
PODSTAWY GENETYKI
DZIEDZICZENIE MENDLOWSKIE
CHOROBY MONOGENOWE
CHOROBY I WADY WIELOGENOWE
KURS Z GENETYKI KLINICZNEJ DLA
LEKARZY RODZINNYCH I LEKARZY
GINEKOLOGÓW
Dr n. med. Magdalena Pasińska

CELE ANALIZY GENETYCZNYCH
UWARUNKOWAŃ DOBROSTANU
CIĄŻY
•Ocena ryzyka wystąpienia u płodu:
- chorób uwarunkowanych
genetycznie
- wad rozwojowych
•Wyodrębnienie ciąż wysokiego
ryzyka i rozpoznanie patologii
genetycznych; współudział w
kształtowaniu dalszych decyzji
diagnostycznych i leczniczych
•Określenie ryzyka genetycznego dla
następnej ciąży
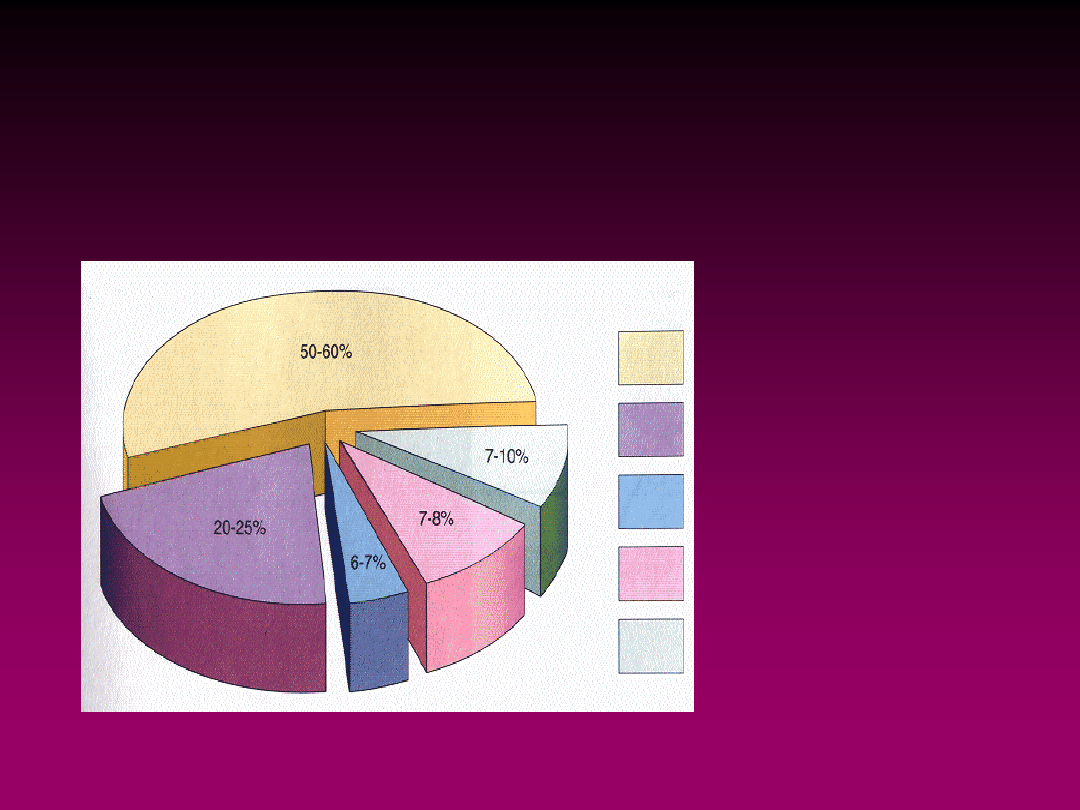
PATOLOGIE GENETYCZNE
I WADY ROZWOJOWE PŁODU
ISTNIEJE 0K. 3000 ZDEFINIOWANYCH CHOROB GENETYCZNYCH I ZESPOŁÓW
WAD
Etiologia nieznana
Ch. wielogenowe
Aberracje
chromosomów
Choroby jednogenowe
Z
definiowane
czynniki środowiskowe

RYZYKO GENETYCZNE
•Jest oceniane jako
prawdopodobieństwo wystąpienia
zdefiniowanego zaburzenia
•Powinno być ocenione ilościowo
•Dotyczy zawsze określonej pary
TEORETYCZNE : WYLICZONE Z PRAW
MENDLA DLA CH. JEDNOGENOWYCH,
RODZINNYCH ABERRACJI STRUKTURALNYCH
CHROMOSOMÓW
ZMODYFIKOWANE: jw. Z UWZGLĘDNIENIEM
DODAT KOWYCH POPRAWEK
EMPIRYCZNE : Z OBSERWACJI POPULACYJNYCH,
DLA CH. WIELOGENOWYCH, ABERRACJI
LICZBOWYCH CHROMOSOMÓW, ZESPOŁÓW WAD
MAŁE
< 5 %
ŚREDNIE
5 - 10
%
DUŻE
> 10 %

TECHNIKI DIAGNOSTYKI
GENETYCZNEJ
• Analiza rodowodowo – kliniczna
• Analiza fenotypu
• Analizy laboratoryjne
- diagnostyka cytogenetyczna
– diagnostyka molekularna DNA i RNA
– diagnostyka biochemiczna

ANALIZA
RODOWODOWO-KLINICZNA
• Dane o zachorowaniach, utratach ciąż,
niepłodności w zakresie wszystkich
krewnych I°,wsparte dokumentacją
lekarską, fotograficzną itp.
• Weryfikacja charakteru dziedziczenia;
niekiedy może być nietypowy w określonej
rodzinie.
• Taki sam fenotyp może wynikać z różnych
uwarunkowań.
• Zapis symboliczny umożliwia lepszą
orientację w uwarunkowaniach.

ZAPIS RODOWODOWY

Dziedziczenia autosomalne
dominujące i recesywne
Wg Mc Kusicka z roku 2000 - 11 000
chorób monogenowych
- 10671 cechy zlokalizowane na
autosomach
- 624 na chromosomie X
- cechy monogeneowe - cechy
mendlowskie
* zasady segregacji
* zasady niezależnego doboru

Dziedziczenia
autosomalne dominujące i
recesywne
• Wg Mc Kusicka z roku 2000 - 11
000 chorób monogenowych
– - 10671 cechy zlokalizowane na
autosomach
– - 624 na chromosomie X
– - cechy monogeneowe - cechy
mendlowskie
– * zasady segregacji
– * zasady niezależnego doboru

Prawa Mendla
Zasada segregacji:
organizmy rozmnażające się
w sposób płciowy posiadają
geny, które występują parami,
ale tylko jeden gen danej
pary jest przekazywany
potomstwu.
Zasada niezależnego
doboru: geny zlokalizowane
w różnych loci są
przekazywane kolejnym
pokoleniom niezależnie.

PRAWDOPODOBIEŃSTWO
GENETYCZNE
PRAWDOPODOBIEŃSTWO
jest to procentowa możliwość
wystąpienia danego wyniku w
serii zdarzeń

Genotyp - genetyczna budowa
jednostki w badanym locus.
Fenotyp - rzeczywisty obraz
fizyczny lub kliniczny danej
jednostki. Jest wynikiem
współdziałania pomiędzy
genotypem i czynnikami
środowiskowymi.
Rodowód - określa związki
pomiędzy członkami rodzin i
pokazuje, którzy z nich są
dotknięci chorobą genetyczną, a
którzy nie.

Dziedziczenie
autosomalne
dominujące
• 1/200 osób
• chory rodzic może przekazać swoim dzieciom
gen „zdrowy” lub „chory”
• prawdopodobieństwo - 1/2
– - osobnicy obu płci wykazują cechę
chorobową w równych proporcjach
– - dana cecha pojawia się w każdym
pokoleniu (pionowy schemat przekazywania
choroby)
– chora heterozygota przekazuje cechę
połowie swoich dzieci
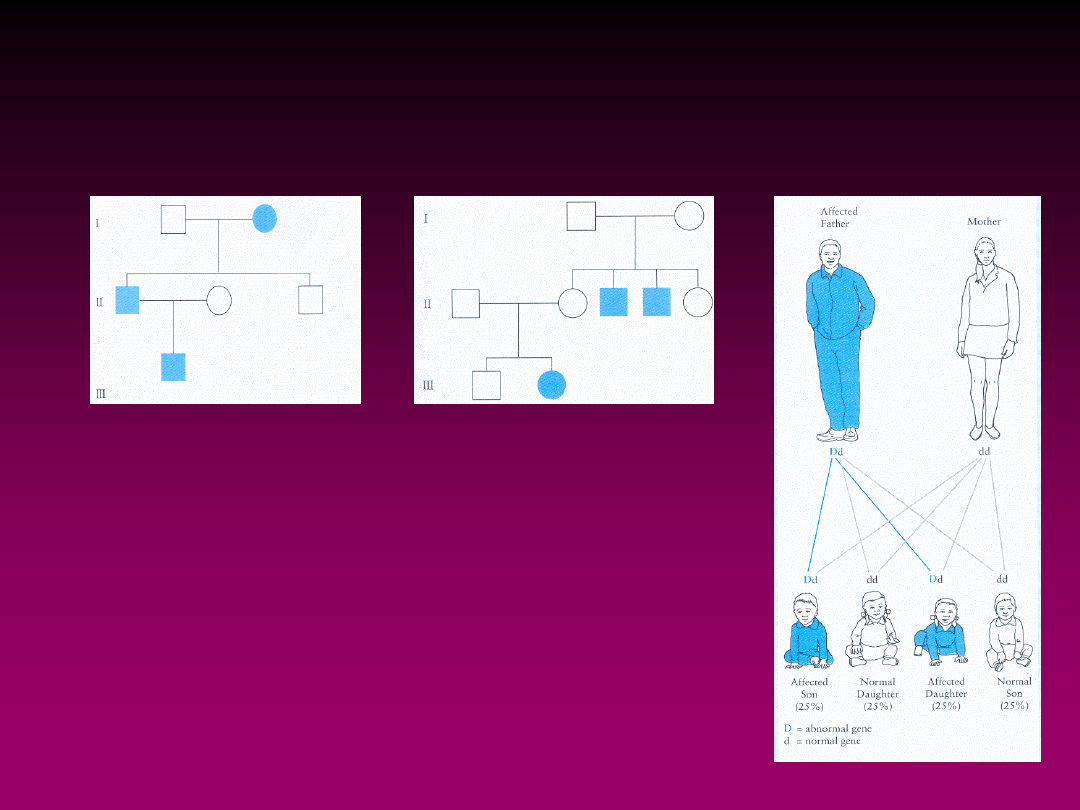
CHOROBY JEDNOGENOWE
AUTOSOMALNE
DOMINUJĄCE
•
manifestuje się „w pionie” niezależnie od płci,
niekiedy częste mutacje de novo ( wtedy izolowana,
ryzyko rośnie z wiekiem ojca), ale zwykle wywiad
dodatni
ryzyko rzeczywiste zwykle mniejsze od teoretycznego
ze względu na niższą penetrację i ekspresję mutacji
• ekspresja zależna od genów modyfikująccyh
i środowiska, penetracja od charakterystyki mutacji
• niekiedy homozygoty nie przeżywają do terminu
porodu
Defekty białek strukturalnych, Predyspozycje do
nowotwrów, Fakomatozy, Pląsawica Huntingtona,
PKD, Otoskleroza i in

Dziedziczenie
autosomalne recesywne
• rodzice osób chorych na choroby
recesywne to heterozygotyczni
nosiciele
• 1/4 potomstwa będzie zdrowymi
homozygotami, 1/2 fenotypowo
zdrowymi heterozygotami, 1/4
homozygotami, u których wystąpi
choroba

Dziedziczenie
autosomalne recesywne
c.d.
• Choroby występują u jednego lub kilku
członków rodzeństwa, ale nie we
wcześniejszych pokoleniach
• mężczyźni i kobiety chorują w równych
proporcjach
• średnio 1/4 potomstwa dwóch
heterozygotycznych nosicieli będzie dotknięta
chorobą
• najczęściej choroba ujawnia się w związkach
pokrewieństwa (większe prawdopodobieństwo
posiadania
wspólnych genów)
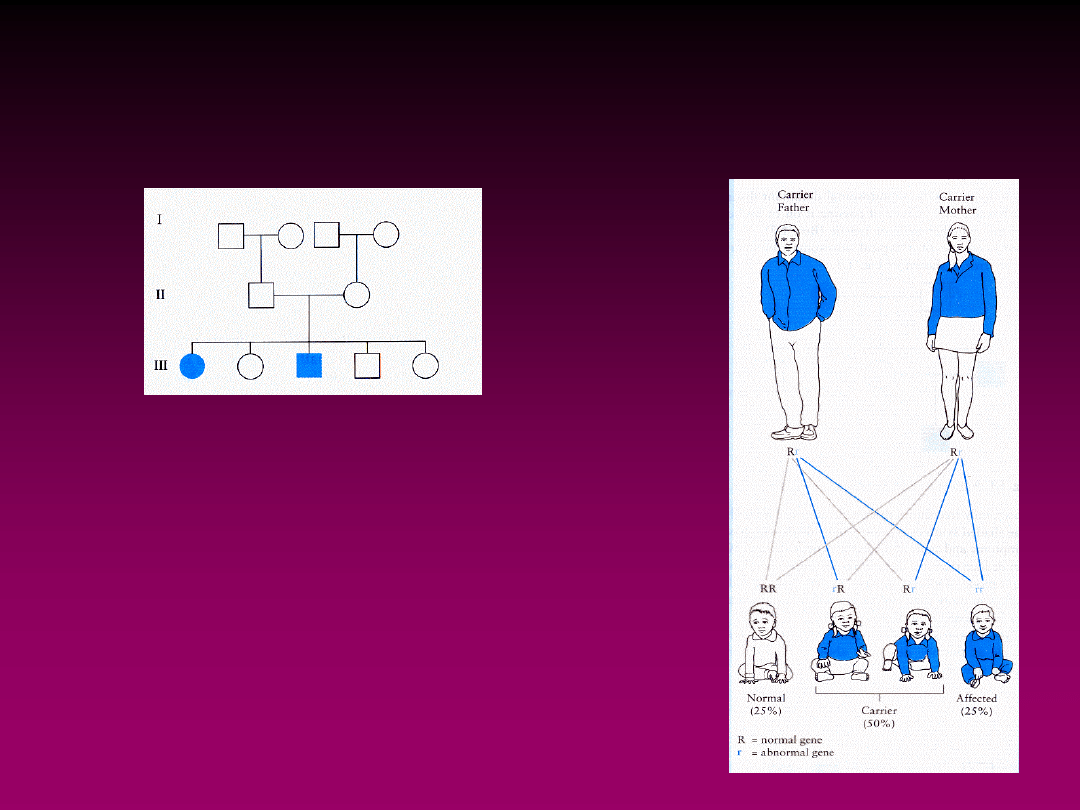
CHOROBY JEDNOGENOWE
AUTOSOMALNE
RECESYWNE
• choroba manifestuje się „w poziomie”, wywiad rodzinny
często ujemny
• występuje równie często u obu płci
• częstsze w małżeństwach krewniaczych
• ryzyko zmodyfikowane zwykle równe teoretycznemu
• często możliwa identyfikacja nosicieli
Mukowiscydoza, Bloki metaboliczne, Retinitis,
Defekty reparacji DNA, SLO, Immunodeficyty i in.
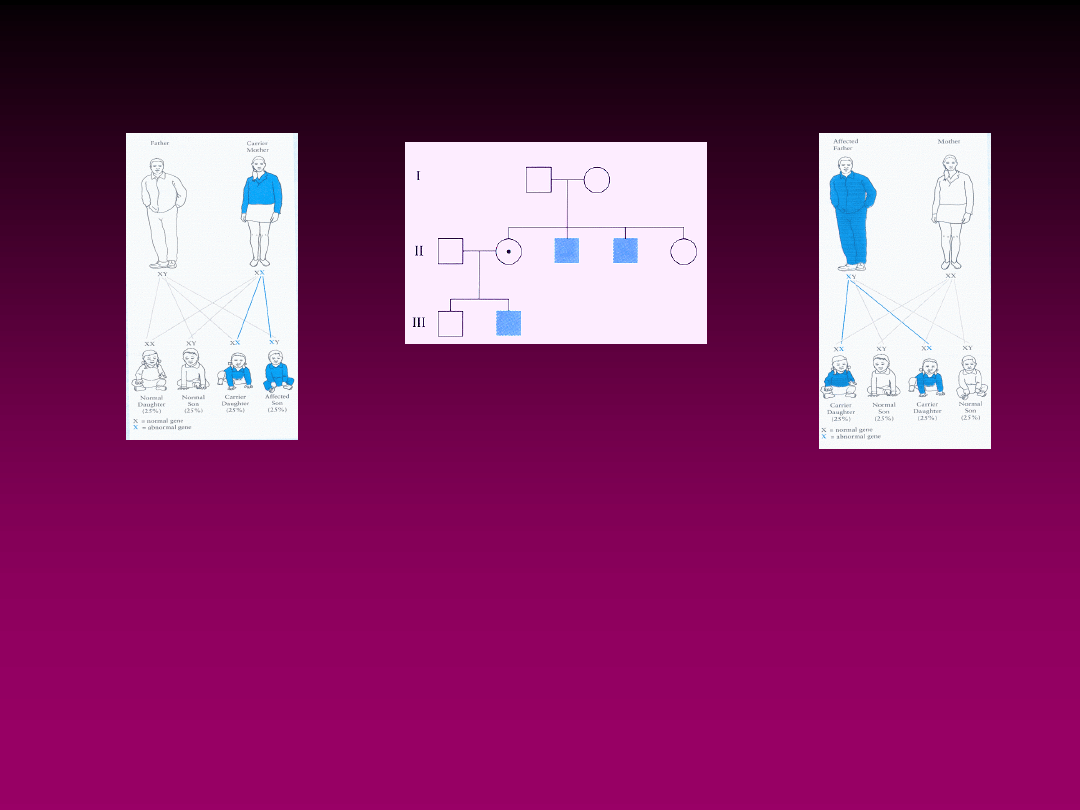
CHOROBY SPRZĘŻONE Z
X
• manifestują się w pionie współwystępując zazwyczaj z płcią
• mogą być dziedziczone recesywnie lub dominująco
• recesywny mogą manifestować się u kobiet (homozygota,
delecje X),
• u kobiet obligatoryjne nosicielstwo, mozaikowość somatyczna
• dominujące manifestują się u kobiet i mężczyzn, męskie
hemizygoty
i żeńskie homozygoty niekiedy nie dożywają do terminu
porodu
Hemofile A i B, DMD/BMD, AIS, FRA-X, nsXMR, SMA, Retinitis,
Alport S., OFDS, Incontinentia pignenti, Krzywica oporna na vit D

NIETYPOWE CHOROBY
JEDNOGENOWE
• CHOROBY SPRZĘŻONE Z
CHROMOSOMEM Y:
TYLKO W LINII MĘSKIEJ, DEFEKTY
SPERMATOGENEZY
• MUTACJE GENOMU
MITOCHONDRIALNEGO:
DZIEDZICZENIE MATCZYNE
• MUTACJE DYNAMICZNE:
ANTYCYPACJA

NIEKTÓRE CHOROBY
JEDNOGENOWE WYMAGAJĄ
SPECYFICZNEGO POSTĘPOWANIA
POŁOŻNICZEGO
• CF
• PKU
• Hemoglobinopatie
• vWD, fVLeydenD
• Marfan S. EDS, inne
defekty kolagenu
• Deficyt MTHFR
• CF
• PKU
• PCKD
• Osteogenesis
imperfecta
• Miopatie
• WPS
• Bloki metabolizmu
sterydów nadnercza
MATKA PŁÓD/ PORÓD/ NOWORODEK

CHOROBY WIELOGENOWE
• WYRAŹNY WPŁYW ŚRODOWISKA
(„WIELOCZYNNIKOWE’’)
• NAJCZĘSTSZE Z CHOROB
”GENETYCZNYCH”
• IZOLOWANE LUB NIEREGULARNE W
RODOWODZIE
• EFEKT PROGU LUB ADDYCYJNY
• RÓŻNA CZĘSTOŚĆ W RÓŻNYCH
POPULACJACH
• RYZYKO EMPIRYCZNE

CHOROBY WIELOGENOWE
• NADCIŚNIENIE
• CUKRZYCA
• MIAŻDŻYCA
• NIEKTÓRE WADY SERCA
• NIEKTÓRE ZESPOŁY WAD

CHOROBY WIELOGENOWE
• NADCIŚNIENIE
• CUKRZYCA
• MIAŻDŻYCA
• NIEKTÓRE WADY SERCA
• NIEKTÓRE ZESPOŁY WAD

RYZYKO EMPIRYCZNE
CHORÓB WIELOGENOWYCH
• WZORCOWE WARTOŚCI RYZYKA
OCENIONE DLA IDENTYCZNYCH
PAR
I W ARUNKÓW EKOLOGICZNYCH
• RYZYKO WYRAŹNIE SPADA ZE
STOPNIEM POKREWIEŃSTWA
• ZNACZENIE PŁCI
UPRZYWILEJOWANEJ
• WYKLUCZYĆ CH. JEDNOGENOWE
O ZBLIŻONYM FENOTYPIE

W MIARĘ ROZWOJU MEDYCYNY
Z CH. WIELOGENOWYCH
WYODRĘBNIAJĄ SIĘ CHOROBY
JEDNOGENOWE
• Wady cewy nerwowej → niedobór
reduktazy MTHFR
• MODY (Maturity Onset Diabetes of
Youth
– MODY 1 : deficyt wątrobowego cz. transkrypcji
HNF-4α
– MODY 2 : deficyt glukokinazy
– MODY 3 : deficyt HNF-1α
– MODY 4 : deficyt insulinowego cz. promotorowego
IPF-1
– MODY 5 : deficyt HNF-1β
– MODY 6 : deficyt czynnika promotorowego β2

DIAGNOSTYKA
PRZEDURODZENIOWA – WSKAZANIA
GENETYCZNE
• Wiek matki < 35 (38), wiek ojca > 55(60) rż.
• Nosicielstwo zrównoważonych rearanżacji
chromosomowych i niektórych mozaikowości
• Poprzednie dziecko z aberracją chromosomów
• Podwyższone ryzyko określonej choroby
jednogenowej
• Podwyższone ryzyko zespołów wad
• ? Poprzedzająca ciążę niepłodność, szczególnie
jeśli nie określono przekonywająco jej przyczyny
i/lub stosowane były techniki wspomagania
rozrodu

DIAGNOSTYKA
PRZEDURODZENIOWA –PODZIAŁ :
• Diagnostyka gamet
• Diagnostyka przedimplantacyjna
• Przedurodzeniowa diagnostyka płodu
- nieinwazyjna
obrazowa
komórek płodu i metabolitów we krwi
matki
- inwazyjna
amniopunkcja
CVS
kordocenteza
fetoskopia

Diagnostyka
przedimplantacyjna
• Badanie I rzadziej II ciałka kierunkowego
- nie dłużej niż 6 godzin, nie ma danych
ojca, nie wyklucza mozaik de novo
powstałych po zapłodnieniu
• Mikrobiopsja 1-2 blastomerów 3-
dniowego zarodka
- standard docelowy (ECC 2003):
FISH 21,13,18, PCR : CFTR, ChH, DMD

CVS? – AMNIOPUNKCJA?-
KORDOCENTEZA?
• CVS? : komórki matczyne u płodów XX,
confined mosaicism wymagający dalszej
weryfikacji, ograniczona liczba możliwych
jednocześnie badań biochemicznych
• Amniopunkcja : późna i długotrwała,
możliwość wyselekcjonowania linii
komórkowych w proporcjach nie
odpowiadających rzeczywistym u płodu
• Kordocenteza : bardzo późna – weryfikacja
confined mosaicism i w przypadku
późnego zgłoszenia się ciężarnej

POSTĘPOWANIE POŁOŻNICZE A
DIAGNOSTYKA POURODZENIOWA
– Wykonanie u ciężarnej immunodiagnostyki
TORCH ; niezbędna dla późniejszego
różnicowania wielu zespołów
– Zapis diagnostyki obrazowej – do powtórnej
oceny w trakcie późniejszej diagnostyki
dziecka
– Precyzyjna ocena z zapisem ilości wód, stanu
łożyska i błon płodowych (zwłaszcza stanu
naczyń pępowinowych)
– Pełna dokumentacja j. w. w posiadaniu rodziny
obciążonego dziecka

Postępowanie diagnostyczne w
przypadku śmierci okołoporodowej
dziecka obciążonego genetycznie
• Kariotyp – możliwe pobranie krwi bezpośrednio
po zgonie; fibroblasty – pobrać 3 -5 mm wycinek
skóry, umieścić w NaCl i 4°C, transport do 12
godz.
• DNA – dowolna zamrożona tkanka
• Dokumentacja fotograficzna wad
• USG czaszki i n. wewnętrznych – także post
mortem
• Babygram – zawsze post mortem, przyżyciowo w
ramach bieżącej diagnostyki rtg
• Zawsze sekcja zwłok, diagnostyka mikroskopowa
łożyska
• Zachować pełną dokumentację, udostępnić do
dalszej diagnostyki genetycznej
• Zawsze skierować rodzinę na konsultację
genetyczną
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
Wyszukiwarka
Podobne podstrony:
Dziedziczenie mendlowskie
Dziedziczenie mendlowskie prawidlowych i patologicznych cech czlowieka, Fizjoterapia, Genetyka
Prelekcja 6 - Dziedziczenie mendlowskie, ROK 1, Genetyka
Dziedziczenie mendlowskie cz III
DZIEDZICZENIE MENDLOWSKIE
Genetyka mendlowska, I prawo Mendla to podstawa korpuskularnej, czyli cząsteczkowej teorii dziedzicz
Rodowody, dziedziczenie, imprinting
Genetyka mendlowska wyklad
dziedziczenie chorob jednogenowych
dziedziny wychowania(1)
Kolonialne dziedzictwo
16 Dziedziczenie przeciwtestamentowe i obliczanie zachowkuid 16754 ppt
078c rozp zm rozp min gosp w spr szkolenia w dziedzinie bhp
Decyzja Rady 90 424 EWG z dnia 26 czerwca 1990 r w sprawie wydatków w dziedzinie weterynarii
CWICZENIA PORZADKOWE[1], Materiały naukowe z różnych dziedzin, Kinezyterapia
więcej podobnych podstron