VII w kaskadzie
koagulacyjnej).
Hamuje szlak fibrynolizy
poprzez stymulowanie
ekspresji PAI-1, inhibitora
aktywatora tkankowego
plazminogenu (t-PA)
osłabiając proces
rozpuszczania skrzepu.
RAA przyczynia się także do aktywacji i
agregacji płytek krwi, które posiadają na
swojej powierzchni receptory dla
angiotensyny II, co prowadzi do ich
uwrażliwienia na działanie płytkowych
agonistów i stymuluje uwalnianie
płytkowych czynników naczyniokurczących
(tromboksan A2) i proliferacyjnych (PDG
F).

Rola Ang II w generacji ROS i oksydacji LDL
Angiotensyna II poprzez zwiększanie aktywności oksydazy NADPH wytwarzającej
reaktywne formy tlenu ma duży udział w powstawaniu i rozwoju miażdżycy
(powstają oxLDL)
•
RFT stymulują m.in. aktywowane-mitogenami kinazy – Akt, szlak JAK/STAT, indukują
czynniki transkrypcyjne (białko aktywatora-1), wpływają na funkcjonowanie kanałów
jonowych, a indukcja powyższych szlaków sygnałowych prowadzi do wzrostu i
migracji komórek mięśni gładkich naczyo, regulacji funkcji śródbłonka, ekspresji
czynników prozapalnych i modyfikacji macierzy zewnątrzkomórkowej
Angiotensyna II zwiększa oksydację cząsteczek LDL nie tylko poprzez stymulację oksydazy
NADPH w makrofagach, ale i lipoksygenazy.
•
oxLDL osłabiają tworzenie NO, zwiększają wytwarzanie reaktywnych form tlenowych
i indukują syntezę endotelialnych cząstek adhezyjnych (E-selektyna, ICAM-1, VCAM-
1), chemokin i czynników wzrostu mięśni gładkich
•
angiotensyna przyczynia się do regulacji „w górę” receptora LOX-1 (lectin-like
oxidized LDL receptor) zlokalizowanego na powierzchni komórek śródbłonka i
makrofagów oraz receptora „scavenger” (CD36) przez co odgrywa istotną rolę w
przyswajaniu cząstek ox-LDL przez makrofagi
Rola angiotensyny II w stymulacji procesu zapalnego
Poprzez receptor AT1 aktywuje ona w komórkach mięśni gładkich naczyo, w
monocytach i komórkach endotelium
prozapalny czynnik transkrypcyjny NF-
B,
który z kolei przyczynia się do zwiększenia syntezy komórkowych czynników
adhezyjnych (ICAM-1, VCAM-1), cytokin prozapalnych (MCP-1, IL-6)
Białko MCP-1 działając lokalnie przyciąga monocyty i limfocyty T zawierające
receptor CCR2 do miejsca uszkodzenia naczynia, natomiast interleukina-6 (IL-6)
uwalniana przez aktywowane monocyty i komórki mięśni gładkich naczyo,
stymuluje proliferację tych ostatnich

Rola angiotensyny II w stymulacji wzrostu komórkowego i
niestabilności blaszki miażdżycowej
Angiotensyna II aktywuje wiele szlaków sygnałowych
kinaz białkowych
(np. kinaza
Janusa/STAT, MAPK),
stymuluje wytwarzanie proto-onkogenów
(c-fos, c-jun, c-
myc) oraz wielu
autokrynnych czynników wzrostu
(TGF-
1, PDGF).
•
Angiotensyna II stymuluje proces proliferacji fibroblastów i syntezę kolagenu
poprzez zwiększanie sekrecji TGF-ß. Działając poprzez receptor AT1 angiotensyna II
powoduje hipertrofię komórek mięśni gładkich naczyo, powstawanie lokalnych
ognisk zapalnych prowadzące do rozwoju miażdżycy i pękania blaszki
miażdżycowej.
•
AngII indukuje ekspresję cytokin prozapalnych (IL-6) w hodowlach komórek mięśni
gładkich naczyo (VSCM) i makrofagów, które indukują proliferację VSMC na drodze
aktywacji PDGF i stymulacji degradacji macierzy przez metaloproteinazy.
•
Także stymulowane przez AngII generowanie reaktywnych form tlenu, które
regulują aktywnośd metaloproteinaz MMP-9 i MMP-2 degradujących kolagen,
przyczynia się do zmniejszenia stabilności blaszki miażdżycowej.
•
Angiotensyna II wywołuje
kompensacyjny przerost miocytów
oraz
komórek
mięśni naczyo krwionośnych
, co poprzez zmniejszenie przepływu krwi może
prowadzid do niedokrwienia mięśnia sercowego. Działając poprzez receptor AT2,
który uaktywnia fosfatazy, a nie kinazy tak jak AT1, angiotensyna II może hamowad
przerost mięśni gładkich naczyo i kardiomiocytów.
Aldosteron
• Aldosteron należy do mineralokortykoidowych hormonów kory nadnerczy.
Wydzielany przez warstwę kłębkowatą
kory nadnerczy
, aldosteron pełni
ważną funkcję w
utrzymywaniu prawidłowej równowagi wodno-
elektrolitowej
poprzez regulację transportu jonów w cewkach dystalnych
(retencja Na
+
i Cl
-
oraz wydalanie H
+
i K
+
) oraz ciśnienia tętniczego krwi.
Zwiększenie syntezy aldosteronu zachodzi pod wpływem takich czynników jak:
angiotensyna II
hormon adrenokortykotropowy
endotelina 1
spadek objętości krążącej krwi
obniżenie ciśnienia tętniczego
zmniejszenie stężenia sodu (Na) w organizmie
wzrost aktywności układu adrenergicznego
Swoje działanie hormon może wywierad bezpośrednio poprzez
receptory
mineralokortykoidowe
(MR) bądź pośrednio na drodze zwiększania ekspresji
angiotensyny II, endoteliny, aktywację COX-2, lub zwiększenie stresu
oksydacyjnego
K
+
kora nadnerczy
(warstwa kłębkowata)
angiotensyna II
ACTH
aldosteron
resorbcji Na
+
Na
+
wydalanie K
+
wydalanie H
+
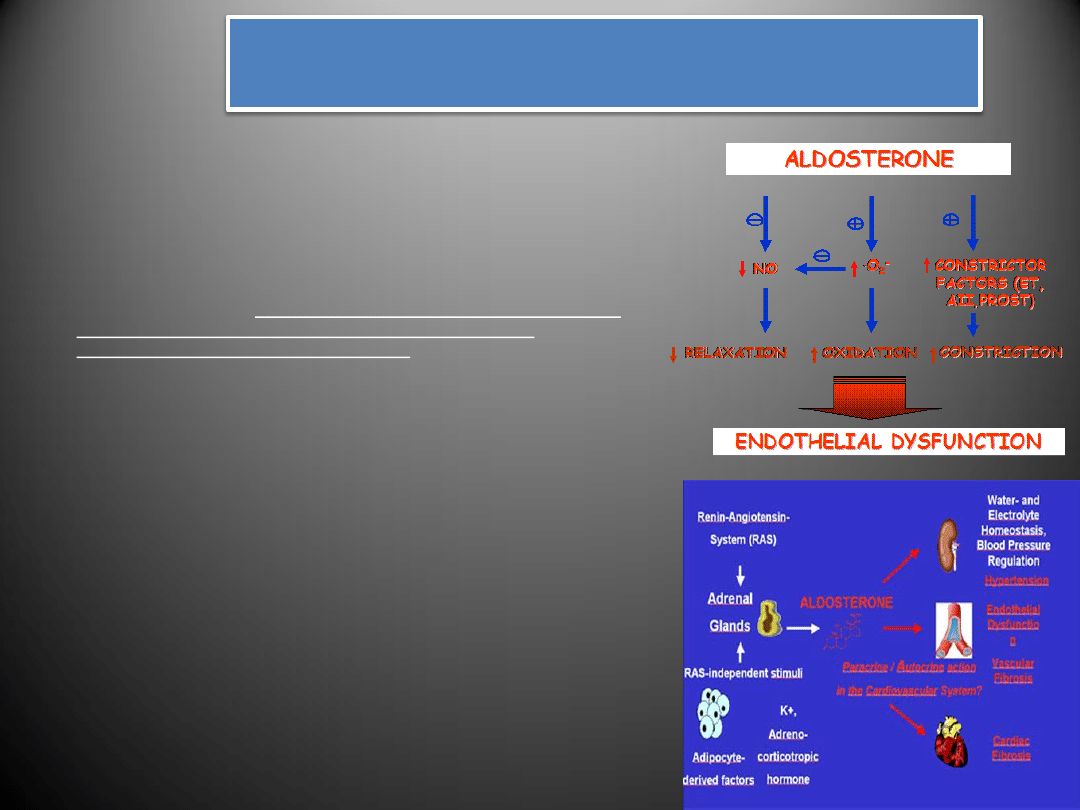
Rola aldosteronu w patogenezie chorób układu
sercowo-naczyniowego
•
Nadmierna synteza aldosteronu, działającego poprzez receptory
mineralokortykoidowe zlokalizowane w sercu, naczyniach
krwionośnych i mózgu przyczyniad się może do
włóknienia serca
i naczyo krwionośnych
poprzez działanie na fibroblasty
naczyniowe, uszkodzeo śródbłonka naczyniowego
oraz ciężkich
uszkodzeo tętnic wieocowych.
•
Uszkodzenia ścian naczyo wywołane przez nadmiar aldosteronu
mogą byd wywołane zwiększonym napływem sodu do miocytów,
zwiększoną aktywacją receptorów dla angiotensyny II oraz
hamowaniem wychwytu katecholamin.
•
Aldosteron nasila także
syntezę czynników wzrostu
, wykazuje
działanie
prozapalne
,
mitogenne
i
prozakrzepowe.
•
Podwyższone stężenie aldosteronu może stymulowad
powstawanie w sercu nacieków zapalnych z komórek
makrofagów, limfocytów T, monocytów oraz białek układu
dopełniacza prowadzących do lokalnych ognisk martwicy lub
niedokrwienia, co sugeruje, że hormon ten może uczestniczyd w
aktywowaniu komórek immunokompetentnych
•
Zwiększenie aktywności oksydazy NADPH przez aldosteron,
wpływa na
zwiększenie stresu oksydacyjnego
, co może
wywierad działanie prozapalne i nasilad syntezę niektórych
czynników wzrostu
•
Poza nasilaniem dysfunkcji śródbłonka i upośledzeniem
zależnego od endotelium rozszerzania naczyo na drodze
zmniejszania wydzielania tlenku azotu
oraz promowaniem
powstawania wtórnych ognisk martwicy, aldosteron zaburza
funkcje baroreceptorów oraz wychwyt norepinefryny przez
miokardium, co prowadzid może do zaburzenia rytmu serca

Endotelina-1
•
Endoteliny są obecne prawie we wszystkich tkankach i narządach, gdzie biorą udział
w regulacji układu krążenia, ośrodkowego i obwodowego układu nerwowego, układu
wewnątrzwydzielniczego i wielu innych
•
Endoteliny pełnią niezwykle ważną rolę w regulacji funkcji układu krążenia,
ośrodkowego i obwodowego układu nerwowego, układu wewnątrzwydzielniczego,
układu oddechowego i pokarmowego, regulacji równowagi kwasowo-zasadowej
ustroju oraz wielu innych narządów i układów
•
Przypisuje im się także udział w patogenezie wielu stanów chorobowych
(nadciśnienie płucne, nadciśnienie tętnicze, samoistne zwłóknienie płuc, astma i
alergie pokarmowe, kardiomiopatia rozstrzeniowa, skurcz naczyo mózgowych)
ET-1
ET-2
ET-3
komórki śródbłonka, mięśni
gładkich naczyo, neurony,
hepatocyty, astrocyty,
komórki mezangialne
kłębuszków nerkowych,
neutrofile, monocyty,
makrofagi, fibroblasty
nerki, jelito, niewielkie
ilości w sercu i komórkach
łożyska
ośrodkowy układ nerwowy,
komórki śródbłonka,
komórki nabłonka
kanalików nerkowych ,
komórki epitelialne jelita

Ekspresja i wydzielanie endoteliny-1
•
Endotelina-1 nie jest magazynowana w komórkach, ale
ulega syntezie de novo pod wpływem takich czynników jak:
rozciąganie komórek mięśniowych, hipoksja,
niedokrwienie, kwasica metaboliczna, angiotensyna II,
trombina, wazopresyna argininowa, cząsteczki HDL i LDL,
cytokiny (IL-1, IL-2, IL-6, TNF-α, THF-β), oraz czynniki
wzrostu (EGF, FGF, IGF-1)
•
Wydzielanie endoteliny zapoczątkowuje aktywacja
fosfolipazy C
prowadząca do zwiększenia stężenia
zewnątrzkomórkowego wapnia
i
stymulacji kinazy
białkowe
j.
•
Indukcja wewnątrzkomórkowej syntezy cGMP wywołana
działaniem przedsionkowego peptydu natriuretycznego
(ANP), tlenku azotu (NO), prostacyklin (PGI2), endoteliny-3,
heparyny, glikokortykosteroidów powoduje obniżenie
produkcji endoteliny-1.
•
Stężenie endoteliny-1 w osoczu jest zazwyczaj niewielkie,
gdyż jest ona głównie wydzielana (przez błonę komórkową
stykającą się bezpośrednio ze ścianą naczynia), a jej reszta
jest szybko wychwytywana przez swoiste receptory
(ETA i
ETB).
•
Działanie endoteliny w warunkach fizjologicznych jest
niewielkie i obejmuje głównie rozszerzanie naczyo działając
przez śródbłonkowe receptory ETB. Jednakże w sytuacjach
patologicznych takich jak: stymulacja mięśnia sercowego
przez neurohormony i cytokiny lub wzrost obciążenia
wstępnego obserwuje się zwiększenie stężenia peptydu,
pobudzenie ich receptorów na komórkach mięśni gładkich
oraz skurcz tych ostatnich.

Wpływ endotelin na układ krążenia
Endoteliny w zdrowym sercu wpływają na kurczliwośd miocytów, inotropię, chronotropię
mięśnia sercowego oraz powstawanie arytmii. Mogą one także działad mitogennie na
fibroblasty i wywoływad przerost kardiomiocytów.
ET-1 w komórkach śródbłonka i komórkach mięśni gładkich naczyo bierze udział w
neowaskularyzacji i proliferacji mięśni gładkich
Endotelina może brad udział w patogenezie nadciśnienia tętniczego, nie tylko dlatego, że
zwiększa całkowity opór obwodowy, ale i poprzez upośledzenie prawidłowego
funkcjonowania nerek
Oddziałuje ona także na układ renina-angiotensyna-aldosteron, wazopresynę i peptydy
natriuretyczne, przez co wpływa na homeostazę wodno-elektrolitową.
Zwiększa ona także wydzielanie aldosteronu w wyniku pobudzenia receptorów
zlokalizowanych na powierzchni warstwy kłębuszkowej kory nadnerczy.
Endotelina wpływa także na uwalnianie innych czynników kurczących naczynia takich jak
wazopresyna, angiotensyna II, adrenalina, noradrenalina, tromboksan A2.
Sprzyja ona powstawaniu stanów zapalnych i zwiększa infiltrację makrofagów i leukocytów
poprzez nasilanie produkcji cytokin m.in. czynnika martwicy guza, IL1, IL6, IL8, czynnika
stymulującego kolonie granulocytów i makrofagów.
Pobudza proliferację komórek mięśni gładkich poprzez aktywację receptorów ET-A oraz
adhezję granulocytów obojętnochłonnych i agregację płytek.
Ekspresja ET-1 może przyczyniad się także do pękania blaszki miażdżycowej i powstania
ostrych incydentów niedokrwiennych. Stężenie endoteliny jest podwyższone u pacjentów z
ostrymi zespołami wieocowymi, zawałem mięśnia sercowego i samoistnym nadciśnieniem
.
Działanie tlenku azotu i endoteliny na komórki mięśni
gładkich naczyo
Mięśni gładkich naczyń
SKURCZ
Śródbłonek
NOS
ECE
ET
B
ET
B
ET
Duża-ET-1
ET-1
PIP
IP
NO
C
GMP
+
+
+
-
-
L-arginina
G
A
3
q
Peptydy natriuretyczne
W skład tej grupy wchodzą następujące hormony:
przedsionkowy peptyd natriuretyczny ANP (atrial natriuretic peptide)
mózgowy peptyd natriuretyczny BNP (brain natriuretic peptide)
peptyd natriuretyczny C CNP (C-natriuretic peptide)
urodilatina i DNP
Do biologicznych działao peptydów natriuretycznych należy
natriureza, rozszerzanie naczyo,
supresja systemu RAA
.
Peptydy natriuretyczne są syntetyzowane i magazynowane w postaci prohormonów
•
ANP
jest wytwarzany głównie w
przedsionkach serca
•
BNP
syntetyzowany jest głównie w
komorach
pomimo, iż po raz pierwszy został odkryty w
mózgu
(stąd nazwa mózgowy peptyd natriuretyczny).
Peptydy ANP i BNP, a także receptory dla nich występują także w rdzeniu kręgowym,
przysadce mózgowej, nerkach, nadnerczu
•
CNP
wytwarzany jest głównie w
komórkach śródbłonka
w
naczyniach
, w mniejszej ilości w
sercu
•
Urodilatyna
jest produkowana głównie w
nerkach
, natomiast obecnośd
DNP
stwierdzono
w ludzkim
osoczu
i
przedsionkach serca
.

Funkcje peptydów natriuretycznych
Peptydy natriuretyczne wpływają na prawidłowe funkcjonowanie śródbłonka poprzez
rozkurcz mięśniówki gładkiej naczyo, działanie antagonistyczne w stosunku do układu
renina-angiotensyna-aldosteron
oraz hamowanie aktywności presyjnej układu
noradrenergicznego.
ANP poprzez rozszerzenie naczyo, zwiększenie filtracji kłębuszkowej i wydzielania
sodu oraz poprzez inhibicję systemu RAA odgrywa istotną rolę w regulacji wodno-
elektrolitowej oraz w kontroli ciśnienia.
ANP zwiększa także przepuszczalnośd naczyo, co umożliwia przepływ płynów z
przestrzeni wewnątrznaczyniowej do tkanki śródmiąższowej.
CNP w ścianach naczyo i fibroblastach wpływa na napięcie i proliferację przyległych
komórek mięśni gładkich sąsiadujących fibroblastów. Nie wykazuje on działania
natriuretycznego, posiada jednak właściwości wazodylatacyjne.
Peptyd DNP obecny w ludzkim osoczu i przedsionkach serca
wykazuje natomiast
działanie natriuretyczne i rozszerza on naczynia krwionośne.
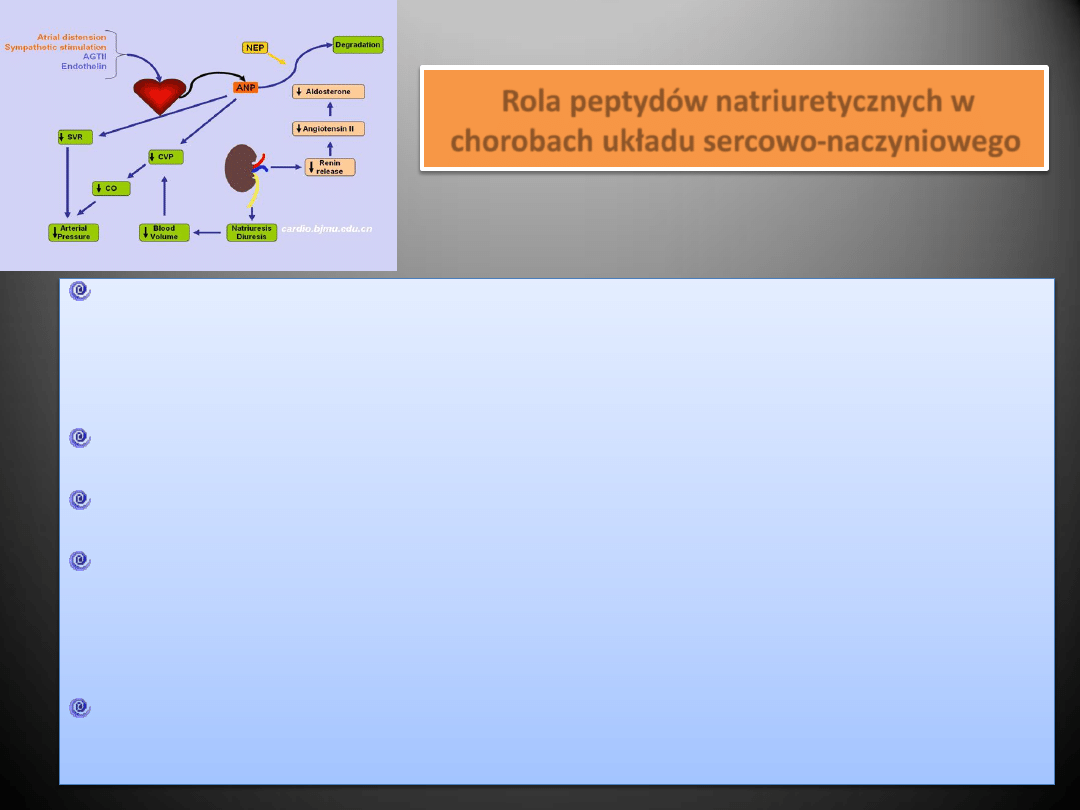
Rola peptydów natriuretycznych w
chorobach układu sercowo-naczyniowego
Peptydy natriuretyczne osłabiają aktywnośd systemu RAA (ograniczenie przebudowy mięśnia
sercowego), hamują wydzielanie aldosteronu, ograniczają syntezę kolagenu, hamują stymulowaną
przez AngII, ET-1, FGF (czynnik wzrostu fibroblastów) i IGF (insulinowy czynnik wzrostu) syntezę DNA.
Hamowanie wydzielania reniny, aldosteronu i endoteliny, obniżanie aktywności ACE oraz rozkurcz
mięśni gładkich naczyo przez peptydy natriuretyczne powoduje zmniejszenie obciążenia następczego i
wstępnego serca oraz obniżenie ciśnienia tętniczego krwi.
Peptydy natriuretyczne wykazują działanie antymitogenne w stosunku do komórek mięśni gładkich
naczyo, komórek śródbłonka i miocytów sercowych.
Peptydy natriuretyczne chronią mięsieo sercowy modulując uszkodzenia niedokrwienno-reperfuzyjne,
włóknienie i przerost.
Wzrost wewnątrzkomórkowego cGMP i aktywacja kinazy białkowej C spowodowane działaniem
peptydów hamują wewnątrzkomórkowe sygnały wzrostowe, przez co ograniczają martwicę w mięśniu
sercowym. Inhibicja przerostu i włóknienia, hamowanie wytwarzania angiotensyny II i endoteliny-1
przez peptydy natriuretyczne są zjawiskami korzystnymi dla niewydolnego serca, jednakże procesy
pro-apoptotyczne stymulowane także przez te peptydy mogą prowadzid do utraty komórek
kardiomiocytów i dalszego uszkadzania serca
Peptydy natriuretyczne są także zaangażowane w regulację systemu koagulacyjnego i fibrynolitycznego
m.in. poprzez hamowanie ekspresji PAI-1 indukowanej angiotensyną II i TF
Peptydy natiuretyczne
Poziom osoczowych peptydów natriuretycznych jest podwyższony u
osób zastoinową
niewydolnością serca
i we
wczesnej fazie zawału mięśnia sercowego
.
Zwiększona sekrecja ANP i BNP u pacjentów z niewydolnością krążenia korelowała z
„ciężkością” choroby, a także z przerostem serca. Utrzymujący się przez dłuższy czas
podwyższony poziom BNP koreluje z
powiększeniem lewej komory
i z jej
zmniejszoną
kurczliwością
, u pacjentów z zastoinową niewydolnością serca i zawałem. Z badao klinicznych
wynika, że BNP może służyd jako specyficzny marker dysfunkcji lewej komory
Stężenie osoczowe
NTpro-peptydów BNP i CNP
może byd wykorzystywane jako marker
diagnostyczny.
Poziom
NTpro-BNP
w osoczu u pacjentów z chorobą mózgowo-naczyniową jest dobrym
markerem kolejnego incydentu niewydolności serca, a poprawnośd prognozy jeszcze wzmacnia
jednoczesne oznaczenie białka C reaktywnego.
Jednakże przy wyciąganiu wniosków dotyczących funkcji lewej komory i rokowania pacjenta na
podstawie pomiaru NTpro-BNP lub BNP należy uwzględnid takie parametry jak m.in.
funkcjonowanie nerek, nadwaga itp.
Oznaczenie poziomu NTpro-CNP może wnieśd dodatkowe informacje dotyczące patofizjologii
niewydolności serca i odpowiedzi krążenia obwodowego na ten stan.

Receptory adrenergiczne
Podstawowymi regulatorami rzutu minutowego serca, zwłaszcza w czasie wysiłku fizycznego i/lub
stresu psychicznego (tzw. sytuacje „walki i ucieczki”, ang. fight-or-flight), są współczulny układ
nerwowy i mechanizm Franka Starlinga. Stymulacja układu sympatycznego powoduje uwolnienie
noradrenaliny ze współczulnych zakooczeo nerwowych w sercu oraz adrenaliny z rdzenia
nadnerczy. Mediatory te, działając poprzez błonowe receptory beta-adrenergiczne (ang. beta-
adrenergic receptor, β-AR) zlokalizowane na komórkach sercowych, powodują przyspieszenie
akcji serca oraz wzrost kurczliwości kardiomiocytów. Oba te mechanizmy są odpowiedzialne za
wzrost rzutu minutowego serca pod wpływem stymulacji współczulnej.
W skład rodziny receptorów adrenergicznych wchodzi dziewięd typów receptorów:
•
receptory
1A
-,
1B
-,
1D
- adrenergiczne oddziałujące głównie na białko Gq (stymulacja
fosfolipazy C)
•
receptory
2A
-,
2B
-,
2C
- adrenergiczne oddziałujące głównie na białko Gi (inhibicja cyklazy
adenylowej)
•
receptory
1
-,
2
- i
3
-adrenergiczne wiążące głównie białko Gs (stymulacja cyklazy adenylowej).
Receptory
adrenergiczne
1
2
1
2
Lokalizacja Komórki presynaptyczne serce mięśnie
docelowe zakończenia
gładkie
nerwowe
Efekt skurcz rozkurcz siły skurczu relaksacja
naczyń naczyń
częstości akcji wydzielania NP
kontrola serca
uwalniania NA przewodzenia
agregacja przez węzeł PK
płytek
rozkurcz naczyń
wydzielania ADH
i reniny

Receptory adrenergiczne
W strukturze receptorów adrenergicznych sprzężonych z białkami G występuje
charakterystycznych
siedem hydrofobowych
obszarów o długości 20-25
aminokwasów przedzielonych ośmioma hydrofilowymi regionami o zmiennej
długości.
Każda z hydrofobowych domen tworzy
transmembranową helisę
, a łączące je
hydrofilowe pętle wystają po stronie zewnątrz- i wewnątrzkomórkowej.
Region N-koocowy receptora znajduje się po stronie zewnątrzkomórkowej błony,
natomiast fragment C-koocowy po stronie cytoplazmatycznej. Sekwencje regionów
hydrofobowych tworzących helisę transmembranową wykazują duże podobieostwo
Receptory sprzężone z
białkami G
są glikoproteinami, wszystkie posiadają
konserwatywne sekwencje (od 1 do 3) miejsc N-glikozylacji w pobliżu N-kooca
receptora.
Aktywacja receptorów
-adrenergicznych
Receptory adrenergiczne występują na powierzchni komórek, gdzie aktywowane wiązaniem
agonistów wywołują uruchomienie szlaków sygnałowych. Transdukcja sygnału rozpoczyna się
od przyłączenia agonisty (aminy katecholowej) do receptora w błonie komórkowej, co
stymuluje interakcje receptorów z białkami G
Wszystkie białka G składają się z trzech podjednostek:
G
, G
i G
i są zlokalizowane po
stronie cytozolowej błony komórkowej.
Kiedy białko
G jest połączone z cząsteczką GDP
znajduje się ono w stanie
nieaktywnym.
W wyniku oddziaływao białko-receptor następuje uwolnienie GDP z miejsca wiążącego GTP
w białku G, i przyłączenie do tego miejsca GTP.
Aktywowane białko G
następnie
oddysocjowuje
od receptora i stymuluje enzym efektorowy. Hydroliza GTP wprowadza białko
G z powrotem w stan nieaktywny. Przyłączenie liganda do receptora wywołuje poprzez
stymulację białka G aktywację lub inhibicję enzymu generującego przekaźnik II typu.
Białka G mogą stymulowad
cyklazę adenylową
i
guanylową
,
fosfolipazy C i A2
,
fosfodiesterazy
oraz
kanały wapniowe i potasowe
.
Rodzaj aktywowanego przekaźnika i uruchomionego szlaku zależy od rodzaju białka, np.
białko Gs stymuluje cyklazę adenylową i przyczynia się do zwiększenia stężenia cAMP, białko
Gq/11 uruchamia szlak fosfolipazy C (PLC) i stymuluje metabolizm fosfoinozytydów, a białko
Gi/o hamuje aktywnośd cyklazy adenylowej i obniża stężenie cyklicznego AMP.
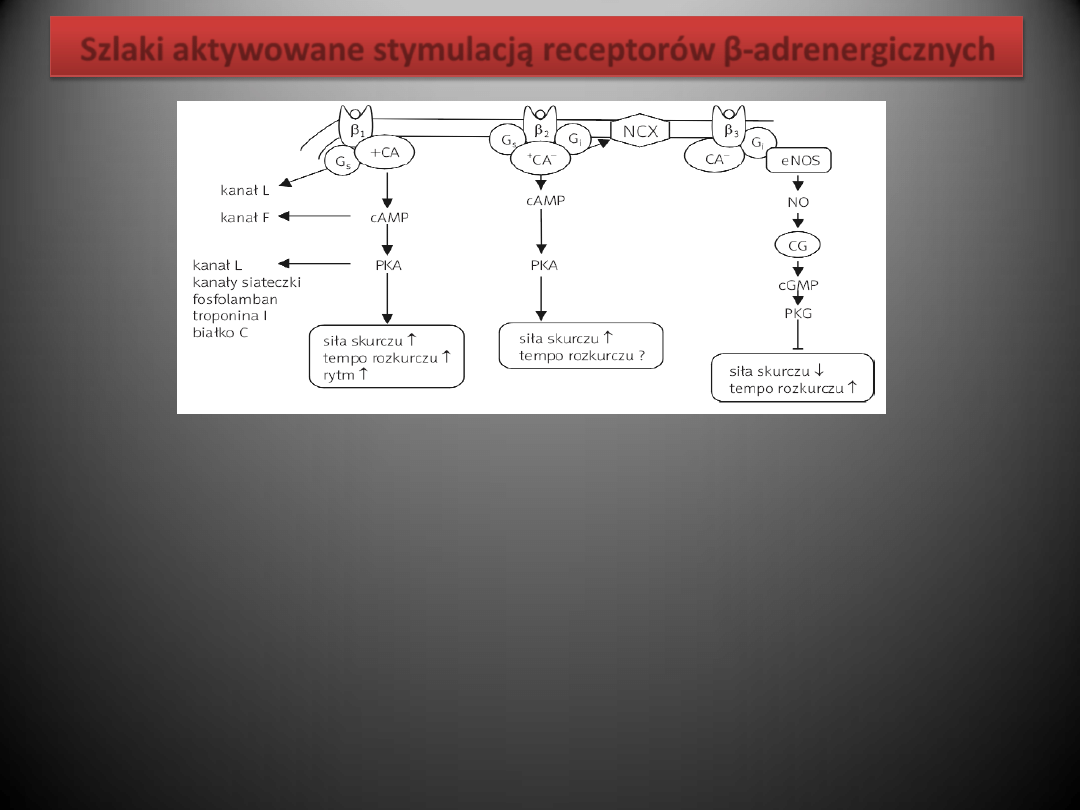
Szlaki aktywowane stymulacją receptorów β-adrenergicznych
Aktywacja β1-AR powoduje aktywację cyklazy adenylowej, wzrost komórkowej produkcji cAMP i stymulację
przez cAMP kinazy białkowej typu A. Enzym ten, poprzez przenoszenie reszt kwasu fosforowego z
cząsteczki ATP na różne białka (fosforylacja), powoduje zmianę właściwości tych białek. Efektem
czynnościowym fosforylacji białek przez kinazę białkową A (ang. protein kinase A, PKA) w sercu jest
przyspieszenie akcji serca i przewodzenia p-k, wzrost siły skurczu i przyspieszenie rozkurczu mięśnia
sercowego oraz szereg efektów metabolicznych.
W sercu ssaków i w sercu ludzkim β2-AR są sprzężone zarówno z białkiem Gs, jak i z białkiem Gi. Jednakże w
zdrowym sercu w sumie przewagę mają efekty związane z aktywacją cyklazy adenylowej i zwiększoną
produkcją cAMP.
NO aktywuje cyklazę guanylową, co prowadzi do wzrostu komórkowego stężenia cyklicznego GMP (cGMP). Z
kolei cGMP aktywuje kinazę białkową G (PKG) oraz fosfodiesterazę typu drugiego (PDEII). Kinaza białkowa
G fosforyluje: a) kanał wapniowy typu L, co przeciwnie do fosforylacji przez PKA zmniejsza prąd
wapniowy i siłę skurczu, oraz b) troponinę I, co – podobnie jak fosforylacja przez PKA – przyspiesza
rozkurcz. PDEII jest enzymem rozkładającym cAMP i tym samym prowadzącym do zmniejszenia
aktywności PKA i stopnia fosforylacji jej substratów.

Znaczenie receptorów
-adrenergicznych w chorobach układu
sercowo-naczyniowego
•
Dysfunkcja lewej komory prowadzi do aktywacji neurohormonalnej (systemu RAA, układu
współczulnego, uwalniania cytokin), co jest korzystne w ostrej fazie, jednakże przedłużająca
się stymulacja prowadzi do stopniowego pogarszania funkcjonowania serca. Zwiększona
stymulacja β-adrenergiczna jest główną przyczyną progresji niewydolności serca. Uważa się,
że stopieo aktywacji współczulnej odwrotnie koreluje z przeżywalnością pacjentów HF.
Główne zmiany obserwowane w układzie receptorów β-AR w niewydolności serca to
:
1) wzrost ekspresji i aktywności kinazy β-ARK;
2) spadek wrażliwości receptorów β1 i β2 na katecholaminy;
3) spadek gęstości β1 (zmniejszenie ilości mRNA o ok. 50%);
4) wzrost gęstości receptorów β3;
5) wzrost ekspresji białka Gi;
6) wzrost powinowactwa receptora β2 do białka Gi.
•
Stymulacja receptorów
1-adrenergicznych w sercu prowadzi do dodatniego efektu
inotropowego, luzitropowego i chronotropowego, w którym pośredniczy białko Gs i wzrost
stężenia cAMP, natomiast aktywacja receptorów β2-adrenergicznych wywołuje relaksację
mięśni gładkich naczyo, a przez to przyczynia się do zmniejszenia całkowitego oporu
obwodowego i obniżenia ciśnienia krwi. W zdrowym sercu ekspresja receptorów
2-
adrenergicznych jest niewielka, natomiast w niewydolnym sercu receptory te mogą stanowid
nawet 40% całkowitej puli receptorów ze względu na drastyczny spadek ilości receptorów
1.

Znaczenie receptorów
-adrenergicznych w chorobach układu
sercowo-naczyniowego
Ciągła aktywacja receptorów adrenergicznych stymuluje remodelling lewej komory,
obumieranie komórek na drodze nekrozy lub apoptozy oraz retencję wody i soli.
Pobudzenie układu adrenergicznego stymuluje uwalnianie reniny, a przez to i
wytwarzanie angiotensyny II, co jeszcze przyspiesza progresje niewydolności serca.
Aktywacja receptorów a1 i
-adrenergicznych przez noradrenalinę może, poprzez
stymulowanie syntezy kolagenu, prowadzid do zwłóknienia oraz przerostu mięśnia
sercowego.
Stymulacja receptorów
1 powoduje przyspieszenie akcji serca i zwiększoną jego
kurczliwośd oraz pobudzenie tworzenia reniny, co z kolei może przyczyniad się do
wystąpienia zaburzeo przepływu krwi i wzrostu napięcia ścian naczyo, prowadząc
do uszkodzenia śródbłonka i pękania blaszki miażdżycowej. W przeciwieostwie do
receptorów
1, receptory
2 mogą mied kardioprotekcyjne działanie, ich
pobudzenie w badaniach na modelach zwierzęcych hamowało apoptozę w
kardiomiocytach przypuszczalnie za pośrednictwem białka inhibitorowego Gi
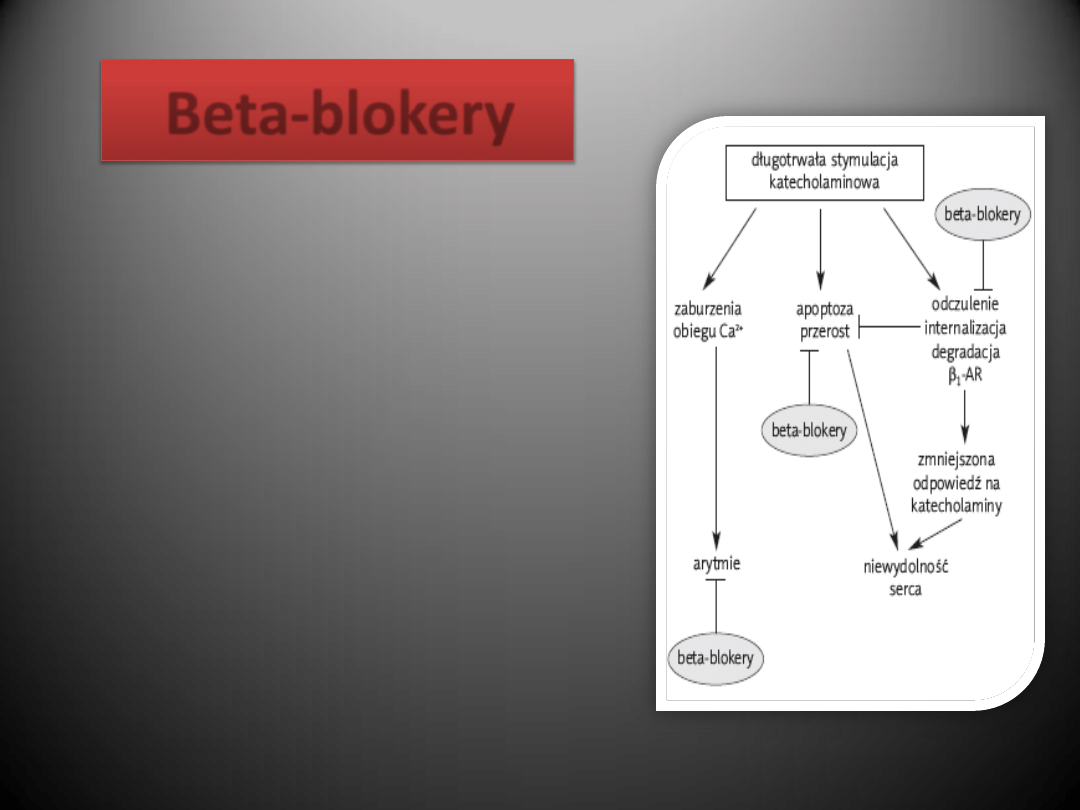
Beta-blokery
Przewlekła zwiększona stymulacja sercowego
układu
-adrenergicznego jak wykazano
jest toksyczna dla serca i może leżed u
podstaw patogenezy zastoinowej
niewydolności serca.
Stosowanie leków blokujących te receptory w
leczeniu osób po zawale zmniejsza ich
śmiertelnośd i ryzyko wystąpienia
powtórnego zawału.
Wykazano, że blokada receptora
1-
adrenergicznego w znacznym stopniu
poprawia stan pacjenta z niewydolnością
serca.

PODZIAŁ, MECHANIZMY POWSTAWANIA,
POWIKŁANIA.
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ.
NADCIŚNIENIE TĘTNICZE

PRZESZŁOŚD A DZIŚ
- U przodków Homo Sapiens poważnym problemem było ryzyko spadku ciśnienia, co
wiązało się z ograniczonym dostępem do wody i soli. Ryzyko to tworzyło presję ewolucyjną,
selekcjonującą osobniki wyposażone w geny, sprzyjające mechanizmom przeciwdziałającym
zmniejszaniu, (a nie zwiększaniu!) ciśnienia tętniczego.
-
Czynniki ryzyka nadciśnienia tętniczego pojawiły się dopiero w warunkach cywilizacji.
-
Nadciśnienie tętnicze znacznie częściej występuje u rasy czarnej, prawdopodobnie jako
skutek genetycznie zaprogramowanej wzmożonej aktywności enzymów syntezy amin
katecholowych, niezbędnych w łaocuchu syntez melaniny.

Ciśnienie tętnicze
– Ciśnienie wywierane przez krew na ściany tętnic.
Ciśnienie skurczowe
(maksymalne, górne) - Ciśnienie krwi
w tętnicach w czasie skurczu serca – gdy mięsień sercowy wpompowuje
krew do naczyń.
Ciśnienie rozkurczowe
(dolne) – Najniższa wartość ciśnienia
w chwili, gdy mięsień sercowy jest rozkurczony, a zastawki
półksiężycowate są zamknięte.
Nadciśnienie tętnicze
– Przewlekła choroba układu krążenia, podlegającą
długotrwałemu leczeniu, w której występuje (utrwalone – wykazane w kilku
pomiarach) zwiększone (zgodnie z zaleceniami WHO powyżej 139 mm HG
dla ciśnienia skurczowego i 89 mm Hg dla ciśnienia rozkurczowego)
ciśnienie krwi w naczyniach tętniczych.
DEFINICJE
CZYNNIKI RYZYKA
CHOROBY NADCIŚNIENIOWEJ
ŚRODOWISKOWE
GENETYCZNE
INNE
Nadwaga i otyłośd
Wiek i płed
Alkohol i papierosy
Napięcie psychiczne
(stres)
Mała aktywnośd fizyczna
Nadmierne spożycie soli
Zaburzenia metaboliczne
Cukrzyca typu 2.
Zaburzenia lipidowe
Insulinoopornośd
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADWAGA I OTYŁOŚĆ
- Otyłość jest bardzo ważnym czynnikiem, predysponującym
do wystąpienia nadciśnienia tętniczego, dlatego poszukiwane
są coraz bardziej skuteczne sposoby jej zapobiegania.
Waga ciała obecnie jest najczęściej mierzona i klasyfikowana wg tzw.
Wskaźnika Masy Ciała BMI
(ang. Body Mass Index), którego wartość oblicza
się z następującego wzoru:
BMI = masa ciała (kg) / wzrost
2
(m
2
)
Według WHO interpretacja wyniku jest następująca:
BMI <18,5 kg/m
2
- niedowaga
BMI 18,5–24,9 kg/m
2
- masa prawidłowa
BMI 25,0–29,9 kg/m
2
- nadwaga
BMI >30,0 kg/m
2
- otyłość
Podstawowe ogniwa łączące otyłość i nadwagę z patogenezą nadciśnienia
tętniczego:
- Zaburzenia hemodynamiczne
- w porównaniu z nadciśnieniem
tętniczym u osób szczupłych, w otyłości nadciśnienie tętnicze
charakteryzuje się zwiększoną objętością krwi i zwiększonym
rzutem serca.
-
Insulinooporność
- komórki tłuszczowe wydzielają substancję
o nazwie TNF-
powodującą insulinooporność.
-
Zwiększenie aktywności układu renina-angiotensyna-aldosteron (RAA)
-
Wzrost aktywności układu współczulnego
- skurcz naczyń obwodowych i
związany z tym wzrost oporu naczyniowego
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADWAGA I OTYŁOŚD
Tkanka tłuszczowa składa się z komórek tłuszczowych (adipocytów).
- Wśród substancji produkowanych przez adipocyty szczególną rolę
w patogenezie chorób układu sercowo-naczyniowego przypisuje się
leptynie.
- Efekt hipertensyjny leptyny dokonuje się przede wszystkim poprzez
pobudzenie układu współczulnego oraz pobudzenie mięśni gładkich
naczyń do rozrostu.
Komórka tłuszczowa – adiopocyt
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADWAGA I OTYŁOŚD
Błona komórki
Trójglicerydy
Jadro komórki
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
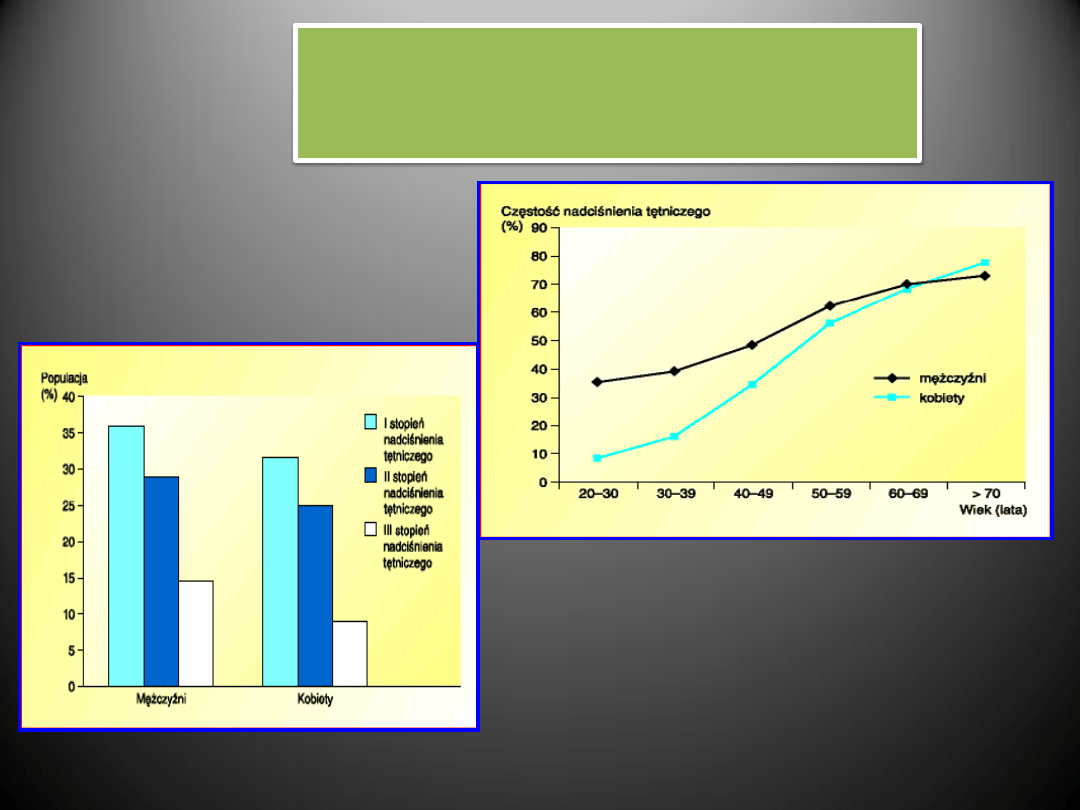
WIEK I PŁEĆ
Przybliżona liczba osób w Polsce chorujących
na nadciśnienie tętnicze
Częstośd występowania nadciśnienia tętniczego w Polsce
w zależności od wieku i płci
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
PALENIE TYTONIU
Nikotyna i jej metabolit powstający w organizmie palacza – kotynina – przyczyniają się do
wzrostu stężenia substancji odpowiedzialnych za zwężanie naczyo – angiotensyny II i
endoteliny I. Skutkiem tego jest upośledzone rozszerzania naczyo krwionośnych i
predyspozycja do występowania stanów zakrzepowych w naczyniach.
Nikotyna
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
PALENIE TYTONIU
Substancje utleniające, pochodzące z dymu tytoniowego ułatwiają proces utleniania
cholesterolu LDL. Powstający w ten sposób związek jest czynnikiem silnie uszkadzającym
komórki śródbłonka.
1
Obniżone
HDL:LDL
Złogi
LDL
Utleniony
LDL
Miejsce
zapalne
Proliferacja
komórek
mięśniowych
Miażdżyca
2
Zmniejszenie
elastyczności
ściany
naczynia
Nadciśnienie
Udar
Zawał
Działanie utlenionego LDL

CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADMIERNE SPOŻYCIE SOLI
Na podstawie badao epidemiologicznych przeprowadzonych na licznej populacji udowodniono, że
nadmiar spożywanej soli powoduje wzrost ciśnienia tętniczego. Zjawisko to wiąże się z pojęciem
sodowrażliwości.
Ze względu na nadmierne spożycie soli w przeważającej części populacji ludzkiej, celowe jest
ograniczenie jej spożycia.
Wg zaleceo dieta powinna zawierad < 6g soli dziennie (co odpowiada < niż 100 mmol Na)
Zmniejszenie spożycia soli o 50 mmol/dobę powoduje zmniejszenia:
-o 50% liczby osób wymagających leczenia farmakologicznego
-o 22% liczby zgonów związanych z udarem mózgu
-o 16% liczby zgonów spowodowanych chorobą wieocową
- Szacuje się, że w 50%, na wartość ciśnienia tętniczego wpływają czynniki
genetyczne.
- Okazało się, że u chorych z nadciśnieniem pierwotnym częściej występują
odmiany genów, które u nosiciela zwiększają wchłanianie sodu w nerkach.
Dochodzi wówczas do nadmiernego gromadzenia sodu w organizmie, co
sprzyja podwyższeniu ciśnienia. Przypuszcza się, że taki mechanizm
powoduje wzrost ciśnienia tętniczego u osób spożywających duże ilości soli
kuchennej (chlorku sodu).
- Współczesny stan badań nad pierwotnym nadciśnieniem tętniczym
dowodzi, że niezbędnym warunkiem do jego powstania jest poligenowe
uwarunkowanie genetyczne. Osoby z pierwotnym nadciśnieniem tętniczym
wykazują w 1 chromosomie polimorfizm alleli genu kodującego
angiotensynogen (AGT). Występuje także polimorfizm insercyjno-delecyjny
genu kodującego konwertazę angiotensyny w 17 chromosomie.
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
CZYNNIKI GENETYCZNE
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
ZABURZENIA METABOLICZNE
Cukrzyca
Przyczynami rozwoju nadciśnienia tętniczego towarzyszącego cukrzycy są:
- Zwiększenie oporu obwodowego (stymulacja układu współczulnego),
- Zwiększenie objętości krwi i wzrost resorpcji sodu w nerkach,
- Zaburzenie czynności układu renina-angiotensyna-aldosteron (RAA),
- Uszkodzenie śródbłonka naczyniowego,
- Rozwój miażdżycy (wzrost oporu obwodowego) i angiopatii cukrzycowej,
- Produkty tkanki tłuszczowej wywołujące insulinooporność i zwiększone
wydzielanie insuliny,
- Glomerulopatia cukrzycowa (nadciśnienie nerkowe) jako powikłanie
cukrzycy.

Insulinooporność
- Osoby z opornością na insulinę obciążone są większym ryzykiem
rozwoju nadciśnienia niż pozostała część populacji.
- Insulinooporność może wpływać na rozwój nadciśnienia, między
innymi poprzez zmiany gospodarki jonami wapnia
w komórkach mięśni gładkich naczyń.
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
ZABURZENIA METABOLICZNE
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
ZABURZENIA METABOLICZNE
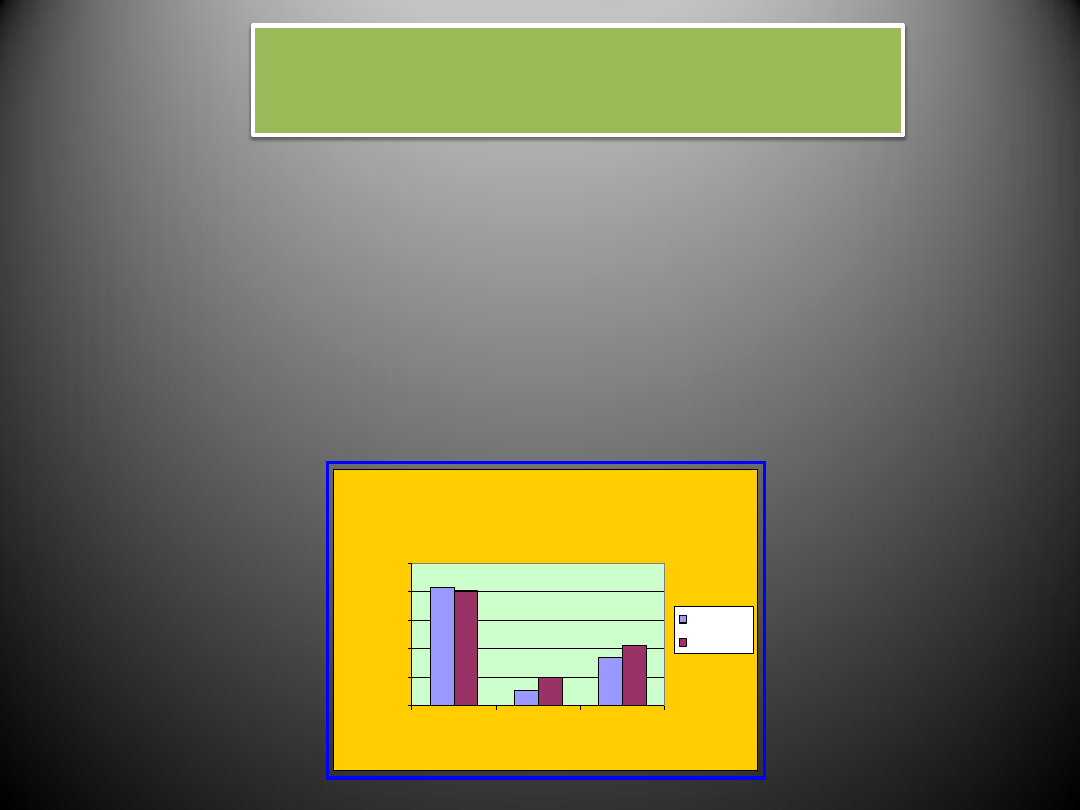
Zaburzenia lipidowe
Patogeneza nadciśnienia tętniczego w przypadku zaburzeń lipidowych wiąże się z następującymi
zjawiskami:
- Cząsteczki lipidów o niskiej gęstości (LDL) u chorych z nadciśnieniem tętniczym łatwiej wnikają w
ścianę naczynia, uszkadzając ją.
- Rozrost mięśni gładkich naczyń utrwala nadciśnienie tętnicze.
- Hipercholesterolemia i nadciśnienie tętnicze są przyczyną dysfunkcji śródbłonka naczyń, co
niekorzystnie zmienia wydzielanie substancji naczynioskurczających i naczyniorozszerzających.
Zaburzenia lipidowe u osób z nadciśnieniem
tętniczym
0%
20%
40%
60%
80%
100%
cholesterol
>200 mg/d
HDL < 40
mg/d
TG > 150
mg/d
%
Kobiety
Mężczyźni

Objawy
NT
Krwawienie
z nosa
Szum
w uszach
Nerwowośd
i większa
potliwośd
Ból i zawroty
głowy
Najczęściej
bezobjawowo!
Zaczerwienienie
skóry
OBJAWY
RODZAJE NADCIŚNIENIA TĘTNICZEGO
N A D C I Ś N I E N I E
T Ę T N I C Z E
P I E R W O T N E (idiopatyczne)
- brak bezpośredniej przyczyny
(podłoże genetyczne, menopauza,
zła dieta, stres)
- do 95% przypadków nadciśnienia
- po 40. roku życia
W T Ó R N E
- towarzyszy innym chorobom
(gł. przewlekłym chorobom nerek,
zaburzeniom hormonalnym,
miażdżycy)
- do 30. i po 50. roku życia
- z reguły usunięcie lub wyleczenie
przyczyny nadciśnienia prowadzi
do jego wyeliminowania
MECHANIZM NADCIŚNIENIA TĘTNICZEGO
- Utrwalone nadciśnienie tętnicze charakteryzuje się takim
zwiększeniem całkowitego obwodowego oporu naczyniowego (TPR),
że spoczynkowy przepływ krwi w narządach organizmu pozostaje
prawidłowy.
- Bezpośrednie przyczyny nadciśnienia polegają więc na zmienionej
czynności i strukturze obwodowych tętnic dużego krążenia.
- Występuje nadmierna ekspresja czynników sprzyjających zwężeniu
i przebudowie ściany tętnic lub niedostateczna ekspresja czynników
rozszerzających naczynia obwodowe.
- Istotną rolę w mechanizmie nadciśnienia odgrywa czynnik
neurogenny: spoczynkowa aktywność współczulna skierowana do naczyń
krwionośnych i do serca jest podwyższona, większe jest także uwalnianie
noradrenaliny zwężającej tętnice i neuropeptydu Y (NPY, związek
wzmagający naczyniozwężające działanie noradrenaliny),
przebudowującego jej ściany.
- Tlenek azotu hamuje aktywację genu egr-1 (odpowiedzialny
za proliferacja miocytów i komórek śródbłonka) wywierając tym samym
korzystny wpływ antyproliferacyjny na miocyty, zmniejszając
przebudowę ściany tętnic. Dzięki tym mechanizmom tlenek azotu
przeciwdziała rozwojowi nadciśnienia tętniczego.
- Jednym z mechanizmów nadciśnienia tętniczego jest przestawienie
na wyższy poziom i zwiększona reaktywność odruchu
z chemoreceptorów tętniczych. Wzmaga on aktywność współczulną
adresowaną do naczyń krwionośnych.
MECHANIZM NADCIŚNIENIA TĘTNICZEGO

MECHANIZM NADCIŚNIENIA TĘTNICZEGO
TEORIE MACHANIZMU
POWSTAWANIA
NADCIŚNIENIA TETNICZEGO
T E O R I A F O L K O W ’A
T E O R I A G U Y T O N ’A
MECHANIZM NADCIŚNIENIA TĘTNICZEGO
TEORIA FOLKOW’A
-
Podstawowym mechanizmem nadciśnienia są zgrubiałe ściany tętnic.
-
Podwyższenie stosunku grubości ściany naczyniowej
do wewnętrznego promienia naczynia zmienia ich geometrię tak,
że zwiększa się ich odpowiedź zwężająca na wszystkie bodźce.
-
Powtarzające się stresowo – emocjonalne wzrosty ciśnienia
tętniczego, wywołane aktywnością współczulną i osłabieniem odruchu
z baroreceptorów, hamującym w prawidłowych warunkach nadmierną
aktywność współczulną, powodują po pewnym czasie adaptacyjną
przebudowę i zgrubienie warstwy mięśni gładkich w ścianach naczyń
tętniczych oporowych.
MECHANIZM NADCIŚNIENIA TĘTNICZEGO
TEORIA GUYTON’A
Podwyższenie progu
wydalania Na
Zwiększenie stężenia Na
w płynach ustrojowych
Rozciągnięcia zbiornika
tętniczego
Zwiększenie objętości krwi
(woda podąża za Na)
Autoregulacja – ogólnoustrojowe
zwężenie naczyo
Wzrost ciśnienia tętniczego
Zwiększenie przepływu
i filtracji kłębuszkowej
Zwiększenie ilości Na w moczu
pierwotnym (zw. ze wzrostem
objętości moczu pierwotnego)
Nadmiar Na napływający do
kanalików ułatwia jego
prawidłowe wydalanie
Zachowanie homeostazy objętości krwi kosztem
zwiększenia ciśnienia tętniczego
-
Nadciśnienie jest reakcją fizjologiczną utrzymującą homeostazę
płynów ustrojowych, adaptującą organizm do zmniejszonej zdolności
usuwania sodu przez nerki.
CO WPŁYWA NA WARTOŚĆ CIŚNIENIA
TĘTNICZEGO
- Ciśnienie w układzie tętnic zależy od siły i częstości skurczów serca,
oporu, jaki stawiają ściany naczyń napływającej fali krwi oraz od lepkości
krwi. Wykazuje ono charakterystyczną zmienność, związaną z pracą serca.
- Wartości ciśnienia tętniczego nie są stale jednakowe.
- W nocy ciśnienie tętnicze jest najczęściej nieco niższe niż w ciągu dnia
(spada o co najmniej 10%), co wiąże się ze spadkiem aktywności
i zmniejszeniem pobudzenia układu nerwowego.
- Praca fizyczna lub stres psychiczny mogą zwiększyć wartości ciśnienia
tętniczego. Również wysoka temperatura otoczenia, chłód i ból mogą mieć
wpływ na jego wartość.
- U człowieka zdrowego ciśnienie tętnicze zwiększa się jednak zawsze tylko
na krótki czas, np. w trakcie wysiłku fizycznego, po czym normalizuje się
bardzo szybko.
- „Zespołu białego fartucha" - chwilowego wzrostu ciśnienia podczas
badania przez lekarza.

POMIAR CIŚNIENIA TĘTNICZEGO
- Najczęstszą metodą pomiaru ciśnienia
tętniczego jest użycie sfigmomanometru
(metoda Riva-Rocci - stąd skrót RR
określający ciśnienie tętnicze)
z zastosowaniem metody osłuchowej
(Korotkowa).
- Holter (Ambulatoryjne całodobowe
monitorowanie ciśnienia krwi - ABPM) -
automatyczne, całodobowe mierzenie
ciśnienia tętniczego krwi przez specjalny
aparat składający się z mankietu
do zakładania na ramię oraz sprzężonego
z nim rejestratora.
Ciśnienie
skurczowe
Ciśnienie
rozkurczowe
Rozpoznanie
Komentarz
< 120
< 80
Ciśnienie optymalne
- Najlepsze dla zdrowia
120 - 129
80 - 84
Ciśnienie prawidłowe
- Nie powoduje szkód
130 - 139
85 - 89
Ciśnienie wysokie prawidłowe
- Zmiana stylu życia
- U osób z czynnikami ryzyka
leczenie farmakologiczne
140 - 159
90 - 99
Nadciśnienie 1. stopnia - łagodne
- Zmiana stylu życia
- Konsultacja lekarska
(wdrożenie leczenia)
160 - 179
100 - 109
Nadciśnienie 2. stopnia - umiarkowane
180
110
Nadciśnienie 3. stopnia - ciężkie
KLASYFIKACJA NADCIŚNIENIA TĘTNICZEGO
120
/
80
Ciśnienie skurczowe
Ciśnienie rozkurczowe
LECZENIE
FARMAKOLOGICZNE
NIEFARMAKOLOGICZNE
POLITERAPIA
(częściej)
MONOTERAPIA
Diuretyki tiazydowe
i tiazydopochodne
-blokery
Blokery kanału
wapniowego
Inhibitory ACE
Antagoniści receptora
angiotensynowego
ZMIANA STYLU ŻYCIA
beta
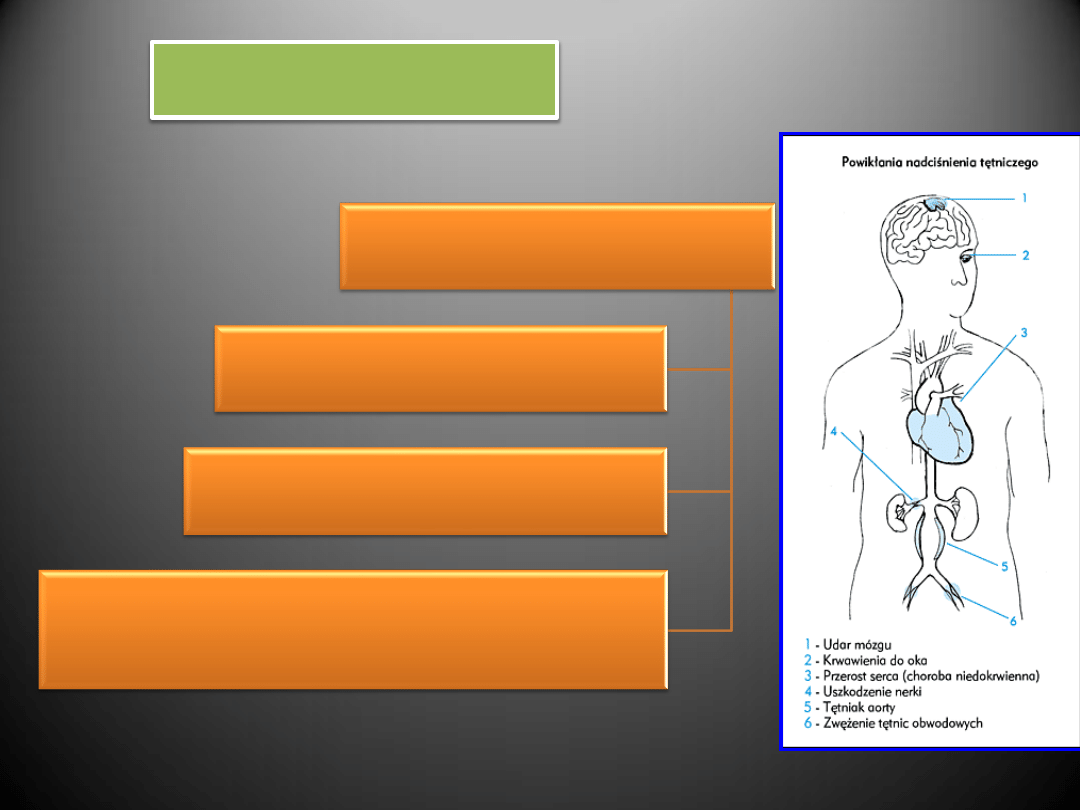
POWIKŁANIA
STADIA NADCIŚNIENIA TĘTNICZEGO
WG WHO
I stadium - brak zmian w narządach
II stadium – przerost lewej komory serca,
retinopatia nadciśnieniowa, białkomocz
III stadium – nadciśnieniowe uszkodzenia: serca
(niewydolność lewokomorowa), nerek (niewydolność nerek),
mózgu i oka (retinopatia)
- UDAR MÓZGU (APOPLEKSJA)
– Uszkodzenie mózgu na skutek zbyt
wysokiego ciśnienia krwi. Objawia się nagłym pogorszeniem lub utratą
możliwości poruszania kończynami (niedowład, paraliż), zwykle po jednej
stronie ciała. Mogą towarzyszyć zaburzenia mowy i czucia. Niekiedy objawy
nie są nasilone, a czasami ustępują same. Udar może być wynikiem
zamknięcia tętnicy (dopływ krwi, a tym samym tlenu, jest odcięty - udar
niedokrwienny) lub przerwania jej ściany i krwawienia do mózgu (udar
krwotoczny). Prawidłowe leczenie nadciśnienia tętniczego zmniejsza ryzyko
wystąpienia udaru w obydwu tych mechanizmach.
- KRWAWIENIA DO OKA
- Do struktur oka krew dociera przez bardzo małe
i delikatne tętniczki. Podwyższone ciśnienie bardzo łatwo uszkadza ich
ściany. Niewielkie krwotoki, które gdzie indziej nie miałyby znaczenia,
w siatkówce oka mogą nawet prowadzić do utraty wzroku. Prawidłowe
leczenie nadciśnienia może więc zapobiec powikłaniom w strukturach oka.
POWIKŁANIA
-
CHOROBA NIEDOKRWIENNA SERCA
- zespół objawów chorobowych będących następstwem
przewlekłego stanu niedostatecznego zaopatrzenia komórek mięśnia sercowego w tlen i
substancje odżywcze. Zaburzenie równowagi pomiędzy zapotrzebowaniem a możliwością ich
dostarczenia, doprowadza do niedotlenienia - niewydolnośd wieocowa, a nawet do zawału
mięśnia sercowego.
-
NEFROPATIA
– ok. 1/5 objętości krwi pompowanej podczas jednego skurczu serca przepływa
przez nerki. Jeśli dzieje się to pod zbyt dużym ciśnieniem, dochodzi do uszkodzenia także i tego
narządu. Nerki coraz słabiej oczyszczają więc organizm ze szkodliwych substancji. Uczestniczą w
regulowaniu poziomu ciśnienia krwi, toteż ich uszkodzenie może prowadzid do dalszego wzrostu
ciśnienia. Podwyższone ciśnienie prowadzi do niewydolności nerek.
- TĘTNIAK AORTY
– poszerzenie aorty o ponad 50% w stosunku
do jej prawidłowej szerokości.
POWIKŁANIA
Wyszukiwarka
Podobne podstrony:
Niewydolność serca mechanizmy
Niewydolno¶ć serca
Niewydolnosc serca
PRZEWLEKLA NIEWYDOLNOSC SERCA 2009wer 1 1
Przewlekła niewydolność serca
przewlekła niewydolnosc serca
Niewydolność serca i nadciśnienie tętnicze klinika i pielęgnowanie
Niewydolność serca suma
Niewydolność Serca IV rok
Niewydolność serca, Studia - ratownictwo medyczne, 3 rok, Zawansowane procedury ratunkowe
Kardiologia praktyczna 5 Niewydolność serca (1)
Kardiologia praktyczna 5 Niewydolność serca (2)
niewydolnosc serca u dzieci i niemowlat
15. NIEWYDOLNOŚĆ SERCA, Anatomia, ukł. krążenia
definicja objętości wyrzutowe?finicja pojemność minutowej jakie są fizjologiczne kompensacje niewydo
więcej podobnych podstron