KINETYKA
CHEMICZNA
Szybkość =
Zmiana
stężenia
czas
v =
±
c
t
v = – =
dc
dt
dx
dt
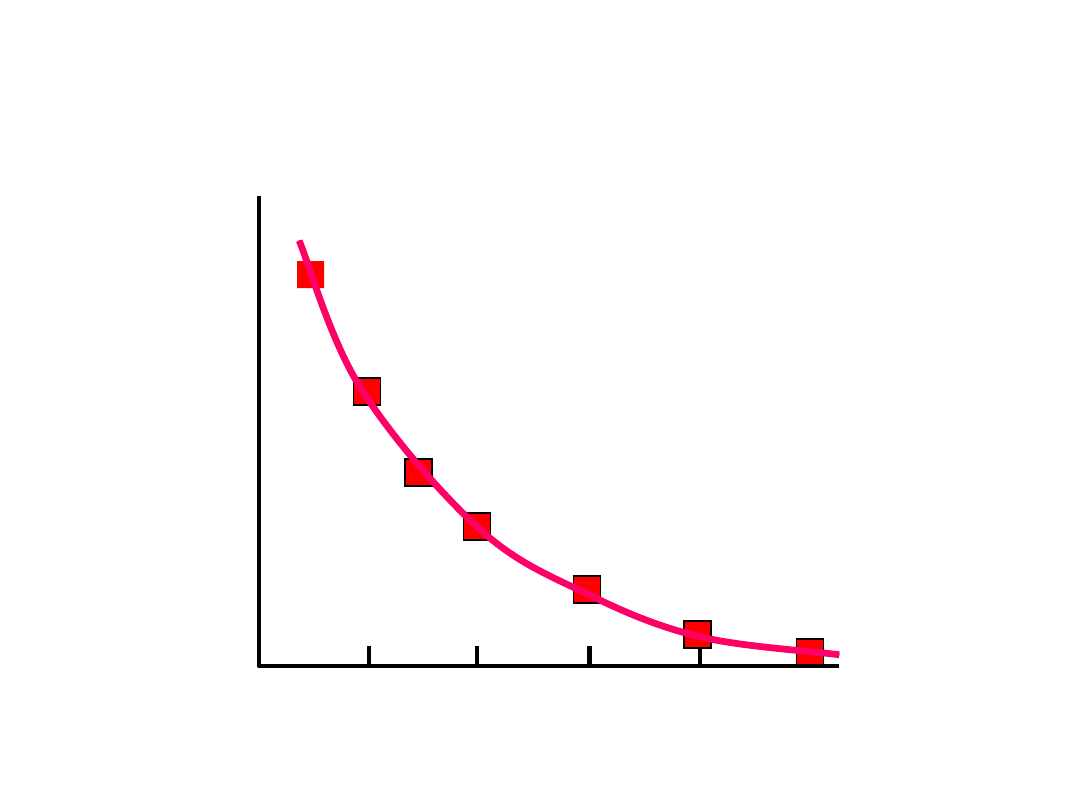
S
tę
że
n
ie
(
g
/m
l)
80
60
40
20
0
0 2 4 6 8
Czas (h)
A B
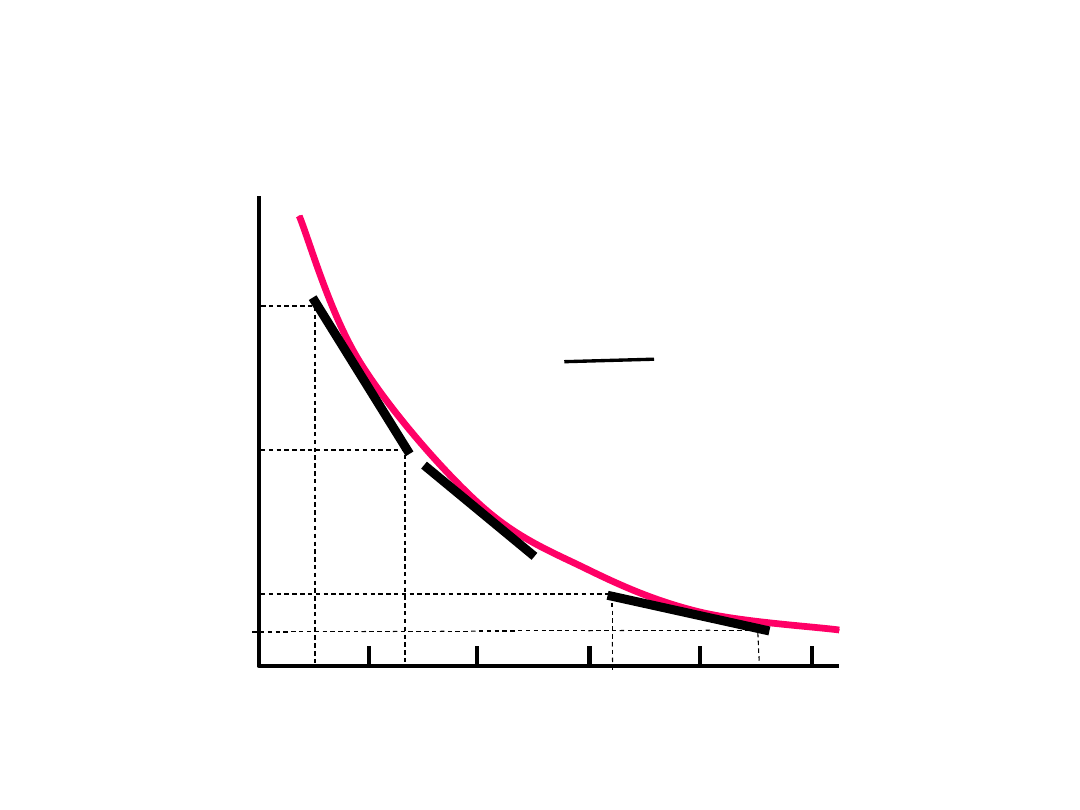
S
tę
że
n
ie
(
g
/m
l)
80
60
40
20
0
0 2 4 6 8
Czas (h)
v =
–
dc
dt

Przykład:
Jeżeli stężenie jonów wodorowych maleje o
0,01 M w ciągu 1 ns, to jaka jest średnia
szybkość reakcji? (mol/dm
3
)
Szybkość = 0,01 M / 1* 10
-9
s = 1,00 * 10
7
M/s
Szybkość =
Zmiana
stężenia
czas

v
n
i
i
C
v
Rzędowość reakcji
Br
2
= Br + Br
v = k * [Br
2
]
reakcja pierwszego
rzędu
H
2
+ I
2
= 2HI
v = k * [H
2
] * [I
2
]
całkowity rząd
reakcji 2
2NO + Cl
2
= 2NOCl
v = k * [NO]
2
* [Cl
2
]
całkowity rząd
reakcji 3

H
2
+ Br
2
= 2HBr
Całkowity rząd reakcji nieokreślony
]
[
]
[
]
][
[
'
2
2
/
3
2
2
HBr
k
Br
Br
H
k
v
Natomiast, gdy [Br
2
] >> k’ * [HBr]:
2
/
1
2
2
]
][
[
Br
H
k
v
Równania kinetyczne
Reakcja zerowego rzędu
A B
v = k [A]
0
k
x
a
k
dt
dx
v
0
)
(
Po scałkowaniu:
kt
x
k
A
2
0
2
/
1
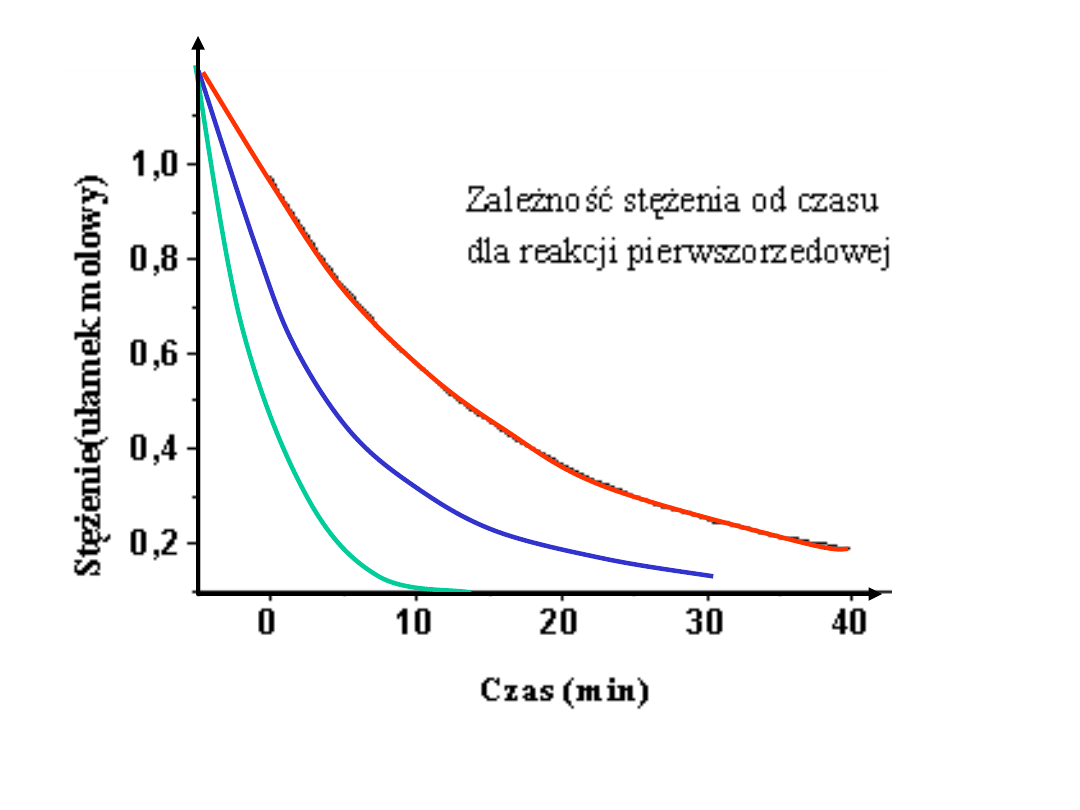
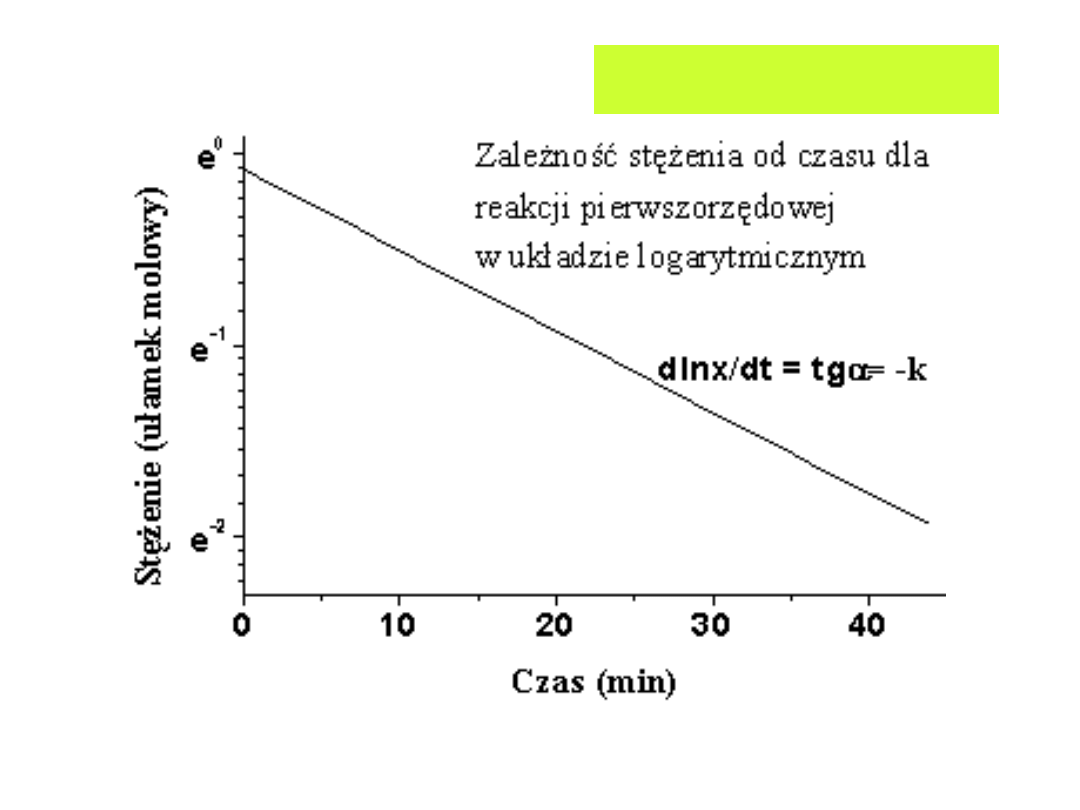
Reakcja pierwszego rzędu:
)
(
x
a
k
dt
dx
v
Po scałkowaniu:
x
a
a
t
k
ln
1
x
a
a
t
k
log
303
,
2
kt
a
x
a
ln
)
ln(

Okres półtrwania (połowicznej przemiany)
reakcji rzędu pierwszego:
k
693
,
0
2
/
1

Kinetyka rozpadu promieniotwórczego
Przykład:
Nuklid
226
88
Ra
222
86
Rn +
4
2
He
Wiedząc, że 1 g radu emituje w ciągu 1 s
3,70*10
10
cząstek oblicz:
a) Stałą szybkości rozpadu,
b) Czas połowicznego rozpadu (okres
półtrwania),
c) Procent początkowej ilości radu, który
pozostaje w próbce po upływie 1000 lat,
d) Czas po którym rozpadnie się 99,9%
początkowej ilości radu.

a) Wyznaczane stałej szybkości rozpadu
1 g radu zawiera 6,023*10
23
/206,05 =2,664
*10
21
atomów.
Liczbie rozpadów odpowiada - N.
k = 3,70*10
10
/2,664*10
21
*1s = 1,388*10
-11
s
-1
Ponieważ 1 rok = 3,156*10
7
s,
to k = 4,38*10
-4
rok
-1
kN
t
N
)
*
/(
t
N
N
k

b) Czas połowicznego rozpadu obliczamy ze
wzoru:
1/2
= 0,6931 / 4,38*10
-4
rok
-1
= 1580 lat
k
693
,
0
2
/
1

c) Liczbę atomów, która nie uległa rozpadowi,
obliczamy ze wzoru:
W próbce pozostanie 64,5% początkowej ilości
(liczby atomów) radu.
645
,
0
)
/
(
438
,
0
1000
*
10
*
38
,
4
)
/
ln(
*
)
/
ln(
0
1
4
0
0
N
N
lat
rok
N
N
t
k
N
N

d) Obliczamy czas, po którym rozpadnie
się 99,9% początkowej ilości radu:
lat
t
rok
k
N
N
t
158000
10
*
38
,
4
/
)
999
,
0
1
ln(
/
)
/
ln(
1
4
0

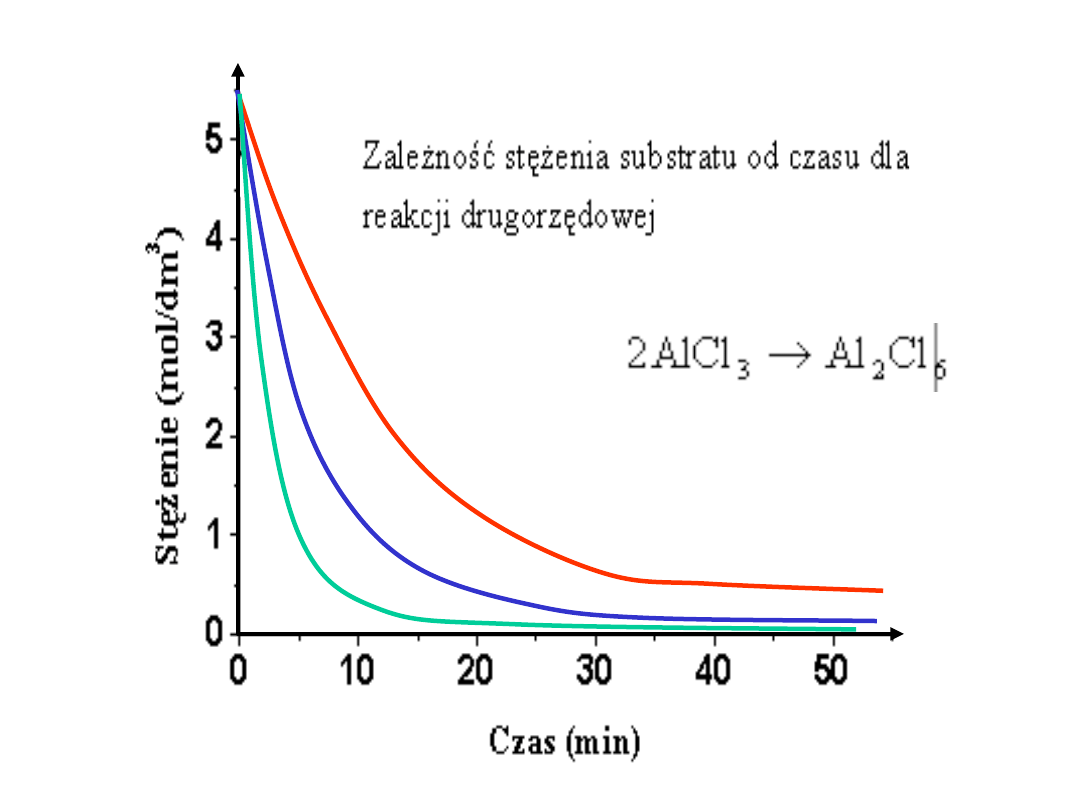
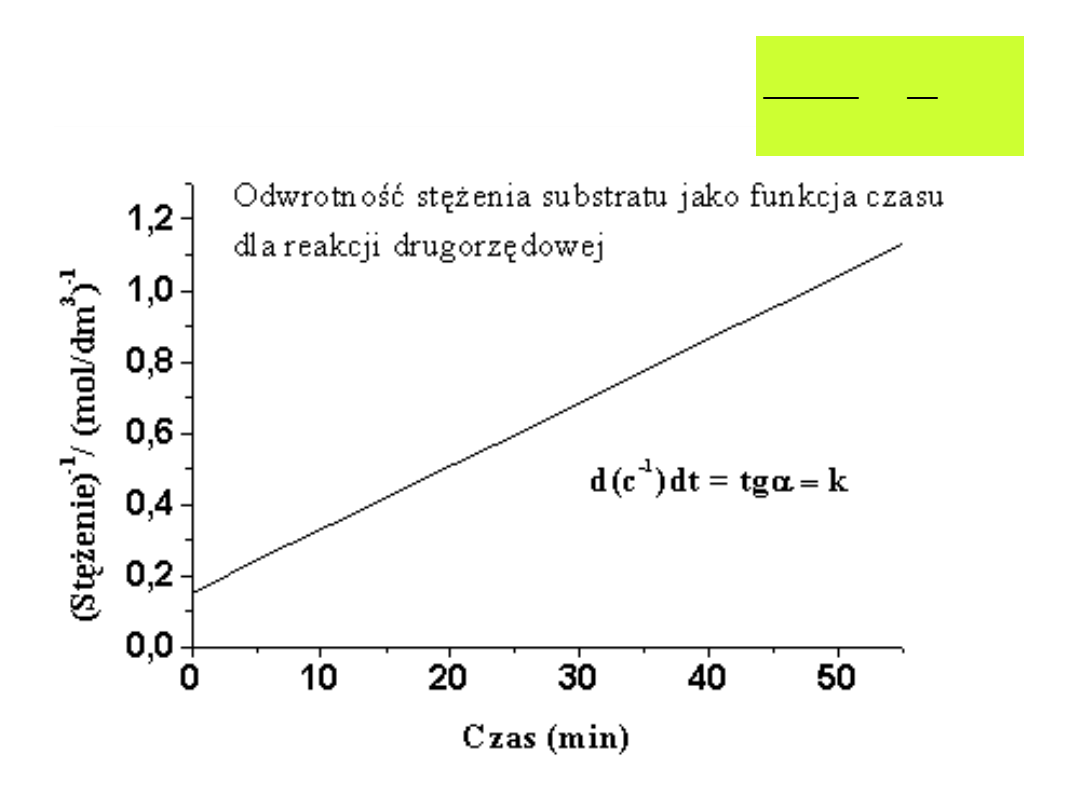
Reakcja drugiego rzędu (przypadek, gdy
stężenia obu reagentów są jednakowe)
Po scałkowaniu:
Okres półtrwania:
2
)
(
x
a
k
dt
dx
v
kt
a
x
a
x
a
a
x
t
k
1
1
)
(
1
ka
1
2
/
1

Reakcja trzeciego rzędu (przypadek, gdy
stężenia obu reagentów są jednakowe)
Po scałkowaniu:
lub:
Okres półtrwania:
3
)
(
x
a
k
dt
dx
v
2
2
1
)
(
1
2
1
a
x
a
t
k
kt
a
x
a
2
1
)
(
1
2
2
2
2
/
1
2
1
ka
Metody wyznaczania rzędu reakcji
1) Podstawiania do wzoru:
t
x
k
x
a
a
t
k
log
303
,
2
)
(
1
x
a
a
x
t
k
2
2
1
)
(
1
2
1
a
x
a
t
k
Rząd:
0
1
2
3
2 Graficzna:
kt
x
kt
a
x
a
ln
)
ln(
kt
a
x
a
1
1
kt
a
x
a
2
1
)
(
1
2
2

3) Ostwalda
1
2
/
1
1
n
a
A B
Energi
a
Postęp
reakcji
K
L
M
E
1
H
E
2
Teoria zderzeń aktywnych

Liczba cząsteczek o energii równej lub
większej niż energia aktywacji dana jest
rozkładem
statystycznym
Maxwella-
Boltzmanna:
kT
E
e
N
N
0
*
N* jest liczbą cząsteczek o energii
większej niż energia aktywacji E
a
,
N
0
całkowitą liczbą cząsteczek,
k stała Boltzmanna
T temperatura w skali bezwzględnej
(Kelvina)
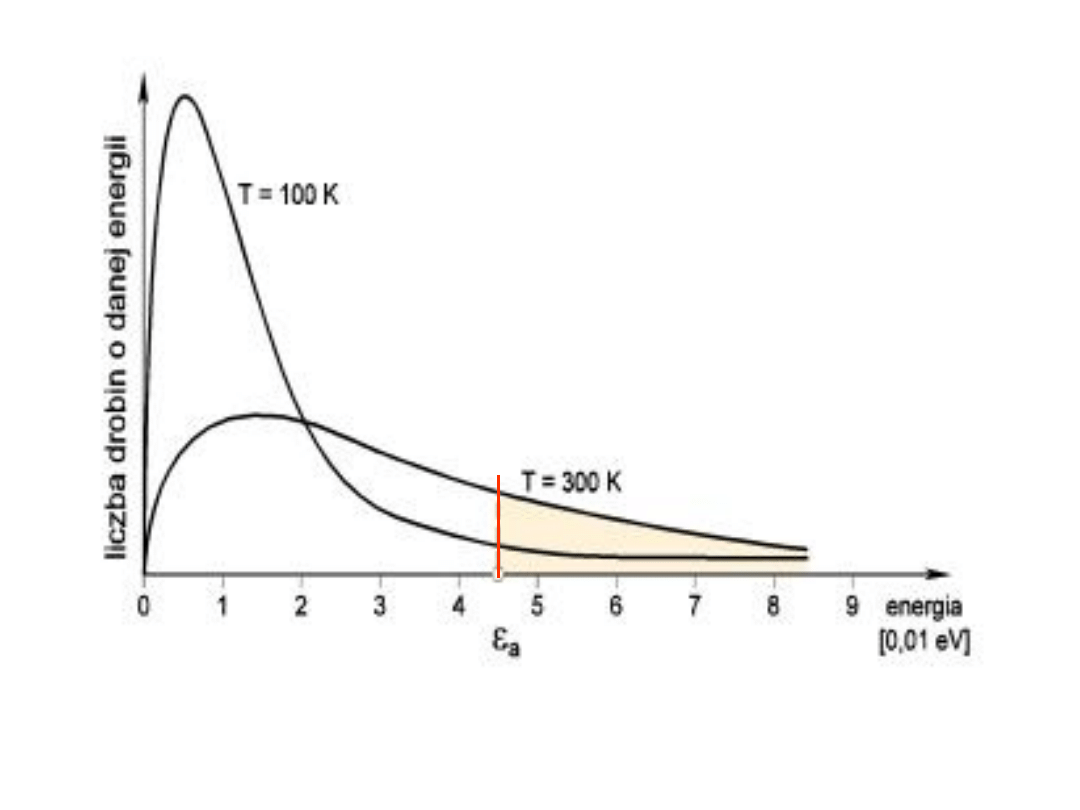
Rozkład prawdopodobieństwa występowania
cząsteczek o energii E
Szybkość
reakcji
chemicznej
jest
proporcjonalna do ilości zderzeń aktywnych,
a więc do ilości cząsteczek o energii równej
lub większej od energii aktywacji E
a
:
kT
E
a
e
N
k
N
k
v
0
0
0
*
Jeżeli liczbę cząsteczek zastąpimy liczbą moli
(n
0
) to:
RT
E
a
e
n
k
n
k
v
0
0
0
*
Gdzie:
A
N
N
n
/
A
kN
R
RT
E
a
e
n
k
n
k
v
0
0
0
*
A
B
RT
E
a
e
v
]
[A
]
[A
k
v
RT
E
a
e
k
RT
E
a
e
k
k
0
RT
E
a
e
k
k
0
RT
E
k
k
a
0
ln
ln
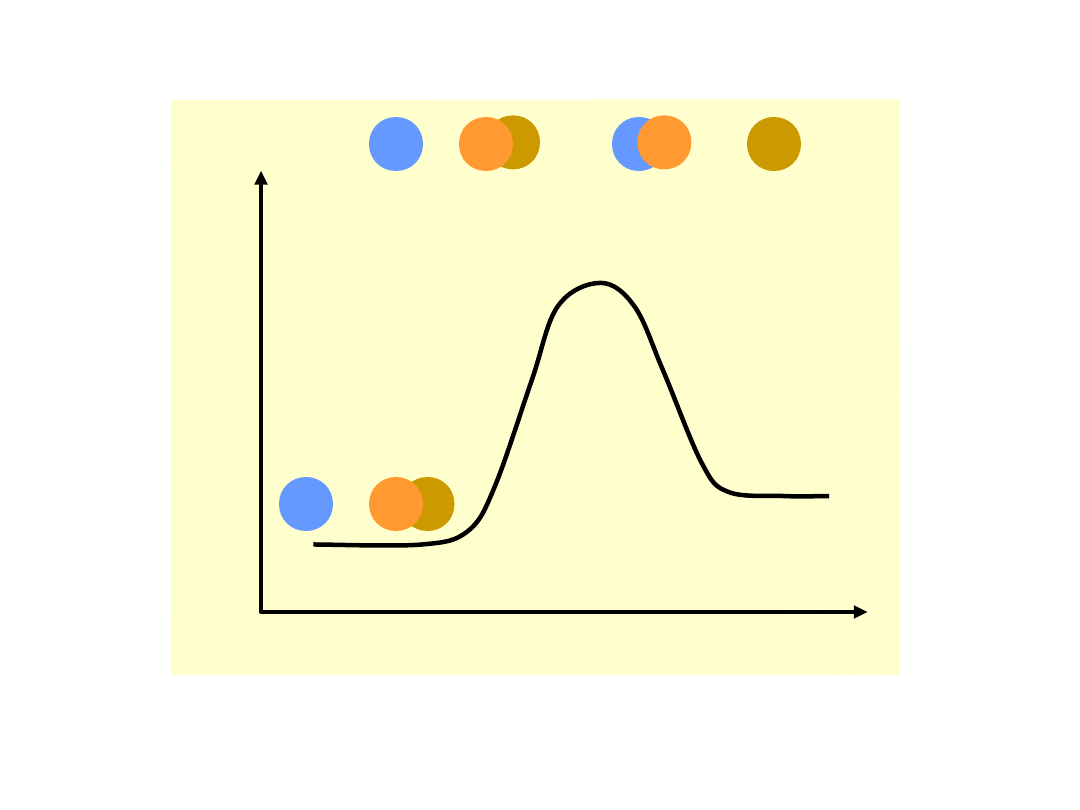
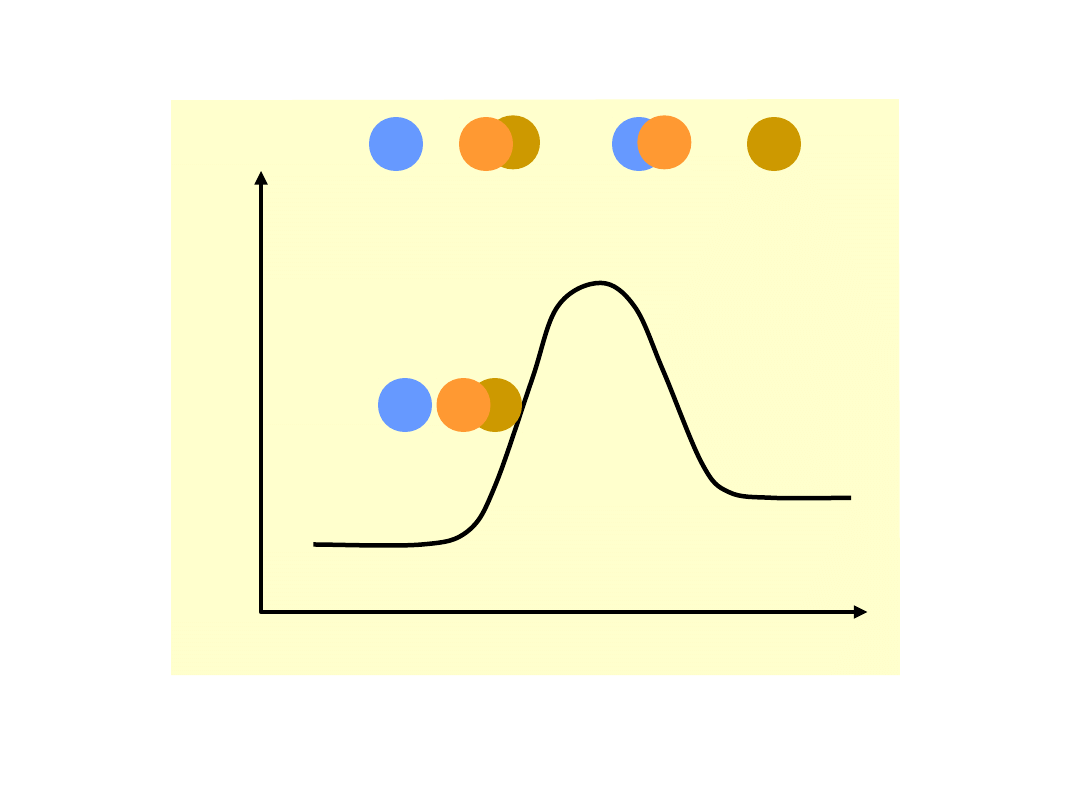
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
Teoria kompleksu aktywnego (stanu
przejściowego)
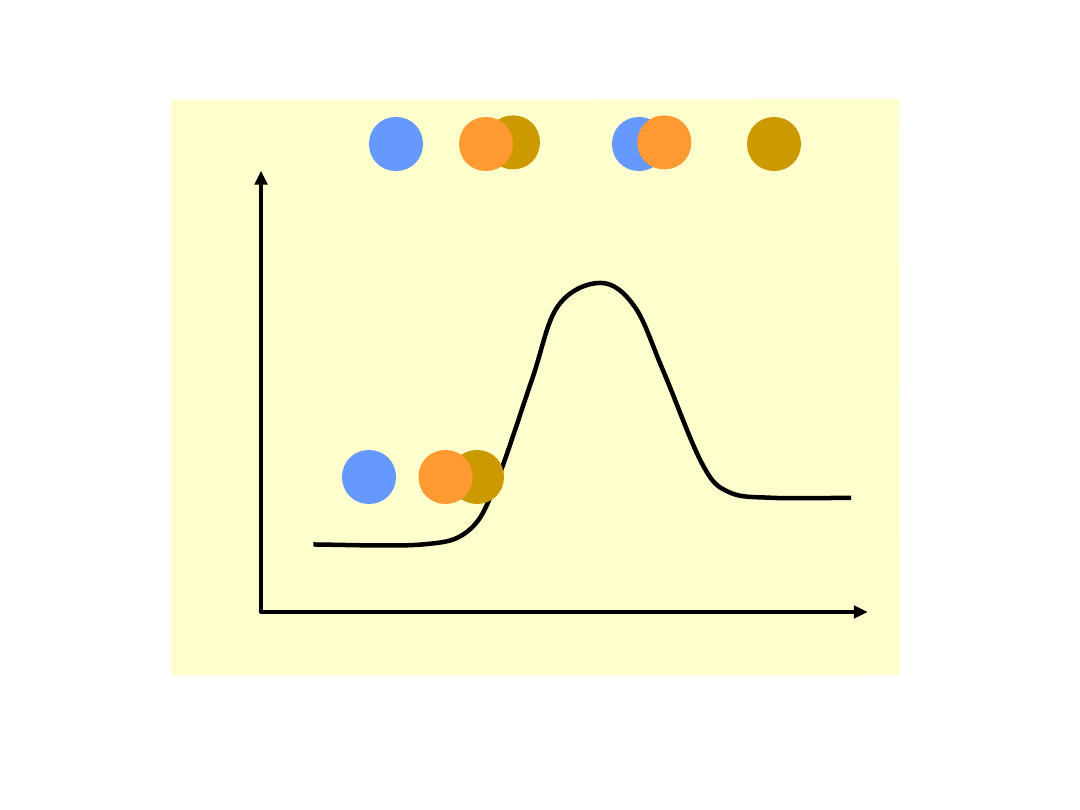
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
Energia
potencja
lna
Bieg reakcji
Komple
ks
aktywn
y
A + BC AB + C
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
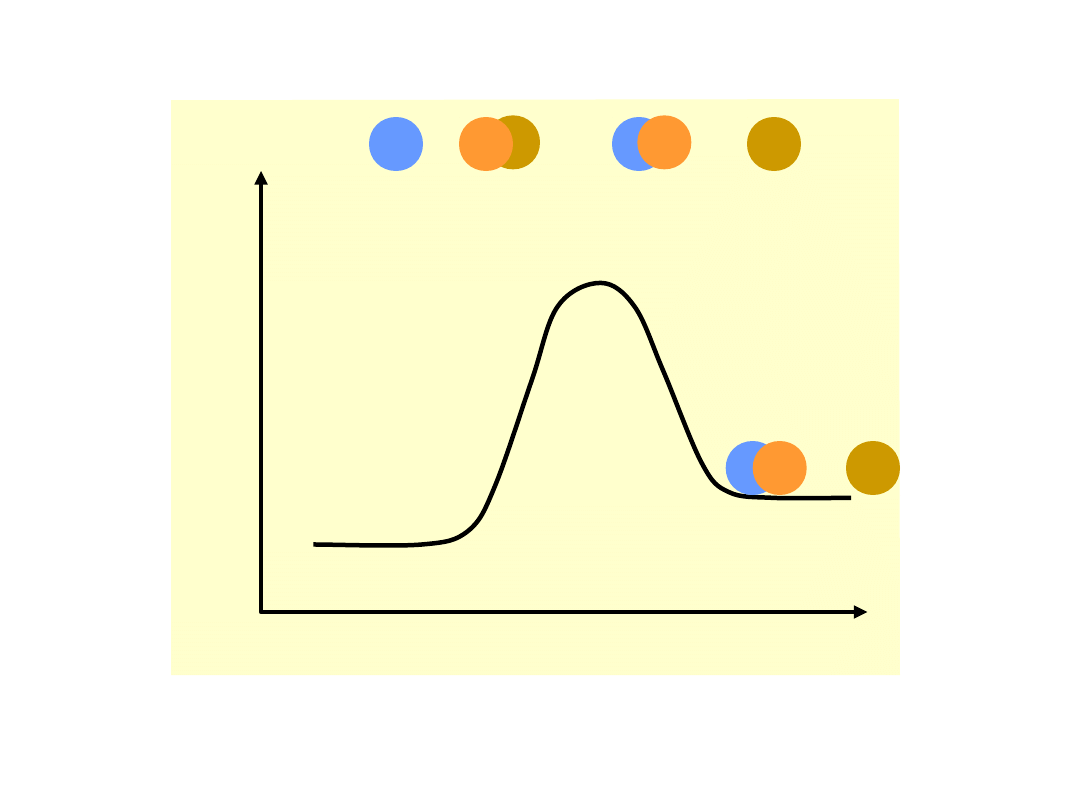
Energia
potencja
lna
Bieg reakcji
A + BC AB + C
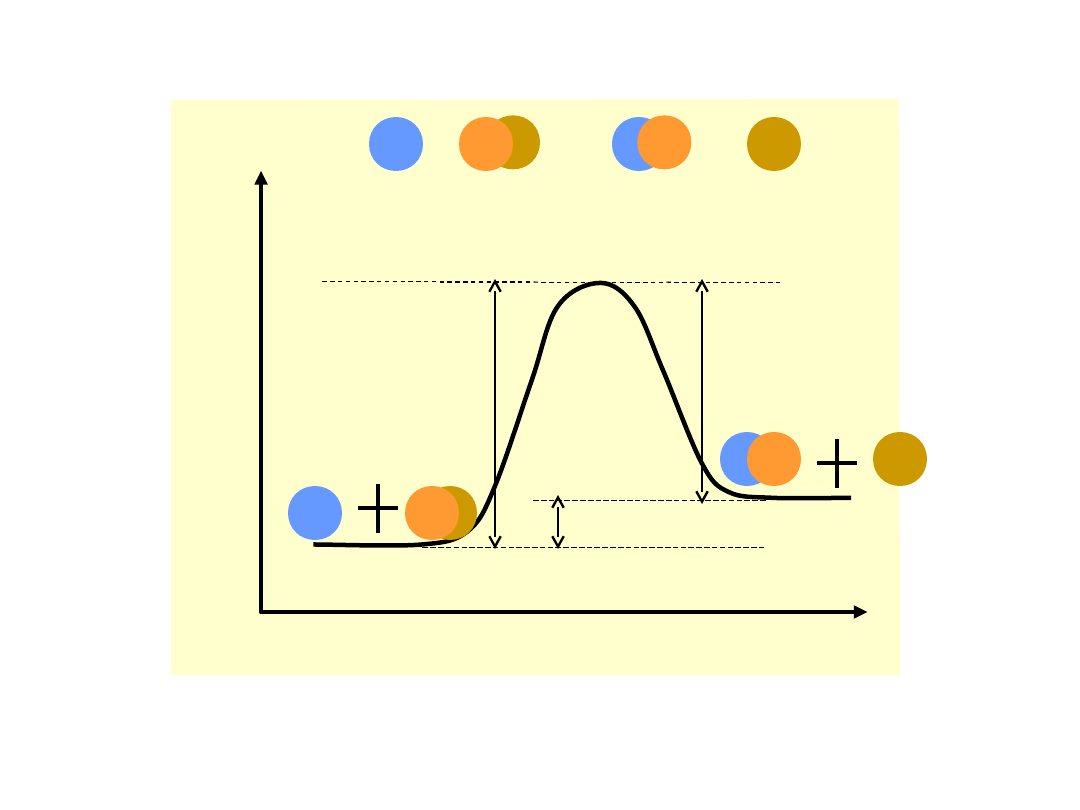
Energia
potencja
lna
Bieg reakcji
H
E
a1
E
a2
A + BC AB + C
Energia
aktywacji
E
a1
jest
równa
wysokości bariery energetycznej, którą
muszą pokonać reagujące cząsteczki na
drodze reakcji, E
a2
– energia aktywacji
reakcji odwrotnej. Różnica pomiędzy
stanem
energetycznym
produktów
i
substratów E jest równa ciepłu reakcji
H.
Koncepcja kompleksu aktywnego pozwala
zdefiniować energię aktywacji,
cząsteczkowość reakcji, oraz wyprowadzić
wyrażenie opisujące wpływ temperatury na
szybkość reakcji .

A + BC AB + C
A + BC ABC* AB + C
]
][
[
]
[
BC
A
ABC
K
]
][
[
]
][
[
]
[
BC
A
k
v
BC
A
K
v
ABC
v
PROD
PROD
PROD
Z ostatnich dwóch zależności wynika
proporcjonalność stałej równowagi tworzenia
kompleksu aktywnego i stałej k.

Stała równowagi K
jest związana z entalpią
swobodną równaniem:
RT
H
R
S
k
e
k
e
K
S
T
H
G
e
K
K
RT
G
RT
S
T
H
RT
S
T
H
RT
G
ln
ln
/
)
(
/
)
(
/

Mechanizm reakcji chemicznej
Cząsteczkowość reakcji
Mechanizm reakcji jest bardziej
skomplikowany niż to wynika z zapisu
stechiometrycznego
H
2
+ Br
2
= 2HBr
Przebieg reakcji:
1) Br
2
= 2Br*
2) Br* + H
2
= HBr + H*
3) H* + Br
2
= HBr + Br* itd..
4) H* + Br* = HBr

Każdy akt elementarny następuje w wyniku
efektywnych zderzeń dwóch lub najwyżej
trzech cząsteczek.
Przykładowo szybkość reakcji:
opisuje równanie kinetyczne:
2HI + H
2
O
2
I
2
+
2H
2
O
v = k · c
HI
·
c
H2O2

Taką postać równania kinetycznego, w którym
stężenie jodowodoru jest w potędze pierwszej,
można wyjaśnić przyjmując dwu-etapowy
mechanizm reakcji:
etap I – powolny
etap II – szybki
HI + H
2
O
2
HIO +
H
2
O
HIO + HI H
2
O
+ I
2

Reakcja jednoetapowa:
H
2
+ I
2
= 2HI
Reakcje mające co najmniej po jednym
produkcie pośrednim (czyli składające się co
najmniej z dwóch aktów elementarnych) są
nazywane reakcjami wieloetapowymi lub
złożonymi.
Tylko
na
drodze
badań
doświadczalnych możemy stwierdzić czy
dana reakcja jest prostą czy złożoną.
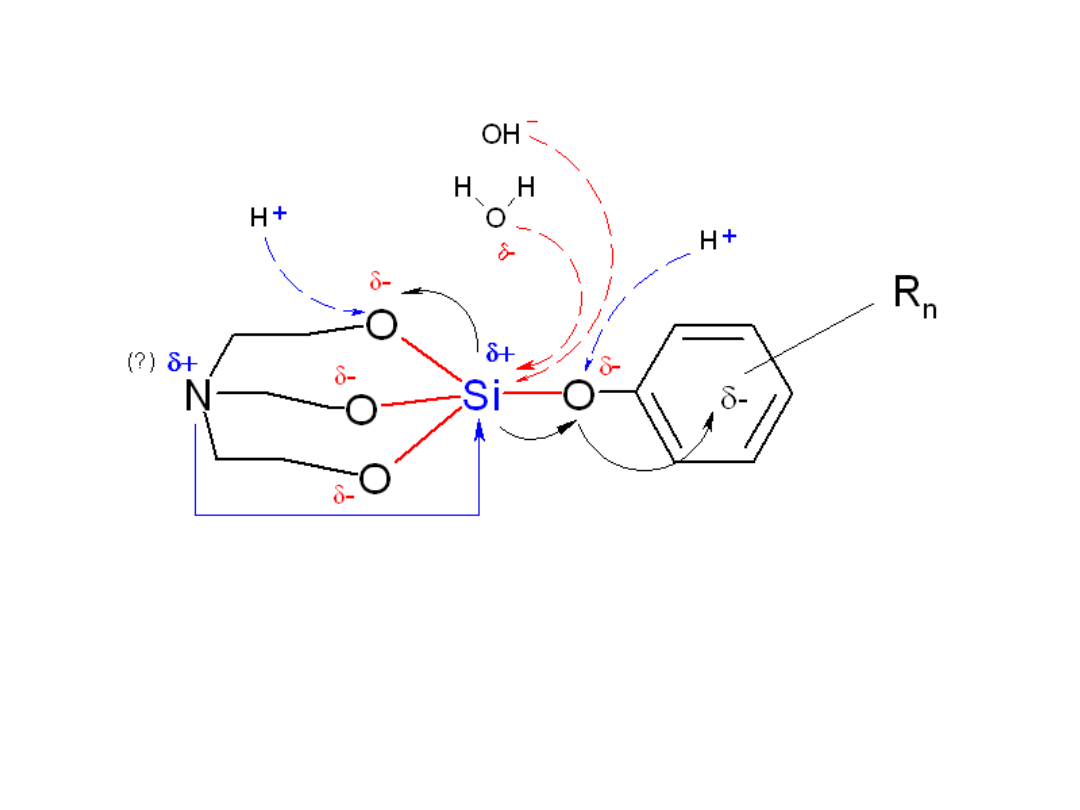
Struktura cząsteczki i
różnice w elektroujemnościach atomów
poszczególnych pierwiastków pozwalają
zaproponować podany powyżej rozkład
ładunków w cząsteczce związku.

Reakcje złożone
1) równoległe,
2) sprzężone,
3) następcze,
4) odwracalne.
v
1
v
2
x
a
a
t
k
k
ln
1
2
1
Ad 1) r-cje równoległe:
B
A
C
2
1
v
v
v

v = v
1
+ v
2
+ ........ + v
i
= Σv
i

Ad. 2) r-cje sprzężone:
a) A + B M
b) A + C N

Ad. 3) r-cje następcze:
A B C
v
1
v
2
2
1
1
1
1
v
v
v
2
1
1
1
1
v
v
v
2
1
2
1
1
v
v
v
v
v
2
1
2
1
v
v
v
v
v
i
v
v
v
v
1
......
1
1
1
2
1

Inna reakcja następcza:
1) A + B AB powoli
2) AB + B AB
2
szybko
3) A + 2B AB
2
r-cja
sumaryczna
v
2
>> v
1
2
1
2
1
v
v
v
v
v
v
= v
1
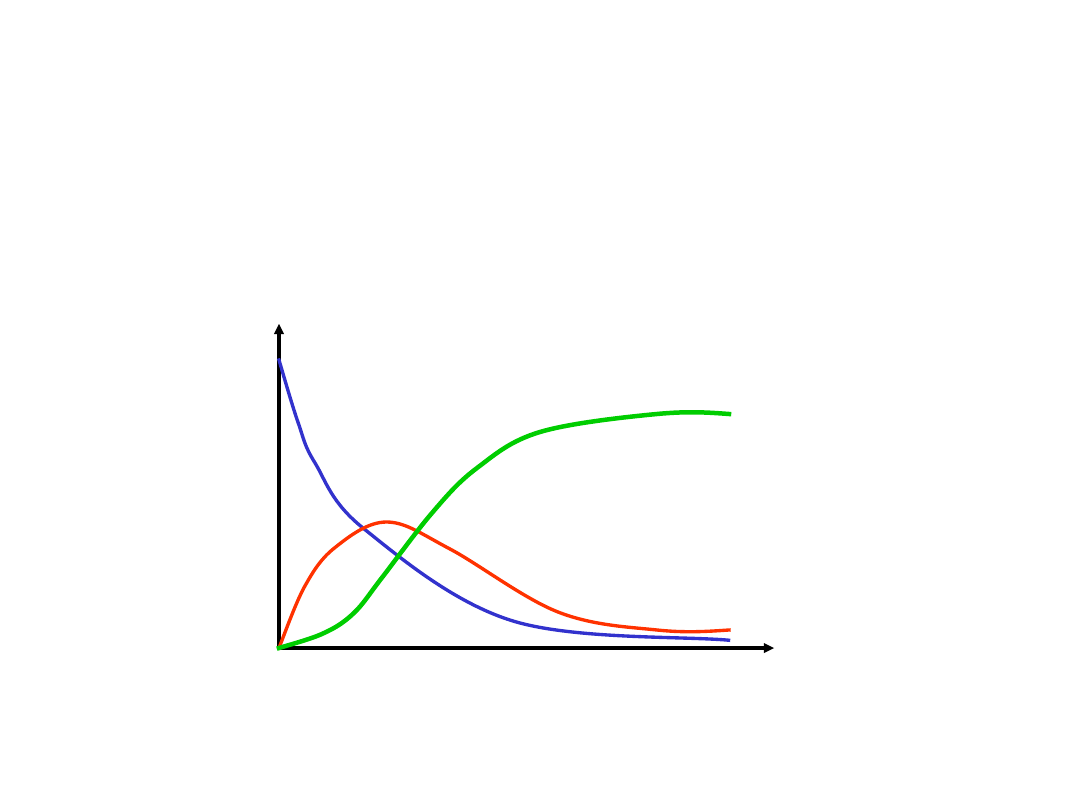
stężenie
czas
A
B
C
Zmiana stężeń reagentów w reakcji
następczej
A B C

cd. Ad. 3) r-cje następcze:
reakcje łańcuchowe
Ad. 4) r-cje odwracalne
rozważmy przypadek, gdy obie
reakcje są pierwszego rzędu:
A B
f
r
k
k
]
[
]
[
]
[
B
k
A
k
dt
A
d
r
f
Po scałkowaniu:
eq
eq
o
r
f
A
A
A
A
k
k
t
log
303
,
2
r
f
eq
eq
eq
eq
k
k
A
A
A
B
K
1

Szybkość reakcji jest cechą charakterystyczną
dla danego zespołu reagentów i zależy od
wielu czynników. Najważniejsze z nich to:
· stężenie reagentów,
· temperatura,
· ciśnienie (gdy reakcja przebiega w fazie
gazowej),
· promieniowanie elektromagnetyczne (r-
cje
fotochemiczne),
· rozwinięcie powierzchni (reakcje
powierzchniowe)
· obecność katalizatora lub inhibitora,
. środowisko reakcji (pH, elektrolity,
rozpuszczalniki
organiczne)
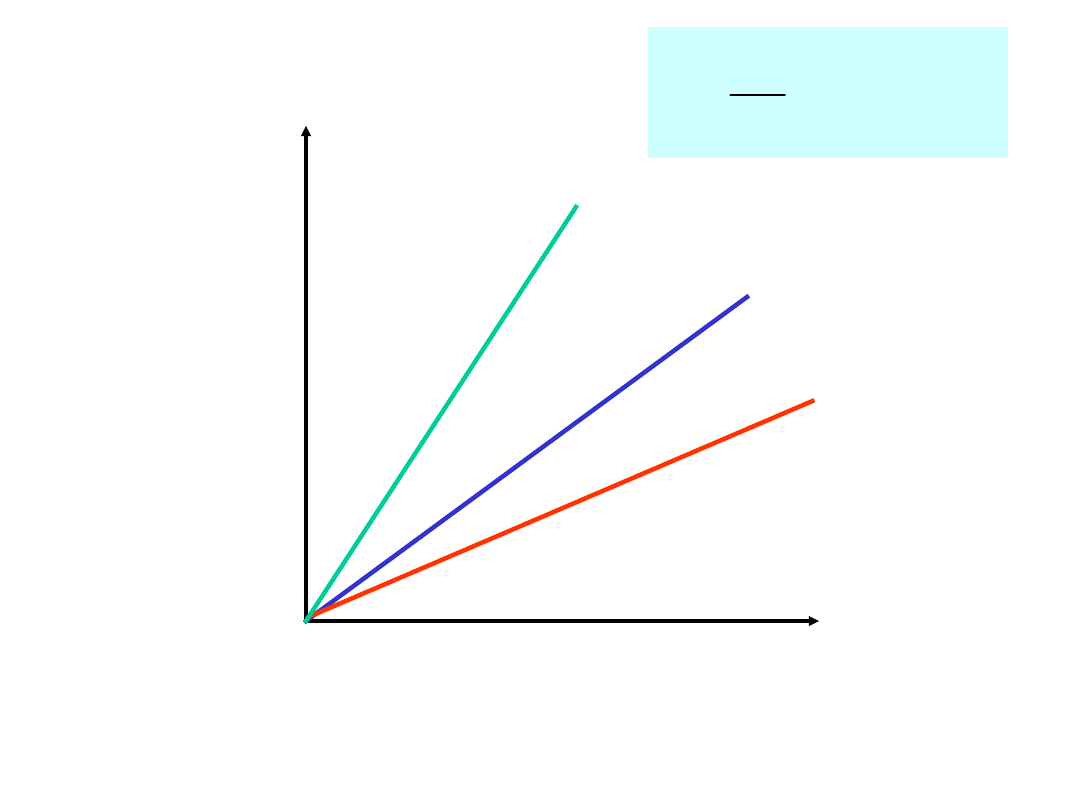
Wpływ
stężenia
k małe
k
średnie
k
duże
kt
a
x
a
ln
)
ln(
Wpływ
stężenia
Szybkoś
ć
Stężenie (a –
x)
Wpływ
stężenia
)
(
x
a
k
dt
dx
v
k małe
k
średnie
k
duże
k małe
k
średnie
k
duże
Wpływ
stężenia
kt
a
x
a
1
1
Wpływ
stężenia
2
)
(
x
a
k
dt
dx
v
Szybkoś
ć
Stężenie (a – x)
Wpływ
stężenia
k małe
k
średnie
k
duże

Wpływ temperatury na szybkość reakcji
chemicznej
zwykle opisuje się poprzez
przedstawianie
zależności stałej k od
temperatury
Empiryczna reguła van’t Hoffa:
T
K
T
T
K
T
k
k
v
v
Q
10
10
10

Równanie Arrheniusa
RT
E
a
a
a
e
k
k
RT
E
k
k
k
A
R
E
B
T
B
A
k
/
0
0
0
ln
ln
ln
'
'
ln

Równanie Arrheniusa
RT
E
a
a
a
e
k
k
RT
E
k
k
k
A
R
E
B
T
B
A
k
/
0
0
0
ln
ln
ln
'
'
ln
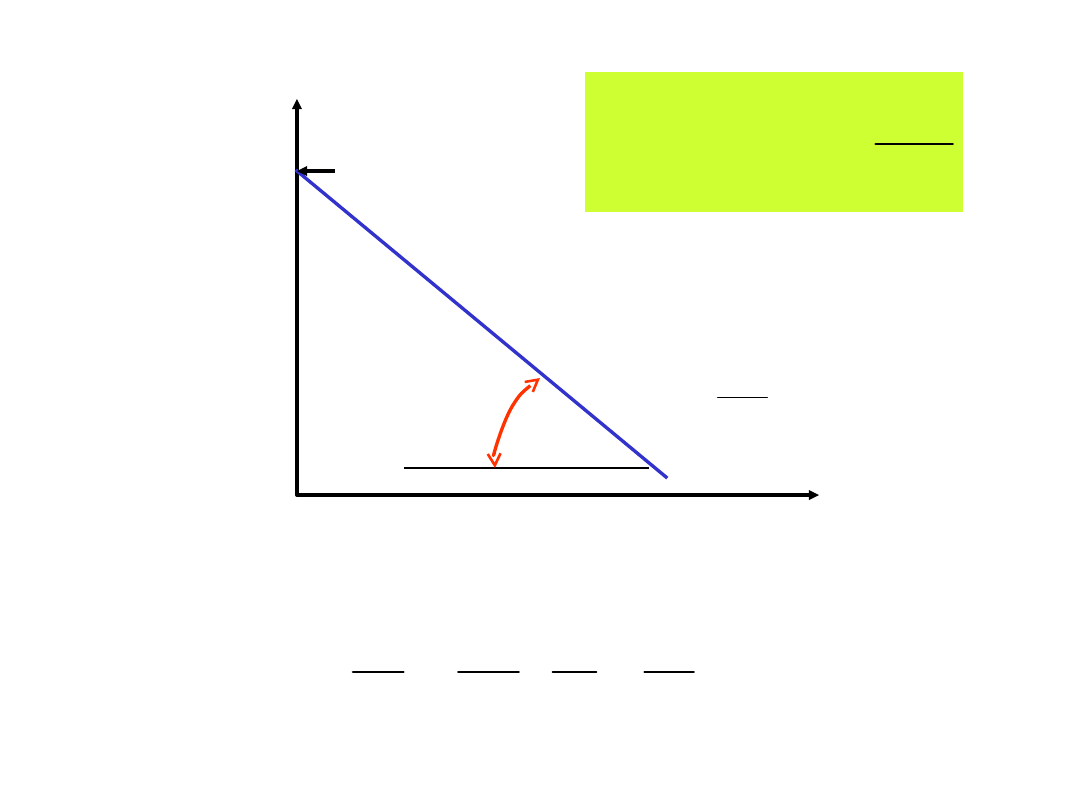
ln k
1/T
2
1
1
2
1
1
ln
T
T
R
E
k
k
a
R
E
a
tg
RT
E
k
k
a
0
ln
ln
ln k
0

Przykład:
Obliczyć ile razy wzrośnie stała szybkości
reakcji w wyniku ogrzania układu reagującego
od temperatury 0C do temp. 100C, jeżeli
energia aktywacji reakcji jest równa : a) 50
kJ/mol, oraz b) 200 kJ/mol.
ln k = ln k
o
– E
A
/ RT
ln (k
T2
)/(k
T1
) = = – E
A
/ R ( 1/T
2
– 1/ T
2
)
ln (k
T2
)/(k
T1
) = (E
A
/ R) ( T
2
– T
1
) / (T
1
* T
2
)
ln (k
373,2 K
)/(k
273,3 K
) = (50000/8,314) * 100 /
(373,2 * 273,2) = 5,898
czyli (k
373,2 K
)/(k
273,3 K
) = 365
Dla E
A
= 200 kJ/ mol (k
373,2 K
)/(k
273,3 K
) = 1,78 *
10
10
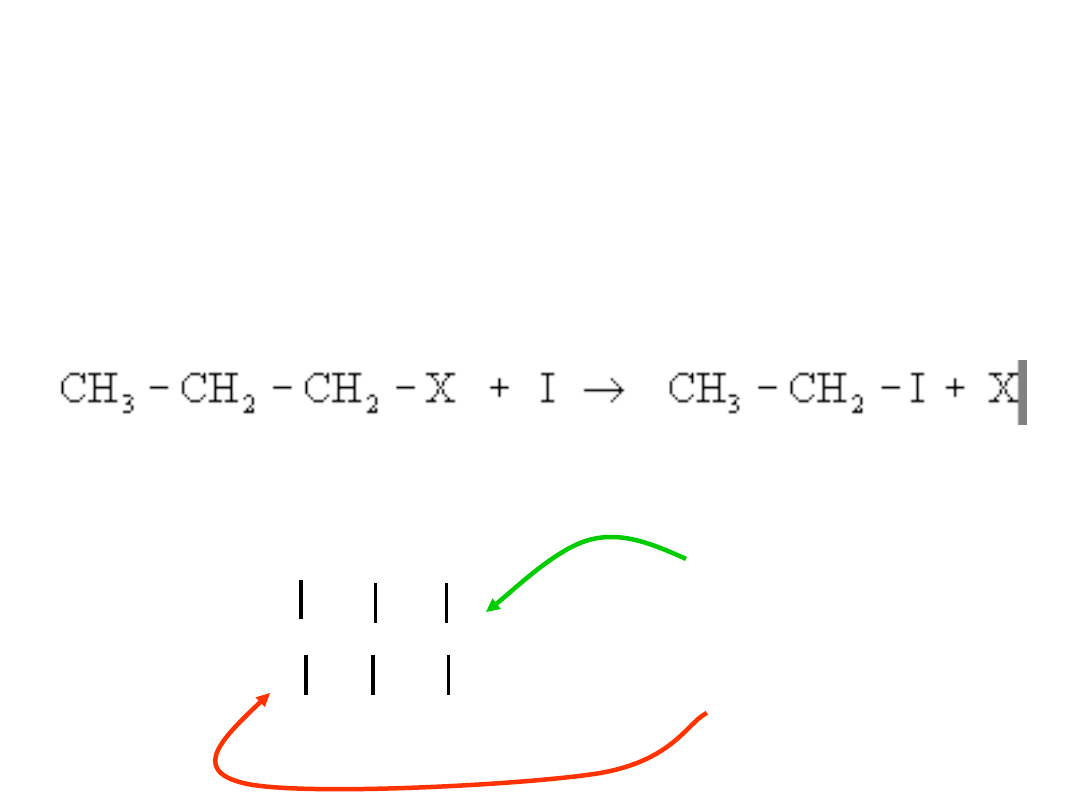
Wpływ orientacji cząsteczek na szybkość
reakcji chemicznej
Aby zderzenie było efektywne cząsteczki
muszą się zderzyć odpowiednimi miejscami,
np. w reakcji:
drobina I musi uderzyć atom węgla związany
z grupą X:
H H H
I
H – C – C – C – X
H H H
Zderzenie
efektywne
Zderzenie
nieefektywne

Katalizatorem danej reakcji nazywamy
substancję, która wprowadzona do układu
reagującego
zwiększa
szybkość
tej
reakcji, a sama nie ulega przemianom
chemicznym
w
ostatecznym
wyniku
reakcji.
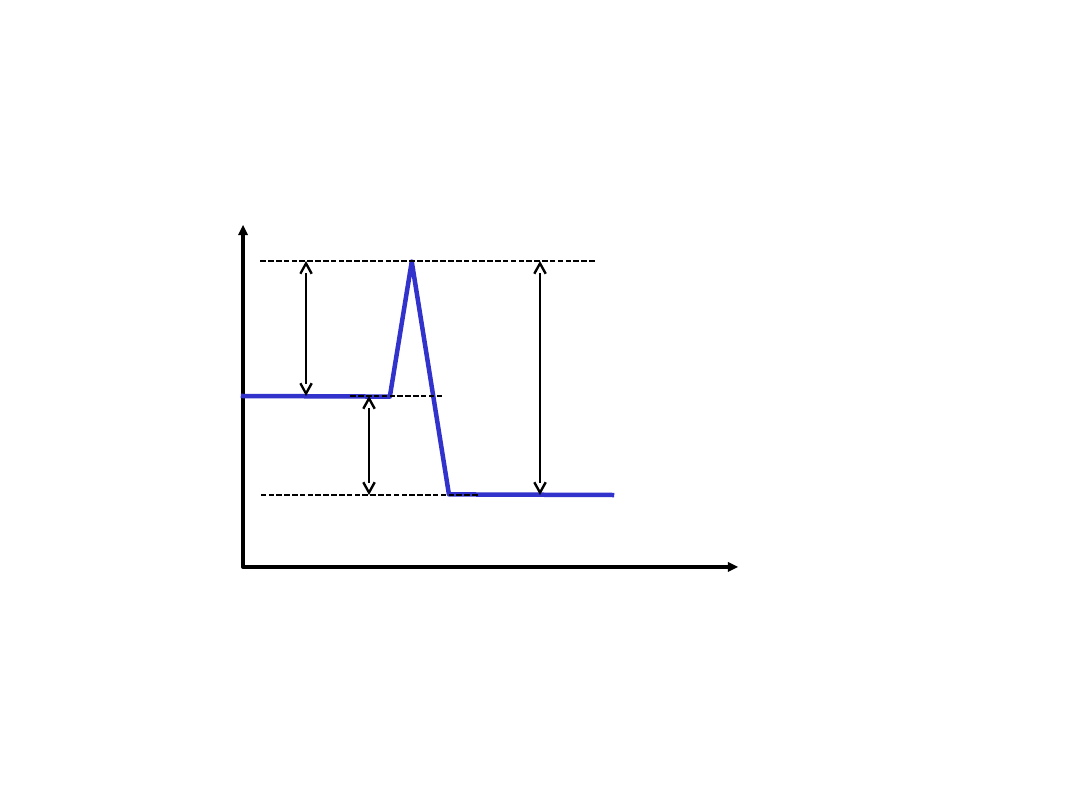
Wpływ katalizatora
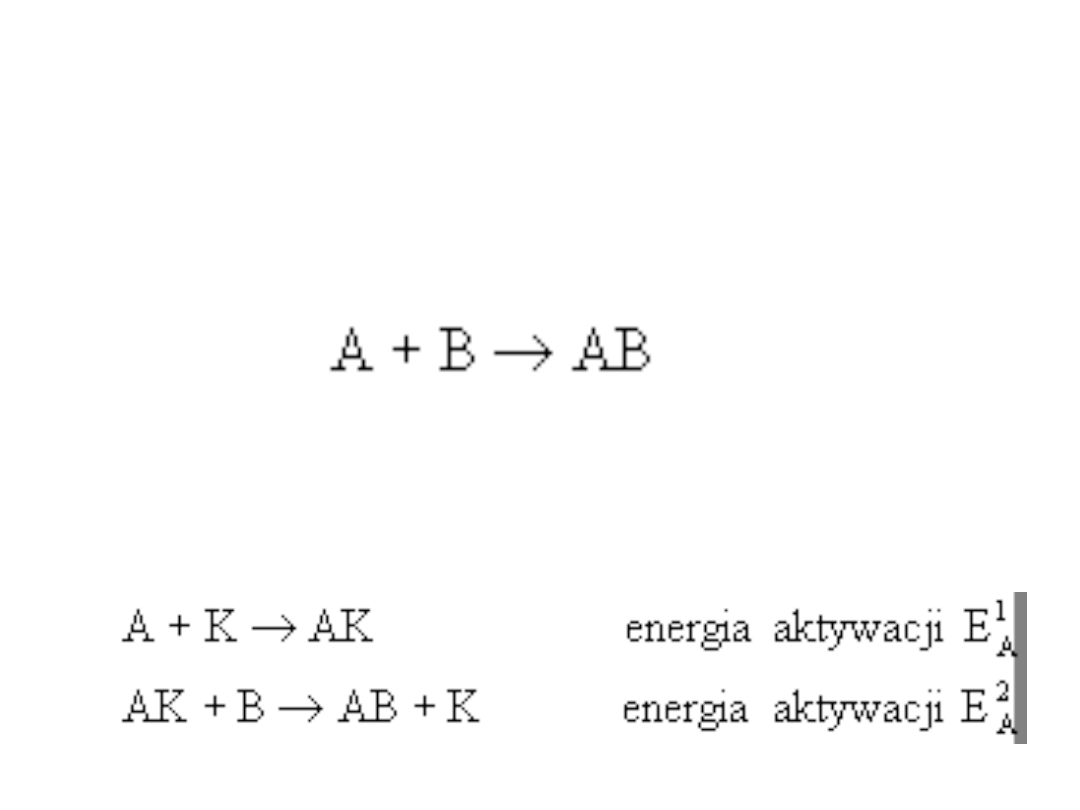
Przyśpieszenie reakcji przez katalizator
polega na zmniejszeniu energii aktywacji w
porównaniu z reakcją nie katalizowaną. Dla
reakcji zachodzącej bez katalizatora:
mamy energię aktywacji E
A
. Tą samą
reakcję
przebiegającą
przy
udziale
katalizatora K można opisać równaniami:
Wpływ katalizatora
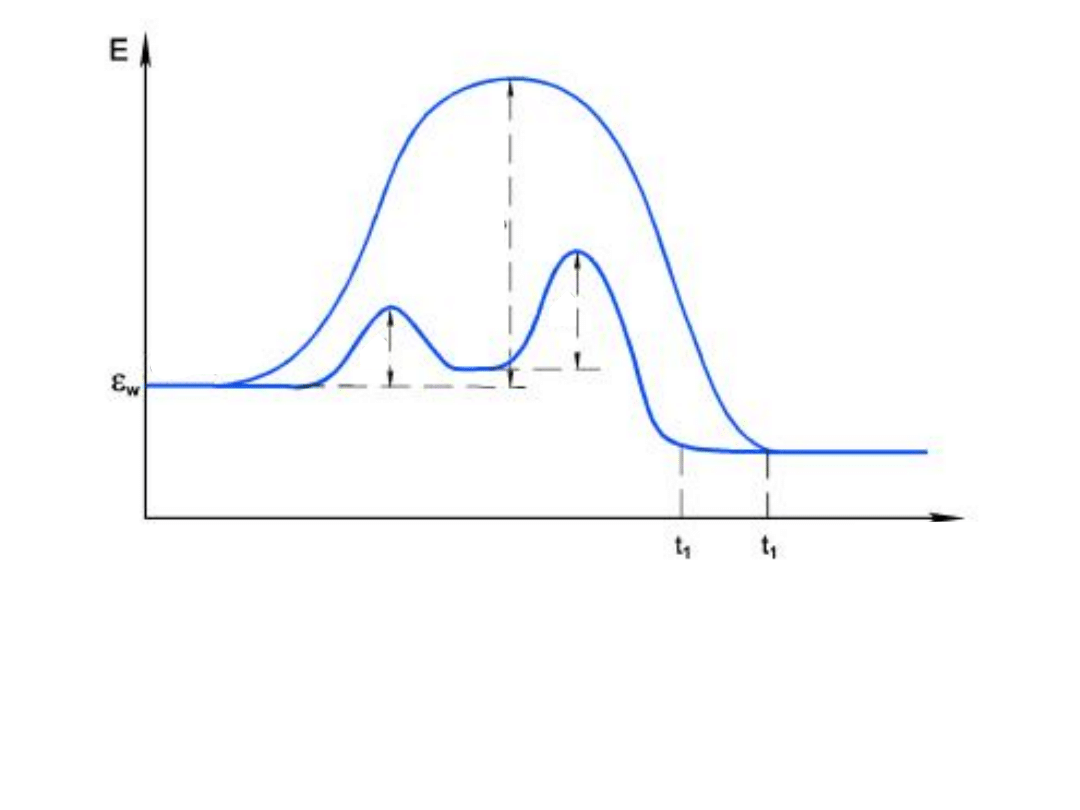
Jeżeli reakcja bez katalizatora wymaga energii
aktywacji E
A
to w obecności katalizatora K,
dwie reakcje wykazują energię aktywacji E
1
i
E
2
przy czym każda z nich jest mniejsza od E
A
AB, K
E
A
A, B,
K
E
1
E
2
Czas
Wpływ katalizatora
Kataliza homogeniczna
Katalizator
może
stanowić
jeden
ze
składników jednorodnej fazy (gazowej lub
ciekłej) w której zachodzi reakcja - mówimy
wówczas
o
katalizie
homogenicznej,
jednorodnej, jednofazowej.

Kataliza heterogeniczna
Jeśli katalizator stanowi odrębną fazę w
układzie reagującym, to katalizowana reakcja
przebiega na granicy faz i wówczas mamy do
czynienia
z
katalizą
niejednorodną
(heterogeniczną, wielofazową).
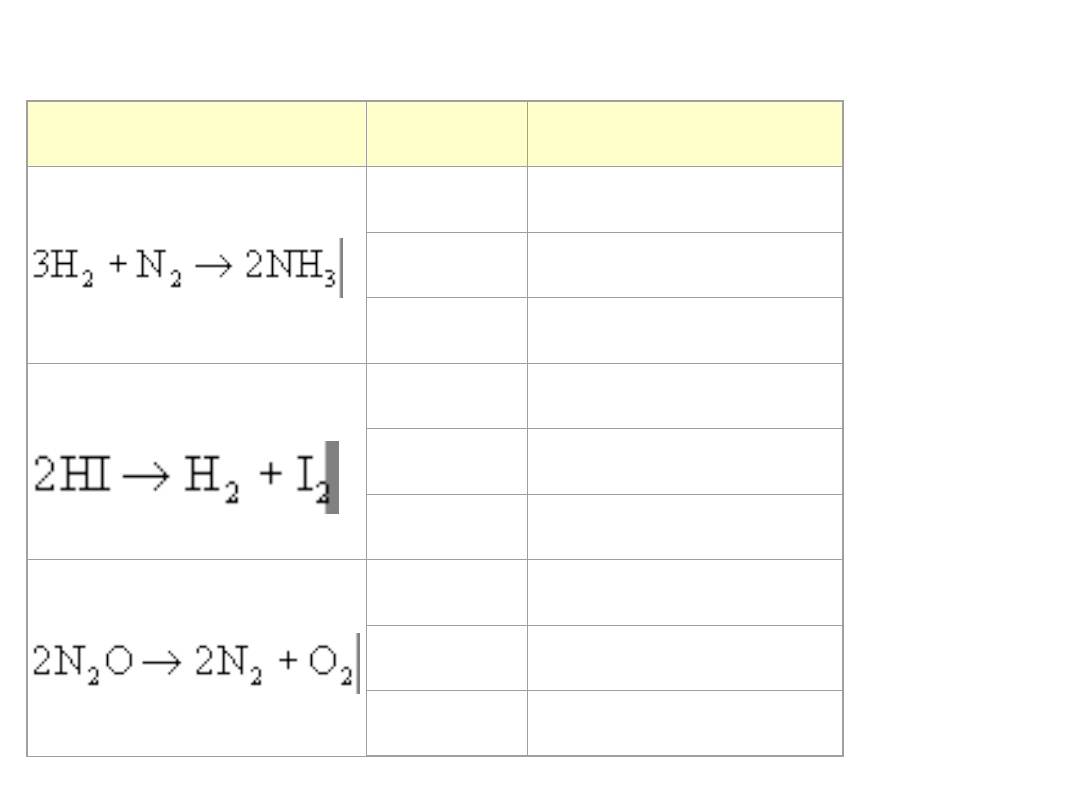
Wartości Ea (energii aktywacji) dla wybranych reakcji
Reakcja
Kat.
Ea [kJ/mol]
brak
335
wolfra
m
163
osm
197
brak
184
platyn
a
105
złoto
59
brak
247
platyn
a
138
złoto
121

Mechanizm katalitycznego uwodorniania
etylenu
C
2
H
2
zgodnie
z
reakcją:
Przebieg procesu odbywa się w czterech
etapach
CH
2
=CH
2
+ H
2
CH
3
-
CH
3
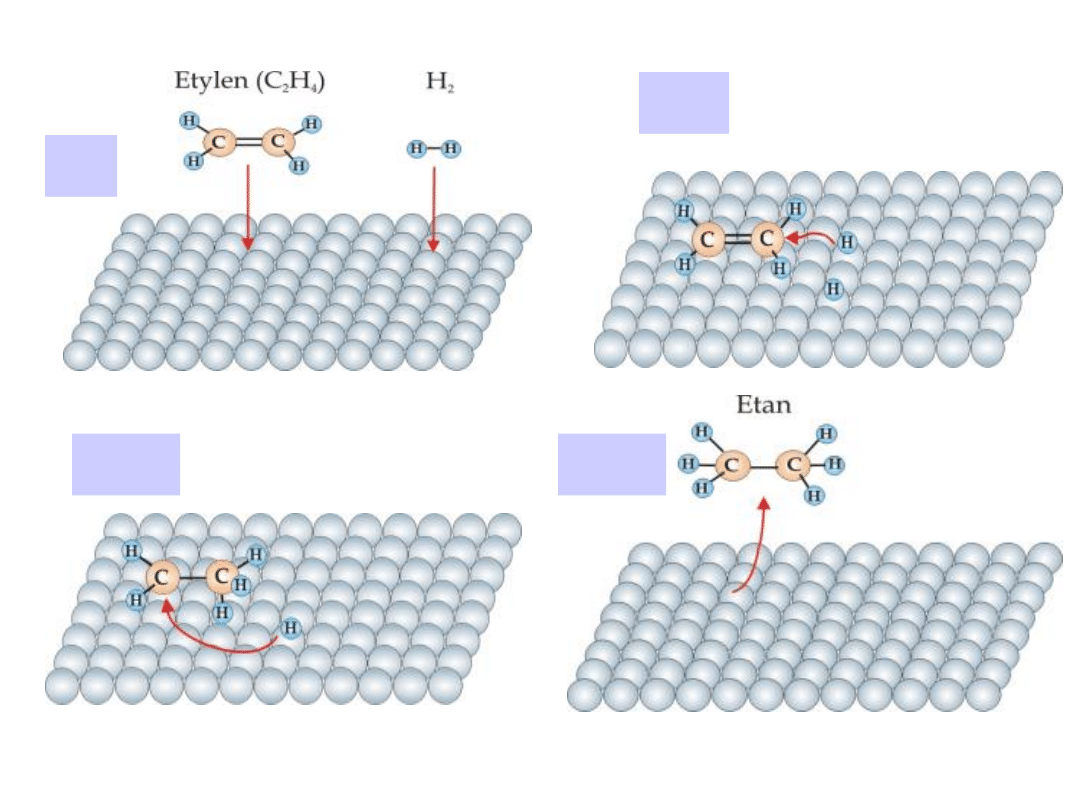
(I)
(II)
(III)
(IV)
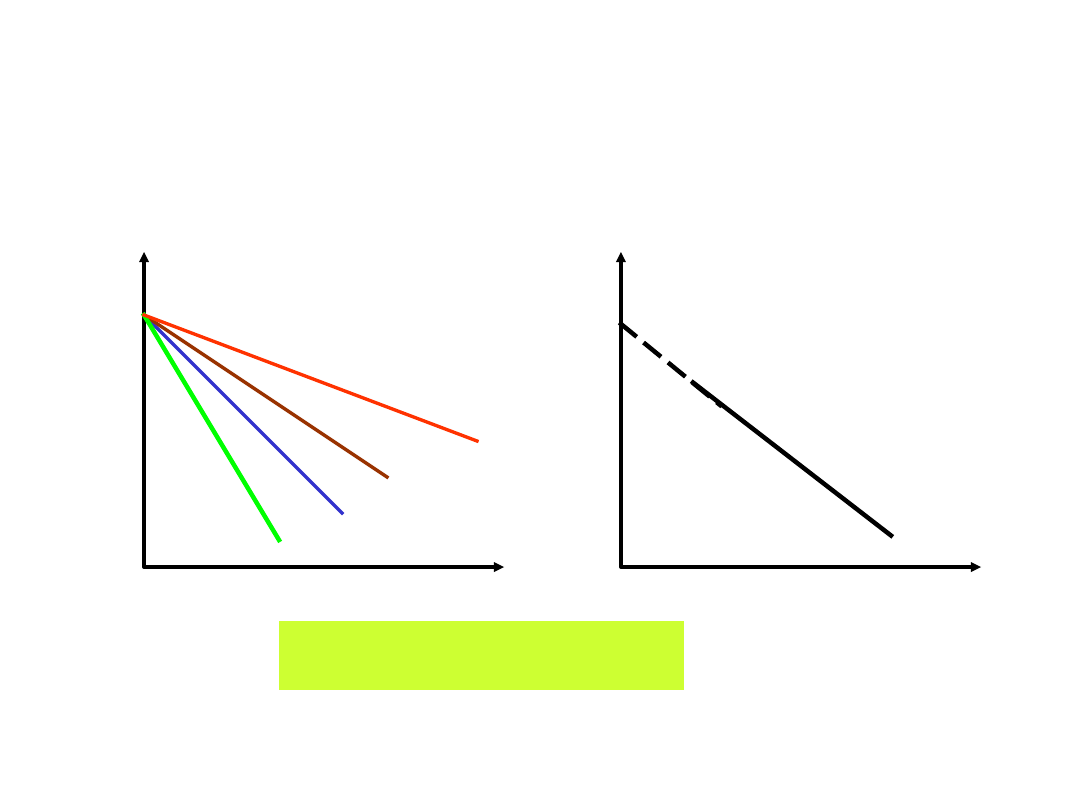
Badanie trwałości leków metodą
przyśpieszonego starzenia
log
c
czas
T
4
T
3
T
2
T
1
log k
1/T
kt
a
x
a
ln
)
ln(
Znając k można wyznaczyć czas
przechowywania leku

Kinetyka reakcji enzymatycznych
]
[
]
[
]
[
1
1
3
1
2
S
ES
E
k
v
P
E
ES
S
E
k
k
k
[E] – [ES] - stężenie wolnego enzymu
[S] - stężenie substratu
]
[
]
[
3
3
2
2
ES
k
v
ES
k
v

W stanie równowagi mamy:
3
2
1
v
v
v
Po podstawieniu:
]
[
]
[
]
[
]
[
]
[
3
2
1
ES
k
ES
k
S
ES
E
k
m
K
k
k
k
ES
S
ES
E
1
3
2
]
[
]
[
]
[
]
[
Jeżeli stan równowagi tworzenia kompleksu
ES ustala się bardzo szybko, wtedy k
2
>> k
3
i wzór upraszcza się:
oraz:
Jeżeli stężenie substratu jest odpowiednio
duże, wtedy wszystkie cząsteczki enzymu są
związane w kompleks ES, a szybkość reakcji
osiąga wartość maksymalną:
1
2
k
k
K
m
]
[
3
3
ES
k
v
v
]
[
3
max
E
k
v

Ostatnie dwa równania pozwalają na
podstawienie wyrażeń za [E] i [ES] w
równaniu na K
m
:
]
[
]
[
max
S
K
S
v
v
m
Stała K
m
(stała Michaelisa) jest marą
powinowactwa enzymu do substratu.
Stałą K
m
można wyznaczyć z powyższego
równania.
Równanie
Michaelisa
–
Menten
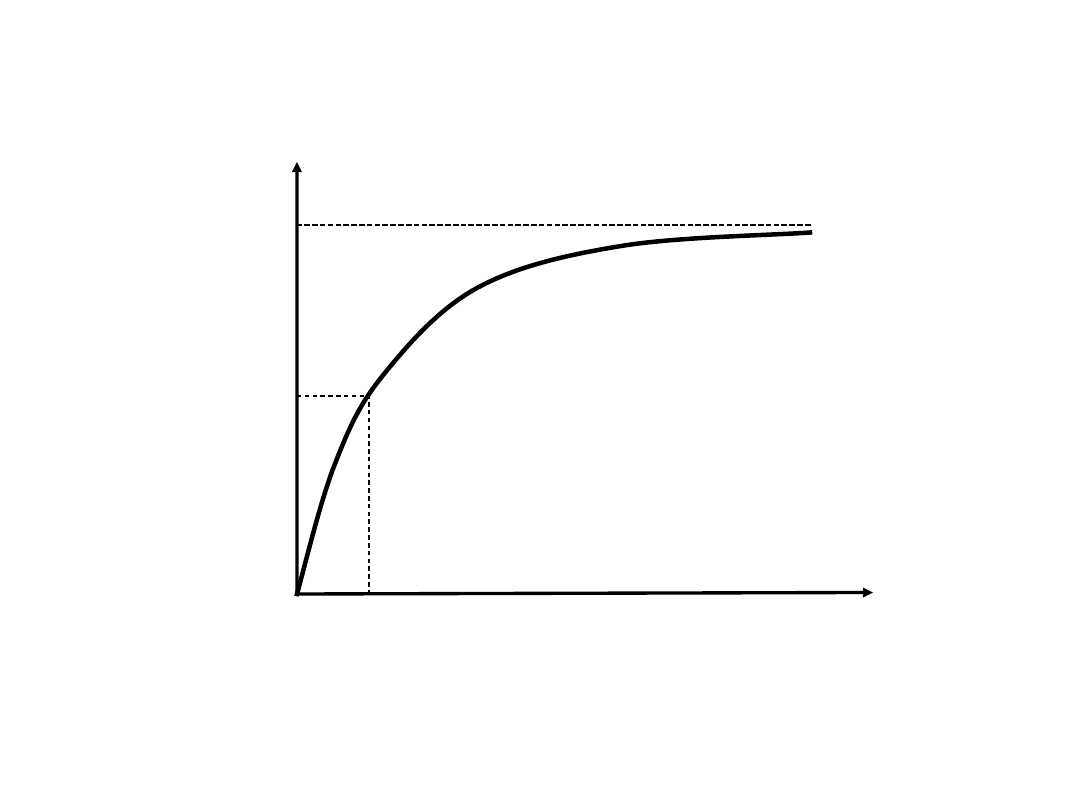
v
v
max
v
max
/
2
K
m
[S]
Graficzne
przedstawienie
równania
Michaelisa – Menten
]
[
]
[
1
max
S
v
S
K
v
m
max
max
1
]
[
1
v
S
v
K
v
m
Równanie Lineweavera –
Burka
]
[
]
[
max
S
K
S
v
v
m
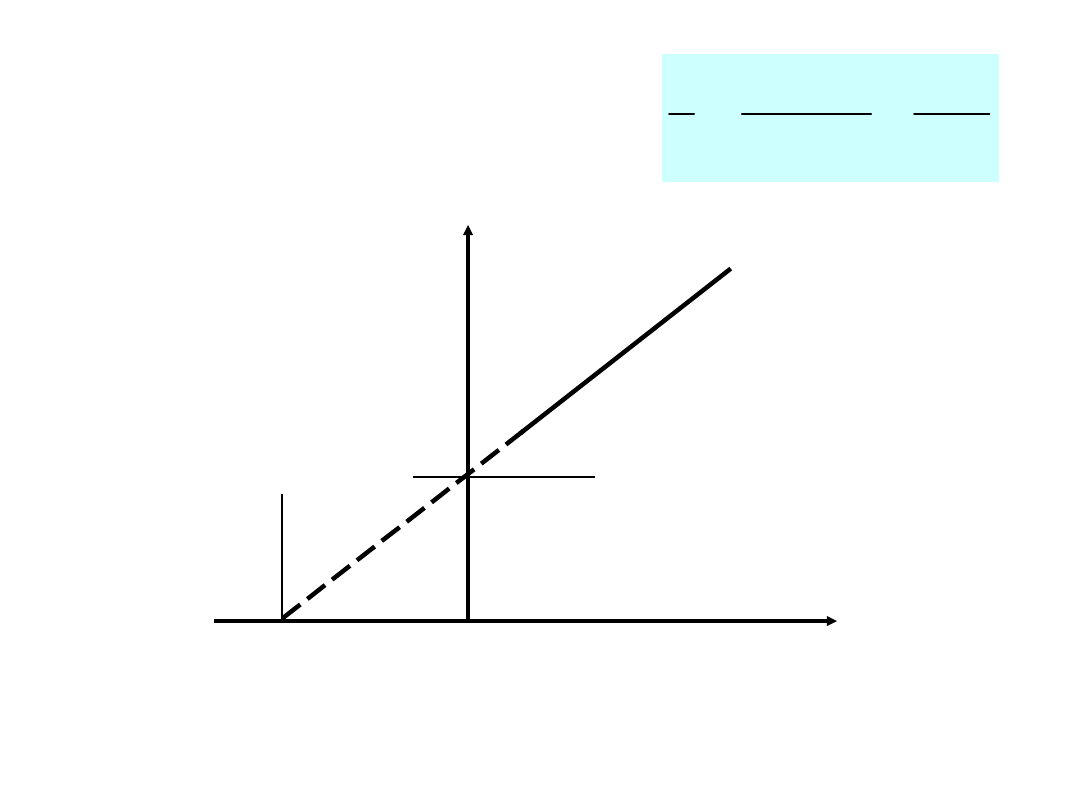
1/v
1/
[S]
1/v
ma
x
-
1/K
m
max
max
1
]
[
1
v
S
v
K
v
m
Graficzne przedstawienie równania
Lineweavera – Burka
Fotochemia
AB +h
+ A +
B
Prawo Starka – Einsteina:
Każda cząsteczka biorąca udział w
pierwotnym procesie fotochemicznym
absorbuje jeden foton.

Wydajność kwantowa pierwotnego procesu
fotochemicznego,
:
liczba cząsteczek substratu tworzących
określony produkt pierwotny na każdy
zaabsorbowany kwant.
Wydajność kwantowa całkowitego procesu:
liczba cząsteczek substratu
przeprowadzonych w produkt w wyniku
absorpcji jednego kwantu promieniowania.
=
Liczba cząsteczek ulegających przemianie
fotochemicznej
Liczba pochłoniętych kwantów
promieniowania
Przykłady reakcji fotochemicznych:
HI +h
H + HI H
2
+ I
I + I I
2
O
2
+ h

Cl
2
+ h
Cl + Cl
Cl + H
2
HCl + H
H + Cl
2
HCl + Cl
Cl + H
2
HCl + H itd.
Br
2
= 2Br
Br + H
2
= HBr + H
H + Br
2
= HBr + Br itd.
H* + Br* = HBr

CH
2
-COOH + H
2
O CH
2
-COOH + HCl
Cl OH
6CO
2
+ 6H
2
O C
6
H
12
O
6
+
6O
2
h

Szybkość rozpuszczania się
ciał stałych
Równanie
Boguskiego:
)
(
x
a
kS
dt
dx
V
V - objętość cieczy,
S – powierzchnia rozpuszczanego ciała,
a – początkowe stężenie roztworu,
x – stężenie substancji rozpuszczonej w
chwili t.
Po scałkowaniu w przedziałach od t
1
do t
2
oraz x
1
do x
2
otrzymujemy:
2
1
1
2
log
)
(
303
,
2
x
a
x
a
S
t
t
V
k
Przy uwzględnieniu stężenia roztworu
nasyconego zamiast stężenia początkowego,
równanie może przyjąć postać:
)
(
'
C
C
S
k
dt
dC
nas
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
Wyszukiwarka
Podobne podstrony:
10 Kinetyka chemiczna i kataliza
10 Kinetyka chemiczna
10 wykad Statyka kinetyka chemiczna[F 2006]
10 Hist BNid 10866 ppt
10 badania niereaktywneid 10789 ppt
wlasciwosci chemiczne alkenow 1 ppt
Kinetyka Chemiczna
10 Uklad pokarmowyid 11141 ppt
10 XML appsid 11206 ppt
kinetyka chemiczna
4) Kinetyka chemiczna
10 PODLOGI I POSADZKIid 11002 ppt
więcej podobnych podstron