
DZIEDZICZENIE JEDNOGENOWE
DZIEDZICZENIE JEDNOGENOWE
AUTOSOMALNE
AUTOSOMALNE

DZIEDZICZENIE
JEDNOGENOWE
AUTOSOMALNE
FENYLOKETONURIA
ALBINIZM
ALKAPTONURIA
MUKOWISCYDOZA
NIEDOKRWISTOŚĆ
SPIERPOWATOKRWINKOWA

Ogólne zasady
dziedziczenia
jednogenowego
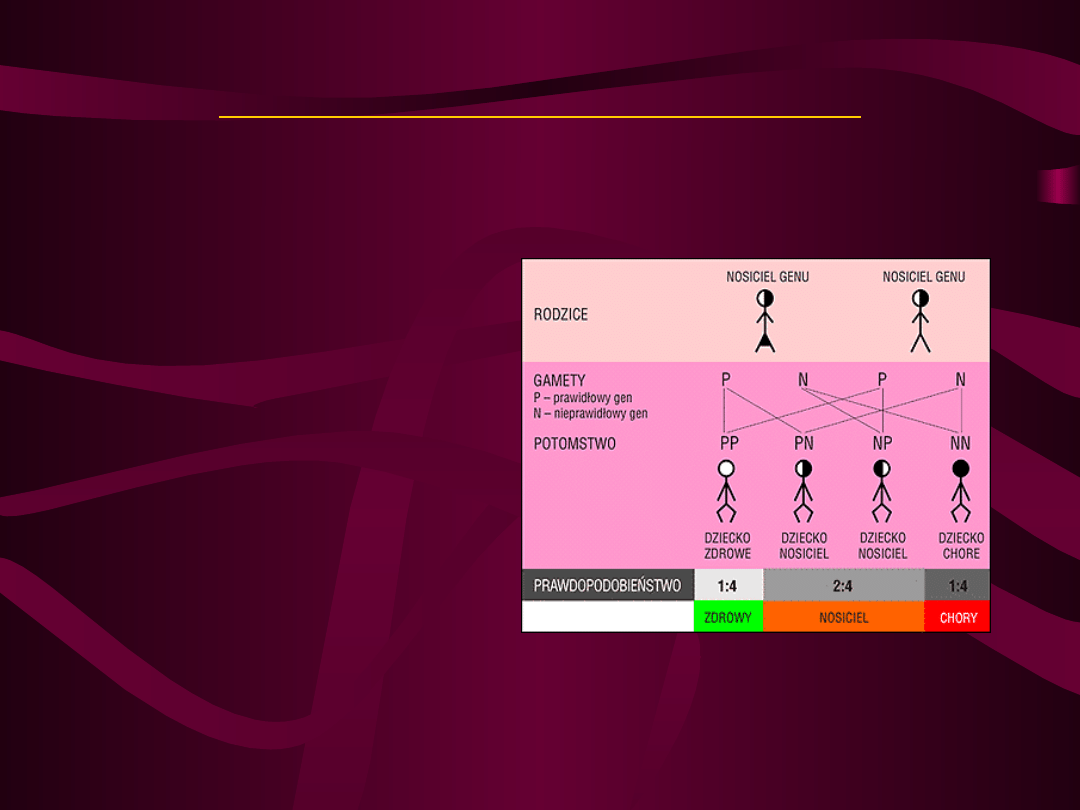
• Geny występują parami – jeden
dziedziczymy od ojca, drugi od matki.
• Geny jednej pary mogą występować w
różnych odmianach (allele), które
funkcjonują w sposób dominujący lub
recesywny (geny dominujące/recesywne).
• W czasie mejozy allele ulegają segregacji
pomiędzy gametami w ten sposób, że
każda gameta otrzymuje jeden gen z
pary (gamety są haploidalne).

Dziedziczenie
autosomalne
recesywne
Choroby które będą tu opisane dziedziczą się w sposób
autosomalny recesywny który charakteryzuje się
następująco:
1.Choroba ujawnia się u homozygot recesywnych, niezależnie
od płci pod względem zmutowanego genu.
2.Heterozygoty z jednym nieprawidłowym genem są zdrowymi
nosicielami.
3.Prawdopodobieństwo wystąpienia choroby recesywnej jest
takie samo dla obu płci.
4.Jeśli oboje rodzice są heterozygotycznymi nosicielami mają
25% ryzyka urodzenia dziecka chorego (przy każdej ciąży)
– pojawienie się choroby w rodzinie jest związane z
nosicielstwem rodziców.
5.Osoba chora (homozygota) przekazuje zmutowany gen
wszystkim swoim dzieciom, a wiec wszystkie jej dzieci będą
nosicielami (przy założeniu, że partner jest osobą zdrową).
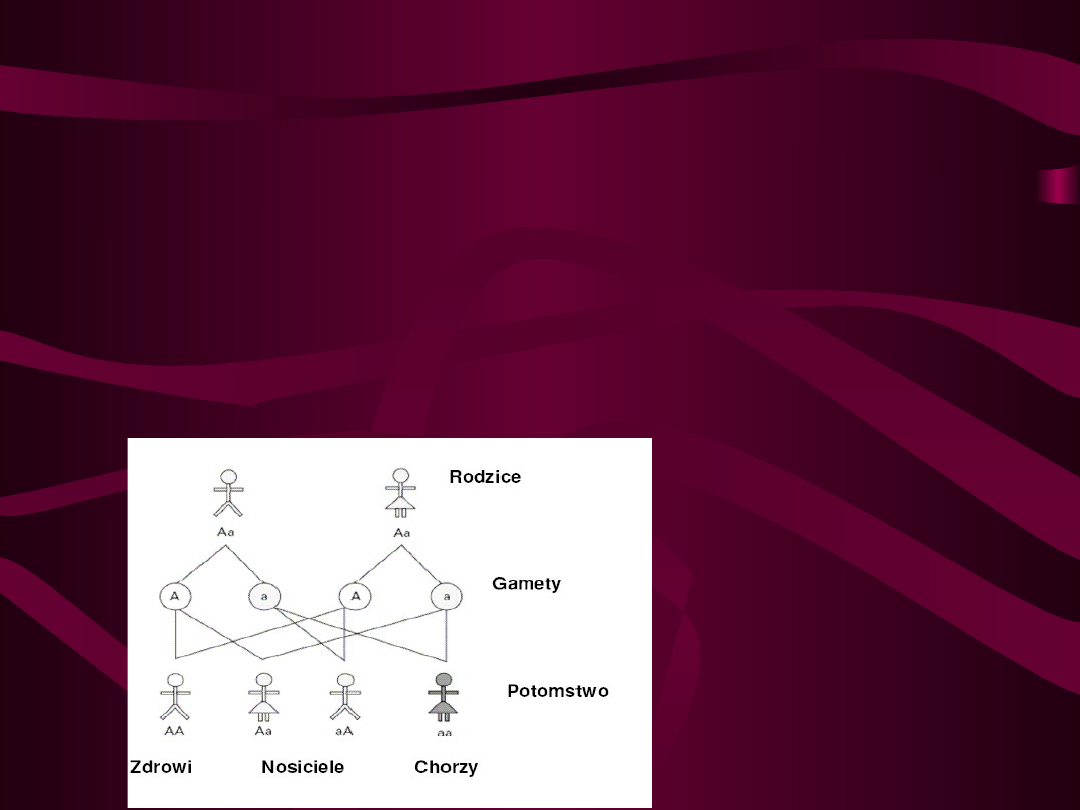
6.Zdrowe rodzeństwo osoby chorej ma 66% ryzyka, że jest
heterozygotycznym nosicielem.
7.W przypadku małżeństwa krewniaczego (np. kuzynostwo) ryzyko
urodzenia dziecka z chorobą autosomalną recesywną wzrasta.
8.Choroby dziedziczone autosomalnie recesywnie są najczęściej
wynikiem mutacji genów strukturalnych, kontrolujących syntezę
białek enzymatycznych, co prowadzi do zaburzeń
metabolicznych ustroju a przez to do zaburzeń jego procesów
życiowych

Fenyloketonuria
PKU
Jest najczęstszą wrodzoną wadą w metabolizmie
aminokwasów. Choroba pojawia się z częstotliwością
1/ 7000-8000 urodzeń, co oznacza że rocznie rodzi
się w naszym kraju 60-70 chorych dzieci
Na obecność tej choroby robi się badania
noworodkom w 3-4 dniu życia, za pomocą metody
kolorymetrycznej test Guthriego. Choroba jest
recesywna i aby się ujawniła dziecko musi być
homozygotą . Heterozygoty są tylko nosicielami.

Choroba polega na całkowitym braku lub
znacznym ograniczeniu aktywności enzymu
hydroksylazy fenyloalaninowej (PAH).
Efektem mutacji jest brak enzymu
przekształcającego fenyloalaninę w tyrozynę;
przemiana fenyloalaniny przebiega więc
bocznym torem, w wyniku czego powstaje
kwas fenylopirogronowy > fenylooctowy >
fenylomlekowy. Powoduje to nagromadzenie
się we krwi tych metabolitów co powoduje
uszkodzenie układu nerwowego. Powoduje to
różne formy niepełnosprawności
intelektualnej i zaburzenia neurologiczne,
które stanowią główne objawy nie leczonej lub
leczonej niewystarczająco PKU.

Do objawów choroby należą uporczywe
„chlustające wymioty „ , „mysi zapach”
moczu spowodowany obecnością kwasu
fenylooctowego, wzrost napięcia mięśni oraz
napady drgawek. Chore dzieci są jasnowłose,
jasnookie, o jasnej skórze, wykazują
zahamowanie rozwoju psychoruchowego,
mają obniżony iloraz inteligencji
Dzieci u których choroba zostaje wykryta w
pierwszych dniach życia i które następnie są
wcześnie, systematycznie i długotrwale
leczone,(dieta pozbawiona fenyloalaniny)
rozwijają się prawidłowo, wykorzystując w
pełni swój potencjał intelektualny.
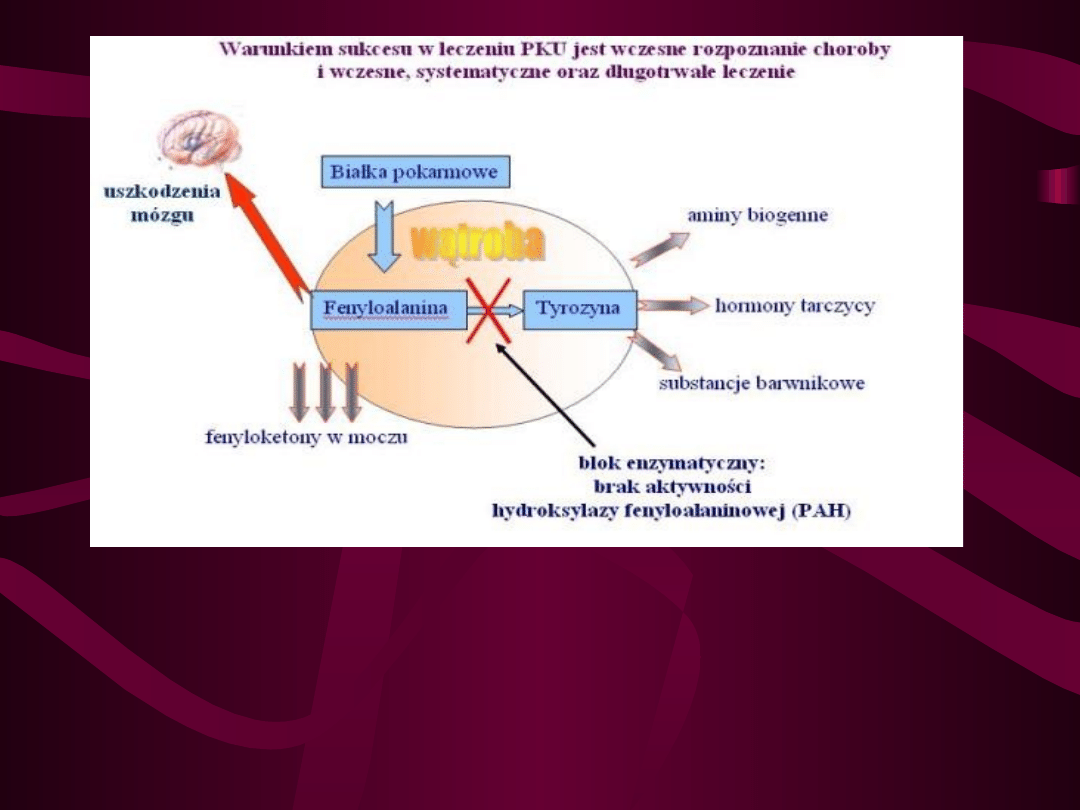

Dziedziczenie fenyloketonurii
Jest to choroba o podłożu genetycznym,
przekazywana dziecku przez oboje rodziców.
Dziedziczy się w sposób autosomalny
recesywny co oznacza, że chore dziecko
musi otrzymać po jednym nieprawidłowym
genie wywołującym PKU od każdego z
rodziców. Do pełnego wystąpienia choroby
potrzebne są dwa „chore” geny. Rodzice są
najczęściej tylko bezobjawowymi nosicielami
nieprawidłowego genu i zwykle nie zdają
sobie sprawy z tego, że mogą go przekazać
swojemu potomstwu.

W populacji polskiej co 46 osoba jest
zdrowym nosicielem nieprawidłowego
genu PKU. W rodzinie, gdzie mama i tata
są takimi bezobjawowymi nosicielami
istnieje 25% ryzyko, że każde dziecko
urodzi się chore. Urodzenie jednego
chorego dziecka nie oznacza, że kolejne
na pewno będą zdrowe – ryzyko
urodzenia następnego chorego dziecka
jest stałe i dla każdej ciąży wynosi 25%.
Jeżeli jedno z rodziców jest chore na PKU,
a drugie jest bezobjawowym nosicielem,
ryzyko urodzenia chorego dziecka z
każdej ciąży wynosi 50%. Z kolei
potomstwo dwójki osób chorych na PKU
będzie chore w 100%
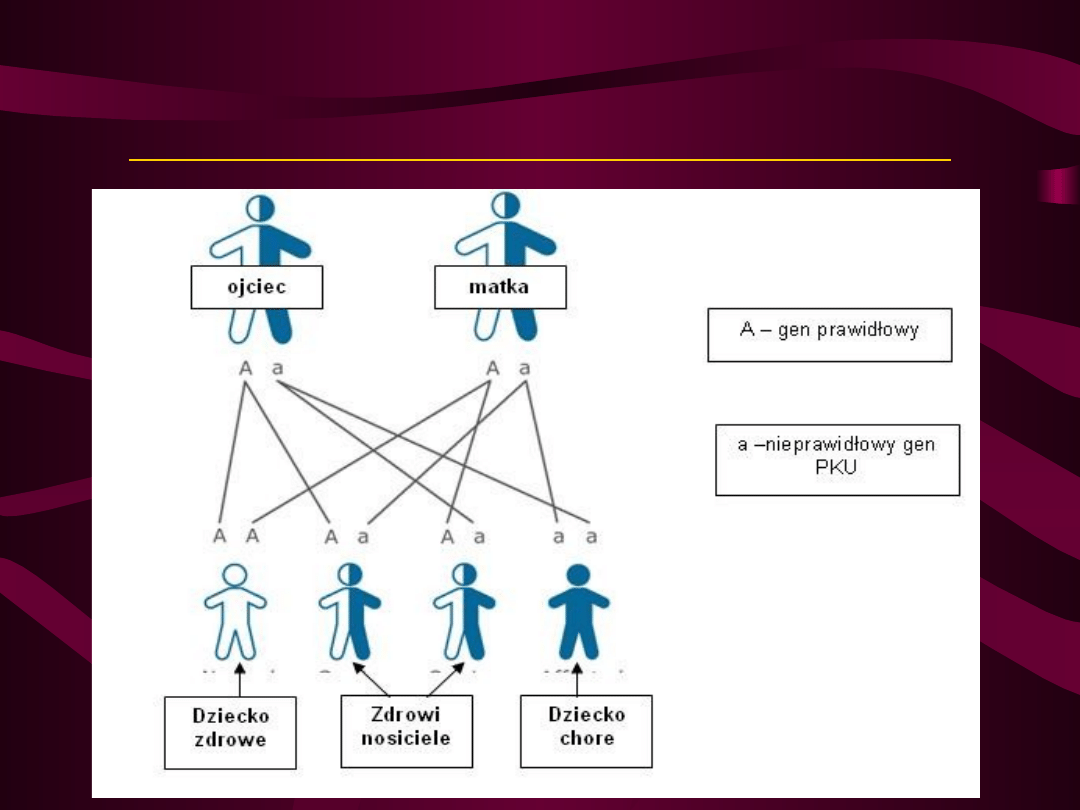
Schemat dziedziczenia fenyloketonurii

Albinizm
•
Częstość występowania 1:10 000.
•
Jest to defekt przemiany tyrozyny,
spowodowany mutacją genu strukturalnego,
kontrolującego syntezę tyrozynazy.
•
Brak enzymu jest odpowiedzialny za
zahamowanie syntezy melanin w
melanocytach naskórka, cebulek włosowych i
siatkówce.
•
Albinosi mają więc różową i cienką skórę
przez, którą prześwitują naczynia krwionośne,
włosy są białe, tęczówki niebieskie lub
różowe, z zaznaczonym czerwonym połyskiem.

• Zmniejszona jest ostrość wzroku.
• Chorujący na albinizm człowiek jest
nadwrażliwy na światło słoneczne, bowiem
melanina jest barwnikiem produkowanym w
melanocytach skóry właściwej, naskórka,
tęczówki oraz cebulek włosa i chroni przed
działaniem promieni słonecznych. Prowadzi
to do stanów zapalnych poparzeń i
zrogowacenia skóry.

• Wyróżniamy dwa rodzaje bielactwa wrodzonego:
uogólnione i częściowe.
W przypadku bielactwa uogólnionego w skórze chorego
występują prawidłowe melanocyty, zakłócony jest jednak
sam proces powstawania melaniny.
Typowe objawy to: rumienie, nadżerki, pęcherze,
nadmierne rogowacenie skóry.
Melaniny pozbawione są również włosy, dlatego też
są one białe lub biało-żółte, tęczówki oczu są różowe,
a źrenice czerwone.
Częściowe bielactwo jest również dziedziczne ale
dziedziczenie w tym przypadku jest dominujące.
Dziecko od urodzenia nie ma melanocytów lub są
one uszkodzone
Dotyczy tylko niektórych części skóry i włosów.
Najczęściej spotyka się je na linii środkowej czoła,
a bardzo rzadko zmiany widoczne są na rękach i
nogach.
Oczy są często różnokolorowe.

Alkaptonuria
(ochronoza) AKU
Częstość występowania choroby 1:200 000.
Alkaptonuria jest spowodowana mutacją
genu oksydazy kwasu homogentyzynowego
(HGD), zlokalizowanego na 3 chromosomie.
W efekcie tego dochodzi do niedoboru
enzymu oksydazy kwasu
homogentyzynowego, co zaburza rozkład,
produktu ubocznego przemiany tyrozyny
oraz fenyloalaniny kwasu
homogentyzynowego (ciemniejącego na
powietrzu) do kwasu fumaryloacetooctowego
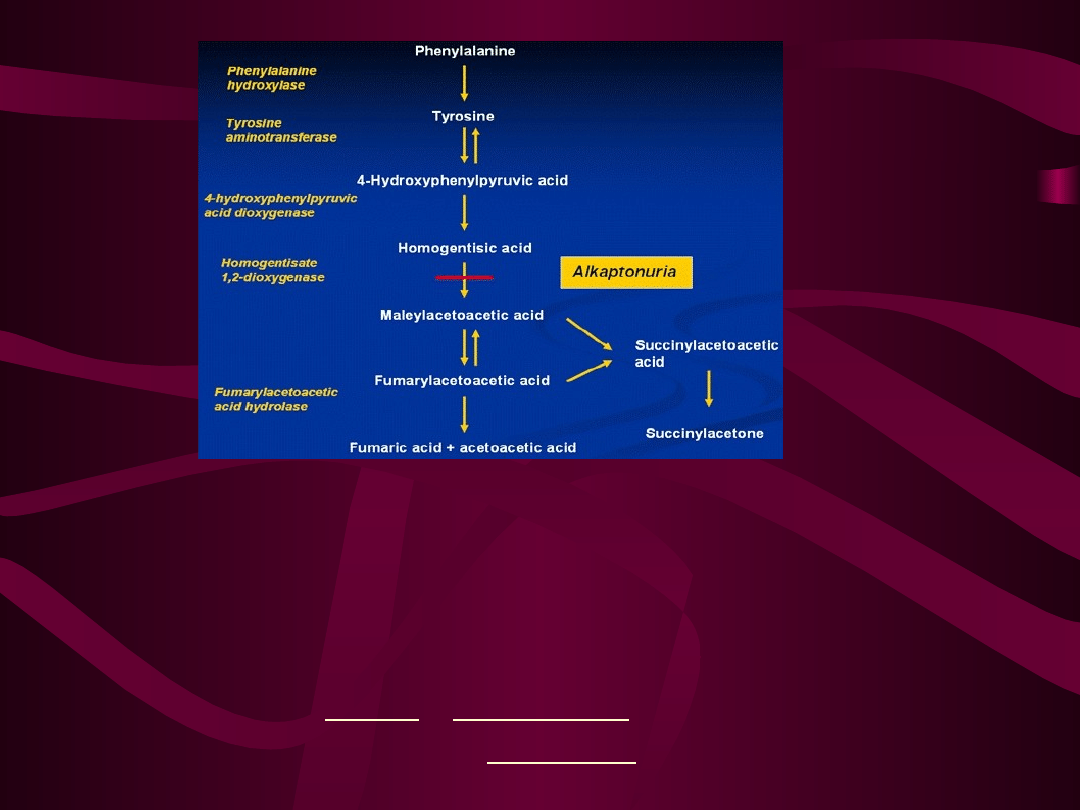
Nadmiar kwasu homogentyzynowego oraz
produktów jego dalszego metabolizmu
gromadzi się we krwi, wywierając szkodliwy
wpływ na tkankę łączną różnych narządów,
szczególnie kości i chrząstek, a wydalany jest
w dużych ilościach z moczem

Objawy :
W niemowlęctwie objawem alkaptonurii
może być ciemne zabarwienie pieluszek
przez mocz zawierający duże ilości kwasu
homogentyzynowego. Jednak często taki
objaw nie występuje, szczególnie jeśli mocz
ma kwaśny odczyn. Stąd najczęściej przez
dzieciństwo, młodość i wczesny okres
dojrzałości przebieg choroby jest
bezobjawowy. Czasem daje się zauważyć
ciemne zabarwienie pod pachami lub
woskowiny.
U dorosłych, zazwyczaj w 3 lub 4 dekadzie
życia, wcześniej u mężczyzn, na pierwszy
plan wysuwają się objawy zmian
zwyrodnieniowych stawów (artropatia
ochronozowa), głównie bóle, zniekształcenia,
ograniczenia ruchomości oraz funkcji czy
wysięki.

Dotyczą one przede wszystkim dużych
obciążonych stawów: biodrowych, kolanowych,
barkowych, a także stawów kręgosłupa. Ich
przyczyną jest wieloletnie gromadzenie się
polimerów kwasu homogentyzynowego w
kościach i chrząstkach stawowych, które
powodują ich ochrowe zabarwienie. Dość często
stopień uszkodzenia stawów wymaga
wszczepienia endoprotez.
Do częstych objawów należą także
uszkodzenia i naderwania ścięgien.
W zakresie układu krążenia dochodzi do
występowania zwapnień w tętnicach
wieńcowych oraz uszkodzeń zastawech
mitralnych i aortalnych, wymagających niekiedy
wykonania zabiegu operacyjnego.
W nerkach dochodzi do powstawania złogów
W prostacie dochodzi do zwapnień

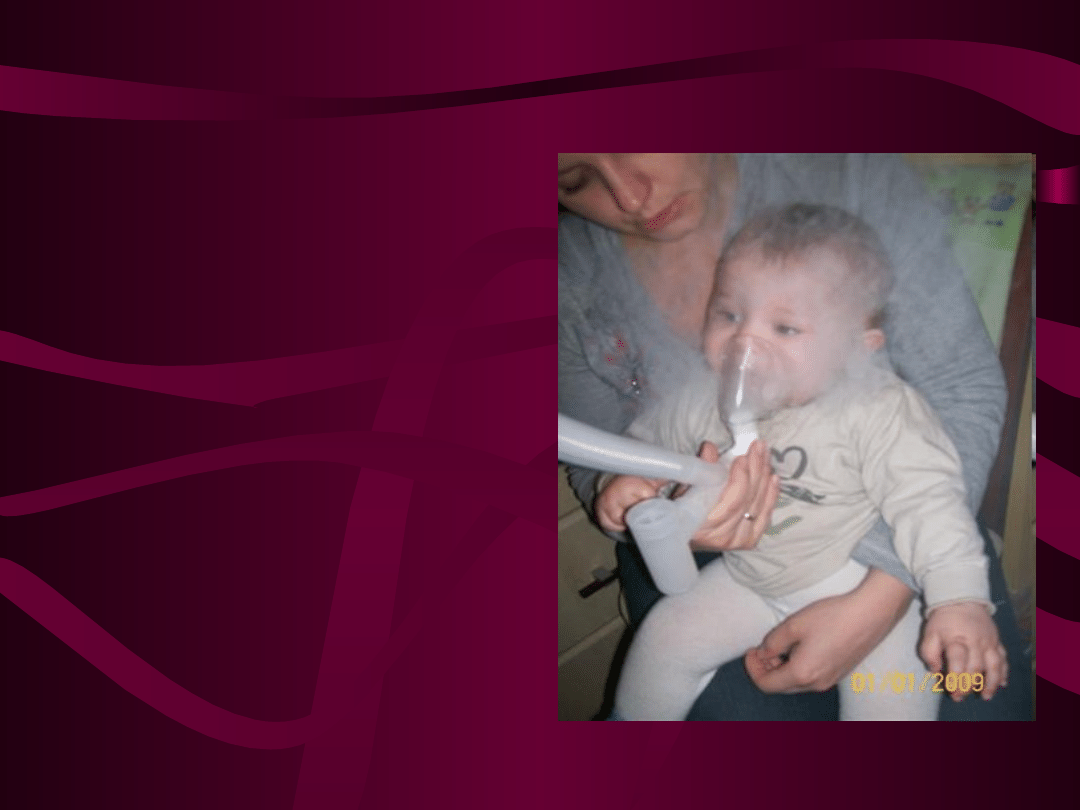
Mukowiscydoza
• Mukowiscydoza – torbielowate zwyrodnienie
trzustki należy do najczęstszych w Polsce i na
świecie wrodzonych chorób metabolicznych,
występujących u rasy białej. Częstość
występowania 1:2000 żywo urodzonych dzieci.
Częstość heterozygot (nosicieli) w populacji wynosi
5%.
• Choroba występuje u osób posiadających
nieprawidłowy gen w 7 chromosomie
odpowiedzialny za syntezę błonowego kanału
chlorkowego CFTR
(cystic fibrosis transmembrane
conductante regulator)
•
Homozygoty charakteryzują
się zmianami w wielu
narządach i licznymi
objawami klinicznymi.
Heterozygoty nie wykazują
objawów chorobowych.
•
Jest to najczęstsza letalna
choroba wieku dziecięcego.
Dochodzi do uogólnionego
uszkodzenia gruczołów
wydzielania zewnętrznego.
•
Nadmiernie gęsta i lepka
wydzielina upośledza drożność
przewodów wyprowadzających
gruczołów wydzielania
zewnętrznego w trzustce,
przewodzie pokarmowym,
płucach, wątrobie, śliniankach
i narządach płciowych.
•
Drugim czynnikiem
patogennym

• Drugim czynnikiem patogennym w
mukowiscydozie są zmienione właściwości
wydzieliny w gruczołów. Zawiera ona większe ilości
sodu, wapnia a także białka. Wykazano, że w
wydzielinie gruczołów występuje czynnik hamujący
resorpcję sodu w przewodach wyprowadzających,
a także czynnik hamujący ruch rzęsek w drogach
oddechowych.
•
Objawy ze strony układu oddechowego:
Wystepują u ponad 90% chorych
Przewlekły i napadowy kaszel,
Nawracające i przewlekłe zapalenie płuc,
Zapalenie oskrzelików,
Obturacyjne zapalenie oskrzeli,
Krwioplucie,
Zmiany w płucach widoczne w Rtg: nawracająca
niedodma, rozdęcie i rozstrzenie oskrzeli,
Polipy nosa,
Przewlekłe zapalenie zatok przynosowych.

•
Objawy ze strony przewodu pokarmowego:
Występują u około 75% chorych
Występują obfite, nieuformowane, cuchnące,
tłuszczowe stolce od wczesnego dzieciństwa,
Powiększenie objętości brzucha, niekiedy
wypadanie odbytnicy,
Niedrozność smółkowa jelit w okresie
noworodkowym, spowodowana czopem gęstej
smółki zatykającym jelito grube i jej ekwiwalenty:
zespół korka smółkowego, nawracające epizody
bólu brzucha z objawami niedrożności
przepuszczającej określane jako dystalna
niedrożność jelit,
Może wystąpić (4-5% przypadków) wtórna
marskość żółciowa wątroby z powodu
niedrożności kanalików żółciowych,
Kamica żółciowa,
Zaczopowanie przewodów ślinianek gęstą
wydzieliną śluzową,
Skret jelit w okresie płodowym,

Gęsty i lepki śluz blokuje przewody trzustkowe,
przyjmowane pokarmy nie są odpowiednio
trawione doprowadzając do objawów zespołu
złego wchłaniania,
Nawracające zapalenie trzustki.
Inne objawy:
U starszych dzieci pojawia się szybkie uczucie
zmęczenia, osłabienia po niewielkim wysiłku oraz
opóźnienie rozwoju fizycznego
mukowiscydoza może powodować niepłodność. U
kobiet ze względu na wzrost gęstości śluzu
szyjkowego, co implikuje trudności w przechodzeniu
plemników w kierunku komórki jajowej; u mężczyzn
związana z oligospermią lub azoospermią
spowodowana niedrożnością i aplazją
nasieniowodów
W wyniku zmian płucnych i zwiększonego oporu
krążenia płucnego może dojść do powstania serca
płucnego
Dziedziczenie Mukowiscydozy
Czy dzieci rodziców będących nosicielami będą
również chore?
Przypadek 1.
Obydwoje rodzice są nosicielami. Każdy
z nich posiada tylko jedną nieprawidłową
"część" genu (allel), dlatego
prawdopodobieństwo wystąpienia dwóch
nieprawidłowych "części" genu i co za
tym idzie wystąpienie mukowiscydozy u
ich potomstwa wynosi 25% (rys. 1.).
Przypadek 2.
Jeden z rodziców jest nosicielem, drugi jest
zdrowy, wówczas szanse na urodzenie
dziecka zdrowego i nosiciela są równe.
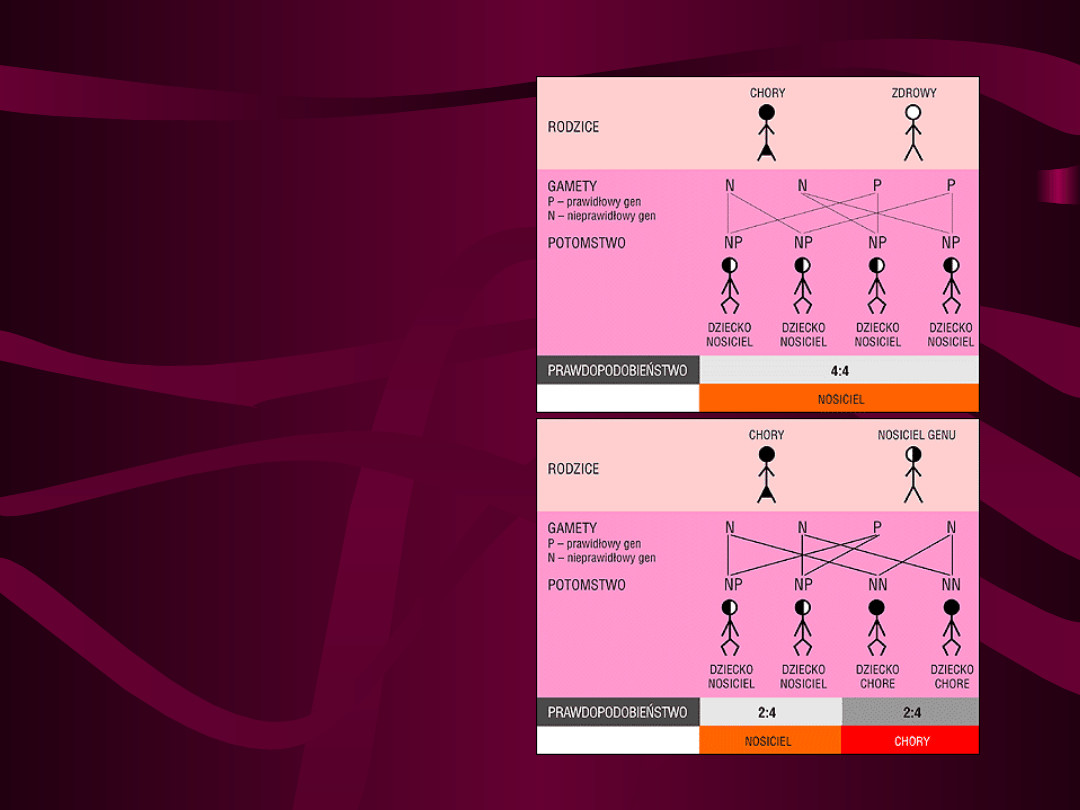
Przypadek 3.
Jeden z rodziców jest chory na
mukowiscydozę, a drugi zdrowy i nie jest
nosicielem, wówczas wszystkie dzieci będą
nosicielami, lecz nie ma obawy, aby
dziecko było chore na mukowiscydozę
(rys. 2.)
Przypadek 4.
Jeden z rodziców jest chory na
mukowiscydozę, a drugi jest nosicielem.
W tym przypadku 50% dzieci może być
chorych na mukowiscydozę, a 50% - być
nosicielami (rys. 3)

Niedokrwistośc
sierpowatokrwinkowa
(anemia sierpowata)
•
Jest to choroba spowodowana wadliwą budową
białka hemoglobiny. Zamiast normalnej
hemoglobiny powstaje tzw. hemoglobina typu S
(HbS), która zawiera w łańcuchu zamiast
aminokwasu zwanego kwasem glutaminowym,
aminokwas walinę.
•
W efekcie mutacji, krwinki przybierają kształt
sierpowaty stąd nazwa choroby. Mutacja
powoduje, że cząsteczki hemoglobiny łatwo łączą
się ze sobą, a takie skupisko hemoglobiny
powoduje odkształcenie erytrocytu, który zamiast
normalnego kształtu biszkopta przybiera kształt
sierpa

• Wadliwa budowa
krwinek prowadzi do ich
rozpadu (hemolizy)
• Na anemię sierpowatą
choruje około 5 mln
ludzi na świecie.
• Heterozygoty (nosiciele)
nie wykazują objawów
chorobowych, w ich krwi
występuje tylko 1% a u
homozygot około 50%
krwinek sierpowatych.
Heterozygoty są
bardziej odporne na
zakażenie przez
pasożyty malarii

• Objawy anemii sierpowatej:
Łatwa hemoliza erytrocytów prowadzi do
obniżonej odporności osmotycznej,
Większa lepkość krwi,
Krwinki o zmienionym kształcie łatwiej
przylegają do ściany naczyniowej, co sprzyja
tworzeniu się skrzeplin, a w rezultacie również
zatorów zatykających naczynia,
Zatory zamykające światło naczyń i
uniemożliwiające dostateczny przepływ krwi,
wiążą się z nawracającymi silnymi bólami oraz
obrzękiem dłoni i stóp,
Występuje zażółcenie skóry i błon śluzowych,
Wątroba i śledziona są powiększone, a w
pęcherzyku żółciowym obecne są kamienie,
Owrzodzenie goleni,

Niewydolność nerek, krwiomocz,
białkomocz,
Bóle głowy
Zatory w naczyniach są przyczyną zawału
serca, udaru mózgu i niewydolności wielu
narządów
Uszkodzeniu ulega wzrok może nawet
rozwinąć się jaskra
DZIĘKUJEMY
DZIĘKUJEMY
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
Wyszukiwarka
Podobne podstrony:
Dziedziczenie jednogenowe autosomalne i sprzężone z płcią
DZIEDZICZENIE JEDNOGENOWE U CZLOWIEKA, PWSZ, Genetyka
DO TEL! 7 Dziedziczenie jednogenowe (27 05 2014 r )
Dziedziczenie jednogenowe i sprzężone z płcią
DO TEL! 7. Dziedziczenie jednogenowe (27.05.2014 r.)
3= Dziedziczenie jednogenowe id Nieznany (2)
7 Dziedziczenie jednogenowe Analiza rodowodow 01
Cechy monogenne i patologiczne dziedziczące się autosomalnie
3= Dziedziczenie jednogenowe
DO TEL! 7 Dziedziczenie jednogenowe (27 05 2014 r )
więcej podobnych podstron