
Choroby krwi i układu
krwiotwórczego

Wstęp
Choroby krwi i układu krwiotwórczego
są mniej znaną, ale bardzo ważną
częścią nauk medycznych.
Stykamy się z nimi na co dzień,
począwszy od stosunkowo "łagodnej"
niedokrwistości z niedoboru żelaza do
bardzo złośliwych i niebezpiecznych
białaczek.

Niedokrwistość (anemia)
Właśnie niedokrwistości są najczęstszą z chorób
hematologicznych. Nazywamy tak zmniejszenie stężenia
hemoglobiny (czerwonego barwnika krwi), hematokrytu
(stosunku objętości krwinek czerwonych do objętości
pełnej krwi) lub liczby erytrocytów poniżej wartości
prawidłowych. Biorąc pod uwagę najczęściej używany
parametr, czyli stężenie hemoglobiny, niedokrwistość
(anemię) rozpoznaje się przy wartości tego stężenia
poniżej 13,5 g/dl (gram na decylitr) u mężczyzn i poniżej
12,0 g/dl u kobiet.

Podział niedokrwistości jest dosyć złożony i
przebiega według różnych kryteriów.
niedokrwistości można dzielić w zależności od etapu, w
którym zaczyna brakować krwinek.
Anemia może powstać w wyniku zmniejszenia
wytwarzania (erytrocytów lub hemoglobiny), bądź w
wyniku zwiększenia rozpadu krwinek czerwonych, bądź z
powodu ich utraty.
Do pierwszej grupy należą między innymi niedokrwistości
niedoborowe (niedobór żelaza czy witaminy B12), do
drugiej niedokrwistości hemolityczne, a do trzeciej
niedokrwistości pokrwotoczne.

Inny popularny podział wykorzystuje średni ciężar
hemoglobiny w krwince (MCH).
Im jest on większy, tym krwinki są "bardziej czerwone".
Rozróżniamy niedokrwistości:
niedobarwliwe, hipochromiczne (niskie MCH, np.
niedokrwistości z niedoboru żelaza),
niedokrwistości normochromiczne (prawidłowe MCH, np.
niedokrwistości hemolityczne)
niedokrwistości nadbarwliwe, hiperchromiczne (wysokie
MCH, np. niedokrwistości z niedoboru witaminy B12 i
kwasu foliowego).

Anemie możemy też podzielić w
zależności od wielkości krwinek
na;
normoblastyczne (krwinki prawidłowe),
megaloblastyczne i mikrocytarne (krwinki małe).
W podziale tym wykorzystuje się inny spotykany
na wynikach badania krwi parametr:
MCV, czyli średnią objętość krwinki.

Anemia z niedoboru żelaza
Najważniejszą i najczęściej występującą
grupą niedokrwistości są anemie z
niedoboru żelaza.
Stanowią prawie 80% wszystkich
niedokrwistości i dotyczą głównie kobiet.

Przyczyny:
najczęściej utrata krwi (i związanego z nim
żelaza) w wyniku przewlekłych krwawień z
dróg rodnych (krwawienia miesiączkowe),
z przewodu pokarmowego (spowodowane
głównie chorobą wrzodową), z dróg
moczowych i innych narządów.

Przyczyny:
Rzadziej zdarza się, że nie dostarczamy
organizmowi żelaza w odpowiedniej ilości (np. w
przypadku ścisłej diety wegetariańskiej) albo że
zapotrzebowanie na ten makroelement wzrasta
(w ciąży, podczas karmienia).
Niedobór żelaza (związanego z utraconymi
erytrocytami) jest przyczyną niedokrwistości
niedoborowej, która jest jednocześnie
mikrocytarna (krwinki są małe) i niedobarwliwa.

Objawy niedokrwistości możemy
uszeregować w dwóch grupach.
W I objawy wspólne dla wielu anemii, takie jak
bladość skóry i błon śluzowych, uczucie
osłabienia, duszność po wysiłku, bóle głowy czy
szmer skurczowy wysłuchiwany przez lekarza nad
sercem.
II grupę stanowią symptomy charakterystyczne
dla poszczególnych niedokrwistości.

W przypadku anemii z niedoboru
żelaza
jest to na przykład łamliwość włosów i
paznokci, zmiany na błonie śluzowej
języka, gardła i przełyku będące
przyczyną bólu i uczucia pieczenia przy
przełykaniu oraz zajady w kącikach ust.

Badanie laboratoryjne krwi
ujawnia zmniejszenie stężenia hemoglobiny (jest to
wspólne dla wszystkich niedokrwistości), liczby
erytrocytów i hematokrytu. Krwinki są mniejsze niż
zwykle i niedobrawione. Stężenie żelaza w surowicy jest
zmniejszone.
Te objawy i badania wystarczają do postawienia
rozpoznania niedokrwistości z niedoboru żelaza. Ważne
jest także poszukiwanie przyczyn tego stanu: u kobiet
trzeba wykonać badanie ginekologiczne, u wszystkich
zbadać kał na tzw. krew utajoną (znacznik krwawienia z
przewodu pokarmowego).

Jeśli znane przyczyny, to należy
je zwalczać.
niedokrwistość leczy się podając preparaty żelaza, najlepiej
doustnie, po jedzeniu. Takie uzupełnianie zapasów żelaza
należy stosować co najmniej 3 miesiące.
Wskaźnikiem prawidłowo prowadzonej terapii jest wzrost
stężenia hemoglobiny i zawartości "młodych" krwinek
czerwonych - retikulocytów.
Jeśli stwierdza się upośledzenie wchłaniania lub choroby
zapalne żołądka i jelit, to żelazo należy podawać w postaci
zastrzyków dożylnych (zastrzyki domięśniowe są bardzo
bolesne). Niestety, należy się wówczas liczyć z odczynami
alergicznymi i innymi oznakami złej tolerancji tego leku.

Niedokrwistości megaloblastyczne
Inną dużą i rozpowszechnioną grupą anemii są
niedokrwistości megaloblastyczne, w których
krwinki czerwone są większe od prawidłowych.
Najczęstszą przyczyną tych niedokrwistości jest
niedobór witaminy B12 (zdecydowana
większość przypadków) i kwasu foliowego
(rzadziej).

Witamina B12 i kwas foliowy
odgrywają ważną rolę w tworzeniu DNA, substancji, w
której zapisana jest informacja o naszych genach.
Jesteśmy uzależnieni od dowozu tych substancji wraz z
produktami żywieniowymi.
"Magazyn" witaminy B12 w wątrobie zapewnia jej
dostarczanie przez 3 lata, bez dowozu z zewnątrz; w
przypadku kwasu foliowego ten okres wynosi 3 miesiące.
Do wchłonięcia witaminy B12 wymagany jest także tzw.
czynnik wewnętrzny produkowany w błonie śluzowej
żołądka.

Właśnie niedobór tego
czynnika
jest jedną z najczęstszych przyczyn niedokrwistości
niedobarwliwych. Dzieje się tak przy usunięciu (resekcji)
części lub całości żołądka podczas operacji z powodu choroby
wrzodowej bądź raka żołądka albo w tzw. niedokrwistości
złośliwej, kiedy wytwarzane przez organizm chorego
przeciwciała niszczą komórki produkujące czynnik
wewnętrzny.
Inną przyczyną anemii z niedoboru witaminy B12 są
schorzenia jelit z upośledzeniem wchłaniania czy
niewystarczający dowóz witaminy w diecie ściśle jarskiej.

Niedobory dietetyczne
w przewlekłym alkoholizmie są za to częstą
przyczyną niedokrwistości spowodowanej
brakiem kwasu foliowego. Innymi przyczynami
są także: zwiększone zapotrzebowanie na tę
substancję w ciąży, choroby z upośledzonym
wchłanianiem i terapia niektórymi lekami, np.
pochodnymi hydantoiny czy metotreksatem.

W niedokrwistości z niedoboru
witaminy B12
obok objawów typowych dla anemii w ogóle (bladość - w tym
przypadku z odcieniem słomkowym, osłabienie, duszność)
spotykamy często ze zmianami w układzie pokarmowym
(zanikowe zapalenie błony śluzowej żołądka w niedokrwistości
złośliwej, piekący i czerwony język) oraz z objawami
neurologicznymi (niepewny chód, objawy niedowładu,
mrowienie dłoni i stóp i uczucie stąpania po filcu).
W przypadku niedoboru kwasu foliowego brak jest objawów
neurologicznych.
We krwi obserwujemy niedokrwistość (zmniejszone stężenie
hemoglobiny) z dużymi i nadbarwliwymi krwinkami
czerwonymi, często zmniejszona jest liczba krwinek białych i
płytek krwi.

W rozpoznaniu
konieczne jest też badanie szpiku, w którym widać próby
organizmu zrekompensowania występującej we krwi
niedokrwistości. Niestety są to próby nieefektywne.
W surowicy można również zmierzyć bezpośrednio stężenie
witaminy B12 i kwasu foliowego (w tym przypadku oczywiście
zmniejszone). Dla potwierdzenia ewentualnych zaburzeń
wchłaniania witaminy B12 przeprowadza się także tzw. test
Schillinga, a w przypadku podejrzenia niedokrwistości
złośliwej oznacza się wspomniane już przeciwciała przeciwko
komórkom błony śluzowej żołądka.

W terapii tych
niedokrwistości
ważne jest zwalczanie ich przyczyn.
Oprócz tego musimy uzupełnić niedobory
witaminy B12 i kwasu foliowego. Tą pierwszą
podajemy w formie zastrzyków domięśniowych.
W przypadku niedokrwistości złośliwej takie
leczenie prowadzi się do końca życia.
W niedoborze kwasu foliowego uzupełnia się
zawartość tej substancji, podając ją w
tabletkach.

Niedokrwistości hemolityczne
Hemoliza oznacza dosłownie rozpuszczenie
krwinek; w medycynie tym pojęciem określa się
skrócenie czasu przeżycia erytrocytów
(prawidłowo "żyją" około 120 dni) do kilku
tygodni lub dni.
Jeśli organizm nie nadąża z produkcją nowych
krwinek, to pojawia się niedokrwistość.

Przyczyny niedokrwistości
hemolitycznych
Ogólnie wyróżnia się anemie spowodowane
czynnikami wewnątrzkrwinkowymi i
zewnątrzkrwinkowymi.
Do tych pierwszych zaliczamy defekty błony
erytrocytów (np. sferocytoza), defekty enzymów
tych krwinek (np. niedobór G-6-PD) lub
zaburzenia tworzenia hemoglobiny - tzw.
hemoglobinopatie, do których należą między
innymi talasemie.

Wszystkie te z pozoru odlegle od siebie
choroby łączy skrócenie czasu przeżycia
krwinek czerwonych spowodowane
jakimiś wewnętrznymi usterkami
związanymi z samym erytrocytem.

Przyczyny:
Przyczyną są najczęściej przeciwciała, pochodzące z innego
"obcego" organizmu (np. konflikt serologiczny u noworodków),
bądź produkowane przez samego chorego - tzw.
niedokrwistości autoimmunohemolityczne.
Niedokrwistości hemolityczne zewnątrzkrwinkowe mogą być
też spowodowane niektórymi lekami, chorobami zakaźnymi
(występują np. w malarii) czy też czynnikami chemicznymi (jad
żmii) lub fizycznymi (sztuczne zastawki serca).
Rzadziej pojawiają się anemie hemolityczne w zaburzeniach
przemiany materii (np. zespół Zievego), czy też bardzo ciężkie
postaci, w których hemoliza następuje w małych naczyniach
krwionośnych (zespół Gassera, zespół Moschkowitza).

Objawy kliniczne niedokrwistości
są podobne do tych, które występują w innych anemiach.
Poza tym pojawiają się objawy hemolizy: żółtaczka,
powiększenie śledziony, często kamica żółciowa.
Powyższe symptomy są związane ze wzmożonym
rozpadem krwinek, z których tworzy się m.in. bilirubina
odpowiedzialna za żółte zabarwienie skóry i powstawanie
kamieni. Czasami rozpad krwinek następuje bardzo
gwałtownie. Nazywamy to przełomem hemolitycznym.
Przebiega z gorączką, dreszczami, żółtaczką, bólami
brzucha, głowy i pleców.

Badania diagnostyczne
{kind=link}
We krwi osoby chorej na niedokrwistość hemolityczną obok
zmniejszonych wartości parametrów związanych z samą
anemią (stężenie hemoglobiny, liczba krwinek
czerwonych) często są nieprawidłowe kształty erytrocytów
(np. sferocyty, które są po prostu krwinkami czerwonymi
przybierającymi kształt kuli, czy też erytrocyty
sierpowate).
We krwi wzrasta również zawartość enzymu LDH oraz
żelaza i bilirubiny (jest to związane z rozpadem krwinek i
przemianami hemoglobiny), wzrasta też liczba
retikulocytów ("młodych" krwinek czerwonych), co jest
świadectwem wzmożonej odnowy erytrocytów w szpiku.

Granulocytopenia i agranulocytoza
Jednymi z najgroźniejszych chorób
dotyczących najliczniejszych z krwinek
białych - granulocytów
obojętnochłonnych, są granulocytopenia i
agranulocytoza. Końcówka -penia oznacza
tutaj brak, niedobór.

Granulocytopenią
nazywamy zmniejszenie liczby granulocytów obojętnochłonnych
(neutrofili) poniżej dolnego zakresu normy, czyli 2500 w 1 mm3.
Dzieje się tak najczęściej przy uszkodzeniu szpiku, gdzie tworzą
się i dojrzewają te krwinki. Uszkodzenie może nastąpić na skutek
działania czynników chemicznych (np. benzen), leków (np.
cytostatyki, związki złota), naświetlania i działania szkodliwych
przeciwciał. Również zmiany nowotworowe, wypierając
prawidłowe komórki ze szpiku, są przyczyną granulocytopenii.
W skrajnych przypadkach, na skutek działania przeciwciał
niszczących granulocyty i ich prekursorów, w ogóle brakuje
granulocytów. Mamy wówczas do czynienia z bardzo groźną
chorobą zwaną agranulocytozą.

Objawy granulocytopenii i
agranulocytozy
są związane z upośledzeniem odporności i rozwojem zakażeń,
najczęściej w jamie ustnej i migdałkach (owrzodzenia). Dzieje się tak
zwykle, kiedy liczba neutrofilii spadnie poniżej 500 w 1 mm3.
W leczeniu próbuje się określić i opanować przyczynę zaburzeń. Może
to być na przykład odstawienie "podejrzanych" leków.
Ważna jest dodatkowa ochrona przed zakażeniami, w której
przestrzega się ściśle zasad jałowości. Kiedy już pojawi się infekcja,
stosuje się oczywiście silne antybiotyki, podawane w postaci
zastrzyków. Osiągnięcia współczesnej medycyny pozwalają na
pobudzenie tworzenia w szpiku nowych granulocytów. Dzieje się tak
dzięki podaniu czynników wzrostowych dla neutrofilii znanych pod
skrótami angielskimi: G-CSF i GM-CSF.

Nowotwory
Należą do nich między innymi
chłoniaki złośliwe, które dzielimy na
dwie duże grupy: ziarnicę złośliwą
(chłoniak Hodgkina) i chłoniaki
nieziarnicze (nie-Hodgkin).

Ziarnica złośliwa, czyli choroba
Hodgkina,
jest głównie chorobą młodych ludzi (szczyt
występowania - 20-30 r.ż.), przeważnie mężczyzn.
Jej zasadniczą cechą jest nowotworowy rozrost
komórek, początkowo w węzłach chłonnych, a
później (w zaawansowanych stadiach) w innych
narządach. Biorąc pod uwagę to zaawansowanie,
przebieg choroby dzieli się na cztery okresy: od
zajęcia jednej grupy węzłów chłonnych (okres I)
do rozsianego zajęcia wątroby, śledziony, płuc,
szpiku kostnego i innych narządów (okres IV).

Objawy:
Głównym objawem jest powiększenie węzłów
chłonnych, przeważnie karkowych, rzadziej pachowych
lub pachwinowych. Węzły są charakterystycznie
niebolesne i zbite w pakiety.
Może być również powiększona wątroba albo śledziona.
Często pojawiają się także objawy zwane ogólnymi:
gorączka, poty nocne, utrata masy ciała.
W badaniach laboratoryjnych obserwujemy zwiększenie
OB, niedokrwistość, zmniejszenie ilości limfocytów.

Rozpoznanie
Aby rozpoznać ziarnicę, potrzebne jest
potwierdzenie histologiczne. Pobiera się próbkę
tkanki na drodze biopsji powiększonych lub
podejrzanych z innych przyczyn węzłów chłonnych.
Charakterystyczna jest obecność tak zwanych
komórek Sternberga. W celu ustalenia
zaawansowania choroby wykonuje się też badania
radiologiczne, ultrasonografię, tomografię
komputerową, bada się szpik kostny (po
wcześniejszej biopsji) i wykonuje scyntygrafię
kośćca.

W oparciu o dane
uzyskane z tych badań grupuje się chorych według okresów
zaawansowania choroby i stosuje odmienne metody leczenia.
w okresie I i II stosuje się naświetlania (radioterapię) zajętych
i sąsiednich obszarów węzłowych.
W okresie III bez objawów ogólnych stosuje się chemioterapię
łączoną z radioterapią; a w okresie III z objawami ogólnymi i w
okresie IV stosuje się już tylko chemioterapię.
Przy nawracającej ziarnicy złośliwej można również próbować
przeszczepu szpiku.
W grupie o dobrym rokowaniu (okres I i II bez objawów
ogólnych) uzyskuje się aż 80% wyleczeń.

chłoniaki nieziarnicze.
Niestety gorszymi prognozami co do wyleczenia
charakteryzuje się druga grupa chłoniaków - chłoniaki
nieziarnicze. Są to nowotwory złośliwe wywodzące się z
limfocytów (T lub B), zlokalizowane w tkance chłonnej.
Najczęściej chorują osoby w starszym wieku, mężczyźni
częściej niż kobiety. W ich powstawaniu odgrywają rolę
zakażenia wirusowe i czynniki genetyczne.
Podział chłoniaków nieziarniczych jest dosyć
skomplikowany. Istnieją różne jego kryteria. I tak
wyróżniamy chłoniaki o mniejszej i o większej złośliwości;
chłoniaki typu B i typu T. Istnieje także podział według
kryteriów morfologicznych, m.in.: limfocytowe,
plazmocytowe, centrocytowe.

Chłoniaki
stopniuje się również w zależności od
okresu zaawansowania. Tutaj podział
jest bardzo zbliżony do tego, któremu
podlega ziarnica złośliwa - również
wyróżniamy okresy od I do IV.

Objawy:
Chorzy zauważają zwykle powiększenie węzłów
chłonnych i z tym objawem zgłaszają się do lekarza.
Często występują objawy ogólne (gorączka, poty nocne,
utrata masy ciała).
We krwi może wystąpić niedokrwistość, zmniejszenie
ilości białych krwinek i płytek krwi.
Do postawienia rozpoznania potrzebne jest badanie
mikroskopowe "podejrzanego" węzła. Ważne jest także
określenie zaawansowania choroby dokonane za pomocą
przedstawionych przy okazji ziarnicy metod obrazowych.

Leczenie zależy od rodzaju
chłoniaka.
W chłoniakach o mniejszej złośliwości usuwamy
chirurgicznie zajęte węzły albo w okresie zmian
uogólnionych czekamy, aż zaistnieją wskazania do
chemioterapii.
w chłoniakach o mniejszej złośliwości chemioterapia nie
wyleczy choroby, może jedynie zmniejszyć uciążliwość
objawów.
Przebieg choroby jest tutaj długoletni, a czas przeżycia
wynosi od 2 do 10 lat.
Chłoniaki o dużej złośliwości przebiegają dużo szybciej,
rozpoznaje się je głównie w okresie uogólnionym, rozsianym.
Jeśli jednak zastosuje się chemioterapię, to istnieje szansa
(50%) na wyleczenie.

Jednym z chłoniaków nieziarniczych jest
szpiczak mnogi
.
Rozrostowi ulegają tu uzłośliwione komórki
plazmatyczne - rodzaj limfocytów, który produkuje
przeciwciała. Szpiczak rozwija się, jak wskazuje sama
nazwa, w szpiku, niszcząc kość i produkując duże ilości
takich samych przeciwciał lub ich części, które wcale nie
pomagają w walce z drobnoustrojami. Przeciwnie,
hamują wytwarzanie prawidłowych przeciwciał. Do tego
nowotworowe komórki szpiczaka wypierają normalne
komórki szpiku, co jest przyczyną zmniejszenia
erytrocytów, krwinek białych i płytek we krwi.

Szpiczak
występuje głównie około 60. r.ż. Podobnie jak w innych
nowotworach krwi przyczyna jego rozwoju jest nieznana.
Chorzy skarżą się najczęściej na bóle kości, uczucie
zmęczenia, stany podgorączkowe i poty nocne. We krwi
wykrywa się duże ilości białka, na które składają się
wytwarzane przez szpiczak przeciwciała. Zdjęcia
rentgenowskie ujawniają charakterystyczne "dziury" w
kości, głównie w czaszce, kręgach kręgosłupa, kości
miednicy, ramion i ud. Są to tzw. ogniska osteolityczne,
czyli miejsca, w którym szpiczak zniszczył tkankę kostną.

Badania laboratoryjne:
W badaniach laboratoryjnych spotyka się
bardzo przyspieszone OB (ponad 100 mm/h),
a w moczu tzw. białko Bence'a-Jonesa, na
które składają się przesączone razem z
moczem części przeciwciał.
Charakterystyczne jest także podwyższenie
poziomu wapnia we krwi.
Później obserwuje się również niedokrwistość
i zmniejszenie liczby płytek krwi.

Rozpoznanie:
W postawieniu rozpoznania najważniejsze jest
badanie szpiku, które ujawnia obecność
nacieków złożonych z komórek
plazmatycznych. Diagnozowanie jest jednak
trudne i wiąże się z częstymi błędami. W
rozpoznaniu bierze się bowiem pod uwagę
choroby reumatyczne, złamania po urazie czy
osteoporozę, nie myśląc wcale o szpiczaku.

Powikłania
Ze szpiczakiem, który należy do chłoniaków o mniejszej
złośliwości, wiąże się występowanie wielu powikłań.
Do najważniejszych należą złamania kości, głównie
kręgów kręgosłupa, tzw. nerki szpiczakowe, uszkodzone
przez duże ilości napływających części przeciwciał, co
doprowadza w końcu do niewydolności tego organu;
niedobór przeciwciał, który prowadzi do rozwoju
zakażeń, oraz skrobiawica, czyli gromadzenie się
nieprawidłowych białek w różnych narządach (m.in. w
wątrobie, nerkach).

Postęp choroby dzieli się na
okresy (podobnie jak klasyczne
chłoniaki);
w ostatnim (trzecim) pojawia się znaczna
niedokrwistość, zaawansowane zmiany w
kościach i duże stężenia nieprawidłowych
przeciwciał. Oprócz klasycznej formy
choroby istnieje również tzw. szpiczak
tlący się o powolnym przebiegu,
trwającym latami bez powikłań.

W leczeniu
wykorzystuje się chemioterapię (od II okresu choroby)
czasami w połączeniu z podawaniem interferonu.
Ważne jest również zwalczanie i łagodzenie objawów, nawet
wtedy, kiedy nie widzi się szans na zupełne wyleczenie
szpiczaka. Stosuje się wtedy miejscowe naświetlanie, środki
przeciwbólowe, leczy się niedokrwistość, obniża nadmierne
stężenie wapnia we krwi.
Czas przeżycia jest zróżnicowany w zależności od okresu
choroby, w trzecim (najbardziej zaawansowanym) chorzy
przeżywają zwykle 6-12 miesięcy.

Leukemia
Białaczka jest chorobą nowotworową,
polegającą na rozroście w całym organizmie
leukocytów (białych krwinek) jednego typu.
Te nieprawidłowe, rozrastające się białaczkowe
komórki zajmują ostatecznie prawie cały szpik
kostny, naciekają inne pozaszpikowe narządy i
zmieniają obraz krwi oglądanej pod
mikroskopem.

Białaczki dzielimy na:
ostre (częstsze) i przewlekle (rzadsze).
Drugim podziałem jest rozróżnienie białaczek
limfatycznych (rozrostowi ulegają limfocyty) i szpikowych
(rozrost innych rodzajów białek krwi).
Te dwa podziały dają w rezultacie cztery typy białaczek:
ostrą limfatyczną (angielski skrót ALL), ostrą szpikową
(angielski skrót AML), przewlekłą szpikową (CML) i
przewlekłą limfatyczną (CLL). U dzieci występuje głównie
ostra białaczka limfatyczna, u dorosłych (raczej w
starszym wieku) przeważa ostra białaczka szpikowa.

Ostre białaczki
omawiane są zwykle wspólnie, mają, bowiem podobne
objawy kliniczne, choć zupełnie inne rokowanie.
Przyczyny, tak jak w przypadku innych nowotworów,
są nieznane. Przypuszcza się, że znaczenie ma
zakażenie niektórymi typami wirusów, uszkodzenia
szpiku przez substancje chemiczne (benzen, niektóre
leki stosowane w onkologii), promienie jonizujące,
czynniki genetyczne (np. częstsze występowanie
białaczki w zespole Downa).

Objawy ostrej białaczki wynikają między
innymi z wyparcia przez komórki
białaczkowe (blasty) prawidłowych
komórek szpiku
Pojawia się więc niedokrwistość (z bladością, dusznością i
osłabieniem), krwawienia związane z małą ilością płytek krwi,
podatność na zakażenia w wyniku małej ilości prawidłowych
białych krwinek chroniących nas przed drobnoustrojami.
Inną grupą są objawy ogólne: osłabienie, gorączka, poty nocne.
Wreszcie, w wyniku naciekania przez komórki białaczkowe
innych niż szpik narządów pojawia się obrzmienie węzłów
chłonnych, powiększenie śledziony, objawy neurologiczne (przy
naciekaniu układu nerwowego), bóle kostne czy zmiany na
skórze.

Badania nlaboratoryjne:
W badaniu krwi często występuje niedokrwistość,
małopłytkowość i granulocytopenia. Wbrew obiegowym
opiniom liczba krwinek białych nie zawsze jest
podwyższona, może być wręcz zmniejszona.
Najważniejsze w rozpoznaniu jest wykrycie
niedojrzałych, nieprawidłowych krwinek we krwi i w
szpiku. W celu zbadania szpiku wykonuje się w tym
wypadku jego punkcję. Kiedy już wykryje się
nieprawidłowe komórki, przeprowadza się dalsze
badania cytochemiczne i immunologiczne, które
pozwalają na dokładne określenie typu białaczki.

Leczenie ostrych białaczek
polega głównie na chemioterapii, której pierwotnym celem jest
osiągniecie remisji - czyli doprowadzenie obrazu krwi i szpiku do
normy i przynajmniej tysiąckrotne zmniejszenie liczby komórek
nowotworowych; później stosuje się leczenie podtrzymujące
uzyskaną remisję. Leczenie jest długie i trudne. Trwa czasem
latami.
Wiąże się z występowaniem objawów niepożądanych (nudności,
działanie toksyczne na serce, płuca, nerki i inne narządy,
zahamowanie pracy szpiku, wypadanie włosów, toksyczne
działanie na rozrodczość).
U dorosłych te wyniki są gorsze, ponieważ dominuje tutaj ostra
białaczka szpikowa. Pięć lat przeżywa około 25% chorych. Dlatego
tak dużą wagę przywiązuje się do innych niż chemioterapia metod
leczenia.

przeszczep szpiku.
Wyróżniamy dwa główne typy tej
metody: przeszczep od innej osoby
(allogeniczny) i przeszczep własnego
szpiku (autologiczny)

Przeszczep allogeniczny
stosuje się u chorych poniżej 45 r.ż. i
pozostających w okresie remisji. Dawca szpiku
musi być zgodny antygenowo z biorcą. Dlatego
zostaje nim najczęściej ktoś z rodzeństwa.
Przeszczep szpiku, tak jak każdy inny przeszczep,
może bowiem zostać odrzucony, jeśli organizm
wykryje jego "obcość". Im bardziej tkanka (w tym
przypadku szpik) jest "podobna" do organizmu
biorcy, tym większe szanse na przyjęcie
przeszczepu. O "obcości" czy "swojskości"
decydują małe cząsteczki, znaczniki zwane
antygenami.

cd
Zasadą tej metody jest agresywne zniszczenie komórek
białaczkowych przez leczenie chemioterapią i
intensywne naświetlania całego ciała, a później
przeszczep szpiku. Transplantację kojarzy się zwykle z
operacją, tymczasem tutaj jest to po prostu dożylny
wlew szpiku dawcy; komórki tego szpiku same znajdują
swoje właściwe miejsce w nowym "gospodarzu". Wyniki
tych przeszczepów są coraz lepsze, najlepiej dokonać
takiej transplantacji w pierwszej remisji, w przypadku
AML 3 lata przeżywa wtedy ponad 50% chorych.

Przeszczep autologiczny
to druga metoda, w której paradoksalnie na koniec
intensywnej chemioterapii niszczącej komórki białaczkowe
podaje się szpik pobrany od samego chorego przed
rozpoczęciem tego leczenia. Znika tutaj problem z
szukaniem dawcy, ale pojawia się inna trudność. Szpik jest
przecież "zanieczyszczony" komórkami nowotworowymi,
które przez przeszczepem trzeba jakoś usunąć. Takie
"czyszczenie" jest trudnym zadaniem. Jeśli u chorego po
przeszczepie autologicznym uzyskano remisję, to 5 lat
przeżywa przeciętnie 40% chorych.
Odmianą przeszczepu szpiku są też transplantacje
komórek pnia z krwi obwodowej.

Leczenie:
Oprócz chemioterapii i przeszczepów szpiku
stosuje się także naświetlania: całego ciała jak
przy przygotowaniach do przeszczepu szpiku
albo jednego obszaru, np. ośrodkowego układu
nerwowego u dzieci. Nie mniej ważna jest
odpowiednia opieka nad chorym: staranna
higiena i maksymalna jałowość pomieszczeń.
Istotne jest odkażanie przewodu pokarmowego i
jamy ustnej za pomocą leków oraz uzupełnianie
brakujących krwinek czerwonych i płytek krwi.

Objawy:
Bardzo często na początku choroba nie daje
żadnych objawów.
W połowie przypadków występuje (na
początku) niebolesne powiększenie węzłów
chłonnych. Później ten objaw pojawia się już u
prawie wszystkich chorych.
Obserwuje się też powiększenie śledziony i
wątroby oraz świąd i różne zmiany skórne.

Badania laboratoryjne:
We krwi spotyka się duże liczby krwinek
białych, pośród których przeważają limfocyty.
Obowiązkowe jest też badanie szpiku (punkcja),
w których duży odsetek komórek (większy niż
zwykle) również stanowią limfocyty.
Później, podobnie jak w ostrych białaczkach,
przeprowadza się dalsze badania pozwalające
na dokładniejsze określenie typu
nieprawidłowych komórek.

Przebieg przewlekłej białaczki
limfatycznej dzieli się
- podobnie jak przebieg ziarnicy złośliwej i chłoniaków
nieziarniczych - na okresy. Najbardziej zaawansowane
stany przebiegają z niedokrwistością, w której stężenie
hemoglobiny spada poniżej 11 g/dl i trombocytopenią
(zmniejszeniem ilości płytek krwi).
Obrazuje to wypieranie prawidłowych komórek szpiku
przez nadmiernie rozrastające się limfocyty. Prowadzi to
do zakażeń, które występują także z powodu niedoboru
przeciwciał.

W leczeniu obowiązuje zasada, że leczy się najpóźniej jak
można, ze względu na to, że pełne wyleczenie nie jest
obecnie możliwe, a terapia niesie za sobą
niebezpieczeństwo objawów ubocznych. Wskazaniami do
rozpoczęcia leczenia jest niedokrwistość, mała liczba
płytek krwi i takie powiększenie węzłów chłonnych albo
śledziony, które daje nasilone objawy. W terapii stosuje się
chemioterapię i naświetlania. Czasami usuwa się również
śledzionę, co pomaga w powikłaniu przewlekłej białaczki
limfatycznej, jakim jest niedokrwistość hemolityczna.
Stosuje się również próby z podawaniem interferonu.

Przewlekła białaczka
szpikowa
należy do grupy chorób zwanych przewlekłymi
chorobami mieloproliferacyjnymi, w której rozrostowi
ulegają komórki szpiku będące prekursorami różnych
linii komórkowych (krwinek czerwonych, białych, płytek
krwi). Przyczyny przewlekłej białaczki szpikowej są
nieznane; czasami do jej rozwoju przyczynia się
narażenie na promienie jonizujące i benzen. W
powstawaniu choroby niebagatelne znacznie ma
również obecność nieprawidłowego chromosomu (części
jądra komórkowego przenoszącego geny), zwanego
chromosomem Philadelphia.

Przebieg choroby
Przewlekła białaczka ma podstępny, wieloletni przebieg.
Na początku chory może nie odczuwać objawów, a choroba
jest odkrywana przypadkowo na podstawie badania krwi
(zwiększenie liczby krwinek białych) czy badania brzucha
przez lekarza, najczęściej z innych przyczyn, w którym
ujawnia się powiększenie śledziony. To powiększenie
śledziony jest bardzo charakterystyczne i sprawia czasami
dolegliwości w postaci ucisku w nadbrzuszu.
Taki wstępny, przewlekły okres może trwać latami,
przechodząc następnie w okres przyspieszenia
(akceleracji). Wtedy pojawia się niedokrwistość, narasta
zwiększona liczba krwinek białych, a spada liczba płytek,
powiększa się śledziona i ewentualnie dołącza się gorączka

cd
W ostatnim okresie (przełomie blastycznym) obraz choroby
przypomina ostra białaczkę, a ona sama kończy się zgonem.
W badaniu krwi obserwujemy dużą liczba krwinek białych ze
zwiększoną liczbą granulocytów obojętnochłonnych i ich
prekursorów, niedokrwistość, początkowo zwiększoną, a następnie
zmniejszoną liczbę płytek krwi. Liczba białych krwinek może być na
tyle duża, że dochodzi do zatorów białaczkowych, będących
przyczyną zawału (martwicy) śledziony czy siatkówki. W szpiku
kostnym pobranym za pomocą punkcji obserwuje się wzmożenie
produkcji białych krwinek, a często również płytek krwi. Jednak tutaj
badanie szpiku nie jest rozstrzygające w rozpoznaniu. W postawieniu
diagnozy ważne jest połączenie danych z obrazu klinicznego i obrazu
krwi oraz obecność wspomnianego chromosomu Philadelphia.

Leczenie:
W leczeniu przewlekłej białaczki szpikowej stosuje się
interferon alfa, chemioterapię i allogeniczny przeszczep
szpiku kostnego. Spośród tych metod całkowite wyleczenie
jest możliwe tylko dzięki allogenicznemu (od innej osoby)
przeszczepowi szpiku. Warunkiem jego przeprowadzenia
jest obecność odpowiedniego dawcy i nieprzekroczony
wiek 50 lat. Pięć lat po przeszczepie przeżywa około 60%
chorych, a 10 lat około 40%. Leczenie interferonem, aby
było skuteczne, musi być prowadzone przez cały czas.
Odstawienie leku doprowadza do nawrotu nowotworu.
Najmniej skuteczna, choć również szeroko wykorzystywana
jest chemioterapia głównie za pomocą hydroksymocznika.

Do innych, rzadszych już chorób
mieloproliferacyjnych należą: czerwienica
prawdziwa, nadpłytkowość samoistna i
zwłóknienie szpiku kostnego.
W czerwienicy prawdziwej rozrostowi (proliferacji) ulegają
głównie prekursorzy krwinek czerwonych, doprowadzając do
ich zwiększonej ilości we krwi. Pojawia się zaczerwienienie
twarzy, zawroty i bóle głowy, krwawienia z nosa, nadciśnienie.
We krwi obserwuje się podwyższenie hematokrytu i stężenia
hemoglobiny oraz zwiększenie liczby krwinek czerwonych.
Choroba ta jest przykładem, że zwiększenie liczby krwinek
czerwonych jest również (jeśli nie bardziej) szkodliwe niż ich
niedobór (czyli niedokrwistość). Pojawiają się wówczas
powikłania w postaci zakrzepów i bardzo niebezpiecznych
zatorów, ostatecznie może rozwinąć się zwłóknienie szpiku lub
ostra białaczka. Zaburzeniu ulega czynność płytek krwi -
pojawiają się krwawienia.

Rozpoznanie
W rozpoznaniu ważne jest wykluczenie tzw.
czerwienicy wtórnej, czyli zwiększenia stężenia
hemoglobiny związanego ze zbyt małą ilością
tlenu we krwi (choroby serca i płuc) lub
nieprawidłową produkcją erytropoetyny
(nowotwory i niektóre choroby nerek). W
ostatecznym rozpoznaniu istotna jest również
biopsja i badanie szpiku.

Leczenie:
W leczeniu stosuje się metodę znaną już od
wieków, czyli upusty krwi.
Próbuje się także chemioterapii i radioterapii,
szczególnie przy dużym niebezpieczeństwie
zatorów.
Przy stosowaniu leczenia średni czas przeżycia
wynosi 10-15 lat.

Nadpłytkowość samoistna,
zgodnie z nazwą, polega na nadmiernym
rozroście komórek produkujących płytki.
Następstwem ich zwiększonej liczby są zatory i
zakrzepy (najczęstsza przyczyna zgonu) oraz
krwawienia (z racji upośledzonej funkcji płytek).
We krwi obserwuje się zwiększona liczbę płytek,
a w szpiku rozrost megakariocytów. W leczeniu
stosuje się interferon alfa lub hydroksymocznik.
Średni czas przeżycia wynosi 10-15 lat.

Osteomieloskleroza(zwłóknienie
szpiku )
Zgodnie z nazwą włóknieniu ulega tutaj
szpik, doprowadzając do "ogołocenia" z
komórek wytwarzających krew.
Komórki prekursorowe "wyprowadzają"
się wtedy do śledziony i wątroby, gdzie
"zastępczo" tworzą się krwinki, jednak
ta produkcja jest bardzo nieefektywna.

Objawy:
Początek choroby jest bardzo podstępny.
Charakterystyczne powiększenie śledziony
łatwo można przeoczyć.
Pojawia się również utrata masy ciała i
(ewentualnie) gorączka.
We krwi występuje najpierw zwiększenie, a
potem zmniejszenie liczby krwinek.

Rozpoznanie:
W rozpoznaniu ważna jest biopsja szpiku,
w którym obserwuje się zwłóknienie.
W leczeniu prowadzone są próby z
interferonem alfa. Poza tym podaje się
brakujące krwinki czerwone. Średnio
chorzy przeżywają 5 lat.

Skazy krwotoczne
Jest to stan, w którym różne defekty hemostazy
(utrzymywania krwi w naczyniach krwionośnych) powodują
nieprawidłową, patologiczną skłonność do krwawień.
Wyróżniamy trzy rodzaje skaz krwotocznych.
Są to skazy związane z naczyniami (inaczej waskulopatie),
skazy związane z płytkami (skazy krwotoczne płytkowe)
oraz skazy związane z czynnikami krzepnięcia
występującymi w osoczu (tzw. koagulopatie).

Skazy osoczowe
Najważniejszymi skazami osoczowymi są koagulopatie
niedoborowe z najbardziej znaną hemofilią. Inne grupy
koagulopatii to tzw. koagulopatie ze zużycia (czynników
krzepnięcia), immunokoagulopatie (związane z tworzeniem
przeciwciał przeciwko czynnikom krzepnięcia) oraz
hiperfibrynolizy (związane z nadmierną aktywacją układu
przeciwstawnego do kaskady krzepnięcia krwi, czyli fibrynolizy
rozpuszczającej zakrzepy).
Najbardziej znaną i najczęściej występującą koagulopatią jest
hemofilia, czyli krwawiączka. Jej przyczyną jest wrodzony
niedobór VIII (hemofilia A) lub IX (hemofilia B) czynnika
krzepnięcia.

Hemofilia
jest chorobą dziedziczną. Każdy człowiek ma
23 pary chromosomów, małych ciałek
przenoszących geny, w których zakodowana
jest informacja o naszym rozwoju i o
niektórych chorobach. Wynika z tego, że geny
również są zdublowane i ustawione w pary.
Ostatnia para chromosomów to tzw.
chromosomy związane z płcią określane jako X
i Y. Kobieta ma dwa chromosomy X, a
mężczyzna jeden chromosom X i jeden Y.

Geny mogą być dominujące - wtedy wystarczy tylko jeden z
pary, aby mógł wywołać odpowiedni efekt, albo recesywne -
oba geny w parze muszą być wtedy recesywne, aby pojawił
się właściwy im skutek.
Nieprawidłowy gen odpowiedzialny za rozwój hemofilii
znajduje się na chromosomie X i jest recesywny. Wynika stąd,
że chorują w przeważającej mierze chłopcy. Bardzo rzadko
zdarza się, aby zachorowały kobiety - wtedy spotkać się ze
sobą muszą dwa nieprawidłowe geny na chromosomach X.
Kobiety mogą być natomiast przenosicielkami choroby i
przenosić ją na swoich synów.

W dziedziczeniu hemofilii A ważne są
również następujące stwierdzenia:
• wszystkie córki chorego na hemofilię są nosicielkami
(mają jeden gen prawidłowy, a drugi nieprawidłowy)
• wszyscy synowie chorego na hemofilię są zdrowi
• szansa przekazania przez nosicielkę chorego
chromosomu X wynosi 50%
Krwawienia, podstawowy objaw hemofilii i innych skaz
krwotocznych, mogą pojawić się już u noworodka
(krwawienie z pępowiny). Najczęściej jednak choroba
ujawnia się później w postaci krwawień do mięśni i
stawów, a także krwawień po operacjach, krwotoków z
nosa czy z dróg rodnych.

Badania laboratoryjne:
W rozpoznaniu oprócz obrazu klinicznego ważne są
badania laboratoryjne.
W skazach krwotocznych mierzy się czas krwawienia i
krzepnięcia.
W przypadku hemofilii czas krwawienia (za który
odpowiadają płytki krwi i skurcz naczyń krwionośnych)
jest prawidłowy, a czas krzepnięcia, z racji braku jednego
z czynników potrzebnych do tworzenia skrzepu jest
przedłużony. Później ocenia się również stężenie czynnika
VIII (w przypadku hemofilii A) lub IX (w przypadku
hemofilii B) we krwi.

Objawy:
W zależności od stężenia czynników krzepnięcia we
krwi wyróżniamy różne stopnie ciężkości hemofilii.
Objawy pojawiają się zwykle dopiero w postaci
lekkiej (są jeszcze: subhemofilia, postać średnio
ciężka i ciężka), w której to stężenie wynosi 5-15
jednostek międzynarodowych w jednym decylitrze
krwi (jm./dl). Wtedy krwiaki pojawiają się po
wyraźnych urazach. W postaci ciężkiej krwawienia
mogą występować bez urazów, samoistnie.

Leczenie:
Zasadniczym leczeniem jest podawanie brakujących czynników krzepnięcia: VIII w przypadku
hemofilii A, a IX w przypadku hemofilii B. U chorych z rzadkimi krwawieniami można podawać
te czynniki jedynie w koniecznych przypadkach (krwawienie samoistne, przed operacją). W
ciężkiej hemofilii koncentraty odpowiednich czynników podaje się bardziej systematycznie. W
hemofilii A o lekkim przebiegu zamiast czynnika VIII podaje się niekiedy związek o nazwie
DDAVP (desmopresynę). Powoduje ona uwalnianie czynnika VIII z "magazynów" znajdujących
się w śródbłonku naczyń.
Dawne ryzyko zakażeń (wirus HIV, wirusy żółtaczki czy cytomegalii) wyeliminowano poprzez
podawanie preparatów uzyskanych dzięki inżynierii genetycznej.

choroba von Willebranda.
Inną skazą osoczową (koagulopatią) jest
choroba von Willebranda.
W tym schorzeniu brakuje jednego ze
składników czynnika VIII, zwanego właśnie
czynnikiem von Willebranda. Chorują równie
często chłopcy (mężczyźni), jak i dziewczynki
(kobiety). Dziedziczenie jest bardziej złożone niż
w hemofilii dlatego, że wyróżniamy aż trzy typy
tej choroby.

Objawy:
Obraz choroby jest kombinacją objawów spotykanych w
hemofilii (krwawienie do mięśni, stawów, podbiegnięcia
krwawe) i symptomów charakterystycznych dla skaz
płytkowych (krwawienia z nosa i innych błon śluzowych,
drobne wybroczyny). Przedłużony jest zarówno czas
krwawienia (upośledzona funkcja płytek krwi), jak i czas
krzepnięcia.
W leczeniu stosuje się DDAVP, a w cięższych przypadkach
koncentraty czynnika VIII z aktywnością czynnika von
Willebranda.

Koagulopatie
mogą być również nabyte, powstają
wtedy najczęściej w wyniku niedoboru
witaminy K, niezbędnej do prawidłowego
tworzenia się niektórych czynników
krzepnięcia krwi.

Przyczyną jest:
uszkodzenie wątroby u noworodków,
upośledzenie wchłaniania w jelitach (w tym
witaminy K) występujące w wielu chorobach,
antybiotykoterapia (antybiotyki zabijają bakterie
jelitowe, które tworzą witaminę K) oraz żółtaczka
mechaniczna (żółć jest niezbędna do wchłaniania
tej witaminy).
nną dużą grupę przyczyn stanowi leczenie
środkami, które są antagonistami witaminy K,
czyli wykazują przeciwstawne do niej działanie
(np. kumaryna).

Skazy płytkowe
Najczęstszymi skazami krwotocznymi są jednak
nie skazy osoczowe, ale skazy płytkowe. Spośród
nich najważniejsze są te wywołane małą liczbą
płytek, czyli małopłytkowości (trompocytopenie).
Mała liczba tych elementów krwi może być
spowodowana zaburzeniami ich wytwarzania w
szpiku kostnym oraz przyspieszonym
niszczeniem płytek we krwi.

Przyczyny:
Przyczynami zmniejszonego ich
wytwarzania jest uszkodzenie szpiku
przez leki (np. cytostatyki), substancje
chemiczne (np. benzen), zakażenia (np.
HIV), a także wypieranie prawidłowych
komórek przez komórki białaczkowe czy
chłoniakowe albo zwłóknienie szpiku.

trombocytopenie
Spośród małopłytkowości uwarunkowanych
przyspieszonym niszczeniem płytek sporą grupę
stanowią trombocytopenie spowodowane przez
własne przeciwciała organizmu. Tak dzieje się
między innymi w samoistnej plamicy
małopłytkowej, jednej z najczęstszych i najbardziej
znanych małopłytkowości. Inne przyczyny
obejmują niszczenie płytek przez powiększoną
śledzionę (hipersplenizm) oraz sztuczne zastawki
serca (uszkodzenia mechaniczne).

Objawy:
Krwawienia w skazach płytkowych różnią
od obrazu hemofilii. Chorzy krwawią z
błon śluzowych (z nosa, dziąseł).
Na ich skórze pojawiają się drobne
wybroczyny.
Nie ma tutaj charakterystycznych dla
koagulopatii (m.in. hemofilia) wylewów
do stawów i mięśni.

Badania laboratoryjne:
W badaniach laboratoryjnych obserwuje się
przede wszystkim zmniejszoną liczbę płytek
(poniżej 140 tys w 1 mm3) Przedłużony jest też
czas krwawienia przy prawidłowym czasie
krzepnięcia (odwrotnie niż przy skazach
osoczowych). W rozpoznaniu ważne jest
poszukiwanie ewentualnych chorób będących
przyczyną zmniejszonej liczby płytek (np.
białaczki) oraz badanie szpiku kostnego.

Leczenie:
Leczy się przede wszystkim leżącą u podłoża
małopłytkowości chorobę.
W razie krwawień lub przed spodziewanym leczeniem
cytostatykami podaje się choremu koncentraty płytek
krwi uzyskanych od dawców (optymalnie jednego
dawcy).
Oprócz ryzyka zakażenia (obecnie bardzo małego)
istnieje niebezpieczeństwo uczulenia organizmu
chorego przez podawane "obce" preparaty. Wtedy przy
następnych przetoczeniach pojawia się groźba
szybkiego zniszczenia podanych płytek.

Skazy naczyniowe (waskulopatie)
Ostatnią, rzadszą grupą skaz krwotocznych są skazy
naczyniowe. Wtedy krwawienie jest uzależnione od
kruchości naczyń krwionośnych.
W tej grupie chorób, podobnie jak w skazach
płytkowych, wydłużony jest czas krwawienia, a
prawidłowy czas krzepnięcia.
Do wrodzonych skaz naczyniowych zaliczamy między
innymi chorobę Rendu-Oslera, w której na wargach,
języku, koniuszkach palców i innych częściach ciała
występują teleangiektazje - rozszerzenia małych naczyń
krwionośnych. Miejsca te mają skłonność do krwawień.
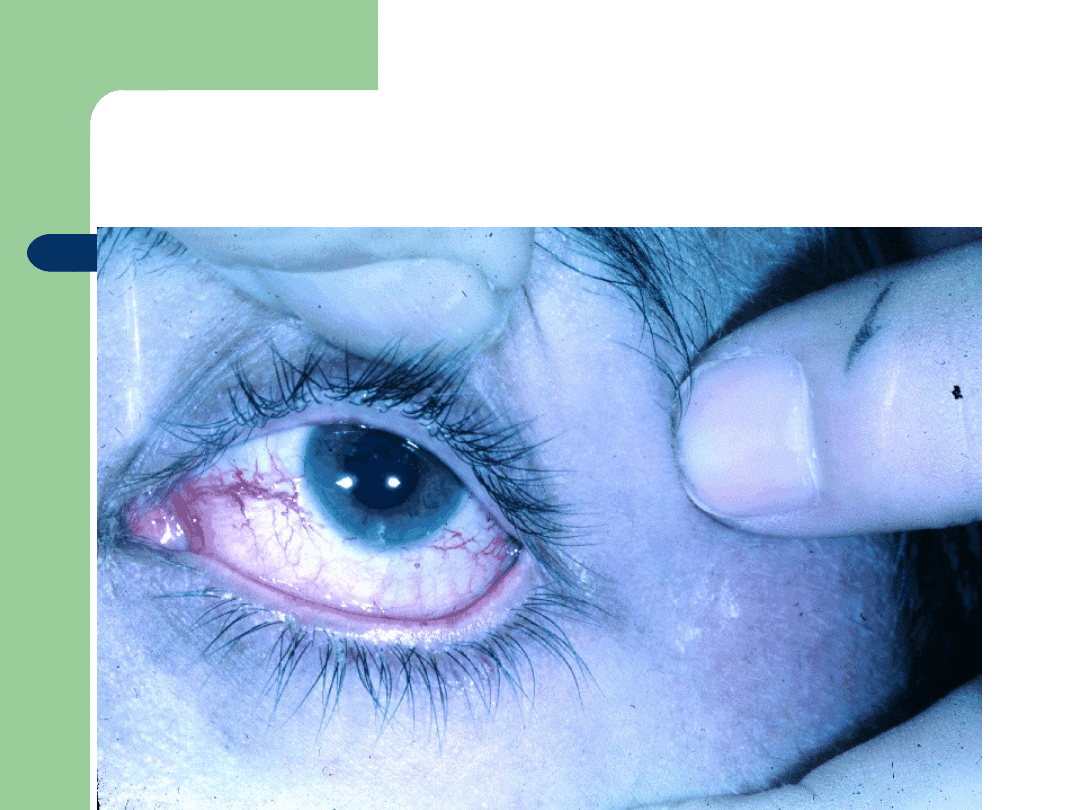
Poszerzone naczynia spojówki u
pacjenta z zespołem ataksja-
teleangiektazja

Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
Wyszukiwarka
Podobne podstrony:
LEKI STOSOWANE W?RMAKOTERAPII CHORÓB KRWI I UKŁADU KRWIOTWÓRCZEGO
Choroby krwi i układu krwiotwórczego, Rat med rok 2, Choroby wewnętrzne
Farmakoterapia chorób krwi i układu krwiotwórczego
Choroby krwi i układu krwiotwórczego 2
Choroby krwi i układu krwiotwórczego
choroby rozrostowe układu krwiotwórczego
w7.FARMAKOLOGIA KRWI I UKŁADU KRWIOTWÓRCZEGO, wykłady PMWSZ w Opolu - Pielęgniarstwo, Farmakologia
patofizjologia chorób krwi i narządów krwiotwórczych
MASKI KLINICZNE CHORÓB ROZROSTOWYCH UKŁADU KRWIOTWÓRCZEGO
choroby rozrostowe układu krwiotwórczego, studia pielęgniarstwo
choroby rozrostowe układu krwiotwórczego
Choroby układu krwiotwórczego i krwi, Podst pielegniarstwa
Farmakologia Leki w chorobach układu krwiotwórczego
Farmakologia, Leki w chorobach układu krwiotwórczego
więcej podobnych podstron