
Choroba von
Willebranda

Jest najczęstszym wrodzonym zespołem
zaburzeń krzepnięcia.
Dziedziczenie autosomalne
Częstość jego występowania ocenia się
na 1-2% w populacji ogólnej.
Charakteryzuje się mniejszą skłonnością
do samoistnych krwawień niż hemofilia.
U osób homozygotycznych chorobę
stwierdza się rzadko, ale ma ona u nich
znacznie cięższy przebieg.

Czynnik von Willebranda
Jest osoczowym białkiem adhezyjnym i
nośnikowym.
Jego biosynteza zachodzi w komórkach śródbłonka
i w megakariocytach (gen na 12 chromosomie).
Występuje w:
osoczu
ziarnistościach α
błonach megakariocytów i płytek
ciałkach Weibela i Palade’a komórek
śródbłonków
pod śródbłonkiem

Należy do białek ostrej fazy.
Wydzielanie pobudza: trombina , adrenalina,
estry forbolu, interleukina 1, kachektyna(TNF), a
także wysiłek fizyczny i emocje.
W osoczu występuje w kompleksie z czynnikiem
VIII.
Transportuje go i ochrania przed degradacją we
krwi (przez proteolityczne białko C) oraz
zagęszcza na powierzchni przylegających płytek.
Poziom w osoczu to wskaźnik pobudzenia lub
uszkodzenia komórek śródbłonka.
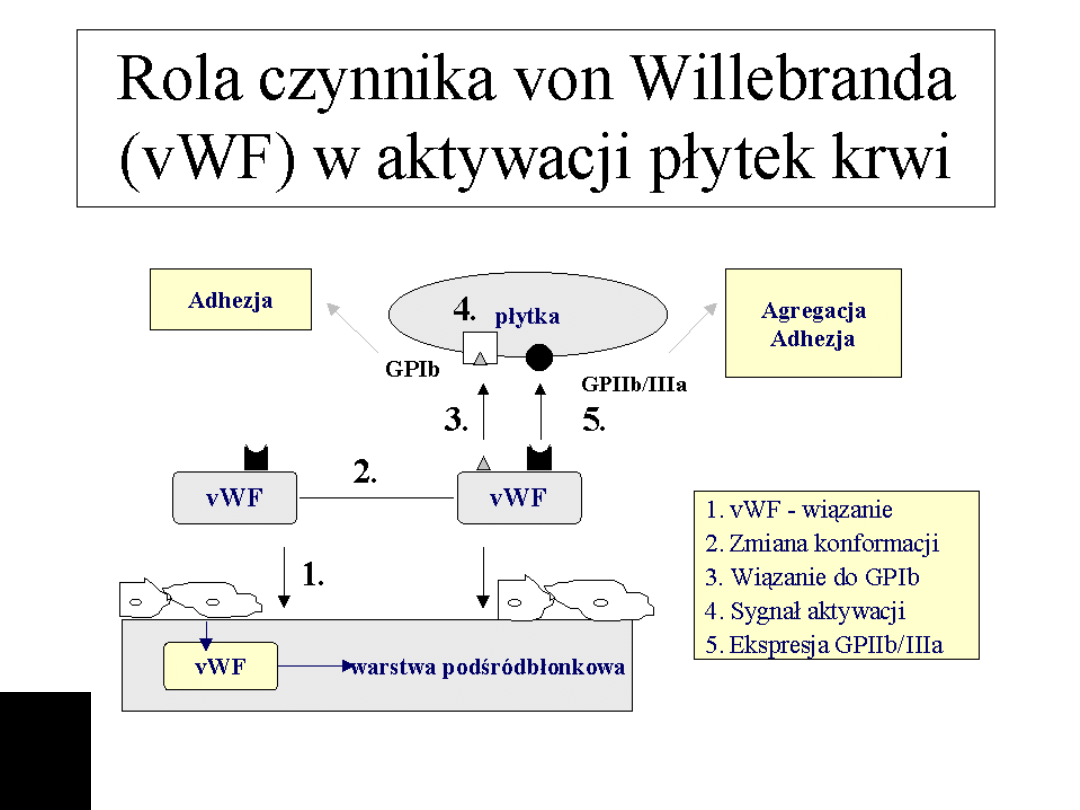

Adhezja - rola vWF.
Płytki krwi rozpoznają odsłoniętą warstwę podśródbłonkową.
Specyficzne receptory dla kolagenu głównie GPIa/GPIIa i GPIV
odpowiadają za adhezję płytek do odsłoniętego kolagenu.
Pierwotne zakotwiczenie jest niewystarczające, szczególnie w
naczyniach tętniczych w warunkach przepływu o wysokim module
ścinającym. W takim przypadku niezbędny jest cz. vW.
Produkowany głownie przez komórki śródbłonka, uwalniany jest
także z a-granul płytek. W czasie kontaktu z warstwą
podsródbłonkową vWF zmienia konformację i jest rozpoznawany
przez specyficzny receptor na płytkach - kompleks GPIb/GPV/GPIX
a także przez receptor dla fibrynogenu. Kluczową rolę odgrywa
glikoproteina GPIb, niedobór tej glikoproteiny jest przyczyną
choroby Bernarda-Souliera, natomiast niedobory i zaburzenia
funkcji cz vW są przyczyną choroby von Willebrand'a.
Podwyższenie poziomu we krwi stężenia cz.vW świadczy o
uszkodzeniu śródbłonka, wysoki poziom vWF jest związany z kolei
ze zwiekszonym ryzykiem rozwoju ChNS.
Metodą umożliwiającą pośrednio badanie zdolności płytek do
adhezji jest pomiar czasu okluzji w analizatorze funkcji płytek, np.
PFA-100.
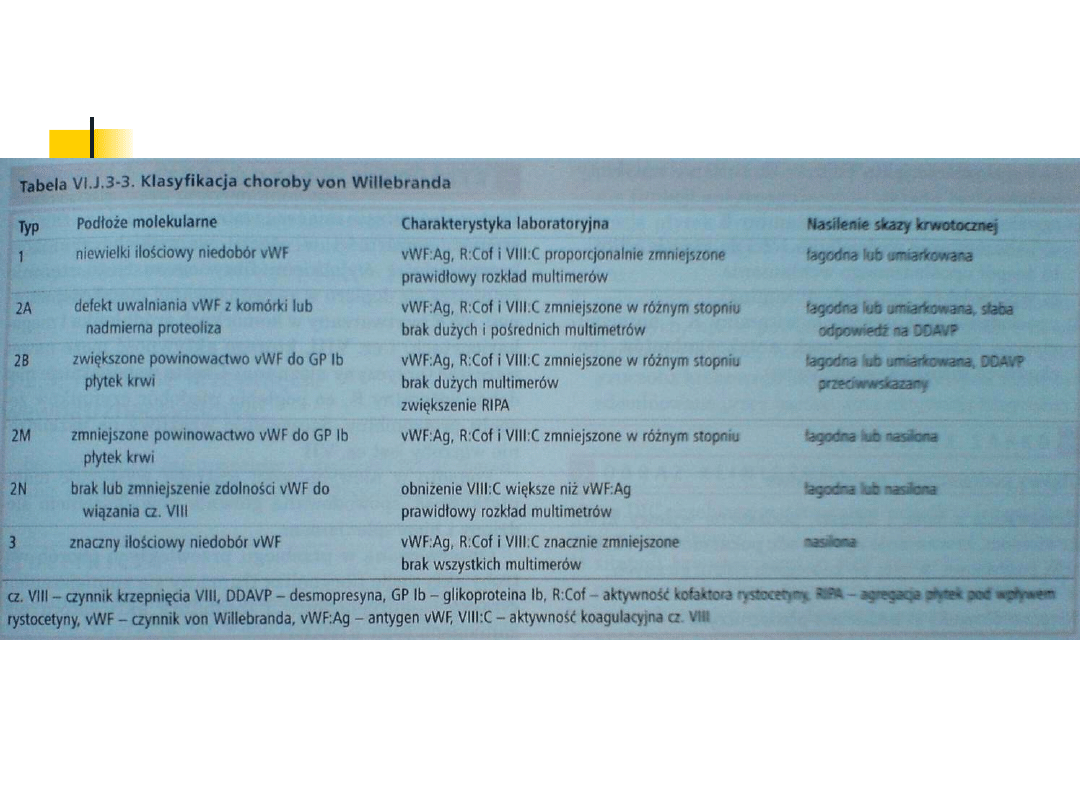

Klasyfikacja choroby:
Postacie wrodzone:
vWs typu 1
(80% chorych): charakteryzuje się
niedoborem vWF i czynnika VIII. Dziedziczy się w
sposób dominujący, genem autosomalnym.
vWs
typu 2A
typu 2A (15% chorych): charakteryzuje się
zaburzeniem budowy i czynności vWF,
polegającym na niezdolności do jego polimeryzacji.
Steżenie vWF i aktywność czynnika VIIIC mogą być
zmniejszone lub prawidłowe
U chorych z
typem 2B
typem 2B stwierdza się zwiększone
powinowactwo nieprawidłowego vWF do receptora
glikoproteinowego Ib płytek krwi (GPIb). Jest to
choroba autosomalna dominująca.

Rzadkie odmiany choroby:
vWs
typu 3
typu 3 (<5%chorych); jest to najcięższa
postać choroby von Willebranda. Stwierdza się
brak vWF i znaczne zmniejszenie czynnika
VIIIC w surowicy krwi. Dziedziczy się
autosomalnie recesywnie.
vWs
2M
2M (polega na upośledzonej intrakcji vWF
z płytkami krwi, ale zachowana jest zdolność
do polimeryzacji cząsteczek)
vWs
2N
2N
( jest to defekt vWF polegający na
zmniejszonym powinowactwie do czynnika VIII)

Postacie nabyte:
występujące jako objaw choroby podstawowej,
np. gammapatii monoklonalnej, chłoniaków
złośliwych, chorób autoimmunologicznych,
jako powikłanie leczenia kwasem
walproinowym.

Patogeneza:
Niedobór vWF – glikoproteiny osoczowej –
która występuje w kompleksie z czynnikiem
VIII i bierze udział w pierwotnej hemostazie,
warunkując prawidłową adhezję płytek do
uszkodzonej ściany naczyniowej.
Mutacja genu na chrom.12 → synteza vWF
upośledzona lub nieprawidłowe białko.
Wtórnym zjawiskiem jest niedobór cz.VIII (bez
ochrony vWF jest inaktywowany).
Zaburzeniu ulega hemostaza pierwotna i
wtórna

Objawy :
Krwawienia z błon śluzowych jamy ustnej i
przewodu pokarmowego
Krwotoki z nosa
Menorrhagia i metrorrhagia
Wybroczyny na skórze i błonach śluzowych
Siniaki
Krwotoki po urazach i w czasie zabiegów
chirurgicznych
Skłonność do krwawień, nasilająca się po
lekach zawierających kwas
acetylosalicylowy

Występujące objawy mają cechy
pośrednie między krwawieniem chorego
na hemofilię a wybroczynami krwawymi.
Często pierwszym poważnym objawem
są nadmiernie obfite krwawienia w
czasie pierwszych miesiączek u
dziewczyn.

Rozpoznanie:
Wywiad – krwawienia samoistne i po zabiegach u
chorego i rodziny
Badania przesiewowe:
APTT
Czas krwawienia
Czas okluzji w PFA-100 (plateled function analyzer)
Badania potwierdzające rozpoznanie:
Stężenie antygenu i aktywność vWF
Analiza multimerów vWF
Agregacja płytek pod wpływem rystocetyny

Badania laboratoryjne:
Wydłużony czas krwawienia przy
prawidłowej liczbie płytek krwi
Zmniejszone stężenie vWF i czynnika
VIII:C
Wydłużone czasy krzepnięcia i
kaolinowo-kefalinowy
Brak agregacji płytek przy obecności
rystocetyny

Różnicowanie:
Hemofilia A
Nabyta choroba von Willebranda
wywołana:
Autoprzeciwciałami neutralizującymi vWF
Adsorpcją vWF na komókach
nowotworowych
Nadmierną degradacją lub zmniejszeniem
syntezy vWF (np. niedoczynność tarczycy)

Leczenie:
1. desmopresyna
2. koncentrat cz.VIII z vWF
3. krioprecypitat
4. kw. traneksamowy
5. doustne środki antykoncepcyjne
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
Wyszukiwarka
Podobne podstrony:
CHOROBA VON WILLEBRANDA
Choroba von Willebranda
choroby von willebranda stom otg
skazy, 1 zdanie falszywe o chorobie von Willebranda
Choroba von Willebranda 2
CHOROBA VON WILLEBRANDA
Choroba von Willebranda
Diagnostyka i leczenie choroby von willebranda u psow
choroby von willebranda stom otg
Zespol Lesch-Nyhana i choroba von Girkego
Patogeneza i objawy ch von Willebranda
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
więcej podobnych podstron