
CHOROBY
CHOROBY
GENETYCZNE
GENETYCZNE
Podstawowe pojęcia,
Podstawowe pojęcia,
przyczyny, obraz
przyczyny, obraz
kliniczny, możliwości
kliniczny, możliwości
leczenia
leczenia

Choroby genetyczne
Choroby genetyczne
(
(
gr.
gr.
genetes
genetes
= rodzi
= rodzi
ć
ć
, zrodzony)
, zrodzony)
to grupa
chorób
wywołana
mutacjami w obrębie genu
lub chromosomów,
mających znaczenie dla
prawidłowej budowy i
czynności organizmu.

Podstawowe pojęcia
Podstawowe pojęcia
GEN- to jednostka dziedziczenia.
Zbudowane z DNA i mieszczące się w
określonych miejscach chromosomów
CHROMOSOM- pręcikowe struktury
mieszczące się w jądrze każdej
komórki organizmu, zawierające
DNA, z którego zbudowane są geny
Allel – wersja genu, kodującego daną
cechę, umiejscowiona w konkretnym
miejscu chromosomu

Mutacja jest to zmiana
Mutacja jest to zmiana
powstająca wskutek:
powstająca wskutek:
zmiany genu w jego nowy allel –
mutacja genowa – zmienia informację
kodowaną przez gen, tak że produkuje
on uszkodzone białko lub w ogóle
zaprzestaje produkowania białek.
zmiany struktury chromosomu –
mutacja chromosomowa strukturalna
zmiana liczby chromosomów – mutacja
liczbowa

Rodzaje mutacji genowych
Rodzaje mutacji genowych
Mutacje punktowe
tranzycja –polega na zamianie
jednej zasady purynowej na
drugą purynową czy tez zasady
pirymidynowej na pirymidynową
tranwersja – zamiana zasady
purynowej na pirymidynową lub
pirymidynowej na purynową
delecja – wypadnięcie
pojedynczej lub większej liczby
par nukleotydów z danego genu
insercja – wstawienie
pojedynczej lub większej liczby
par nukleotydów do danego genu.
Mutacje
chromosomowe
1.
strukturalne –dotyczą komórek z
prawidłową liczbą, ale zaburzoną
budową chromosomów
Inwersja – odwrócenie fragmentu chromosomu o 180
Translokacja – przemieszczenie fragmentu chromosomu w
obrębie tego samego lub innego chromosomu
Duplikacja – podwojenie tych samych odcinków
chromosomów
Delecja – utrata odcinka chromosomu
Chromosom kolisty – powstaje na skutek pęknięcia,
następnie połączenia końców chromosomu
Izochromosom – powstaje w wyniku nieprawidłowego,
poprzecznego podziału centromeru chromosomu
2. liczbowe – dotyczą komórek,
które zawierają nieprawidłową
liczbę chromosomów
Monosomia – brak jednego chromosomu
Nulisomia – brak w komórce jakiejkolwiek kopii danego
chromosomu
Trisomia – dodatkowy chromosom

Czynniki mutagenne
Fizyczne – promieniowanie jonizujące X,
alfa, beta, gamma, protony i neutrony
emitowane przy rozpadzie pierwiastków
oraz promieniowanie niejonizujące –
ultrafioletowe
Biologiczne – wirusy
Chemiczne – kwas azotawy,
hydroksylamina, barwniki akrydynowe,
nadtlenki, niektóre leki (cytostatyki), środki
konserwujące (azotyn sodowy)

Rodzaje
Rodzaje
dziedziczenia
dziedziczenia
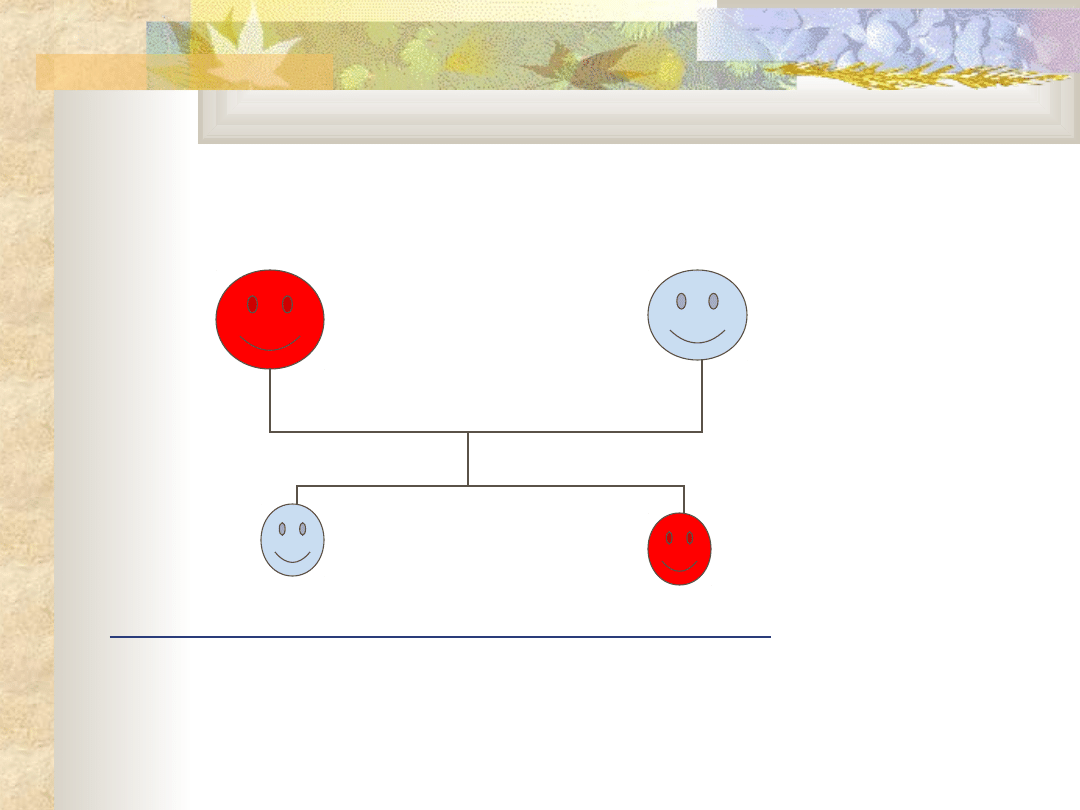
Dziedziczenie autosomalne dominujące
Odziedziczenie pojedynczego allelu jest
wystarczające, aby osobnik zachorował.
Dziecko chorej jednostki będzie miało 50%
prawdopodobieństwa że zachoruje.
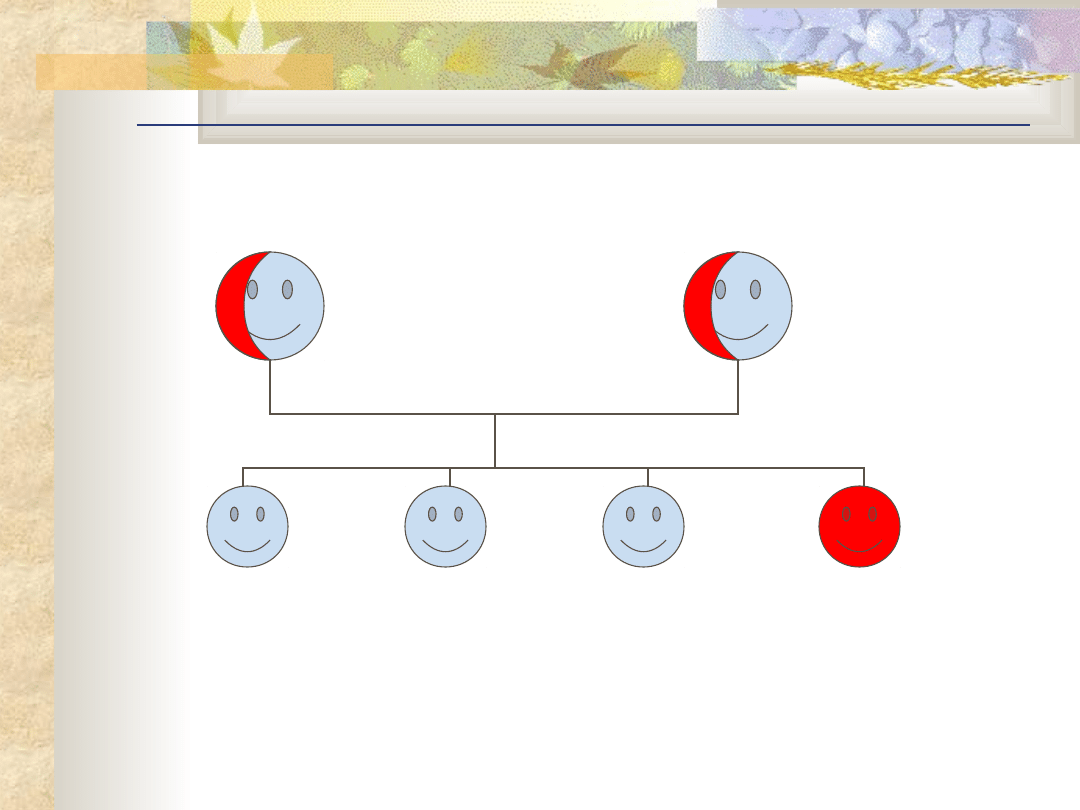
Dziedziczenie autosomalne recesywne
aby zachorować trzeba odziedziczyć dwa zmutowane
allele po jednym od każdego rodzica.
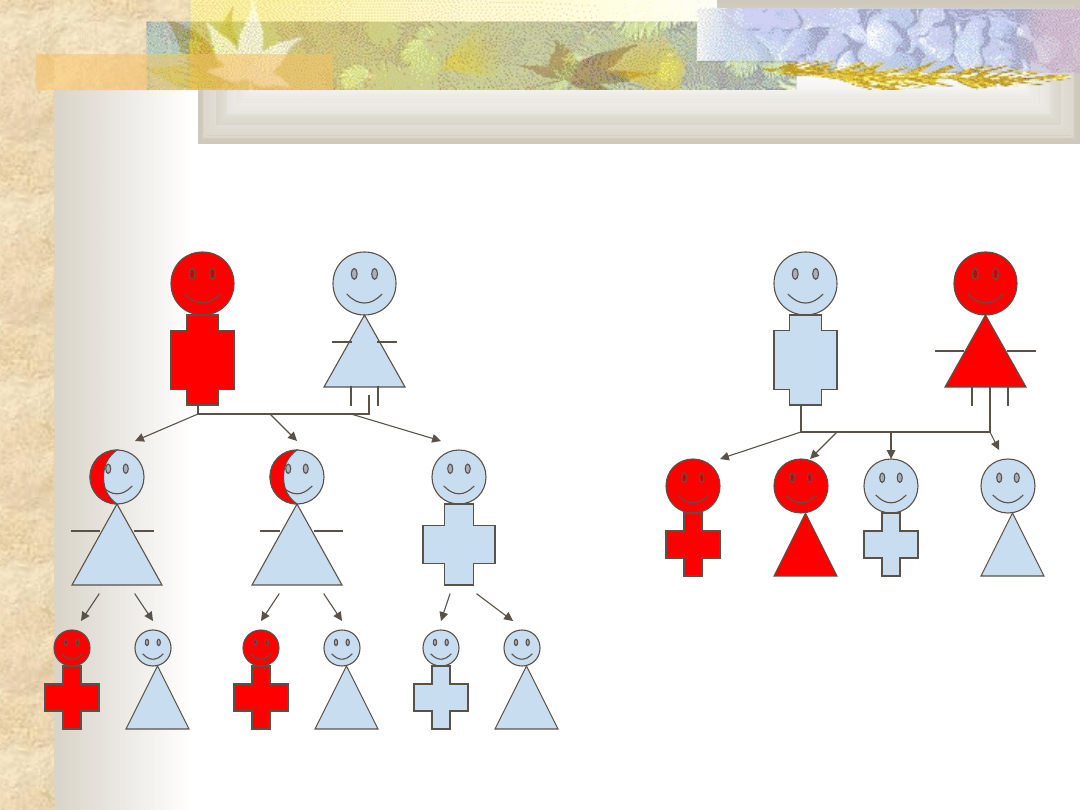
Dziedziczenie recesywne
sprzężone z
chromosomem X
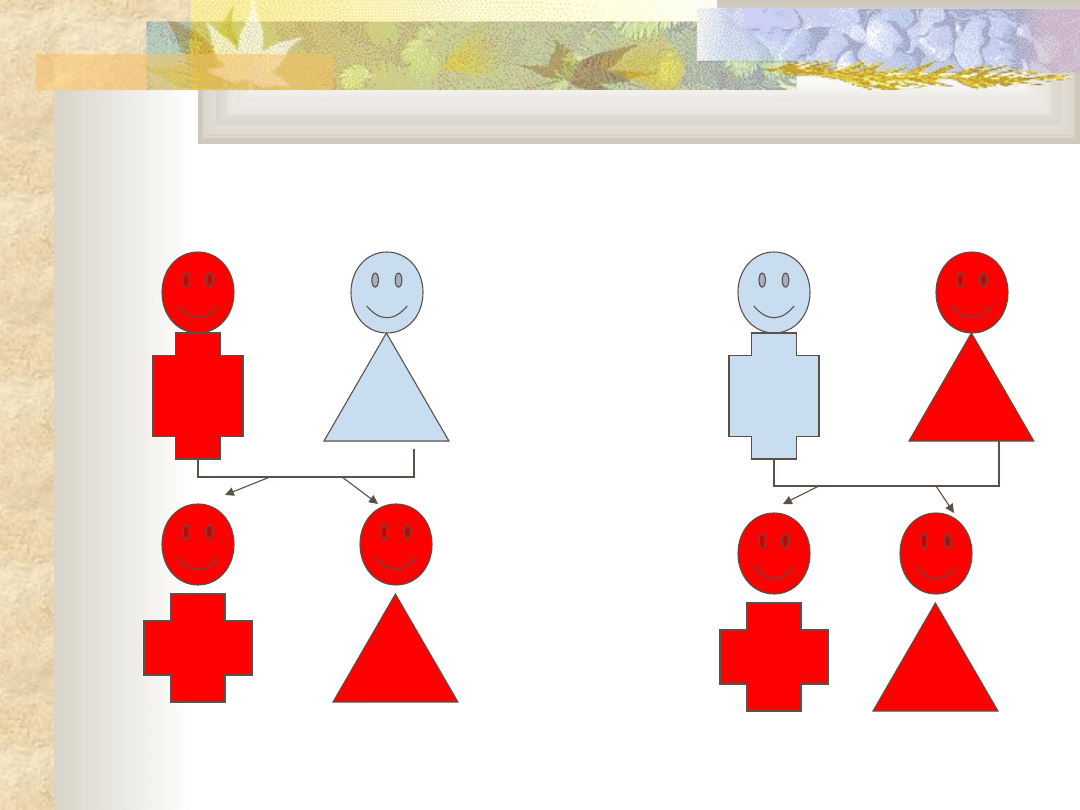
Dziedziczenie dominujące
związane z chromosomem
X

Choroby dziedziczone w sposób
Choroby dziedziczone w sposób
autosomalny dominujący
autosomalny dominujący
Achondroplazja
Achondroplazja
Zespół Marfana
Zespół Marfana
Choroba Recklinghausena
Choroba Recklinghausena
Choroby dziedziczone w sposób
Choroby dziedziczone w sposób
autosomalny recesywny
autosomalny recesywny
fenyloketonuria
fenyloketonuria
albinizm
albinizm
mukowiscydoza
mukowiscydoza

ACHONDROPLAZJA
ACHONDROPLAZJA
chondrodystrofia
chondrodystrofia
płodowa
płodowa
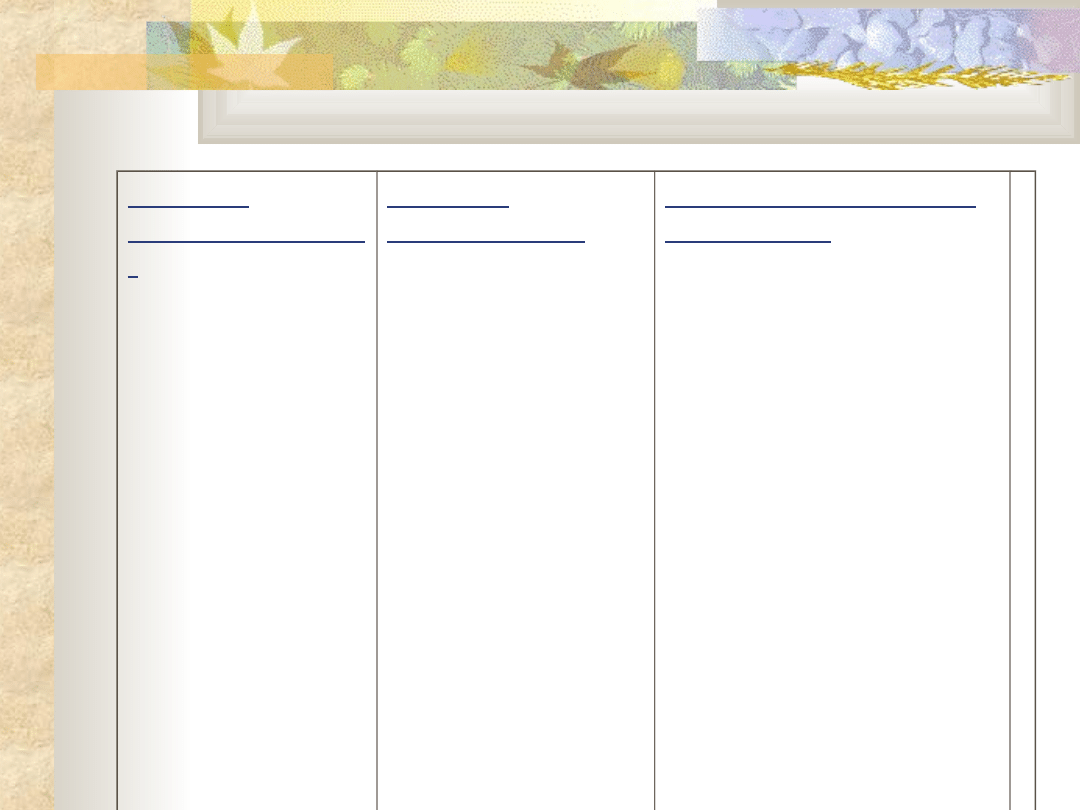
Obraz kliniczny
niski wzrost (karłowatość)
zaburzenia proporcji ciała
Skrócenie kończyn, małe
dłonie
Duża głowa
Twarz z wypukłym czołem
i zapadniętą nasadą nosa
Częstość występowania
choroby ocenia się na 1 :
25000 urodzeń
dotyczy w równym stopniu
obu płci i wszystkich ras
Leczenie
Hormon wzrostu
metoda Ilizarowa

Achondroplazja leczenie
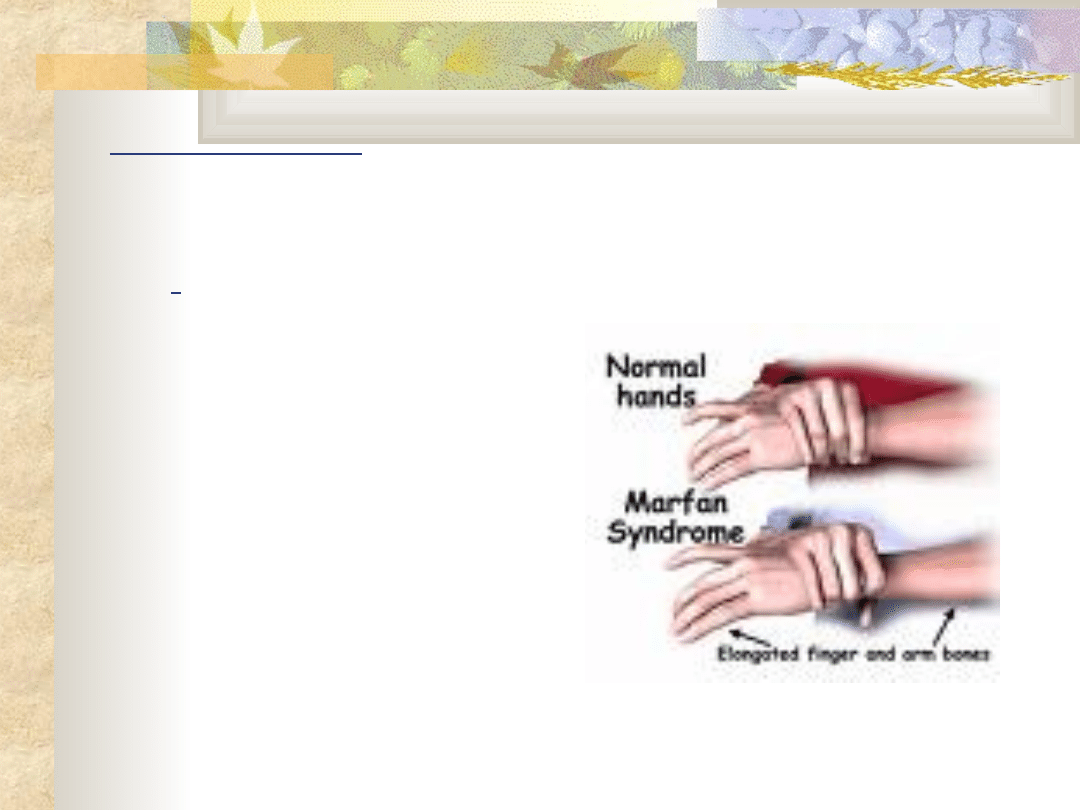
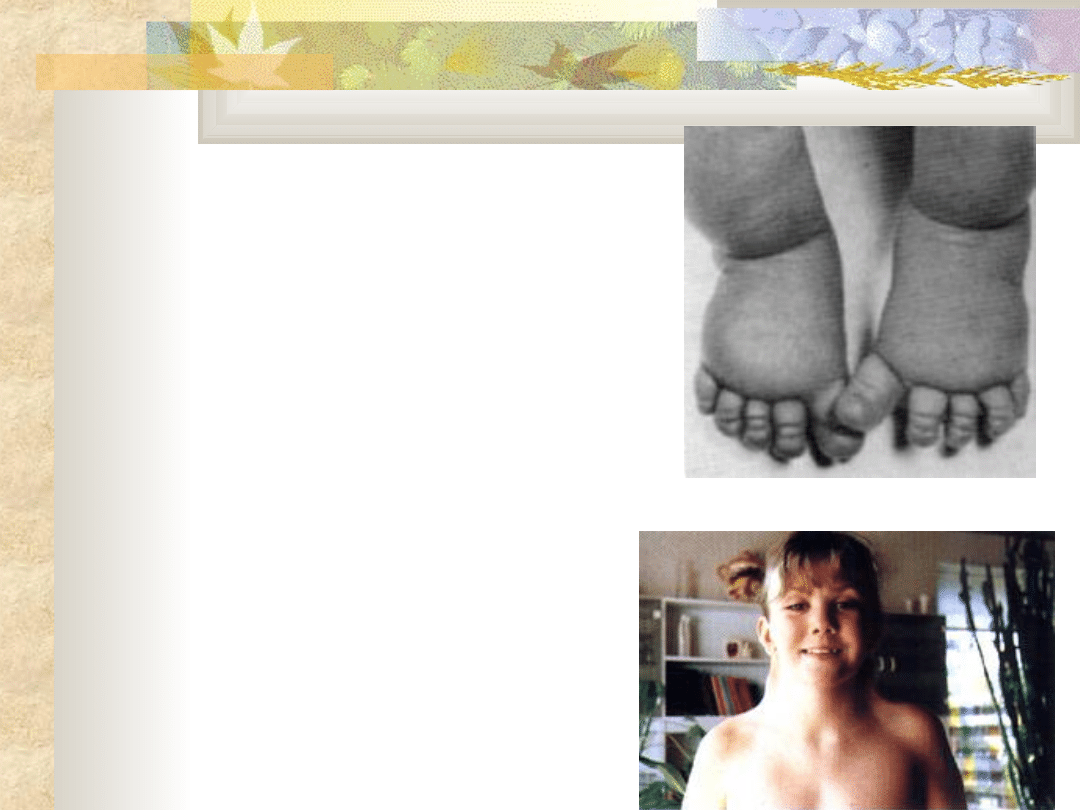
Zespół Marfana
Zespół Marfana
Q 87.4
Obraz kliniczny
wąska długa czaszka
wysoko wysklepione
"gotyckie„ podniebieni
e
klatka piersiowa
lejkowata
skrzywienie kręgosłupa
nadmierny wzrost kości
długich
wysoki wzrost
nadmiernie długie
palce rąk i stóp
(pająkowatość palców)
wady wrodzone serca
wady narządu wzroku
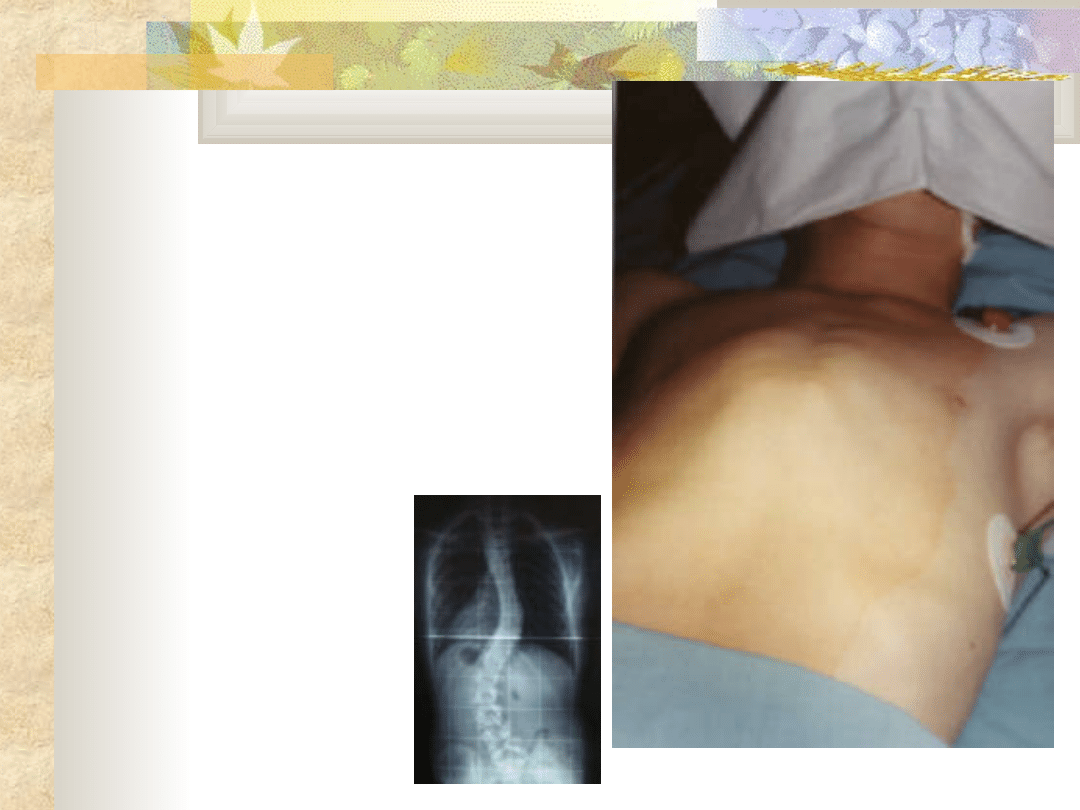
Zespół
Marfana
Leczenie
zachowawcze
opiera się na wykonywaniu
ćwiczeń poprawiających
sprawność życiową płuc,
wzmacniające mięśnie
grzbietu i
brzucha
Leczenie operacyjne
serca
i narządu wzroku.
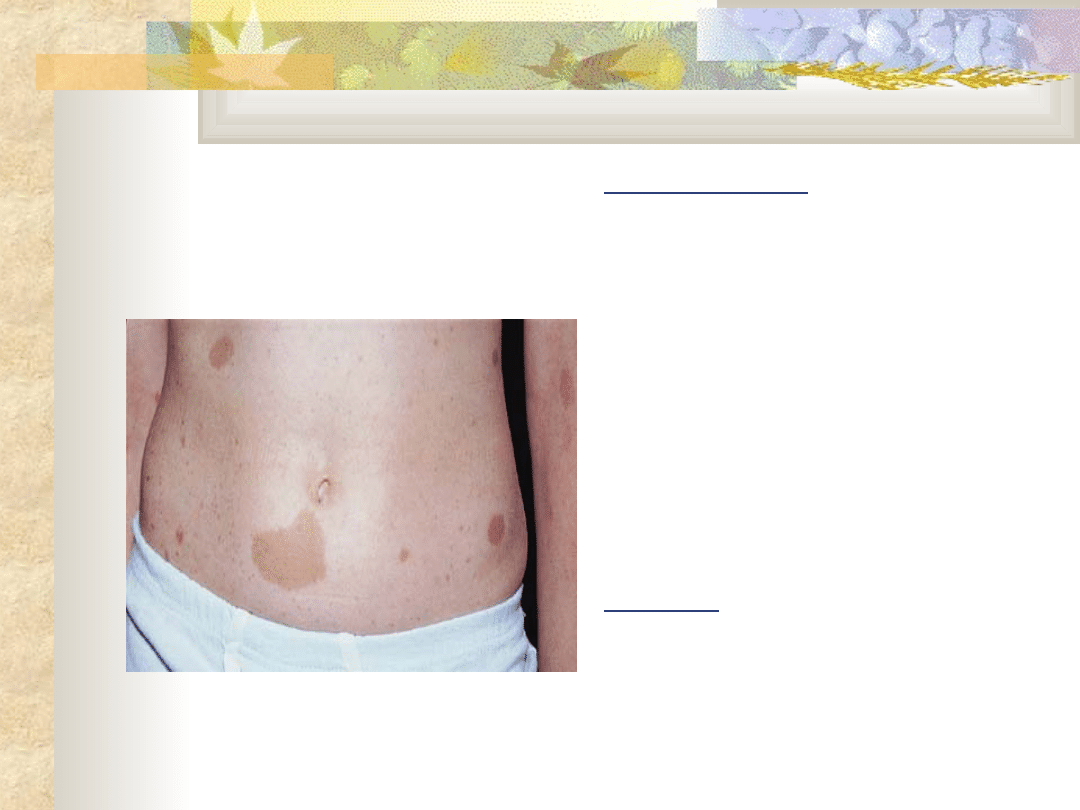
Choroba
Choroba
Recklinghausena
Recklinghausena
nerwiakowłókniakowato
nerwiakowłókniakowato
ść
ść
Obraz kliniczny
plamy barwnikowe na
skórze (plamy kawowe)
Piegi w okolicach pach i
pachwin
guzki skórne i podskórne
wywodzące się z nerwów
obwodowych i ich zakończeń
Guzki w obrębie kanału
kręgowego mogą być
przyczyną niedowładów i
zaburzeń czucia
obniżenie sprawności
intelektualnej
padaczka
Leczenie
Zaopatrzenie ortopedyczne i
rehabilitacja
Właściwe postępowanie
edukacyjne

Fenyloketonuria E70.0
Fenyloketonuria E70.0
Obraz kliniczny przy nie
stosowaniu diety
opóźnienie rozwoju
psychoruchowego
niepełnosprawność
intelektualna
mysi zapach moczu
padaczka
zaburzenia chodu i postawy
ruchy ateotyczne
zesztywnienie stawów
Leczenie
zastosowanie diety
zapobiega uszkodzeniu OUN

Albinizm E70.3
Albinizm E70.3
bielactwo
bielactwo
Obraz kliniczny
Brak pigmentu w
skórze, cebulkach
włosów i tęczówce
Jasna skóra
Białe włosy, brwi, rzęsy
Czerwone oczy, rzadziej
niebieskawe zabarwienie
Leczenie
Unikanie działania
promieni słonecznych
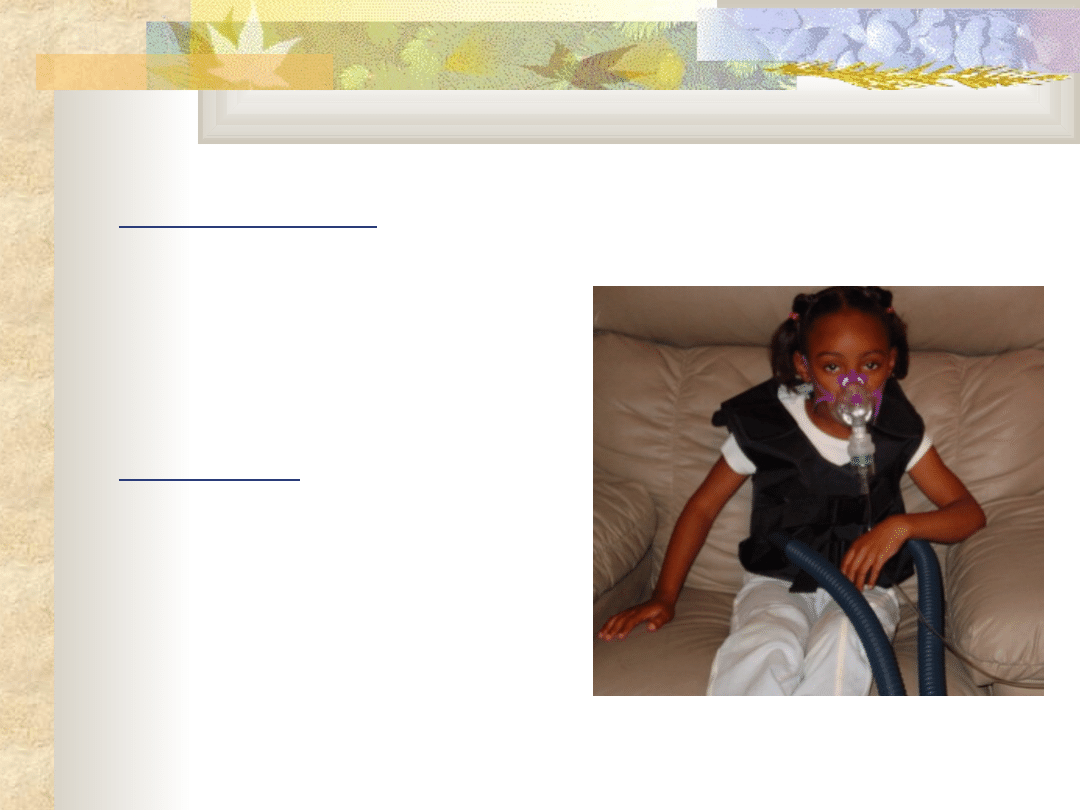
Mukowiscydoza E.84
Mukowiscydoza E.84
Obraz kliniczny
Nadmierna produkcja
śluzu
Nawracające zapalenia
oskrzeli, płuc, zatok
Niewydolność
enzymatyczna trzustki
Powikłania
Niepłodność
Cukrzyca
Niewydolność serca
Marskość wątroby

Mukowiscydoza
Mukowiscydoza
leczenie
leczenie
Leczenie objawów ze
strony układu
oddechowego:
Antybiotykoterapia
inhalacje
Fizykoterapia
(drenaże)
sport
mukolityki
Terapia tlenowa
Przeszczep płuc
Leczenie objawów ze
strony układu
pokarmowego
substytucja
enzymatyczna
dieta
wysokokaloryczna
suplementacja
witaminowa

Choroby
Choroby
sprzężone z
sprzężone z
chromosomem X
chromosomem X

Zespół Retta F84.2
Zespół Retta F84.2
Okres normalnego wczesnego
rozwoju, po którym następuje
częściowa lub całkowita utrata
sprawności manualnej i mowy oraz
zahamowanie tempa wzrostu głowy,
czas ten przypada na okres ok. 7
m.ż. do 24 m.ż. dziecka
W ICD 10 klasyfikowany jako
całościowe zaburzenie rozwojowe

Zespół Retta
Zespół Retta
Obraz kliniczny
Utrata zdolności
mowy
Stereotyp w postaci
kręcenia palcami
Hiperwentylacja
Utrata celowych
ruchów rąk
Zahamowaniu ulegają
rozwój zabawy i
społeczny (pierwsze
2-3 lata życia)
W połowie
dzieciństwa rozwija
się ataksja tułowia i
apraksja połączone
ze skoliozą i
kifoskoliozą
Czasami ruchy
choreoatetotyczne
Niepełnosprawność
intelektualna
Napady padaczkowe
(zwykle przed 8 r.ż.)

Hemofillia D66
Hemofillia D66
Hemofilia A -
niedobór VIII
czynnika krzepnięcia
krwi (czynnika
antyhemolitycznego),
"klasyczna hemofilia"
Hemofilia B -
niedobór IX czynnika
krzepnięcia krwi
(czynnika
Christmasa),
"choroba Christmasa
"
Postacie hemofilii:
postać ciężka - poziom
czynnika poniżej 1%
normy. Charakteryzuje się
częstymi wylewami
pourazowymi i
występowaniem tzw.
krwotoków samoistnych
(bez wyraźnej przyczyny
urazowej)
postać umiarkowana -
poziom czynnika poniżej
5% normy
postać lekka - poziom
czynnika powyżej 5%
normy

Hemofilia
Hemofilia
Objawy to krwotok nawet przy
niewielkim urazie, skaleczeniu oraz
wylewy dostawowe, które prowadzą
do zniekształceń stawów i
przykurczów
Leczenie profilaktyczne polega na
uzupełnianiu dożylnie czynnika
krzepnięcia

Choroby nerwowo-
Choroby nerwowo-
mięśniowe
mięśniowe
Dystrofie, miopatie
Neuropatie
Zanik rdzeniowy mięśni
Objawy wspólne:
1.
Wiotkość mięśniowa
2.
Osłabienie siły mięśni
3.
Zanik mięśni
4.
Ból mięśni, sztywność, kurcze, przykurcze

Dystrofia mięśniowa
Duchenna (DMD)
Upośledzenie ruchu
Paraliż kończyn dolnych
Symetryczne osłabienie
mięśni obręczy biodrowej
i barkowej
„kaczkowaty chód”
Trudności ze wstawaniem
z pozycji leżącej i
poruszaniem po schodach
Beckera (BMD)
Objawy takie same jak DMD
ale przy łagodniejszym
przebiegu

Miopatie
Typu central core
Miopatia nitkowata
Miopatia miotubularna
Inne: glikogenozy, miopatie tłuszczowe i
mitochondrialne
Leczenie
Rehabilitacja ruchowa
Ćwiczenia oddechowe
Ew. zabiegi korekcyjne

Rdzeniowy zanik mięśni
Typ 1 ostra postać dziecięca –
choroba Werdinga Hoffmanna
Typ 2 postać pośrednia
Typ 3 postać młodzieńcza
Kugelberga-Welander
Zaburzenie
rozpoznawania barw
Rodzaje
dichromatyzmu
:
protanopia – nie
rozpoznawanie
barwy czerwonej
(lub myleniu jej z
barwą zieloną)
deuteranopia
(tzw. daltonizm)
– nie
rozpoznawanie
barwy zielonej
(lub myleniu jej z
barwą czerwoną)
tritanopia – nie
rozpoznawanie
barw żółtej i
niebieskiej
Rodzaje
trichromacji:
protanomalia
obniżona
percepcja
nasycenia i
jaskrawości
czerwieni
deuteranomalia
obniżona
percepcja
nasycenia (ale nie
jaskrawości)
zieleni
tritanomalia
obniżona
percepcja barwy
niebieskiej
Monochromatyzm w
3 formach:
Monochromacja
czopków, nie ma
możliwości rozróżniania
barw, ale wizja jest w
miarę normalna
Achromatopsja poza
niezdolnością do
rozpoznawania barw
występują również
trudności w widzeniu
przy normalnym świetle
Agnozja barw

Zaburzenia w
Zaburzenia w
liczbie
liczbie
chromosomów
chromosomów

Zespół Downa Q90
Zespół Downa Q90
spłaszczona potylica lub
małogłowie
spłaszczona nasada nosa
drobne, skośnie, szeroko
rozstawione szpary
powiekowe nisko osadzone,
małe małżowiny uszne
mikrognacja i związana z nią
rzekoma makroglosia (język
nie mieści się w jamie
ustnej)
bruzdy na języku tzw. język
mosznowy
Wady serca, narządu wzroku
i słuchu
Obniżone napięcie
mięśniowe

Zespół Pataua Q91
Zespół Pataua Q91
rozszczep warg i podniebienia
wady oczu (mikroftalmia, anoftalmia)
wady uszu (niskie osadzenie, zniekształcenie małżowin)
anomalie kończyn (polidaktylia, ustawienie palców w
kształcie "kurka od strzelby", pojedyncza bruzda
zgięciowa)
wady sercowo-naczyniowe
wady nerek (torbielowatość nerek, wodonercze)
anomalie mózgu
hipotonia mięśni
napady drgawek
wnętrostwo
wady rozwoju macicy

Zespół Edwardsa
Zespół Edwardsa
niska masa urodzeniowa noworodka (dystrofia
wewnątrzmaciczna)
deformacje czaszki: małogłowie, łódkogłowie
wąskie szpary powiekowe
wady rogówki i tęczówki
nisko osadzone, dysplastyczne małżowiny uszne
anomalie szkieletu:
krótki mostek
zwichnięcie stawów biodrowych
deformacje zgięciowe palców, nakładające się na siebie palce
zgięcie podeszwowe paluchów ("młotkowaty paluch„)
wady serca
wady rozwojowe nerek
wady narządów płciowych
głębokie upośledzenie umysłowe

Zaburzenia w liczbie
Zaburzenia w liczbie
chromosomów płci
chromosomów płci
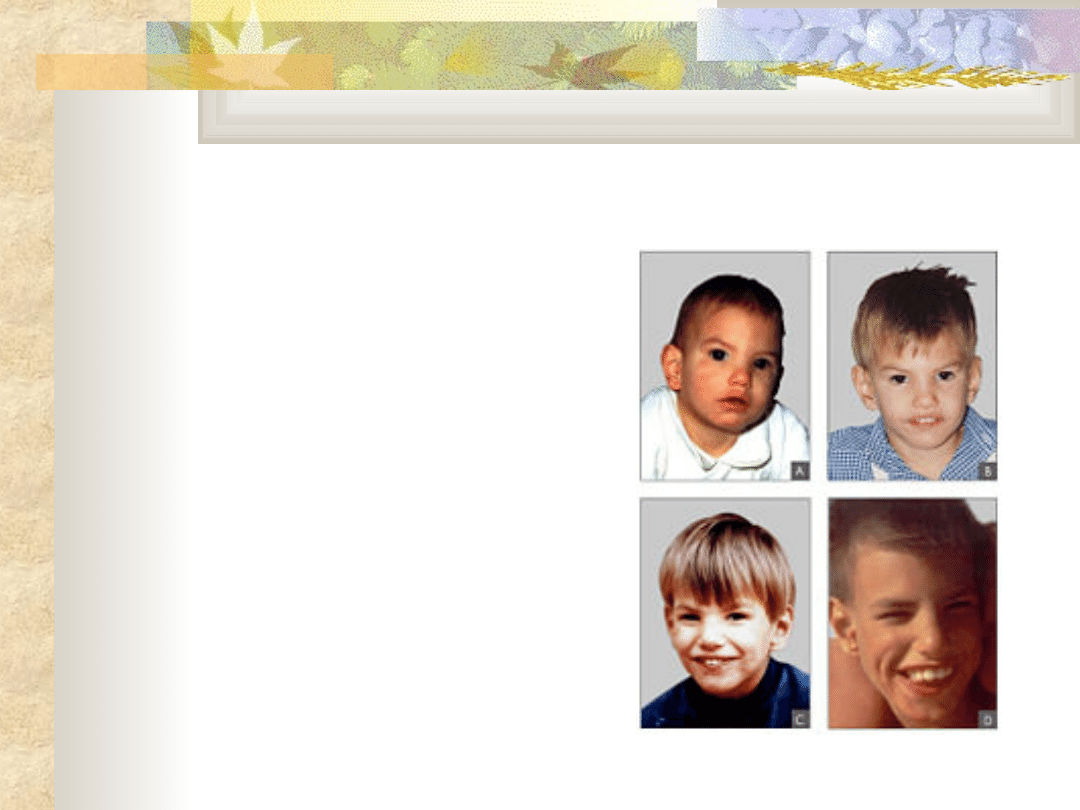
Zespół Turnera Q96
Zespół Turnera Q96
poduszeczkowate obrzęki
limfatyczne dłoni i stóp w
okresie noworodkowym i
niemowlęcym
nadmiar skóry na karku,
płetwiasta szyja
nisko osadzone,
zrotowane ku tyłowi uszy,
wady układu krążenia
wady układu moczowego
niepełnosprawność
intelektualna, zaburzenia
rozwoju emocjonalnego

Zespół Klinefeltera Q
Zespół Klinefeltera Q
98
98
Słabe umięśnienie
Kobieca sylwetka
Dłuższe niż przeciętnie kończyny
górne i dolne
Zmniejszenie rozmiaru jąder i
brak spermatogenezy
Zmniejszenie ilości testosteronu

Trisomia chromosomu
Trisomia chromosomu
X Q 97
X Q 97
zespół XXX, nadkobieta
zespół XXX, nadkobieta
Kobieta posiada 47 chromosomy
Dobrze zaznaczone
trzeciorzędowe cechy płciowe
Niepełnosprawność intelektualna
w stopniu lekkim
Czasami obniżona płodność

Delecje
Delecje
Zespół kociego krzyku
Zespół kociego krzyku
Q 93.4
Q 93.4
Zaburzenia w budowie
krtani powodują
charakterystyczny
płacz dziecko w
okresie pierwszych
miesięcy życia,
przypominający
miauczenie kota,
Zmiany kostno-
stawowe, koślawość
kończyn, nadmierna
ruchomość stawów,
Widoczne cechy
dysmorficzne twarzy

Zespół Angelmana
Zespół Angelmana
Q95.3
Q95.3
Niepełnosprawno
ść intelektualna,
Ataksja
Padaczka
Wybuchy
śmiechu bez
przyczyny
Liczne
sensoryzmy

Zespół Wolfa-
Zespół Wolfa-
Hirschhorna Q 93.3
Hirschhorna Q 93.3
Niska masa urodzeniowa
Spadziectwo
Małogłowie
Dysmorfia twarzy (twarz
przypominająca hełm grecki)
Wady układu kostnego

Zespół Pradera- Williego
Zespół Pradera- Williego
Q87.1
Q87.1
Niski wzrost
Niepełnosprawność
intelektualna
Hyperfagia- niepohamowane
uczucie głodu przy
zmniejszonym energetycznym
zapotrzebowaniu organizmu

Zespół Di George a D
Zespół Di George a D
82.1
82.1
Pierwotny niedobór odporności
spowodowany aplazją grasicy
Wrodzone wady serca
Zaburzenia w rozwoju
podniebienia
Trudności w uczeniu

Profilaktyka
Profilaktyka
możliwości leczenia
możliwości leczenia
Poradnictwo genetyczne
Centrum Genetyki Medycznej
Poradnia Genetyczna
ul. Grudzieniec 4, Poznań
Stowarzyszenie na Rzecz Dzieci z
Zaburzeniami Genetycznymi GEN
os. Lecha 14 (Przedszkole nr 53) , 61-293
Poznań
www.gen.org.pl

Profilaktyka i możliwości
leczenia
Badania prenatalne
1.
Badania inwazyjne
2.
Badania nieinwazyjne
Terapia genowa

Dziękuję za
Dziękuję za
uwagę
uwagę

Bibliografia
Bibliografia
Dziecko niepełnosprawne
ruchowo.Red.Z. Łosiowski,
WSP,W-wa 1997r
Międzynarodowa Statystyczna
Klasyfikacja Chorób i
Problemów Zdrowotnych,
rewizja dziesiąta, Vesalius W-
wa 2000r
www.gentyk.pl
www.genetyka.hg.pl
www.marfan.sprint.pl
www.akson.pl
www.orto.med..pl
www.allsengoz.net
www.gdansk.sprint.pl
www.gen.org.pl
www.
allaboutar.ritis.com
www.mukowiscydoza.pl
www.muko.med..pl
www.rettsyndrom.gd.pl
www.wikipedia.pl
www.portalwiedzy.onet.
pl
www.prader-willi.pl
www.spellcoder.com
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
Wyszukiwarka
Podobne podstrony:
Szkol Choroby genetyczne mutacje
Szkol Choroby Ból głowy
choroby genetyczne zespoly meta Nieznany (2)
choroby genetyczne tabelka, I rok, I rok, gieldy, pen, medycyna, 1 semestr, Biologia medyczna, Genet
Choroby genetyczne
ROZDZIAL VII CHoroby genetyczne
Choroby genetyczne zadania z kolokwium
wykłady choroby, genetyka, moja prezentacja
4 Charakterystyka chorób genetycznych człowieka
Choroby genetyczne człowiekai ich diagnostyka Biologia
Choroby genetyczne. Zaburzenia w liczbie chromosomów płci, Biologia
Biologia część V, Zespół Turnera choroba genetyczna
Szkol Choroby Prionowe
BIOLOGIA Prezentacja choroby genetyczne
więcej podobnych podstron