
Komórki B produkują immunoglobuliny
zwane przeciwciałami

Każdy rodzaj przeciwciał spełnia określone
funkcje
Immunoglobuliny biorą udział w
niszczeniu
bakterii, wirusów i grzybów

IgM
pierwsza rozpoczyna
akcję
UWAGA
Wróg przed nami
Do dzieła ! ...

Łapać go !
Lecę za
tobą !
Idę z
wami !
Już po
tobie !
Załatwion
e
Zarazek !!
Łapać !
Trzymać !
Czasem przeciwciała podejmują
wspólne akcje.

Limfocyty T są odpowiedzialne za swoistą
odporność komórkową


T zabójca zabija zakażone komórki
Koniec z tobą
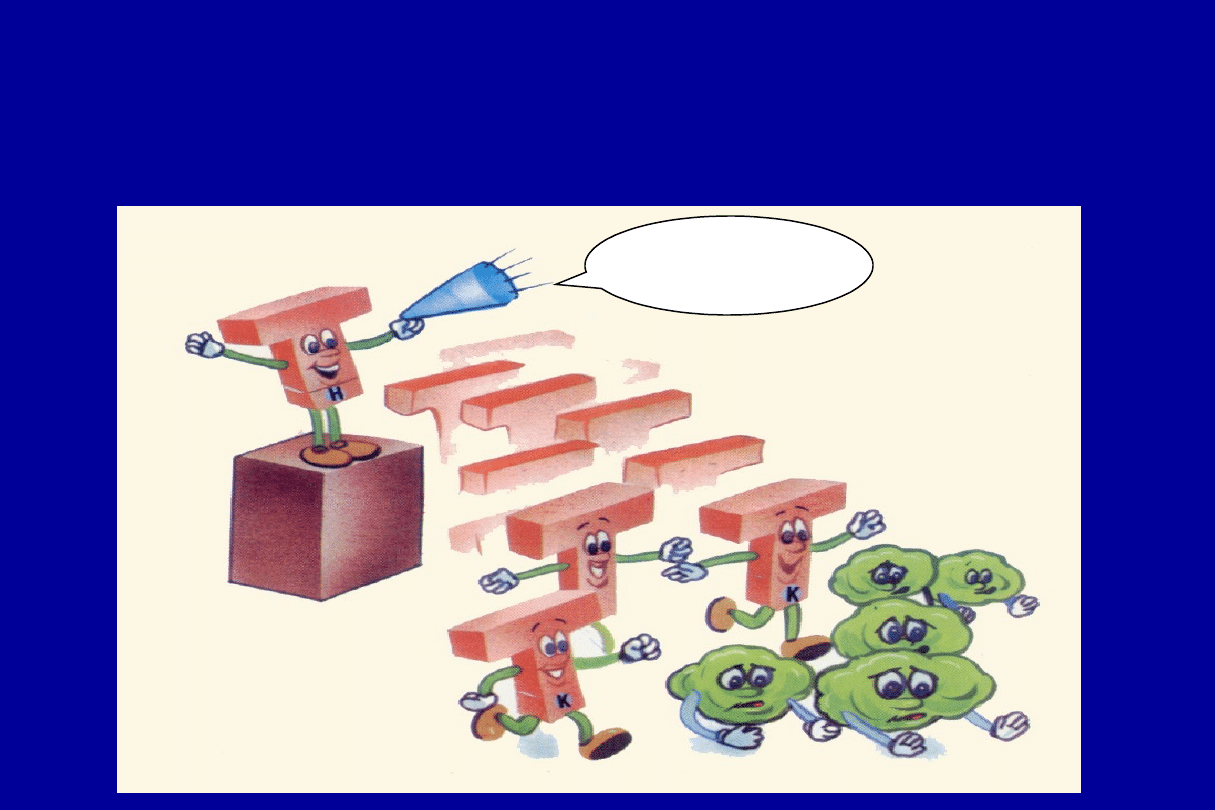
Limfocyty T-pomocnicze zwołują
komórki T-zabójcy, aby zniszczyć
zarazki ...
Wzywamy
wszystkie
komórki T-
zabójcy.
Potrzebujemy
Was !
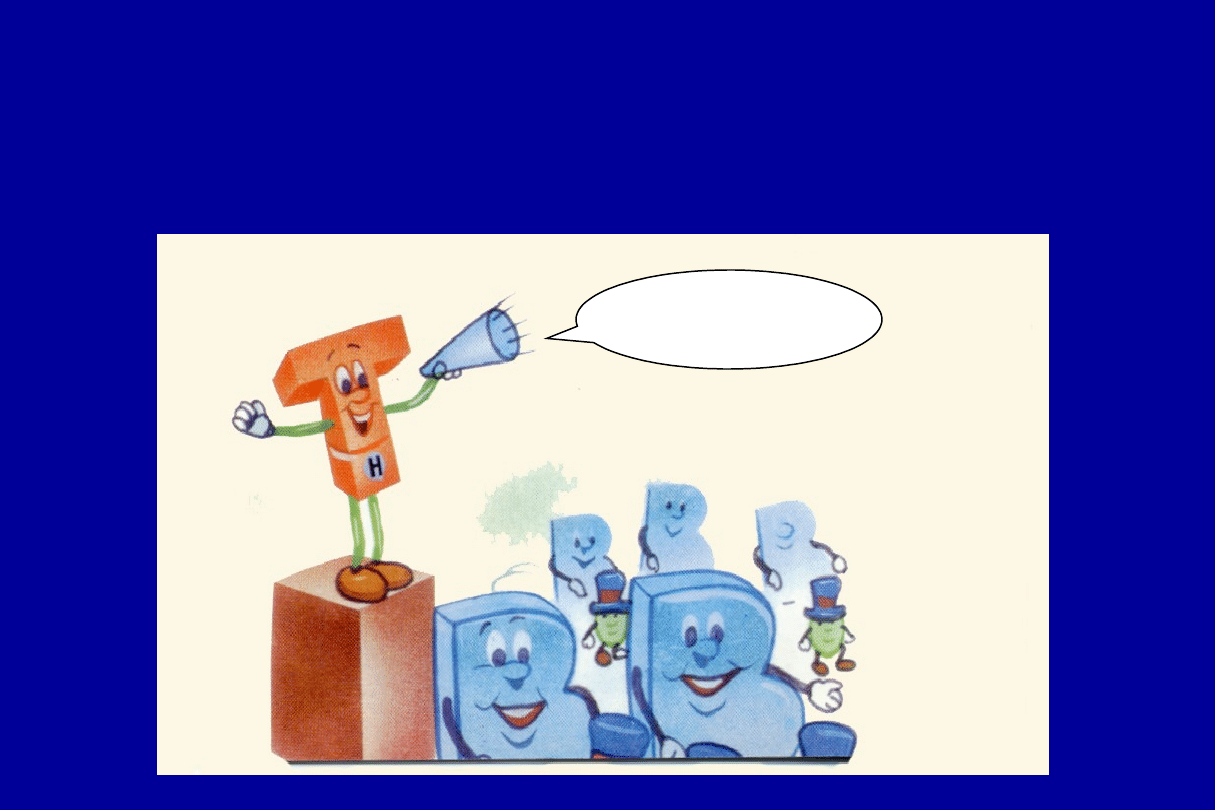
i powiadamiają komórki B kiedy
rozpocząć wytwarzanie przeciwciał
Komórki B
rozpocząć
wytwarzanie
immunoglobuliny

Limfocyty T-supresyjne
powiadamiają komórkę B o
zaprzestaniu produkcji przeciwciał
OK Kom
órki B!
Kończym
y !

Inny obrońca to fagocyt .
Fagocyty niszczą zarazki
poprzez fagocytozę
i zabijanie wewnątrzkomórkowe

Potrafią też wysyłać sygnał
wzywający inne fagocyty do
pomocy.
Wzywam
wszystkie
fagocyty
Akcja!
Ostatni z naszych obrońców to
komplement.
Składa się z 18 różnych
cząsteczek.

Układ dopełniacza współdziała z
IgG
i fagocytami aby szybciej
wyeliminować zarazki.

A ZATEM MAMY:
PRZECIWCIAŁA
czyli immunoglobuliny
produkowane przez
komórki B
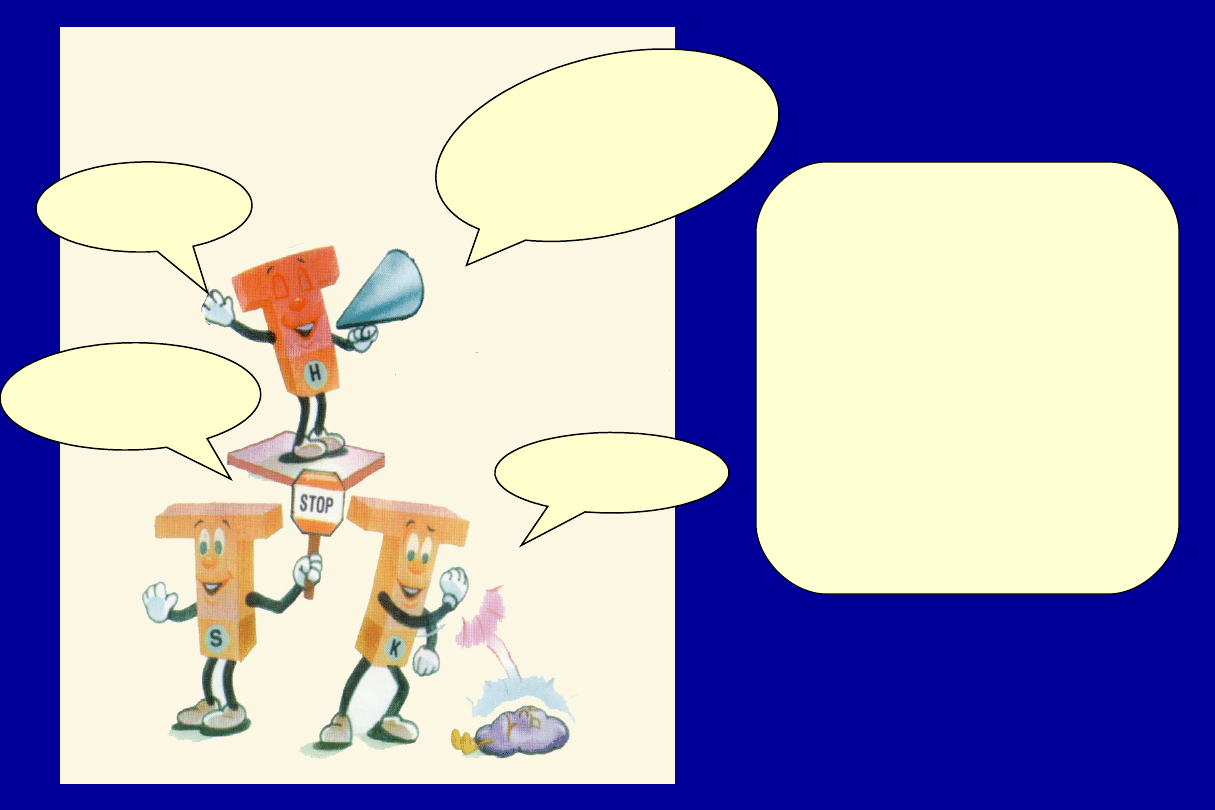
Ja m
ówię
kom
órko
m
T-zab
ójcom
by
atak
ował
y zar
azki
i kom
órko
m B
kied
y ma
ją za
czyn
ać
prod
ukcję
prze
ciwc
iał
Ja
rozpoczynam
akcję !
Ja daję
znak komórkom B
kiedy mają skończyć
Ja zabijam
zarazki !
3 rodzaje
komórek typu T

FAGOCYTY

i KOMPLEMENT

WSKAZANIA DO BADAŃ ODPORNOŚCI
(a)
obciążający wywiad rodzinny – stwierdzony wcześniej
pierwotny niedobór odporności,
wady serca i dużych naczyń oraz obniżony poziom
wapnia w okresie noworodkowym (cechy
charakterystyczne dla zespołu DiGeorge’a),
choroby o których wiadomo, że często towarzyszą
im niedobory odporności (zespół Downa, rybia
łyska, małogłowie i ptasi wygląd twarzy),

WSKAZANIA DO BADAŃ ODPORNOŚCI (b)
przewlekle zakażenia, biegunki, brak przyrostu
masy ciała i zahamowanie wzrostu u dzieci,
zakażenia drobnoustrojami oportunistycznymi lub
o słabej wirulencji,
wysypki o niejasnej etiologii
nawracające, przewlekłe, źle leczące się
zakażenia, ropnie
stany zapalne tkanki okołozębowej,
nieprawidłowe gojenie się ran
autoimmunizacyjna anemia, małopłytkowość,
neutropenia

DO PODSTAWOWYCH BADAŃ
IMMUNOFENOTYPOWYCH POZWALAJĄCYCH
OCENIĆ STAN ODPORNOŚCI CHOREGO NALEŻY:
ocena ilości limfocytów T, ich subpopulacji,
ocena ilości limfocytów B,
ocena ilości komórek NK,
badanie ekspresji antygenów MHC klasy II
ocena ekspresji CD40L na aktywowanych limfocytach
T
badanie ekspresji cząsteczek adhezyjnych CD11/CD18

KLASYFIKACJA SCID
1. T
-
B
-
(we krwi obwodowej brak limfocytów T i B)
a) niedobór dezaminazy adenozyny
b) niedobór RAG1 lub RAG2
c) dyzgenezja siateczki
2. T
-
B
+
(brak limfocytów T, obecne B) najczęstsza forma
SCID - 80%
a) sprzężony z chromosomem X
b) autosomalny recesywny
3. T
+
B
+-
(limfocyty T obecne, B obecne, często obniżone)
a) zespół Omenna
b) niedobór IL-2 R
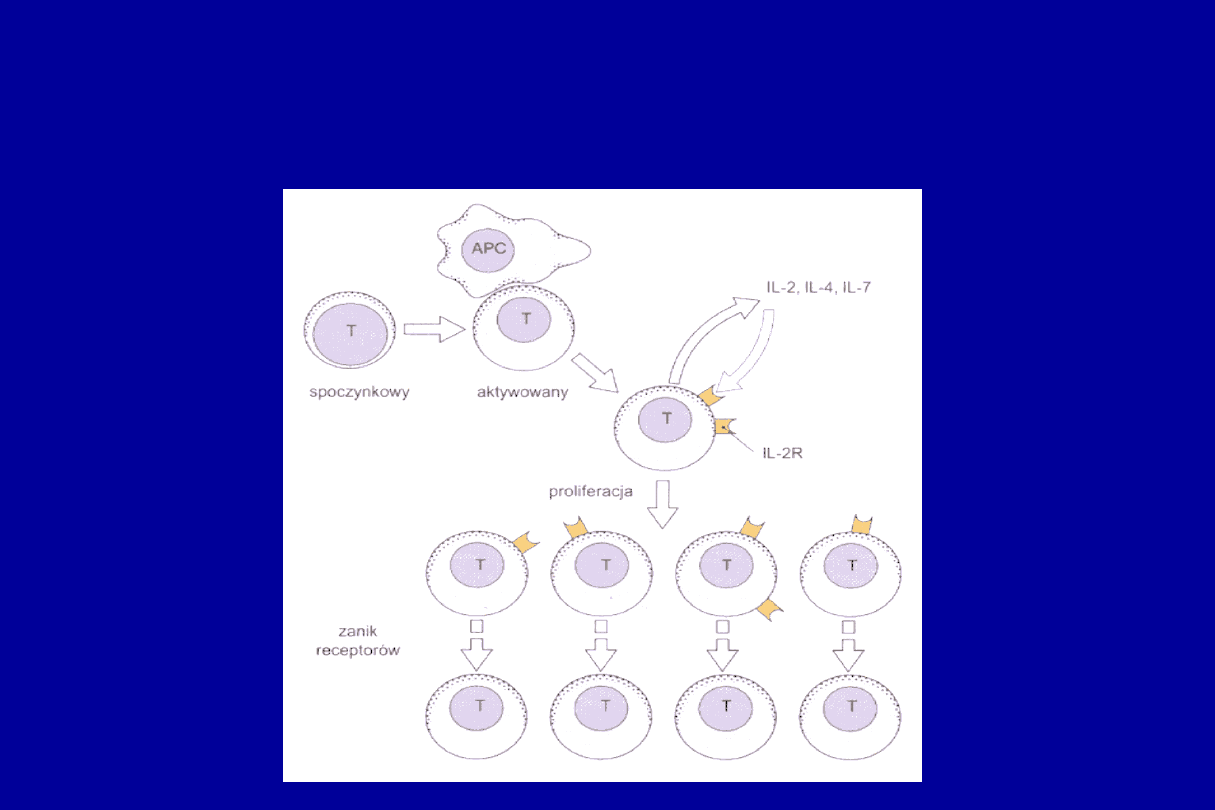
PROLIFERACJA KOMÓREK T
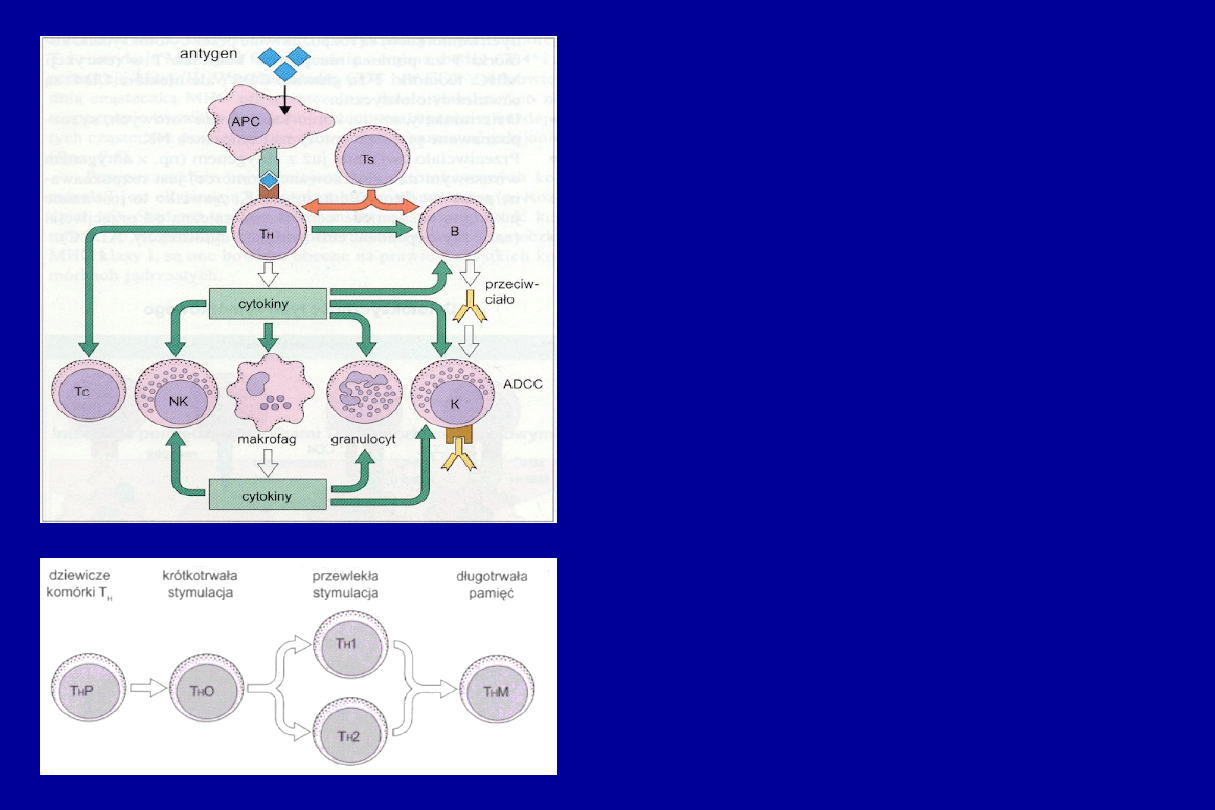
CENTRALNA ROLA
KOMÓREK T
H
W ODPORNOŚCI TYPU
KOMÓRKOWEGO
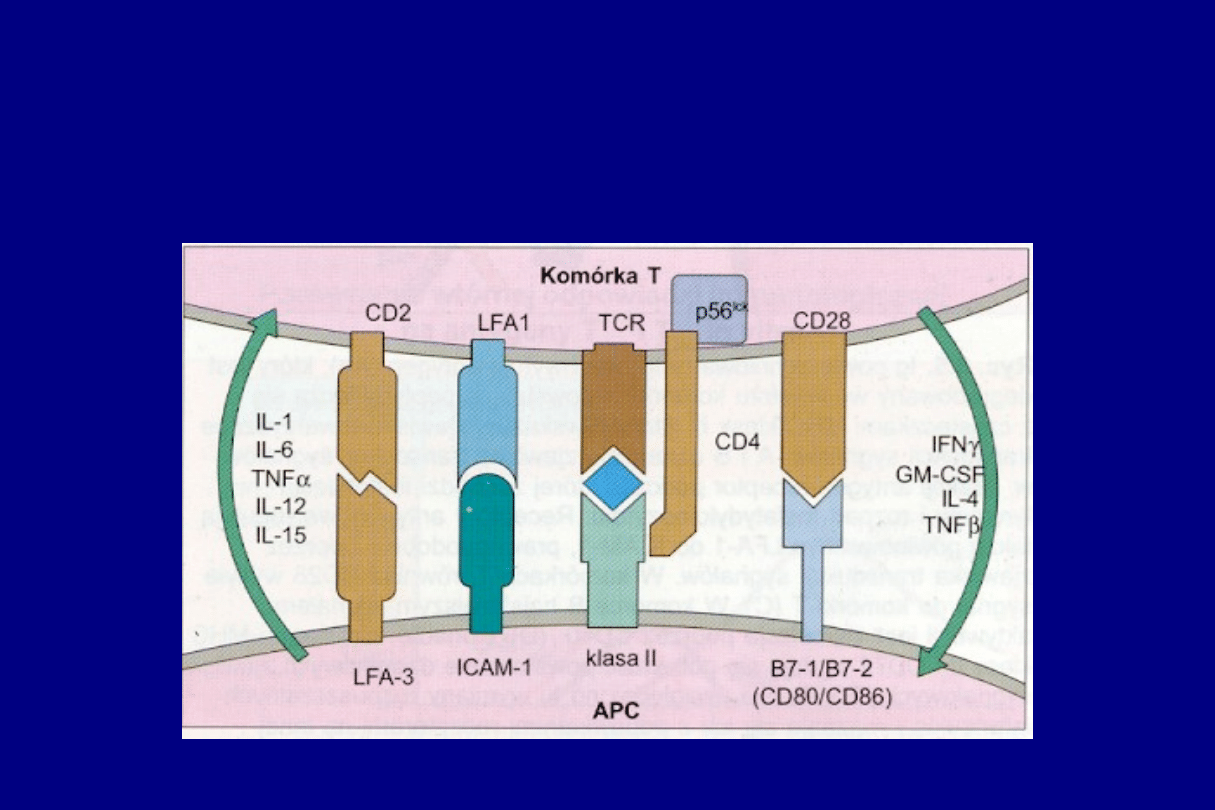
CZĄSTECZKI BIORĄCE UDZIAŁ W
PREZENTACJI ANTYGENU
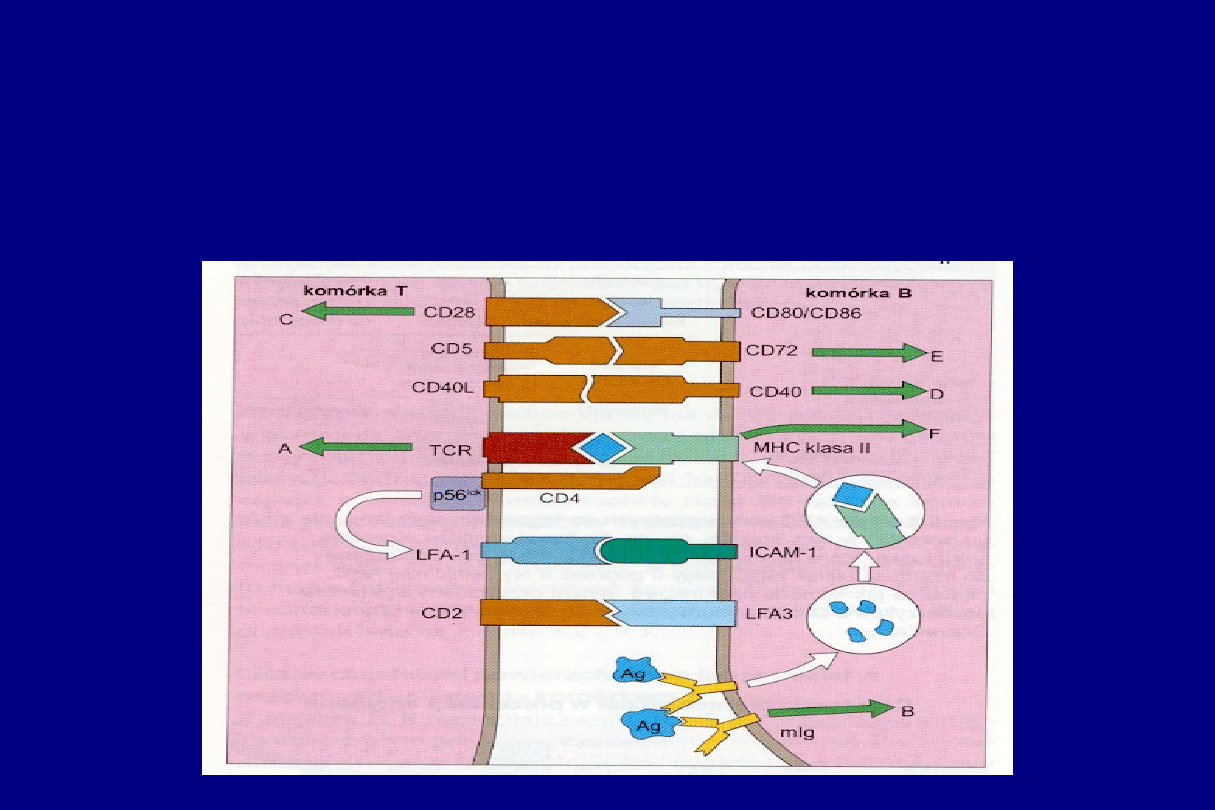
CZĄSTECZKI POWIERZCHNIOWE KOMÓREK
BIORĄCYCH UDZIAŁ W INTERAKCJACH
POMIĘDZY KOMÓRKAMI B I T
H

NIEKTÓRE CYTOKINY W REGULACJI
ODPORNOŚCI HUMORALNEJ
IL-1:
indukuje dojrzewanie i proliferację limfocytów B,
zwiększa syntezę immunoglobulin
IL-4:
reguluje zmianę izotypu i syntezę podklas
immunoglobulin
IL-6:
odpowiedzialna za końcowe różnicowanie
limfocytów B w komórki produkujące Ig
IL-10:
stymuluje proliferacje limfocytów B, indukuje
uwalnianie
IgG, IgA, IgM przez aktywne
limfocyty B
TNFa i TNFb
wzmagają proliferację i różnicowanie limfocytów
B, rola regulacyjna w syntezie immunoglobulin

TESTY OCENIAJĄCE ODPORNOŚĆ
ODPORNOŚĆ HUMORALNA
pomiar stężenia imunoglobulin - IgG, IgA, IgM w surowicy oraz
płynach ustrojowych
1.
dyfuzja radialna
2.
turbidymetria
3.
nefelometria: próg pomiaru dla
IgG – 0,07 g/l
IgA – 0,06 g/l
IgM – 0,04 g/l
badania podklas IgG
ocena syntezy swoistych przeciwciał
po czynnej immunizacji standardowymi szczepionkami
(przeciwciała przeciwtężcowe, przeciw błonicze)
ocena ilości limfocytów B
ekspresja determinant powierzchniowych CD19, CD20 i CD22

TESTY OCENIAJĄCE ODPORNOŚĆ
ODPORNOŚĆ KOMÓRKOWA
określenie liczby limfocytów T i ich subpopulacji
ekspresja determinant charakterystycznych dla
limfocytów T: CD2, CD3, CD4, CD8
ocena funkcji limfocytów in vitro (stymulacja mitogenami,
antygenami i alloantygenami)
ocena aktywacji na podstawie:
proliferacji komórek
ekspresji „antygenów aktywacji” (receptor dla IL-2
(CD25), receptor dla transferyny (CD71), HLA-DR
uwalniania mediatorów np. cytokin
ocena ekspresji antygenów zgodności tkankowej (HLA) klasa I
i II

TESTY OCENIAJĄCE ODPORNOŚĆ
GRANULOCYTY
test chemiluminescencji
ilościowy test redukcji barwnika
NBT
ocena obecności cząsteczek
adhezyjnych CD11a, CD18

CIĘŻKI ZŁOŻONY NIEDOBÓR
ODPORNOŚCI
Dziecko poniżej drugiego roku życia, u którego można wykazać w krążeniu matczyne limfocyty T; odsetek
limfocytów CD3 jest nie większy niż 20%, a liczba bezwzględna limfocytów nie przekracza 3000 l i
określono jedno z poniższych zaburzeń:
mutacja genu łańcucha gamma dla receptora cytokin (
c
),
mutacja w JAK3,
mutacja w RAG1 lub RAG2
mutacja w IL-R7,
aktywność deaminazy adenozyny mniejsza niż 2% w stosunku do kontroli lub mutacja obu alleli tego
enzymu.
ROZPOZNANIE DEFINITYWNE
Dziecko poniżej drugiego roku życia, u którego odsetek
limfocytów CD3 jest nie większy niż 20%, a liczba
bezwzgledna limfocytów nie przekracza 3000/ l; odpowiedź
proliferacyjna limfocytów wynosi mniej niż 10 % kontroli;
można wykazać matczyne limfocyty T w krążeniu.
ROZPOZNANIE PRAWDOPODOBNE

CIĘŻKI ZŁOŻONY NIEDOBÓR ODPORNOŚCI
SPRZĘŻONY Z CHROMOSOMEM X
Chłopiec, u którego można wykazać w krążeniu
matczyne limfocyty T; odsetek limfocytów CD3 jest nie
większy niż 10%, komórek NK (CD16
+
/56
+
) mniejszy od
2%, limfocytów B większy niż 75% i można stwierdzić
jedno z poniższych zaburzeń:
mutacja genu łańcucha gamma dla receptora
cytokin (g
c
),
brak mRNA dla g
c
w limfocytach techniką Northern
blot,
brak g
c
na powierzchni limfocytów lub komórkach
linii limfocytów,
kuzyni ze strony matki: wuj, siostrzeniec, bratanek
z rozpoznaniem X-SCID
ROZPOZNANIE
DEFINITYWNE

CIĘŻKI ZŁOŻONY NIEDOBÓR ODPORNOŚCI
SPRZĘŻONY Z CHROMOSOMEM X
Chłopiec, u którego odsetek limfocytów CD3 jest nie
większy niż 10%, komórek NK (CD16
+
/56
+
) mniejszy od 2%,
limfocytów B większy niż 75% i można stwierdzić wszystkie z
poniższych zaburzeń:
występowanie groźnych dla życia, zaburzających rozwój,
zakażeń poniżej 1 rż,
stężenie IgG i IgA poniżej 2 SD normy dla wieku,
przewlekła lub nawracająca biegunka, pleśniawki,
zakażenia dróg moczowych
ROZPOZNANIE PRAWDOPODOBNE
Chłopiec, u którego odsetek limfocytów B we krwi obwodowej
jest większy niż 40% i można stwierdzić jedno z poniższych
zaburzeń:
obecność matczynych limfocytów T w krążeniu,
kuzyni ze strony matki: wuj, siostrzeniec, bratanek z
rozpoznaniem XSCID
ROZPOZNANIE MOŻLIWE

ZESPÓŁ Di GEORGE’A
Dziecko ze zmniejszoną liczbą limfocytów CD3 (mniej niż
500/ml) i dwoma z trzech poniższych
charakterystycznych objawów:
wrodzona wada serca (przetrwały przewód tętniczy,
tetralogia Fallota, przerwany łuk aorty, nietypowe
odejście prawej tętnicy podobojczykowej),
hipokalcemia trwająca dłużej niż 3 tygodnie,
wymagająca leczenia,
delecja w obrębie chromosomu 22q11.2.
ROZPOZNANIE
DEFINITYWNE

ZESPÓŁ Di GEORGE’A
Dziecko ze zmniejszona liczba limfocytów CD3 (mniej niż
1500/ml) i delecją w obrębie chromosomu 22q11.2
ROZPOZNANIE PRAWDOPODOBNE
Dziecko ze zmniejszoną liczbą limfocytów CD3 (mniej niż
1500/ml) i przynajmniej jednym z poniższych zaburzeń:
wada serca,
hipokalcemia trwajaca dłużej niż 3 tygodnie,
wymagająca leczenia,
cechy dysmorficzne twarzy lub nieprawidłowości
podniebienia
ROZPOZNANIE MOŻLIWE

ATAKSJA-TELEANGIEKTAZJA
Dziecko, u którego stwierdza się postepującą ataksję
móżdżkową, znaczną łamliwość chromosomów pod
wpływem promieniowania w warunkach hodowli
komórek, mutacje obu alleli genu ATM
ROZPOZNANIE DEFINITYWNE
Pacjent płci męskiej lub żeńskiej, u którego stwierdza się
postępującą ataksję móżdżkową i trzy z czterech
poniższych zaburzeń :
teleangiektazje gałki ocznej lub twarzy,
stężenie IgA w surowicy poniżej 2 SD normy dla
wieku,
stężenie a-fetoproteiny powyżej 2 SD normy dla
wieku,
znaczną łamliwość chromosomów pod wpływem
promieniowania w warunkach hodowli komórek
ROZPOZNANIE PRAWDOPODOBNE

ATAKSJA-TELEANGIEKTAZJA
Pacjent płci męskiej lub żeńskiej, u którego
stwierdza się postępującą ataksję móżdżkową i
przynajmniej jedno z czterech poniższych zaburzeń :
teleangiektazje gałki ocznej lub twarzy,
stężenie IgA w surowicy poniżej 2 SD normy dla
wieku,
stężenie a-fetoproteiny powyżej 2 SD normy dla
wieku,
znaczną łamliwość chromosomów pod wpływem
promieniowania w warunkach hodowli komórek
ROZPOZNANIE MOŻLIWE

ZESPÓŁ WISKOTTA-ALDRICHA
Chłopiec z wrodzoną małopłytkowością (mniej niż
70000 płytek/ml), małymi płytkami i przynajmniej
jednym z poniższych zaburzeń:
mutacja w obrębie genu WASP,
brak mRNA dla WASP w limfocytach techniką
Northern blot,
brak białka WASP w limfocytach,
kuzyni ze strony matki: wuj, siostrzeniec,
bratanek z małymi płytkami i
małopłytkowością
ROZPOZNANIE DEFINITYWNE

ZESPÓŁ WISKOTTA-ALDRICHA
Chłopiec z wrodzoną małopłytkowością (mniej niż 70
000 płytek/ml), małymi płytkami i przynajmniej
jednym z poniższych zaburzeń:
wyprysk,
brak/słaba synteza przeciwciał w odpowiedzi
na antygeny polisacharydowe,
nawracające infekcje bakteryjne lub wirusowe,
choroba autoimmunizacyjna,
chłoniak, białaczka, guz mózgu.
ROZPOZNANIE PRAWDOPODOBNE

ZESPÓŁ WISKOTTA-ALDRICHA
Chłopiec z wrodzoną małopłytkowością (mniej niż 70
000 płytek/ml), małymi płytkami lub chłopiec, u
którego wykonano splenektomię z powodu
małopłytkowości, który wykazuje przynajmniej
jedno z poniższych zaburzeń:
wyprysk,
brak/słaba synteza przeciwciał w odpowiedzi
na antygeny polisacharydowe,
nawracające infekcje bakteryjne lub wirusowe,
choroba autoimmunizacyjna,
chłoniak, białaczka, guz mózgu.
ROZPOZNANIE MOŻLIWE

AGAMMAGLOBULINEMIA SPRZĘŻONA Z
CHROMOSOMEM X
Chłopiec z < 2% limfocytów B (CD19) i
stwierdzonym przynajmniej jednym z poniższych
zaburzeń:
mutacja w obrębie genu Btk,
brak Btk mRNA w badaniu metodą Northern
blot neutrofili lub monocytów,
brak białka Btk w monocytach lub płytkach
krwi,
kuzyni ze strony matki: wuj, siostrzeniec,
bratanek z < 2% limfocytów B
ROZPOZNANIE DEFINITYWNE

AGAMMAGLOBULINEMIA SPRZĘŻONA Z
CHROMOSOMEM X
Chłopiec z < 2% limfocytów B (CD19), u którego
występują wszystkie poniższe zaburzenia:
początek nawracających zakażeń bakteryjnych w
pierwszych pięciu latach życia,
stężenie IgG, IgA, IgM w surowicy poniżej 2 SD
normy dla wieku,
brak izohemaglutynin i (lub) słaba odpowiedź na
szczepionki,
wykluczenie innej przyczyny
hipogammaglobulinemii.
ROZPOZNANIE PRAWDOPODOBNE

AGAMMAGLOBULINEMIA SPRZĘŻONA Z
CHROMOSOMEM X
Chłopiec z < 2% limfocytów B (CD19), u którego
zostały wykluczone inne przyczyny
hipogammaglobulinemii (j.w.) i stwierdza się
przynajmniej jedno z poniższych zaburzeń:
początek nawracających zakażeń bakteryjnych
w ciągu pierwszych pięciu lat życia,
stężenie IgG, IgA, IgM w surowicy poniżej 2 SD
normy dla wieku,
brak izohemaglutynin.
ROZPOZNANIE MOŻLIWE

ZESPÓŁ HIPER-IgM SPRZĘŻONY
Z CHROMOSOMEM X
Chłopiec ze stężeniem IgG w surowicy poniżej 2
SD normy dla wieku i spełniający jedno z
poniższych kryteriów zaburzeń:
mutacja genu CD40L,
kuzyn ze strony matki, wuj, siostrzeniec
z rozpoznaniem XHIM
ROZPOZNANIE DEFINITYWNE

ZESPÓŁ HIPER-IgM SPRZĘŻONY Z
CHROMOSOMEM X
Chłopiec ze stężeniem IgG w surowicy poniżej 2 SD
normy dla wieku i spełniający wszystkie z poniższych
kryteriów zaburzeń:
prawidłowa liczba limfocytów T i ich prawidłowa
odpowiedź proliferacyjna na mitogeny,
prawidłowa lub zwiększona liczba limfocytów B przy
braku swoistych przeciwciał klasy IgG
jedno lub więcej z następujących typów zakażeń lub
zaburzeń: naracające zakażenia bakteryjne w ciagu
pierwszych pięciu lat życia, zakażenie Pneumocitus
carinii w pierwszym roku życia, neutropenia, biegunka
powiązana z Cryptosporidium, stwardniające
zapalenie dróg żółciowych, niedokrwistość
aplastyczna wyindukowana przez Parvovirus,
brak CD40L na pobudzonych limfocytach CD4
ROZPOZNANIE PRAWDOPODOBNE

ZESPÓŁ HIPER-IgM SPRZĘŻONY
Z CHROMOSOMEM X
Chłopiec ze stężeniem IgG w surowicy poniżej 2 SD
normy dla wieku, prawidłowa liczba limfocytów T i
B oraz jednym lub więcej z poniższych zaburzeń:
stężenie IgM w surowicy powyżej 2 SD normy dla
wieku,
zakażenie Pneumocystis carinii w pierwszym roku
życia,
niedokrwistość aplastyczna wyindukowana przez
Parvovirus
biegunka powiązana z Cryptosporidium,
choroba wątroby o ciężkim przebiegu
(stwardniające zapalenie dróg żółciowych).
ROZPOZNANIE MOŻLIWE

SELEKTYWNY NIEDOBÓR IgA
Dzieci powyżej czwartego roku życia, u których stężenie IgA w
surowicy jest mniejsze niż 7 mg/dl (0.07 g/l), przy prawidłowych
stężeniach IgG i IgM, prawidłowej odpowiedzi poszczepiennej w
klasie IgG i wykluczeniu innych przyczyn hipogammaglobulinemii.
ROZPOZNANIE DEFINITYWNE
Dzieci powyżej czwartego roku życia, u których stężenie
IgA w surowicy jest poniżej 2 SD normy wieku, przy
prawidłowych stężeniach IgG i IgM w surowicy,
prawidłowej odpowiedzi poszczepiennej w klasie IgG i
wykluczeniu innych przyczyn hipogammaglobulinemii.
ROZPOZNANIE PRAWDOPODOBNE

POSPOLITY ZMIENNY NIEDOBÓR
ODPORNOŚCI
Dziecko, u którego stężenie dwóch z trzech głównych klas Ig w
surowicy jest poniżej 2 SD normy dla wieku i spełnia wszystkie z
poniższych kryteriów:
początek choroby po 2 roku życia,
brak izohemoglutynin i (lub) słaba odpowiedź na
szczepionki,
wykluczenie innych przyczyn hipogammaglobulinemii
ROZPOZNANIE PRAWDOPODOBNE
Dziecko, u którego stwierdza się znaczne zmniejszenie
(poniżej 2 SD normy dla wieku) stężeń surowiczych jednego z
trzech głównych izotypów immunoglobulin (IgG, IgA, IgM) i
które spełnia wszystkie poniższe kryteria:
początek choroby po 2 roku życia,
brak izohemaglutynin i (lub) słaba odpowiedź na
szczepionki,
wykluczenie innych przyczyn hipogammaglobulinemii.
ROZPOZNANIE MOŻLIWE

PRZEWLEKŁA CHOROBA ZIARNINIAKOWA
Dziecko z nieprawidłowym NBT lub „wybuchem
oddechowym” aktywowanych neutrofili (mniej niż
5% kontroli), u którego wykazano jedno z poniższych
kryteriów:
mutacja w gp91, p22, p47 lub p67
phox
,
brak mRNA dla jednego z powyższych wykazany
w analizie Northen blot,
kuzyni ze strony matki: wuj, siostrzeniec,
bratanek z nieprawidłowym NBT lub „wybuchem
oddechowym” neutrofili.
ROZPOZNANIE DEFINITYWNE

PRZEWLEKŁA CHOROBA ZIARNINIAKOWA
Dziecko z nieprawidłowym NBT lub „wybuchem
oddechowym” aktywowanych neutrofili (mniej niż 5%
kontroli), u którego stwirdzono jedno z poniższych
kryteriów:
ropnie wątroby, okołoodbytnicze, płuc, węzłów
chłonnych, osteomyelitis o etiologii Stapylococcus
aureus, Serratia marcescens, Candida lub
Aspergillus,
ziarniniaki w układzie oddechowym, pokarmowym
lub moczowym,
brak przyrostu masy ciała i hepatosplenomegalia
lub limfadenopatia,
ROZPOZNANIE PRAWDOPODOBNE

NIEDOBÓR ADHEZJI LEUKOCYTÓW
Dziecko ze zmniejszoną ekspresją CD18 na krwinkach
białych (mniej niż 5% kontroli) i wykazujące jedno
z poniższych zaburzeń:
mutacja genu dla integryn b2,
brak mRNA dla integryn b2 w krwinkach białych.
ROZPOZNANIE DEFINITYWNE
Dziecko ze zmniejszoną ekspresją CD18 na krwinkach
białych (mniej niż 5% kontroli) i wykazujące wszystkie
z poniższych zaburzeń:
nawracające lub przewlekające sie zakażenia
bakteryjne lub grzybicze,
leukocytoza większa niż 25 000 ml,
opóźnione oddzielanie się pępowiny i/lub
zaburzenia gojenia sie ran.
ROZPOZNANIE PRAWDOPODOBNE

NIEDOBÓR ADHEZJI LEUKOCYTÓW
Niemowlę z leukocytozą powyżej 25 000/ml
wykazujące jedno z poniższych zaburzeń:
nawracajace zakażenia bakteryjne,
ciężkie zakażenia narządowe,
brak treści ropnej w miejscu zakażenia
.
ROZPOZNANIE MOŻLIWE
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
Wyszukiwarka
Podobne podstrony:
ZDROWIE PUBLICZNE -seminarki, Lekarski, Zdrowie publiczne
3.10..15- seminaria lekarski 2015 n 2016 (6) (1), V rok, Psychiatria, 2015-16
Biologia seminarium 7, Lekarski I rok ŚUM, biologia
Protezy, IV rok Lekarski CM UMK, Rehabilitacja, Seminaria
program nauczania, program seminariów, PROGRAM SEMINARIÓW DLA WYDZIAŁU LEKARSKIEGO
program nauczania, program seminariów, PROGRAM SEMINARIÓW DLA WYDZIAŁU LEKARSKIEGO
Psychologia Lekarska seminaria-zagadnienia do kolokwiumgr5, 1.Lekarski, I rok, Psychologia, Wykłady
Seminaria IV ROK 08-09 WYDZIAL LEKARSKI, pediatria
etyka seminaria, Medycyna, Etyka lekarska ŚUM Katowice
Przelomy metaboliczne- do druku, IV rok Lekarski CM UMK, Endokrynologia, Seminaria, Seminarium 3
Udar mózgu, IV rok Lekarski CM UMK, Rehabilitacja, Seminaria
Skale pomiaru depresji i l¦Öku, IV rok Lekarski CM UMK, Psychiatria, Seminaria, Prezentacje
GENETYKA seminaria rok VI, VI rok Lekarski CM UMK, Genetyka CMUMK 2015 VI rok, Genetyka, GENETYKA VI
Porażenia, IV rok Lekarski CM UMK, Rehabilitacja, Seminaria
Seminaria 2011, IV rok Lekarski CM UMK, Rehabilitacja, Seminaria
więcej podobnych podstron