
Choroby genetyczne
Choroby genetyczne
spowodowane
spowodowane
mutacjami
mutacjami
chromosomowymi
chromosomowymi

Choroby genetyczne
Choroby genetyczne
Związane z zaburzeniami liczby
Związane z zaburzeniami liczby
chromosomów płciowych
chromosomów płciowych
Związane z aneuploidią autosomów
Związane z aneuploidią autosomów

Choroby związane z liczbą
Choroby związane z liczbą
chromosomów płciowych
chromosomów płciowych
Zespół Turnera
Zespół Turnera
Zespół Klinefeltera
Zespół Klinefeltera
Zespół nadkobiety
Zespół nadkobiety
Zespół nadmężczyzny
Zespół nadmężczyzny

Zespół Turnera
Zespół Turnera
Monsomia heterosomów – zespół Turnera
Monsomia heterosomów – zespół Turnera
.
.
wystąpi wtedy, gdy w garniturze człowieka będzie
wystąpi wtedy, gdy w garniturze człowieka będzie
tylko jeden chromosom płciowy (X-ponieważ bez
tylko jeden chromosom płciowy (X-ponieważ bez
niego nie powstanie żywy organizm). Osobniki
niego nie powstanie żywy organizm). Osobniki
taki posiada 45 chromosomów (44
taki posiada 45 chromosomów (44
autosomy+X).Osoba z tym zespołem ma płeć
autosomy+X).Osoba z tym zespołem ma płeć
chromosomową żeńską (brak chromosomu Y), nie
chromosomową żeńską (brak chromosomu Y), nie
ma także ciałek Barra. Fenotypowo są to kobiety,
ma także ciałek Barra. Fenotypowo są to kobiety,
bezpłodne (niedorozwinięte jajniki), przeważnie
bezpłodne (niedorozwinięte jajniki), przeważnie
upośledzone umysłowo, maja niski wzrost i krępą
upośledzone umysłowo, maja niski wzrost i krępą
budowę ciała, płetwiastą szyję
budowę ciała, płetwiastą szyję

Zewnętrzne i wewnętrzne cechy
Zewnętrzne i wewnętrzne cechy
zespołu TURNERA !
zespołu TURNERA !
Specyficzne cechy morfologiczne
Specyficzne cechy morfologiczne
dysmorfia (zniekształcenia) twarzy,
dysmorfia (zniekształcenia) twarzy,
płetwistość szyi, obrzęki rąk i nóg,
płetwistość szyi, obrzęki rąk i nóg,
niska linia włosów na karku, koślawość
niska linia włosów na karku, koślawość
łokci, skrócenie IV kości śródręcza,
łokci, skrócenie IV kości śródręcza,
smukłość palców, hipoplazja paznokci,
smukłość palców, hipoplazja paznokci,
niskie osadzenie i (lub) odstawanie
niskie osadzenie i (lub) odstawanie
małżowin usznych szeroki rozstaw
małżowin usznych szeroki rozstaw
brodawek sutkowych, puklerzowata
brodawek sutkowych, puklerzowata
klatka piersiowa itp.
klatka piersiowa itp.
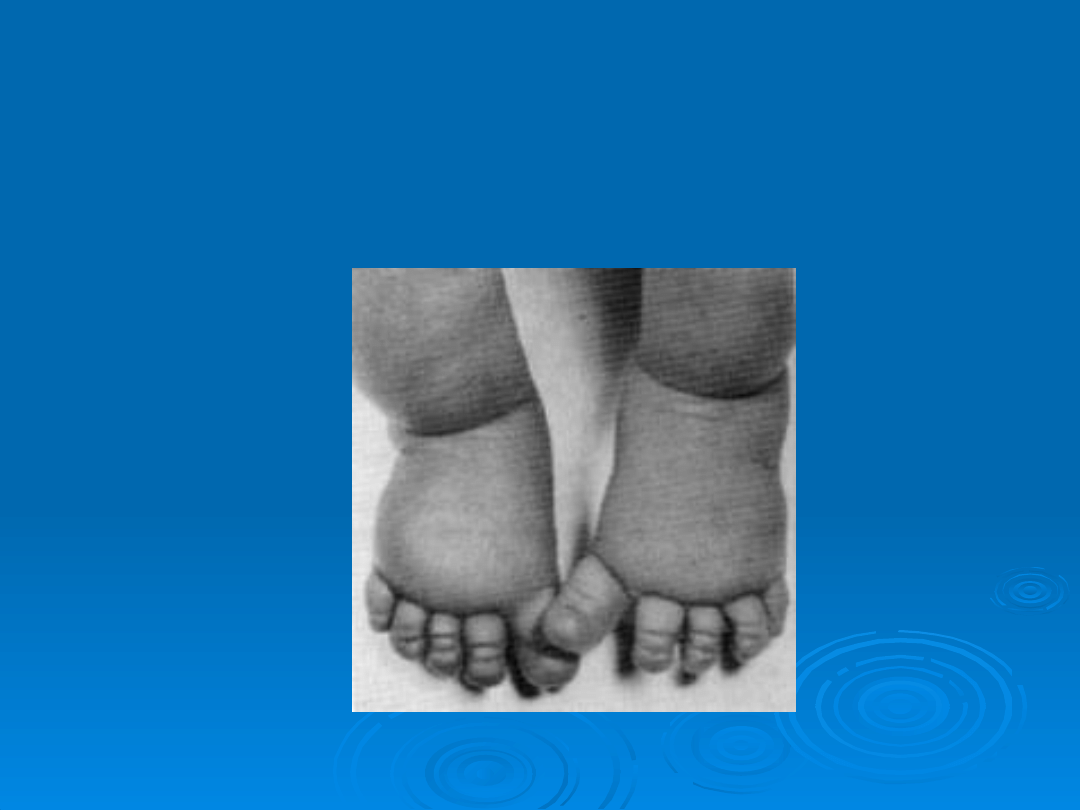
Poduszeczkowaty obrzęk
Poduszeczkowaty obrzęk
stóp
stóp
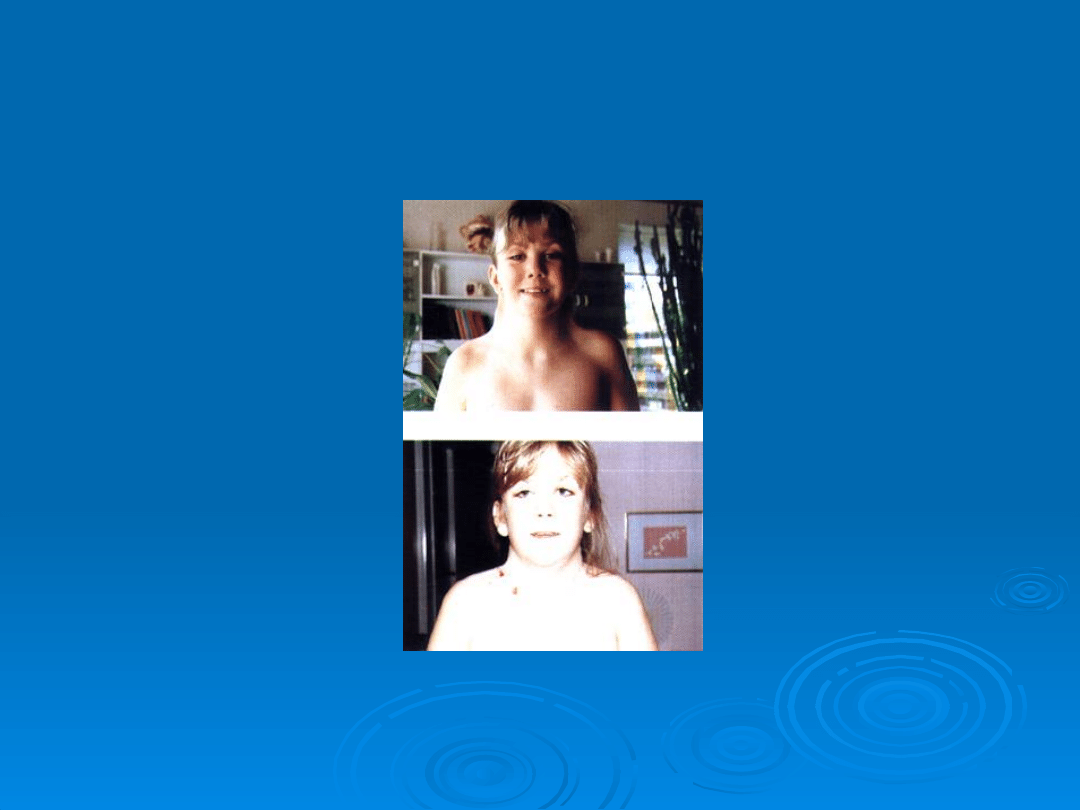
Płetwiasta szyja
Płetwiasta szyja
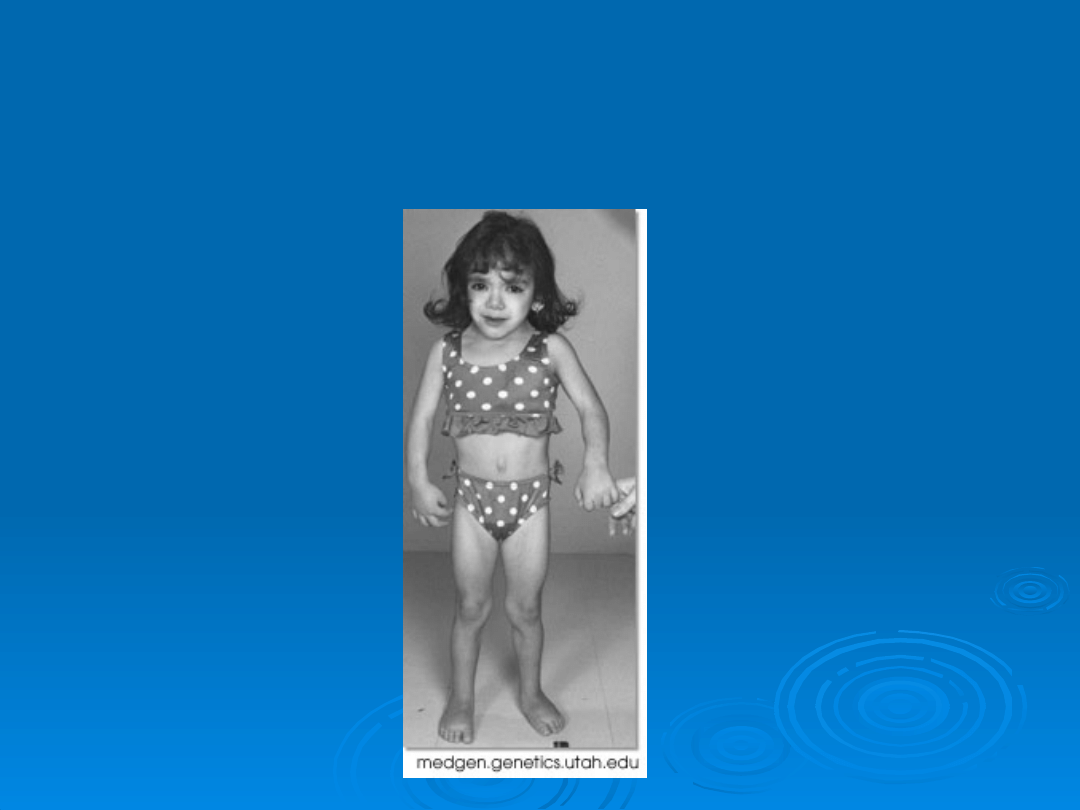
Dziewczynka z Zespołem
Dziewczynka z Zespołem
Turnera
Turnera

Zespól Klinefeltera
Zespól Klinefeltera
Kariotyp - 44 autosomy + trzy chromosomy płciowe (XXY).
Kariotyp - 44 autosomy + trzy chromosomy płciowe (XXY).
Płeć chromosomowa jest męska, płeć chromatynowa
Płeć chromosomowa jest męska, płeć chromatynowa
żeńska. Fenotypowo są to mężczyźni, zwykle wyżsi i słabiej
żeńska. Fenotypowo są to mężczyźni, zwykle wyżsi i słabiej
umięśnieni niż inni mężczyźni w ich rodzinach (średni
umięśnieni niż inni mężczyźni w ich rodzinach (średni
wzrost chorych to 182 cm, podczas gdy średni wzrost w
wzrost chorych to 182 cm, podczas gdy średni wzrost w
populacji męskiej wynosi 175 cm). u których cechy
populacji męskiej wynosi 175 cm). u których cechy
patologiczne pojawiają się w okresie dojrzewania: są
patologiczne pojawiają się w okresie dojrzewania: są
bezpłodni (niedorozwój jąder), wykazują objawy
bezpłodni (niedorozwój jąder), wykazują objawy
ginekomastii (rozwój sutek w typie kobiecym), występują
ginekomastii (rozwój sutek w typie kobiecym), występują
także inne cechy kobiece takie jak brzmienie głosu,
także inne cechy kobiece takie jak brzmienie głosu,
charakter owłosienia, układ tkanki tłuszczowej. Przeważnie
charakter owłosienia, układ tkanki tłuszczowej. Przeważnie
maja normalny poziom inteligencji, ale mogą wykazywać
maja normalny poziom inteligencji, ale mogą wykazywać
zaburzenia i anomalie seksualne a także agresywność.
zaburzenia i anomalie seksualne a także agresywność.
Osoby z zespołem Klinefeltera stanowią 0,1% populacji
Osoby z zespołem Klinefeltera stanowią 0,1% populacji
męskiej.
męskiej.
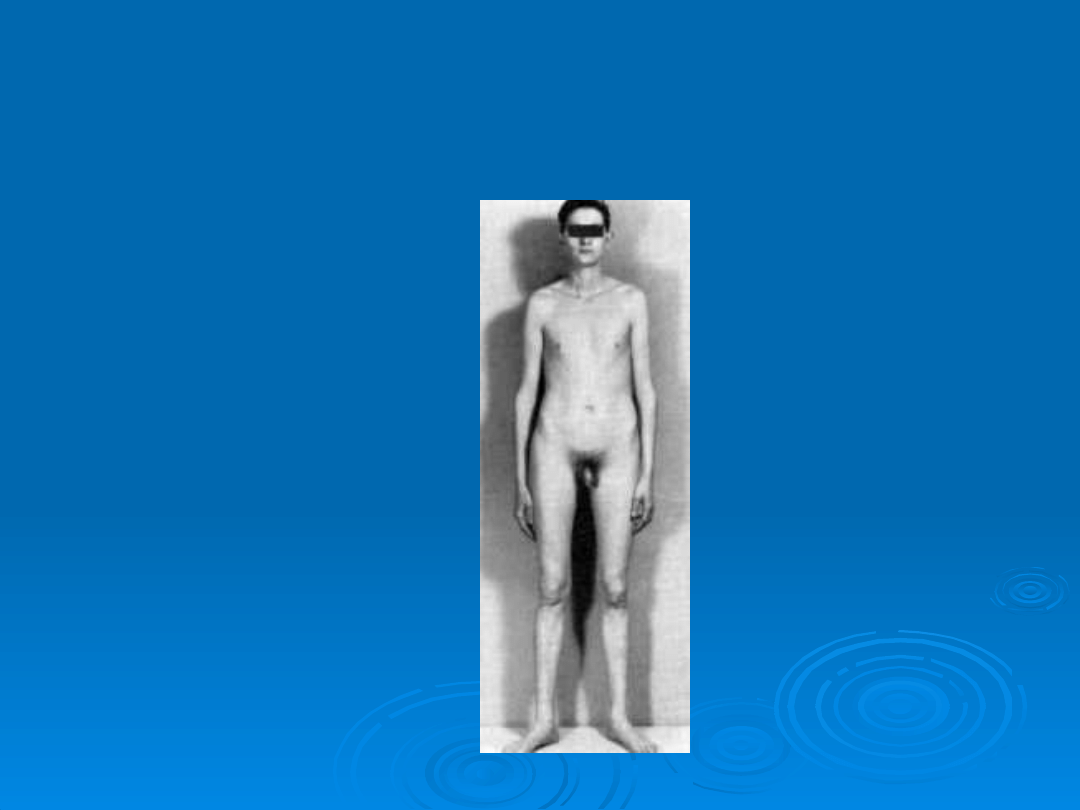
Mężczyzna z zespołem
Mężczyzna z zespołem
Klinefeltera
Klinefeltera

Zespół nadkobiety
Zespół nadkobiety
Trisomia chromosomu X
Trisomia chromosomu X
(
(
zespół XXX
zespół XXX
,
,
nadkobieta
nadkobieta
,
,
metakobieta
metakobieta
,
,
nadsamica
nadsamica
) -
) -
. Kobieta dotknięta tym zespołem posiada
. Kobieta dotknięta tym zespołem posiada
dodatkowy
chromosomów w
chromosomów w
.
W znaczeniu
W znaczeniu
nadsamicy
nadsamicy
- osobnik o cechach
- osobnik o cechach
żeńskich, często z wyjątkowo dobrze zaznaczonymi
żeńskich, często z wyjątkowo dobrze zaznaczonymi
trzeciorzędowymi cechami płciowymi.
trzeciorzędowymi cechami płciowymi.
Wada ta jest silnie związana z wiekiem matki, im większy wiek
Wada ta jest silnie związana z wiekiem matki, im większy wiek
matki, tym większe prawdopodobieństwo trisomii XXX u córki.
matki, tym większe prawdopodobieństwo trisomii XXX u córki.
Występuje z częstością 1/1000 urodzeń (szacuje się, że 0,1%
Występuje z częstością 1/1000 urodzeń (szacuje się, że 0,1%
populacji kobiet ma ten zespół) i zazwyczaj przebiega bez
populacji kobiet ma ten zespół) i zazwyczaj przebiega bez
istotnych cech fenotypowych lub klinicznych choroby. Trisomii X
istotnych cech fenotypowych lub klinicznych choroby. Trisomii X
może towarzyszyć wysoki wzrost. Większość pacjentek rozwija się
może towarzyszyć wysoki wzrost. Większość pacjentek rozwija się
normalnie i nie ma problemów z płodnością, choć zdarzają się
normalnie i nie ma problemów z płodnością, choć zdarzają się
przypadki zaburzeń miesiączkowania i obniżenia płodności.
przypadki zaburzeń miesiączkowania i obniżenia płodności.
Trisomii X towarzyszy też zwiększone ryzyko wystąpienia trudności
Trisomii X towarzyszy też zwiększone ryzyko wystąpienia trudności
w uczeniu się, a w niektórych przypadkach lekkie upośledzenie. U
w uczeniu się, a w niektórych przypadkach lekkie upośledzenie. U
kobiet z zespołem XXX występują dwa ciałka Barra.
kobiet z zespołem XXX występują dwa ciałka Barra.

Zespół nadmężczyzny
Zespół nadmężczyzny
Kariotyp - 44 autosomy +XYY.
Kariotyp - 44 autosomy +XYY.
Są to mężczyźni o wysokim
Są to mężczyźni o wysokim
wzroście, z intensywnym
wzroście, z intensywnym
trądzikiem, jednak są oni płodni i
trądzikiem, jednak są oni płodni i
ich potomstwo jest bez skazy.
ich potomstwo jest bez skazy.
Stanowią około 0,1% populacji
Stanowią około 0,1% populacji
męskiej.
męskiej.

Choroby związane z
Choroby związane z
aneuploidią autosomów
aneuploidią autosomów
Zespół Downa
Zespół Downa
Zespół Patau
Zespół Patau
Zespół Edwardsa
Zespół Edwardsa

Zespół Downa
Zespół Downa
Trisomia chromosomu 21.
Osoby z tą wadą mają mongoidalne rysy twarzy,
szczególny układ
bruzd na dłoniach – małpie
dłonie, niezborność ruchów i niedorozwój
umysłowy. Inne częste problemy to przepukliny i
brak jąder w worku
mosznowym u chłopców (jądra
niezstąpione). Zwykle w dalszej perspektywie
obserwuje się opóźnione dojrzewanie płciowe i
niski wzrost (u obu płci). Mężczyźni
przeważnie są
bezpłodni. Dziewczynki mogą miesiączkować, a
niektóre zajść w ciążę. Obecnie niemal 80%
chorych z zespołem Downa przeżywa
więcej niż 30
lat. Niemal wszyscy z trisomią 21 po czterdziestym
roku życia
zapadają na chorobę Alzheimera.
Dzieci z
zespołem Downa z reguły są pogodne, ciepłe, bardzo
przywiązane do rodziców.

Zespół Downa
Zespół Downa
Zespół Downa wykrywa się metodami
Zespół Downa wykrywa się metodami
diagnostyki prenatalnej. Zaobserwowano,
diagnostyki prenatalnej. Zaobserwowano,
że starsze kobiety w większym stopniu są
że starsze kobiety w większym stopniu są
narażone na ryzyko urodzenia dziecka z
narażone na ryzyko urodzenia dziecka z
tą wadą niż kobiety młodsze. U matek,
tą wadą niż kobiety młodsze. U matek,
które nie przekroczyły 28. roku życia, z
które nie przekroczyły 28. roku życia, z
zespołem Downa rodzi się jedno dziecko
zespołem Downa rodzi się jedno dziecko
na tysiąc, u matek czterdziestoletnich
na tysiąc, u matek czterdziestoletnich
częstotliwość ta wynosi jeden do stu, a u
częstotliwość ta wynosi jeden do stu, a u
starszych nawet jeden do pięćdziesięciu.
starszych nawet jeden do pięćdziesięciu.

Dziecko z zespołem Downa
Dziecko z zespołem Downa

Zespół Edwardsa
Zespół Edwardsa
trisomia chromosomu 18.
trisomia chromosomu 18.
Częstotliwość występowania tej wady wynosi
Częstotliwość występowania tej wady wynosi
3/10000 żywo urodzonych niemowląt, z tym że u
3/10000 żywo urodzonych niemowląt, z tym że u
dziewczynek występuje on 3 razy częściej. Wiek
dziewczynek występuje on 3 razy częściej. Wiek
matki wpływa na częstotliwość występowania tego
matki wpływa na częstotliwość występowania tego
zespołu. Większość dzieci przeżywa 3-4 miesięcy,
zespołu. Większość dzieci przeżywa 3-4 miesięcy,
wyjątkowo do 2 lat. Dzieci z tym zespołem
wyjątkowo do 2 lat. Dzieci z tym zespołem
charakteryzują się m.in. wzmożonym napięciem
charakteryzują się m.in. wzmożonym napięciem
mięśniowym, przykurczem kończyn, nisko
mięśniowym, przykurczem kończyn, nisko
osadzonymi uszami, pojawiają się wady stóp (tzw.
osadzonymi uszami, pojawiają się wady stóp (tzw.
suszkowate stopy), niemowlęta zaciskają
suszkowate stopy), niemowlęta zaciskają
charakterystycznie piąstkę. Często zespołowi
charakterystycznie piąstkę. Często zespołowi
Edwardsa towarzyszą wady serca bądź ośrodkowego
Edwardsa towarzyszą wady serca bądź ośrodkowego
układu nerwowego.
układu nerwowego.
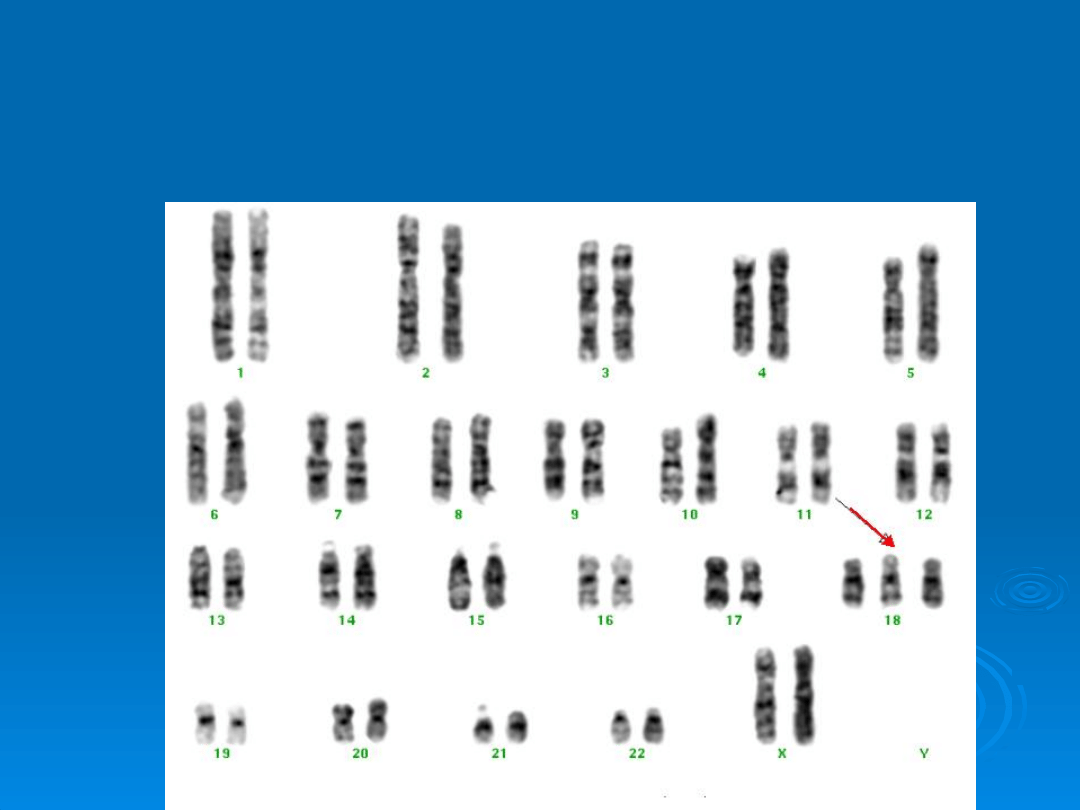
Kariotyp Zespołu Edwardsa
Kariotyp Zespołu Edwardsa

Zespół Patau
Zespół Patau
trisomia chromosomu 13.
trisomia chromosomu 13.
Częstotliwość występowania tej wady wynosi 2/10000 żywo
Częstotliwość występowania tej wady wynosi 2/10000 żywo
urodzonych niemowląt. Wiek matki, choć w niewielkim zakresie,
urodzonych niemowląt. Wiek matki, choć w niewielkim zakresie,
wpływa na częstotliwość występowania tego zespołu.
wpływa na częstotliwość występowania tego zespołu.
Dzieci z zespołem Patau umierają zazwyczaj w kilka godzin lub
Dzieci z zespołem Patau umierają zazwyczaj w kilka godzin lub
dni po urodzeniu. Tylko 1 dziecko z 20 przeżywa dłużej niż 6
dni po urodzeniu. Tylko 1 dziecko z 20 przeżywa dłużej niż 6
miesięcy. Jednakże niektóre dzieci przeżywają okres dojrzewania.
miesięcy. Jednakże niektóre dzieci przeżywają okres dojrzewania.
Osoby dorosłe z tym zespołem są rzadkością. Chorzy z Zespołem
Osoby dorosłe z tym zespołem są rzadkością. Chorzy z Zespołem
Patau charakteryzuje się ubytkami owłosienia na głowie, oczy są
Patau charakteryzuje się ubytkami owłosienia na głowie, oczy są
osadzone blisko siebie (może dojść nawet do ich "połączenia").
osadzone blisko siebie (może dojść nawet do ich "połączenia").
Zespołowi temu towarzyszy często rozczep wargi i podniebienia,
Zespołowi temu towarzyszy często rozczep wargi i podniebienia,
polidaktylia czyli dodatkowy palec, najczęściej od strony palca
polidaktylia czyli dodatkowy palec, najczęściej od strony palca
małego, zdarzają się wytrzewienia.
małego, zdarzają się wytrzewienia.
Z chorób towarzyszących liczne są wady serca, występują one w
Z chorób towarzyszących liczne są wady serca, występują one w
80% przypadków. Zazwyczaj obejmują one takie zaburzenia jak :
80% przypadków. Zazwyczaj obejmują one takie zaburzenia jak :
przetrwały przewód tętniczy, defekt przegrody
przetrwały przewód tętniczy, defekt przegrody
międzyprzedsionkowej i międzykomorowej, prawostronne
międzyprzedsionkowej i międzykomorowej, prawostronne
położenie serca).
położenie serca).
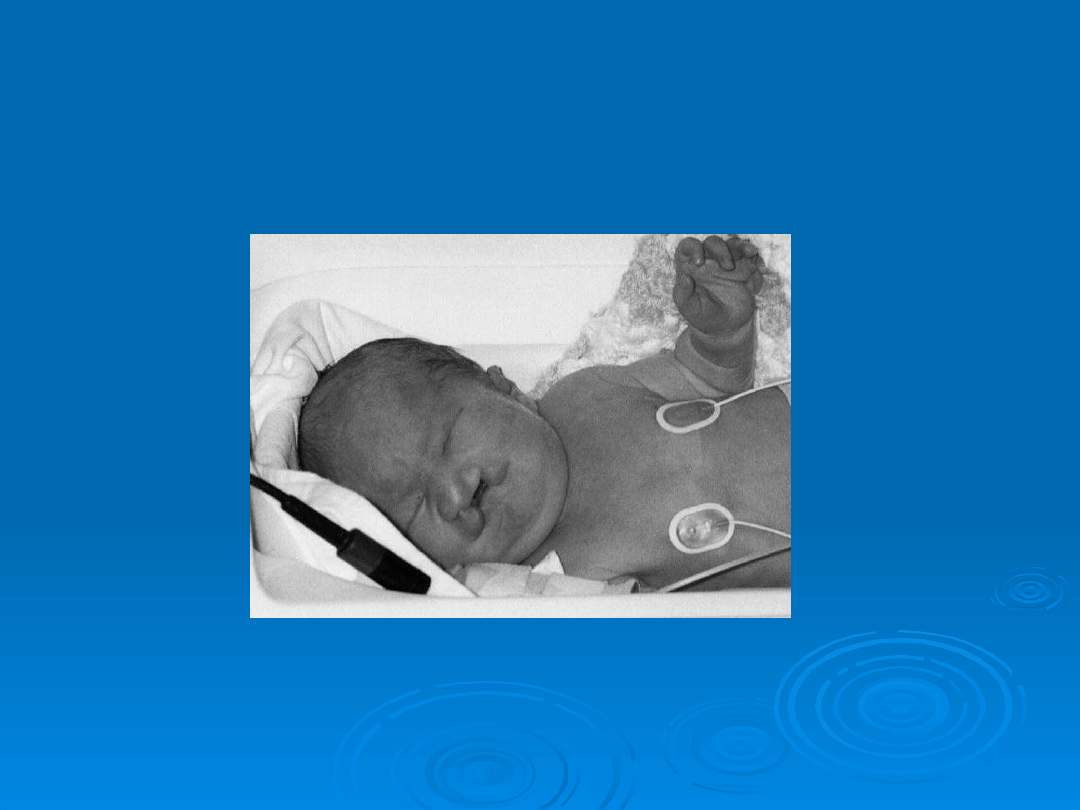
Dziecko z Zespołem Patau
Dziecko z Zespołem Patau

Zespół kociego krzyku
Zespół kociego krzyku
Zespół kociego krzyku (
Zespół kociego krzyku (
cri du chat
cri du chat
) powoduje delecja krótkiego
) powoduje delecja krótkiego
ramienia chromosomu 5 (kariotyp 46,XX,del 5p lub 46,XY,del 5p).
ramienia chromosomu 5 (kariotyp 46,XX,del 5p lub 46,XY,del 5p).
Wielkość delecji może być różna, od 5p15.2 do całego krótkiego
Wielkość delecji może być różna, od 5p15.2 do całego krótkiego
ramienia. Przypuszcza się,
ramienia. Przypuszcza się,
że do genów, których delecja wpływa
że do genów, których delecja wpływa
na fenotyp zespołu kociego krzyku należą położone na krótkim
na fenotyp zespołu kociego krzyku należą położone na krótkim
ramieniu chromosomu 5 geny kodujące białka biorące udział w
ramieniu chromosomu 5 geny kodujące białka biorące udział w
rozwoju mózgu - semaforynę F i delta-kateninę, a także gen
rozwoju mózgu - semaforynę F i delta-kateninę, a także gen
kodujący telomerazę. W okresie noworodkowym i pierwszych
kodujący telomerazę. W okresie noworodkowym i pierwszych
miesiącach okresu niemowlęcego stwierdza się
miesiącach okresu niemowlęcego stwierdza się
charakterystyczny płacz dziecka, zwykle prowadzący do
charakterystyczny płacz dziecka, zwykle prowadzący do
prawidłowej diagnozy i od którego zespół wziął swoją nazwę.
prawidłowej diagnozy i od którego zespół wziął swoją nazwę.
Płacz przez wysoką tonację dźwięków i monotonię przypomina
Płacz przez wysoką tonację dźwięków i monotonię przypomina
miauczenie kota. Wiązane jest to z nieprawidłową budową krtani
miauczenie kota. Wiązane jest to z nieprawidłową budową krtani
(krtań jest mała, wąska, romboidlanego ksztłtu) i nagłośni
(krtań jest mała, wąska, romboidlanego ksztłtu) i nagłośni
(wiotka, mała, hipotoniczna), a także zaburzeniami
(wiotka, mała, hipotoniczna), a także zaburzeniami
czynnościowymi i strukturalnymi układu nerwowego. Płacz zanika
czynnościowymi i strukturalnymi układu nerwowego. Płacz zanika
w ciągu kilku miesięcy
w ciągu kilku miesięcy
Spotyka się ponadto zmiany kostno-stawowe, takie jak koślawość
Spotyka się ponadto zmiany kostno-stawowe, takie jak koślawość
kończyn, nadmierna ruchomość stawów
kończyn, nadmierna ruchomość stawów

Dziecko z zespołem kociego
Dziecko z zespołem kociego
krzyku
krzyku
8 miesięcy, 42 lata, 4 lata, 9,5 roku
8 miesięcy, 42 lata, 4 lata, 9,5 roku
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
Wyszukiwarka
Podobne podstrony:
Szkol Choroby genetyczne
Choroby genetyczne. Mutacje punktowe, Biologia
Choroby genetyczne. Mutacje dynamiczne. Inne choroby, Genetyka
Choroby genetyczne. Mutacje punktowe, Genetyka
Choroby genetyczne. Mutacje dynamiczne. Inne choroby, Biologia
Choroby genetyczne spowodowane mutacją punktową
Choroby genetyczne u człowieka spowodowane są mutacjami
ĆW Mutacje Choroby genetyczne 3 10 09r
Mutacje, choroby genetyczne
Choroby genetyczne człowieka wynikajAce z mutacji chromosomowych strukturalnych
II Mutacje, choroby genetyczne, inżynieria genetyczna
Genetyka MUTACJE I WYBRANE CHOROBY GENETYCZNE (inf prez )
Materiał genetyczny, mutacje, systemy naprawy DNA, test Amesa
Szkol Choroby Ból głowy
choroby genetyczne zespoly meta Nieznany (2)
choroby genetyczne tabelka, I rok, I rok, gieldy, pen, medycyna, 1 semestr, Biologia medyczna, Genet
więcej podobnych podstron