
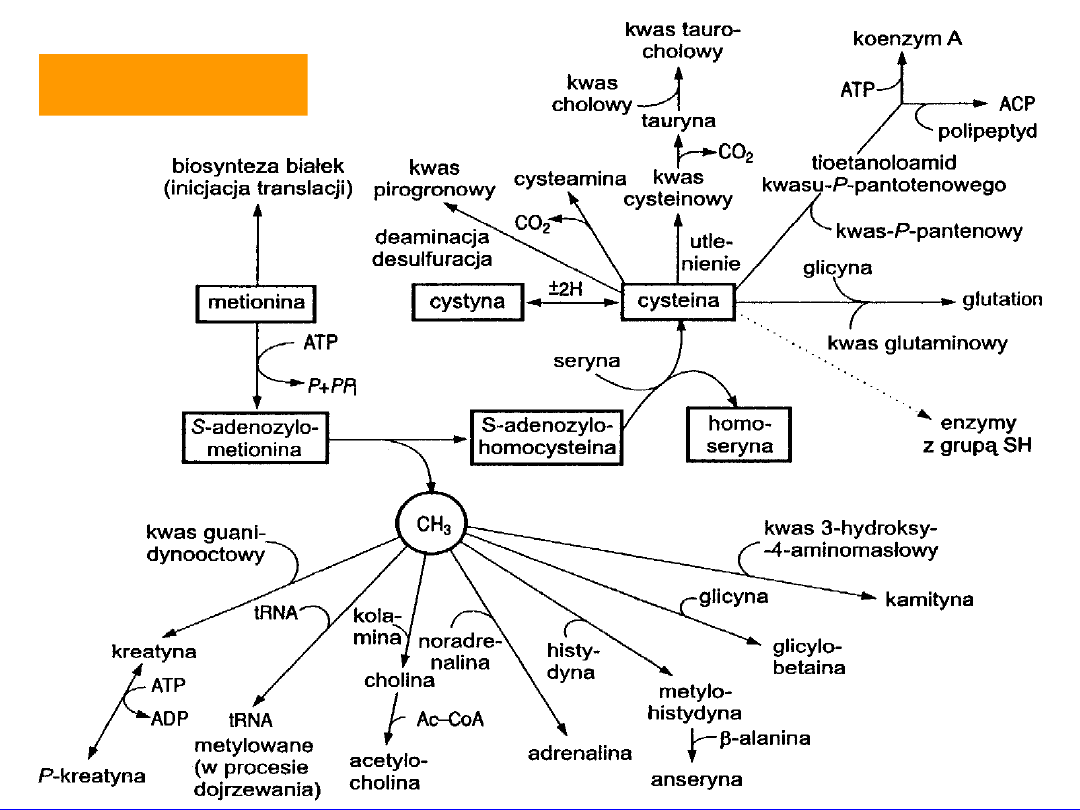
METIONINA
Egzogenna
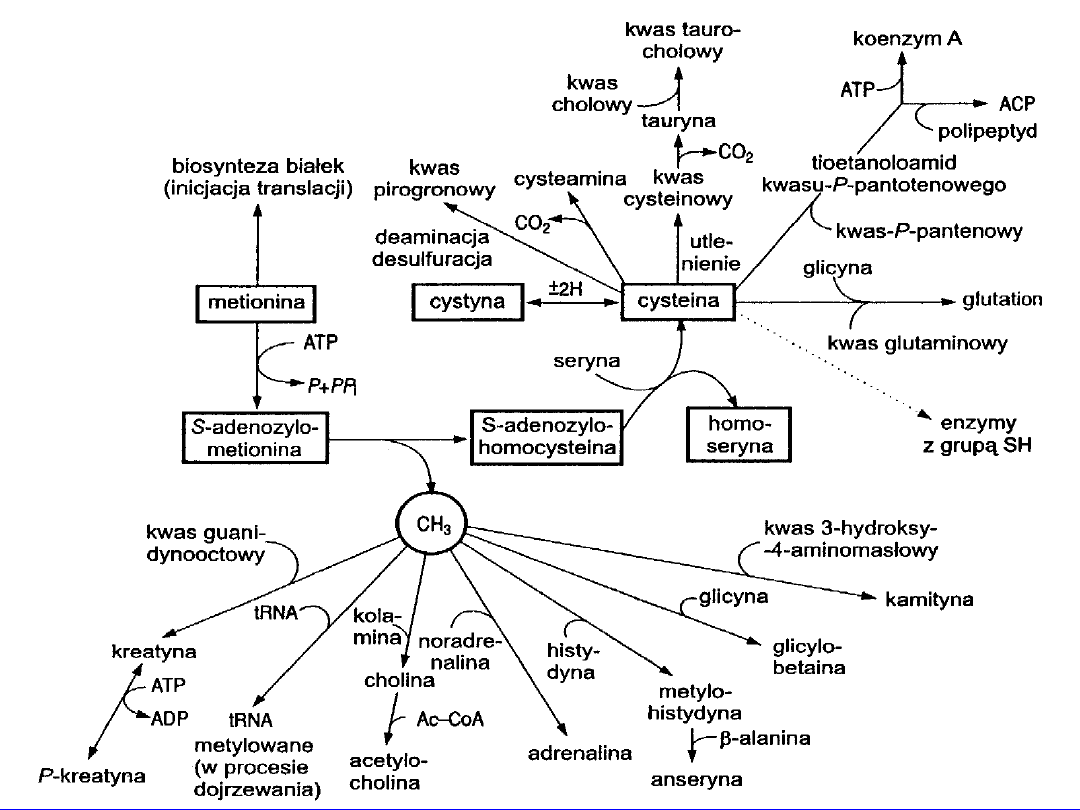
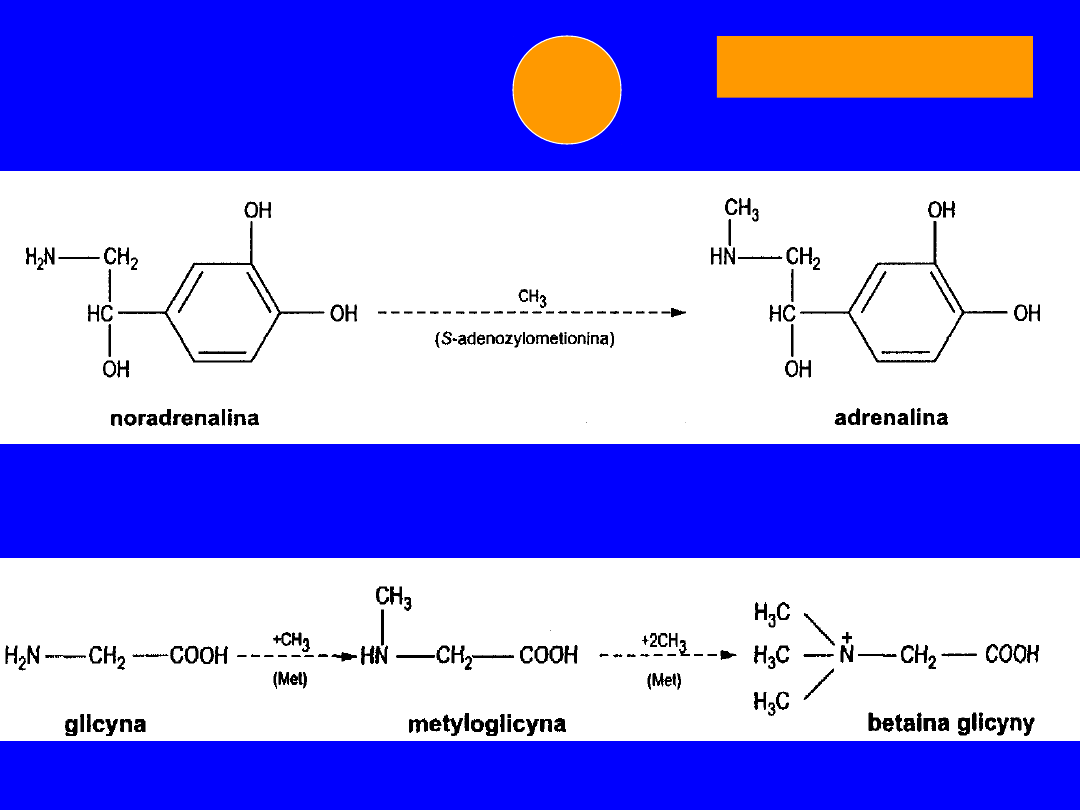
metylacje
inicjuje biosyntezę białka
metylacja w „dojrzewaniu” hnRNA
tworzenie homoseryny i cysteiny
Forma aktywana
S-adenozylometionina
dawca grup
metylowych
w reakcjach:
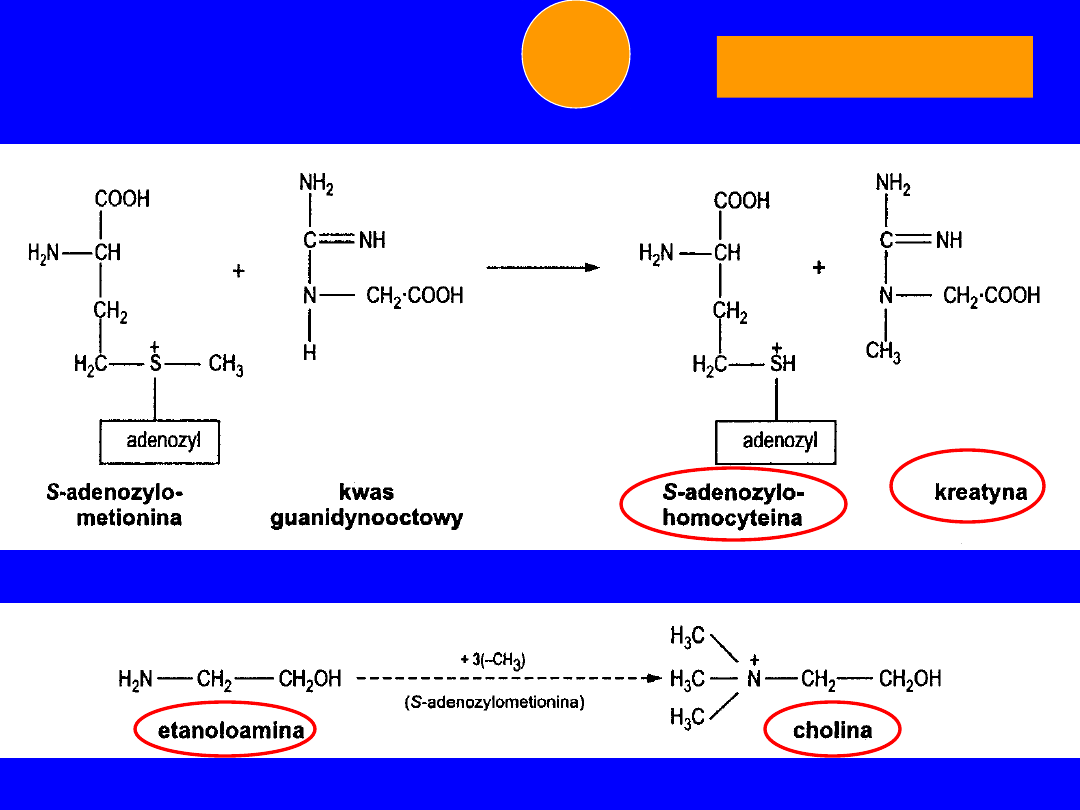
• kwas guanidynooctowy -
kreatyna
• etanolamina(kolamina) -
cholina
acetylacja acetylocholina
• noradrenalina -
adrenalina
• histydyna -
metylohistydyna
+ -alanina anseryna
Główny szlak przemian - powstawanie SAM
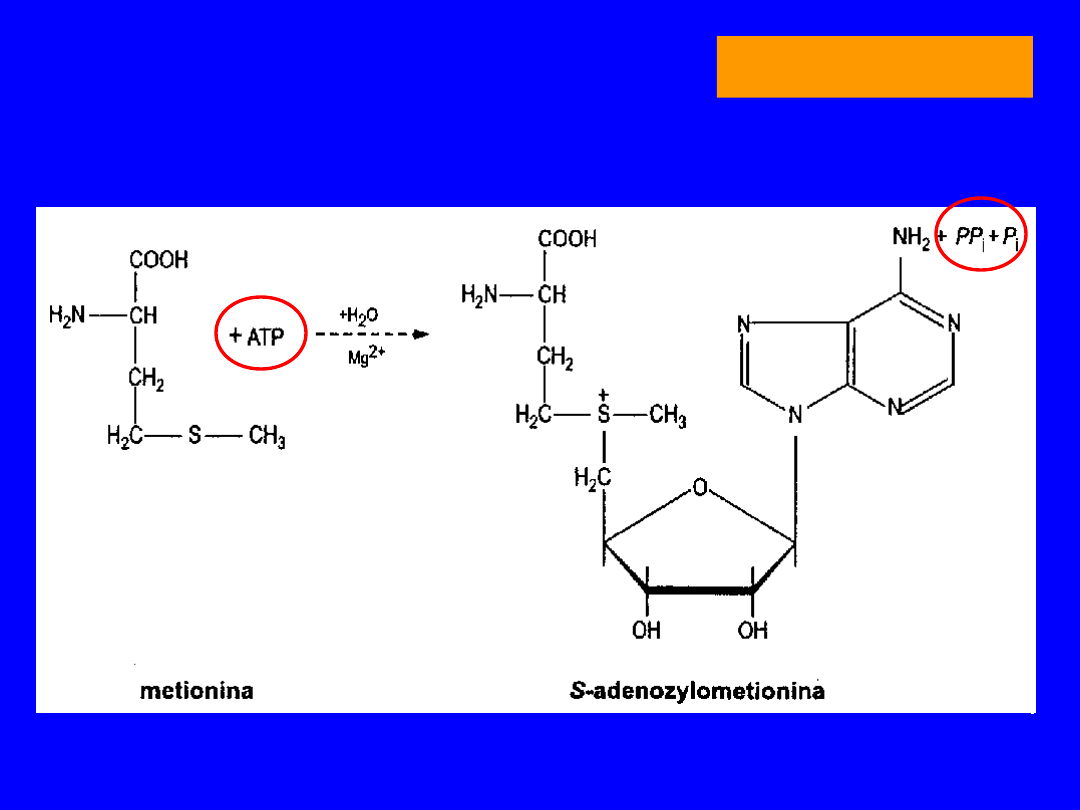
METIONINA
Główny szlak przemian - powstawanie SAM
METIONINA
+CH
3
METIONINA
+CH
3
METIONINA
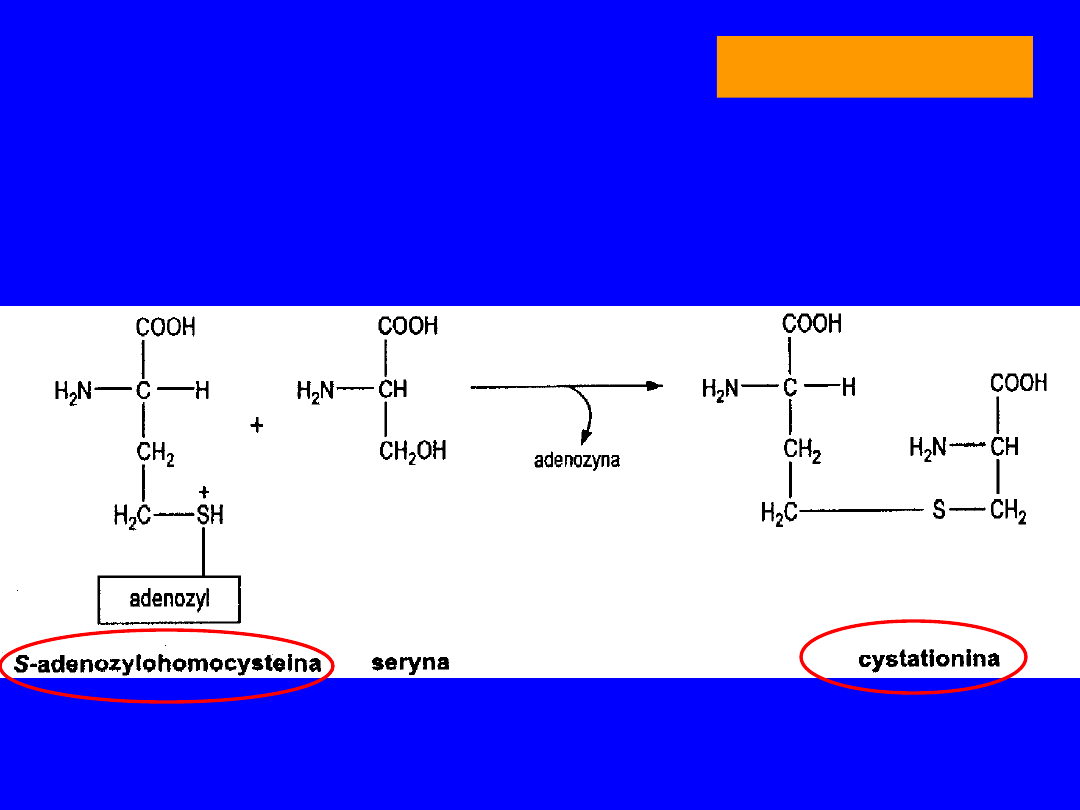
Po demetylacji powstaje S-adenozylohomocysteina
• z seryną nowe tioaminokwasy -
homoseryna
i
cysteina
• metabolitem pośrednim
cystatotionina
METIONINA
Po demetylacji powstaje S-adenozylohomocysteina
• z seryną nowe tioaminokwasy -
homoseryna
i
cysteina
• metabolitem pośrednim
cystatotionina

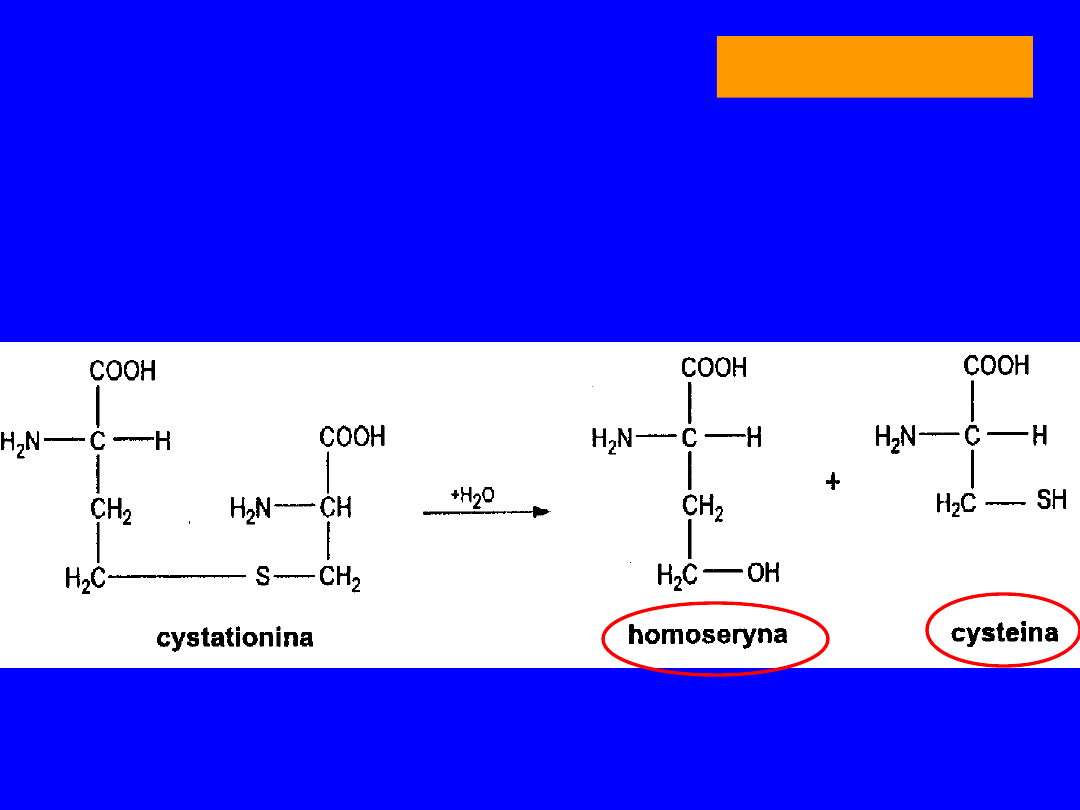
METIONINA
Homoseryna
-liaza cystatotioninowa
2-oksomaślan
NAD+
oksydacyjna dekarboksylacja
HS-CoA
propionylo-CoA
+ CO
2
+ NADH+H
+

Homocysteina (HCY)
Produkt metabolizmu metioniny
Uwalniana do osocza; krąży w postaci utlenionej jako
homocystyna
i
dwusiarczek
cystyna-HCY
w
większości związana z białkami
Nie może być wbudowana w strukturę białek
Metabolizowana do cystyny lub metioniny
(enzymatycznie)

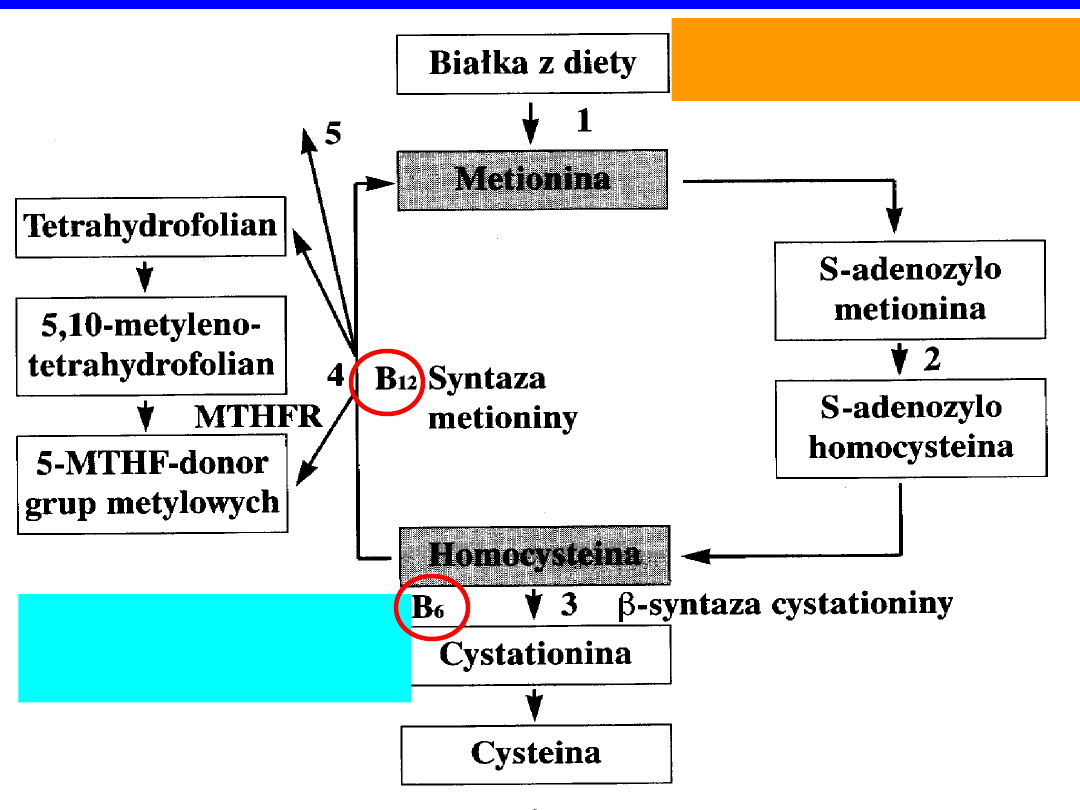
PRZEMIANA METIONINY I HOMOCYSTEINY
1. Metionina z białek pokarmowych wnika do ustroju
2. Demetylacja SAM „metioniny”
homocysteina
3. Transsufuracja homocysteiny cysteina;
koenzymem
-syntazy cystatotioniny
jest wit.
B
6
4. Remetylacja homocysteina metionina; enzymy
–
syntaza
metioniny
i
reduktaza
metylenotetrahydrofolianu
(
MTHFR
); koenzymy – wit.
B
12
i kwas foliowy
5. Nagromadzanie homocysteiny przy upośledzonym
metabolizmie
Homocysteina (HCY)
Reduktaza
metylenotetrahydrofolianu
(MTHFR);
PRZEMIANA METIONINY
I HOMOCYSTEINY
Nagromadzanie
homocysteiny
przy
upośledzonym
metabolizmie
Demetylacja
Transsulfuracja
Remetylacja

Hiperhomocystynemia
Norma
< 16 mmol/l
zależy
od metody
Umiarkowana 16-30 mmol/l
Pośrednia
30-100 mmol/l
Ciężka
> 100 mmol/l
Homocysteina (HCY)

Homocysteina (HCY)
Hiperhomocystynemia
PRZYCZYNY
1. Niedobory (lub brak) enzymów metabolizmu
metioniny:
reduktazy
metylenotetrahydrofolianu lub -syntazy
cystatotioniny
Uwarunkowane genetycznie występowanie
termolabilnej formy
MTHFR
(mutacja C677T – zamiana cytozyny na
alaninę w pozycji 6777 co daje
zamianę ala na wal); częstość
występowania homozygot 10-13% populacji
rasy białej
umiarkowana hiperohomocystynemia
, szczególnie przy
obniżonej ilości kwasy foliowego
Brak
syntazy cystationiny
(
ciężka hiperhomocyteinemia
;
1:200000), brak
MTHFR lub syntazy metioniny
Objawy – zmiany kośćca (nadmierne wydłużanie),
osteoporoza,
miażdżycowe zmiany naczyniowe, skłonność
do zakrzepów i zatorów
częstość występowania heterozygot z niedoborem -syntazy
cystationiny i
umiarkowaną hiperohomocysteinemią –
1:200

Homocysteina (HCY)
Hiperhomocystynemia
PRZYCZYNY
2. Nabyte niedobory kwasu foliowego i wit. B
6
,
B
12
(koenzymów przemiany
homocysteiny)
Jako wynik niskiej podaży w stosunku do zapotrzebowania w
diecie lub
upośledzone wchłanianie
Odwrotne korelacja między stężeniem homocysteiny i
spożyciem tych
witamin oraz ich stężeniem we krwi
Prawdopodobnie nieoptymalne stężenie witamin z grupy B
odgrywa rolę w
2/3 wszystkich przypadków
hiperhomocysteinemii

Homocysteina (HCY)
Hiperhomocystynemia
PRZYCZYNY
3. Hiperhomocysteinemia wtórna (w przebiegu
chorób np. w
niewydolności nerek, w
chorobach nowotworowych)
Przewlekła niewydolność nerek – upośledzenie
wydalania oraz zaburzenia metabolizmu
Dializa – wymywany kwas foliowy
łuszczyca, chemioterapia nowotworów (>
obrotu
komórkowego, wyczerpanie B
12
i
kwasu foliowego
Niektóre nowotwory np. sutka, jajnika, trzustki
Umiarkowana w niedoczynności tarczycy
ekspozycja na CS
2

Homocysteina (HCY)
Hiperhomocystynemia
PRZYCZYNY
4
. Jatrogenne (wywołane lekami) oraz
związane ze stosowaniem odżywek
Leki wpływające na przemiany kwasu foliowego
(metotreksat,
fenytoina) lub
wit. B
6
(np. teofilina)
barbiturany (cyt.P450)
Nadmierne spożywanie alkoholu, kawy, palenie
papierosów
duże dawki estrogenów
niedobory wit. B
6
, B
12
, kwasu foliowego
TRISOMIA - zespół Downa -
nadekspresja
syntetazy
;
b. niskie stężenie homocysteiny - rzadko chorują na
miażdżycę

Homocysteina (HCY)
Hiperohomocysteinemia
czynnik ryzyka chorób układu krążenia
1969 r McCully itd.
MECHANIZM – ATEROGENNY I
TROMBOGENNY
Autooksydacja
homocysteiny
reaktywne
rodniki
tlenowe i nadtlenek wodoru
bezpośrednie
uszkodzenie komórek
śródbłonka i utlenianie
lipoprotein LDL
Powstaje także w tej reakcji
tiolakton
homocysteiny
; wchodzi w reakcję z LDL (z apoB)
agregaty LDL-
tiolakton homocysteiny
wychwytywanie przez makrofagi komórki
piankowate dalsze
powstawanie rodników
tlenowych i postępujące
utlenienie LDL;
zmodyfikowane lipoproteiny są
immunogenne
.

Homocysteina (HCY)
Hiperohomocysteinemia
czynnik ryzyka chorób układu krążenia
ogranicza biologiczną
dostępność NO
dla
komórek
śródbłonka hamuje efekty
wazodilatacyjne
Nasila tworzenie nadtlenków lipidowych hamują
syntazę NO
w śródbłonku.
Interakcja homocysteiny ze składnikami
hemostazy:
nasila aktywność
cz. VII i V
a
obniża aktywność
białka C
Nasila
proliferację SMC
– działanie
mitogenne
Zwiększa produkcję
kolagenu
przez SMC

CYSTEINA
• Częściowo egzogenny
• keratyna (włosy i paznokcie)
• stabilizacja struktury białka
• prekursor tauryny
• synteza CoA i glutationu

CYSTEINA
KATABOLIZM
• bezpośrednie utlenienie sulfinian cysteiny
dioksygenaza cysteinowa (Fe
2+
, NAD(P)H)
• transaminacja 3-merkaptopirogronian redukcja
przez
DH-mleczanową 3-merkaptomleczan
desulfuracja
pirogronian +
H
2
S
• 3-merkaptomleczan > w moczu
Cysteina pirogronian
na kilka sposobów:
atom siarki pojawia się w H
2
S, SO
3
-2
lub SCN
-
cystynuria
- wzrost wydalania cystyny z moczem
(20-30X); wytrącanie kamieni cystynowych

CYSTEINA
Synteza koenzymu-A
cysteina + kwas fosfopantotenowy
pantotenylocysteina
-CO
2
tioetanolamid kwasu fosfopantotenowego)
+2ATP
PP
i
+ ADP
białko
przenoszące acyl (ACP)
koenzym A
Synteza glutationu
• syntetaza
-glutamylocysteinowa
• syntetaza gluationowa

CYSTEINA
•
Redukcja
- cystyna
•
Utlenienie
- kwas cysteinowy
• dekarboksylacja kwasu cysteinowego -
tauryna sprzęga kwasy żółciowe kwasy
taurocholowe
•
desulfuracja
(-H
2
S) kwas aminoakrylowy
kwas iminopropionowy -NH
3
kwas pirogronowy
•
transaminacja
kwas merkaptopirogronowy
•
dekarboksylacja
cysteamina
CYSTEINA

TREONINA
• Jedyny egzogenny hydroksy-aminokwas
TREONINA
•
aldolaza treoninowa
glicyna + aldehyd octowy
- cykl aminoacetonowy
- + FH4 seryna
aldehyd octowy dehydrogenaza aldehydowa (FAD)
kwas octowy syntetaza acetylo-CoA (Mg)
+ HS-CoA
acetylo-CoA
•
dekarboksylacja
hydroksypropyloamina
• utlenienie aminoaceton druga droga wejścia
treoniny do cyklu aminoacetonowego
• nie ulega transaminacji
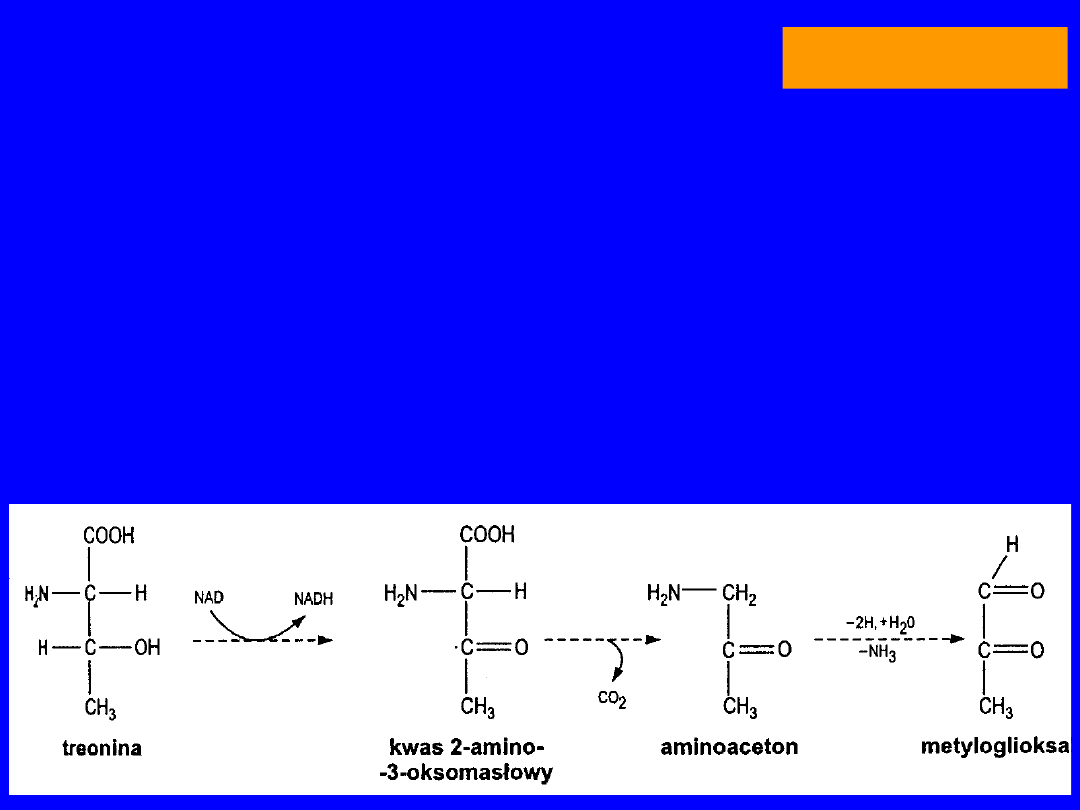
TREONINA
•
główne źródło metyloglioksalu
•
Szent-Gyorgyi - rola w regulacji podziałów komórkowych
METYLOGLIOKSAL
powstaje w
cyklu
aminoacetonowym
treonina
utlenienie
kwas 2-amino-3-oksomasłowy
(acetoaminooctowy)
dekarboksylacja
aminoaceton
aminooksydaza
metyloglioksal
metyloglioksal
utlenienie
kwas pirogronowy
oksydacyjna dekarboksylacja
acetylo-CoA +
glicyna odtworzenie kwasu acetoaminooctowego
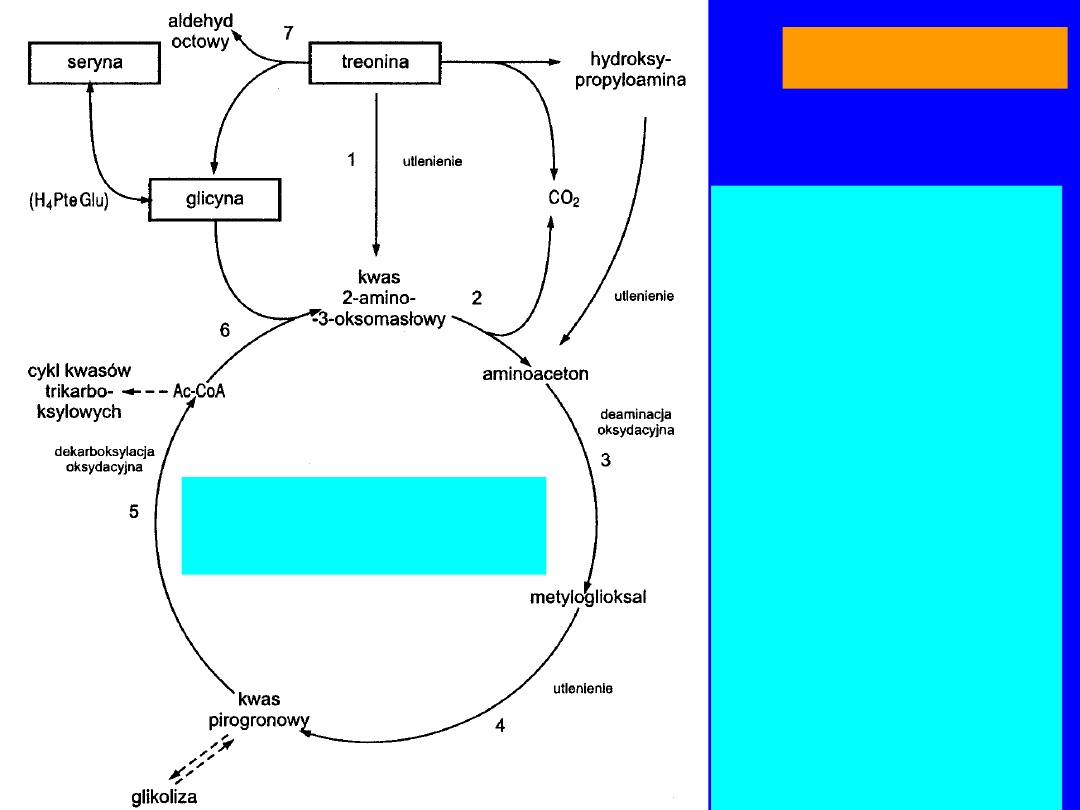
TREONINA
1. DH treoninowa
2. Dekarboksylaza
acetoaminoctanowa
3. Monoaminooksydaza
4. Oksydaza
metyloglioksylowa
5. Układ
wieloenzymowy
dekarboksylacji
oksydacyjnej
6. Syntaza
acetoaminooctowa
7. Aldolaza treoninowa
Cykl
aminoacetonowy

FENYLOALANINA
Egzogenny
•
dekarboksylacja
- enzymy bakteryjne
fenyloetyloamina
• Zasadnicze przemiany - utlenienie pierścienia
aromatycznego przy pomocy O
2
FENYLOALANINA + O
2
+
tetrahydrobiopteryna
(kofaktor)
hydroksylacja
hydroksylaza fenyloalaninowa
(monooksygenaza -
oksydaza o funkcji
mieszanej
(jeden O w produkcie a
drugi w H
2
O)
TYROZYNA + chinoid dihydrobiopteryny
Sumarycznie
fenyloalanina + O
2
+ NADH+H
+
tyrozyna + NAD
+
+
H
2
O

FENYLOALANINA
Tetrahydrobiopteryna
- przenośnik elektronów
• powstaje przez redukcję dihydrobiopteryny przez
NADPH
reduktaza dihydrofolianowa
tetrahydrobiopteryna .
• Podczas hydroksylacji fenyloalaniny forma
chinonidowa
dihydrobiopteryny ponownie
redukowana przez
reduktazę
dihydropterynową
tetrahydrobiopteryna
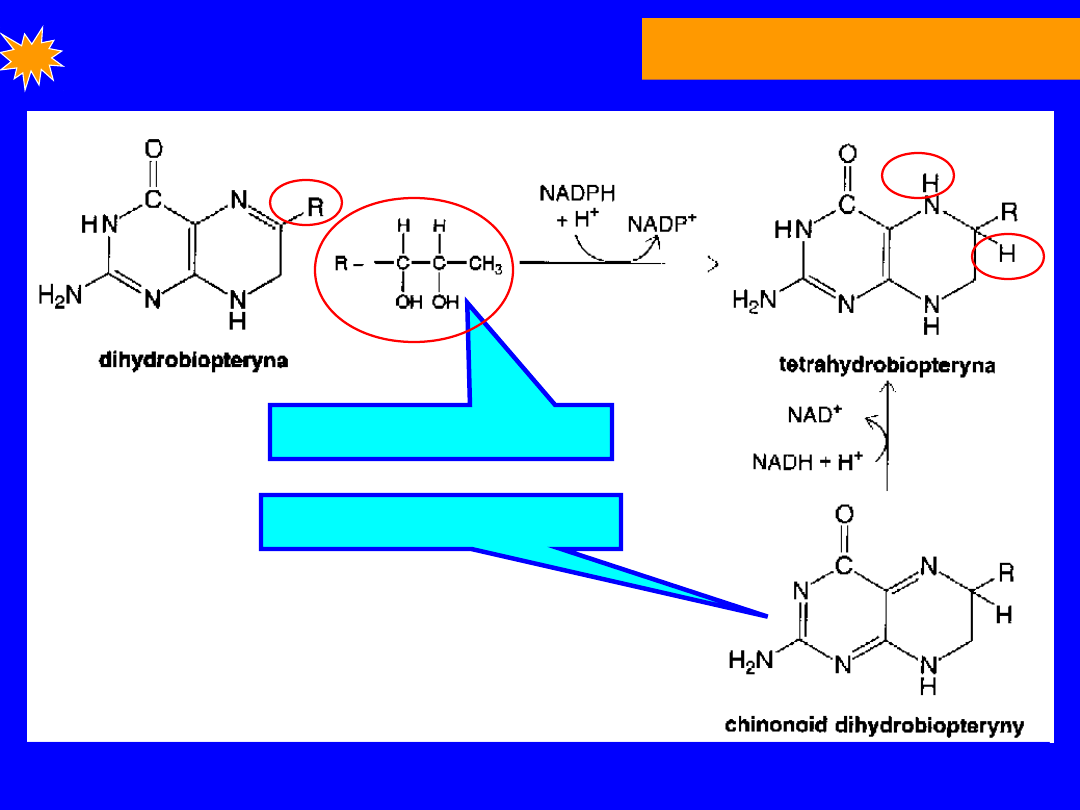
FENYLOALANINA
Reduktaza dihydrofolianowa
Reduktaza dihydropterynowa

FENYLOALANINA
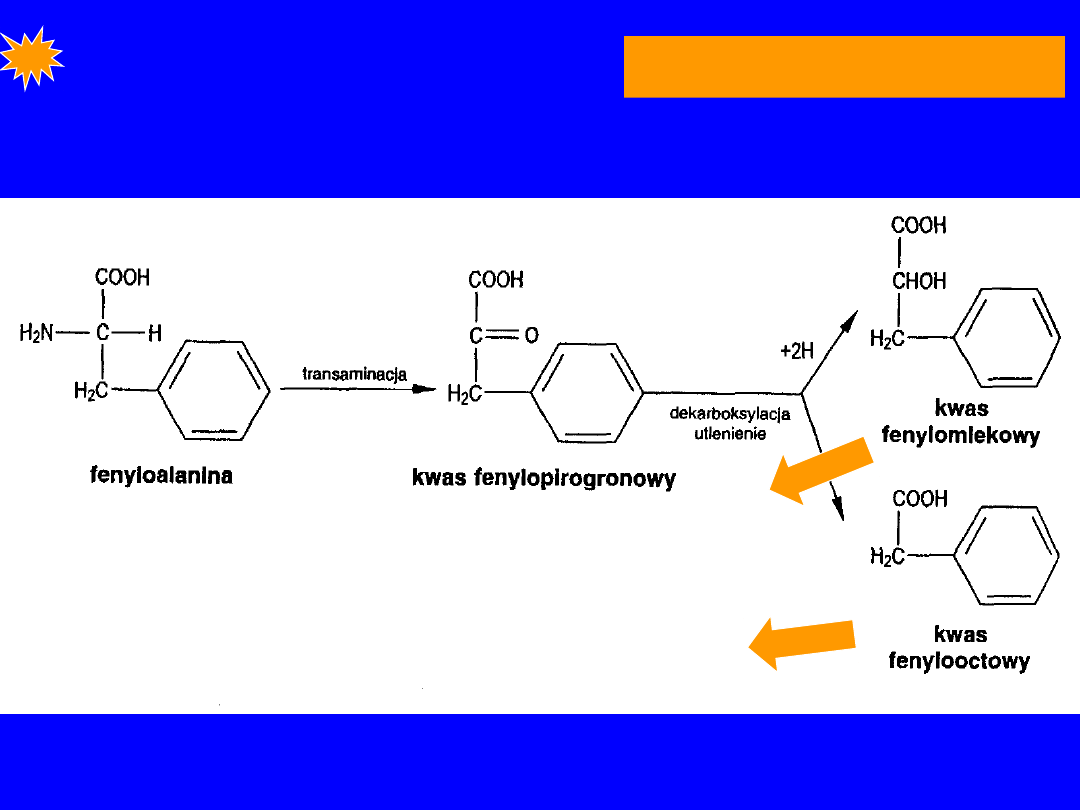
FENYLOKETONURIA;
choroba Fohlinga; niedorozwój
fenylopirogronowy
1:10 000 ; postać klasyczna; autosomalna, recesywna
• niedobór
4-monooksygenazy
(hydroksylazy
fenyloalninowej)
• homozygoci - całkowity niedobór w wątrobie
• na początku fenyloalaniny we krwi (10-20x)
• od 4 miesiąca inna droga przemiany fenyloalaniny
FENYLOALANINA
• od 4 miesiąca inna droga przemiany
fenyloalaniny
Pojawiają się w moczu
gdy fen > 1 mM
> krążenie > OUN

FENYLOALANINA
FENYLOKETONURIA
TYPY HYPERFENYLOALANINEMII
I. Fenyloketonuria - brak 4-monooksygenazy
II. Uporczywa hiperfenyloalaninemia - niedobór 4-
monooksygenazy
III. Przejściowa hiperfenyloalaninemia - opóźnienie
w
dojrzewaniu 4-
monooksygenazy
IV. Niedobór reduktazy dihydrobiopterydyny -
niedobór lub
brak
enzymu
V. Anormalna funkcja dihydrobipteryny - wada w
syntezie
dihydrobiopteryny
Typ I + II + III = 97%
Typ IV i V = 3%
Typ I 1:10 000; Irlandia 1:5000; Polska 1:10000

FENYLOALANINA
• Zaburzenia neurologiczne; zahamowanie rozwoju
umysłowego
OUN
-
zaburzenia transportu aminokwasów
utylizacji glukozy
biosyntezy cholesterolu
(tkanka mózgowa małych
dzieci
syntetyzuje cholesterol!!;
dorosłych NIE)
• wtórne zaburzenie przemian
tyrozyny
-
fenyloalanina
hamuje syntezę dopaminy
w moczu wydalany kwas
wanilinomigdałowy
(postać klasyczna)
• „mysi zapach” moczu (kwas fenylooctowy)
postać nieklasyczna fenyloketonurii
• niedobór
reduktazy dihydropterydyny
-
tetrahydropteryna
• kofaktor hydroksylazy fenyloalaninowej
FENYLOKETONURIA

TYROZYNA
TYROZYNA
transaminacja;
aminotransferaza tyrozynowa
p-HYDROKSYFENYLOPIROGRONIAN
utlenienie
hydroksylaza p-hydroksyfenylopirogronianowa
dioksygenaza
-
obydwa O przyłączane do produktu
(pierścień i -COOH)
HOMOGENTYZYNIAN
rozerwanie
pierścienia
[O
2
]
oksygenaza homogentyzynianowa
4-MALEILOACETOOCTAN
izomeryzacja;
izomeraza maleiloacetooctowa
4-FUMARYLOACETOOCTAN
hydroliza:
hydrolaza fumaryloacetooctowa
FUMARAN + ACETOOCTAN
Tyrozynemia
Alkaptonuria
Tyrozynuria
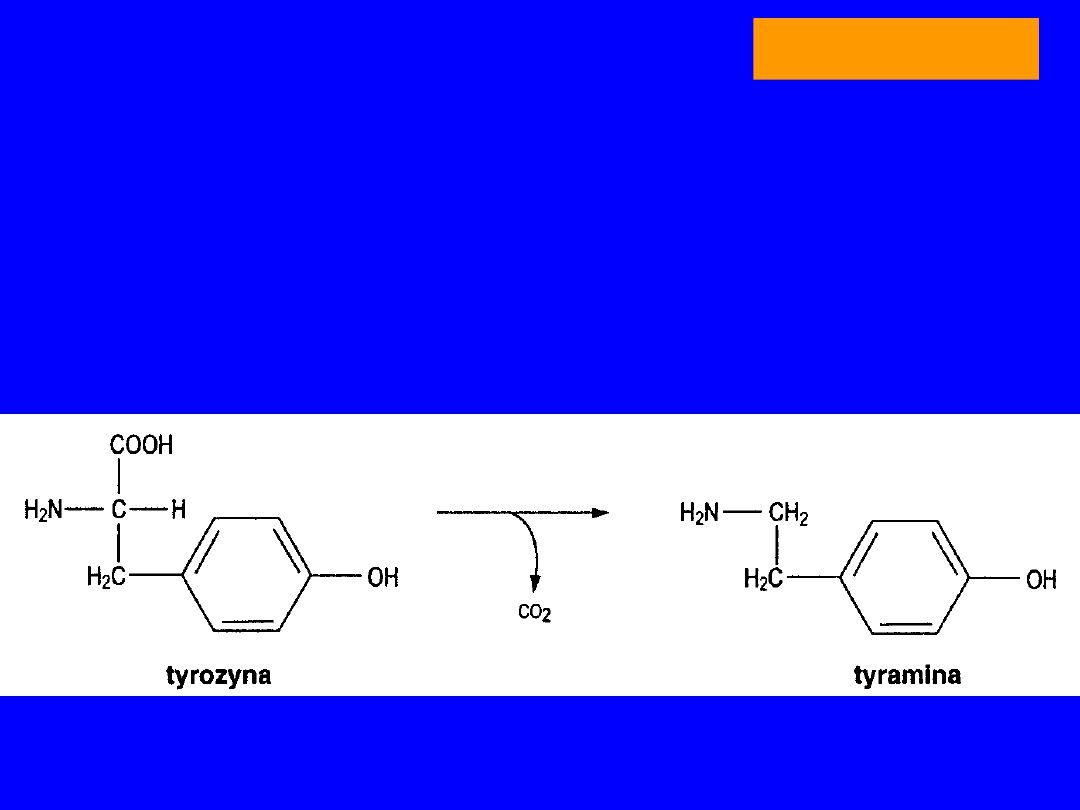
TYROZYNA
Częściowo egzogenna
• prekursor
hormonów
tyraminy, dopaminy, adrenaliny, noradrenaliny,
trójodotyroniny, tyroksyny
Tyramina
dekarboksylacja tyrozyny
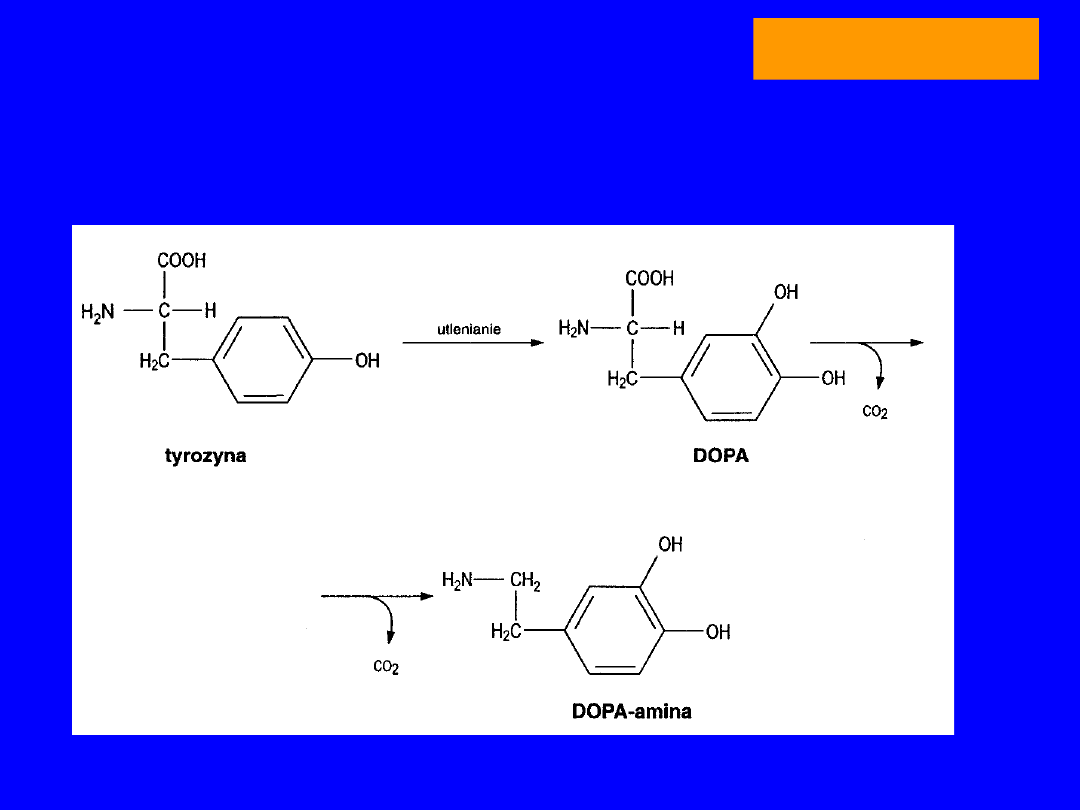
TYROZYNA
utlenienie i dekarboksylacja
dopamina

TYROZYNA
Utlenienie, hydroksylacja DOPA-aminy (
kwas
askorbinowy)
noradrenalina
metylacja
(adenozylometionona)
adrenalina
Rola - pobudzanie receptorów adrenergicznych
NA - głównie alfa -; A - alfa- i beta-adrenergiczne;
Katcholaminy - !! Inhibitorami allosterycznymi
hydroksylazy tyrozyny
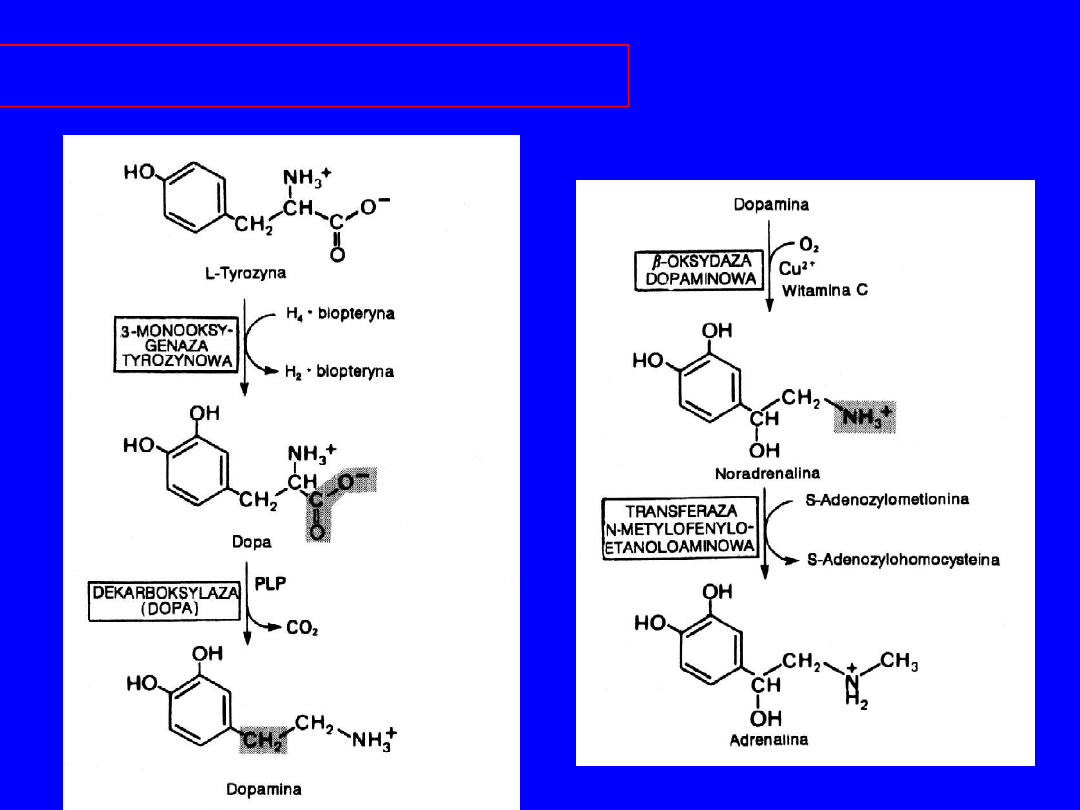
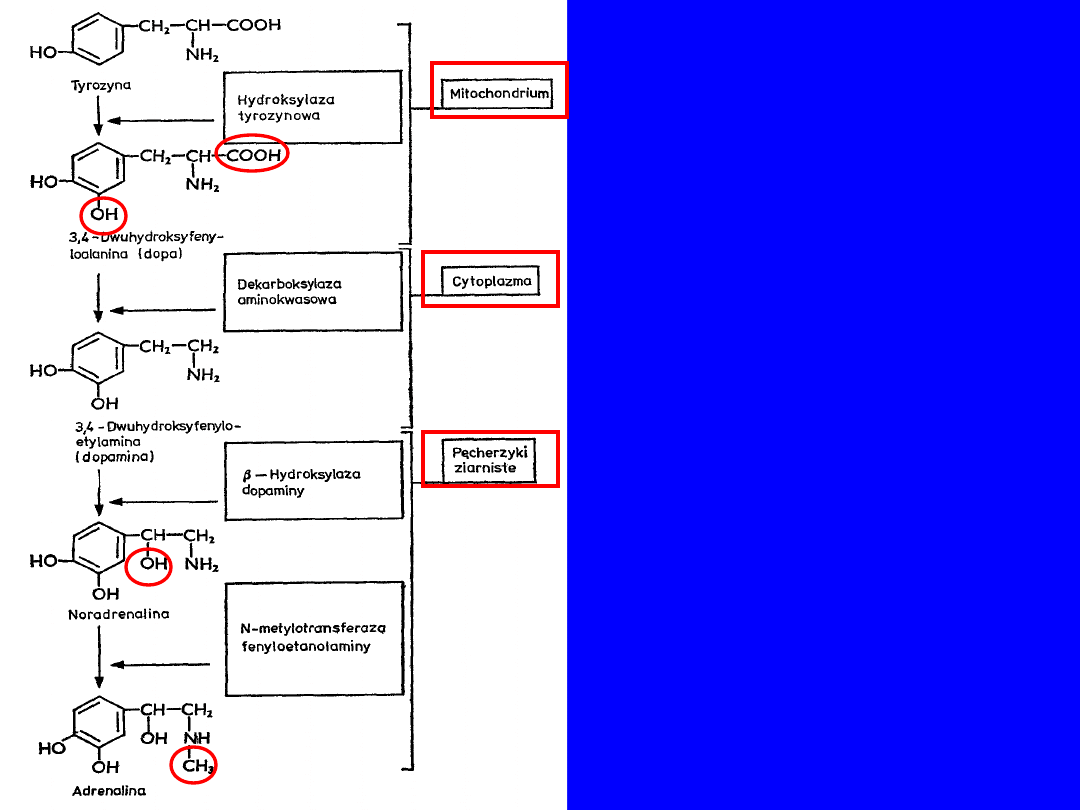
BIOSYNTEZA ADRENALINY
I NORADRENALINY
• Komórki rdzenia nadnerczy - adrenalina
• zakończenia nerwów sympatycznych - noradrenalina
BIOSYNTEZA ADRENALINY
I NORADRENALINY
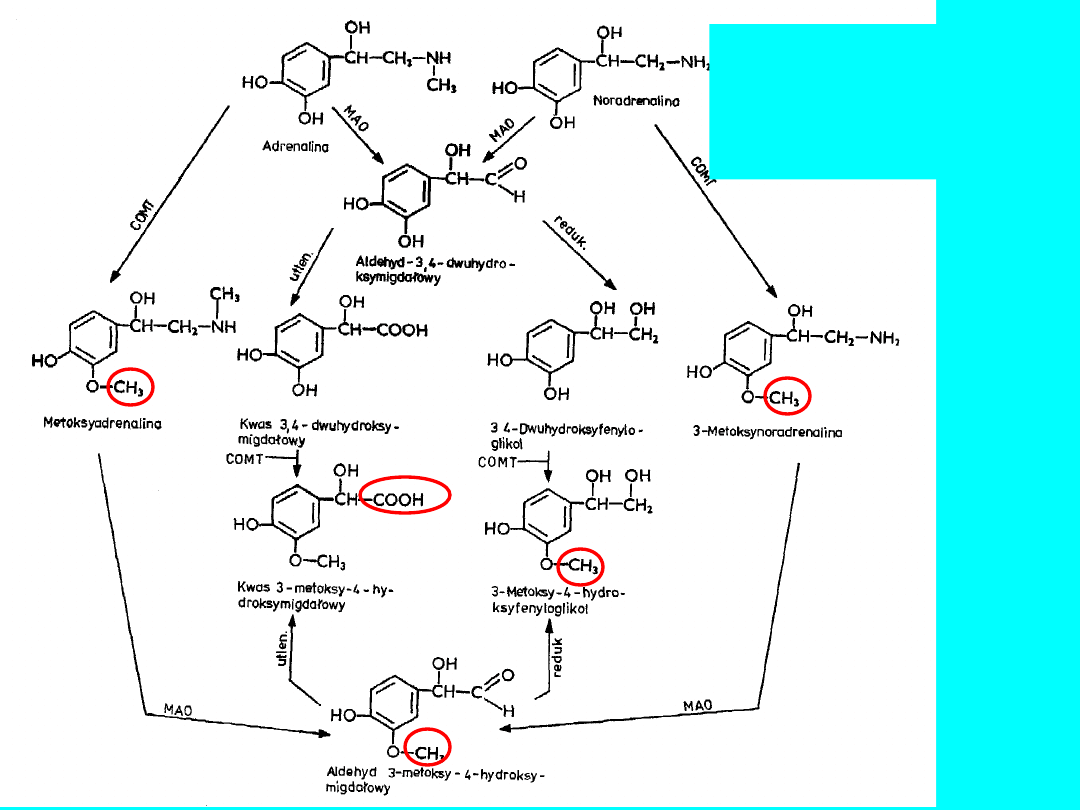
DEGRADACJA
ADRENALINY
I
NORADREANALINY
Metabolity wydalane z
moczem jako siarczany
lub połączenia z
kwasem
glukuronowym
80% wszystkich metabolitów to:
• kwas metoksy-hydroksymigdałowy
(MHM)
• metoksynoradrenalina (MNA)
• metoksyadrenalina
COMT - metylotransferaza tlenowo-katecholowa
MAO - oksydaza monoaminowa

TYROZYNA
SYNTEZA MELANINY
melanosomy w melanocytach
TYROZYNA
oksydaza o-dwufenylowa (tyrozynaza
)
DOPA
oksydaza katecholowa
DOPA-chinon
DOPA-chrom
indochinon
polimeryzacja
MELANINA (EUMELANINY, FEOMELANINY)
Albinizm

TYROZYNA
ALKAPTONURIA
• brak
oksygenazy homogentyzynowej
• nagromadzanie
kwasu
homogentyzynowego
mocz
• zawiera w pierścieniu 2 grupy -OH; jest
hydrochinonem
• który się utlenia ketony zabarwienie
niebieskie
• mocz ciemnieje na powietrzu
skutek metaboliczny niewielki
• niemożność wytwarzania produktów katabolizmu
Fen i Tyr i straty energetyczne
• brak syntezy glukozy z am. aromatycznych
• w późniejszym okresie artretyzm
Alkaptonuria

TYROZYNA
ALBINIZM
• brak tyrozynazy w melanocytach; w innych
tkankach
aktywność prawidłowa
• synteza amin katecholowych (adrenaliny i
dopaminy)
prawidłowa
• upośledzona pigmentacja
• jest 10 postaci albinizmu oczno-skórnego
• tyrozynazo ujemni - całkowity brak widocznego
barwnika
• tyrozynazo dodatni - mają nieco barwnika
• Bielactwo oczne i zabarwienie włosów i skóry
normalne
• U wszystkich albinosów - prawidłowe pole widzenia
lecz
brak widzenia stereoskopowego;
światłowstręt
Albinizm
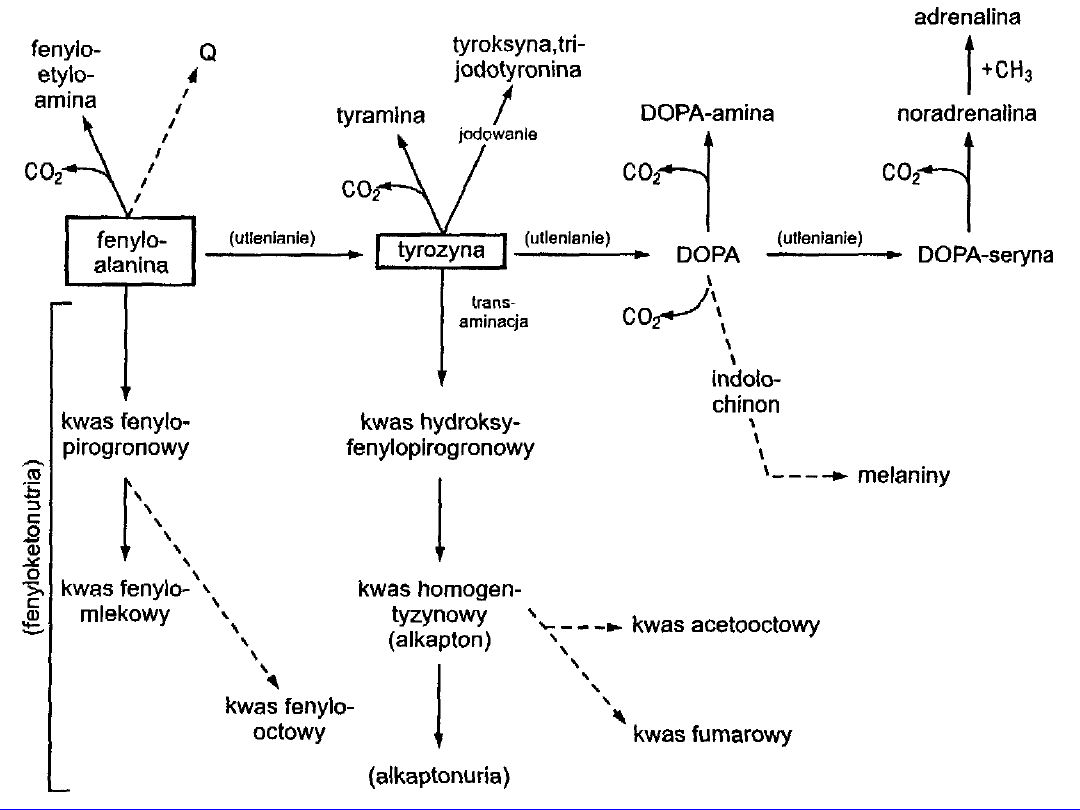

TRYPTOFAN
• egzogenny
• transaminacja „ślepa uliczka” -ketokwas
musi ulec ponownej aminacji
•
główny
szlak degradacji rozerwanie pierścienia
-
dioksygenaza
tryptofanowa
alanina
glukogenny
• kwas nikotynowy (wit.PP)
• hormony -
tryptamina
,
serotonina
,
melatonina
• w jelicie i bakterie
skatol
i
indol
- toksyczne -
zaparcia
• w śluzówce jelita grubego - utlenianie do
indoksylu
- oksydaza indolowa
• w wątrobie - estryfikacja przez aktywny siarczan
kwas indonylosiarkowy -
indykan
-
diagnostyka
• u roślin -
auksyny
np.kwas indoilooctowy
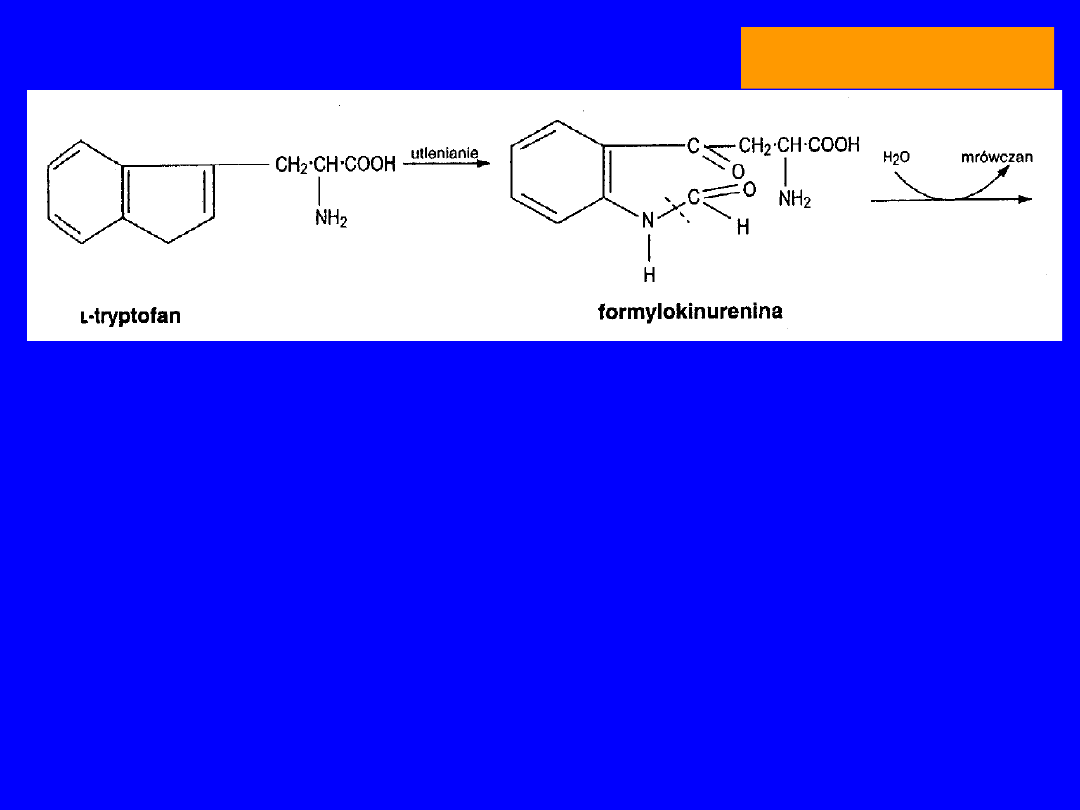
Katabolizm
Szlak kinureninowo-antranilowy
TRYPTOFAN
Dioksygenaza
oksydaza tryptofanowa
(Cu)
FORMYLOKINURENINA
+ H
2
O mrówczan HCO-H4folian
KINURENINA kwas kiniureninowy
monooksygenaza
3-HYDROKSYKINURENINA kwas ksanturenowy
kinureinaza
(zależna od wit. B
6
; niedobór B
6
- test
obciążeniowy Trp wzrost kwasu
ksanturenowego
KWAS 3-HYDROKSYANTRANILOWY +
ALANINA
rozerwanie pierścienia, -CO
2

TRYPTOFAN
Kwas -AMINO--KARBOKSYMUKONOWY
-CO
2
KWAS
PIKOLINOWY
KWAS 2-AMINOMUKONOWY
NAD
+
+NADH+H
+
KWAS 2-OKSOADYPINOWY
-CO
2
GLUTARYLO-CoA
Ligand wiążący Zn

KWAS
PIKOLINOWY
• ligand wiążący Zn (Zn-LB)
• niedobór zaburzone wchłanianie Zn
• szlak syntezy kwasu pikolinowego egzokrynna
cześć
trzustki wydzielenie do przewodu
pokarmowego
• niewydolność trzustki synteza kwasu
pikolinowego
• u dzieci do 4 m życia - niska synteza
• drugie miejsce syntezy - gruczoł mleczny -
karmienie piersią
mleko kobiece 300 M
mleko krowie < 20 M
• potrzebna wit.B
6
objaw niedoboru B
6
Objawy niedoboru
• Acrodermatitis enteropathica
• biegunka, zmiany skórne w dystalnych częściach
kończyn (dłonie, stopy), łysienie, zaburzenia rozwoju
fizycznego
Takie same objawy - niedobór Trp, B6, kwasu
pikolinowego
TRYPTOFAN

TRYPTOFAN
SYNTEZA NAD
Kwas -AMINO--KARBOKSYMUKONOWY
KWAS CHINOLINOWY
+ fosforybozylodifosforan (PRPP)
mononukleotyd kwasu nikotynowego
• W warunkach diety normalnobiałkowej 1/60
Trp może być przekształcona do niacyny;
pokrywa
zapotrzebowanie
Pelagra - u ludzi odżywiających się małą ilością
białka
lub białkiem niepełnowartościowym
• przetworzona kukurydza ma b. mało Trp
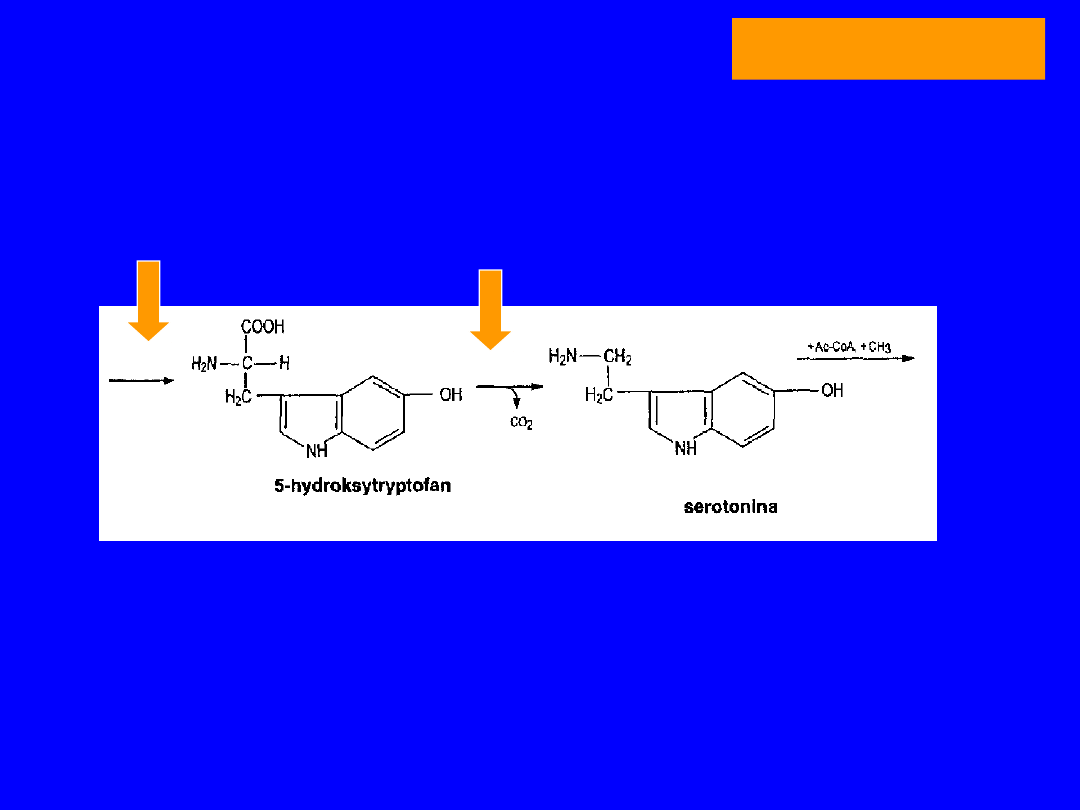
TRYPTOFAN
SYNTEZA SEROTONINY
POCHODNE 5-HYDROKSYTRYPTOFANU
•Przemiana o dużym znaczeniu ilościowym
• w wielu tkankach; ważna w mózgu i szyszynce
• Hydroksylaza - enzym o wysokim Km dla tryptofanu
Degradowana nieodwracalnie
oksydaza monoaminowa MAO
(flawina)
aldehyd 5-OH-indolooctowy + amoniak
utlenienie
kwas 5-OH indolooctowy
usuwany z moczem - oznaczanie !!
hydroksylaza
dekarboksylaza

TRYPTOFAN
SEROTONINA
• Wytwarzanie: komórki APUD (przewód pokarmowy, głównie
jelito cienkie, drzewo oskrzelowe, szyszynka
• Przekaźnik w OUN
• zwęża naczynia krwionośne, stymuluje skurcz m. gładkich
• w rakowiaku (srebrzak) guz chromochłonny jelita -
zwiększone wytwarzanie serotoniny
• zamiast 1%, 60% tryptofanu zamienia się w serotoninę
• następstwem brak tryptofanu do przekształcenia w kwas
nikotynowy
POCHODNE 5-HYDROKSYTRYPTOFANU
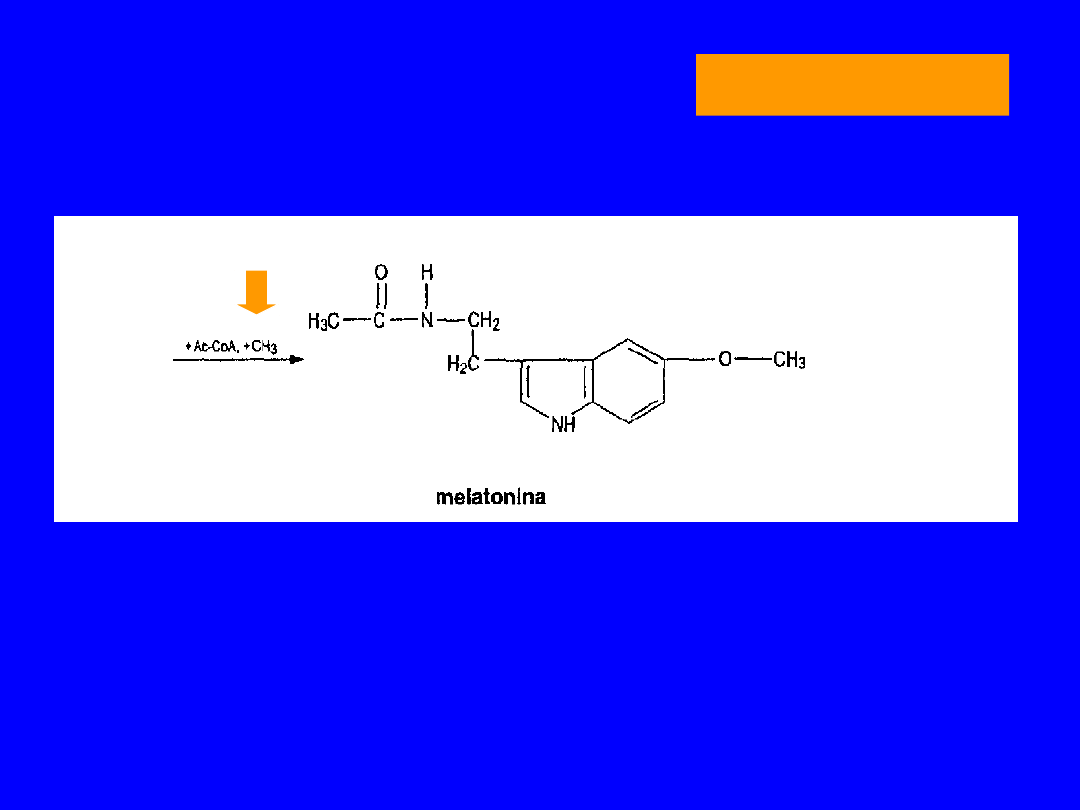
TRYPTOFAN
SYNTEZA MELATONINY
5-HYDROKSYTRYPTOFAN
POCHODNE 5-HYDROKSYTRYPTOFANU
• Wydzielana przez szyszynkę
• działanie antygonadotropowe
antymelanotropowe
antykortykotropowe
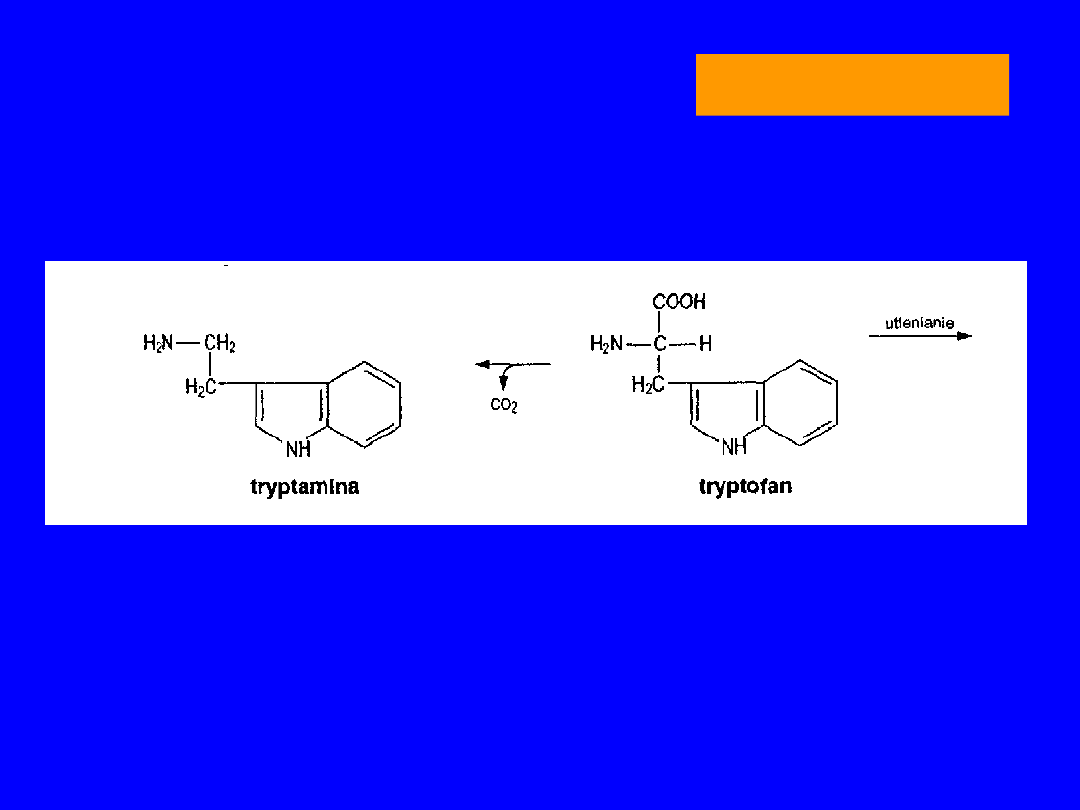
TRYPTOFAN
DEKARBOKSYLACJA TRYPTAMINA
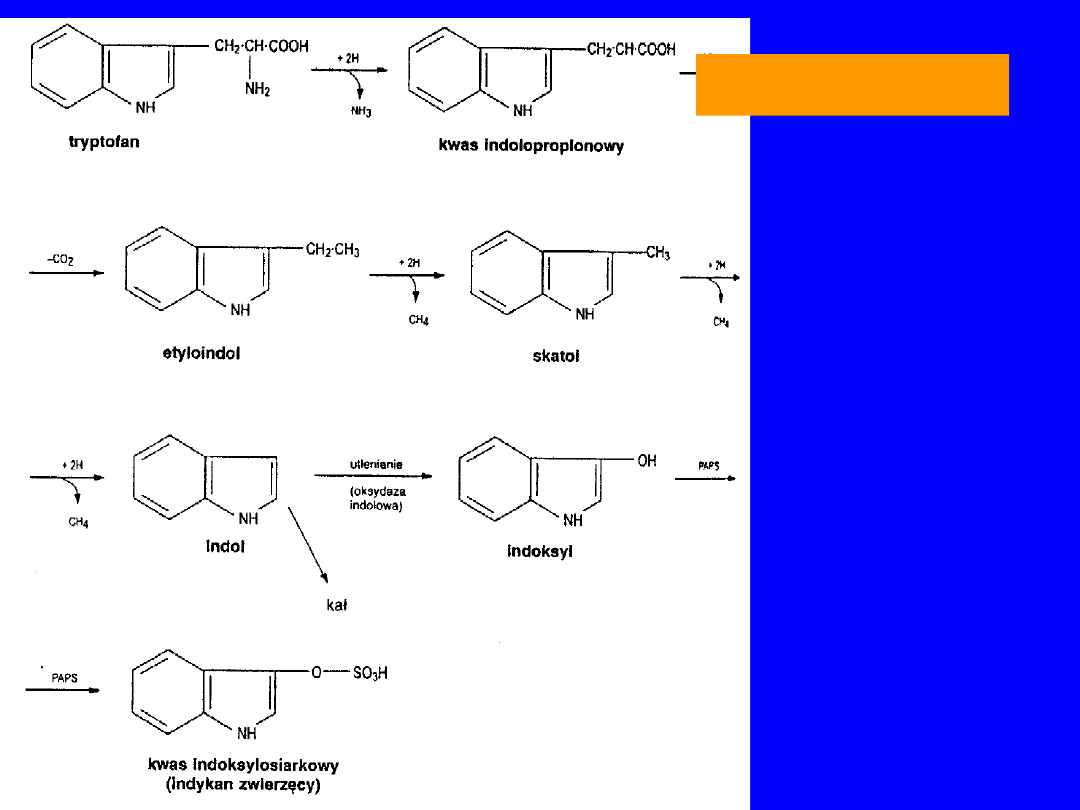
TRYPTOFAN
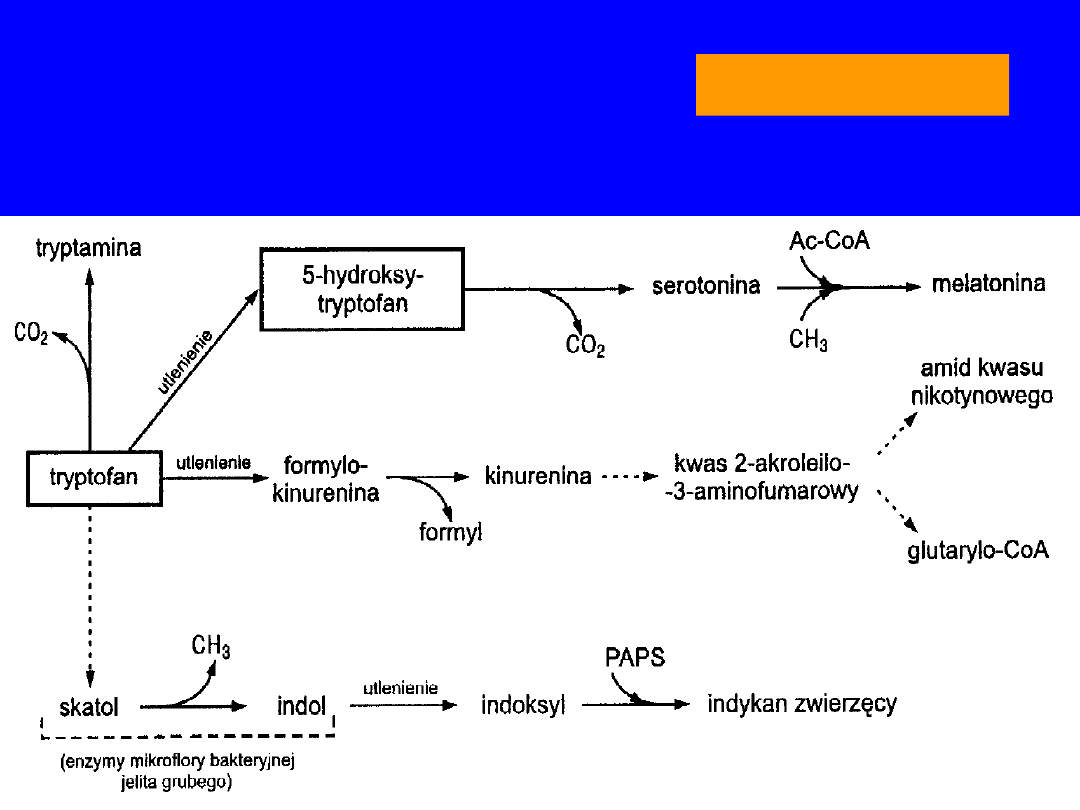
TRYPTOFAN

HISTYDYNA
• Częściowo egzogenna; głównie do syntezy białek
Katabolizm
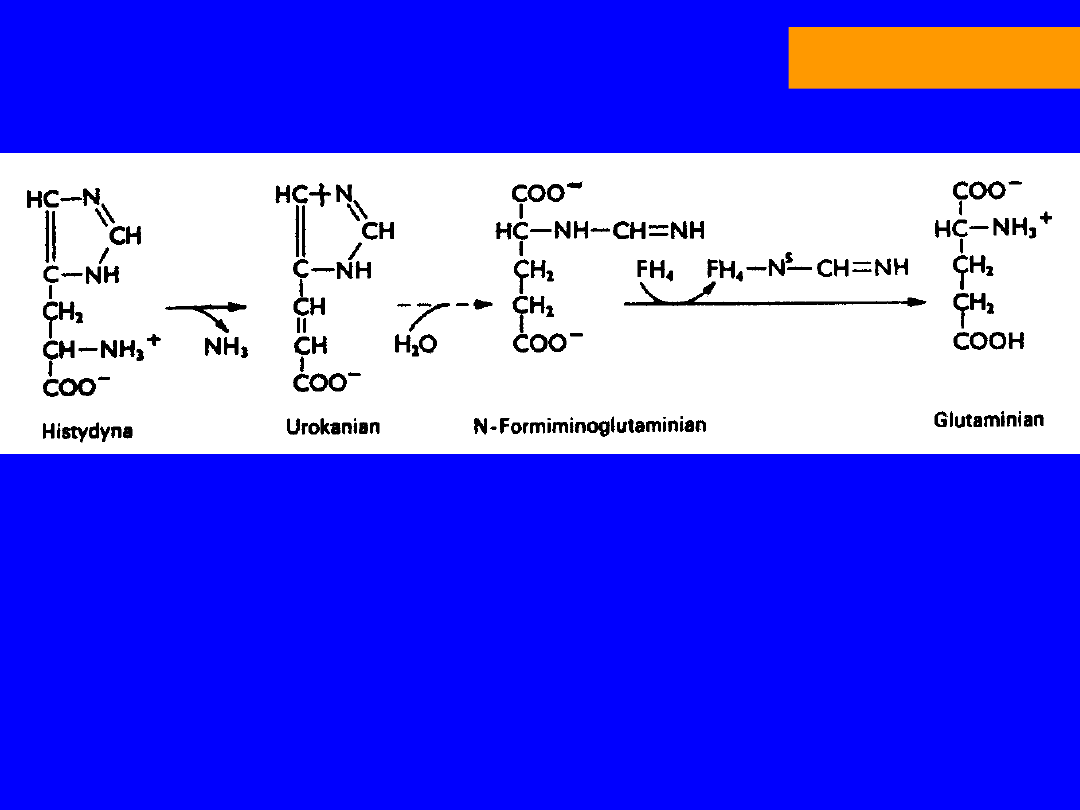
HISTYDYNA
deaminacja; amoniako liaza histydynowa
KWAS UROKANOWY
hydrataza
urokanianowa
-H
2
O; wewnątrzcząsteczkowa oksydoredukcja
4-IMIDIAZOLONO-5-PROPIONIAN
hydroliza (imidazolonopropionaza)
N-FORMININOGLUTAMINIAN (FIGLU)
formininotransferaza
N-5-formininotetrahydrofolian
KWAS GLUTAMINOWY -ketoglutaran
niedobór kwasu foliowego wzrost FIGLU
HISTYDYNA
katabolizm
Histydynemia
• brak amoniako-liazy histydynowej
• autosomalna, recesywana
• w moczu dużo imidiazolopirogronianu
• opóźnienie rozwoju umysłowego, wady wymowy
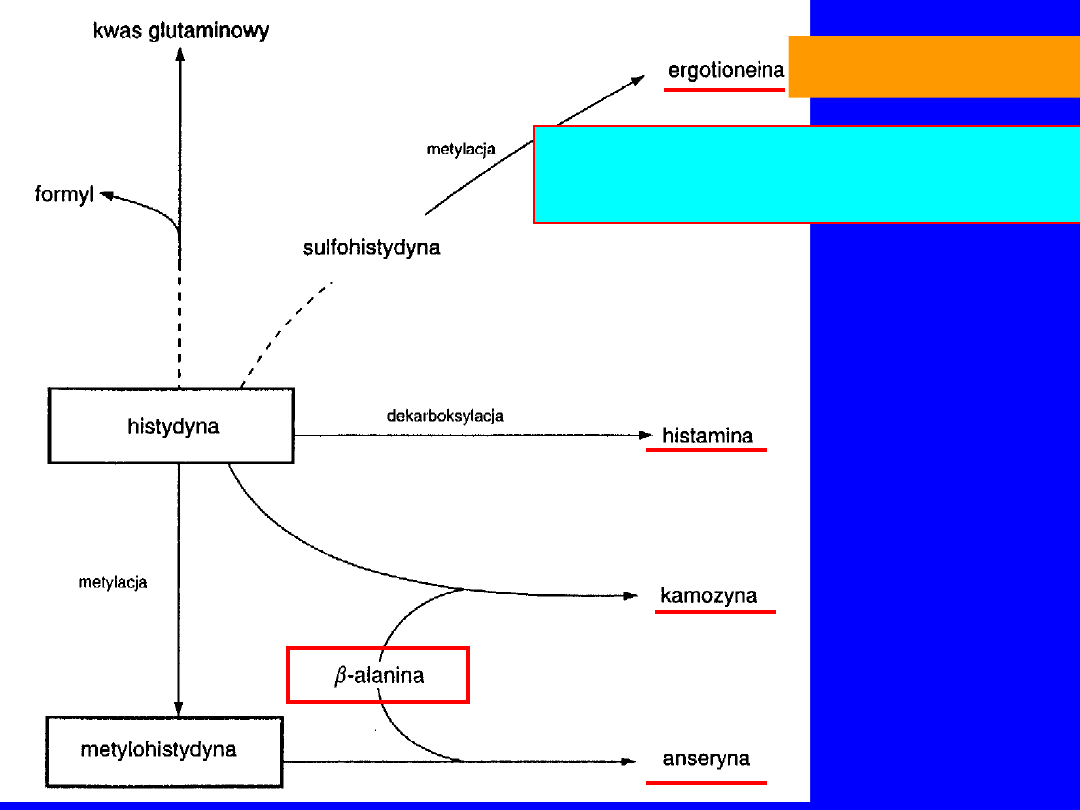
HISTYDYNA
Ergotioneina
• krwinki, wątroba
• dzięki -SH czynnik przeciwutleniajacy

KWAS GLUTAMINOWY
Źródła: pokarm i transaminacja -ketoglutaranu
pośrednio - z histydyny, argininy, proliny
• aminacja -
syntaza
glutaminowa
glutamina
• dekarboksylacja kwas
-aminomasłowy
(4-
aminomasłowy)
transaminacja kwas pirogronowy
kwas bursztynowy + alanina
• kwas glutaminowy i glicyna + kwas glutaminowy
cykl - glutamylowy
• glutaminian sodu dodatek do potraw; podnosi
smak
GLUTAMINA
• rezerwa azotu aminowego do syntez
nukleotydy
purynowe i pirymidynowe,
aminosacharydy
• łatwo przenika do pmr
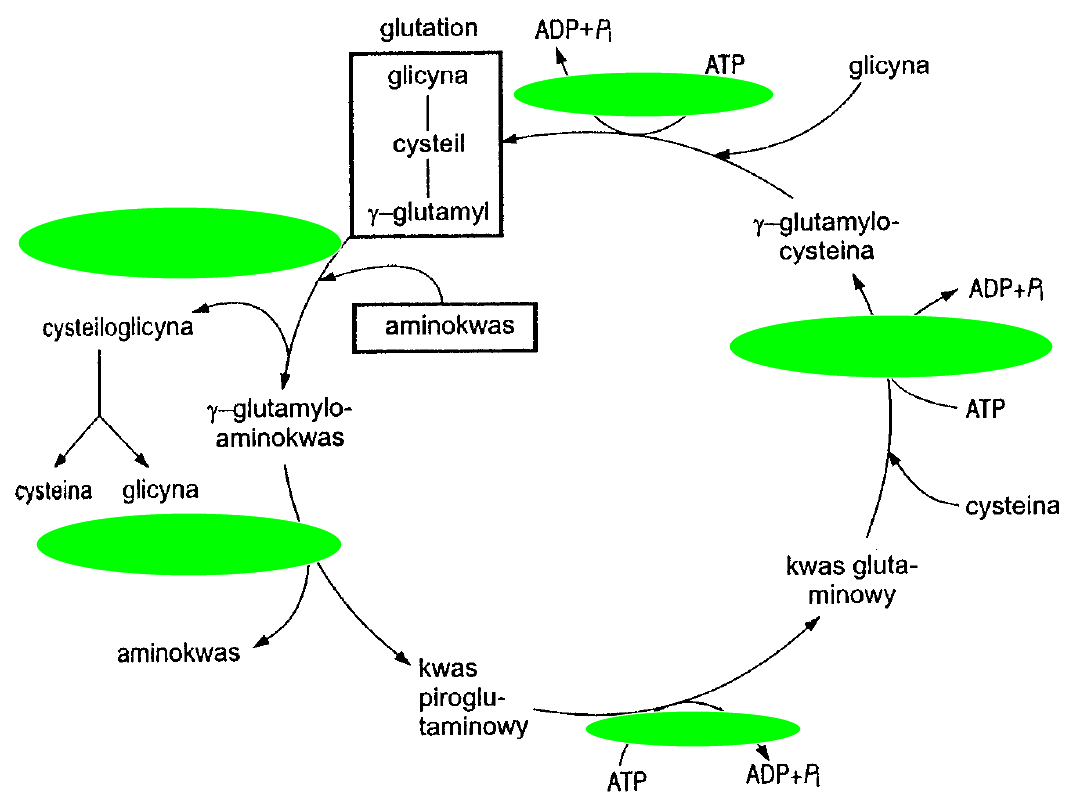
CYKL -GLUTAMYLOWY
-glutamylotranspeptydaza
-glutamylocyklotransferaza
5-oksoprolinaza
syntetaza
-glutamylocysteiny
syntetaza glutationu
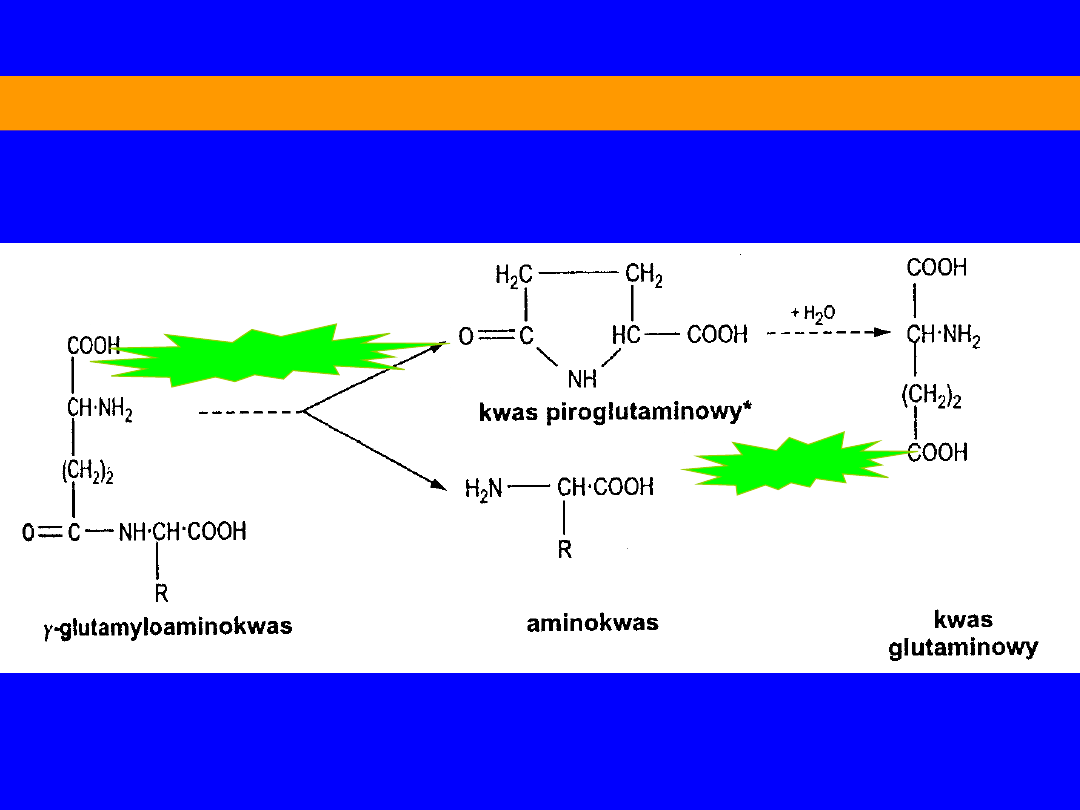
-glutamylocyklotransferaza
5-oksoprolinaza
UDZIAŁ CYKLU -glutamylowego w transporcie aminokwasów
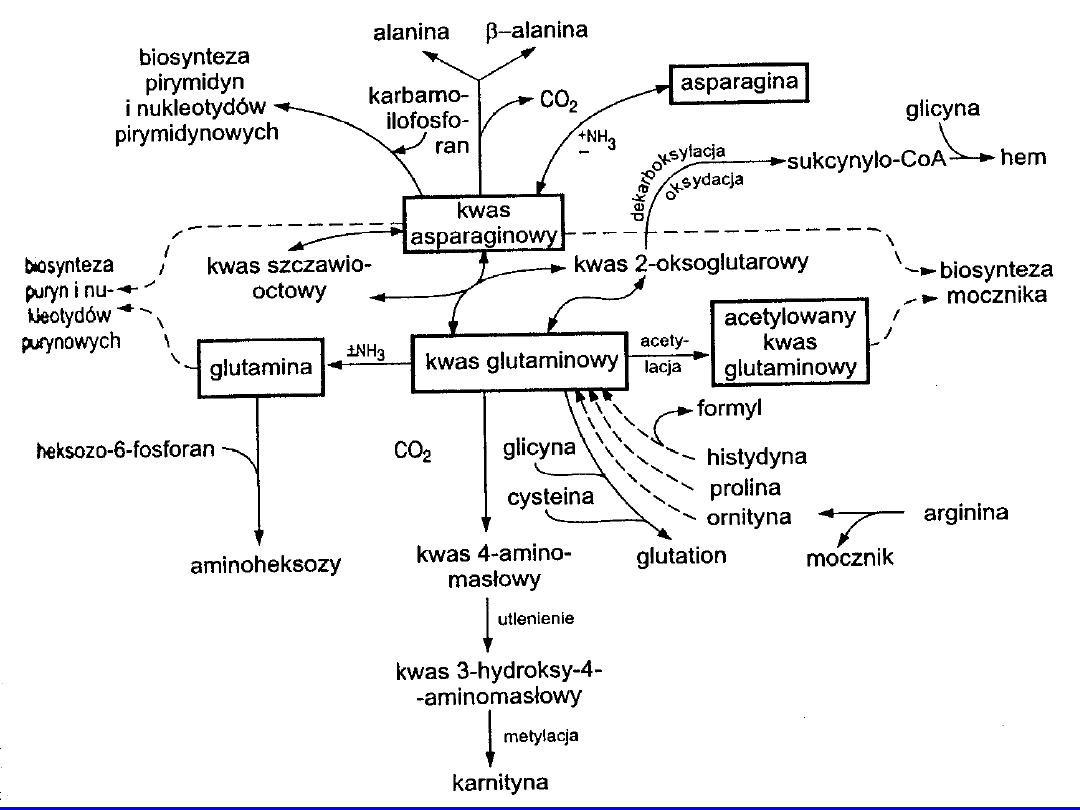
Przemiana pośrednia
aminokwasów
monoaminodikarboksylo
wych

KWAS ASPARAGINOWY
• Transaminacja szczawiooctan
• aminacja -
syntaza asparaginowa
asparagina
• dekraboksylacja -alanina (1-dekarboksylaza)
alanina (4-dekraboksylaza)
• biosynteza mocznika + cytrulina kwas arginobursztynowy
• biosynteaza nukleotydów purynowych i pirymidynowych

ALANINA
Powstaje:
• transaminacja kwasu pirogronowego z kwasem
glutaminowym; asparaginowym; 4-
aminomasłowym
• dekarboksylacja kwasu asparaginowego
• pośrednia przemiana kwasu glutaminowego i
asparaginowego
Alanina + oksykwas
pirogronian
Dekarboksylacja
etanolamina

-ALANINA
• powstawanie -
dekarboksylacja
kwasu
asparaginowego (1-
dekarboksylaza)
• składnik koenzymu A, karnozyny i anseryny
-alanina + histydyna
karnozyna
syntetaza karnozynowa
metylacja karnozyny
anseryna
N-metylotransferaza
(S-metylometionina)
karnozyna - ludzkie mięśnie szkieletowe; anseryna - u gatunków o
szybkim skurczu mięśni (królik-kończyny; ptaki-mięśnie piersiowe)
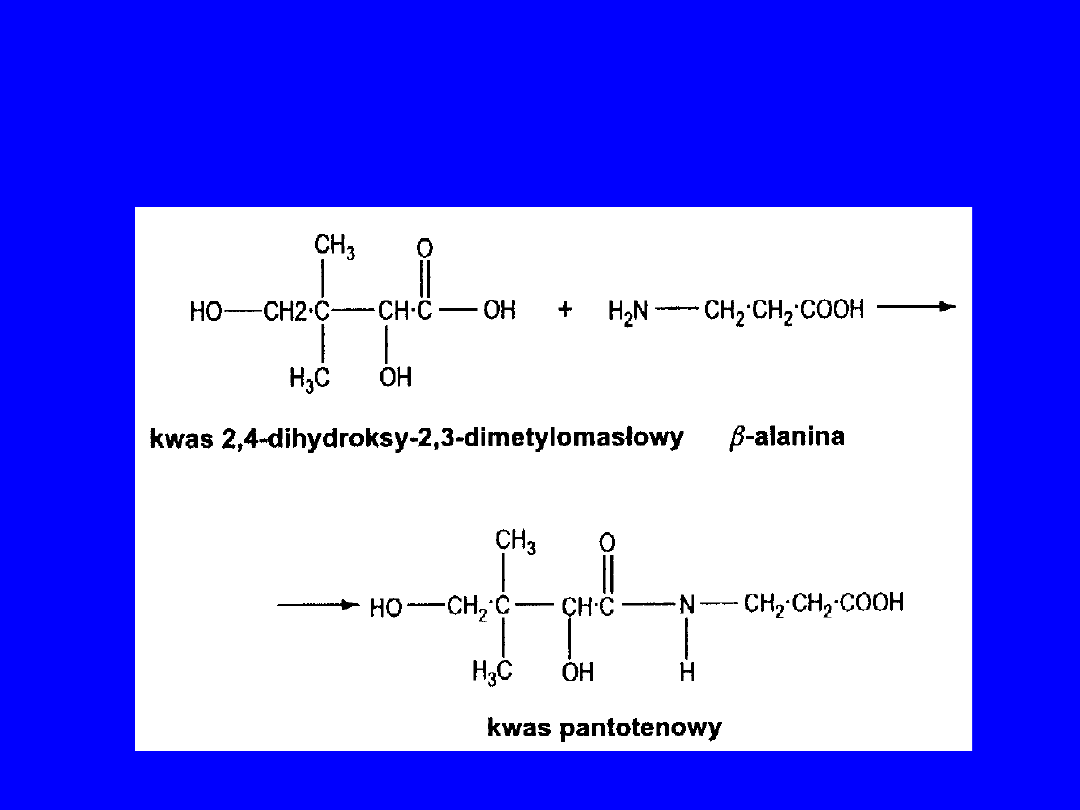
U roślin i mikroorganizmów -alanina
bierze udział w biosyntezie kwasu
pantotenowego
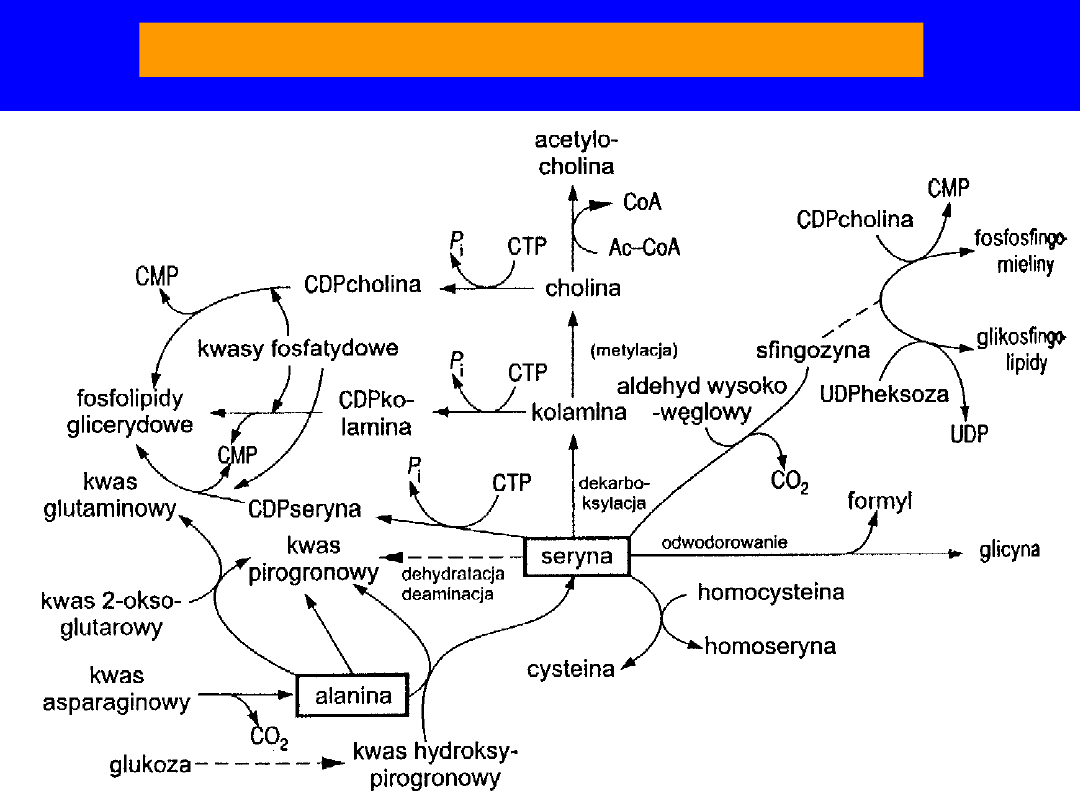
Przemiana pośrednia seryny i alaniny

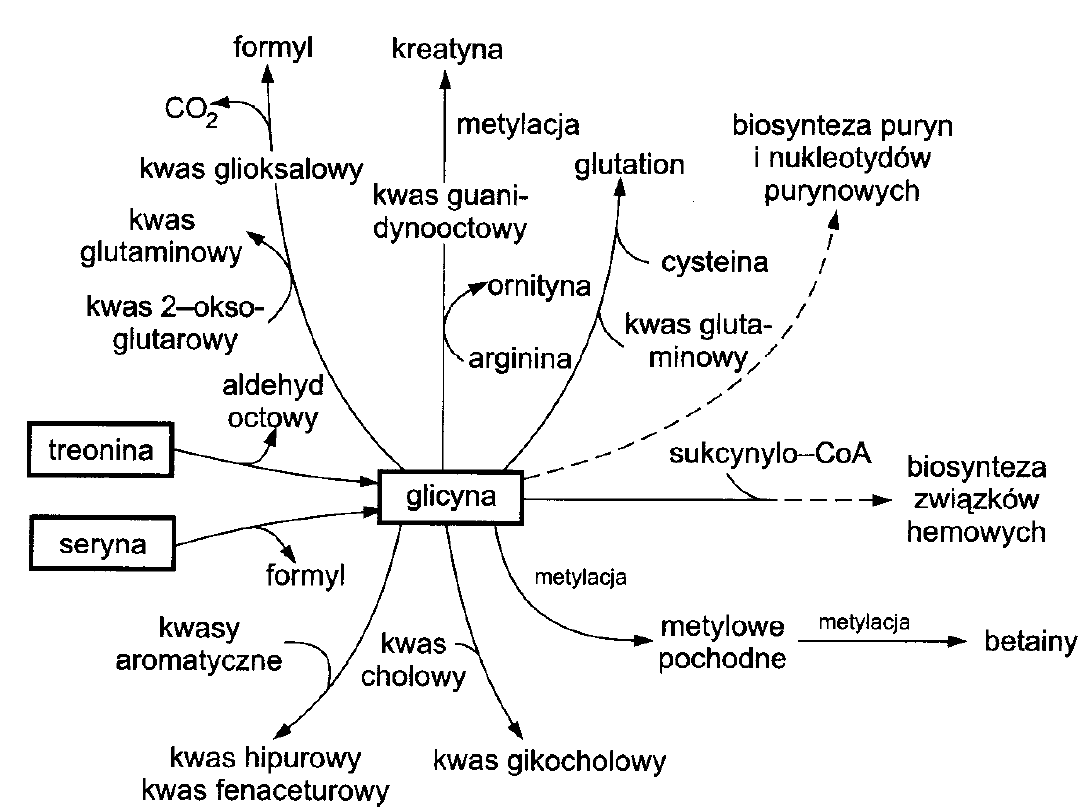
GLICYNA
• Transaminacja (-ketoglutarnan) kwas
glioksylowy +
kwas glutaminowy
• sprzęganie ze związkami aromatycznymi
glicyna + kwas benzoesowy kwas hipurowy
glicyna + kwas fenylooctowy kwas
fenaceturowy
• sprzęganie z kwasami żółciowymi
kwas cholowy + glicyna kwas glikocholowy
• glicyna +
sukcynylo-CoA
biosynteza pierścienia
hemowego
• biosynteza glutationu
• biosynteza kreatyny
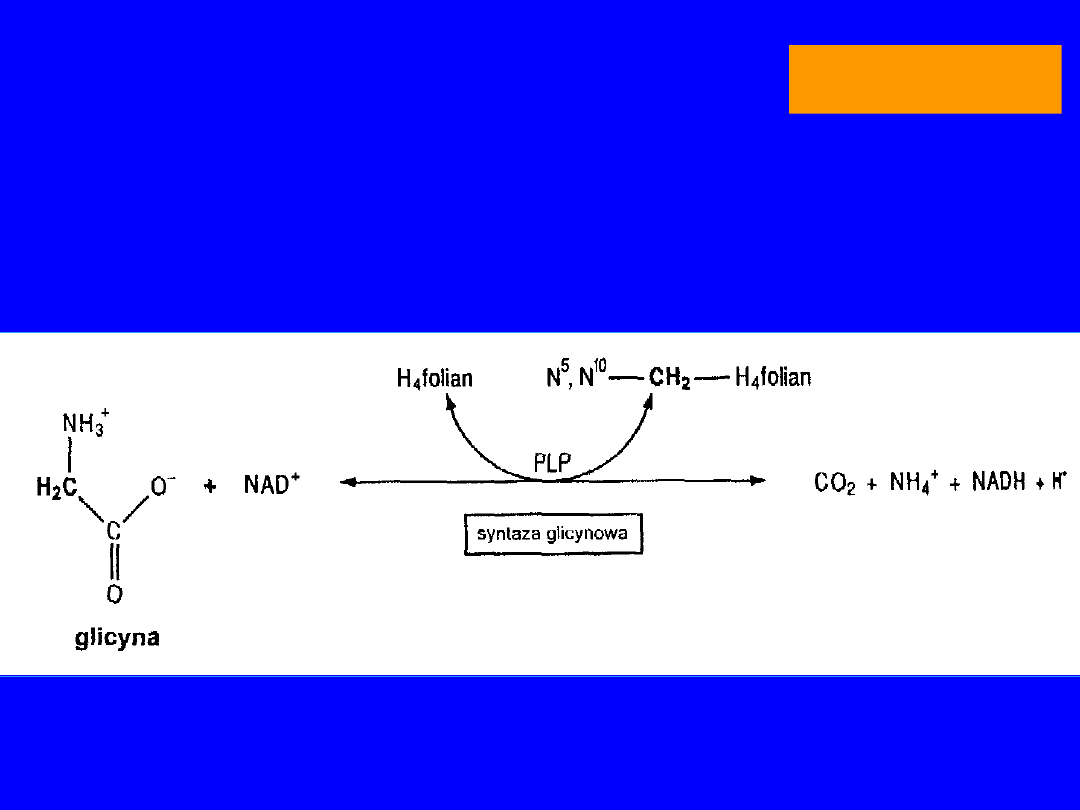
Główny szlak przemian to
Odwracalna przemiana glicyny z
udziałem
mitochondrialnego
kompleksu
syntazy glicynowej
wątroba
GLICYNA
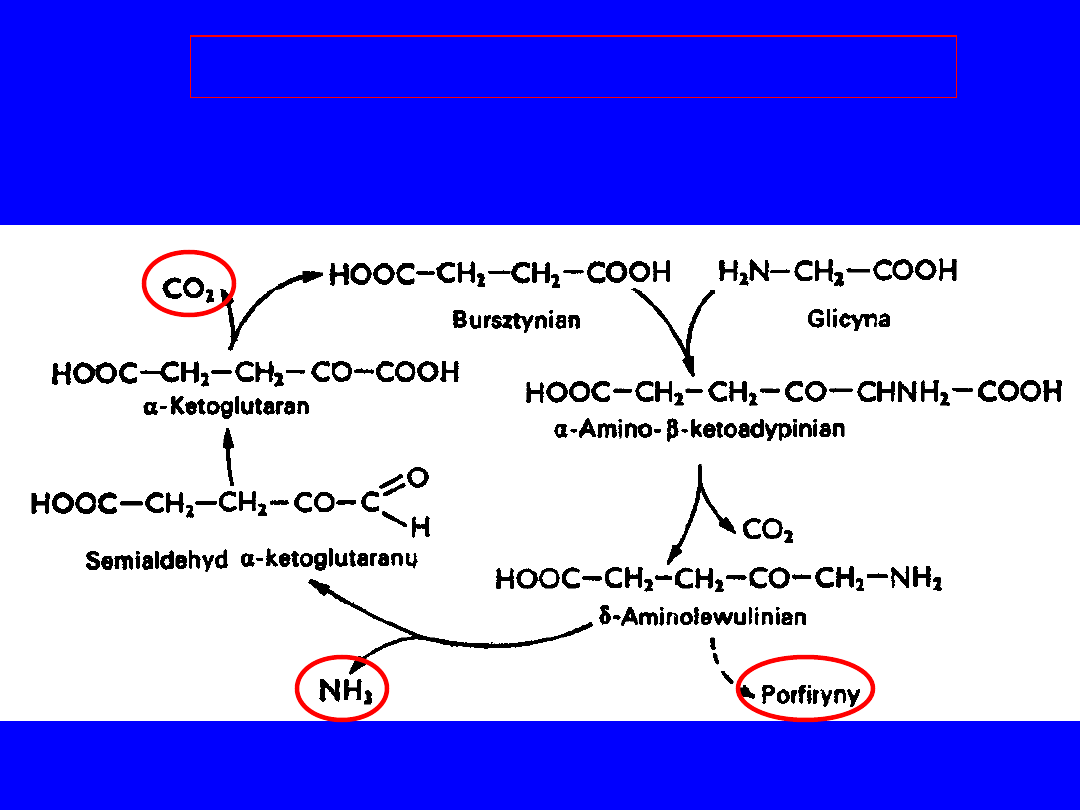
Cykl bursztynianowo-glicynowy
• rozkład glicyny do CO
2
i NH
3
• metabolity pośrednie - kwas -aminolewulinowy PORFIRYNY

SERYNA
• Udział w biosyntezie : sfingozyny, homoseryny,
cysteiny,
metioniny i tryptofanu
• seryna + palmitoilo-CoA
sfingozyna
• -OH - centrum aktywne wielu enzymów,
szczególnie
hydrolaz
• produkty
dekarboksylacji
(kolamina) i metylacji
kolaminy (cholina) - skład fosofolipidów
i
sfingomieliny
• dekarboksylacja etanolamina (kolamina)
metylacja monometylo-,
dimetylo-,
trójmetyloetanolamina -
cholina
• cholina + acetylo-CoA
acetylocholina
•
dehydratacja seryna kwas pirogronowy + NH
3
; u człowieka
nie ma
dehydratazy
SERYNA
Hydroksymetylotransferaza serynowa
SERYNA
GLICYNA
H
4
folian
N
10
-OCH
2
-H
4
folian
-H
2
O
N
5
,N
10
-CH
2
-H
4
folian
Główna droga u człowieka

SERYNA
Metabolit glikolizy
3-fosfoglicerynian
utlenienie
dehydrogenaza
(NAD+)
3-fosfohydroksypirogronian
transaminacja
z glutaminianem
3-fosfoseryna
hydroliza
,
fosfataza
SERYNA

WALINA, LEUCYNA, IZOLEUCYNA
Przeniesienie grupy aminowej
• aminotransferazy swoiste dla leucyny i
izoleucyna, inna dla waliny; AA + kwas -
ketoglutarowy glutamina + odpowiedni -
ketokwas
•
odwracalne
Dekarboksylacja
• ketoizokapronian i ketometylowalerian - ten sam
enzym
izowalerylo-CoA i metylobutyrylo-CoA
• ketoizowalerian - swoisty enzym izobutyrylo-
CoA
•
jednokierunkowe
,
nieodwracalne
•
mechanizm podobny do oksydacyjnej dekarboksylacji
pirogronianu
ketoizokapronian + CoA + NAD+
izowalerylo-CoA + CO
2
+
NADH+H
+

WALINA, LEUCYNA, IZOLEUCYNA
Aminotransferazy
• Wątroba pozbawiona aktywności
• Mózg i mięsień sercowy maja największa aktywność
•
I etap przemian poza wątrobą
Dekarboksylazy
• największa aktywność w
wątrobie
• Całość przemian wewnątrz mitochondrium
Końcowe produkty
leucyna
acetylo-CoA i acetooctan
izoleucyna
propionylo-CoA i acetylo-CoA
walina
metylomalonylo-CoA

WALINA
transaminacja
2-OKSOWALERIAN
oksydacyjna dekarboksylacja
IZOBUTYRYLO-CoA
wprowadzenie wiązania podwójnego (FAD
FADH
2
)
METYLOAKRYLOILO-CoA
+H
2
O
3-HYDROKSYIZOBUTYRYLO-CoA
utlenienie (-HSCoA
)
ANION KWASU OKSOMETYLOMALONOWEGO
-CO
2
PROPIONYLO-CoA
WALINA, LEUCYNA, IZOLEUCYNA

WALINA, LEUCYNA, IZOLEUCYNA
LEUCYNA
transaminacja
2-OKSOIZOHEKSANIAN
oksydacyjna dekarboksylacja
IZOWALERYLO-CoA
utlenienie
3-METYLOKROTONYLO-CoA
karboksylacja
3-METYLOGLUTANOLYLO-CoA
uwodnienie
3-HYDROKSY-3-METYLO-G;UTARYLOCoA
ACETOOCTAN ACETYLO-CoA

WALINA, LEUCYNA, IZOLEUCYNA
IZOLEUCYNA
transaminacja
3-METYLO-2-OKSOWALERIAN
oksydacyjna dekarboksylacja
3-METYLOBUTYRYLO-CoA
utlenienie
2-METYLOKROTONYLO-CoA
+H
2
O
3-HYDROKSY-3-METYLOBUTYRYLO-CoA
utlenienie (NAD > NADH+H+)
2-METYLOACETYLO-CoA
PROPIONYLO-CoA+
ACETYLO-CoA
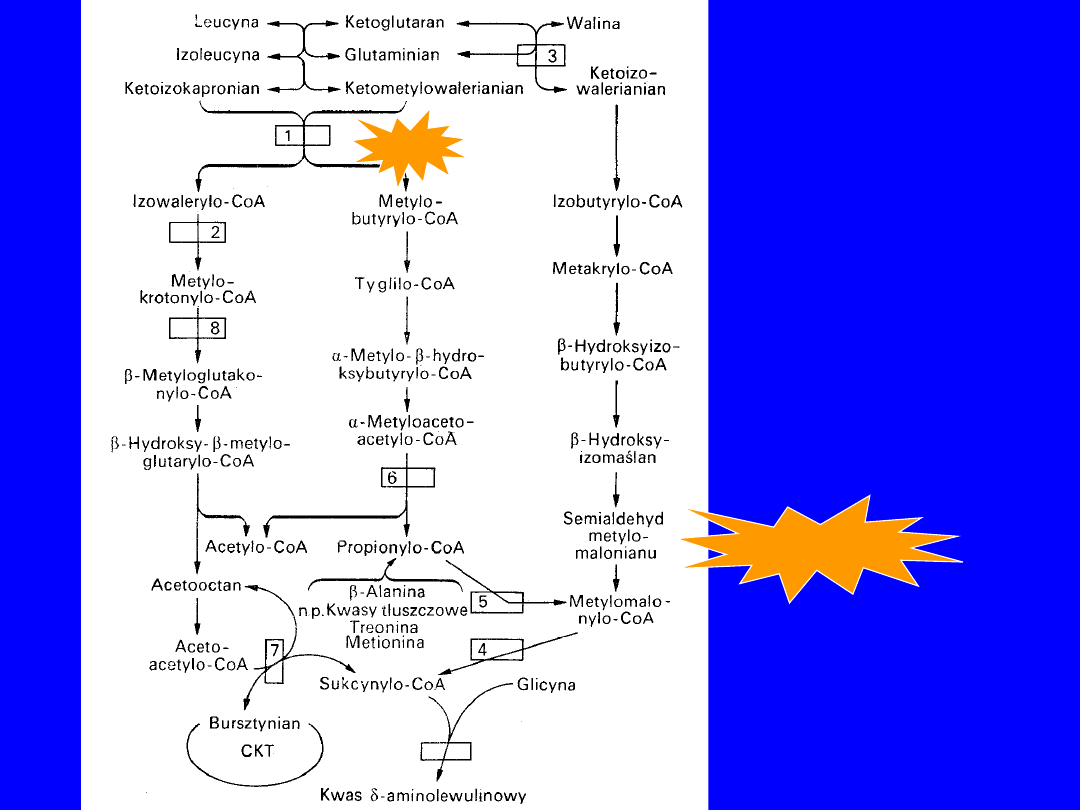
Bloki metaboliczne
MSD
METYLOMALO-
NYLOACYDURIA

• recesywna, autosomalna
• mocz o zapachu syropu klonowego (100
przypadków)
4 postacie
• klasyczna, ciężka, kilka dni po urodzeniu
• zaburzenia neurologiczne, trudności połykania,
drgawki - zgon
w pierwszych tygodniach
• we krwi leu, izo, wal i ketoanalogów
pirogronianu, ketoglutaranu, mleczanu
hydroksykwasów (zapach) powstających
przez redukcję ketoanalogów
• o rozpoznaniu decyduje badanie enzymatyczne;
używa się krwinek białych (metabolizują
wszystkie 3 am i ich
ketoanalogi) lub
fibroblasty skóry w hodowli
KETOAMINOACYDURIA

KETOAMINOACYDURIA
Mechanizm
• nagromadzajacy się we krwi i tkankach kwas
ketoizokapronowy
silny inhibitor
dehydrogenazy
pirogronianowej
• mózg - glukoza - podstawowe źródło pirogronianu
zahamowanie procesów
metabolicznych
• leucyna
hipoglikemizująco
pobudza
wydzielanie insuliny
• alaniny - ketoanalogi leu i izoleu hamują
aminotransfearzę
alaninową
•
leczenie
- dieta eliminująca leu, izoleu i wal
METYLOMALONYLOACYDURIA
PROPIONYLO-CoA
+ATP
+ CO
2
karboksylaza
biotynozależna
D-METYLOMALONYLO-CoA
racemizacja
L-METYLOMALONYLO-CoA
mutaza (koenzym
B
12
)
SUKCYNYLO-CoA
Izoleucyna
uracyl
walina
reszta propylowa
metionina
cholesterolu
treonina
kwasy tłuszczowe
-alanina
nieparzyste

METYLOMALONYLOACYDURIA
W wątrobie fosforylacja substratowa
sukcynylo-CoA + GDP + Pi bursztynian + CoA +
GTP
W tkankach obwodowych
spalanie acetooctanu - mitochondrium
sukcynylo-CoA + acetooctan acetoacetylo=CoA
+
bursztynianan
transferaza sykcynylo-CoA
• CoA ani jego acylopochodne nie przechodzą przez
błony
mitochondrialne - ważne w
patomechanizmie zaburzeń
Na każdym etapie przemian propionylo-CoA
(oprócz transferazy) wykazano niedobory
enzymów
ostre zaburzenia metaboliczne - kwasica
metaboliczna i
hipoglikemia

METYLOMALONYLOACYDURIA
METYLOMALONYLOACYDURIA
• całkowity niedobór
mutazy metylomalonylo-CoA
• ketoacydoza i glicyny
• w moczu metylomalonian; letalna
Mechanizm hipoglikemii
•
błona mitochondrialna nie przepuszcza
metylomalonylo-CoA
• toksyczne działanie wewnatrzmitochondrialnego
• w mitochondrium - glukoneogeneza -
karboksylacja
pirogronianu kwas
szczawiooctowy - aktywowana
przez acetylo-
CoA
• metylomalonylo-CoA znosi pobudzające działanie
acetylo-CoA na karboksylazę pirogronianową
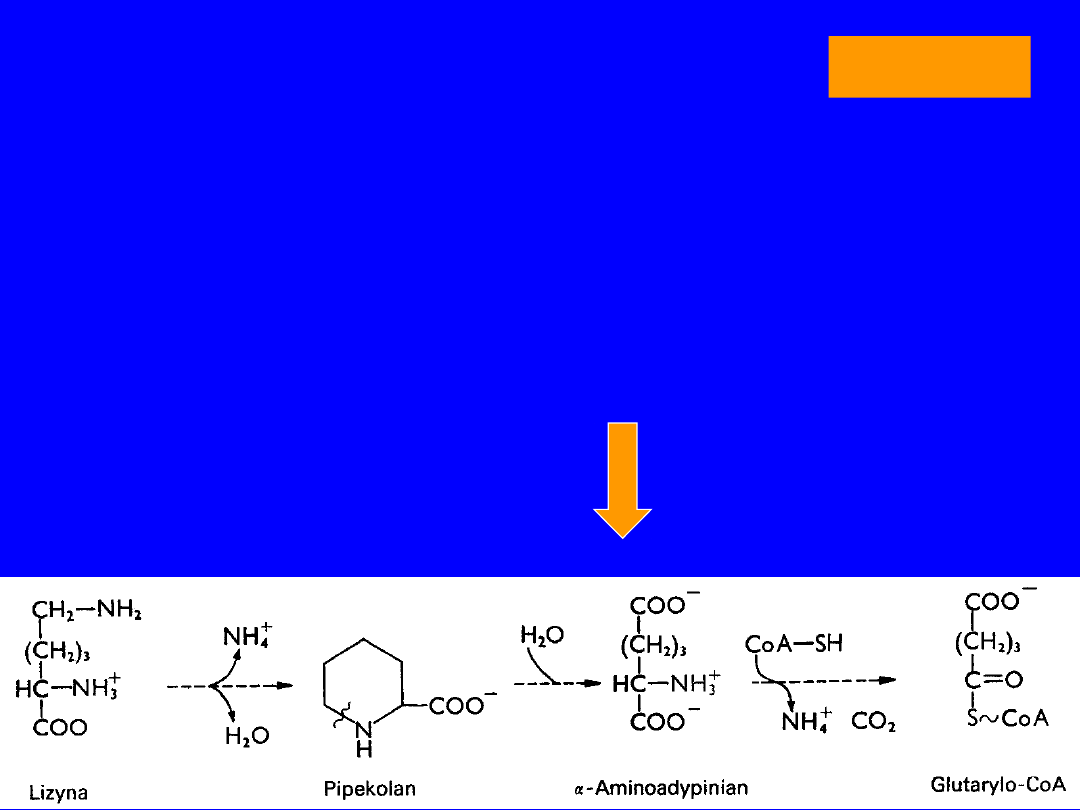
LIZYNA
• Egzogenna > białka
PRZEMIANY
•
oksydacyjna deaminacja
kwas 2-okso-6-
aminokapronowy
•
cyklizacja
kwas dehydropipekolinowy
•
+H
2
O
semialdehyd kwasu 2-aminoadypinowego
•
utlenienie
kwas 2-aminoadypinowy
•
oksydacyjna deaminacja
•
oksydacyjna dekarboksylacja
•
glutarylo-CoA >>> acetoacetylo-CoA

KATABOLIZM
- nietypowy
• reaguje z 2-oksoglutaranem (+ NADPH+H
+
)
zredukowany
kompleks połączony grupą aminową
• oksydacyjne rozerwanie kompleksu (NAD
+
)
glutaminian +
semialdehyd 2-aminoadypinowy
• kolejne przemiany semialdehydu
acetoacetylo-
CoA
Rozkład lizyny jako przykład
prawidłowości w metabolizmie AA
• pierwsza reakcja AA egzogennych -
nieodwracalna
• Wyjątkiem AA rozgałęzione - druga reakcja
nieodwracalna
•
Nieodwracalność pierwszej reakcji - reguluje
tempo
rozkładu AA egzogennych
• Enzymy -
wysokie K
m
;
rozkład dopiero gdy AA w
nadmiarze
LIZYNA

•
transaminacja
- nie znana
•
dekarboksylacja
pentametylenodiamina
(1,5-diaminopentan, kadaweryna)
• oksypochodna -
hydroksylizyna
•
hydroksylaza lizylowa
- oksygenaza o funkcji
mieszanej
• utlenienie w obecności kwasu askorbinowego, O
2
,
Fe2+
i
2-
oksoglutaranu -
LIZYNA

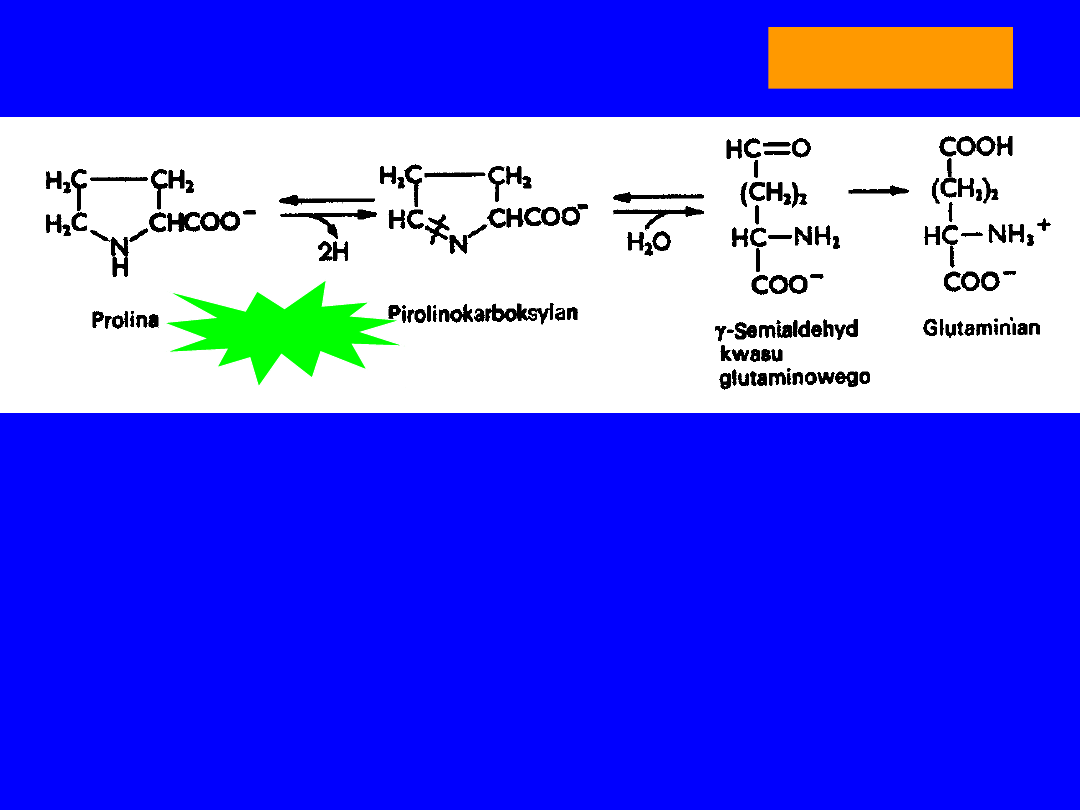
PROLINA
• Endogenna
kwas glutaminowy
dehydrogenaza prolinowa
-semialdehyd kwasu glutaminowego
cyklizacja
DH prolinowa
kwas pirolino-karboksylowy kwas glutaminowy
DH (NAD)
transaminacja
PROLINA
2-oksoglutaran
-transaminacja
ornityna
Hyperprolinemia
Hyperprolinemia
PROLINA
Prolina -ketoglutaran
Hyperprolinemia
•Hydroksylaza prolinowa
4-hydroksyprolina
• kwas askorbinowy, Fe
2+
, -ketoglutaran
• posttranslacyjna modyfikacja kolagenu
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
Wyszukiwarka
Podobne podstrony:
Przemiany poszczególnych aminokwasów
Przemiany poszczególnych aminokwasów
Przemiany białek i aminokwasów
Przemiany aminokwasów w biologicznie ważne, wyspecjalizowane produkty
przemiany, Rozrywka, FILOLOGIA POLSKA, FILOLOGIA POLSKA, PIERWSZY ROK - drugi semestr, Przemiany wsp
biol-cykl mocznikowy, Cykl mocznikowy jest sekwencją reakcji enzymatycznych w toku których grupy ami
OGÓLNA PRZEMIANA AMINOKWASÓW(1)
Biochemia schematy przemian aminokwasów
Ogólna przemiana aminokwasów
Przemiany aminokwasów
od epistoły do smsa, Rozrywka, FILOLOGIA POLSKA, FILOLOGIA POLSKA, PIERWSZY ROK - drugi semestr, Prz
Przemiany aminokawsów genetyka
FIZJOLOGICZNIE WAŻNE PRODUKTY PRZEMIAN AMINOKWASÓW
przemiany aminokwasow, INNE KIERUNKI, biologia
list motywacyjny2, Rozrywka, FILOLOGIA POLSKA, FILOLOGIA POLSKA, PIERWSZY ROK - drugi semestr, Przem
Przemiany aminokwasów w biologicznie ważne, wyspecjalizowane produkty
Biochemia schematy przemian aminokwasów
przemiany aminokwasów
Przemieszczanie produktów między poszczególnymi ogniwami łańcucha logistycznego wiąże się z konieczn
więcej podobnych podstron