
Błędy w przekazywaniu
informacji genetycznej
Trzyna Jakub

Czym jest choroba
genetyczna?
Chorobę genetyczną można zdefiniować
jako pewnego typu odchylenie od stanu
prawidłowego, powstałe na skutek
zmiany w zapisie informacji genetycznej.
Może ono być przekazywane z pokolenia
na pokolenie, może też być wynikiem
świeżej mutacji. Wiele z nich przejawia
się w postaci ciężkiego kalectwa
fizycznego i umysłowego.

Wyróżnia się zazwyczaj:
•
choroby monogenowe – nieprawidłowości w
obrębie
pojedynczych genów, np. mukowiscydoza,
fenyloketonuria, galaktozemia,
• choroby poligenowe (wieloczynnikowe) –
podstawą genetycznej predyspozycji do ich
wystąpienia jest istnienie zmienności
genowej, która decyduje nie o chorobie, a
tylko o skłonności do niej. Skłonność ta
wyzwalana jest przez działanie czynników
środowiskowych. Do chorób tych zaliczamy
m.in. wady serca, wrodzone zwichnięcie
stawów biodrowych, atopię, łuszczycę,
schizofrenię, padaczkę.

•
aberracje chromosomowe – najczęstszym ich
źródłem są zakłócenia w procesie podziału
komórki (zarówno mejotycznym jak i
mitotycznym). Zazwyczaj jest to wynik
nierozdzielenia się określonej pary
chromosomów lub trisomii, czyli aberracji
chromosomowej liczbowej. Możliwe są również
aberracje strukturalne:
- delecja – utrata pewnego fragmentu
chromosomu
- translokacja – przeniesienie odcinka
chromosomu do innego
- duplikacja – podwojenie danego odcinka
chromosomu
- inwersja – odwrócenie fragmentu chromosomu
o 180 ⁰.

Choroby genetyczne

Mukowiscydoza
Monogenowa choroba, dziedziczona w sposób
autosomalny recesywny. Gen mukowiscydozy,
zlokalizowany w długim ramieniu chromosomu
nr 7, koduje białko odpowiedzialne za transport
jonów chlorkowych przez błony komórkowe.
Zakłócenie transportu jonów chlorkowych
prowadzi głównie do uszkodzenia tkanek
układu oddechowego i pokarmowego.
Najpoważniejsze objawy choroby to:
gromadzenie się gęstego śluzu w układzie
oddechowym i niewydolność
zewnątrzwydzielnicza trzustki.
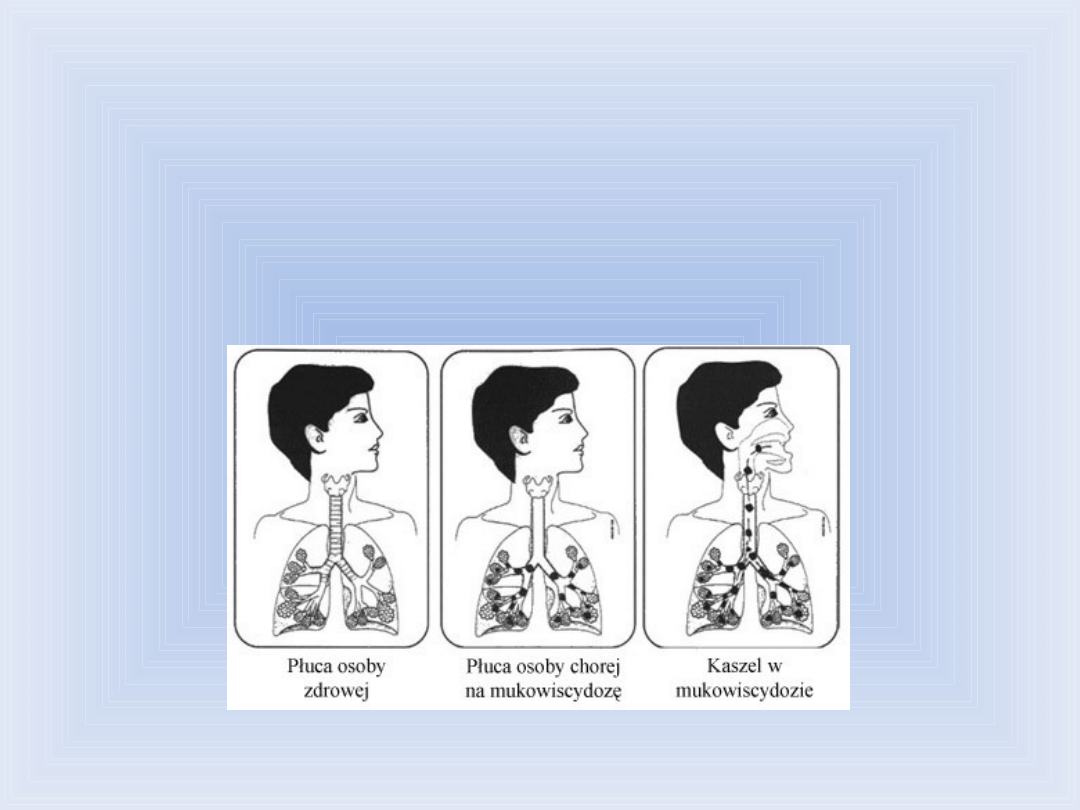
Mukowiscydoza
Leczenie przedłuża życie około 50 %
pacjentów, zawsze jednak kończy się ono
w wieku dwudziestu kilku lat.

Fenyloketonuria
Choroba dziedziczona w sposób autosomalny
recesywny. Schorzenie to jest następstwem
braku enzymu przekształcającego dużą ilość
fenyloalaniny i kwasu fenylopirogronowego,
które uszkadzają rozwijający się układ
nerwowy. Objawy kliniczne choroby
charakteryzują się przede wszystkim
postępującym opóźnieniem rozwoju
psychomotorycznego, a już koło 1 roku życia
nieodwracalnym opóźnieniem rozwoju
umysłowego.
Fenyloketonuria
Przy wczesnym wykryciu choroby jej skutki
można załagodzić stosując dietę o
ograniczonej ilości fenyloaniny, niezbędnej
dla prawidłowej syntezy białek
ustrojowych.
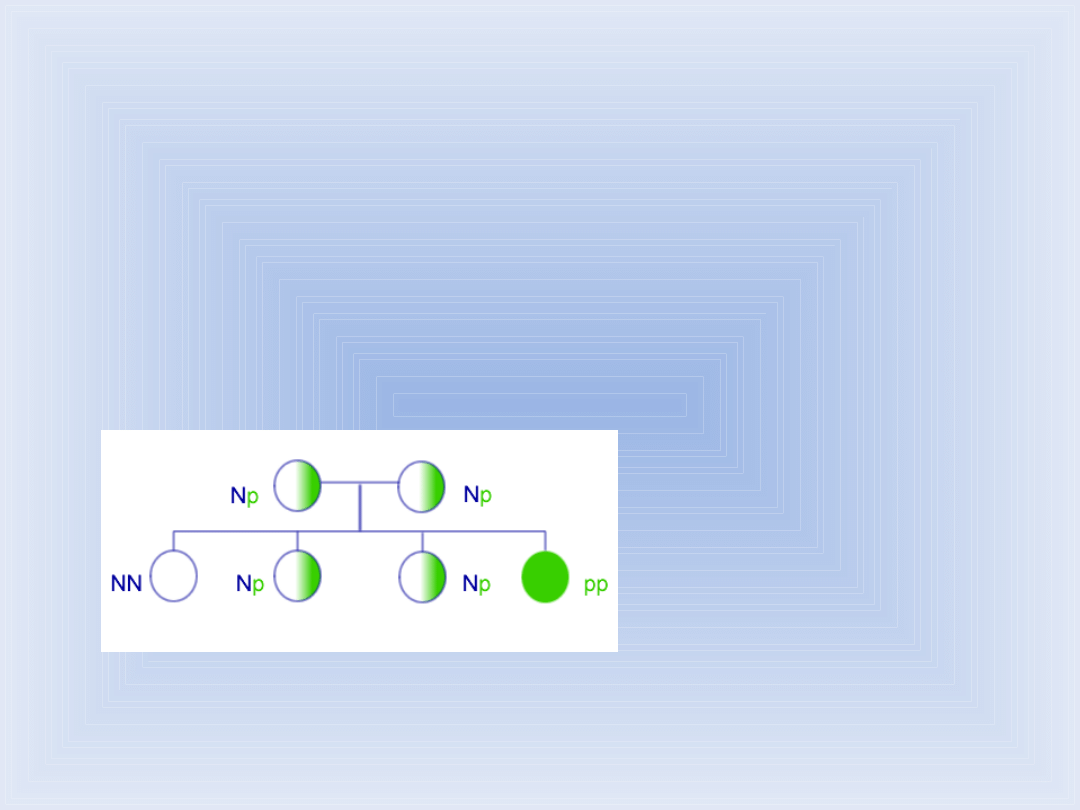
Dziedziczenie fenyloketonurii
Klucz:
(NN) – os. zdrowa
(Np.) – nosiciel
(pp) – os. chora

Galaktozemia
Choroba ta rozwija się na skutek niedoboru lub
braku enzymów odpowiedzialnych za
przekształcanie galaktozy w glukozę.
Prowadzi to do akumulacji galaktozy we krwi,
która w niezmienionej postaci może działać
toksycznie na organizm. W ciężkich
przypadkach galaktozemii opóźnieniom w
rozwoju somatycznym towarzyszy opóźnienie
rozwoju psychicznego. Prawidłowo
prowadzona dieta eliminacyjna warunkuje nie
dopuszczenie do skutków choroby.

Zespół łamliwego
chromosomu X
Jest po zespole Downa najczęściej
występującą, genetycznie uwarunkowaną,
postacią niedorozwoju umysłowego. Jest
chorobą monogenową. Gen będący
przyczyną tego zaburzenia znajduje się w
chromosomie X. Nazwa schorzenia wynika
z faktu, że chromosom zawierający allel
łamliwego X ma tendencję do pękania, co
wykrywane jest badaniem
cytogenetycznym.

Zespół łamliwego
chromosomu X
Tylko połowa dziewcząt z zespołem łamliwego
chromosomu X ujawnia zaburzenie, gdyż jeden z
dwóch chromosomów X pozostaje u
dziewcząt nieaktywny. Wykazują one zazwyczaj
lekki stopień upośledzenia lub jedynie trudności w
uczeniu się.
W przypadku chłopców stwierdza się zwykłe
upośledzenie umysłowe od umiarkowanego do
głębokiego, wydłużoną twarz z wystającą
szczęką, asymetrię twarzy, wydatne guzy
czołowe, duże, odstające uszy, zniekształcenia
kręgosłupa, obniżenie napięcia mięśniowego i
powiększenie jąder po okresie pokwitania.

Zespół Downa
Trisomia 21 pary chromosomów zwana dawniej
mongolizmem. Osobnika z zespołem Downa
cechuje charakterystyczny wygląd twarzy o
płaskim profil, szeroko rozstawione, nieco
skośne oczy z fałdą powiekową podobną do
mongolskiej, mały nos, mała żuchwa, kąciki
ust skierowane ku dołowi, bardzo często
wysunięty język. Małżowiny uszne małe, nisko
osadzone, często zniekształcone. Chorzy są
niscy, cechuje ich długi tułów i krótkie
kończyny.

Zespół Downa
Często występują u nich wady wrodzone
serca, wady przewodu pokarmowego,
układu moczowego, padaczka, zaćma,
białaczka; charakterystyczne jest też
przedwczesne starzenie się. Stałym i
najczęstczym objawem jest upośledzenie
umysłowe, zazwyczaj w stopniu
umiarkowanym.

Zespół Klinefeltera (47,
XXY)
Schorzenie to jest dość rzadko
rozpoznawane w wieku dziecięcym ze
względu na brak charakterystycznych
objawów w tym wieku. Osobnika takiego
cechują zaburzone proporcje długości
tułowia do długości kończyn
dolnych(długie kończyny), szeroka
miednica, niedorozwój gruczołów
płciowych, znaczne zaburzenie
dojrzewania płciowego.

Zespół Klinefeltera (47,
XXY)
Upośledzone wytwarzanie testosteronu
prowadzi do słabego rozwoju wtórnych
cech płciowych i ginekomastii. Większość
chorych wykazuje normalny poziom
inteligencji, jednak u około 20% stwierdza
się cechy upośledzenia.

Zespół Turnera (45, X)
Charakterystycznymi objawami są: stwierdzany
od wczesnego dzieciństwa niedobór wysokości
ciała. Wysokość dorosłych kobiet nie przekracza
150cm, nie obserwuje się przyśpieszania
wzrastania w okresie dojrzewania. Występuje
niedorozwój zewnętrznych narządów płciowych,
wczesny proces degeneracji jajników prowadzi
do ich zaniku i włóknienia. Stanowi to przyczynę
braku miesiączki i bezpłodność. U większości
chorych występuje niska linia owłosienia na
czole i karku, niedorozwój żuchwy i tyłozgryz,
płetwiasta szyja i nisko osadzone małżowiny
uszne, anomalie kostne.

Zespół Turnera (45, X)
Zarówno inteligencja, jak i długość życia nie
odbiegają od normy, jednak u części
chorych stwierdza się cechy upośledzenia
umysłowego w lekkim stopniu. Leczenie
hormonalne (estrogenami) umożliwia
rozwój wtórnych cech płciowych lecz nie
wpływa na wysokość ciała
i bezpłodność.

Zespół miauczenia kota
Jego przyczyna jest delecja fragmentu ramion
krótkich chromosomu nr 5. Stwierdzany w
okresie noworodkowym charakterystyczny
płacz, przypominający miauczenie kota, stał
się przyczyną nazwy zespołu. Cechami
charakterystycznymi są dystrofia
wewnątrzmaciczna, małogłowie, niedorozwój
żuchwy, nisko osadzone małżowiny uszne,
obniżenie napięcia mięśniowego oraz
współistnienie licznych wad rozwojowych
układu krążenia i moczowego. Cechą
dominującą jest głębokie upośledzenie
umysłowe.

Zespół miauczenia kota
Fenotyp zespołu Zespół
miauczenia kota u jednego
pacjenta:
A – w wieku 8 miesięcy,
B – 2 lat,
C – 4 lat,
D – 9,5 lat.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
Wyszukiwarka
Podobne podstrony:
Ekspresja informacji genetycznej-transkrypcja i translacja, NAUKA
4. Przenoszenie informacji genetycznej - mechanizmy, studia-biologia, Opracowane pytania do licencja
kubica, biologia z elementami mikrobiologii, nośniki informacji genetycznej
w sprawie szczegółowego zakresu i trybu przekazywania informacji?nowych z postępowania o zamówi Y3YW
Nośnik informacji genetycznej DNA
DNA jako nośnik informacji genetycznej1
DNA jako nośnik informacji genetycznej
Nowoczesne techniki przekazu informacji
REGULACJA EKSPRESJI INFORMACJI GENETYCZNEJ
Rola cAMP oraz cGMP w przekazywaniu informacji w kom2
Jak dawniej przekazywano informacje
błędy, Technologie Informacyjne, word
W jaki sposób przekazywać informacje zwrotne, absolutnie wszystko o HR
informacja genetyczna
Budowa RNA oraz przekazywanie materiały genetycznego
IT Obraz, jako środek przekazu informacji
Ortodromowy przekaz informacji, II rok, II rok CM UMK, Giełdy, od Joe, FIZJOLOGIA, KOLOKWIA, NEUROFI
Biotechnologia -W, Markery, Inżynieria genetyczna - zespół technik pozwalających na badanie procesów
7 WYKŁAD VII Ekspresja informacji genetycznej
więcej podobnych podstron