
Dziedziczenie
mendlowskie
prawidłowych i
patologicznych cech
człowieka
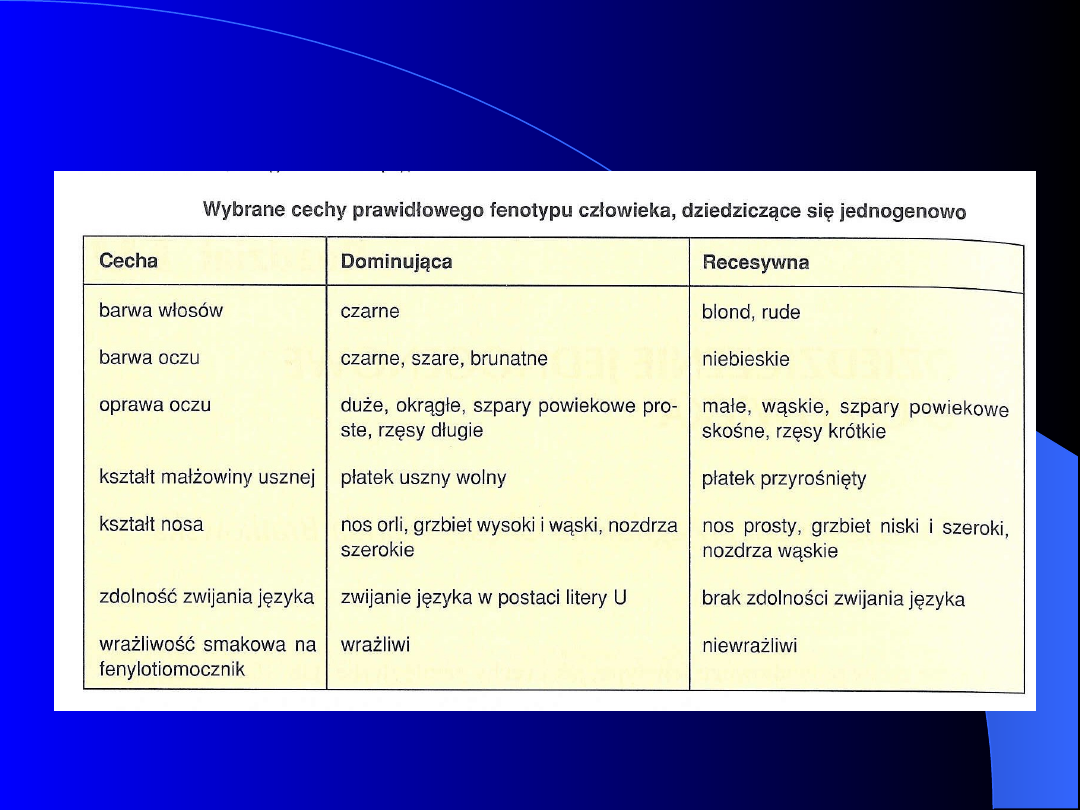

O sposobie dziedziczenia wielu
cech jednogenowych można
wnioskować analizując dane z
wywiadu rodzinnego.
Szczegóły uzyskane z wywiadu
rodzinnego można przedstawić
graficznie w postaci rodowodu.
Poszczególne osoby są
przedstawione w rodowodzie z
uwzględnieniem płci, pokolenia i
ich biologicznego stosunku do
pozostałych członków rodziny.

Ponad połowa opisanych
dotychczas cech jest dziedziczona
dominująco; około 1/3 recesywnie,
a 1/10 jako cechy sprzężone z
chromosomem X.
Pojęcie choroby dziedziczonej
dominująco oznacza, że
pojedynczy allel danej choroby
(jak u heterozygoty) wystarcza do
ujawnienia się jej objawów.

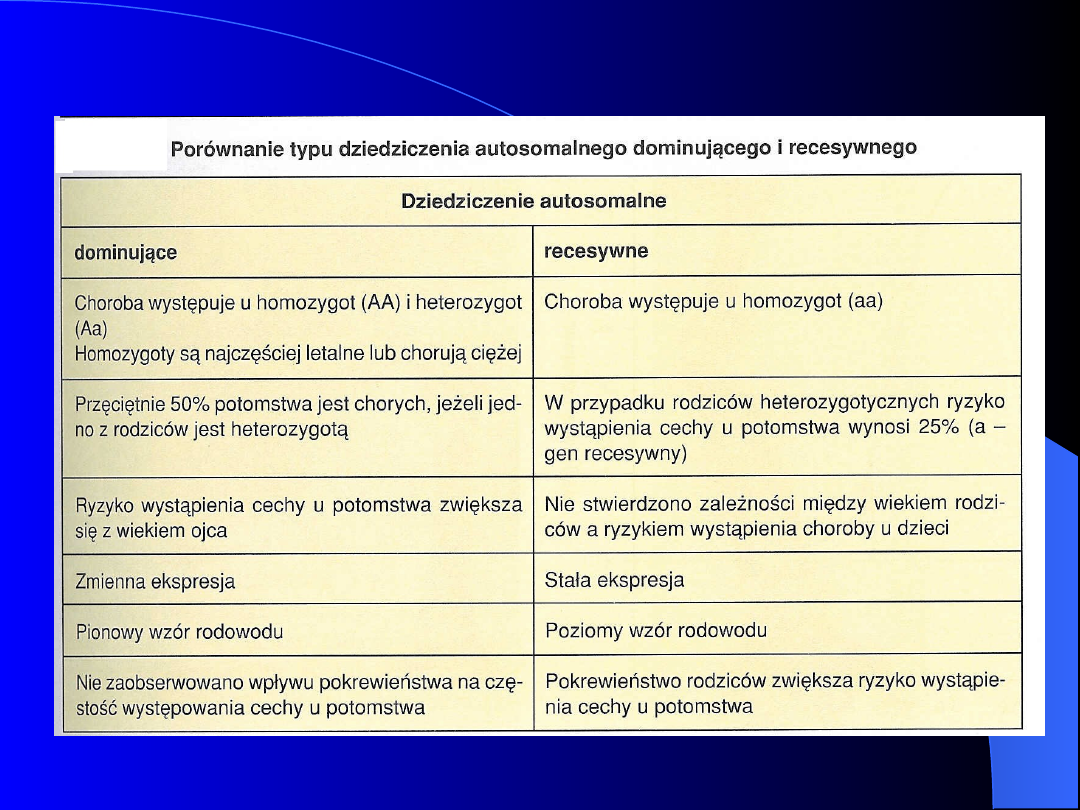
Ogólne cechy dziedziczenia
Ogólne cechy dziedziczenia
autosomalnego dominującego u
autosomalnego dominującego u
ludzi
ludzi
Cecha (choroba) jest przekazywana z pokolenia
na pokolenie „pionowo”.
Niektóre choroby monogenowe ujawniają się w
późnym wieku , np. choroba Huntingtona.
Występowanie
chorób
autosomalnych
dominujących może być wynikiem mutacji de
novo o czym decyduje głównie wiek ojca.
Choroba występuje z tą samą częstością u obu
płci.
Nasilenie objawów choroby (zmienność cechy)
może
zależeć
od
płci
chorego
rodzica
przekazującego cechę.

Obecność lub brak cech klinicznych
oraz ich nasilenie zależy od:
stopnia penetracji patologicznego
genu (pełna penetracja – 100% lub 1;
niepełna penetracja – poniżej 100 %
lub <1),
zmiennej ekspresji genu (chorzy w tej
samej
rodzinie
wykazują
różne
nasilenie objawów choroby),
wieku probanda (penetracja zależna
od wieku).

Geny dominujące wykazują czasem
niepełną penetrację, stąd zjawisko
wyciszania
typowych
objawów
choroby aż do ich zupełnego
zaniku. W wyniku tego zjawiska
może dojść do dziedziczenia z
przeskokiem
pokoleniowym.
Chorują np. dziadkowie i wnuki,
podczas gdy rodzice są zdrowi.

W przypadku kiedy dany gen w populacji
osobników o tym samym genotypie przejawia
się w ten sposób, że badana cecha wykształca
się z różnym nasileniem w fenotypie, to mówi
się o nim, że wykazuje niepełną
ekspresywność.
Jeżeli wśród osobników o tym genotypie tylko
część wykazuje cechę wywołaną posiadanym
genem, a pozostałe osobniki mimo posiadania
tego genu nie przejawiają efektu
fenotypowego, to mówi się, że gen wykazuje
niepełną penetrację.

Jeden gen może wpływać
jednocześnie na powstanie bardzo
różnych cech organizmu. Zjawisko
to określamy jako plejotropowe
działanie genu.

Przykłady chorób dziedziczących się
Przykłady chorób dziedziczących się
autosomalnie dominująco
autosomalnie dominująco
Achondroplazja
(chondrodystrofia, karłowatość
chondrodystroficzna)
Mała zmienność ekspresji, pełna penetracja
Częstość występowania 1 : 15 000 i 1:77 000
Gen – locus 4p 16.3
Gen receptora czynnika wzrostu
fibroblastów (Fibroblast Growth Factor
Receptor 3 – FGFR3)
Mutacja w nukleotydzie 1138 genu FGFR3:
–
tranzycja G A lub transwersja G C

Achondroplazja
Objawy:
- niski wzrost
- skrócenie kończyn, mikromelia
- szpotawe kolana,
- nadmierna lordoza lędźwiowa,
- duża głowa z wypukłym czołem i
zapadniętą nasadą nosa,

Zespół Marfana (arachnodaktylia)
Częstość występowania 1: 10 000
Gen FBN1 (gen fibrylliny) – locus
15q21.1
Wysoki stopień penetracji i zmienna
ekspresja
Fibryllina – białko o masie 350 kD, jest
głównym składnikiem
zewnątrzkomórkowych mikrofibrylli
Jest to defekt tkanki mezenchymalnej
powodujący zmiany w układzie kostno-
stawowym, w układzie krążenia i w
gałkach ocznych

Zespół Marfana
Objawy:
- smukła sylwetka, wysoki wzrost,
- nadmiernie długie palce rąk i stóp,
- „kurza” lub „lejkowata” klatka
piersiowa,
- nadmiernie elastyczna skóra,
- wady wrodzone serca, tętniaki
aorty, wypadanie zastawki mitralnej,
- krótkowzroczność,

Nerwiakowłókniakowatość
(Neurofibromatosis – NF, choroba
Recklinghausena)
Dwie postaci choroby: NF-1 i NF-2
Gen NF-1 (locus – 17q11.2) – pełna
penetracja, zmienna ekspresja
Produkt genu – białko neurofibromina
(obniżony poziom sprzyja rozwojowi
nowotworów)
Częstość 1:3500
Gen NF-2 (locus – 22q12.2) – pełna penetracja
Produkt genu – merlina (białko cytoszkieletu)
Częstość 1:35 000 – 1:40 000

Nerwiakowłókniakowatość
Objawy
mutacja genu NF-1:
- zmiany barwnikowe na skórze (café au lait) -
we wczesnym dzieciństwie
- w okresie dojrzewania rozwijają się liczne
guzki wywodzące się z nerwów obwodowych
(włókniaki, nerwiakowłókniaki, glejaki nerwu
wzrokowego - zwykle łagodne),
- często niedorozwój umysłowy i padaczka,
mutacja genu NF-2:
- nerwiaki nerwu słuchowego,
- oponiaki rdzenia,
- zmętnienie soczewek,
- pierwsze objawy w okresie dojrzewania lub
w drugiej dekadzie życia,

Autosomalna dominująca
wielotorbielowatość nerek (ADWN)
Częstość: 1 : 1000 żywych urodzeń,
Jedną z przyczyn występowania są mutacje genu
PKD1 (16p 13.3) oraz genu PKD2 (4q 13-q23),
PKD (Polycystic Kidney Disease),
Białko – policystyna – 4303 aminokwasy
Mutacja genu powoduje syntezę białka
krótszego, złożonego z 4086 aminokwasów o
zmienionych właściwościach fizykochemicznych.
Objawy kliniczne pojawiają się w 3-4 dekadzie
życia.
Mutacje w genie PKD1 prowadzą do skrajnej
niewydolności nerek w wieku 55-60 lat a u
chorych z mutacją PKD2 w 7 dekadzie życia.

ADWN
Objawy:
- kamica nerkowa,
- krwiomocz,
- nadciśnienie tętnicze,
- bóle brzucha i niewydolność
nerek,
- liczne torbiele w nerkach (u 50%
chorych również w wątrobie),

ARWN (autosomalna recesywna
wielotorbielowatość nerek)
Przyczyna genetyczna nie jest znana.
Częstość: 1: 6000-55000,
Objawy:
- przewlekłe, ropne zapalenia dróg
moczowych,
- liczne torbiele w korze i rdzeniu nerek
oraz w wątrobie,
75 % dzieci z ARWN umiera w ciągu
kilku godzin po urodzeniu a z tych, które
przeżyją
50% - 80% ma szanse dożycia 15 lat,

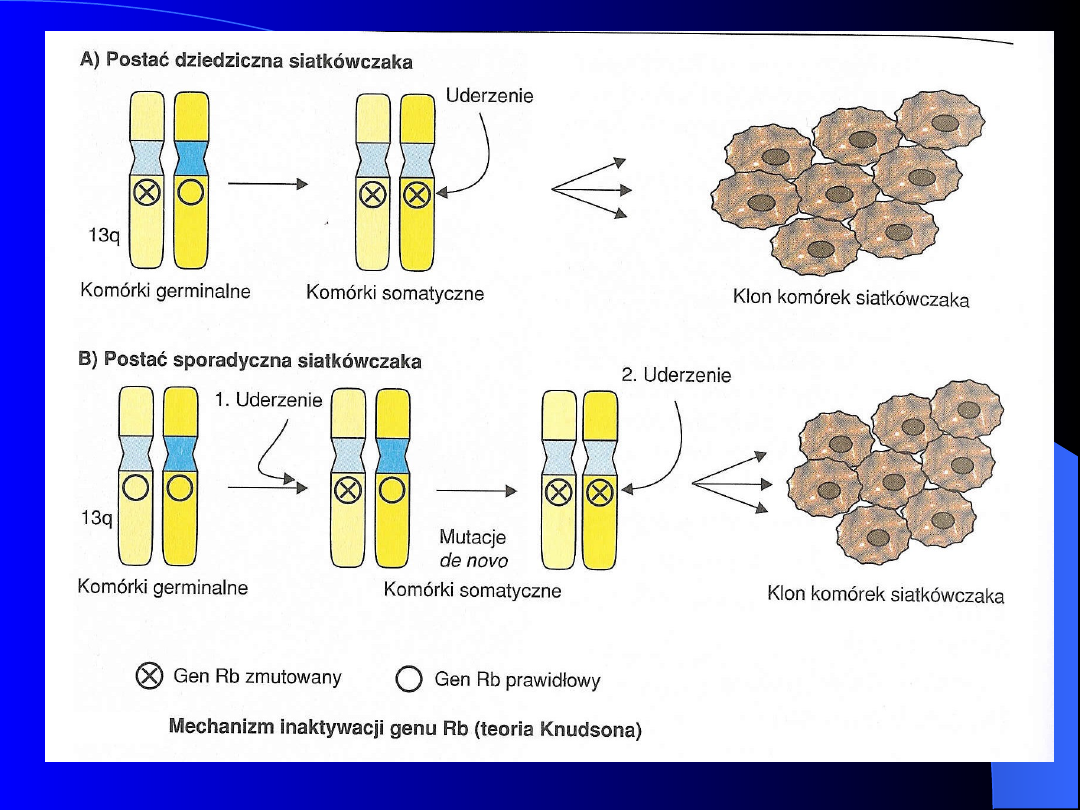
Siatkówczak (Retinoblastoma)
Nowotwór gałki ocznej
Postać sporadyczna i dziedziczna
(rodzinna)
Częstość występowania 1:20 000
Gen Rb – locus 13q 14.1
Przyczyną choroby jest mikrodelecja
regionu 13q 14.1 lub mutacja genu Rb
zlokalizowanego w tym regionie.
Gen Rb wykazuje właściwości
supresorowe, kodowane przez niego białko
wiąże czynnik transkrypcyjny ,
odgrywający rolę w cyklu komórkowym.


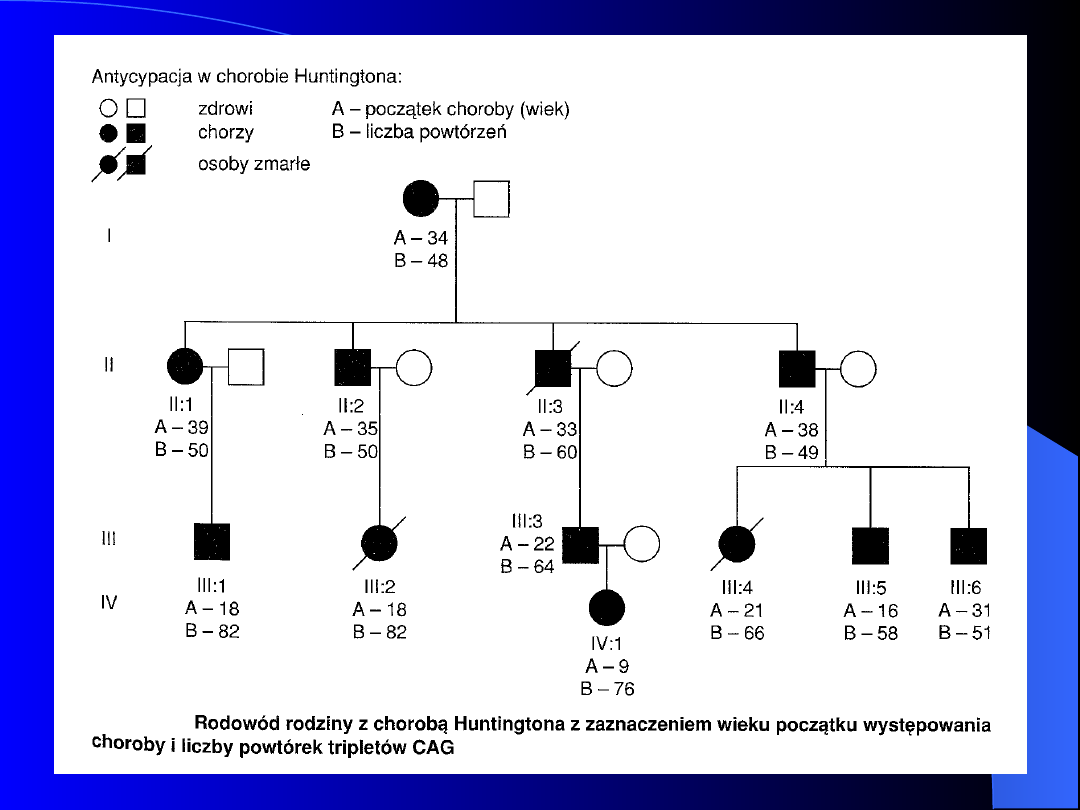
Choroba Huntingtona (Huntington
Disease HD)
Częstość występowania 4 –7 : 100 000
Pełna zależna od wieku penetracja
Gen HD – locus 4p 16.3
Białko – huntingtina
Mutacja dynamiczna: niestabilność
trójnukleotydowych sekwencji powtarzalnych -
(CAG)
n
na końcu 5’ genu kodującego huntingtinę
Osoby zdrowe: 10 –20 powtórzeń CAG
30 – 35 powtórzeń stan przedmutacyjny
Osoby chore: >36 powtórzeń
Antycypacja – ostrzejszy przebieg choroby w
następujących po sobie pokoleniach oraz
występowanie choroby w coraz młodszym wieku
Antycypacja w HD jest mocniej wyrażona, jeżeli
zmutowany gen przekazywany jest przez ojca.

Choroba Huntingtona
Objawy:
- początek choroby zwykle w czwartej
dekadzie życia,
- zmiany neuropatologiczne – selektywne
obumieranie komórek jądra ogoniastego,
- zaburzenia hiperkinetyczne
(przypominające taniec)
- zaburzenia mowy,
- postępująca utrata aktywności
umysłowej,
- charłactwo fizyczne,

Ogólne cechy dziedziczenia
Ogólne cechy dziedziczenia
autosomalnego recesywnego u ludzi:
autosomalnego recesywnego u ludzi:
Choroby o tym typie dziedziczenia
występują głównie u rodzeństwa (poziome
przekazywanie cechy).
Cecha (choroba) ujawnia się tylko u
homozygot
recesywnych
(rodzice
są
heterozygotami
pod
względem
zmutowanego genu).
Częstość
występowania
chorób
autosomalnych
recesywnych
(w
tym
rzadkich) jest zwiększona w małżeństwach
spokrewnionych.

Choroby dziedziczone autosomalnie
recesywnie najczęściej są wynikiem
mutacji genów strukturalnych,
kontrolujących syntezę białek
enzymatycznych, co prowadzi do
zaburzeń metabolicznych ustroju, a
przez to do zaburzeń jego procesów
życiowych.
Większość bloków metabolicznych
dziedziczy się autosomalnie
recesywnie.

Fenyloketonuria
Częstość występowania w populacjach
europejskich średnio 1: 10 000
Locus genu – 12q 24.1
Brak hydroksylazy fenyloalaninowej
(4-monooksygenazy fenyloalaninowej)
Wykrywanie :
test Guthriego – wykrywanie we krwi
zwiększonego stężenia fenyloalaniny
test z FeCl
3
– wykrywanie w moczu kwasu
fenylopirogronowego,
oznaczanie poziomu fenyloalaniny i tyrozyny w
surowicy krwi i ich metabolitów w moczu –
metody enzymatyczne, chromatograficzne i
fluorymetryczne.

Fenyloketonuria
Objawy:
- jasne włosy i jasna karnacja,
- uporczywe wymioty,
- „mysi” zapach moczu,
- wzrost napięcia mięśniowego,
- kiwanie tułowia,
- upośledzenie umysłowe,
Zastosowanie diety pozbawionej
fenyloalaniny umożliwia rozwój
intelektualny zbliżony do normalnego
.

Albinizm (bielactwo uogólnione)
–
częstość średnio 1: 10 000
–
blok metaboliczny przemiany
tyrozyny
–
mutacja genu kontrolującego syntezę
monooksygenazy monofenolowej i
oksydazy katecholowej (tyrozynaza)
–
zahamowanie syntezy melanin w
melanocytach naskórka, cebulek
włosowych, tęczówce i siatkówce

Albinizm
Objawy:
- skóra różowoczerwona,
- włosy białe,
- tęczówki niebieskie lub różowe
(„czerwone źrenice”),
- światłowstręt, zmniejszona
ostrość wzroku,

Alkaptonuria
- częstość 1 : 200 000
- brak 1,2-dioksygenazy
homogentyzynianowej
- kwas homogentyzynowy wydalany z
moczem
Objawy:
- ochronoza – ciemnienie chrząstek,
ścięgien i więzadeł,
- zmiany zapalne i zwyrodnieniowe stawów,
- ciemnienie moczu po zetknięciu z
powietrzem,

Lipidozy
Choroba Gauchera
niedobór beta – glukozydazy,
rozkładającej glukocerebrozyd do glukozy
i ceramidu
Choroba Niemanna-Picka
(sfingomielinoza)
niedobór sfingomielinazy, rozkładającej
sfingomielinę do ceramidu i fosfocholiny
Choroba Tay-Sachsa
niedobór N-acetyloheksozaminidazy,
odszczepiającej N-acetylogalaktozaminę
od glikolipidu

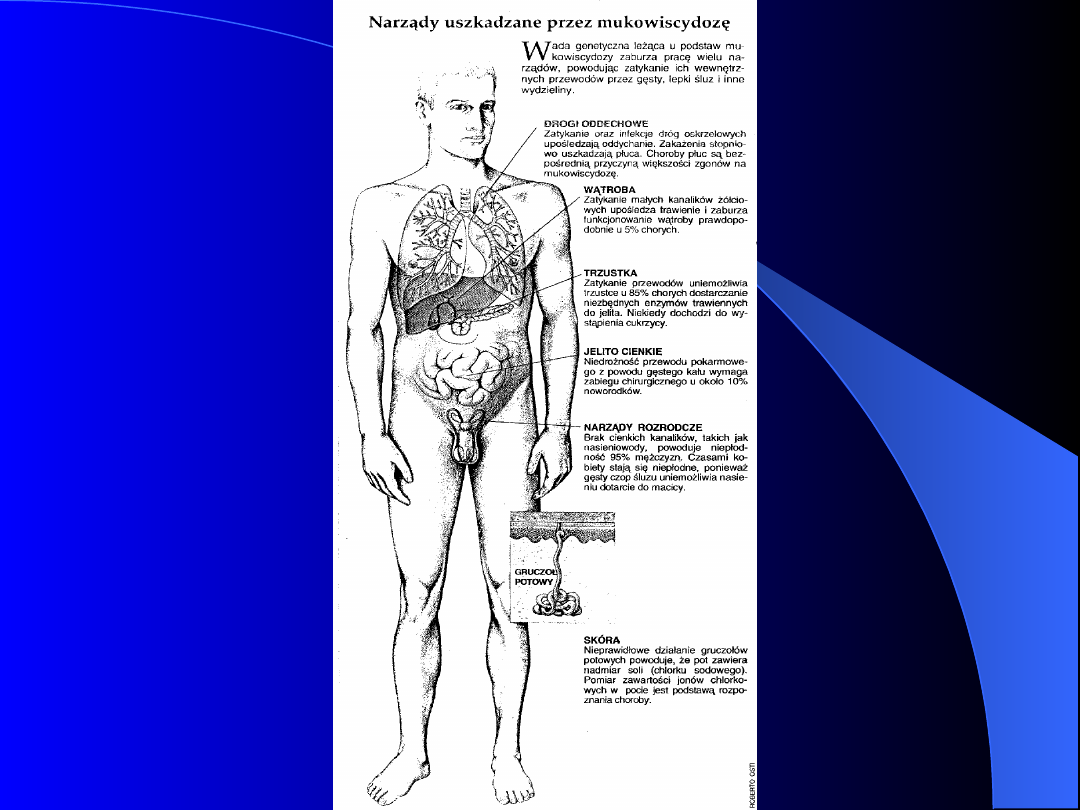
Mukowiscydoza (zwłóknienie
torbielowate trzustki, cystic fibrosis –
CF)
Mucus - śluz, viscid – lepki (łac.)
Częstość :
- Europa 1 :2500
- rasa czarna 1 : 17 000
- rasa żółta 1 : 90 000
Gen CFTR (Cystic Fibrosis Transmembrane
Regulator gene) – locus 7q 31–q32
Wielkość genu 250 kb, 27 eksonów

Mukowiscydoza
Zidentyfikowano ponad 500 różnych
mutacji w genie CFTR
Białko CFTR – błonowy regulator
przewodnictwa specyficzny dla
mukowiscydozy
Charakterystyczny objaw: słony pot
Podwyższone stężenie Na
+
i Cl
-
w pocie
Na
+
> 60 mmol/l (n. 20-25mmol/l)
Cl
-
>70 mmol/l (n. 16-19 mmol/l)

Mukowiscydoza
Objawy:
- słony pot,
- u noworodków : niedrożność smółkowa,
powiększenie brzucha, wymioty, brak
smółki,
- nawracające infekcje dróg
oddechowych, przewlekły kaszel,
- zmiany oskrzelowo-płucne (w 90%
przypadków są przyczyną śmierci)
- zaburzenia procesów trawienia,
- mały przyrost wagi i wzrostu,
- dysfunkcja gruczołów wydzielniczych,
- polipy nosa,
- wrodzony brak nasieniowodów,

Niedokrwistość
sierpowatokrwinkowa
Gen HBB – locus 11p 15.15, koduje
beta-globinę
Hemoglobina S (HbS)
Mutacja punktowa w genie HBB –
zamiana tripletu GAG (kwas
glutaminowy) na GUG (walina)
Heterozygoty HbS/HbA – większa
odporność na zarażenie zarodźcem
malarii
Homozygoty (HbS/HbS) – ciężka
postać niedokrwistości hemolitycznej

Niedokrwistość
sierpowatokrwinkowa
Objawy:
- sierpowaty kształt erytrocytów,
- krwinki łatwiej ulegają hemolizie,
- podwyższona lepkość krwi,
skłonność do tworzenia
zakrzepów,
- niewydolność krążenia,
uszkodzenie wielu organów,

Ogólne cechy dziedziczenia dominującego
Ogólne cechy dziedziczenia dominującego
sprzężonego z chromosomem X
sprzężonego z chromosomem X
Dominujący tor dziedziczenia chorób
sprzężonych z chromosomem X należy
do rzadkości i można go podejrzewać
gdy:
chory mężczyzna ma wyłącznie chore
córki i wyłącznie zdrowych synów,
chore
kobiety
heterozygoty
przekazują
cechę
50%
swego
potomstwa, niezależnie od płci,

Ogólne
cechy
dziedziczenia
dominującego
sprzężonego
z
chromosomem X
chore
kobiety
homozygoty
przekazują
cechę
wszystkim
swoim dzieciom,
w potomstwie chorej kobiety
(heterozygoty)
i
zdrowego
mężczyzny 50% synów i 50%
córek będzie chorych.

Wrodzona hipoplazja skóry
(Incontinentia pigmenti – zespół
Blocha i Sulzbergera)
Częstość 1: 75 000
Locus genu – Xq 27-q28
Chorują głównie dziewczęta
Dla płodów męskich – zespół
letalny

Wrodzona hipoplazja skóry
Objawy:
- dla płodów męskich – zespół letalny,
- po urodzeniu na skórze dziecka
pojawiają się plamy rumieniowe,
zawierające małe pęcherzyki,
- naturalny tatuaż skóry,
- zez,
- wady układu kostnego i serca,
- u 50% chorych – niedorozwój
umysłowy, porażenia, napady
drgawek,

Zespół Retta
Częstość :
Europa 1: 15 000
USA 1 : 20 000
Locus genu – Xp 21.3
Dla płci męskiej - letalny

Zespół Retta
Objawy:
- zaburzenia psychoruchowe –
pojawiają się miedzy 6 a 18
miesiącem życia,
- głębokie upośledzenie umysłowe,
- zaburzenia neurologiczne
(padaczka, spastyczność),
- liczne zaburzenia w rozwoju
fizycznym,

Zespół łamliwego chromosomu X
(s. Zespół Martina-Bella, fragile X
syndrom)
Częstość:
Mężczyźni – 1: 1250
Kobiety – 1 : 2000
Uszkodzenie funkcji genu FMR1 (Fragile
X Mental Retardation)
Locus genu – Xq 27.3 (37kb, 17 eksonów)
Niestabilne sekwencje (CGG)
n
Osoby zdrowe 6 – 52 powtórzeń CGG
Nosiciele bezobjawowi 52 – 200
Osoby chore 230 – 1500 (pełna mutacja)

Zespół łamliwego chromosomuX
Produkt genu FMR1 – białko FMRP – 64
aminokwasy, należy do białek
wiążących RNA.
Kobiety z pełna mutacją – upośledzenie
umysłowe w stopniu lekkim (20-30%)
lub umiarkowanym (1-2%)
Mężczyźni z pełną mutacją ciężki
stopień upośledzenia (max. IQ = 31)

Zespół łamliwego chromosomu X
Objawy:
u noworodków i niemowląt płci męskiej:
- niska waga urodzeniowa,
- mały obwód głowy,
- zwiększona objętość jąder,
- duże małżowiny uszne,
- hipotonia mięśniowa,
u dzieci:
- autyzm, zaburzenia mowy,
- opóźnienie rozwoju psychoruchowego,
u dorosłego mężczyzny:
- deformacje twarzoczaszki (wydatne guzy czołowe,
duże i odstające małżowiny uszne, duża żuchwa,
wysokie podniebienie, hiperteloryzm),
- powiększenie jąder, bladoniebieskie tęczówki,
zniekształcenia kręgosłupa, padaczka, encefalopatia,

Zespół łamliwego chromosomu X
Objawy
- u 1/3 kobiet z pełną mutacją genu
FMR1 występują:
- zmiany w obrębie twarzoczaszki,
upośledzenie umysłowe średniego
stopnia ( IQ 24-41),
- spowolnienie ruchowe,
- trudności wymowy,
- problemy z koncentracją,

Ogólne cechy dziedziczenia recesywnego
Ogólne cechy dziedziczenia recesywnego
sprzężonego z chromosomem X
sprzężonego z chromosomem X
O recesywnym sposobie dziedziczenia
chorób sprzężonych z chromosomem
X mówimy wówczas gdy:
choroba występuje znacznie częściej
u mężczyzn (hemizygota) niż u
kobiet,
kobiety chorują tylko w układzie
homozygotycznym dla zmutowanej
pary alleli,

u kobiet heterozygot (nosicielek
zmutowanego genu) choroba nie
występuje,
chory
mężczyzna
nigdy
nie
przekazuje choroby (cechy) synom, a
wszystkie córki chorego mężczyzny
są nosicielkami zmutowanego genu,
istnieje 50% prawdopodobieństwa,
że
heterozygotyczne
kobiety
przekażą zmutowany gen zarówno
synom, jak i córkom.

Dystrofia mięśniowa Duchenne’a
(DMD)
Częstość u chłopców – 1 : 3500
Locus genu – Xp 21.2 (2,5 mln par zasad,
79 eksonów)
Sekwencje niekodujące stanowią 99,4%
genu.
Brak dystrofiny w mięśniach, zgon w
drugiej dekadzie życia.
Dystrofina – białko strukturalne
zlokalizowane po stronie cytoplazmatycznej
błony komórkowej.
Choroba ujawnia się przed 5 rokiem życia

Dystrofia mięśniowa typu Duchenne’a
Objawy:
symetryczny zanik mięśni obręczy miednicy i
barkowej,
postępująca utrata tkanki mięśniowej
rozpoczyna się we wczesnym dzieciństwie i
prowadzi zwykle do śmierci na skutek
niedomagań oddechowych lub sercowych,
„kaczy” chód,
trudności przy wchodzeniu po schodach i
wstawaniu z pozycji leżącej,
przerost łydek,
zwiększona aktywność w surowicy krwi kinazy
kreatynowej, aldolazy i dehydrogenazy
mleczanowej,

Dystrofia mięśniowa Beckera
(BMD)
Częstość – 1 : 20 000
Nie jest letalna.
Mniejsza ilość dystrofiny (3-10%)
lub zmieniona dystrofina w
mięśniach.
DMD i BMD to są alleliczne formy
tej samej choroby.

Dystrofia mięśniowa typu Beckera
Objawy:
- pojawiają się w drugiej dekadzie
życia,
- zanik mięśni kończyn najpierw
dolnych, a potem górnych,
- przebieg łagodniejszy, ale
prowadzi do inwalidztwa,

Ślepota na barwy
Choruje od 5 do 9% mężczyzn
Locus genu – Xq 28
Protanopia - ślepota na barwę czerwoną
Deuteranopia - ślepota na barwę zieloną
Tritanopia – ślepota na barwę niebieską
Niedowidzenie barw:
- protanomalia
- deuteranomalia
- tritanomalia
Gen odpowiedzialny za percepcję barwy
niebieskiej znajduje się na chromosomie
7.


Hemofilia A i hemofilia B
Częstość występowania hemofilii A
wynosi od 1 :10 000 do 1 : 20 000
Locus genu Xq 28 (186 kb)
Niedobór lub brak VII czynnika
krzepnięcia krwi
Częstość występowania hemofilii B
(choroba Christmasa) wynosi 1 : 30 000
Locus genu – Xq 27.1-q 27.2
Brak czynnika IX krzepnięcia krwi

Hemofilia
Objawy:
- w ciężkiej postaci: samoistne wylewy
dostawowe prowadzące do inwalidztwa
(artropatia dostawowa)
- w umiarkowanej postaci: krwawienia
występują po urazach, wylewy
śródstawowe są mniej ciężkie i występują
rzadziej,
- w postaci łagodnej wylewy występują
tylko po znacznych urazach lub po
operacjach,
- czas krzepnięcia krwi znacznie
wydłużony,
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Ogólne cechy dziedziczenia autosomalnego dominującego u ludzi
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Przykłady chorób dziedziczących się autosomalnie dominująco
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Ogólne cechy dziedziczenia autosomalnego recesywnego u ludzi:
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Ogólne cechy dziedziczenia dominującego sprzężonego z chromosomem X
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Ogólne cechy dziedziczenia recesywnego sprzężonego z chromosomem X
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
Wyszukiwarka
Podobne podstrony:
Dziedziczenie mendlowskie prawidlowych i patologicznych cech czlowieka, Fizjoterapia, Genetyka
Prelekcja 6 - Dziedziczenie mendlowskie, ROK 1, Genetyka
Dziedziczenie mendlowskie cz III
DZIEDZICZENIE MENDLOWSKIE
DZIEDZICZENIE MENDLOWSKIE
Genetyka mendlowska, I prawo Mendla to podstawa korpuskularnej, czyli cząsteczkowej teorii dziedzicz
Rodowody, dziedziczenie, imprinting
Genetyka mendlowska wyklad
dziedziczenie chorob jednogenowych
dziedziny wychowania(1)
Kolonialne dziedzictwo
16 Dziedziczenie przeciwtestamentowe i obliczanie zachowkuid 16754 ppt
078c rozp zm rozp min gosp w spr szkolenia w dziedzinie bhp
Decyzja Rady 90 424 EWG z dnia 26 czerwca 1990 r w sprawie wydatków w dziedzinie weterynarii
CWICZENIA PORZADKOWE[1], Materiały naukowe z różnych dziedzin, Kinezyterapia
więcej podobnych podstron