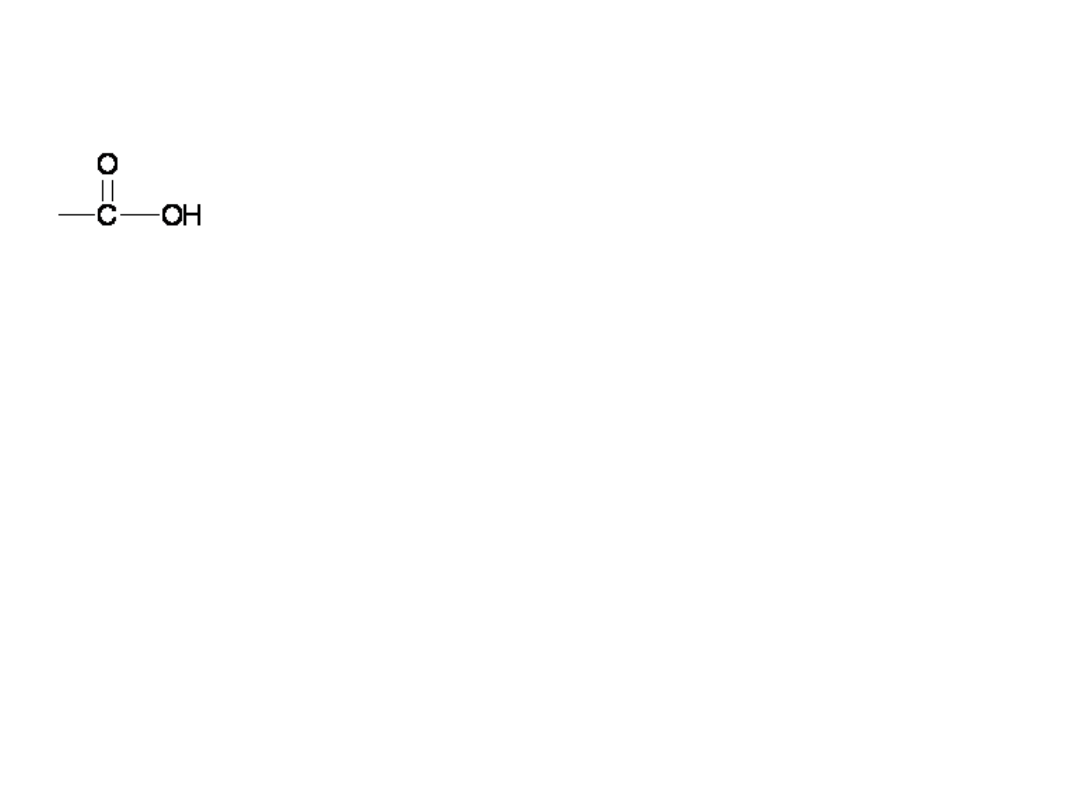
Kwasy Karboksylowe
I Karboksylowe kwasy alifatyczne – tzw. tłuszczowe
grupa karboksylowa, złożona z grup
karbonylowej i hydroksylowej
Alifatyczne kw. karboksylowe znane były od dawna, gdyż wiele z nich
występuje w naturze np. kwas mrówkowy obecny jest w jadzie mrówek.
Związki te mają ogólny wzór CnH2n+1COOH
Kwasy karboksylowe noszą nazwy zwyczajowe.
Oczywiście mają również nazwy systematyczne. Wg. IUPAC tworzenie
nazw kwasów organicznych polega na dodaniu końcówki „owy” lub
„okarboksylowy” do nazwy wyjściowego węglowodoru posiadającego
taką samą ilość atomów C jak kwas.

Szereg homologiczny kwasów alifatycznych:
1. HCOOH – kw. metanowy (mrówkowy)
2. CH
3
COOH – kw. etanowy (octowy)
3. CH
3
CH
2
COOH – kw. propanowy (propionowy)
4. CH
3
CH
2
CH
2
COOH – kw. butanowy (masłowy)
5. CH
3
CH
2
CH
2
CH
2
COOH - kw. pentanowy (walerianowy)
6. CH
3
(CH
2
)
4
COOH - kw. heksanowy (kapronowy)
7. CH
3
(CH
2
)
5
COOH – kw. heptanowy (enantowy)
8. CH
3
(CH
2
)
6
COOH – kw. oktanowy (kaprylowy)
9. CH
3
(CH
2
)
7
COOH – kw. nonanowy (pelargonowy)
10.
CH
3
(CH
2
)
8
COOH – kw. dekanowy (kaprynowy)
11.
CH
3
(CH
2
)
9
COOH – kw. undekanowy (undecylowy)
12.CH
3
(CH
2
)
10
COOH – kw. dodekanowy (laurynowy)
16.CH
3
(CH
2
)
14
COOH – kw. heksadekanowy (palmitynowy)
17.17. CH
3
(CH
2
)
15
COOH – kw. heptadekanowy (margarynowy)
18.
CH
3
(CH
2
)
16
COOH – kw. oktanodekanowy (stearynowy)
W nazwie systematycznej, miejsce przyłączenia podstawnika wskazuje
się cyfrą odpowiadającą kolejnemu numerowi atomu C w łańcuchu. Za
atom C-1 uważa się atom C grupy karboksylowej, np.
C
C
C
C
COOH
5
4
3
2
1
a
b
g
d
Nazwa systematyczna
Nazwa zwyczajowa
Izomeria pojawia się od kwasu butanowego:
CH
3
CH
2
CH
2
COOH (n-butanowy, masłowy)
C
H
3
CH
COOH
CH
3
lub (CH
3
)
2
CH-COOH (2-propanokarboksylowy,
izomasłowy)
CH
3
CH
2
CH
2
CH
2
COOH (n-pentanowy, walerianowy)
C
H
3
CH
CH
3
CH
2
COOH
(3-metylobutanowy, izowalerianowy)
C
H
3
C
CH
3
C
H
3
COOH
(2,2-dimetylopropanowy, piwalonowy,
trimetylooctowy)
C
H
3
CH
2
C
H
CH
3
COOH
(2-metylobutanowy, 2-butanokarboksylowy)
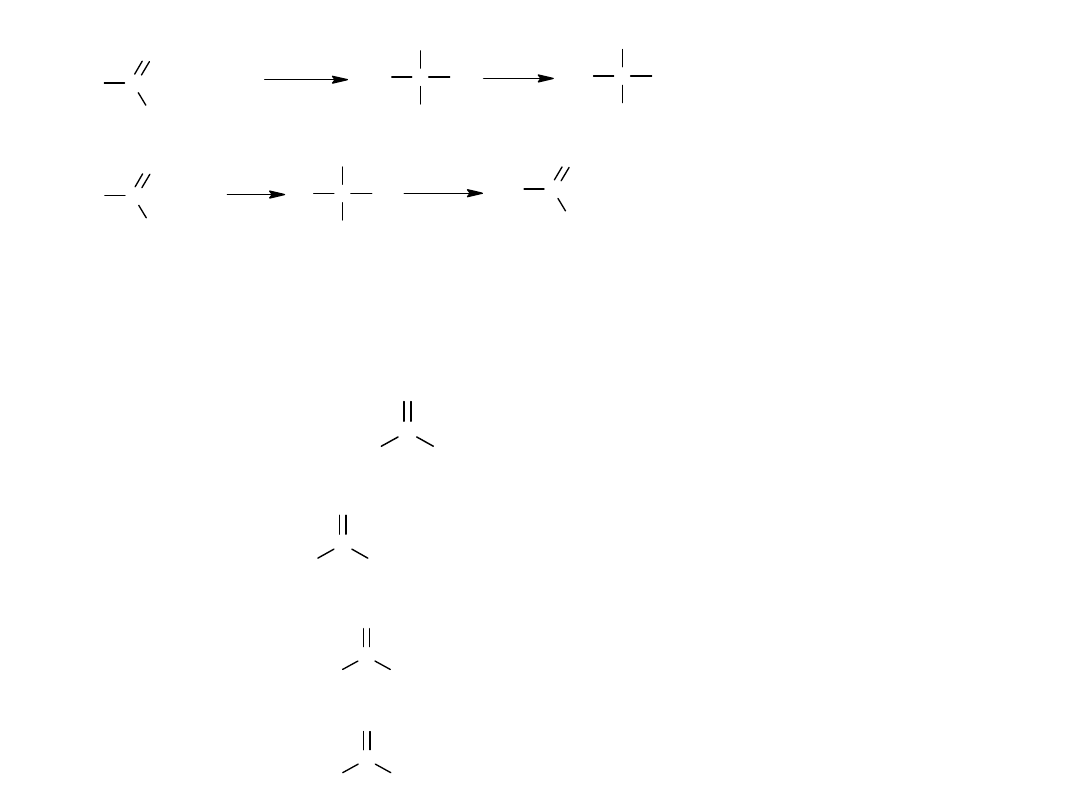
Kwasy karboksylowe zawierające od 1-4 atomów C w cząsteczce są
rozpuszczalne w wodzie. Kwas walerianowy rozpuszcza się częściowo.
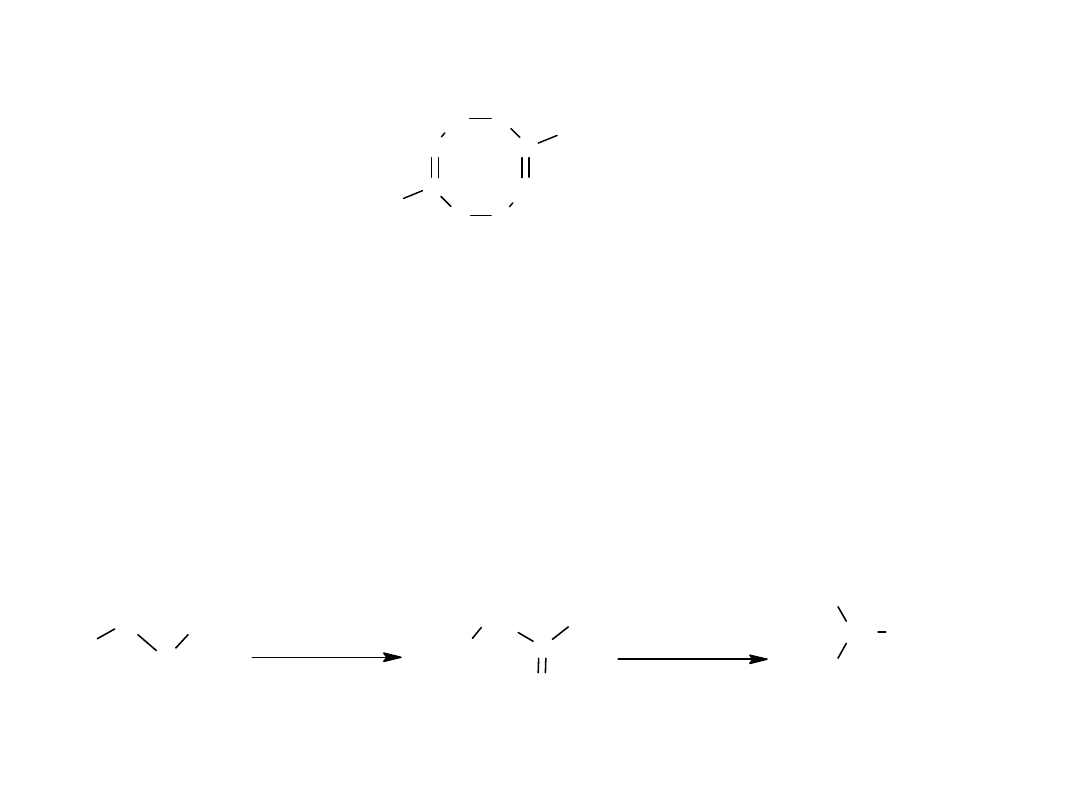
Niższe kw. karboksylowe występują w formie asocjatów (nawet w fazie
gazowej), ponieważ grupa karboksylowa stwarza możliwość powstania 2
wiązań wodorowych.
C
O
O
H
H
H
O
O
C
R
R
Metody otrzymywania kwasów karboksylowych
Istnieje kilka metod ogólnych, oraz pewne specjalne metody np. do
otrzymywania, kwasów aromatycznych.
- utlenianie alkoholi I rz. do aldehydów a następnie kwasów
C
H
3
CH
2
CH
2
OH
propan-1-ol
KMnO
4
C
H
3
CH
2
C
H
O
KMnO
4
C
H
3
C
H
3
CH COOH
2-methylpropanoic acid
kwas izomaslowy
C
H
3
CH
C
H
3
CH
2
OH
KMnO
4
CH
3
C
H
3
CH
COOH
kwas izomaslowy
izobutanal
- hydroliza nitryli
C
H
3
CH
2
Cl
C
H
3
CH
2
CN
K C N
- K C l
cyjanek etylu
C
H
3
CH
2
CN
C
H
3
CH
2
C
OH
O
H
OH
C
H
3
CH
2
C
CH
3
O
- H
2
O
H
2
O , K O H
- N H
3
cyjanek etylu
kw propanowy
(W metodzie tej z niższych związków otrzymuje się kwasy wyższe)
Hydroliza nitryli jest głównie stosowana dla otrzymywania kwasów
aromatycznych.
CN
COOH
kw benzoesowy
benzonitryl
- utlenianie węglowodorów
Metoda ta jest również stosowana dla otrzymania kwasów
aromatycznych:
CH
3
COOH
utl
CH
3
COOH
K
2
Cr
2
O
7,
H
2
SO
4
CH
3
COOH
p-ksylen
kw o-ftalowy
- ze związków Gringarda z suchym lodem (CO2)
C
O
O
C
+
O
-
O
+
RMgI
R
C
O
OMgI
R
C
O
OH
+
MgOHJ
H
2
O
H
3
C
CH
2
HC
CH
3
Cl
M g
s u c h y e t e r
C
H
3
CH
2
CH
C
H
3
MgCl
zw Grignarda
C
O
O
+
C
H
3
CH
2
CH
C
H
3
MgCl
C
H
3
CH
2
CH
CH
3
C
O
OMgCl
H
2
O
C
H
3
CH
2
CH
CH
3
C
O
OH
+
MgOHCl
kw 2 - metylobutanowy
- reakcja Kocha (z alkenów)
C
H
2
CH
2
+
CO
+
O
H
2
C
H
3
CH
2
COOH
C u C l
2
l u b C o C l
2
2 0 0
-
4 0 0 �C , 7 0 0 a t m
Jest to katalityczne przyłączanie wody i tlenku węgla do olefin.
Mechanizm tej reakcji:
R
CH
CH
2
H
+
R
CH
CH
3
addycja protonu
+
R
CH
CH
3
+
+
C=O
(+)(-)
R
CH
C
H
3
C
+
O
karbokation
R
CH
C
H
3
C
O
+
karbokation
hh
H O H
R
CH
CH
3
C
O
O
+
H
H
- H
+
R
CH
CH
3
C
O
OH
kw izotluszczowy
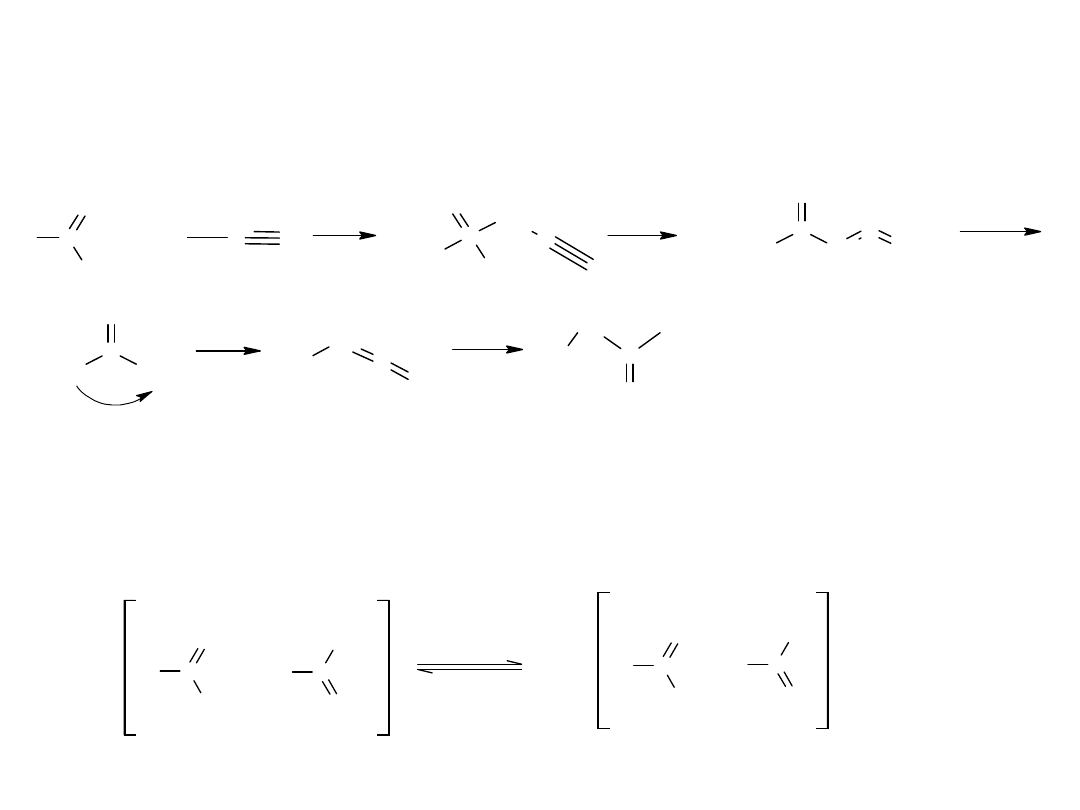
- reakcja Arndt’a i Eistert’a
Reakcja pozwala otrzymać wyższe kwasy z niższych homologów.
Produktem wyjściowym nie są kwasy karboksylowe lecz ich chlorki, na
które działa się diazometanem.
R
C
Cl
O
+
C
H
2
-
N
+
N
R
C
Cl
O
CH
2
N
+
N
- H C l
R
C
O
CH
N
N:
produkt przejsciowy
- N
2
R
C
CH
O
..
R
CH
C
O
keten
H O H
R
CH
2
C
OH
O
Kwasowość kwasów karboksylowych
Dla kwasu karboksylowego i jego anionu, można narysować po 2
struktury:
R
C
O
OH
R
C
O
-
OH
+
I nierownowazne z II
H
+
+
R
C
O
O
-
R
C
O
-
O
III rownowazne z IV
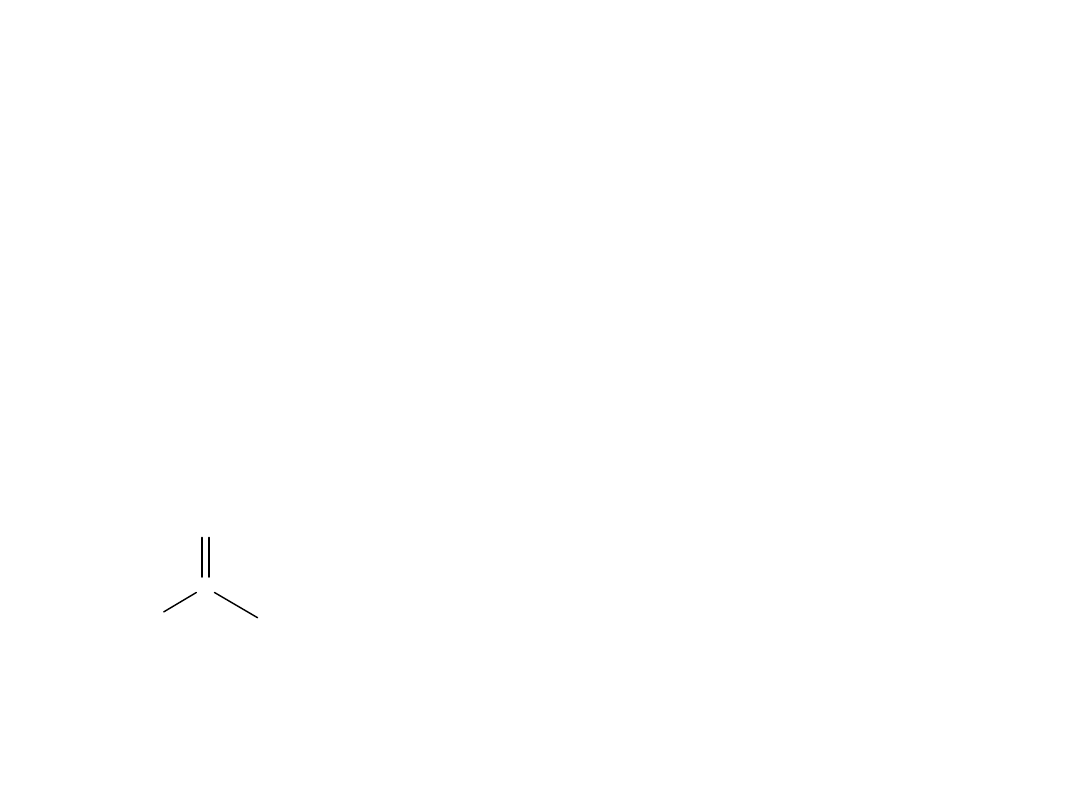
Zarówno cząsteczka kwasu jak i anion, są hybrydami rezonansowymi.
Znacznie bardziej stabilne są równoważne struktury III i IV. W związku z
tym stabilizacja jest większa dla anionu niż dla kwasu, co w konsekwencji
prowadzi do wzrostu jonizacji i zwiększenia wartości Ka.
Oznacza to, że rezonans w przypadku kwasu ma mniejsze znaczenie gdyż
struktury graniczne znacznie różnią się trwałością.
W przypadku jonu równoważne struktury wykazują taką samą trwałość.
Całkowita równocenność tych struktur oznacza, że oba skrajne położenia
elektronów są jednakowo prawdopodobne, czyli że delokalizacja jest
całkowita.
Kwasowość kwasów karboksylowych jest więc spowodowana silną
stabilizacją ich anionu przez rezonans.
Budowa grupy karboksylowej
Grupa karboksylowa jest płaska. (rentgenografia, dyfrakcja elektronów)
C
O
O
H
W niezdysocjowanej cząsteczce kwasu, w grupie karboksylowej istnieją
2 wiązania pomiędzy C i O: –C=O (0,124nm) i –C-OH (0,143nm) o
różnej długości.
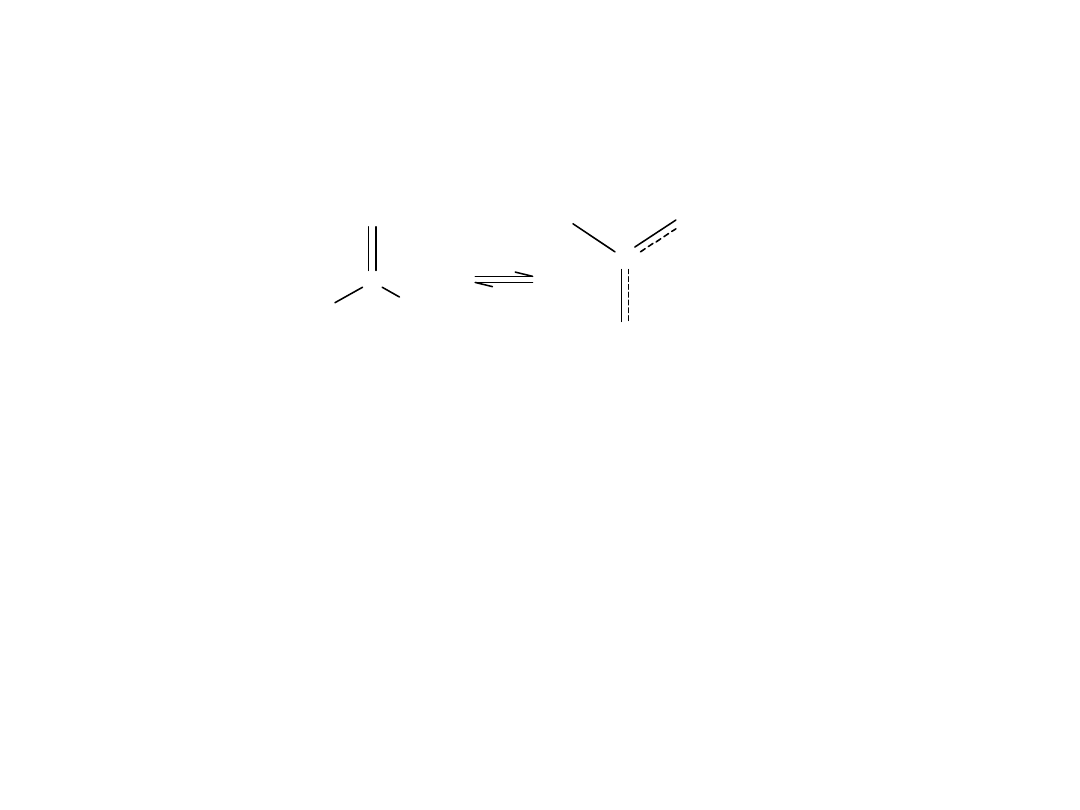
Natomiast w anionach kwasów karboksylowych, wiązania C-O mają
jednakową długość 0,126nm. Wyrównanie długości wiązań oznacza, że
dysocjacja zaciera różnice między atomami O, czyli, że są one połączone
w jednakowy sposób z atomem C. Stan taki jest możliwy, gdy ujemny
ładunek anionu jest jednakowo rozdzielony na obu atomach O.
C
O
OH
0,124nm
0,143nm
C
O
+
O
+
0,126nm
0,126nm
Reakcje chemiczne kwasów karboksylowych
W obrębie grupy funkcyjnej:
I - wymiana H; sole
II - podstawienie grupy hydroksylowej; estry,
amidy, chlorki kwasowe, bezwodniki
III - reakcje w obrębie grupy karbonylowej;
alkohole
IV - wymiana grupy karboksylowej; halogenki
alkilowe

Sole powstają w reakcji kwasów metali lub zasad z kwasami
2CH
3
COOH + Zn (CH
3
COO)
2
Zn + H
2
RCOOH + NaOH RCOONa +H
2
O
RCOOH +NH
4
OH RCOONH
4
+ H
2
O
Wiele soli kwasów organicznych ma praktyczne zastosowanie np. octany
glinu i żelaza hydrolizując w gorącej wodzie, tworzą zasadowe octany,
stosowane jako sole zaprawowe do barwienia tkanin. Benzoesan sodu
służy jako środek konserwujący. Sole sodowe wyższych kwasów
tłuszczowych są mydłami.
C
15
H
31
COOH + NaOH C
15
H
31
COONa +H
2
O (palmitynian sodowy)
Sole sodowe i potasowe są w wodzie rozpuszczalne. Powyżej C=22 stają
się trudno rozpuszczalne. Zasada działania mydła polega na właściwej
orientacji hydrofilowego kationu i hydrofobowego łańcucha alkilowego w
zależności od środowiska.
Mydła otrzymuje się w procesie zmydlania tłuszczów.
Tłuszcze są to estry kwasów tłuszczowych i gliceryny.
CH
2
C
H
CH
2
OOC
COO
COO
C
17
H
35
C
17
H
35
H
35
C
17
+
3NaOH
3C
17
H
35
COONa
+
CH
2
CH
CH
2
O
H
OH
O
H
tluszcz (gliceryd)
reakcja zmydlania
stearynian sodu (mydlo)
gliceryna
h y d r o f o b o w e
w n e t r z e m i c e li
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
COO
-
Na
+
O
H
2
O
H
2
O
H
2
O
H
2
O
H
2
O
H
2
O
H
2
O
H
2
O
H
2
O
H
2
Schematyczny
przekrój miceli
soli sodowej
kwasu
tłuszczowego w
roztworze
wodnym.
W wyniku reakcji podstawienia grupy hydroksylowej powstają chlorki
kwasowe, bezwodniki, estry i amidy.
Charakter zasadowy grup podstawiających grupę –OH w kwasach:
–Cl
–RCOO
–OR
-NH2
Cechą charakterystyczna związków acylowych, (kwasów karboksylowych i
ich
pochodnych) jest substytucja nukleofilowa, podczas której grupy –OH, -Cl,
-RCOO,
-OR i -NH2 zostają zastąpione innymi grupami o charakterze zasadowym.
Związki acylowe wykazują pewne podobieństwo do aldehydów i ketonów,
ponieważ posiadają grupę karbonylową.
Obecność grupy karbonylowej w związkach acylowych powoduje, że są one
podatne na atak nukleofilowy.
Występuje tu jednak pewna różnica:
W reakcji aldehydu lub ketonu tetraedryczny produkt przejściowy
przyjmuje proton, co w rezultacie prowadzi do addycji.
W reakcji zwązków acylowych, tetraedryczny produkt przejściowy,
odszczepia grupę W prowadząc do substytucji. (powstaje związek o
budowie trygonalnej).
R
C
O
R'
+
:Z
R
C
O
-
R'
Z
H
+
R
C
OH
R'
Z ADDYCJA (aldehydy + ketony)
R
C
O
W
:Z
+
R
C
O
-
W
Z
R
C
O
Z
+
:W
SUBSTYTUCJA (zw acylowy)
W= X
-
,ROO
-
,OR
-
,NH
-
2
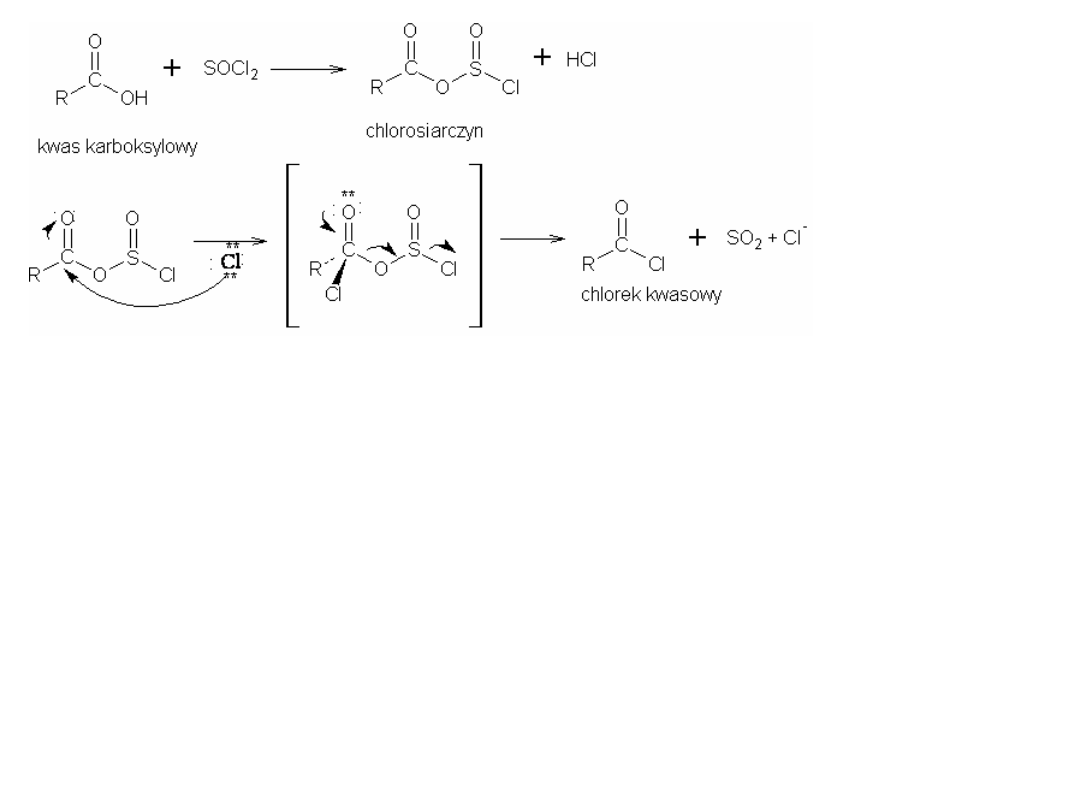
Metody otrzymywania chlorków kwasowych
(bromki i jodki są również znane, ale nie mają znaczenia praktycznego)
3R-COOH + PCl
3
R
C
O
Cl
3
+
H
3
PO
3
R-COOH + PCl
5
R
C
O
Cl
+
POCl
3
+
HCl
R-COOH + SOCl
2
R
C
O
Cl
+
SO
2
+
HCl
R-COOH + SO
2
Cl
2
R
C
O
Cl
+
SO
3
+
Cl
H
Chlorki kwasowe są używane jako środki acylujące (acetylujące), gdyż
reagują z dawcami elektronów.
Są to najbardziej reaktywne pochodne kwasów karboksylowych. Ulegają
reakcji substytucji nukleofilowej, a słabo zasadowy charakter grupy –Cl,
pozwala na natychmiastowe podstawienie jej inna zasadą.
Reaktywność chlorków kwasowych wyraża się w przemianach
zachodzących przy karbonylowym (acylowym) atomie C oraz w jego
bezpośrednim sąsiedztwie.
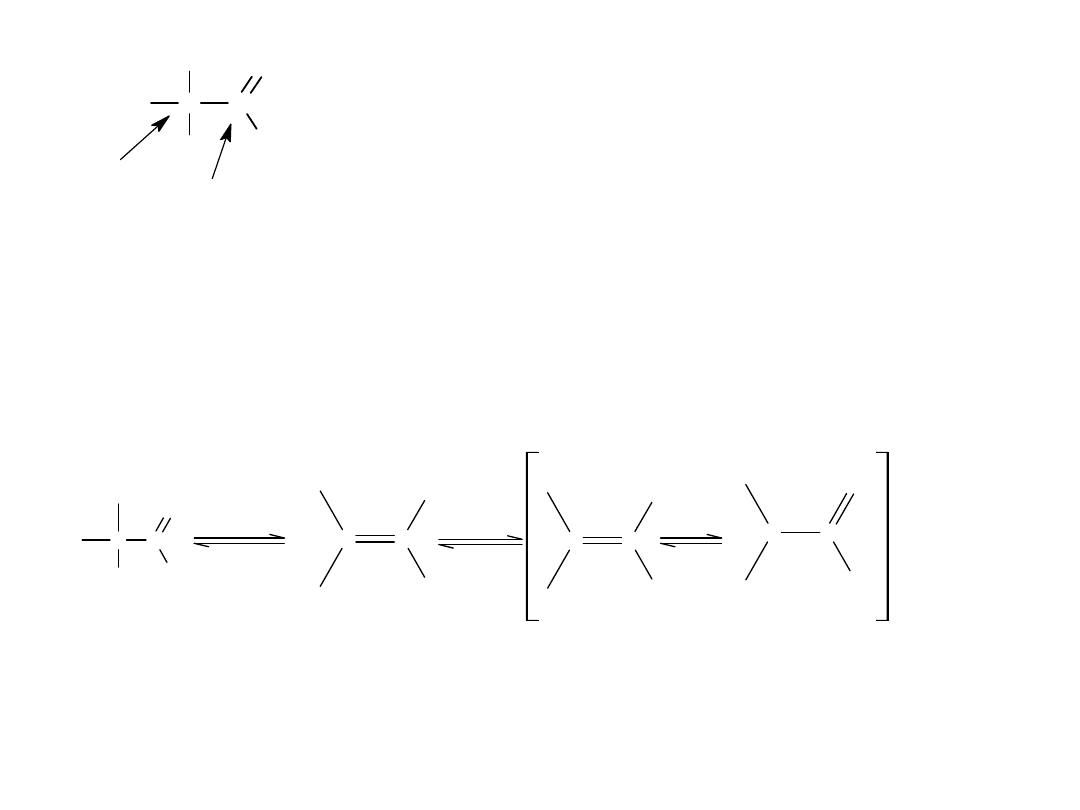
C
C
H
O
Cl
C
C
Cl
OH
- H
+
C
C
Cl
O
-
C
-
C
Cl
O
stabilizowany anion enolu
W przypadku chlorków kwasowych jedyną reakcją wykorzystującą
reaktywność C- α
jest reakcja bromowania metodą Hella, Volharda i Zielińskiego:
Wiązanie C-H w pozycji α chlorków jest reaktywne, ponieważ istnieje
możliwość powstania form enolowych, których aniony są stabilizowane
przez mezomeryczną delokalizację ładunku.
R
C
H
C
H
O
Cl
miejsca reaktywne
(a)
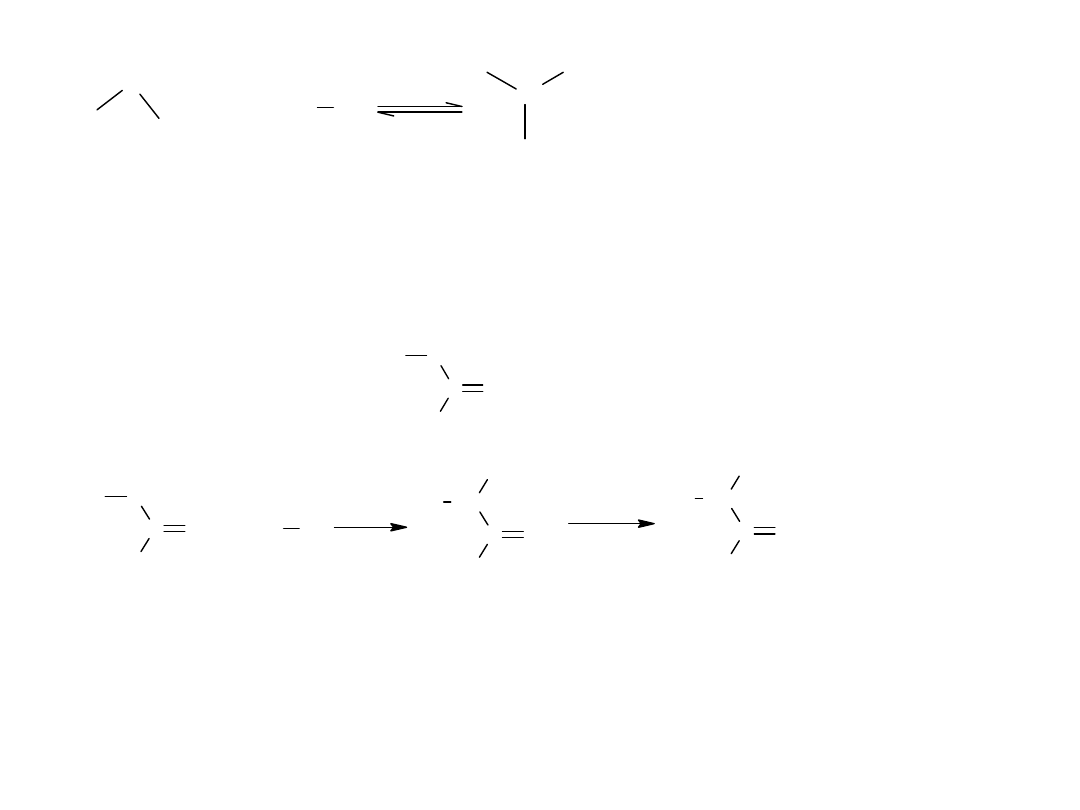
Okazuje się, że szczególną rolę odgrywa tu fosfor, który reagując z Br2
tworzy PBr3 a ten przekształca kwas karboksylowy w jego bromek.
3R-CH
2
-COOH + PBr
3
R
CH
2
C
O
Br
+
H
3
PO
3
3
R
CH
2
C
O
Br
+
Br Br
R
C
H
C
O
Br
Br
H O H
R
C
H
C
O
O
H
Br
R
CH
2
COOH
+
Br Br
P
R
CH
COOH
Br
+
Br
H
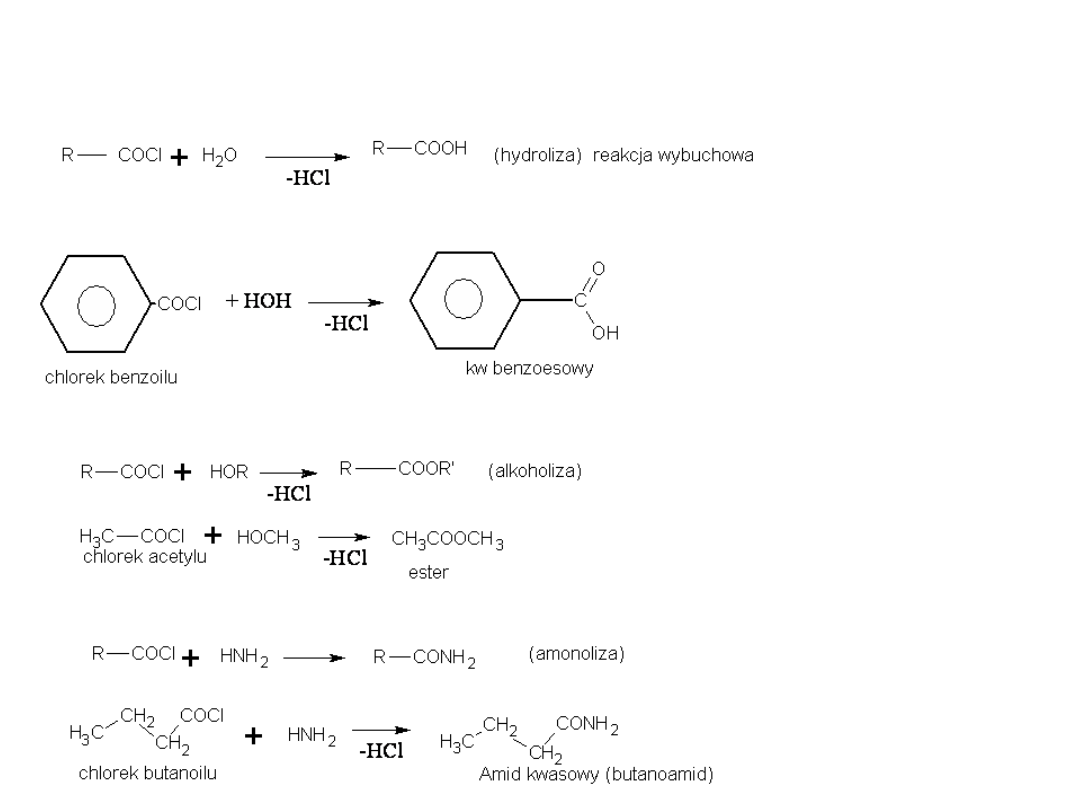
Substytucja nukleofilowa w chlorkach kwasowych
1) Przemiana w kwasy i ich pochodne:
Analogicznie przebiega reakcja z aniliną, w wyniku której powstają anilidy
- H C l
chlorek acetylu
acetanilid
C
H
3
COCl
+
H
2
NC
6
H
5
CH
3
CH
2
CH
2
CONH
2
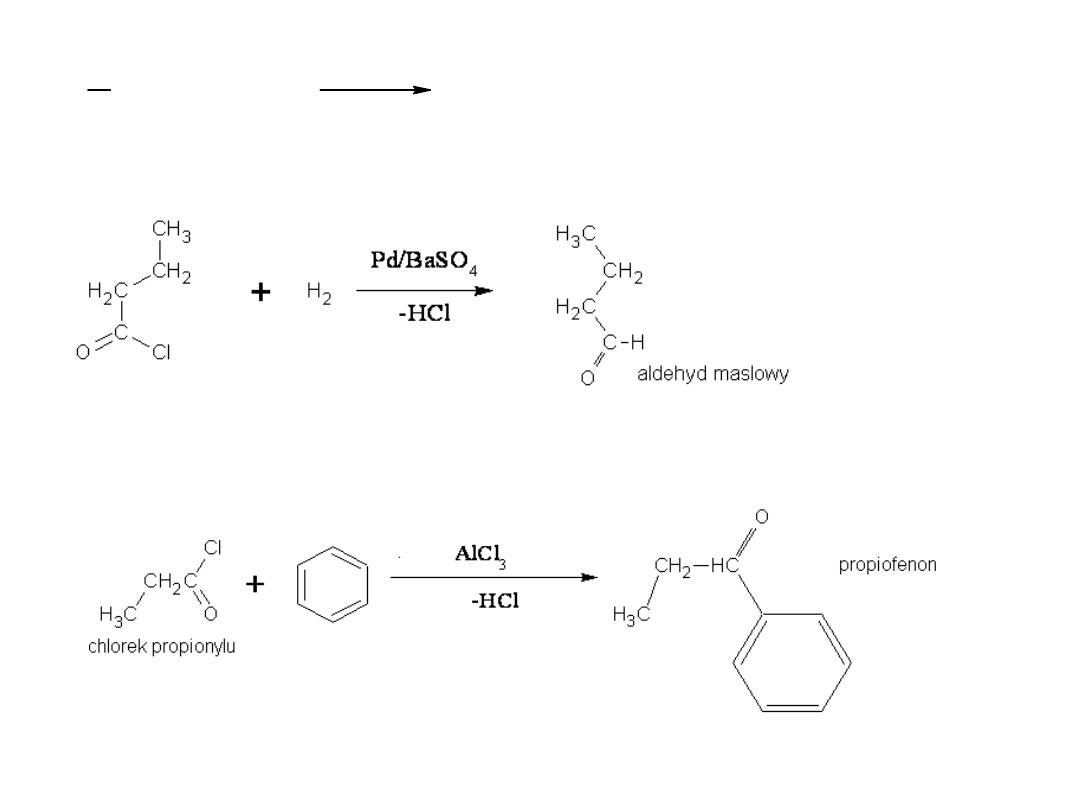
2) Redukcja do aldehydów (reakcja Rosenmunda)
3) Acylowanie metodą Friedela – Craftsa
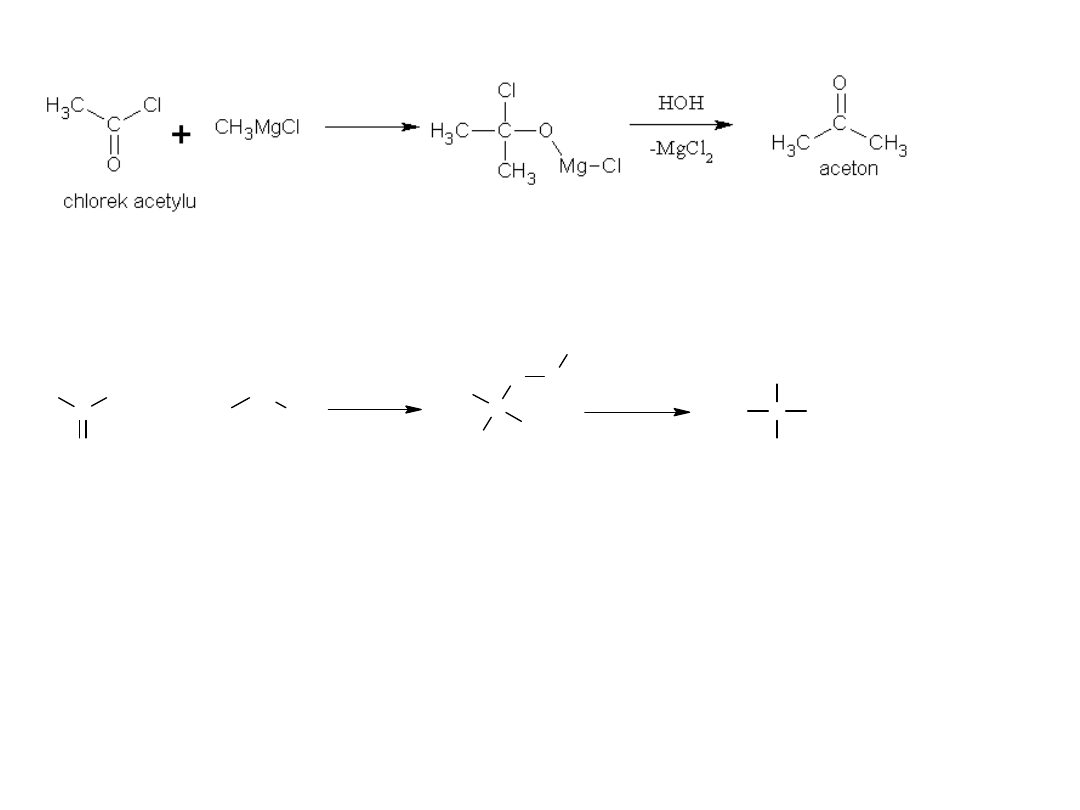
4) Reakcja ze związkami Grignarda
reakcje można prowadzić dalej:
C
H
3
C
O
CH
3
+
C
H
3
Mg
Cl
C
H
3
C
CH
3
C
H
3
O
Mg
Cl
H O H
- M g C l
2
OH
C
H
3
C
CH
3
CH
3
2 - metylopropanol -
2 (alkohol)
Ze względu na dużą reaktywność chlorków kwasowych, częściej
stosuje się słabszy związek metaloorganiczny CH3CdCl, nie
reagujący z ketonami.
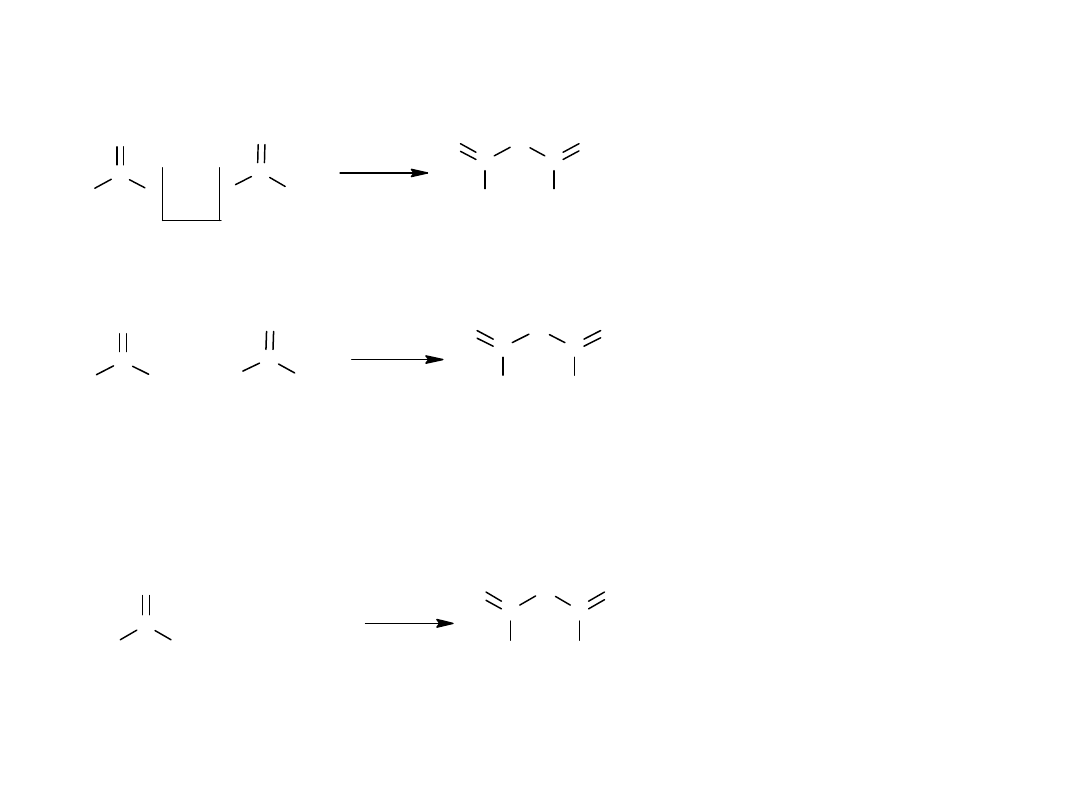
Metody otrzymywania bezwodników kwasowych
Teoretycznie można je otrzymywać przez odszczepienie cząsteczki H
2
O z
kwasu.
O
C
H
3
C
OH
O
O
H
C
CH
3
- H
2
O
O
O
CH
3
C
O
C
CH
3
bezwodnik octowy
ale można :
O
C
H
3
C
OH
O
O
H
C
C
3
H
7
- H
2
O
O
O
CH
3
C
O
C
C
3
H
7
bezwodnik mieszany
Praktyczne metody otrzymywania bezwodników
O
C
H
3
C
Cl
O
O
CH
3
C
O
C
CH
3
bezwodnik octowy
+
NaOOCCH
3
- N a C l
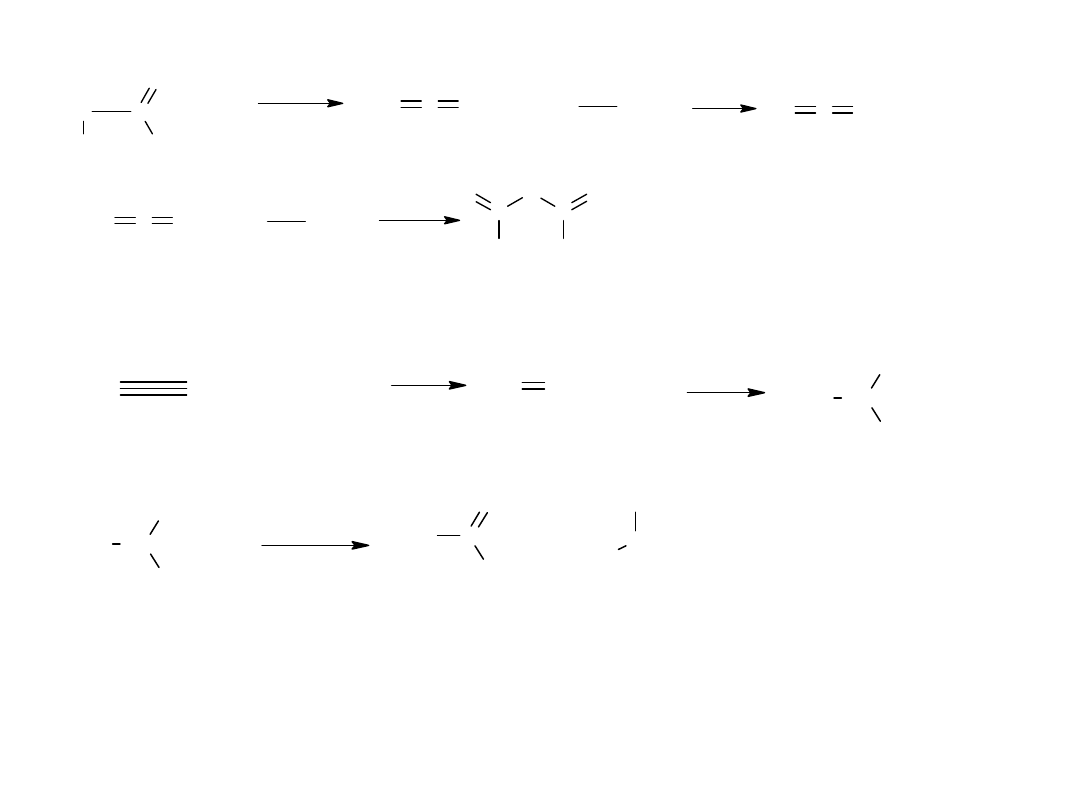
z ketenu, otrzymywanego z bromków bromokwasów
C
H
2
C
Br
O
Br
+
Zn
- Z n B r
2
C
H
2
C
O
lub
keten
C
H
3
COOH
A l P O
4
- H
2
O
C
H
2
C
O
keten
C
H
2
C
O
+
C
H
3
COOH
- H
2
O
O
O
CH
3
C
O
C
CH
3
Przemysł stosuje bardzo ekonomiczne metody otrzymywania
bezwodnika octowego oparte na acetylenie:
C
H
CH
+
HOOCCH
3
s o l e H g
C
H
2
CHOOCCH
3
acetylen
lod kw octowy
octan winylu
C
H
3
C
H
OOCCH
3
OOCCH
3
dioctan etylidenu
C
H
3
C
H
OOCCH
3
OOCCH
3
Z n C l
2
d e s t y l
C
H
3
C
O
H
aldehyd octowy
+
O
COCH
3
CH
3
CO
bezwodnik octowy
Bezwodnik octowy używany jest w przemyśle jako odczynnik do
acylowania. Największe jego ilości zużywa przemysł produkujący
acetylocelulozę, z której otrzymuje się sztuczny jedwab.
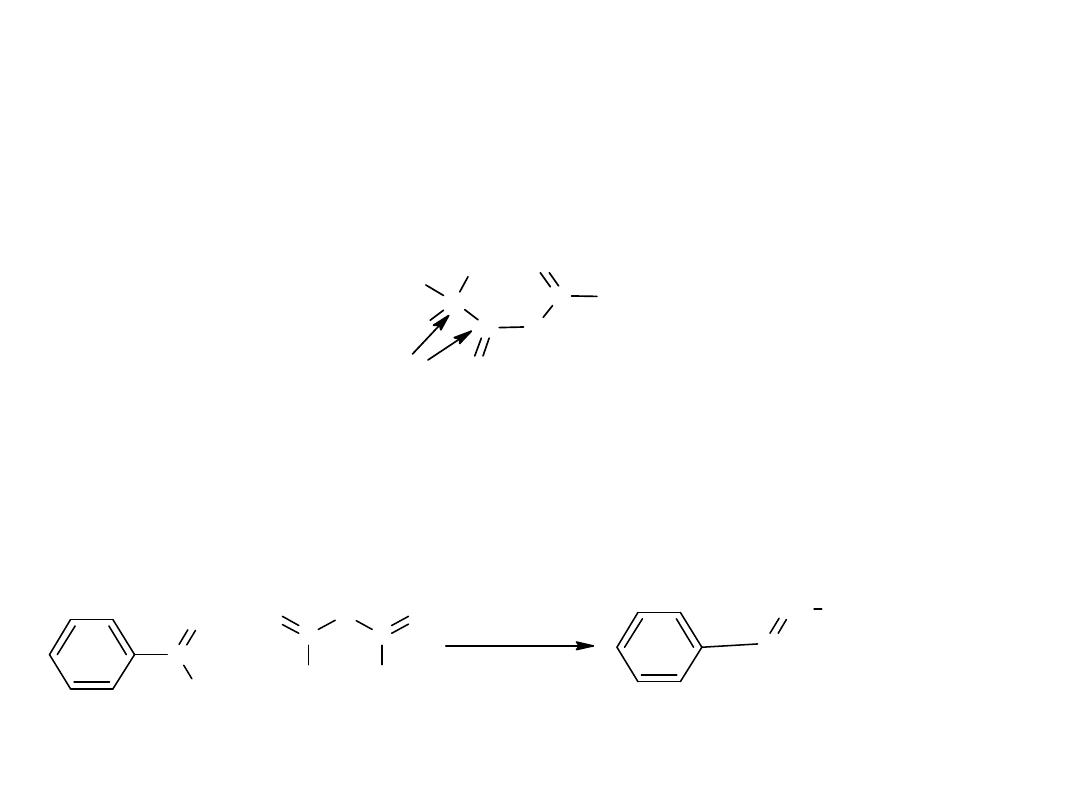
Bezwodniki ulegają takim samym reakcjom jak chlorki kwasowe, choć z
dużo mniejszą szybkością.
Podobnie jak chlorki kwasowe ulegają reakcjom substytucji, a słaby
charakter zasadowy grupy –RCOO pozwala na zastąpienie jej inną zasadą.
Ich reaktywność jest następstwem obecności grupy karbonylowej oraz jej
bliskiego sąsiedztwa.
O
O
H
R
C
H
C
O
C
R
miejsca reaktywne
a
Jedyną reakcją wykorzystującą reaktywność C α w bezwodnikach jest
kondensacja Perkina. Bezwodnik kondensując ze związkiem
karbonylowym daje produkt o rozbudowanym szkielecie węglowym.
C
O
H
+
O
O
CH
3
C
O
C
CH
3
*
a
C H
3
C O O N a
- C H
3
C O O H
CH
CH COOH
aldehyd benzoesowy bezwodnik octowy
kwas cynamonowy

Substytucja nukleofilowa w bezwodnikach kwasowych
- przemiana w kwasy i ich pochodne
(hydroliza)
(RCO)
2
O + HOH 2RCOOH
(CH
3
CO)
2
O + HOH 2CH
3
COOH (kwas octowy)
(alkoholiza)
(RCO)
2
O + R'OH
- R C O O H
RCOOR'
(CH
3
CO)
2
O + HOCH
2
CH
3
CH
3
COOCH
2
CH
3
ester
(amonoliza)
(RCO)
2
O + NH
3
RCONH
2
(CH
3
CO)
2
O + NH
3
- C H
3
C O O H
- R C O O H
CH
3
CONH
2
amid
(CH
3
CO)
2
O
+
N
H
2
C
6
H
5
- C H
3
C O O H
CH
3
CO - NHC
6
H
5
acetanilid
Bezwodniki podobnie jak chlorki reagują z aniliną:
Acylowanie metodą Friedela –Craftsa.
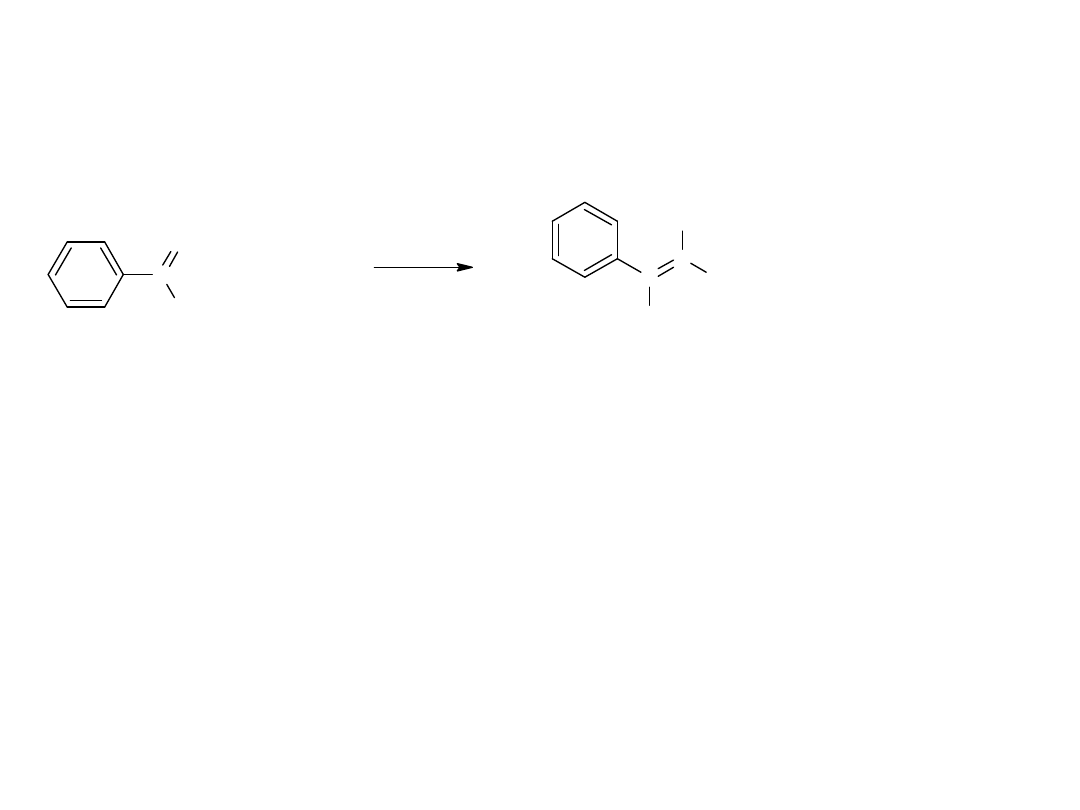
Kondensacja bezwodników ze związkami karbonylowymi
Najprostszym przykładem reakcji Perkina jest otrzymywanie kwasu
cynamonowego z aldehydu benzoesowego i bezwodnika octowego w
obecności octanu potasowego:
C
O
H
+
(CH
3
CO)
2
O
C H
3
C O O K
1 7 0 �C , 5 H
C
C
H
H
COOH
+
CH
3
COOH
kwas trans - cynamonowy (60%)
Jest to reakcja typu kondensacji aldolowej, której pierwszym etapem
jest utworzenie karboanionu z bezwodnika pod wpływem obecnego w
układzie (w postaci soli) anionu kwasu octowego. Karboanion przyłącza
się następnie do grupy karbonylowej aldehydu, po czym następuje
szereg przemian, prowadzących do powstania kwasu cynamonowego.
Pełny mechanizm reakcji Perkina przedstawia schemat:
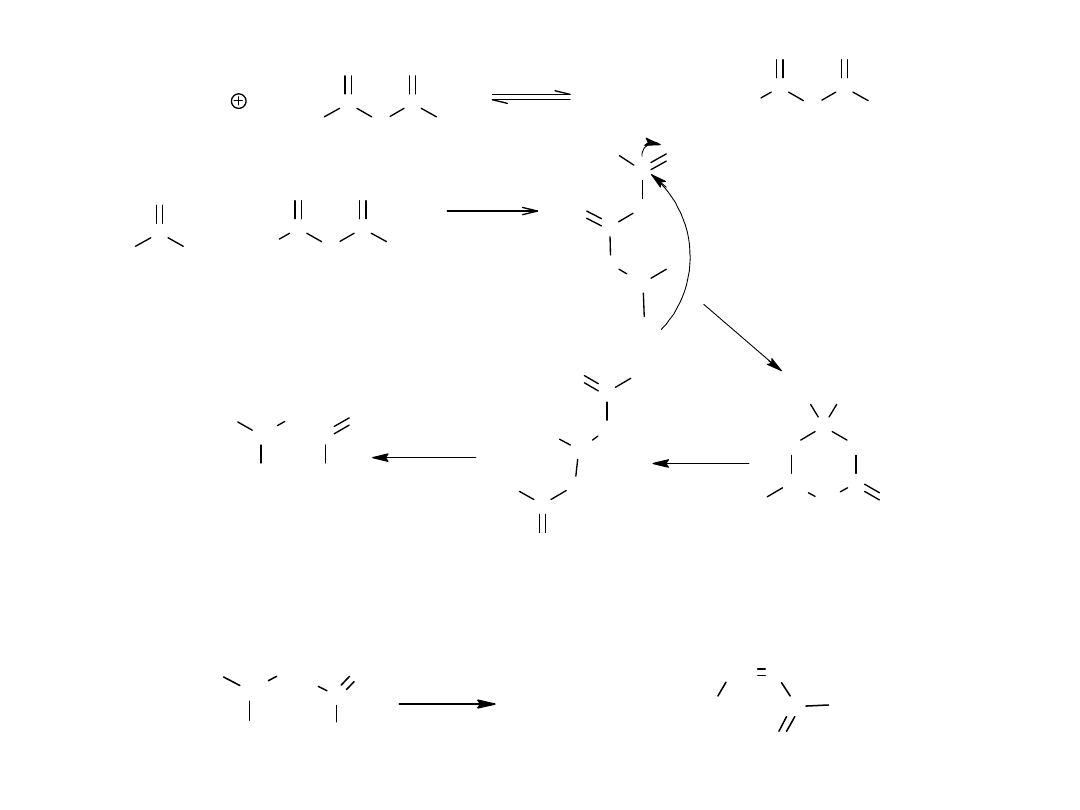
CH
3
COO
+
C
H
3
C
O
C
CH
3
O
O
CH
3
COOH
+
C
H
2
-
C
O
C
CH
3
O
O
O
H
5
C
6
C
H
+
C
H
2
-
C
O
C
CH
3
O
O
C
6
H
5
C
H
C
H
2
C
O
O
C
O
-
O
C
H
3
C
H
3
C
O
-
O
O
CH
C
CH
2
O
H
5
C
6
O
CH
3
C
O
CH
CH
2
C
O
O
-
H
5
C
6
H
+
CH
3
COO
CH
2
C
O
OH
CH
C
6
H
5
Powstały produkt, będący zestryfikowanym przy grupie OH kwasem 2-
fenylo-2-hydroksypropionowym, ulega następnie eliminacji z wydzieleniem
cząsteczki kwasu octowego i utworzenia kwasu cynamonowego:
CH
3
COO
CH
2
C
O
OH
CH
C
6
H
5
CH
3
COOH
+
H
5
C
6
CH CH
C
O
OH

Metody otrzymywania estrów
RCOOH + HOR'
- H
2
O
RCOOR' (estryfikacja)
RCOCl + HOR'
- H C l
RCOOR'
(RCO)
2
O + HOR'
RCOOR'
- R C O O H
I
II
III
Reakcje chlorków i bezwodników prowadzące do estrów zachodzą
również z fenolami.
IV reakcja Arndt’a i Estert’a:
RCOOH + CH
2
N2 RCOOCH
3
+ N
2
(skuteczna metoda otrzymywania estrów metylowych)
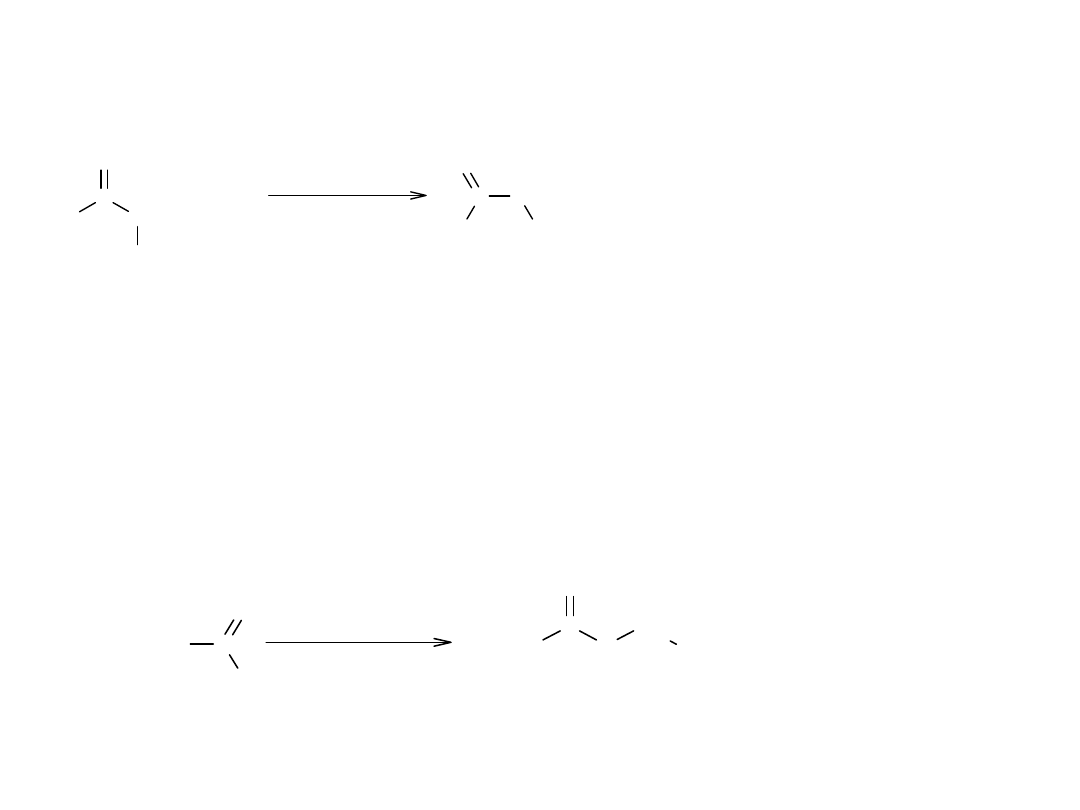
V Transestryfikacja (alkoholiza estrów)
Metoda służy do otrzymywania estrów z estrów bardziej dostępnych.
Jest szeroko stosowana w przemyśle, np. przy produkcji elany.
O
R
C
O
R'
+
HOR"
H
+
l u b O H
-
R
C
O
O
R"
+
R'OH
Mechanizm transestryfikacji jest całkowicie zbieżny z mechanizmem
estryfikacji, ze względu na to, że procesy są podobne. Transestryfikacja
jest procesem odwracalnym, z wyjątkiem transestryfikacji dioli (glikoli).
VI Reakcja Tiszczenki
Reakcja polega na przekształcaniu aldehydów w estry w obecności
alkoholanów glinu. Jest to reakcja analogiczna do reakcji Canizzaro, gdyż
polega na dysproporcjonowaniu aldehydów do alkoholi i kwasów.
C
H
3
C
O
H
( C H
3
C H
2
O )
3
A l
O
C
H
3
C
O
CH
2
CH
3
octan etylu
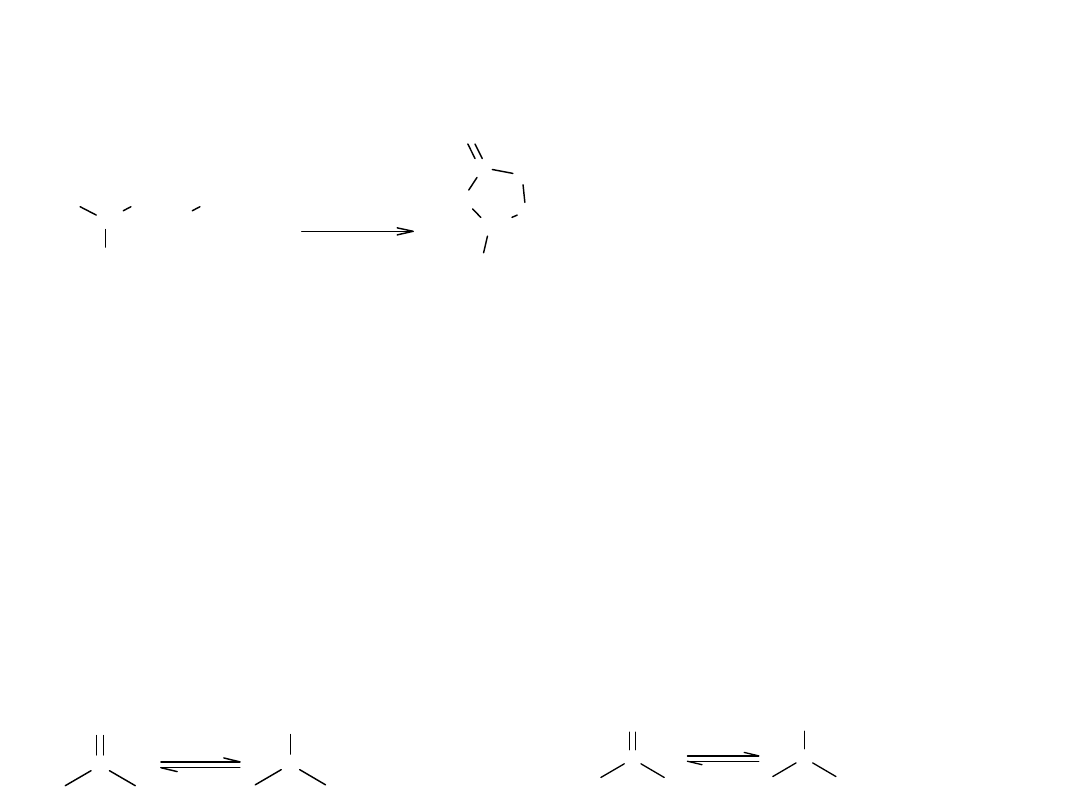
VII Estryfikacja wewnątrzcząsteczkowa
Reakcja charakterystyczna dla hydroksykwasów, które tracąc wodę, tworzą
cykliczny ester – lakton.
R
CH
OH
CH
2
CH
2
COO
-
Na
+
H
+
- H
2
O
O
CH
2
CH
2
CH
O
C
R
p - lakton
sol p - hydroksykwasu
Reakcje estrów
Podobnie jak wcześniej omawiane pochodne kwasów, estry ulegają
reakcji substytucji nukleofilowej.
Wobec odczynników nukleofilowych, estry są mniej reaktywne od
chlorków i bezwodników kwasowych.
Zmniejszona reaktywność grupy karbonylowej estrów, widoczna
zwłaszcza gdy porównuje się estry z ketonami, jest wynikiem sprzężenia
wolnej pary elektronów eterowego atomu O z wiązaniem podwójnym
C=O. Sprzężenie to utrudnia powstanie ładunku dodatniego na acylowym
atomie C.
C
O
C
+
O
-
struktury graniczne
grupy karbonylowej
w aldehydach i ketonach
C
O
C
+
O
-
struktury graniczne grupy estrowej

Dlatego też reakcje substytucji nukleofilowej przeprowadza się niekiedy w
obecności kwasu. W reakcjach katalizowanych przez kwas, proton
przyłącza się do atomu O grupy karbonylowej, wskutek czego atom C tej
grupy staje się bardziej podatny na atak odczynnika nukleofilowego.
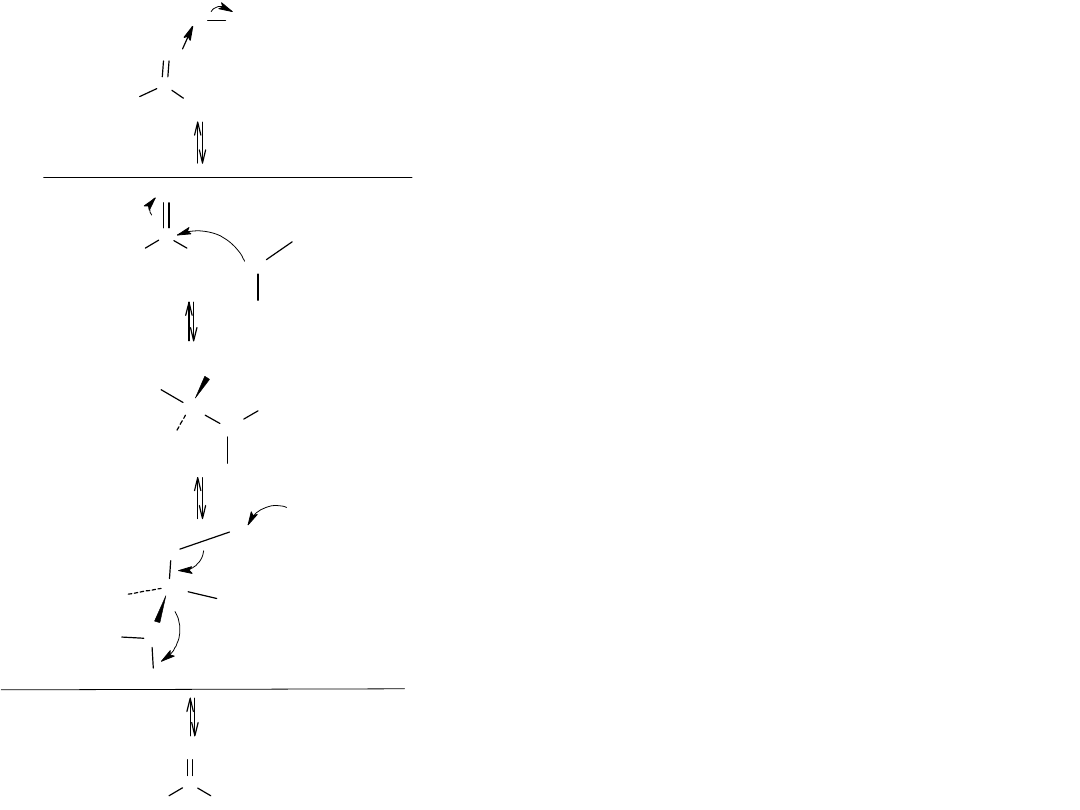
C
O
R
R
O
+
H
+
C
+
O
R
R
OH
C
C
O
C
O
a
a
Miejsca reaktywne
w grupie estrowej
C
R
OH
O
H
Cl
: :
C
R
OH
OH
+
O
H
R'
:**
:
CH
O
H
R
OH
O
+
R'
H'
CH
O
H
R
OR'
O
+
H
H
:OH
2
**
C
R
OR'
O
+
O
H
3
+
Protonowanie karbonylowego atomu
tlenu
Aktywuje kwas karboksylowy...
..na nukleofilowy atak alkoholu, co
prowadzi
do tetraedrycznego produktu pośrednieg.
Przeniesienie protonu z jednego atomu tlenu
na drugi daje drugi tetraedryczny produkt
pośredni
i przekształca grupę OH w grupę łatwo
opuszczającą
Utrata protonu regeneruje katalizator
kwasowy
i prowadzi do produktu estrowego

Rysunek przedstawia mechanizm reakcji estryfikacji Fishera. Reakcja
stanowi katalizowaną kwasem substytucję nukleofilową grupy acylowej
kwasu karboksylowego.
Podczas ogrzewania estru z wodnym roztworem kwasu lub zasady, ester
ulega hydrolizie do kwasu karboksylowego i alkoholu lub fenolu.
W przypadku hydrolizy zasadowej, kwas otrzymuje się w postaci soli, z
której może być wyparty wskutek dodania mocnego kwasu mineralnego.
Hydroliza zasadowa jest procesem zasadniczo nieodwracalnym, gdyż sól
kwasu wykazuje małą skłonność do reakcji z alkoholem.
W środowisku zasadowym, nukleofilem atakującym acylowy atom C, jest
anion –OH.
Mechanizm hydrolizy zasadowej estru:
R
C
O
O
R
+
O
H
-
C
O
H
O
-
O
R
R
C
H
3
C
O
OH
+
R
O
-
R
C
O
-
O
+
R
OH
anion kwasu
Hydroliza stanowi przykład reakcji substytucji
nukleofilowej.
Hydroliza zasadowa zwana jest potocznie zmydlaniem.
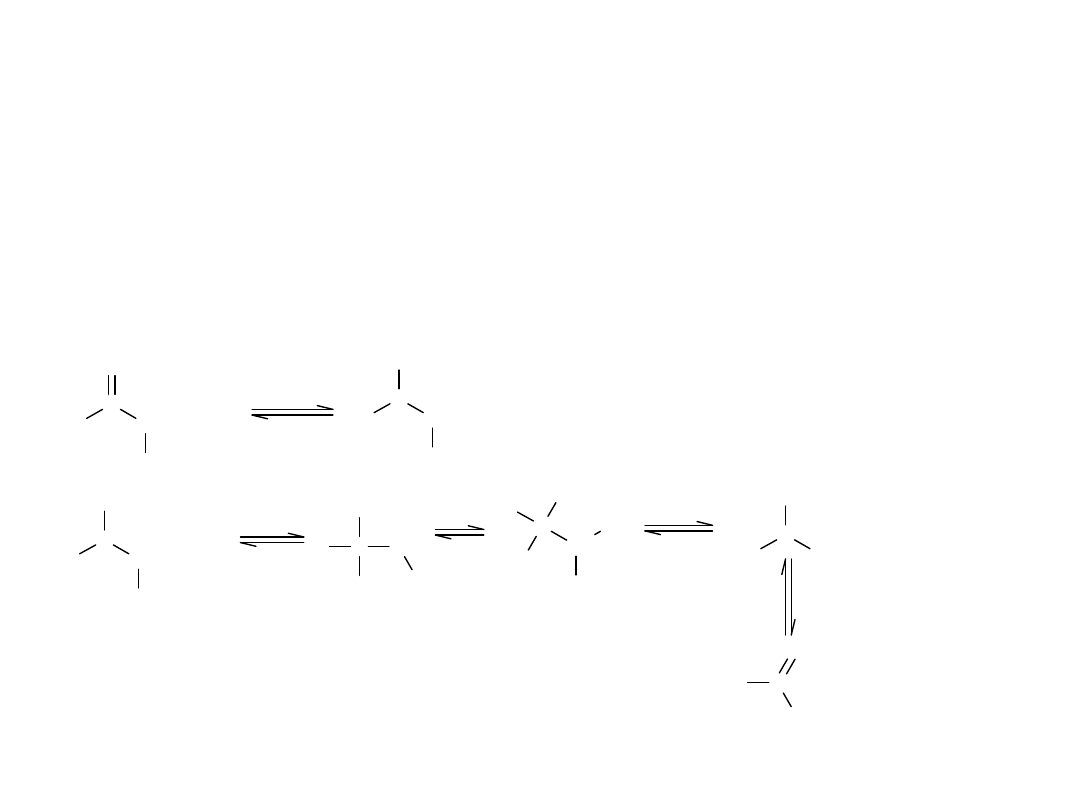
Procesem odwrotnym do estryfikacji jest hydroliza kwasowa. Mocne
kwasy nieorganiczne zwiększają podatność estrów na hydrolizę, na skutek
protonowania atomu O grupy karbonylowej, dzięki czemu zwiększają
podatność atomu C tej grupy na atak nukleofilowy.
W reakcji hydrolizy kwasowej, nukleofilem atakującym acylowy atom C,
jest cząsteczka wody.
Mechanizm hydrolizy kwasowej estrów:
R
C
O
O
R
+
H
+
R
C
+
OH
O
R
z kwasu
R
C
+
OH
O
R
+
O
H
2
R
C
OH
O
R
OH
2
+
R
C
OH
O
+
H
O
H
R
R
C
+
OH
OH
+
ROH
-H
+
R
C
O
OH
kwas
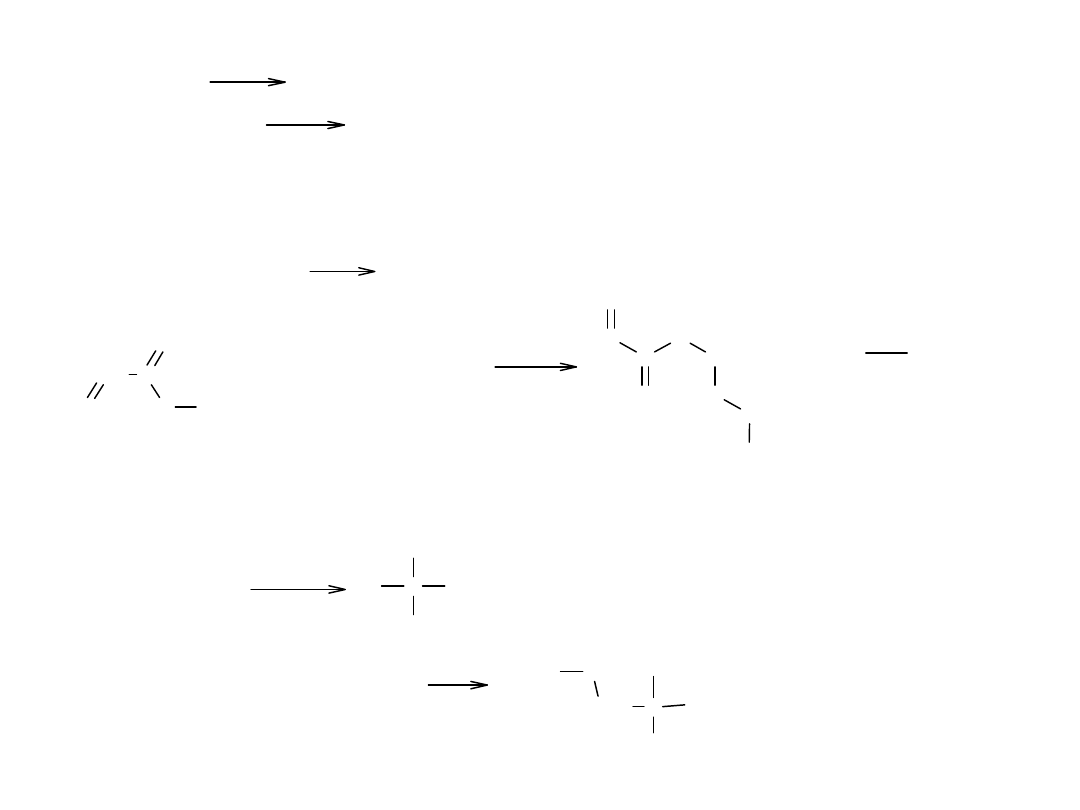
Inne przykłady podstawienia nukleofilowego w estrach:
RCOOR' + NH
3
RCONH
2
+ R'OH (amonoliza)
CH
3
COOC
2
H
3
+ NH
3
- C
2
H
5
O H
CH
3
CONH
2
(acetamid)
- transestryfikacja, czyli alkoholiza
RCOOR' + R"OH
k w l u b z a s
RCOOR" + R'OH
C
H
2
CH C
O
O
CH
3
+
CH
3
CH
2
CH
2
CH
2
OH
O
CH
2
C
H
C
O
CH
2
C
H
2
CH
2
CH
3
+
C
H
3
OH
akrylan metylu
butanol
akrylan butylu
- reakcja ze związkami Grignarda
ROOR' + 2R"MgX
C
OH
R
R'
R"
alkohol III rz
CH
3
CH
2
CH
2
COOC
2
H
5
+
2CH
3
MgJ
C
H
3
CH
2
CH
2
C
CH
3
OH
C
2
H
5
maslan etylu
jodek metylomagnezowy
2 - metylo - 2 pentan
- redukcja estrów do alkoholi.
Metoda służy do otrzymywania wyższych alkoholi z tłuszczów.
CH
3
(CH
2
)
14
COOC
2
H
5
L i A l H
4
CH
3
(CH
2
)
14
CH
2
OH
+
C
2
H
5
OH
palmitynian etylu
1 - heksadekanol
Reakcje z udziałem C
w estrach
Grupa karbonylowa, prócz specyficznych właściwości w jej obrębie,
wpływa na najbliższe sąsiedztwo, zwiększając kwasowość atomów
wodoru przy sąsiednim atomie C. W wyniku jonizacji atomu wodoru a,
powstaje karboanion, który jest hybrydą rezonansową następujących
struktur:
:C
-
C
H
+
:O:
C
C
:O
-
:
**
karboanion
Zwiększona kwasowość atomów H przy węglu a jest przyczyną licznych
reakcji.
Są to reakcje typu kondensacji aldolowej, polegającej na ataku
karboanionu na grupę karbonylową. We wszystkich przypadkach,
karboanion powstaje w ten sam sposób, a mianowicie w wyniku
oderwania przez zasadę atomu H, zajmującego położenie a w stosunku
do grupy karbonylowej.

Typowym przykładem reakcji z udziałem C α w estrach jest kondensacja
Claisena:
Kondensujące ze sobą cząsteczki estru, prowadzą do rozbudowy szkieletu
węglowego
2CH
3
COOC
2
H
5
C
2
H
5
O N a
( z a s )
C
H
3
C
O
CH
2
COOC
2
H
5
+
C
2
H
5
OH
ester eylowy kw acetylooctowego
octan etylu
Mechanizm reakcji jest następujący:
Zasada, którą jest etanolan sodowy, reagując z estrem tworzy anion
estru
C
2
H
5
O
-
+
C
H
3
C
O
O
C
2
H
5
C
2
H
5
OH
+
C
H
2
-
C
O
O
C
2
H
5
C
H
3
C
O
O
C
2
H
5
+
C
H
2
-
C
O
O
C
2
H
5
CH
3
C
O
CH
2
C
O
O
C
2
H
5
+
C
2
H
5
O
-
ketonoester
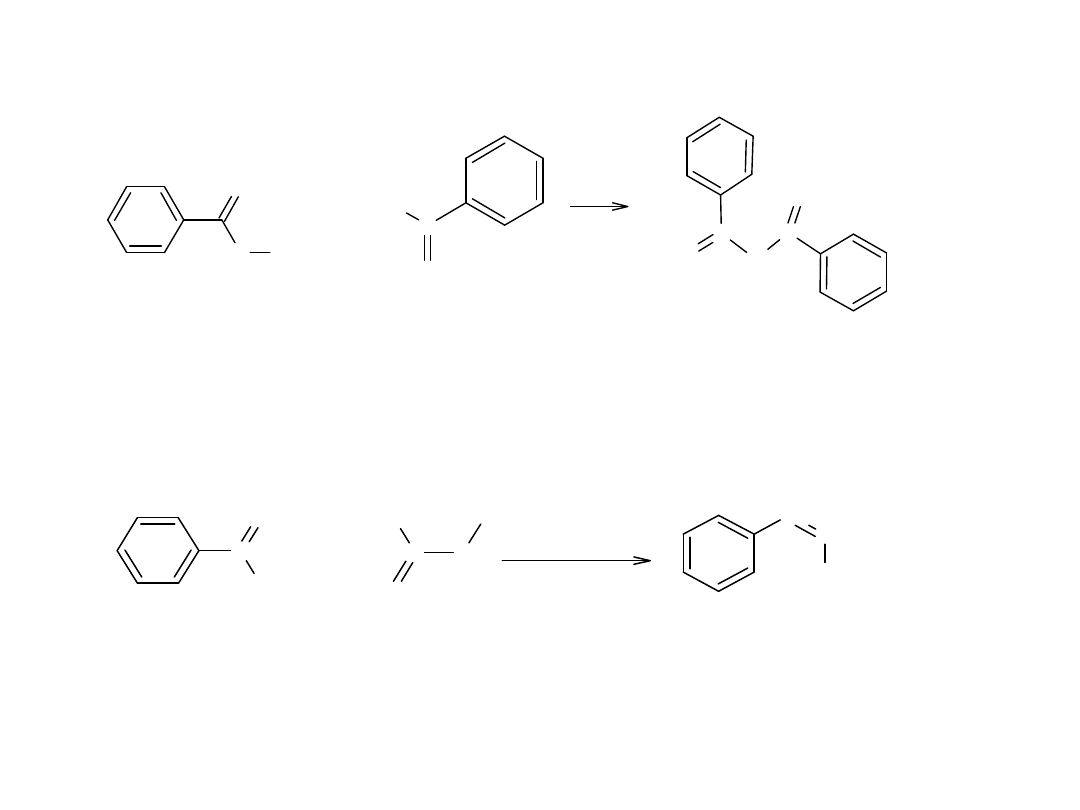
Innym typem kondensacji aldolowej, jest kondensacja estrów z ketonami.
Produktem kondensacji są β-diketony:
O
O
C
2
H
5
+
*CH
3
C
O
(a)
C
2
H
5
O N a
O
C
O
CH
2
C
benzoesan etylu
acetofenon
diketon (dibenzoilometan)
Możliwa jest również, wspomniana przy bezwodnikach kwasowych,
kondensacja Perkina:
C
O
H
*CH
3
C
O
O
C
2
H
5
(a)
+
C
2
H
5
O N a
- H
2
O
CH
CH
COOC
2
H
5
cynamonian etylu
octan etylu
benzaldehyd

Występowanie w przyrodzie i znaczenie estrów
Grupa estrowa jest często spotykanym elementem budowy produktów
naturalnych, pochodzenia roślinnego i zwierzęcego. Obok estrów kwasów
karboksylowych, w przyrodzie występują także estry kwasu siarkowego i
fosforowego. Szczególne znaczenia mają estry kwasu fosforowego, do
których należą kwasy nukleinowe.
Naturalne estry kwasów karboksylowych można podzielić na kilka grup:
są to esencje owocowe i kwiatowe, woski, tłuszcze proste i ich fosforowe
pochodne tzw. fosfoglicerydy (woski, glicerydy i fosfoglicerydy noszą
ogólną nazwę lipidów).
Większość estrów naturalnych charakteryzuje się przyjemnym zapachem
np. octan oktylu – zapach pomarańczy, octan benzylu – zapach jaśminowy,
walerian izoamylu – zapach jabłek.
Przemysł stosuje wiele estrów jako środki zapachowe lub esencje do ciast.
Pewne estry są stosowane jako detergenty, np. proszek IXI, jest estrem
alkoholu laurynowego i kwasu siarkowego.
C
12
H
25
OH + HOSO
3
H
C
12
H
25
OSO
3
H
gdy podstawi sie Na, rosnie rozpuszczalnosc
Triazotan gliceryny znany jest jako nitrogliceryna – materiał wybuchowy.
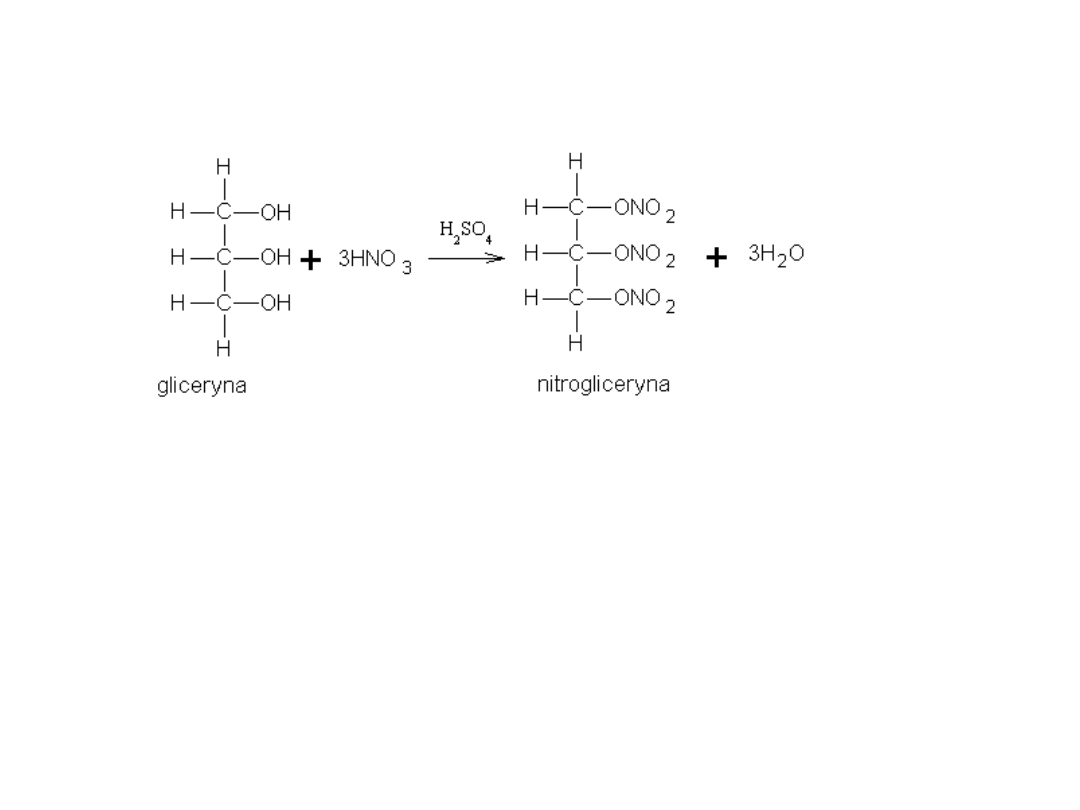
Pierwszym handlowo dostępnym wybuchowym materiałem kruszącym
była nitrogliceryna, otrzymana w 1847 roku w reakcji gliceryny z kwasem
azotowym, w obecności kwasu siarkowego:
Zgodnie z oczekiwaniem, reakcja ta była niezwykle niebezpieczna.
W 1865 roku, szwedzki chemik Alfred Nobel sukcesem zakończył
poszukiwania bezpiecznej metody otrzymywania nitrogliceryny i
wprowadził ją na rynek jako znacznie bezpieczniejszy środek wybuchowy
o nazwie dynamit.
Współczesny dynamit przemysłowy stosowany w kamieniołomach i przy
przygotowaniu podkładów drogowych jest mieszaniną azotanu amonu i
nitrogliceryny zaabsorbowanej na ziemi okrzemkowej.
Wszystkie materiały wybuchowe stosowane w postaci wypełnienia do
bomb lub do amunicji muszą wykazywać małą czułość na wstrząs
spowodowany odpaleniem. Muszą też wykazywać dobrą trwałość w trakcie
długoterminowego przechowywania. TNT (trinitrotoluen), HMX (środek
wybuchowy Jej Wysokości, z ang. His Majesty’s eXplosive) i RDX (środek
wybuchowy ośrodka badawczego – z ang. Research Department eXplosive)
są najpopularniejszymi militarnymi materiałami silnie wybuchowymi. RDX
jest również preparowany z woskami lub syntetycznymi polimerami, dając
tzw. plastikowe materiały wybuchowe, często stosowane przez terrorystów.
CH
3
NO
2
O
2
N
NO
2
N
N
N
N
NO
2
NO
2
O
2
N
O
2
N
N
N
N
NO
2
NO
2
O
2
N
TNT
HMX
RDX
Tłuszcze zwierzęce i roślinne dzieli się na stałe i ciekłe. Do stałych
tłuszczów zwierzęcych należą: smalec i łój a tran rybi stanowi przykład
tłuszczu ciekłego. Wśród stałych tłuszczów roślinnych wymienić należy
oleje palmowy i kokosowy oraz margarynę a wśród ciekłych: oliwę, olej
rzepakowy, olej słonecznikowy, itd.
Margaryna i olej rzepakowy zawierają rakotwórczy kwas erukowy.
CH
3
(CH
2
)
7
CH=CH(CH
2
)
11
COOH

Prócz znaczenia w życiu człowieka, tłuszcze (glicerydy) są surowcami dla
przemysłu. Są one podstawą produkcji pokostu, farb olejnych, lakierów,
mydła, świec. Niektóre gatunki są używane jako smary.
Z mniej energetycznych tłuszczów roślinnych, złożonych głownie z
glicerydów kwasów nienasyconych otrzymuje się tłuszcze stałe w procesie
utwardzania.
C
3
H
5
(OOCC
17
H
33
)
3
H '
2
N i
C
3
H
5
(OOC
17
H
35
)
3
tristearynian
Woski są również estrami wyższych kwasów tłuszczowych i wyższych
alkoholi zawierającymi dodatek wolnych kwasów (tym różnią się od
kwasów), np. wosk pszczeli jest palmitynianem mircylowym
C
17
H
31
COOC
30
H
61
z domieszką kwasu cerotynowego C
25
H
51
COOH.
Wosk służy do wyrobu świec kościelnych i pasty do podłóg.
Innym typem wosku jest lanolina, otrzymywana jako surowiec odpadowy
przy myciu wełny owczej. Stosowana jest w przemyśle kosmetycznym.

Metody otrzymywania amidów kwasowych
1.RCOOH + NH
3
RCONH
2
(m. teoretyczna)
2. RCOOH + R'NH
2
- R 'O H
- H
2
O
3. RCOOCl + NH
3
4. (RCO)
2
O + NH
3
5. RCOOR' + NH
3
6. RCOONH
4
+ NH
3
- H C l
- R C O O H
- R O H
o g r z
- H
2
O
RCONH
2
RCONH
2
RCONH
2
RCONH
2
RCONH
2
Pewne znaczenie przemysłowe posiada synteza amidów z oksymów,
znana jako przegrupowanie Beckmanna.
Reakcja ta zachodzi pod wpływem silnych kwasów protonowych lub
pod wpływem odczynników typu PCl
5
.
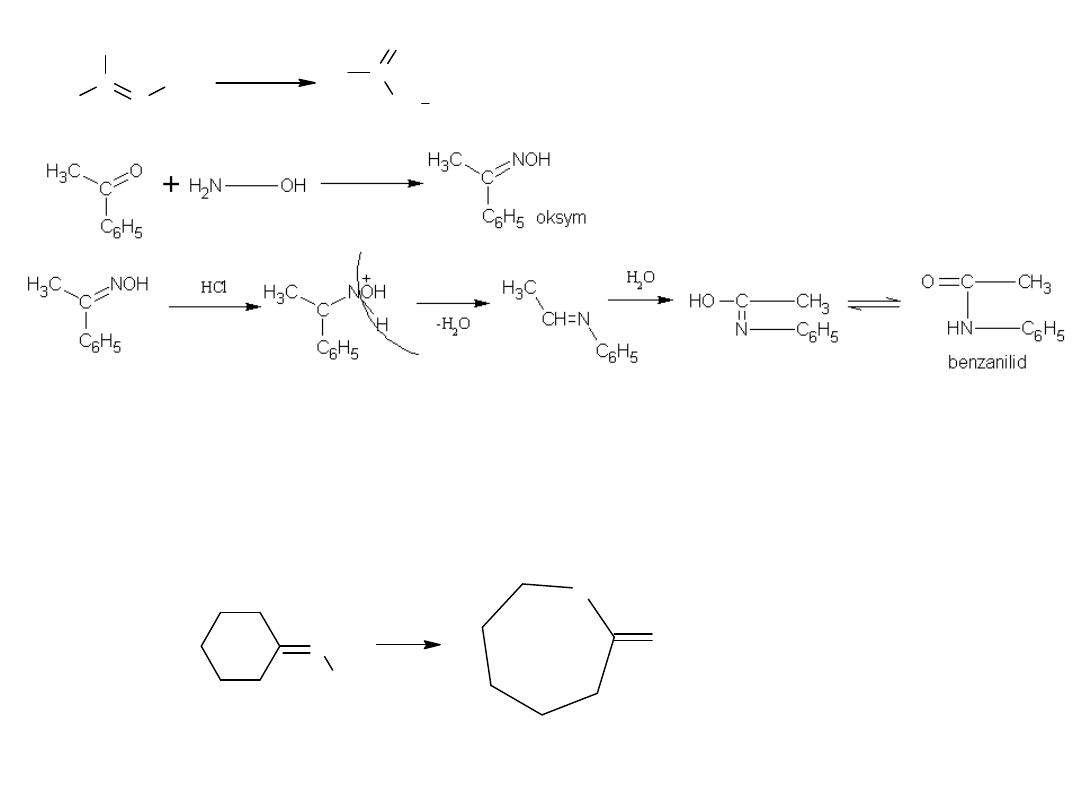
7.
C
N
OH
R
R
H
+
C
NH R
O
R
oksym
przegrupowanie
Metoda wykorzystywana jest do otrzymywania kaprolaktamu, surowca do
syntezy włókien poliamidowych.
Przemysłowa produkcja kaprolaktamu polega na przegrupowaniu
Beckmanna oksymu cykloheksanonu.
N
OH
H
2
S O
4
NH
O
kaprolaktan
oksym cykloheksanonu
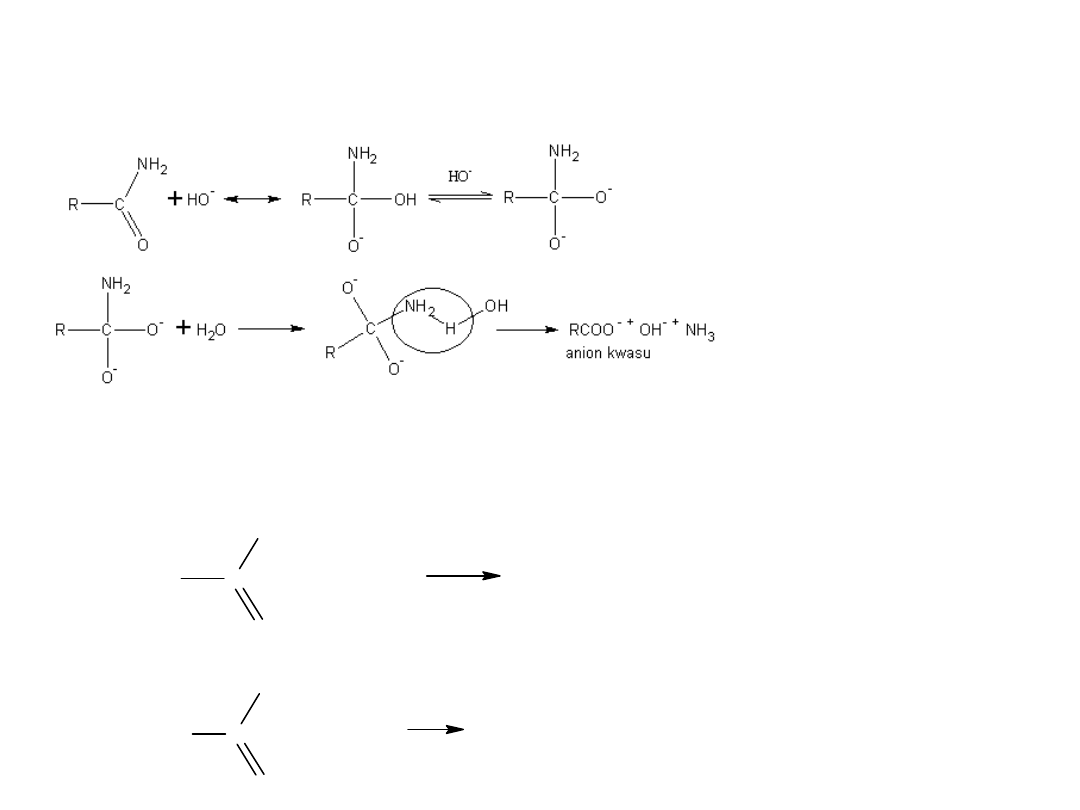
8. Mocznik otrzymuje się z CO
2
i NH
3
CO
2
NH
3
H
2
N - COONH
4
karbaminian amonu
- H
2
O
N
H
2
C
O
NH
2
+
Reakcje amidów kwasowych
Ze względu na zasadowy charakter grupy –NH
2
, amidy kwasowe są
mało reaktywne i tylko w ostrych warunkach zachodzi substytucja
nukleofilowa przy acylowym atomie C. Charakterystyczną cechą
amidów jest brak reaktywności wiązań C-H w położeniu α w
stosunku do grupy –CO oraz pewna reaktywność wiązania N-H.
O
C
N
miejsca reaktywne
W budowie grupy amidowej, znaczną rolę
odgrywa sprzężenie wolnej pary elektronów
atomu N z podwójnym wiązaniem grupy
karbonylowej, wyrażające się w powstawaniu
zdelokalizowanych orbitali, które można
przedstawić strukturami granicznymi:
O
C
N
O
-
C
N
+
Reakcje podstawienia nukleofilowego w amidach kwasowych
1. Hydroliza amidów
Amidy wszystkich typów (RCONH2,RCONHR i RCONR2 oraz imidy), ulegają
hydrolizie kwasowej i zasadowej do amoniaku lub amin i kwasów.
Mechanizm hydrolizy amidów jest analogiczny do hydrolizy estrów.
W hydrolizie kwasowej, nukleofilem atakującym wiązanie amidowe jest
woda.
Hydroliza kwasowa amidów jest nieodwracalna, ponieważ w ostatnim
etapie powstaje jon amoniowy pozbawiony właściwości nukleofilowych.
R
C
O
NH
2
+
H
+
R
C
+
OH
NH
2
R
C
+
OH
NH
2
+
O
H
2
R
C
OH
NH
2
OH
2
+
R
C
OH
NH
3
+
OH
C
H
3
C
O
OH
+
NH
4
+
kwas
W hydrolizie zasadowej amidów, współdziałają ze sobą jony OH- i
cząsteczki H
2
O. Reakcja zaczyna się od ataku nukleofilowego jonu OH- na
acylowy atom C, po czym następuje odłączenie protonu, a powstały
dianion reaguje z H
2
O.
Redukcja amidów za pomocą LiAlH
4.
LiAlH
4
redukuje amidy do amin o tej samej liczbie
atomów C.
R
C
O
NR'
2
+
LiAlH
4
2
2R - CH
2
NR'
2
+ LiAlH
4
H
5
C
2
C
O
NH
2
+
LiAlH
4
C
6
H
5
CH
2
NH
2
+ LiAlO
2
benzyloamina
2. Dehydratacja amidów
Pod wpływem środków odwadniających np. Pb
2
O
5
lub SOCl
2
, amidy tracą
H
2
O i przechodzą w nitryle
R
C
O
NH
2
P b
2
O
5
- H
2
O
R - CN (nitryl)
3. Degradacja amidów Hofmana (reakcja podbrominowa)
Metoda służy do otrzymywania amin I rz. o krótszym łańcuchu
węglowym w stosunku do łańcucha wyjściowego amidu
R
C
O
NH
2
R - NH
2
+ CO
2
( C l O
-
) B r O
-
, O H
-
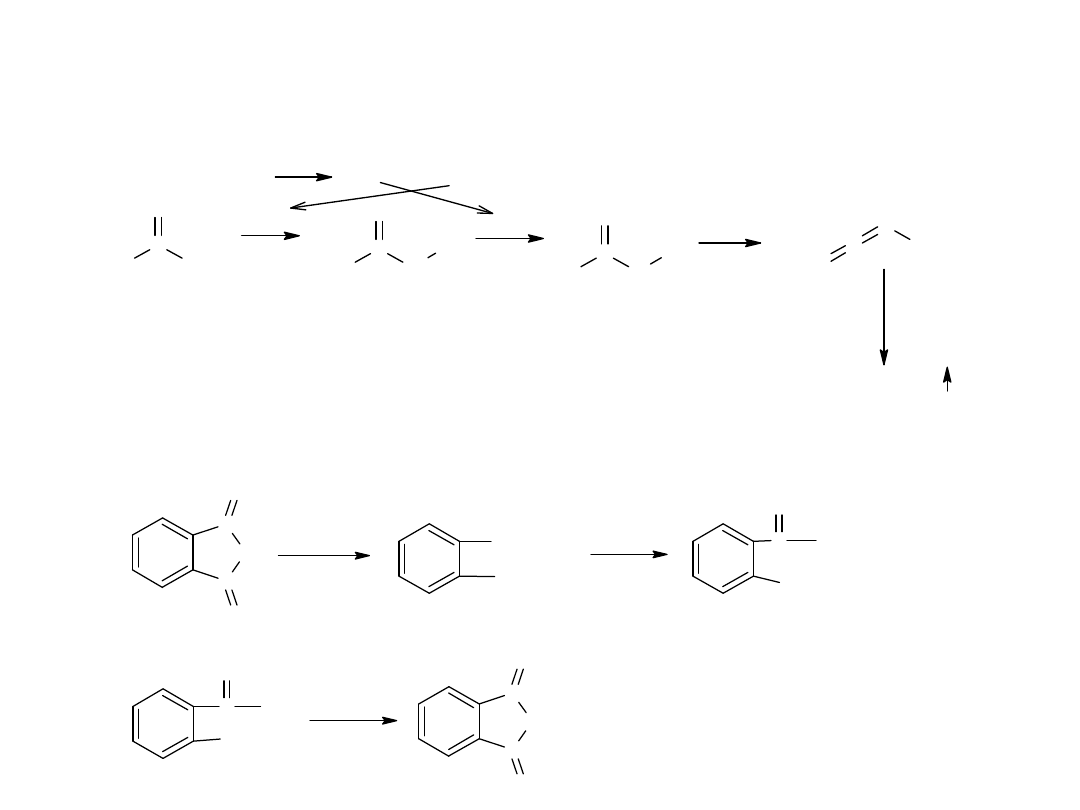
Mechanizm reakcji jest złożony, ponieważ w jej trakcie następuje
przegrupowanie podstawnika R od karbonylowego atomu C do amidowego
atomu N.
Podbromin reaguje w roztworze wodnym jak mieszanina kwasu
podbromowego i wodorotlenku sodu.
NaOBr + HOH
NaOH + HOBr(kwas podbromowy)
O
C
H
3
C
NH
2
H O B r
- H
2
O
O
C
H
3
C
NH
Br
O H
-
- H
2
O
O
C
H
3
C
N
-
Br
- B r
-
O
C
N
R
H
2
O
RNH
2
+ CO
2
amina
Przemiana w imidy
Imidy są to związki, w których atom N połączony jest
z 2 grupami karbonylowymi.
C
C
O
O
O
2 N H
3
CONH
2
COO
-
NH
+
4
(COOH)
C
O
NH
2
COOH
aminokwas
C
O
NH
2
COOH
o g r z
-
H
2
O
C
C
NH
O
O
ftalimid
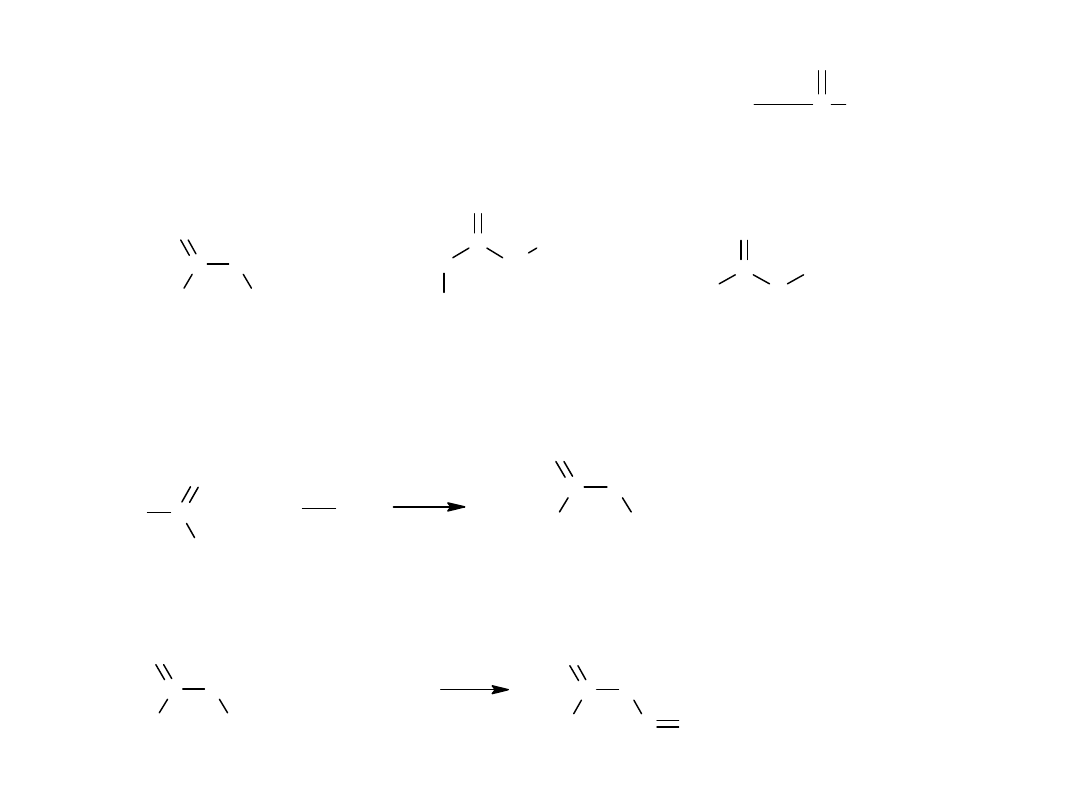
Do amidów należy również zaliczyć kwasy hydroksamowe, hydrazydy i
azydki kwasowe, ponieważ zawierają one charakterystyczną grupę:
C
O
N
C
H
3
C
O
NH
OH
kwas acetohydroksamowy
O
CH
3
C
H
2
C
NH
NH
2
hydrazyd kw propionowego
H
5
C
6
C
O
N
N
2
azydek benzoilu
Hydrazydy powstają w reakcji chlorków kwasowych lub estrów z
hydrazyną.
C
H
3
C
O
Cl
+
N
H
2
NH
2
C
H
3
C
NH
NH
2
O
hydrazyd (wl red)
hydrazyna
Azydki kwasowe powstają w reakcji hydrazydów z kwasem azotowym
(III).
C
H
3
C
NH
NH
2
O
hydrazyd (wl red)
+
HONO
- 2 H
2
O
O
R
C
NH
N
NH
azydek kwasowy
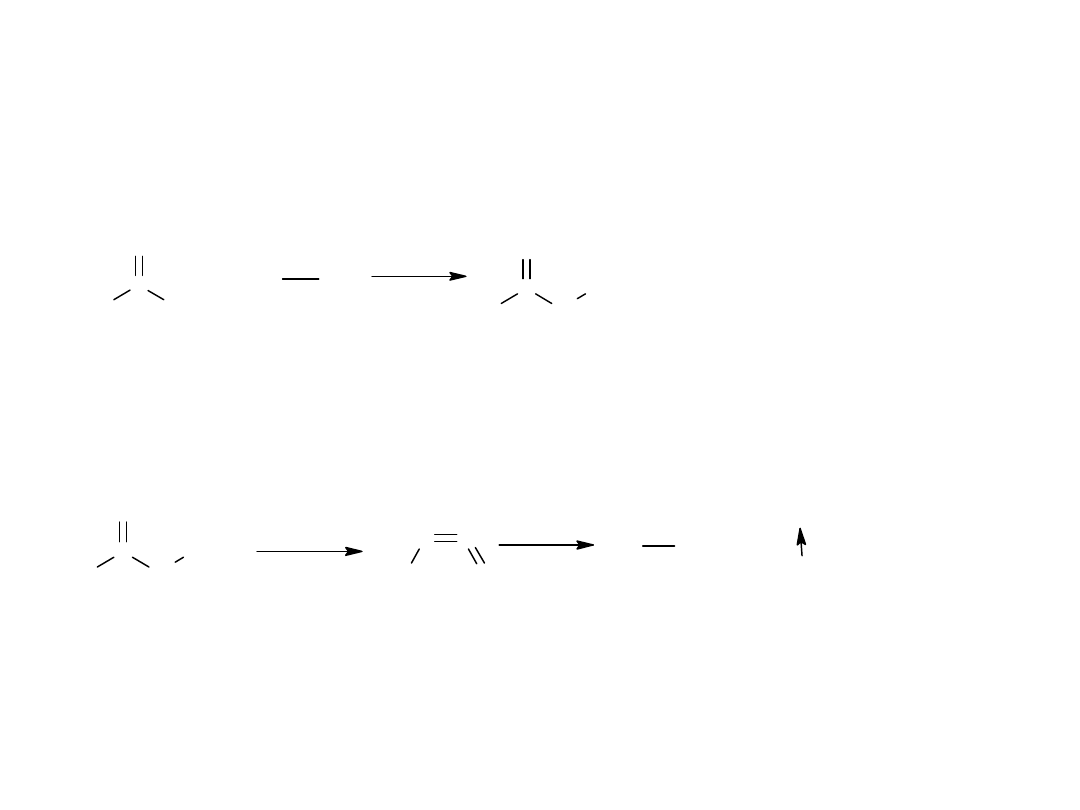
Kwasy hydroksamowe powstają z chlorków kwasowych lub amidów pod
działaniem hydroksyloaminy:
R
C
Cl
O
+
N
H
2
OH
hydroksyloamina
- H C l
R
C
NH
O
OH
kwas hydroksamowy
Kwasy hydroksamowe pod wpływem silnych kwasów ulegają
przegrupowaniu Lossena do amin.
R
C
NH
O
OH
kwas hydroksamowy
k w h i g r o s k o p i j n y
- H
2
O
R
N
C
O
izocyjanian
+ H
2
O
C
H
3
NH
2
+
CO
2
amina
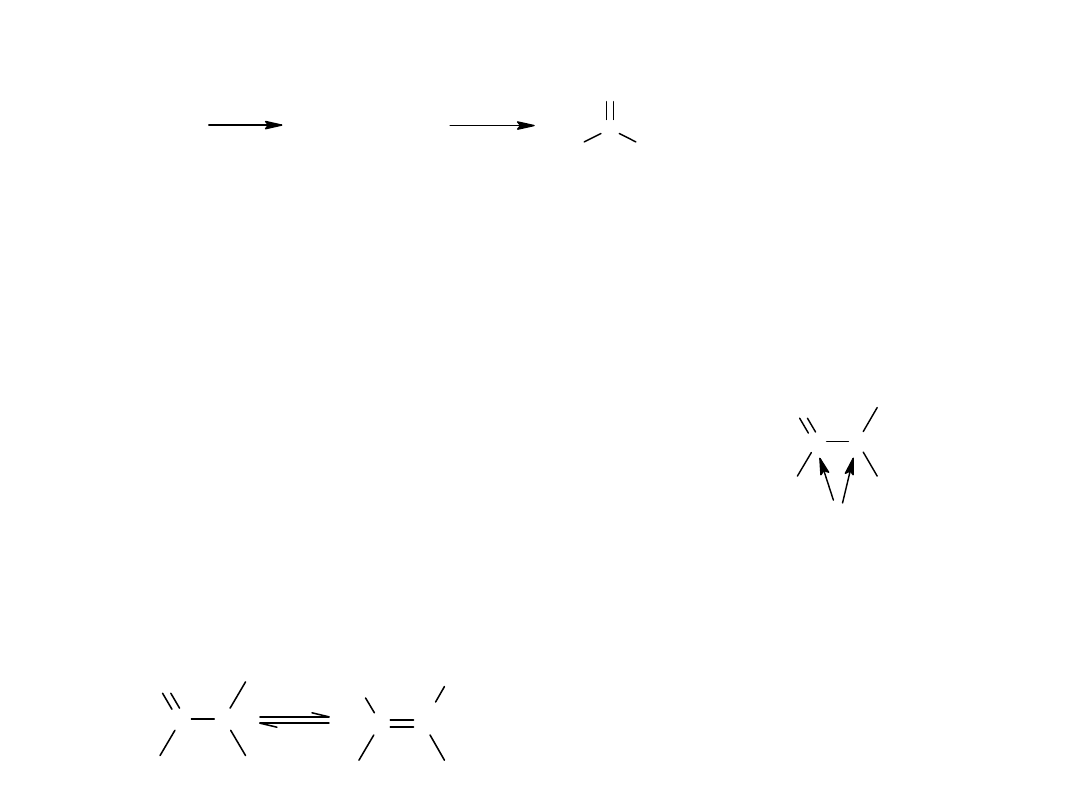
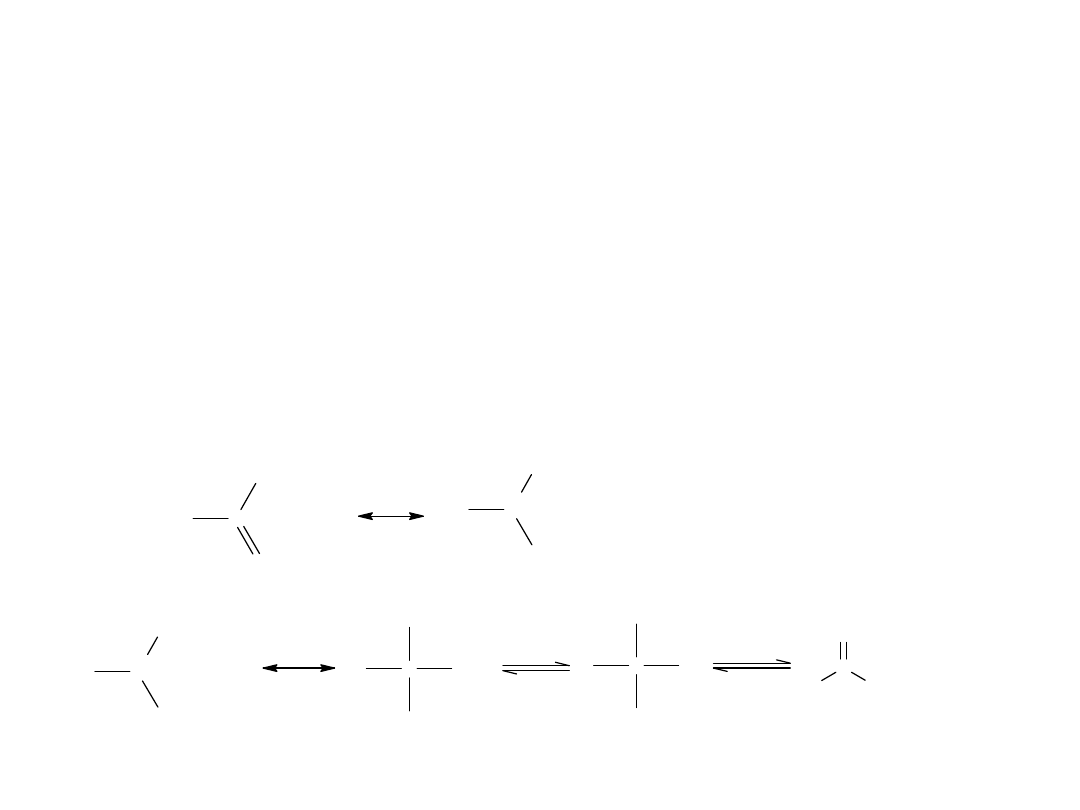
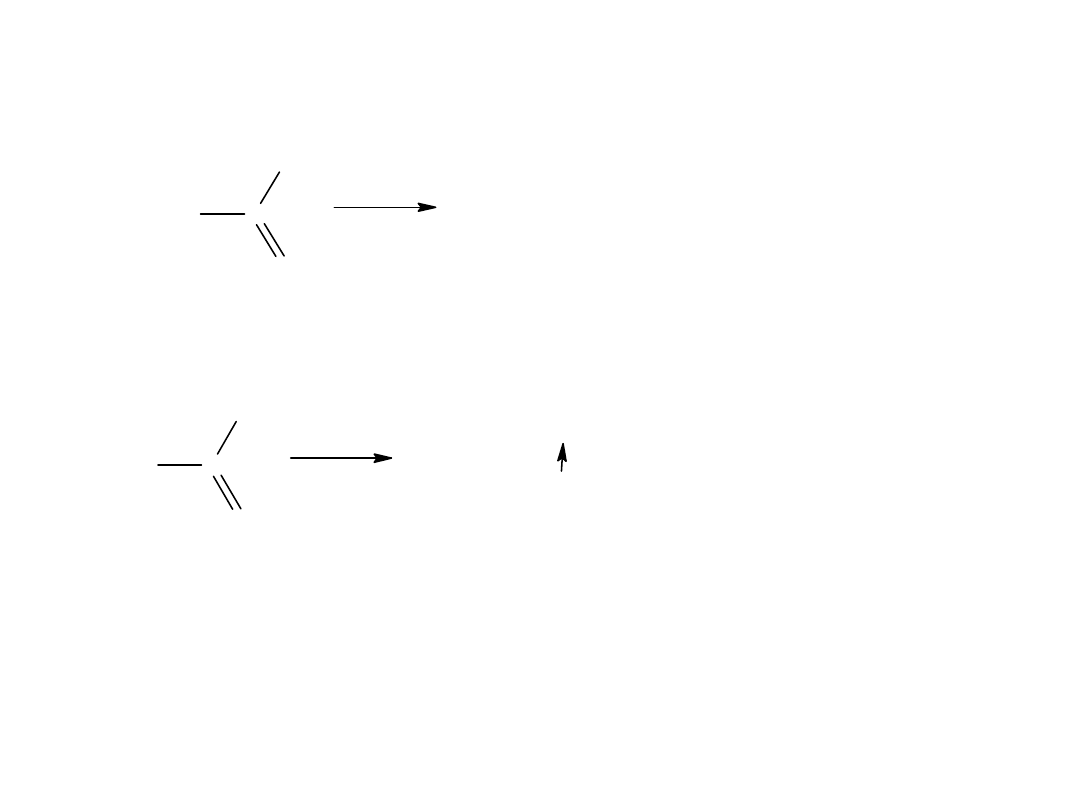
W obrębie grupy karbonylowej kwasu karboksylowego możliwa jest tylko
1 reakcja polegająca na jej redukcji do –CH
2
-.
C
11
H
23
COOH
kw laurynowy
H
2
/ N i
2 0 0 - 3 0 0 A t m
C
11
H
23
CH
2
OH
alkohol laurynowy
Grupę karboksylową można zastąpić grupą karbonylową. Sucha
destylacja soli wapniowych kwasów organicznych prowadzi do ketonów
lub aldehydów:
CH
3
- COO
CH
3
- COO
Ca
octan wpania
o g r z
C
H
3
C
C
H
3
O
+
CaCO
3
aceton
Grupą karboksylową można zastąpić także chlorowcem (Br lub J) w
reakcji Hansdieckera:
R
-
COOAg + Br
2
C C l
4
R - Br + CO
2
+ AgBr

WĘGLOWODANY
Węglowodany ogólnie klasyfikuje się w dwóch grupach: węglowodany
proste i złożone.
Cukry proste, albo monosacharydy, są to węglowodany, takie jak glukoza
i fruktoza, których nie da się przekształcić w procesie hydrolizy w
mniejsze cząsteczki.
Cukry złożone składają się z co najmniej dwóch cukrów prostych
połączonych ze sobą, na przykład, sacharoza - cukier stołowy, jest
disacharydem (dwucukrem), złożonym z jednej cząsteczki glukozy
połączonej z jedną cząsteczką fruktozy. Podobnie, celuloza jest
polisacharydem (wielocukrem) składającym się z kilku tysięcy cząsteczek
glukozy połączonych razem.
1 sacharoza
H
3
O
+
1 glukoza + 1 fruktoza
celuloza
H
3
O
+
~3000 czsteczek glukozy
Monosacharydy można podzielić na aldozy i ketozy.
Przyrostek –oza używany jest w celu oznaczeniu cukru, a przedrostki
aldo- lub keto- oznaczają charakter grupy karbonylowej.
Konfiguracje monosacharydów: projekcje Fischera
Większość z naturalnie występujących cukrów to aldopentozy albo
aldoheksozy.
C
C
C
C
C
CH
3
H
H
H
H
H
O
OH
O
H
OH
OH
C
H
2
C
C
C
C
CH
2
H
H
H
OH
O
OH
OH
OH
O
H
C
C
C
C
H
H
OH
H
O
O
H
H
OH
CH
2
O
H
glukoza (aldoheksoza) fruktoza (ketoheksoza) ryboza (aldopentoza)
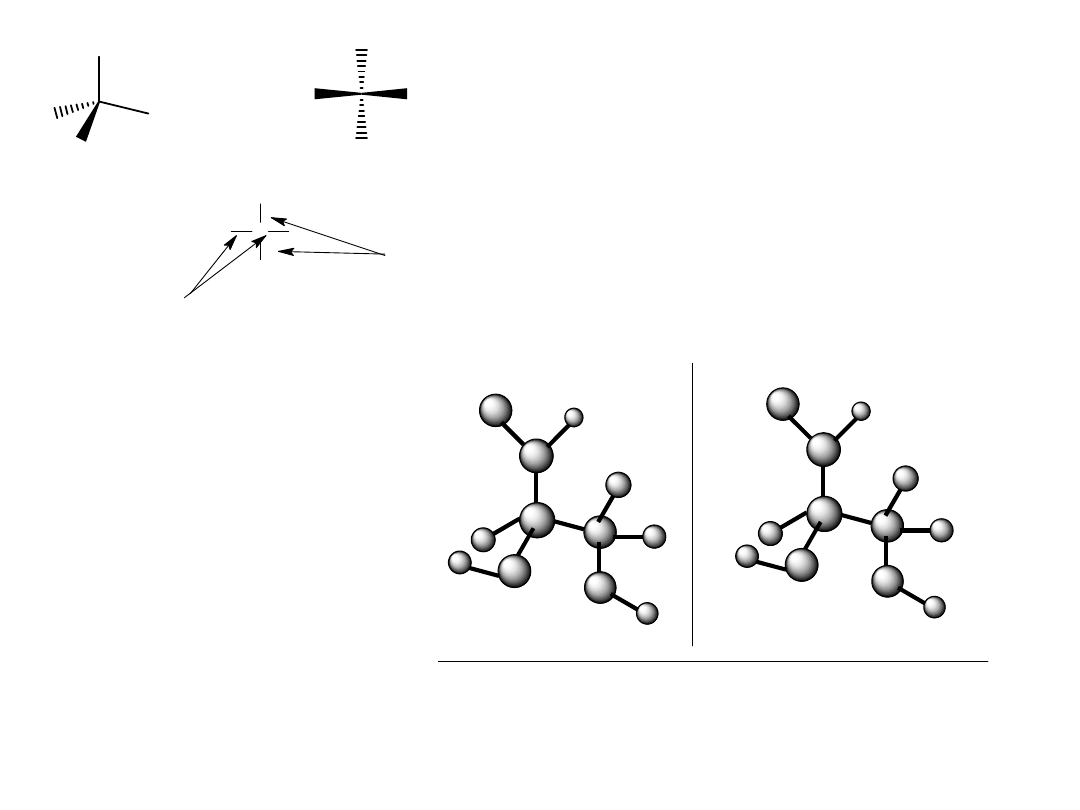
Ponieważ wszystkie węglowodany zawierają stereogeniczne atomy
węgla, już dawno stwierdzono, że konieczna jest standardowa metoda
opisu umożliwiająca przedstawienie stereochemii węglowodanu.
Najczęściej używaną współcześnie metodą jest projekcja Fischera
umożliwiająca przedstawienie struktury centrów stereogenicznych na
płaszczyźnie.
CHO
CH
2
OH
OH
H
=
OH
CHO
CH
2
OH
H
CHO
C
CH
2
OH
OH
H
wiazania wychodzace
ponad plaszczyzne
wiazania wchodzace
pod plaszczyzne
projekcja Fischera (R)-
gliceroaldehydu
obraz trojwymiarowy
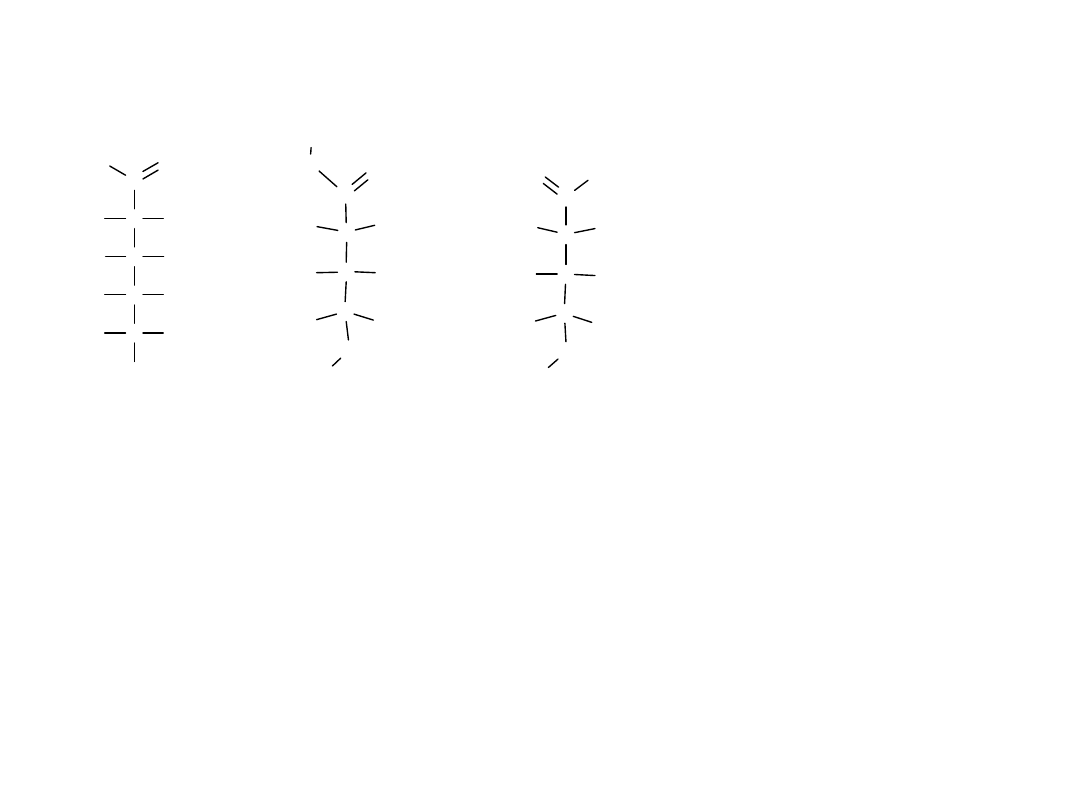
Projekcje Fischera można obracać na płaszczyźnie o 180
o
, nie zmieniając
konfiguracji ich centrum stereogenicznego, podczas gdy obrót o 90 lub o
270
o
powoduje zmianę konfiguracji.
CHO
CH
2
OH
OH
H
CHO
CH
2
OH
H
O
H
taki sam jak
(R)-gliceroaldehyd
Konfiguracje węglowodanów z więcej
niż 1 centrum stereogenicznym
przedstawia się przez układanie
centrów jedno nad drugim, z
karbonylowym atomem węgla
umieszczonym jak najbliżej górnej
części rysunku, np. glukoza ma w
projekcji Fischera 4 centra
stereogeniczne ułożone jedno nad
drugim.
C
C
C
C
C
CH
2
OH
H
H
H
H
H
O
OH
O
H
OH
OH
C
C
C
C
C
CH
2
OH
H
H
H
H
H
O
OH
O
H
OH
OH
=
=
O
H
H
H
H
OH OH
CHO
CH
2
OH
OH
H
glukoza
grupa karbonylowa na
górze

Gliceroaldehyd ma tylko 1 centrum stereogeniczne i w związku z tym
tworzy dwie formy enancjomeryczne (lustrzane).
Z przyczyn historycznych, datujących się na długo przed przyjęciem
systemu R/S, (R)-(+)-gliceroaldehyd nazywany jest też D-
gliceroaldehydem (D oznacza prawoskrętny „dexterus”). Drugi enancjomer
(S)-(-)-gliceroaldehyd, znany jest jako L-gliceroaldehyd (L-oznacza
lewoskrętny „levo”).
Glukoza, fruktoza i niemal wszystkie istniejące naturalnie monosacharydy
mają taką samą konfigurację stereochemiczną jak D-gliceroaldehyd z
centrum stereogenicznym najdalszym od grupy karbonylowej.
Takie związki nazywamy D-cukrami.
Należy zauważyć, że określenie D lub L nie ma związku z obserwacją
doświadczalną, w którą stronę zostaje skręcona płaszczyzna polaryzacji
światła. W ten sposób D-cukier może zarówno być prawo-, jak i
lewoskrętny.
Przedrostek D oznacza jedynie, że stereochemia najniższego
stereogenicznego atomu węgla w projekcji Fischera jest taka sama, jak w
D-gliceroaldehydzie.
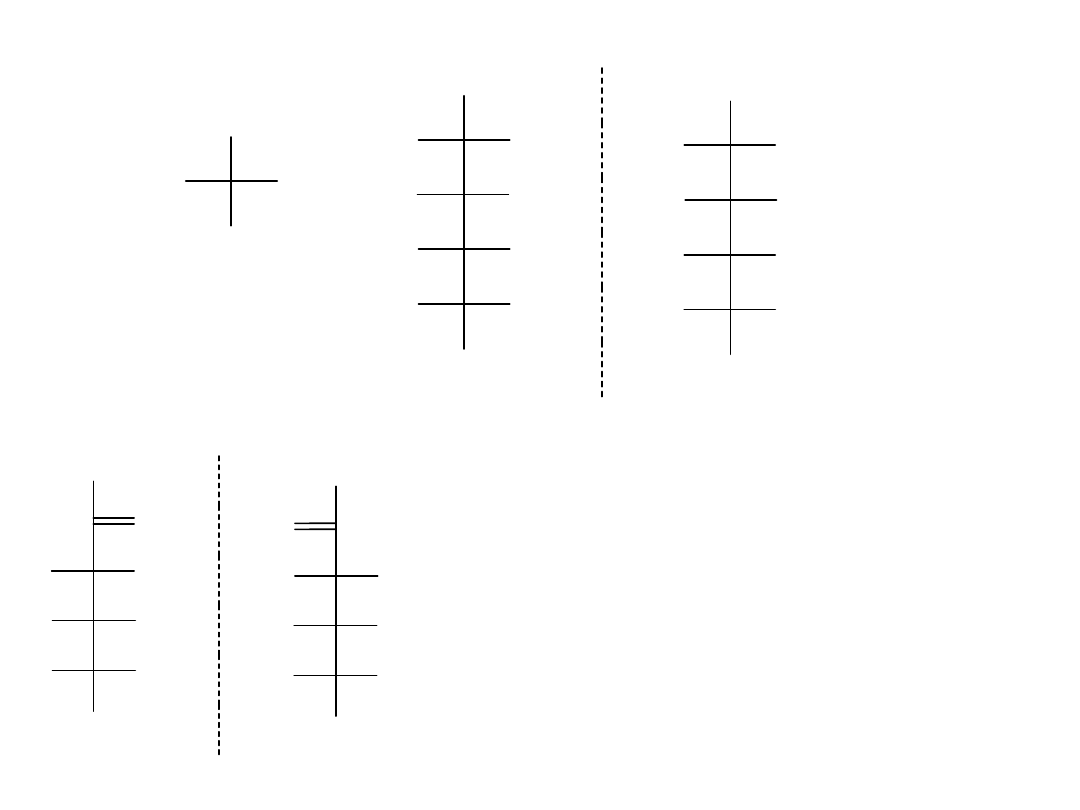
CHO
OH
H
H
HO
OH
H
OH
H
CH
2
OH
CHO
H
HO
OH
H
H
HO
H
HO
CH
2
OH
lustro
L-glukoza
(nie wystepuje w naturze)
D-glukoza
CHO
H
HO
CH
2
OH
L-gliceroaldehyd
[(S)-(-)-gliceroaldehyd]
CH
2
OH
O
H
HO
OH
H
OH
H
CH
2
OH
CH
2
OH
O
OH
H
H
HO
H
HO
CH
2
OH
lustro
L-fruktoza
D-fruktoza

Podział aldoz:
Aldotetrozy to cukry zawierające cztery atomy węgla z dwoma centrami
stereogenicznymi. Istnieje 2
2
= 4 możliwych stereoizomerycznych
aldotetroz albo dwie pary enancjomerów D i L, o nazwach erytroza i
treoza.
Aldopentozy mają trzy centra stereogeniczne, co prowadzi do 2
3
= 8
możliwych stereoizomerów albo czterech par enancjomerów D,L.
Te cztery pary nazywamy rybozą, arabinozą, ksylozą oraz liksozą.
Aldoheksozy mają cztery centra stereogeniczne, czyli razem 2
4
= 16
stereoizomerów albo osiem par enancjomerów D i L. Nazwy ośmiu form to:
alloza, altroza, glukoza, mannoza, guloza, idoza, galaktoza i taloza.
Projekcje Fischera cztero-, pięcio- i sześciowęglowych D-aldoz przedstawia
rysunek.
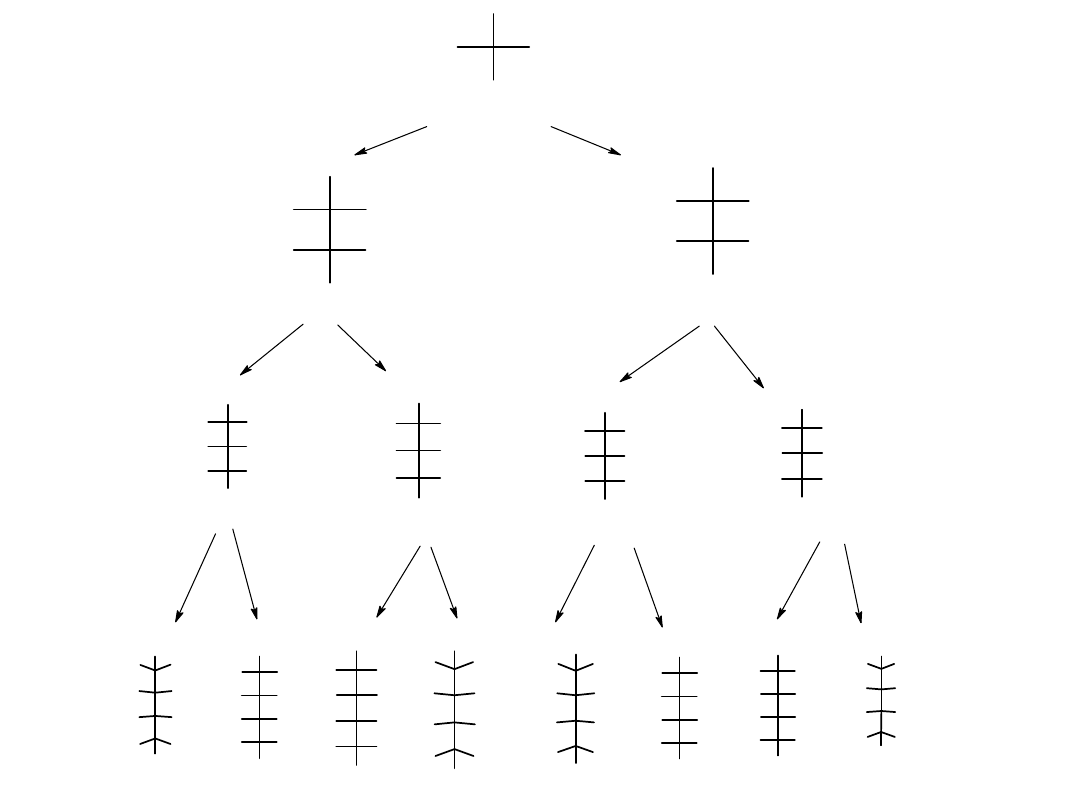
Konfiguracja aldoz:
- g l i c e r o a l d e h y d
D - e r y t r o z a
D - t r e o z a
D - r y b o z a
D - a r a b i n o z a
D - k s y lo z a
D - l i k s o z a
D - a l l o z a
D - a lt r o z a
D - g l u k o z a
D - m a n n o z a
D - g u l o z a
D - id o z a
D - g a l a k t o z a
D - t a l o z a
CHO
O
H
H
O
H
H
O
H
H
OH
H
CH
2
OH
CHO
H
O
H
OH
H
OH
H
OH
H
CH
2
OH
CHO
H
OH
H
OH
O
H
H
OH
H
CH
2
OH
CHO
O
H
H
O
H
H
H
OH
OH
H
CH
2
OH
CHO
OH
H
H
O
H
H
O
H
OH
H
CH
2
OH
CHO
H
OH
H
OH
H
OH
OH
H
CH
2
OH
CHO
H
OH
CH
2
OH
OH
H
OH
H
CH
2
OH
OHC
OH
H
OH
H
CH
2
OH
OHC
OH
H
OH
H
OH
H
CH
2
OH
OHC
H
OH
OH
H
OH
H
CH
2
OH
OHC
OH
H
H
O
H
O
H
H
CH
2
OH
OHC
H
OH
H
O
H
OH
H
CH
2
OH
OHC
CHO
OH
H
H
O
H
OH
H
OH
H
CH
2
OH
CHO
H
O
H
OH
H
H
O
H
OH
H
CH
2
OH
Struktury D-aldoz ustawiono kolejno od prawej tak, że grupy hydroksylowe
na atomie C2 są ułożone naprzemiennie prawa/lewa(R\L). Podobnie grupy
hydroksylowe na atomie C3 są ułożone według zasady dwie po
prawej/dwie po lewej (2R/2L); grupy hydroksylowe na atomie C4 są
ułożone zgodnie z zasadą 4R/4L, a wszystkie grupy hydroksylowe na
atomie C5 znajdują się po prawej stronie (8R).
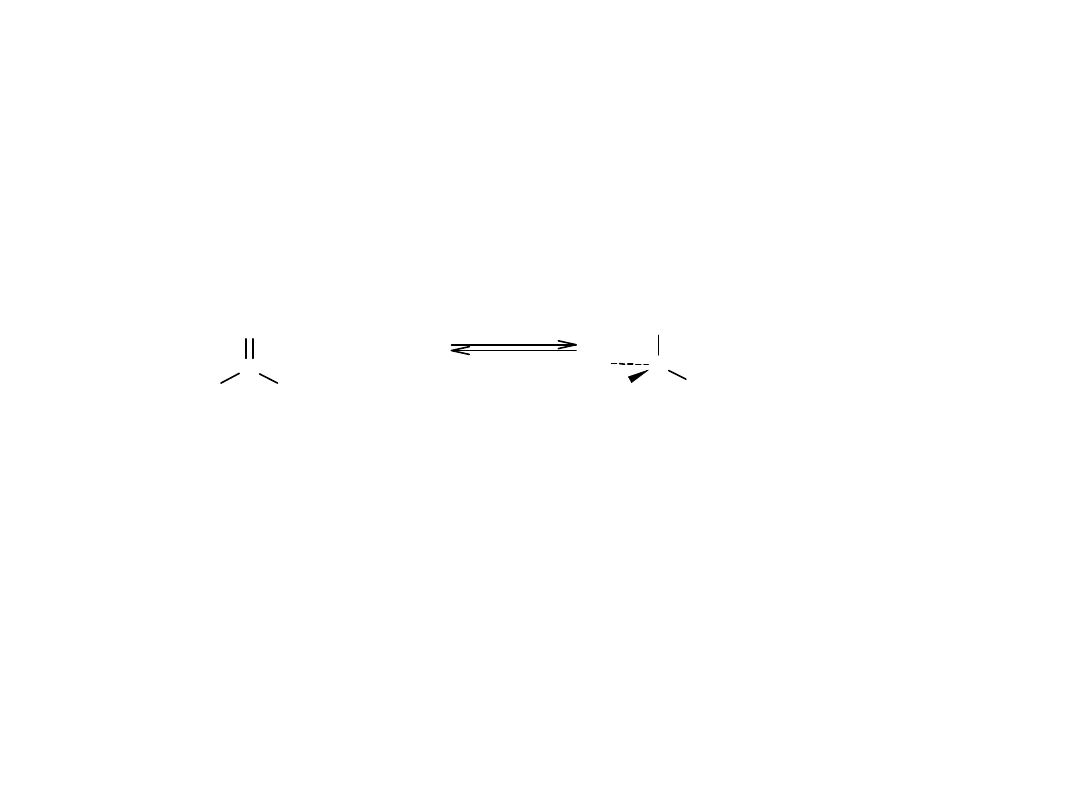
Cykliczna struktura monosacharydów: tworzenie hemiacetali
O
C
R
H
+
R'OH
H
+
k a t a l i z a t o r
OH
C
R
OR'
H
aldehyd
hemiacetal
Gdy w tej samej cząsteczce występuje zarówno grupa karbonylowa, jak i
hydroksylowa, możliwa jest wewnątrzcząsteczkowa reakcja addycji
nukleofilowej, prowadząca do utworzenia cyklicznego hemiacetalu.
Pięcio- i sześcioczłonowe hemiacetale cykliczne są szczegółnie trwałe i z
tego powodu wiele węglowodanów istnieje jako równowagowa
mieszanina form cyklicznych i otwartołańcuchowych. Np. glukoza w
roztworze wodnym istnieje głównie w postaci sześcioczłonowej formy
piranozowej, która jest wynikiem wewnątrzcząsteczkowej addycji
nukleofilowej grupy –OH przy atomie C5 do grupy karbonylowej atomu
C1, natomiast fruktoza istnieje w ok. 80% w formie piranozowej i w ok.
20% jako pięcioczłonowa forma furanozowa, powstała z przyłączenia
grupy –OH z atomu C5 do grupy karbonylowej atomu C2.
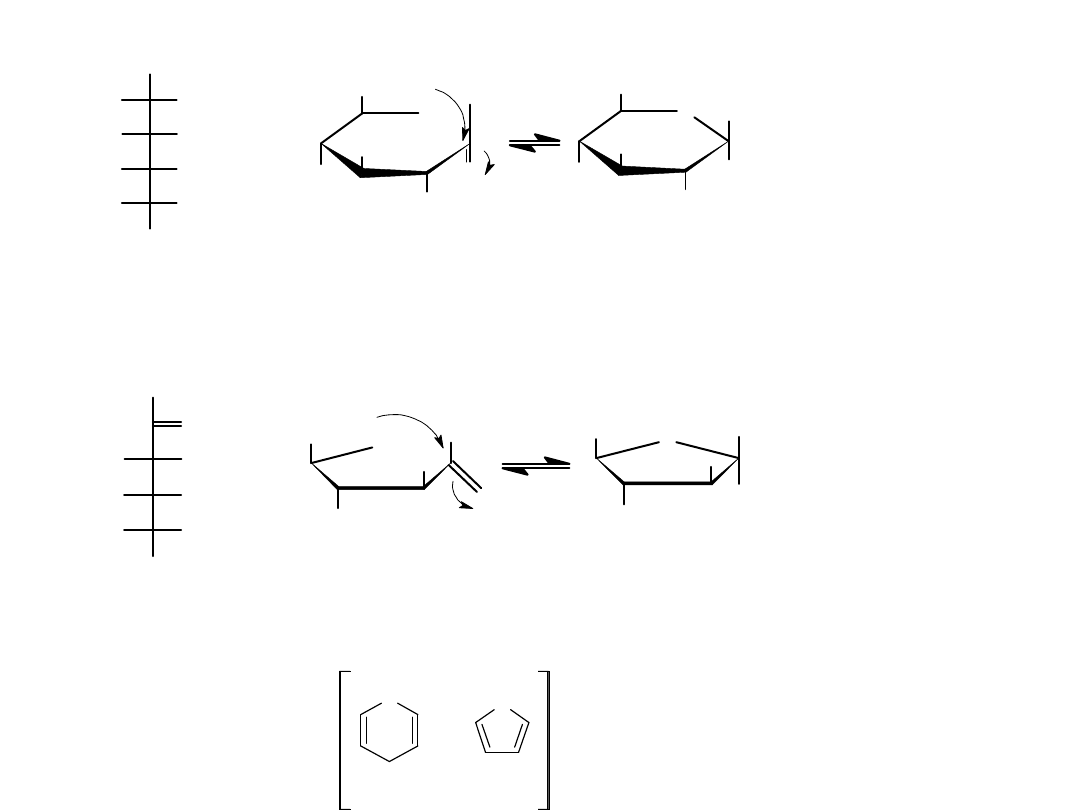
Cykliczna struktura monosacharydów tworzenie hemiacetali
D - g l u k o z a
F is c h e r
D - f r u k t o z a
( F is c h e r )
D - g l u k o z a , f o r m a p i r a n o z o w a
( H a w o r t h )
D - f r u k t o z a , f o r m a f u r a n o z o w a
( H a w o r t h )
O
CH
2
OH
CH
2
OH
OH
OH
OH
OH
OH
H
O
OH
OH
CH
2
OH
CHO
OH
H
H
O
H
OH
H
OH
H
CH
2
OH
CH
2
OH
O
H
O
H
OH
H
OH
H
CH
2
OH
O
OH
OH
H
OH
OH
CH
2
OH
OH
CH
2
OH
CH
2
OH
OH
OH
O
**
**
**
**
O
O
piran
furan
Cząsteczki glukozy i fruktozy
przedstawione w ich
cyklicznych formach:
furanozowej i piranozowej
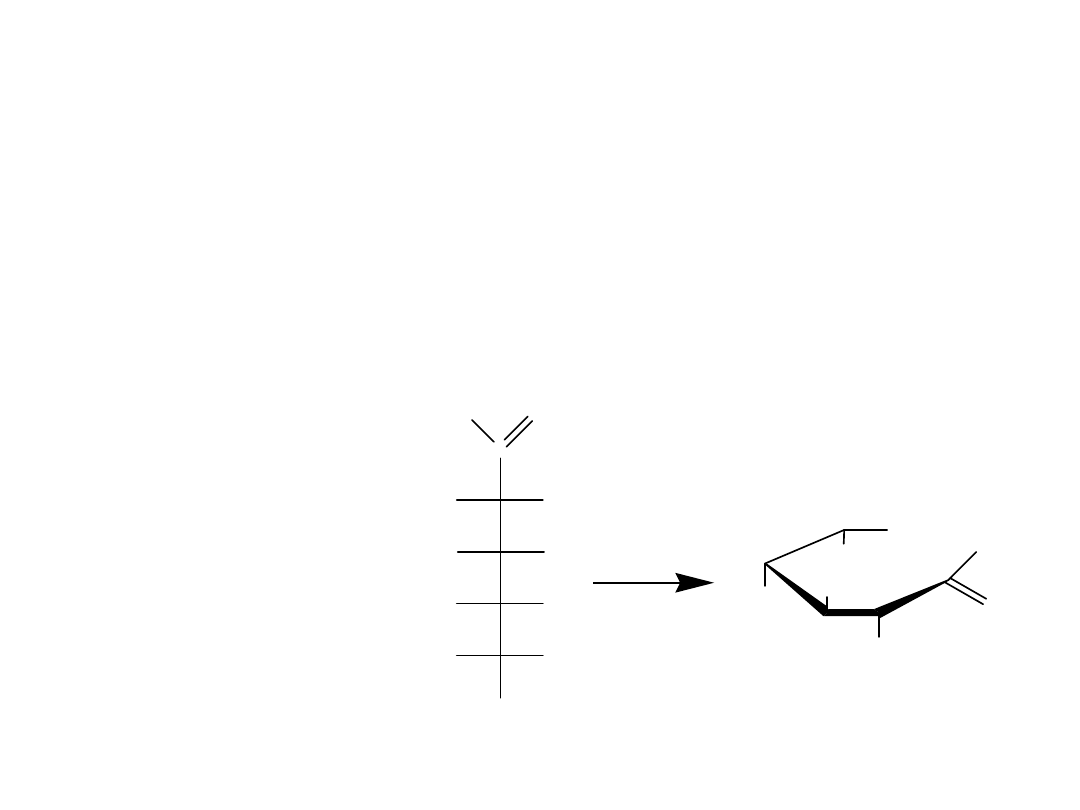
Pierścienie piranozowe i furanozowe są często przedstawiane za pomocą
projekcji Hawortha. W projekcji Hawortha pierścień hemiacetalowy rysuje
się jako strukturę płaską widzianą ukośnie z boku, z atomem tlenu
znajdującym się w prawej górnej części. Mimo, że sposób ten jest
wygodny, nie odzwierciedla dokładnie rzeczywistej struktury, gdyż
pierścienie piranozowe są w rzeczywistości pofałdowane i przyjmują
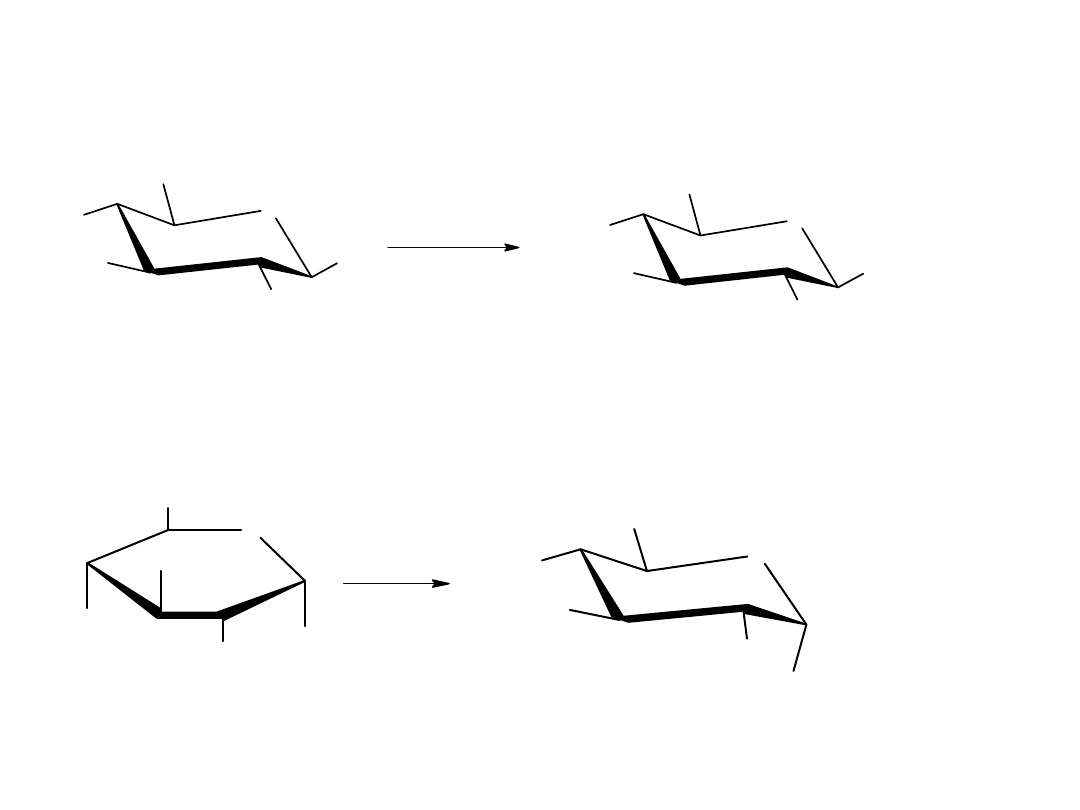
konformację krzesłową, podobnie jak w cykloheksanie.
Gdy przechodzi się od jednej formy projekcji do drugiej, należy pamiętać,
że grupa –OH po prawej stronie w projekcji Fischera jest skierowana na dół
w projekcji Hawortha. Odwrotnie, grupa –OH po lewej stronie w projekcji
Fischera jest skierowana do góry w projekcji Hawortha. Dla D-cukrów
końcowa grupa –CH
2
OH jest zawsze skierowana do góry w projekcji
Hawortha, podczas gdy dla L-cukrów ta grupa –CH
2
OH jest skierowana na
dół.
C
H
OH
HO
H
H
OH
H
OH
CH
2
OH
H
O
D-glukoza
(Fischer)
obróc na bok
CH
2
OH
H
O
OH
OH
OH
OH
CH
2
OH
H
O
OH
OH
OH
OH
obr�� wok�
wi�zania
OH
H
O
OH
OH
OH
CH
2
OH
O
OH
H
OH
OH
OH
CH
2
OH
zamknij
pier�cie�
D-glukoza (Haworth)
Wzajemne przejście między projekcjami Fischera i
Hawortha w przypadku D-glukozy.
Anomery monosacharydów: mutarotacja
Gdy monosacharyd otwartoiłańcuchowy cyklinuje do formy furanozowej
lub piranozowej, tworzy się nowe centrum stereogeniczne na atomie
węgla, który uprzednio był atomem karbonylowey. Dwa
diastereoizometryczne produkty nazywamy anomerami, a chemiacetalowy
atom węgla nazywany jest centrum anomerycznym. Np. w roztworze
wodnym glukoza cyklinuje odwracalnie do mieszaniny dwóch anomerów w
stosunku 36:64. Anomer z grupą –OH w pozycji C1, która znajduje się w
konfiguracj trans do podstawnika
–CH
2
OH na atomie C5 (na dół w projekcji Hawortha), nazywany jest
anomerem a6 . jego pełna nazwa brzmi a-D-glukopiranoza i jest to
anomer, którego jest mniej. główny anomer z grupy –OH na atomie C1, cis
do podstawnika –CH
2
OH na atomie C5 (ku górze w projekcji Hawortha),
nazywany jest anomerem b, a jego pełna nazwa brzmi b-D-glukopiranoza.
OH
OH
OH
OH
OH
CH
2
OH
O
D-glukoza
O
OH
OH
OH
OH
CH
2
OH
Trans
alfa-D-glukopiranoza (
36%)
(alfaanomer: OH i CH
2
OH
s� trans wzgl�dem siebie)
+
O OH
OH
OH
OH
CH
2
OH
Cis
beta-D-glukopiranoza (
64%)
(betaanomer: OH i CH
2
OH
s� cis wzgl�dem siebie)
Mutarotacja przebiega przez odwracalne otwarcie pierścienia każdego z
anomerów do otwartołańcuchowego aldehydu, a następnie zachodzi
ponowne zamknięcie pierścienia. mimo iż to równowagowanie jest
procesem powolnym obojętnym pH, ale jest ono katalizowane zarówno
przez kwas, jak i przez zasadę.
OH
H
OH
OH
OH
CH
2
OH
O
D-glukoza
O
OH
OH
OH
OH
CH
2
OH
alfa-D-glukopiranoza (
36%)
O OH
OH
OH
OH
CH
2
OH
beta-D-glukopiranoza (
64%)
Konformacje monosacharydów
Projekcje Hawortha są łatwe w percepcji, ale nie dają one dokładnego
trójwymiarowego obrazu konformacji cząstkowej. Pierścienie piranozowe,
tak jak pierścienie cykolehsanowe przyjmują kształt podobny do krzesła z
podstawnikami aksjonalnymi i ekwatorialnymi. Każdy podstawnik
skierowany w górę w projekcji Hawortha jest skierowany w górę również w
konformacji krzesłowej, a każdy podstawnik skierowany w dół w projekcji
Hawortha jest skierowany w dół również w konformacji krzesłowej.
O
OH
OH
OH
OH
CH
2
OH
atom tlenu w g�rze po prawej stronie
=
O
HO
HO
HOH
2
C
HO
OH
alfa-D-glukopiranoza
O OH
OH
OH
OH
CH
2
OH
=
O
HO
HO
HOH
2
C
OH
OH
beta-D-glukopiranoza
Reakcje monosacharydów
Ponieważ węglowodany zawierają tylko dwa rodzaje grup funkcyjnych:
grupy hydroksylowe i karbonylowe, większość właściwości chemicznych
monosacharydów to znana już nam chemia alkoholi i związków
karbonylowych.
O
HO
HO
HOH
2
C
OH
OH
(CH
3
CO)
2
O
pirydyna, O�C
O
O
2
CH
3
C
O
2
CH
3
C
H
3
COCOH
2
C
OCOCH
3
OCOCH
3
beta-D-glukopiranoza
penta-O-acetylo-beta-D-glukopiranoza
( 91%)
Węglowodany przekształcane są w etery przez traktowanie
halogenkiem alkilowym w obecności zasady (synteza Williamsona).
O
OH
OH
OH
OH
CH
2
OH
alfa-D-glukopiranoza
Ag
2
O
CH
3
I
O
OH
3
C
OH
3
C
H
3
COH
2
C
H
3
CO
CH
3
O
eter pentametylowy alfa-D-glukopiranozy
( 85%)
Tworzenie glikozydów
O
HO
HO
HOH
2
C
H
OH
OH
CH
3
OH, HCl
H
3
O
+
O
HO
HO
HOH
2
C
H
OH
OCH
3
+
O
H
2
beta-D-glukopiranoza
(hemiacetal)
beta-D-glukopiranozyd metylowy
(acetal)
Nazwy acetyli węglowodanów, zwanych glikozydami, tworzy się przez
zastąpienie końcówki –oza przyrostkiem –ozyd, przyrostkiem następnie
określenie grupy alkilowej.
Redukcja monosacharydów
Traktowanie aldozy lub ketozy NaBH4 redukuje je do pilialkoholu o nazwie
alditol.
C
H
OH
HO
H
H
OH
H
OH
CH
2
OH
H
O
O
HO
HO
HOH
2
C
H
OH
OH
beta-D-glukopiranoza
D-glukoza
NaBH
4
H
2
O
CH
2
OH
H
OH
HO
H
H
OH
H
OH
CH
2
OH
D-glucitol (d-sorbit), alditol
Utlenianie monosacharydów
Aldozy, podobnie jak i inne aldehydy łatwo ulegają reakcji utleniania,
dając odpowiednie kwasy karboksylowe zwane kwasami aldonowymi.
Aldozy reagują z odczynnikiem Tollensa (Ag+ w wodnym roztworze NH
3
) i
odczynnikiem Fehlinga (Cu
2
+ w wodnym roztworze winianu sodu).
D-fruktoza
NaOH, H
2
O
(tautometria
keto-enolowa)
enodiol
NaOH, H
2
O
(tautometria
keto-enolowa)
aldoheksoza
HO
H
H
OH
H
OH
CH
2
OH
C
CH
2
OH
O
HO
H
H
OH
H
OH
CH
2
OH
C
OH
C
OH
H
HO
H
H
OH
H
OH
CH
2
OH
C
C
O
H
OH
H
Choć reakcje Fehlinga i Tollensa służą jako użyteczne testy na cukry
redukujące, nie dają dobrych wydajności kwasów aldonowych, gdyż
warunki zasadowe powodują rozkład węglowodanu. do celów
preparatywnych najlepszym utleniaczem jest buforowany wodny roztwór
Br
2
. Reakcja ta jest specyficzna dla aldoz; ketozy nie ulegają utlenieniu
przez wodny roztwór Br
2
.
beta-D-galaktoza
C
H
OH
HO
H
H
OH
H
OH
CH
2
OH
H
O
Br
2
, H
2
O
pH = 6
C
H
OH
HO
H
H
OH
H
OH
CH
2
OH
HO
O
kwas D-galaktonowy
(kwas aldonowy)
O
OH
HO
CH
2
OH
OH
OH
Gdy w reakcji wykorzystany zostanie silniejszy środek utleniający taki jak
rozcieńczony HNO
3
w podwyższonej temperaturze, aldozy utleniają się do
kwasów dikarboksylowych zwanych kwasami aldarowymi. W reakcji tej
utlenianiu ulegają zarówno grupa –CHO na atomie C1, jak i końcowa grupa
–CH
2
OH.
O
HO
HO
HOH
2
C
OH
OH
beta-D-glukoza
C
H
OH
HO
H
H
OH
H
OH
CH
2
OH
HO
O
HNO
3
C
H
OH
HO
H
H
OH
H
OH
C
HO
O
HO
O
kwas D-glukarowy
(kwas aldarowy)
Przedłużanie łańcucha: synteza
Kilianiego-Fischera.
Jedną z najważniejszych metod
jest synteza Kilianiego-Fischera,
której wynikiem jest wydłużenie
łańcucha aldozy o jeden atom
węgla. Grupa aldehydowa C1
wyjściowego cukru staje się
atomem C2 cukru o wydłużonym
łańcuchu.
HO
H
H
OH
H
OH
CH
2
OH
C
H
O
D-arabinoza
HCN
HO
H
H
OH
H
OH
CH
2
OH
H
OH
C
N
+
HO
H
H
OH
H
OH
CH
2
OH
HO
H
C
N
dwie
cyjanohydryny
H
2
Pd/BaSO
4
katalizator
HO
H
H
OH
H
OH
CH
2
OH
HO
H
C
H
NH
+
HO
H
H
OH
H
OH
CH
2
OH
H
OH
C
H
NH
dwie iminy
H
3
O
+
H
3
O
+
HO
H
H
OH
H
OH
CH
2
OH
H
OH
C
H
O
+
HO
H
H
OH
H
OH
CH
2
OH
HO
H
C
H
O
D-glukoza
D-mannoza
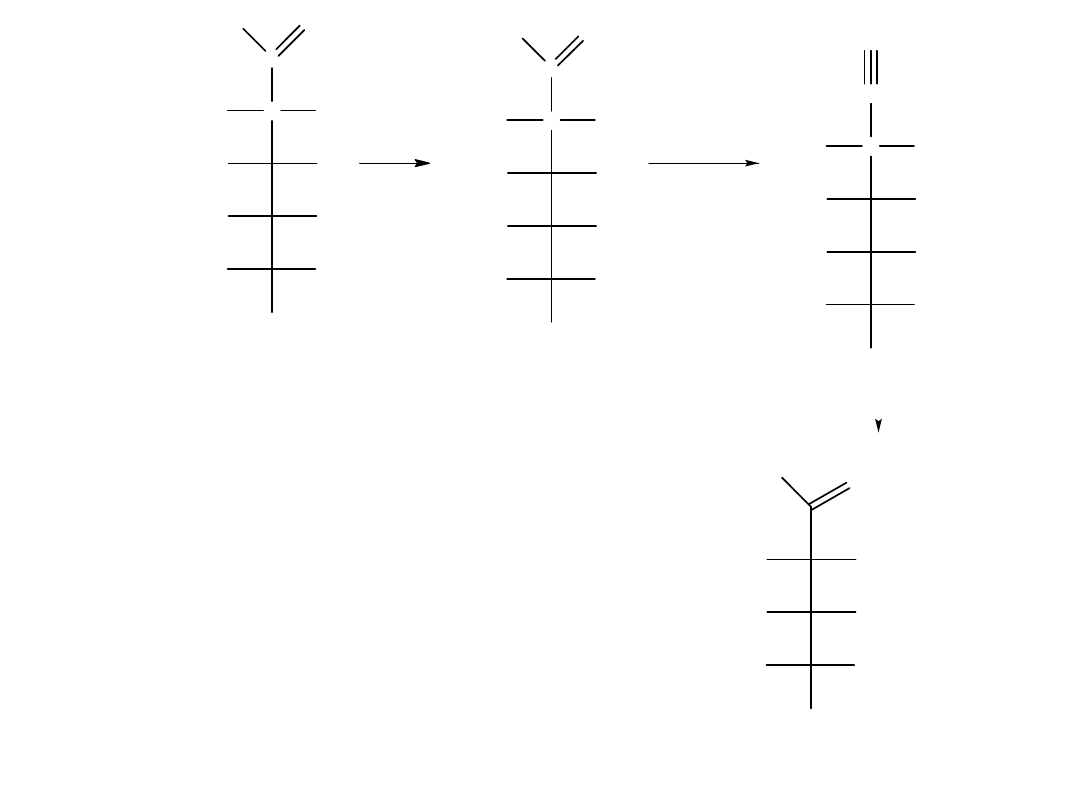
Skracanie łańcucha: degradacja Wohla
Tak jak reakcja syntezy Kilianiego-Fischera,
pozwala wydłużyć łańcuch aldozy o jeden
atom węgla, tak reakcja degradacji Wohla
pozwala ten łańcuch skrócić o jeden atom
węgla. Przekształcenia aldehydu w nitryl
dokonuje się przez traktowanie aldozy
hydroksyloaminą, po czym następuje
odwodnienie (dehydratacja) oksymu
zachodzące pod wpływem bezwodnika
octowego. Np. D-galaktoza ulega
przekształceniu w D-liksozę w degradacji
Wohla:
HO
H
HO
H
H
OH
CH
2
OH
C
C
O
H
OH
H
D-galaktoza
H
2
NOH
HO
H
HO
H
H
OH
CH
2
OH
C
C
NOH
H
OH
H
oksym D-galaktozy
(CH
3
CO)
2
O
CH
3
CO
2
Na
HO
H
HO
H
H
OH
CH
2
OH
C
C
OH
H
N
cyjanohydryna
Na
+
CH
3
O-
HO
H
HO
H
H
OH
CH
2
OH
O
H
+
HCN

Disacharydy
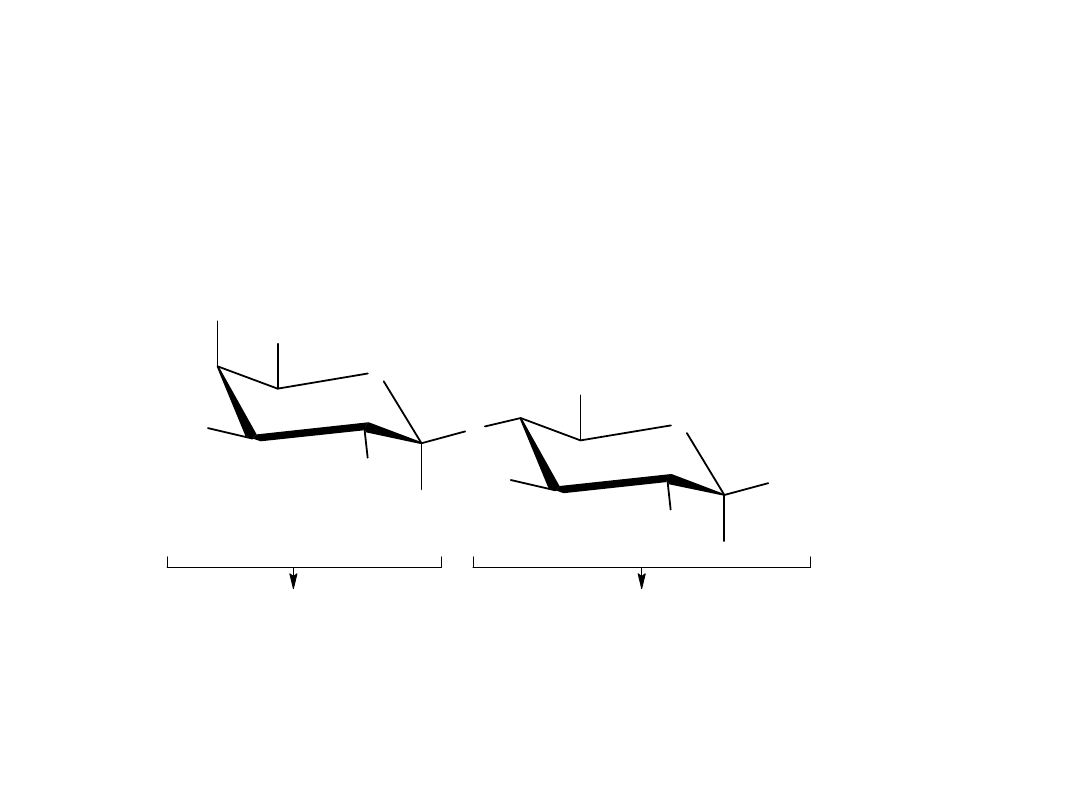
Celobioza i maltoza
Disacharydy są to związki, które zawierają glikozydowe wiązanie
acetalowe między atomem węgla C1 jednej cząsteczki cukru i jakąkolwiek
grupą hydroksylową –OH drugiej cząsteczki cukru. Wiązanie glikozydowe
między atomem C1 pierwszego cukru i grupą hydroksylową –OH na
atomie C4 drugiego cukru jest szczególnie często spotykane w przyrodzie.
Takie wiązanie nazywane jest połączeniem 1,4’. Znak „’ „ wskazuje, że
pozycja 4’ dotyczy innej cząsteczki cukru niż pozycja 1 bez tego znaku.
maltoza, 1, 4'-alf-glikozyd
[ 4-O-(alfa-D-glukopiranozylo)-alfa-D-glukopiranoza]
O
HO
CH
2
OH
OH
H
O
HO
HO
CH
2
OH
OH
OH
1
4'
H
O

O
HO
CH
2
OH
OH
OH
O
OH
HO
CH
2
OH
OH
O
H
H
1
4'
celobioza,
1, 4'-beta-glikozyd
[ 4-O-(beta-D-glukopiranozylo)-beta-D-glukopiranoza]
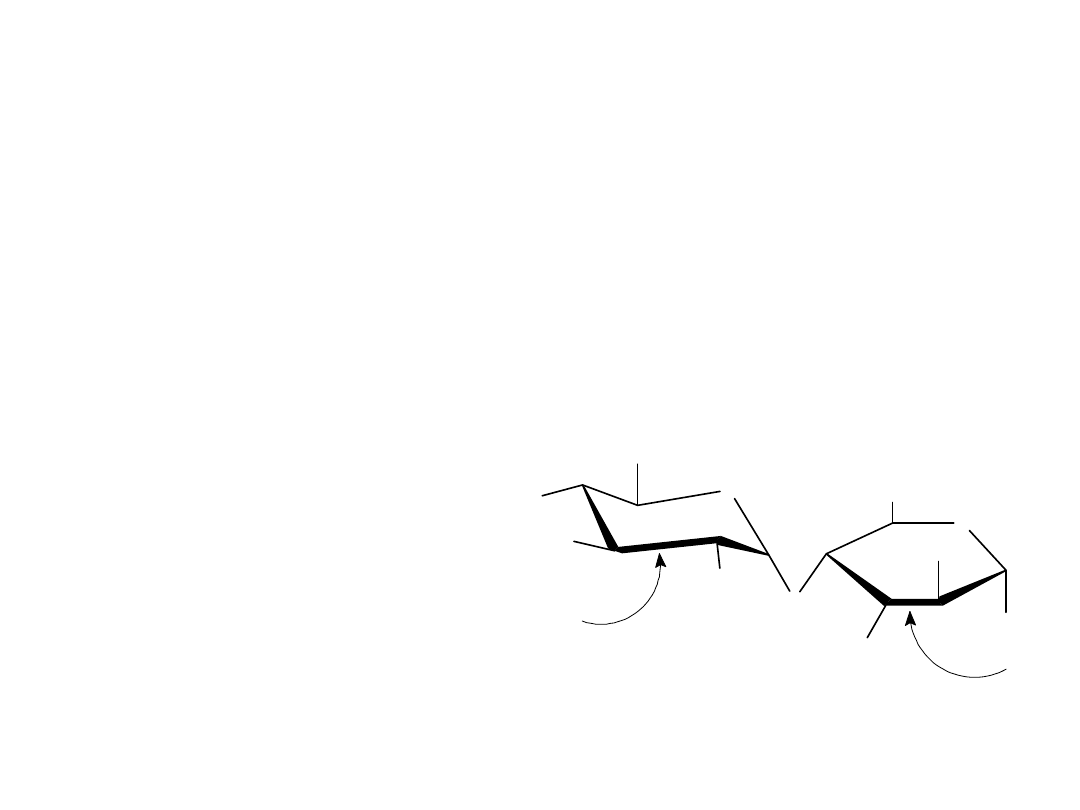
Laktoza
Laktoza jest disacharydem, który występuje naturalnie w mleku ludzkim i
krowim. Jest szeroko stosowana w cukiernictwie i jako składnik
sztucznych odżywek dla niemowląt. Laktoza, podobnie jak celobioza i
maltoza jest cukrem redukującym. Wykazuje mutarotację i jest 1,4’-β-
glikozydem. W odróżnieniu od celobiozy i maltozy, laktoza zawiera dwie
różne jednostki monosacharydowe: D-glukozę i D-galaktozę połączone
wiązaniem ß-glikozydowym między atomami C1 galaktozy i C4 glukozy.
O
HO
CH
2
OH
OH
OH
O
OH
HO
CH
2
OH
OH
H
1
4'
O
H
beta-galaktopiranozyd
beta-glukopiranoza
laktoza, 1, 4'-beta-glikozyd
[ 4-O-(beta-D-galaktopiranozylo)-beta-D-glukopiranoza]
Sacharoza
Sacharoza, czyli cukier stołowy, jest prawdopodobnie
najpowszechniejszym na świecie, czystym związkiem organicznym i także
jednym z najlepiej znanych ludziom, którzy nie są chemikami. Niezależnie
od tego czy pochodzi z trzciny cukrowej (20% masy trzciny cukrowej), czy
z buraków cukrowych (15% masy buraka) i czy jest surowy, czy
rafinowany, cukier ten jest sacharozą. Sacharoza jest disacharydem, który
w wyniku hydrolizy daje równoważne molowo ilości glukozy i fruktozy. Tę
mieszaninę glukozy i fruktozy, często nazywa się cukrem inwertowanym,
ponieważ znak skręcalności optycznej zmienia się (ulega inwersji) podczas
hydrolizy sacharozy. Owady takie jak pszczoły mają enzymy zwane
inwertazami, które katalizują hydrolizę sacharozy do mieszaniny
glukoza+fruktoza.
Miód pszczeli jest w zasadzie mieszaniną
glukozy, fruktozy i sacharozy.
W przeciwieństwie do większości
disacharydów sacharoza nie jest cukrem
redukującym i nie wykazuje mutarotacji.
Obserwacje te oznaczają, że sacharoza
nie ma grup hemiacetalowych, i sugerują,
że zarówno glukoza jak i fruktoza są
glikozydami. Jest to możliwe jedynie
wtedy, gdy te dwa cukry połączone są
ze sobą wiązaniem glikozydowym,
miedzy atomem C1 glukozy i atomem C2 fruktozy.
O
CH
2
OH
OH
OH
CH
2
OH
O
HO
HO
CH
2
OH
OH
O
1
2'
glukoza
fruktoza
sacharoza, 1, 2'-glikozyd
[ 2-O-(alfa-D-glukopiranozylo)-beta-D-fruktofuranozyd]

Polisacharydy
Polisacharydy to węglowodany, w których dziesiątki, setki, a nawet tysiące
cukrów prostych połączone są ze sobą wiązaniami glikozydowymi.
Polisacharydy, ponieważ nie zawierają anomerycznych grup
hydroksylowych (poza jedną na końcu łańcucha), nie są cukrami
redukującymi i nie wykazują mutarotacji. Najpowszechniej występujące
polisacharydy to celuloza i skrobia.
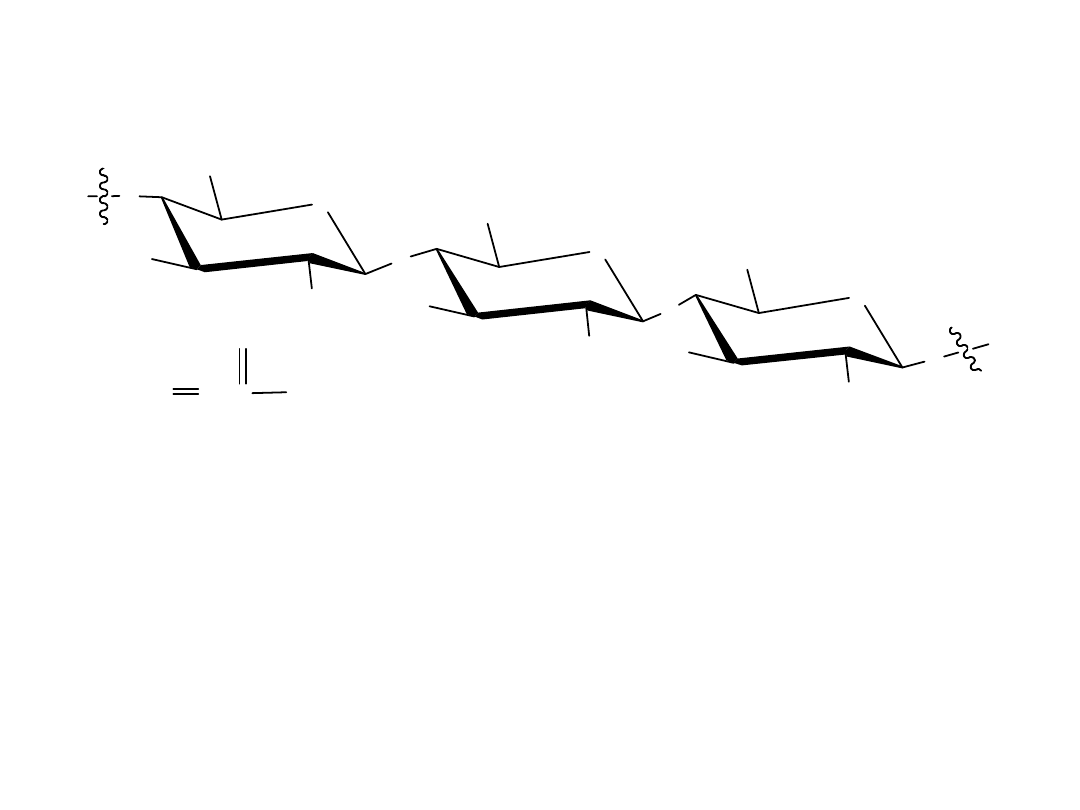
Celuloza
Celuloza składa się z jednostek D-glukozy połączonych ze sobą wiązaniami
1,4’-ß-glikozydowymi, takimi jak w celobiozie. Kilka tysięcy jednostek
glukozy połączonych jest w jedną cząsteczkę i cząsteczki takie
oddziałujące sobą, tworząc wielkie agregaty, których struktura
stabilizowana jest przez wiązania wodorowe.
O
HO
CH
2
OH
OH
O
O
O
HO
CH
2
OH
OH
O
O
HO
CH
2
OH
OH
O
celuloza, polimer
1, 4'-O-(beta-D-glukopiranozydowy)
W naturze celuloza wykorzystywana jest głównie jako materiał budulcowy,
nadający roślinom mechaniczną wytrzymałość i sztywność. Liście, źdźbła
traw oraz bawełna składają się głównie z celulozy. Celuloza jest też
surowcem do produkcji octanu celulozy, znanego pod nazwą jedwabiu
octanowego.
O
AcO
AcO
OAc
O
O
O
AcO
AcO
OAc
O
O
AcO
AcO
OAc
O
Ac
CH
3
C
O
gdzie
fragment �a�cucha octanu celulozy (jedwiabiu octanowego)
Skrobia i glikogen
Ziemniaki, kukurydza i ziarna zbóż zawierają olbrzymi ilości skrobi,
polimeru glukozy, w których jednostki monosacharydowe są połączone
wiązaniem 1,4’-α-glikozydowym, takim jak w maltozie. Skrobię można
rozdzielić na dwie frakcje: frakcję nierozpuszczalną w zimnej wodzie,
zwaną amylazą, oraz frakcję rozpuszczalną w zimnej wodzie, zwaną
amylopektyną. Amyloza stanowi ok. 20% masy skrobi i składa się z
kilkuset jednostek glukozowych połączonych wiązaniami 1,4’-α-
glikozydowymi.

O
HO
CH
2
OH
OH
O
O
HO
CH
2
OH
OH
O
O
HO
CH
2
OH
OH
O
H
H
H
amyloza, polimer
1, 4'-O-(alfa-D-glukopiranozydowy)
Amylopektyna stanowi pozostałe 80% masy skrobi i charakteryzuje się
bardziej złożoną strukturą niż amyloza. Odmiennie, niż celuloza i
amyloza, które są polimerami liniowymi, amylopektyna zawiera
rozgałęzienia 1,6’-α-glikozydowe, powtarzające się co około 25 jednostek.
W rezultacie amylopektyna ma niezwykle złożoną strukturę
trójwymiarową.
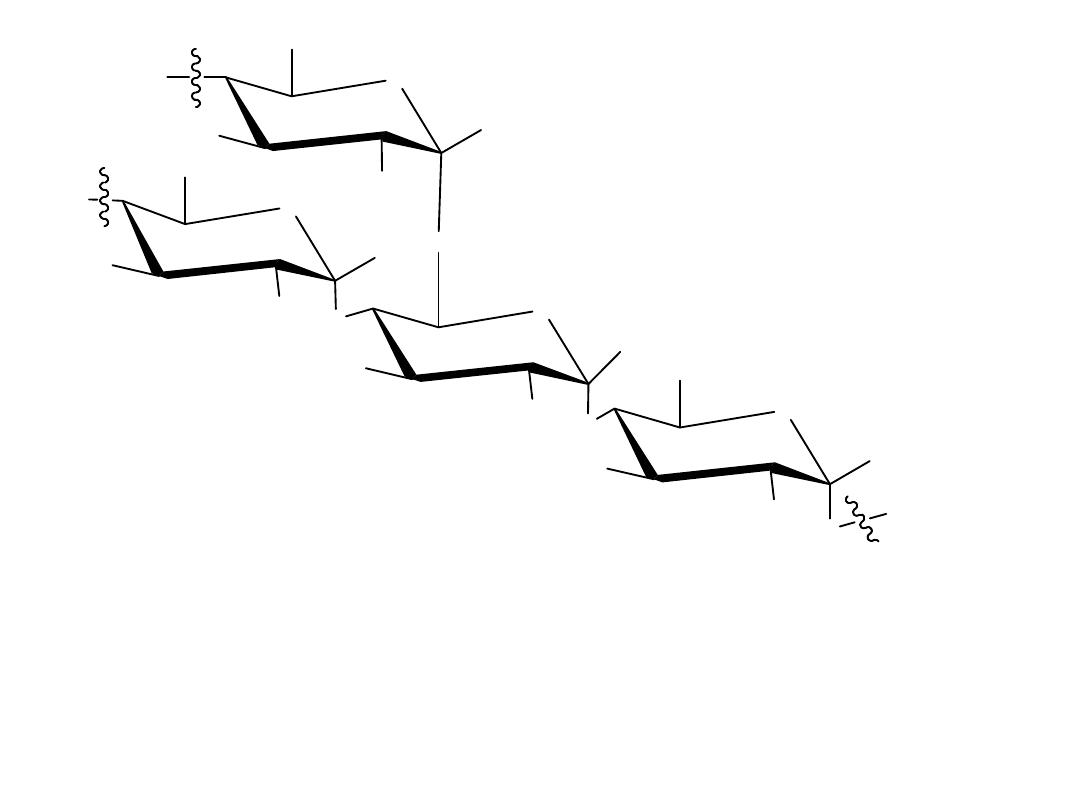
O
HO
CH
2
OH
OH
O
O
HO
O
OH
O
O
HO
CH
2
OH
OH
O
H
H
H
O
CH
2
OH
OH
H
HO
6'
1
amylopektyna
Organiczne związki azotu
Organiczne związki azotu obejmują 4 główne typy połączeń
I związki nitrowe
II aminy
III nitryle i izonitryle (cyjanki i izocyjanki)
IV cyjaniany i izocyjaniany
Ad.I
Związki nitrowe zawierają w swej cząsteczce grupę nitrową –NO
2
.
Atom azotu w grupie nitrowej znajduje się w stanie hybrydyzacji sp2.
Ponieważ badania wykazały jednakową długość wiązań pomiędzy
atomami O i N, przyjmuje się, że rozmieszczenie elektronów w grupie
nitrowej, odpowiada stanowi pośredniemu, między dwoma
strukturami granicznymi.
N
+
O
-
O
N
+
O
-
O
Grupa nitrowa jest więc hybrydą rezonansową (mezomerem). Polarny
charakter grupy nitrowej (rozmieszczenie ładunków pomiędzy N i O)
powoduje, że wszystkie związki nitrowe mają duże momenty dipolowe.
Otrzymywanie związków nitrowych
Istnieje tylko jedna powszechnie stosowana metoda otrzymywania
związków nitrowych, a mianowicie nitrowanie.
W zależności od reaktywności nitrowanego związku, stosuje się
rozcieńczony lub stężony HNO
3
wraz z H
2
SO
4
.
Alifatyczne związki nitrowe nie mają większego zastosowania, natomiast
aromatyczne związki nitrowe są szeroko stosowane jako ostateczne
produkty reakcji oraz półprodukty do dalszych reakcji.
C
H
3
CH
3
H N O
3
, H
2
S O
4
C
H
3
CH
2
NO
2
nitroetan
Nitrowanie związków aromatycznych jest najbardziej typową i najlepiej
poznaną reakcją podstawiania elektrofilowego w układach
aromatycznych. Czynnikiem nitrującym, jest w tej reakcji jon nitroniowy
NO
2+
+
NO
+
2
C
+
NO
2
H
NO
2
+
H
2
Grupa nitrowa jest podstawnikiem II rodzaju, dezaktywującym pierścień
aromatyczny (wyciąga elektrony z najbliższego sąsiedztwa), w związku z
tym atak elektrofilowy następnej grupy nitrowej zachodzi w położenie
meta:
NO
2
NO
2
NO
2
H N O
3
, H
2
S O
4
H N O
3
, H
2
S O
4
NO
2
O
2
N
NO
2
m-dinitrobenzen
(1,3 - dinitrobenzen)
1,3,5 - trinitrobenzen
benzen wybuchowy
p-dinitrobenzen można otrzymać przez utlenianie odpowiedniej diaminy
NH
2
NH
2
u t l
k w . n a d o c t o w y
NO
2
NO
2
p - dinitrobenzen
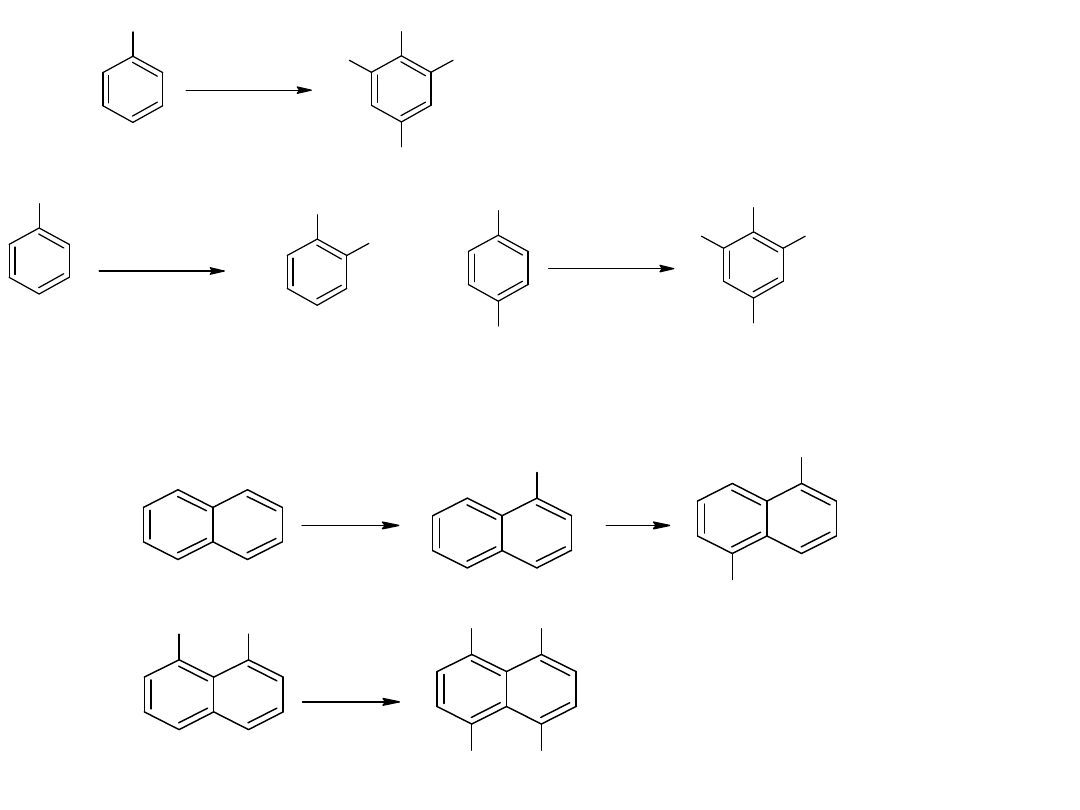
CH
3
CH
3
NO
2
NO
2
O
2
N
2
H N O
3
, H
2
S O
4
toluen
(trotyl) material wybuchowy
OH
fenol
H N O
3
, H
2
S O
4
r o z c
OH
NO
2
i
OH
NO
2
H N O
3
, H
2
S O
4
r o z c
OH
NO
2
NO
2
O
2
N
o - nitrofenol
2
(kw piksynowy) - material wybuchowy
Kwas pikrynowy zawierający 3 grupy –NO
2
jest znacznie silniejszym
kwasem od fenolu i stąd jego nazwa, np. rozkłada węglany z
wydzieleniem CO
2
.
NO
2
NO
2
NO
2
H N O
3
, H
2
S O
4
H N O
3
, H
2
S O
4
1 - nitronaftalen
1,5 - dinitronaftalen
NO
2
NO
2
H N O
3
, H
2
S O
4
NO
2
NO
2
NO
2
NO
2
1,8 - dinitronaftalen
1,81,4,5,8 - tetranitronaftalen
W 1872 r. Meyer, do otrzymywania związków nitrowych, zastosował
chlorowcopochodne alkilowe i azotyn sodu lub srebra. Okazało się jednak,
że w wyniku reakcji powstała mieszanina związku nitrowego i azotynu.
Obecnie wiadomo, że w takiej reakcji zawsze powstaje mieszanina
związków, z tego względu, że jon azotynowy może reagować w dwojaki
sposób:
2CH
3
J + 2AgNO
2
C
H
3
NO
2
C
H
3
O
NO
nitrometan
azotyn metylu
I
R
+
O
-
N
O
O
N
O
R
+
I
-
atak at O
azotyn alkilu
I
I
R
+
O
-
N
O
II
O
-
N
+
O
R
+
I
-
atak at N
zw nitrowy
Azotyny są połączeniami o charakterze estrów. Poddane hydrolizie dają
HNO
2
i odpowiedni alkohol.
Nitrozwiązki nie ulegają hydrolizie.
Prócz budowy przestrzennej cząsteczek, azotyny i związki nitrowe różnią
się produktami redukcji.
R
NO
2
R
O
NO
H
2
- H
2
O
H
2
R
NH
2
R
OH
+
NH
3
amina
nitrozwiazek
azotyn alkilu
alkohol
Właściwości chemiczne związków nitrowych
1. Grupa nitrowa ma wpływ na reaktywność wiązań C-H w położeniu
, w alifatycznych związkach nitrowych. Badania wykazały, że dla
związków nitrowych posiadających H w położeniu zachodzi
przemiana tautomeryczna, polegająca na wędrówce protonu od
atomu C do atomu O grupy nitrowej.
Związek nitrowy posiadający H przy atomie O grupy nitrowej nosi nazwę
związku aci (postać aci nitrozwiązku).
CH
2
R
N
+
O
O
CH
R
N
+
O
O
-
H
CH
R
N
+
O
-
O
-
odmiana nitrowa
postac aci kwasowa
anion postaci aci
-
Okazuje się, że dla związków nitrowych, zawierających wodór przy Cα,
występuje zjawisko desmotropii, które zaobserwowano dla tych związków,
których wzajemne przemiany wymagały dość długiego czasu np.
fenylonitrometan w roztworze NaOH tworzy sól sodową, będącą odmianą
aci, która po kilku dniach przekształca się w związek nitrowy
H
5
C
6
CH
2
N
+
O
-
O
N a O H
H
+
H
5
C
6
CH
2
N
+
O
O
Na
fenylonitrometan (ciecz)
postac aci (zw krystaliczny)
Reaktywność atomów H przy węglu , wyraża się w reakcjach z HNO
2
C
H
3
CH
2
NO
2
+
ON
OH
-H
2
O
R
NO
2
N
O
H
kw azotawy
C
H
3
CH
NO
2
R
+
ON
OH
-H
2
O
R
NO
2
N
O
R
N
OH
kw nitrolowy
gr izonitrozowa
N
O
gr nitrozowa
pseudonitrol
2.
Ze względu na to, że N w grupie nitrowej znajduje się na
najwyższym stopniu utlenienia dla tego pierwiastka, związki nitrowe nie
ulegają utlenianiu. Duża dostępność różnych związków nitrowych
umożliwia natomiast otrzymywanie różnych amin, które są produktami
redukcji związków nitrowych.
C
6
H
5
NO
2
C
6
H
5
NH
2
r e d
nitrobenzen
anilina(amina)
Redukcja związków nitrowych do amin nie jest reakcją jednoetapową,
lecz zachodzi poprzez produkty pośrednie:
C
6
H
5
NO
2
nitrobenzen
C
6
H
5
NO
C
6
H
5
NHOH
C
6
H
5
NH
2
nitrozobenzen
fenylohydroksyloamina
anilina
2 H
+
- H
2
O
2 H
+
2 H
+
- H
2
O
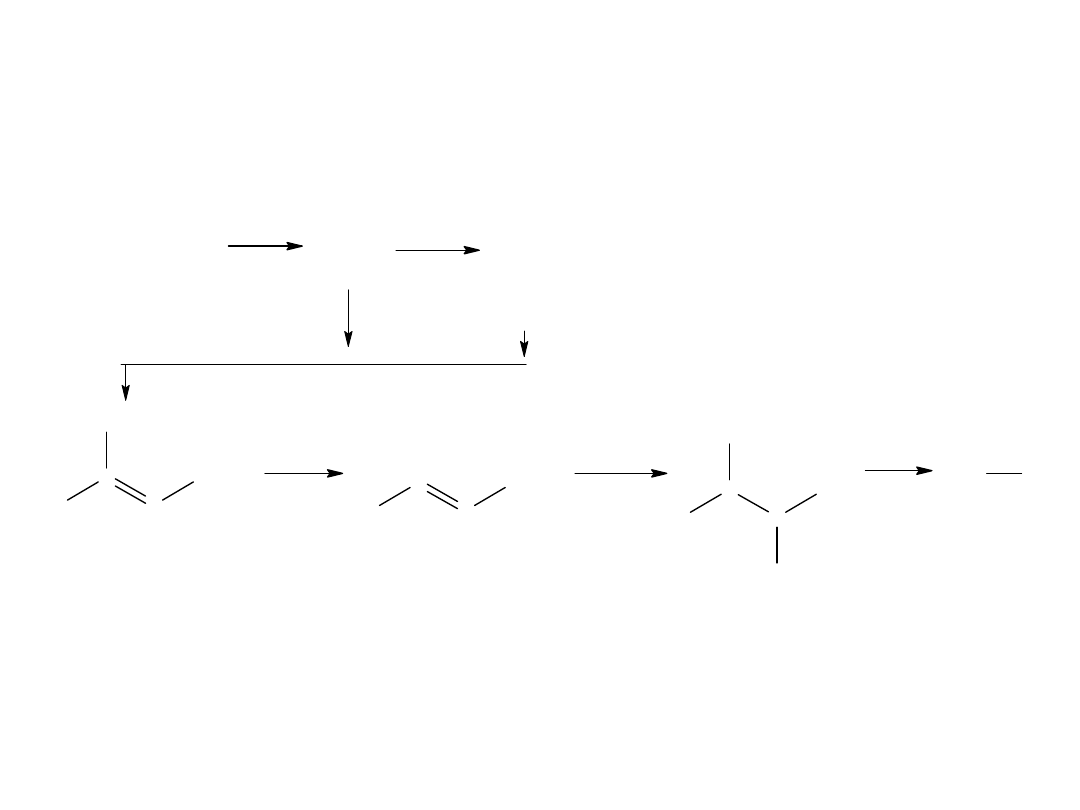
Istnieje również możliwość kondensacji produktów pośrednich między
sobą. Wówczas liczba produktów pośrednich rośnie.
Ich obecność została potwierdzona doświadczalnie, poprzez stosowanie
różnych reduktorów, które pozwalały zahamować redukcję nitrobenzenu
na odpowiednim etapie.
C
6
H
5
NO
2
nitrobenzen
C
6
H
5
NO
C
6
H
5
NHOH
nitrozobenzen
fenylohydroksyloamina
- H
2
O
H
5
C
6
N
+
OH
N
C
6
H
5
azoksybenzen
-H
2
O
2 H
H
5
C
6
N
N
C
6
H
5
azobenzen
2 H
H
5
C
6
N
N
C
6
H
5
H
H
hydrazobenzen
2 H
H
5
C
6
NH
2
anilina
Ad.II
Aminy można traktować jako pochodne amoniaku, powstałe przez
podstawienie atomów H grupami alkilowymi lub arylowymi. Zależnie od
liczny podstawionych atomów H rozróżnia się:
-aminy I rz., o ogólnym wzorze R-NH
2
, zawierające grupę aminową –NH2
- aminy II rz., o wzorze: zawierające grupę liniową
=NH
- aminy III rz., o wzorze:
nie zawierające wodoru
oraz w pewnych uzasadnionionych
wypadkach
- aminy IV rz., o wzorze:
R
1
NH
R
R
1
N
R
2
R
N
R
R
R
R OH
wodorotlenek tetraalkiloamoniowy
Podstawowe właściwości, tj. zasadowość i charakter nukleofilowy, są
podobne u amin różnych typów; wynikają bowiem z obecności wolnej
pary elektronowej przy atomie N.
Atom N w grupie aminowej, znajduje się w stanie hybrydyzacji sp3, lecz ma
tylko 3 niesparowane elektrony, zajmujące 3 orbitale sp3; czwarty
orbital atomu N obsadzony jest przez wolną parę elektronową. Grupa
aminowa ma budowę tetraedru, z wolną parą elektronową na jednym
wierzchołku.
Tylko w aminach czwartorzędowych wszystkie wierzchołki tetraedru
obsadzone są podstawnikami alkilowymi.
Izomeria związków aminowych
a) izomeria łańcuchowa
C
H
3
CH
2
CH
2
CH
2
NH
2
n – butyloamina
2 – metylo – 1aminopropan (2 – metylo –
1propyloamina)
C
H
3
CH
C
H
3
CH
2
NH
2
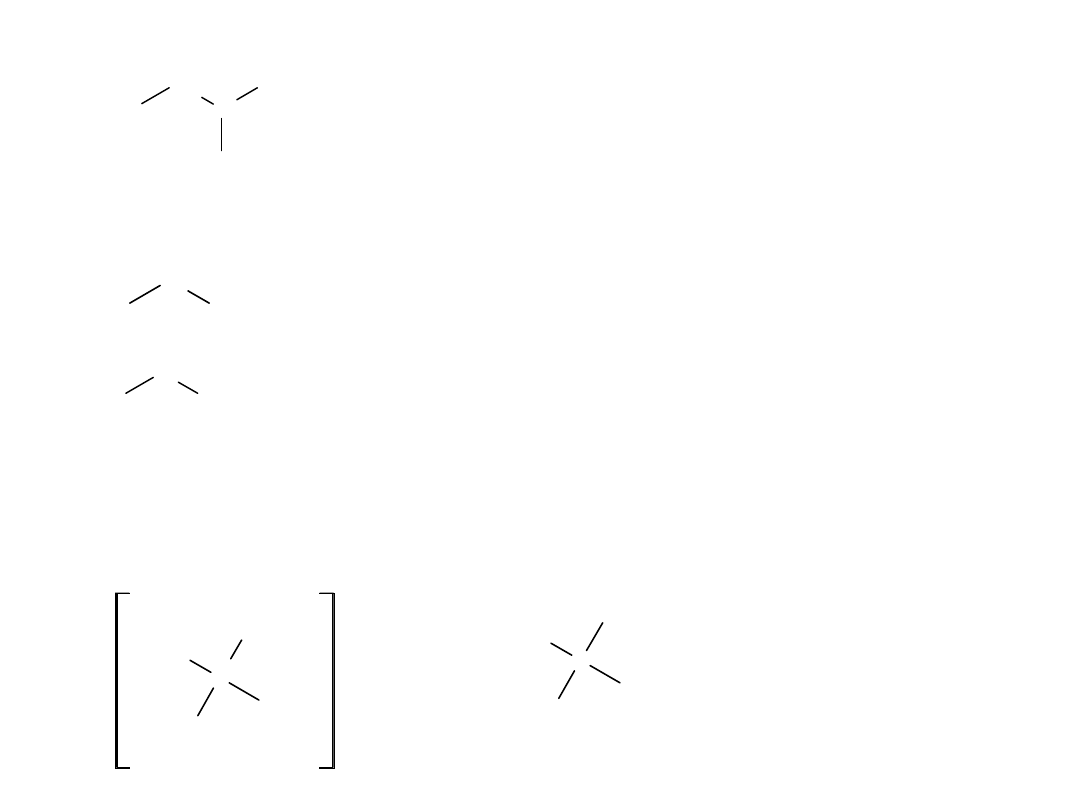
b) izomeria położeniowa
c) metameria
dietyloamina
metylo – n – propyloamina
C
H
3
CH
2
CH
CH
3
NH
2
2-aminobutan
H
5
C
2
NH
C
2
H
5
H
7
C
3
NH
CH
3
d) izomeria optyczna
Ten rodzaj izomerii pojawia się zwłaszcza dla amin czwartorzędowych, w
których atom N związany jest z 4 różnymi podstawnikami, oraz tych
amin, w których atom C związany jest z różnymi podstawnikami, np.
H
5
C
6
N
+
CH
3
H
7
C
3
C
2
H
5
OH
lub
H
5
C
2
C
H
N
H
2
CH
3
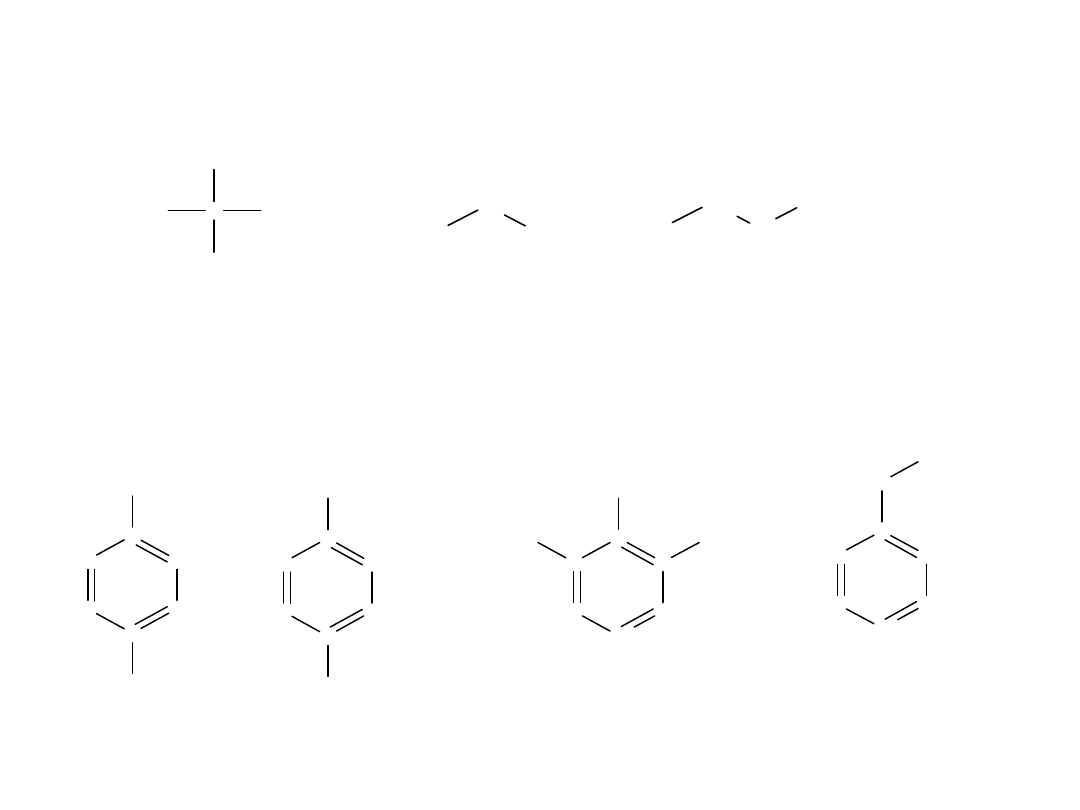
Nazwy amin alifatycznych tworzy się przez dodanie słowa – „amina”, do
nazwy grupy lub grup alkilowych związanych z atomem N.
Bardziej złożone nazwy tworzy się czasem przez dodanie przedrostka –
„amino”
CH
3
C
C
H
3
CH
3
NH
2
tert - butyloamina
(2-dimetyletyloamina)
H
5
C
6
NH
CH
3
N - metylofenyloamina
N
H
2
CH
2
CH
2
OH
2-aminoacetal
(etanoloamina)
Nazwy amin aromatycznych pochodzą często od aniliny. Wyjątek
stanowią toluidyny
CH
CH
C
C
C
H
C
H
NH
2
Br
CH
CH
C
C
C
H
C
H
NH
2
CH
3
C
CH
C
CH
C
C
H
NH
2
CH
3
C
H
3
CH
CH
C
CH
C
H
C
H
N
H
CH
3
p - bromoanilina
p - toluidyna
2,6 - dimetyloanilina
N - metyloanilina
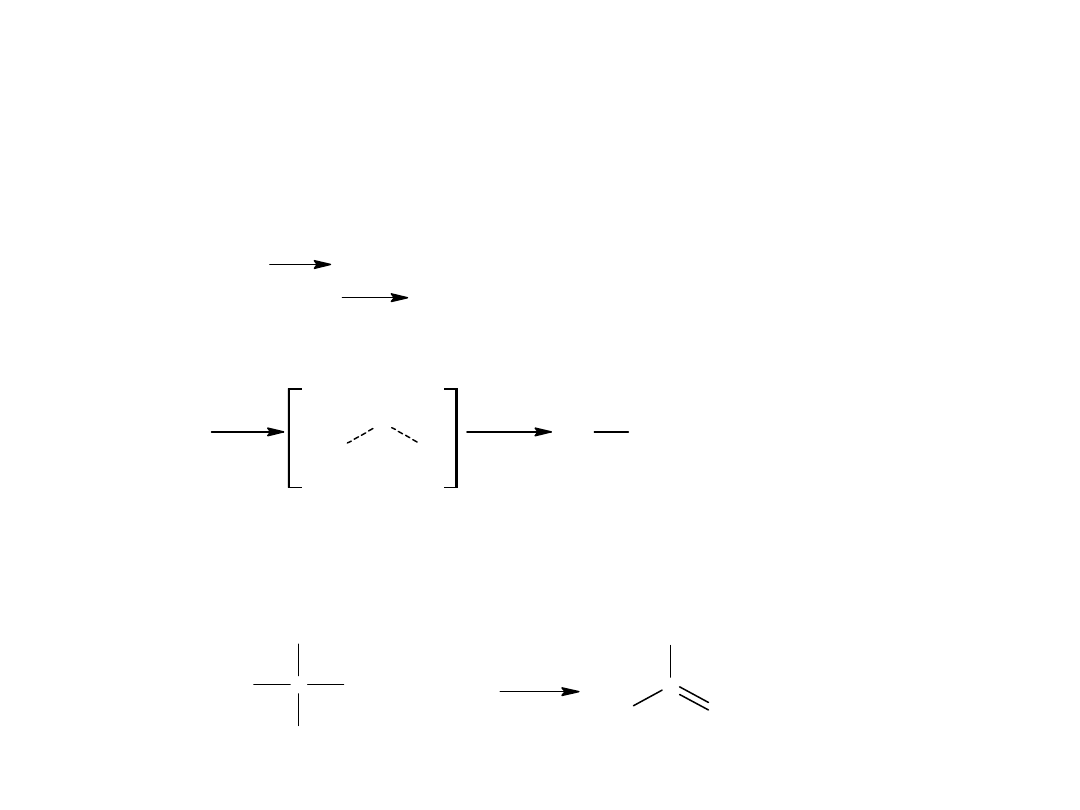
Metody otrzymywania amin
I rz. aminy alifatyczne
1. Amonoliza halogenków – Hoffmanna (alkilowanie amoniaku).
W wyniku zastąpienia chlorowca grupą aminową, powstaje sól aminy, z
której pod wpływem –OH można wyprzeć wolną aminę.
RX + NH
3
RNH
+
3
X
-
RNH
+
3X
-
+ OH
-
RNH
2
+ H
2
O + X
-
Amonoliza jest reakcją substytucji nukleofilowej.
H
3
N: + R -X
N
H
3
R
X
d
+
d
-
N
H
3
+
R
+
X
-
Reakcja nadaje się głównie do otrzymywania amin I rz, gdyż w tych
warunkach dla halogenków II i III rz. przeważa eliminacja np.
C
CH
3
CH
3
C
H
3
Br
+
NH
3
C
H
3
C
CH
2
CH
3
+
NH
4
Br
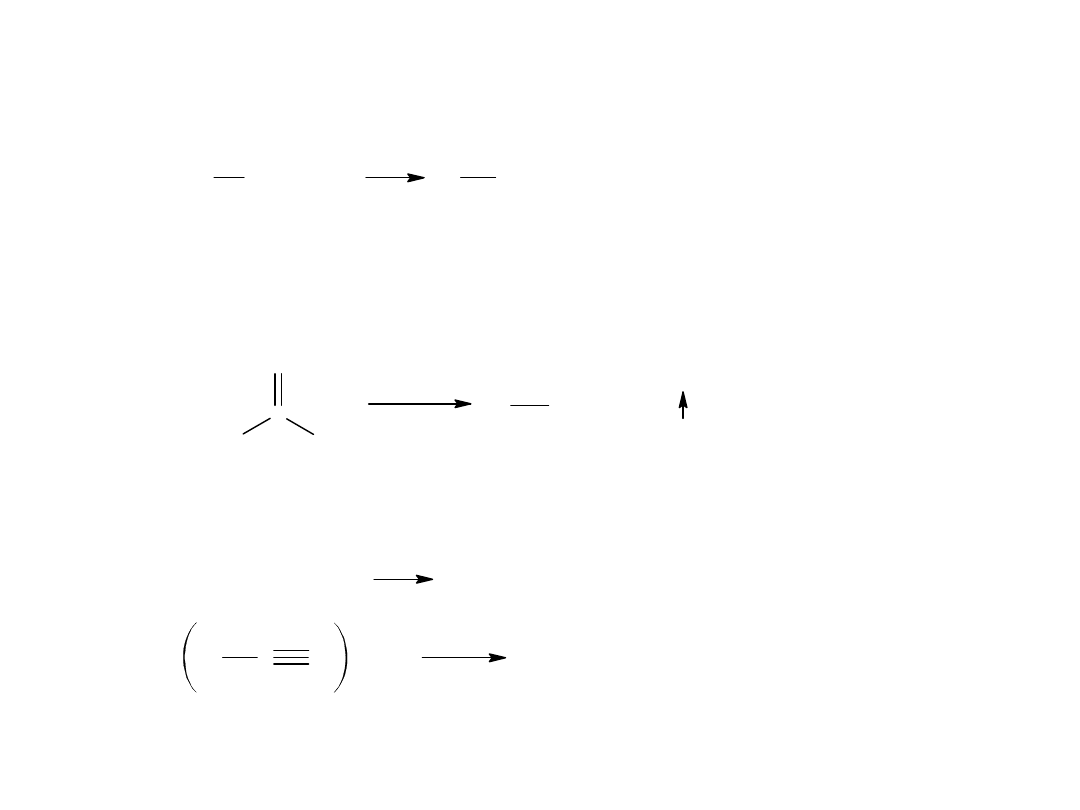
1. Z alkoholu i amoniaku (odmiana metody Hofmanna).
Pary alkoholu przepuszcza się przez rury wypełnione katalizatorem ThO
2
C
H
3
OH
+
NH
3
C
H
3
NH
2
+
O
H
2
2. Degradacja amidów Hofmanna (reakcja podbrominowa)
Metoda prowadzi do otrzymania produktu zawierającego w cząsteczce o
jeden atom węgla mniej niż substrat.
C
H
3
C
NH
2
O
( C l O
-
) B r O
-
, O H
-
R
NH
2
+
CO
2
3. Redukcja nitryli (cyjanków)
R -X + KCN
R -CN + KX
nitryl
R
C
N
R -CN
N a ( a l k o h o l )
R -CH
2
-NH
2
Metoda pozwala na wydłużanie łańcucha węglowego aminy, w stosunku
do wyjściowego halogenku alkilu.
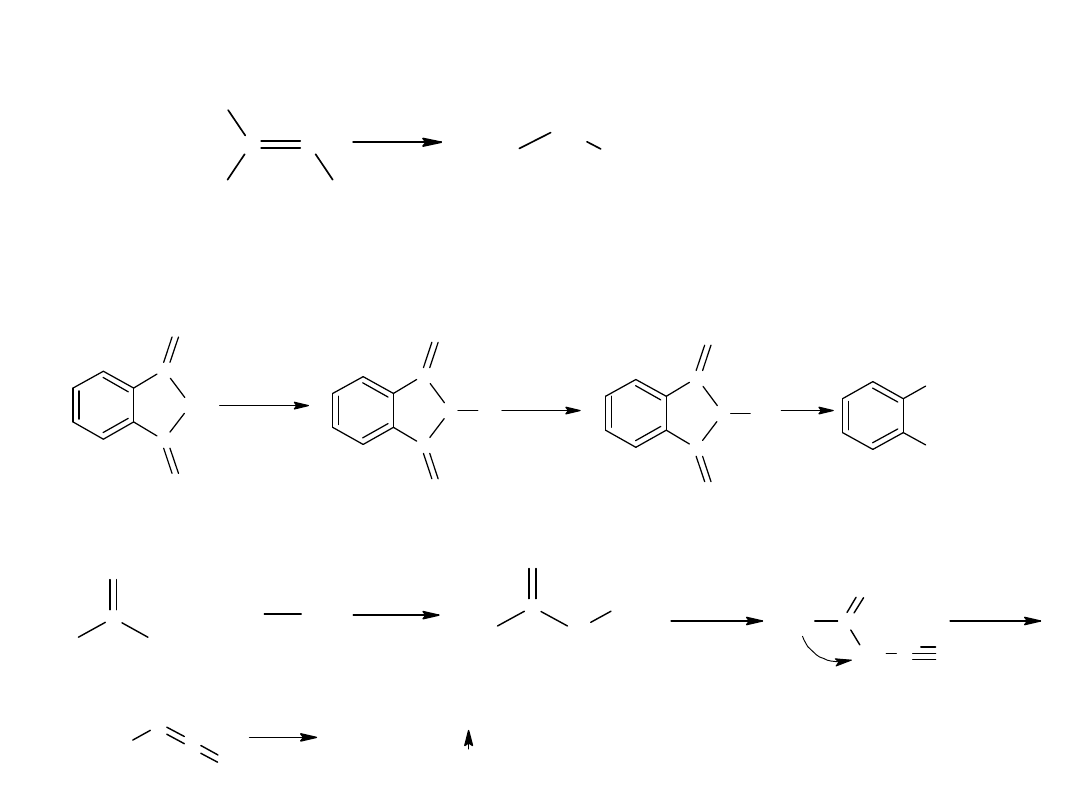
4. Redukcja oksymów.
C
H
3
C
H
N
OH
oksym
P t ( r e d )
R
CH
2
NH
2
5. Reakcja Gabriela – z imidu kwasu ftalowego
NH
C
C
O
O
K O H , C
2
H
5
O H
N
C
C
O
O
K
R - X
D M F
N
C
C
O
O
R
H O H
COOH
COOH
+
R -NH
kw ftalowy
6. Metoda Curtiusa – z azydków kwasowych
R
C
OR'
O
ester
+
N
H
2
NH
2
hydrazyna
O
C
H
3
C
NH
NH
2
hydrazyd
H O N O
k w a z o to w y
R
C
O
NH N
+
N
azydek kwasowy
(przegr)
o g r z
- N
2
C
H
3
N
C
O
izoyjanian
H O H
R -NH
2
+ CO
2
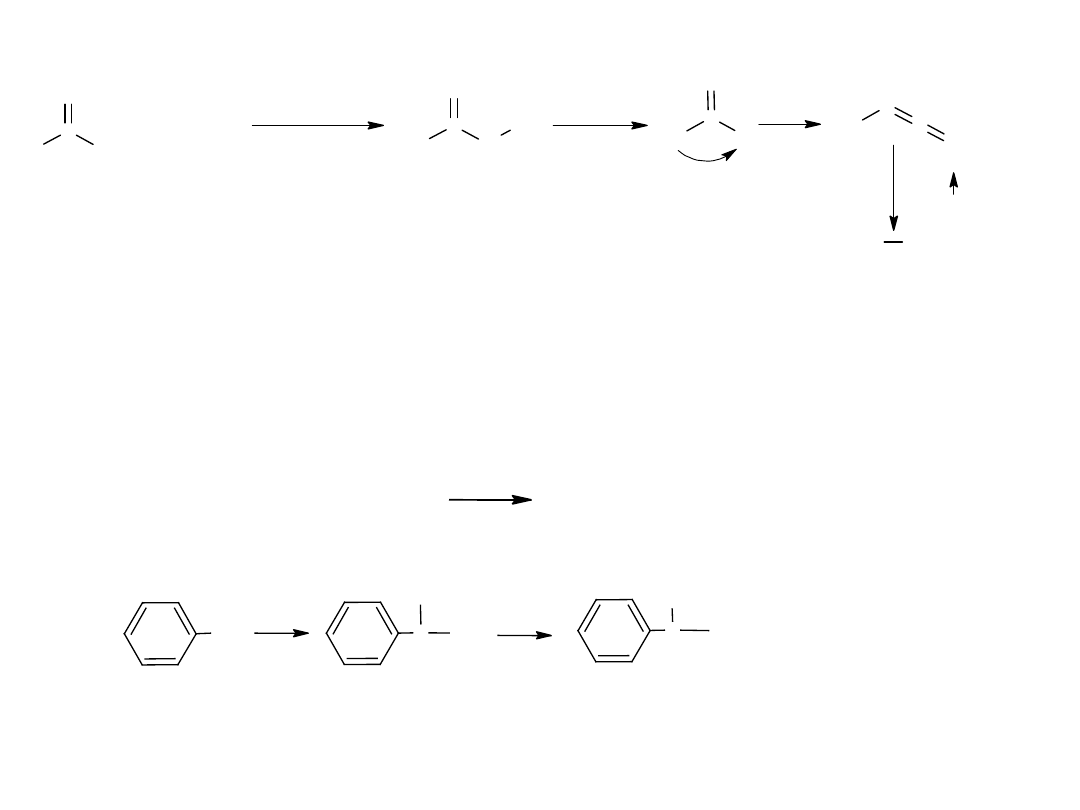
7. Metoda Losena – z kwasów hydroksamowych z SOCl
2
R
C
Cl
O
+
H
2
N -OH
hydroksyloamina
- H C l
R
C
O
NH
OH
kw hydroksamowy
S O C l
2
- H
2
O
R
C
N
O
przegr
R
N
C
O
izocyjanian
-CO
2
R
NH
2
II rz. aminy alifatycze
1. Wyczerpująca amonoliza halogenków.
Przy otrzymywaniu amin II i III rz. nie stosuje się amoniaku, a
odpowiednią aminę.
CH
3
CH
2
CH
2
NH
2
n -propyloamina
+
CH
3
CH
2
Br
CH
3
CH
2
CH
2
NHCH
2
CH
3
N -etylo - n -propyloamina
NH
2
CH
3
Br
N
H
CH
3
N -metyloanilina
CH
3
Br
N
C
H
3
CH
3
N,N - dimetyloanilina

2. Redukcja izocyjanków (izonitryli) alkilowych
C
H
3
CH
2
N
CH
2
CH
3
CH
2
NHCH
3
izocyjanek etylu
N -metylo -etyloamina
III rz. aminy alifatyczne
Wyczerpująca amonoliza halogenków.
Metoda daje dobre rezultaty, w przypadku amin aromatycznych,
natomiast dla amin alifatycznych obserwuje się małe wydajności.
Aminy aromatyczne
Przedstawicielem amin aromatycznych jest anilina, wykryta w produktach
suchej destylacji indyga. Syntetycznie, anilinę otrzymuje się przez
redukcję nitrobenzenu.
1. redukcja nitrozwiązków
NO
2
NH
2
F e \ H C l
2. reakcja Dow – podstawienie chlorowca, grupą aminową, w
odpowiedniej chlorowcopochodnej aromatycznej
Cl
+
NH
3
2 5 0
-
3 0 0 �C
3 5 0 a t m
NH
2
+
NH
4
Cl
Rozróżnianie rzędowości amin
Obecność aminy 1º potwierdza próba izonitrylowa.
1. W wyniku pozytywnej próby izonitrylowej, wydziela się izonitryl
(izocyjanek) o bardzo przykrym zapachu.
R -NH
2
+ CHCl
3
chloroform
�r o d N a O H
R
N
CH
2
+
(NaCl +H
2
O)
2. Z kwasem azotowym (III), aminy zależnie od rzędowości dają
produkty różnego typu
Produktem reakcji amin 1
o
aromatycznych są sole diazoniowe.
NH
2
+
NaNO
2
+
HX
< 1 0 �C
N
N
X
s�l diazoniowa
+
(NaX +H
2
O)
Sole diazoniowe tworzą się również w reakcji 1
o
amin alifatycznych, ale
są tak nietrwałe, że natychmiast rozkładają z wydzieleniem azotu.
R -NH
2
+ NaNO
2
+ HX
< 1 0 �C
R -OH + N
2
+ NaX
alkohol
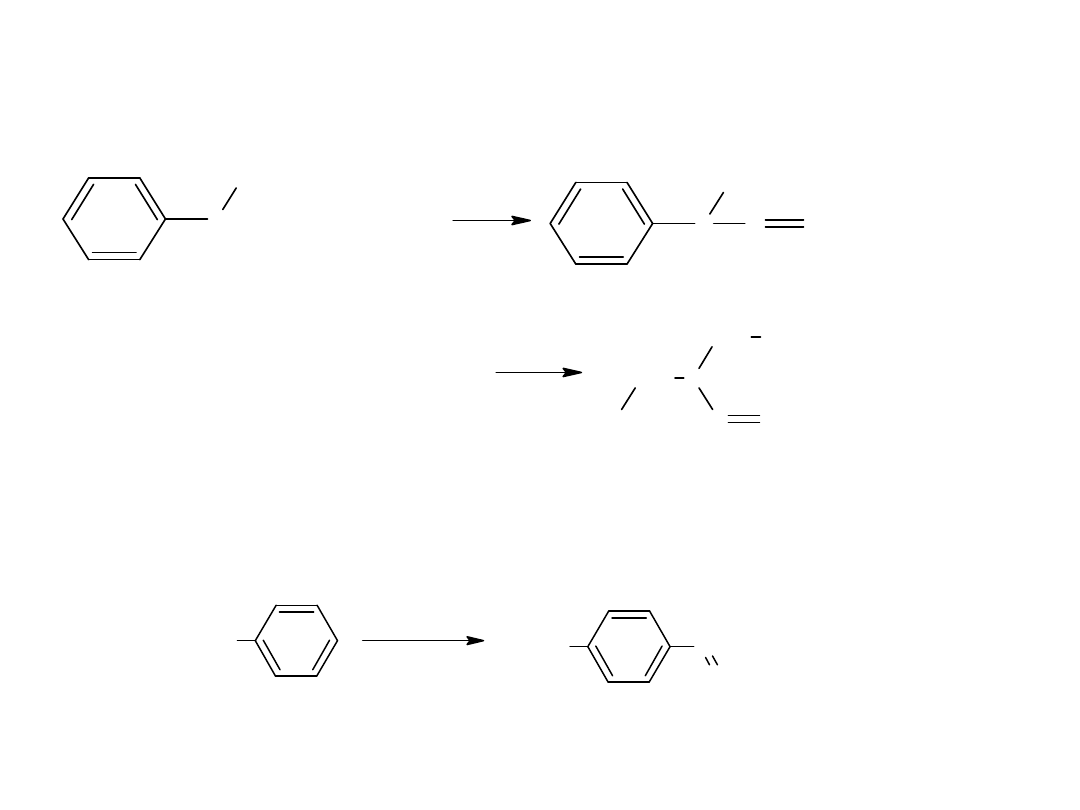
Produktami reakcji amin 2º, zarówno alifatycznych jaki i aromatycznych z
kwasem azotowym (III), są N-nitrozoaminy:
NH
CH
3
+
NaNO
2
+ HX
N
CH
3
N
O
+
NaX + H
2
O
N -metylo -N - nitrozoanilina
N -metyloanilina
CH
3
CH
2
NHCH
2
CH
3
+ NaNO
2
+ HX
C
H
3
CH
2
N
N
CH
2
CH
3
O
N -dietyloamina
N,N -dietylo -N -nitrozoamina
Aromatyczne aminy 3º ulegają substytucji w pierścieniu
(H
3
C)
2
N
N a N O
2
, H X
(H
3
C)
2
N
N
O
N,N -dimetylo -p -nitrozoanilina

3. Skuteczną metodą określania rzędowości amin jest próba Hinsberga.
Próba polega na wytrząsaniu amin z chlorkami kwasów
benzenosulfonowych:
Powstałe pochodne amin 1º, jako zdysocjowane, rozpuszczają się w
wodnym roztworze KOH
Pochodne amin 2
o
są nierozpuszczalne
Aminy 3º nie reagują z chlorkiem kwasu benzenosulfonowego
[C
6
H
5
SO
2
NR
-
]K
+
lub C
6
H
5
SO
2
NR
2
RNH
2
+ ClSO
2
C
6
H
5
RNHSO
2
C
6
H
5
R
2
NH
2
+ ClSO
2
C
6
H
5
R
2
NSO
2
C
6
H
5
Właściwości chemiczne amin
1. Zasadowość.
Aminy alifatyczne są mocniejszymi zasadami od amoniaku, natomiast
aminy aromatyczne są nieco słabsze, ze względu na sprzężenie wolnej
pary elektronowej atomu N z pierścieniem aromatycznym.
Z wodnymi roztworami kwasów, aminy tworzą sole. Swoje właściwości
zasadowe, aminy alifatyczne i aromatyczne wykazują nawet wobec
kwasów Lewisa, co eliminuje kwasy Lewisa jako związki katalizujące
reakcje, w których biorą udział aminy
R -NH
2
H
+
O H
-
NH
3
+
R
Cl
-
NH
2
NH
2
*HCl
chloroodorek aniliny
N:
H
H
+
AlCl
3
NH
2
+
Al
-
Cl
Cl
Cl
kompleks
Sole amin są typowymi związkami jonowymi, dobrze rozpuszczalnymi w
wodzie
2. Nukleofilowść amin
Aminy, podobnie jak amoniak, są dobrymi odczynnikami nukleofilowymi,
przewyższającymi swą reaktywnością wodę, alkohole i estry.
Nukleofilowy charakter amin jest widoczny w reakcjach z
chlorowcopochodnymi alkilowymi, podczas których tworzy się wiązanie
C-N, atom N przyjmuje ładunek dodatni, a chlorowiec oddala się w
postaci anionu.
N:
(R)H
(R)H
+
H
C
H
H
X
R
N
+
C
+
X
-
CH
2
Cl
+
N
H
2
CH
2
NH
np
chlorek benzylu
anilina
N -benzyloanilina
Aminy reagują z aldehydami i ketonami. Z aldehydów powstają
aldoiminy, a z ketonów ketoiminy.
Mechanizm powstawania imin jest następujący: I etapem jest
nukleofilowa addycja aminy do atomu C grupy karbonylowej a II
dehydratacja (eliminacja H
2
O).
Połączenia powstałe w wyniku reakcji amin z aldehydami lub ketonami
noszą nazwę zasad Schiffa
NH
2
+
C
O
H
N
OH
H
H
N
H
aldehyd benzoesowy
hydroksyloamina
benzylideno -anilina (aldoimina)
NH
2
+
C
CH
3
C
H
3
O
keton metylowy
CH
3
N
CH
3
O
H
H
N
C
CH
3
CH
3
metyloetylidenoanilina (ketonina)
Acylowanie – aminy reagują z kwasami karboksylowymi, bezwodnikami i
chlorkami kwasowymi
NH
2
+
C
OH
O
CH
3
kwas octowy
- H
2
O
O
NH
C
CH
3
acetanilid (antyfebryna)
NH
2
+
C
O
Cl
chlorek benzoidylu
- H C l
C
NH
O
benzanilid
Analogicznie
przebiega
reakcja z
bezwodnika
mi
kwasowymi.
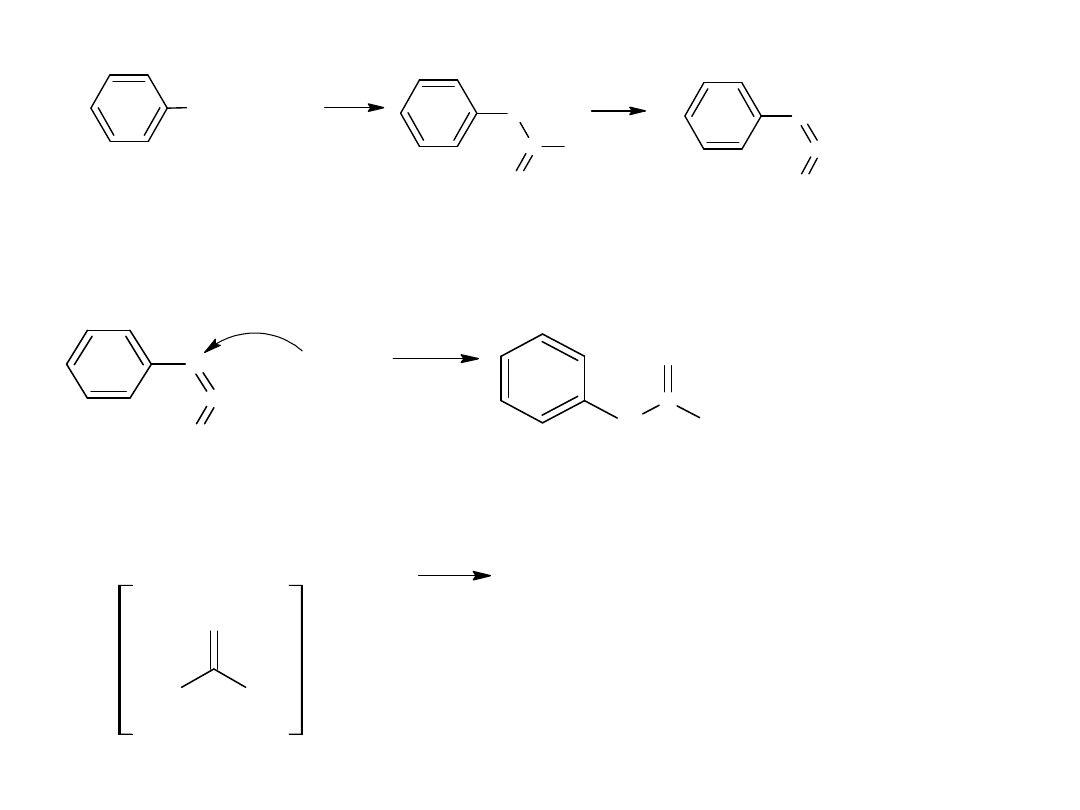
Interesująca jest reakcja aniliny z fosgenem (chlorkiem kwasu
węglowego)
NH
2
+
COCl
2
- H C l
O
NH
C
Cl
chlorek fenylokarbonylu
o g r z
- H C l
N
C
O
izocyjanian
Izocyjaniany z alkoholami tworzą estry kwasu fenylokarbaminowego tzw.
uretany
N
C
O
izocyjanian
+
HOC
2
H
5
O
NH
C
OC
2
H
5
uretan (fenylokarbaminian etylu)
Poliuretany są tworzywami poliaddycyjnymi o dużym znaczeniu
praktycznym, powstającymi z diamin i dioli:
OCN -R -NCO + HO -R -OH
OC -[HN -R -NH -(CO) -O -R -O] -H
poluretan
N
H
2
OH
O
kwas karbaminowy
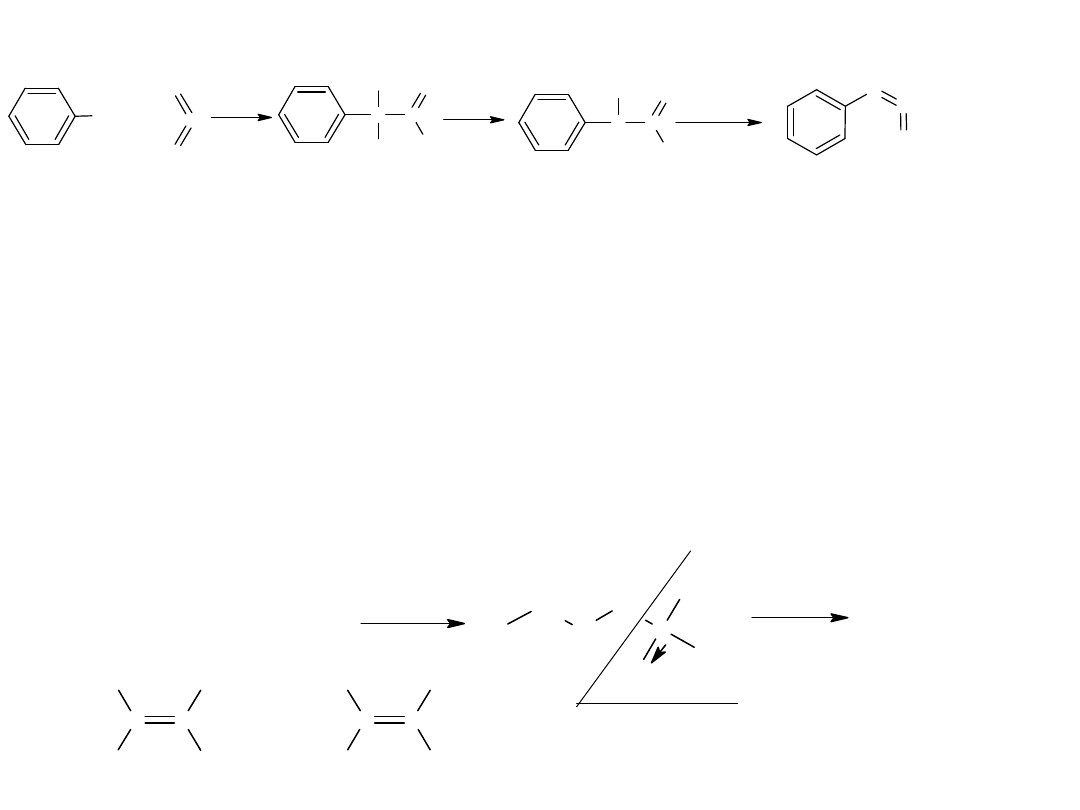
Ważna jest również reakcja aniliny z CS
2
(bezwodnik kwasu tiowęglowego)
NH
2
+
C
S
S
N
+
H
C
S
SH
H
N
H
C
S
SH
kwas fenyloditiokarbaminowy
o g r z
N
C
S
izotiocyjanian (olejek fenylogorczyczny)
Sole i estry kwasu fenyloditiokarbaminowego są surowcami do produkcji
pestycydów, gdyż mają silnie właściwości bakteriobójcze.
3. Utlenianie i redukcja.
Ze względu na to, że atom N w grupie aminowej posiada najniższy stopień
utleniania dla tego pierwiastka, aminy nie ulegają redukcji. Grupy
aminowe można natomiast utleniać do grup nitrowych, co ułatwia
powstawanie odpowiednich nitrozwiązków.
Aminy 3
o
można utlenić do N-tlenków, w wyniku eliminacji Cope’a.
Produktami utleniania są wówczas nietrwałe aminotlenki, które po
ogrzaniu ulegają eliminacji do alkenów
CH
3
CH
2
CH
2
CH
2
N(CH
3
)
2
H
2
O
2
C
H
3
CH
2
CH
2
CH
2
N
CH
3
O
CH
3
o g r z
- ( C H
3
) N O H o k s y n
C
C
C
H
3
CH
3
H
H
+
C
C
C
H
3
CH
3
H
H
+
CH
3
CH
2
CH=CH
2
12%
21%
67%
4. Reakcje podstawiania elektrofilowego w aminach aromatycznych
Grupa aminowa jest podstawnikiem bardzo silnie atakującym pierścień
(oddaje elektrony wywołując zagęszczenie ładunku ujemnego w
najbliższym sąsiedztwie).
Atak grupy elektrofilowej następuje więc w położenie orto- i para.
Olbrzymia reaktywność grupy aminowej powoduje, że reakcje
substytucji elektrofilowej przebiegają poprzez etap podstawienia w
grupie funkcyjnej a nie w pierścieniu
NH
C
H
3
N
N
+
CH
3
NH
2
X
-
moweina Perkina (I syntetyczny
barwnik purpurowy uzyskany podczas
utleniania aniliny K2Cr2O7 w ko�cu XIXw
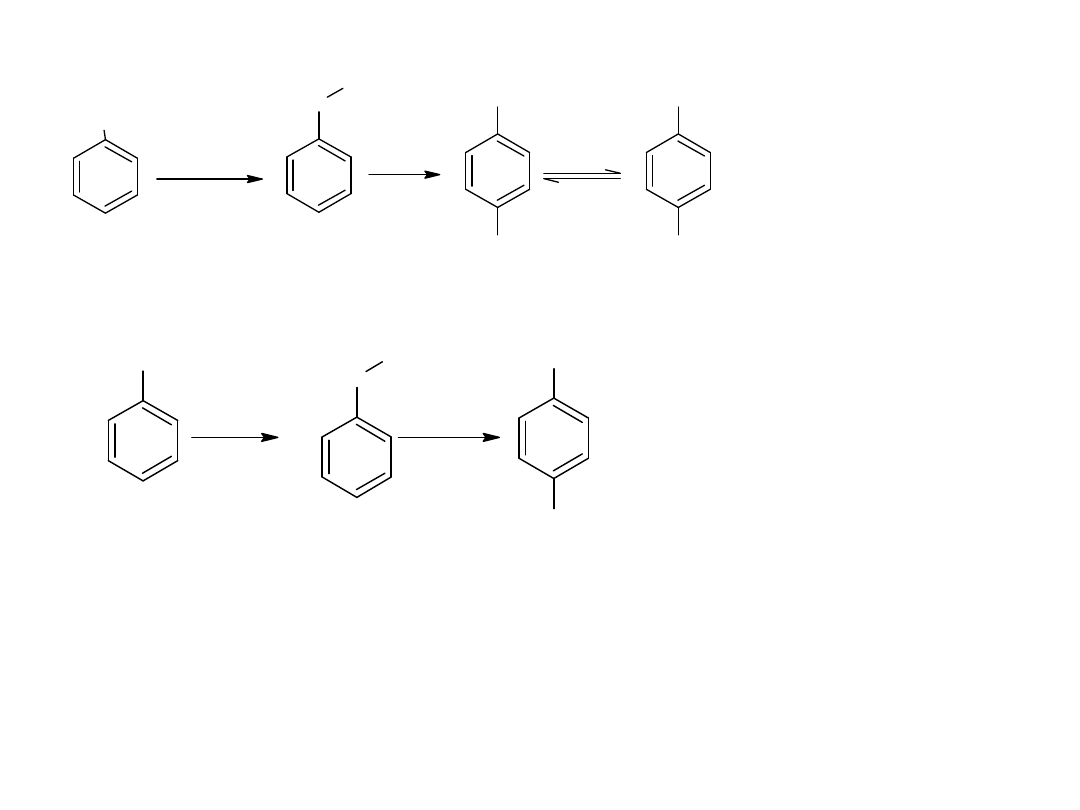
np. sulfonowanie
N
H
2
H
2
S O
4
- H
2
O
N
H
SO
3
H
o g r z
NH
2
SO
3
H
NH
3
+
SO
3
(-)
kw sulfanilowy
kw fenyloaminosulfonowy
bromowanie
NH
2
H
2
S O
4
- H
2
O
N
H
Br
N -bromoanilina
o g r z
NH
2
Br
Ze względu na dużą podatność grupy aminowej na utlenianie,
niemożliwe jest bezpośrednie nitrowanie amin. W związku z tym
zabezpiecza się grupę aminową, przekształcając ją w grupę amidową
(mniej aktywującą pierścień ale również kierującą w położenie orto i
para) w wyniku reakcji acylowania.
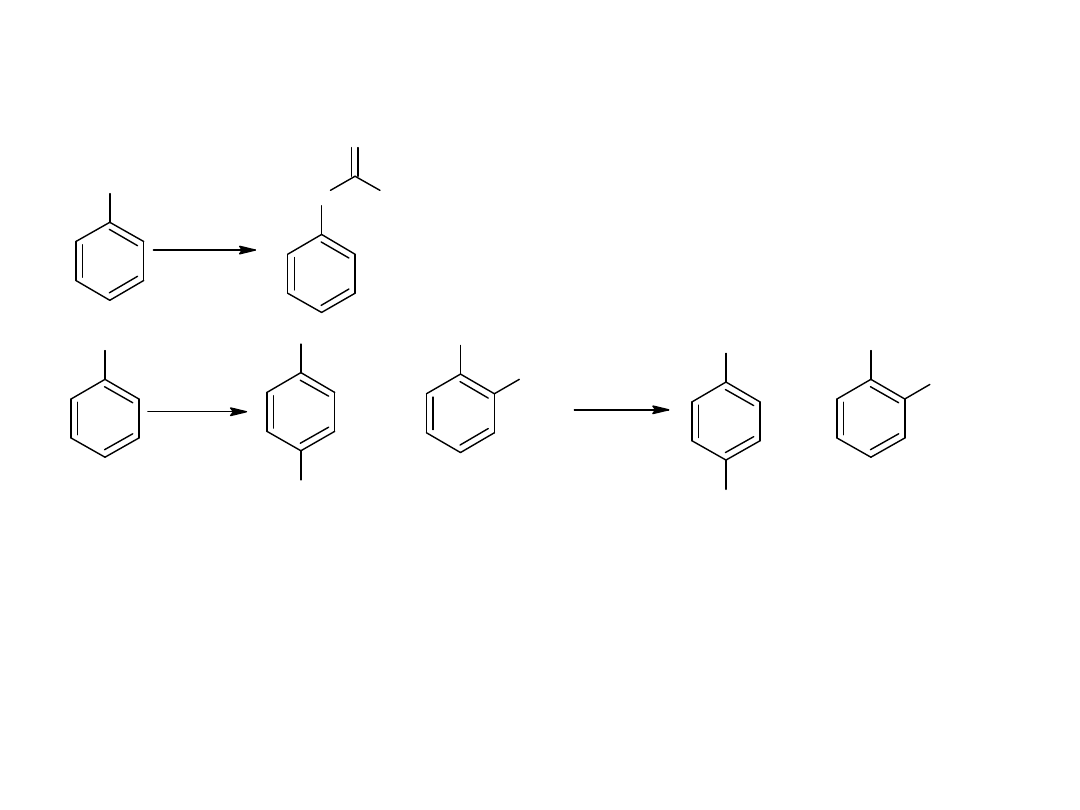
Po zacylowaniu, nitrowanie nie nastręcza trudności. Grupę amidową
usuwa się przez hydrolizę.
NH
2
C H
3
C O O H
N
H
O
CH
3
acetanilid
NHCOCH
3
H N O
3
, H
2
S O
4
NHCOCH
3
NO
2
+
NHCOCH
3
NO
2
H
2
O
NH
2
NO
2
NH
2
NO
2
+
p i orto nitroacetanilidy
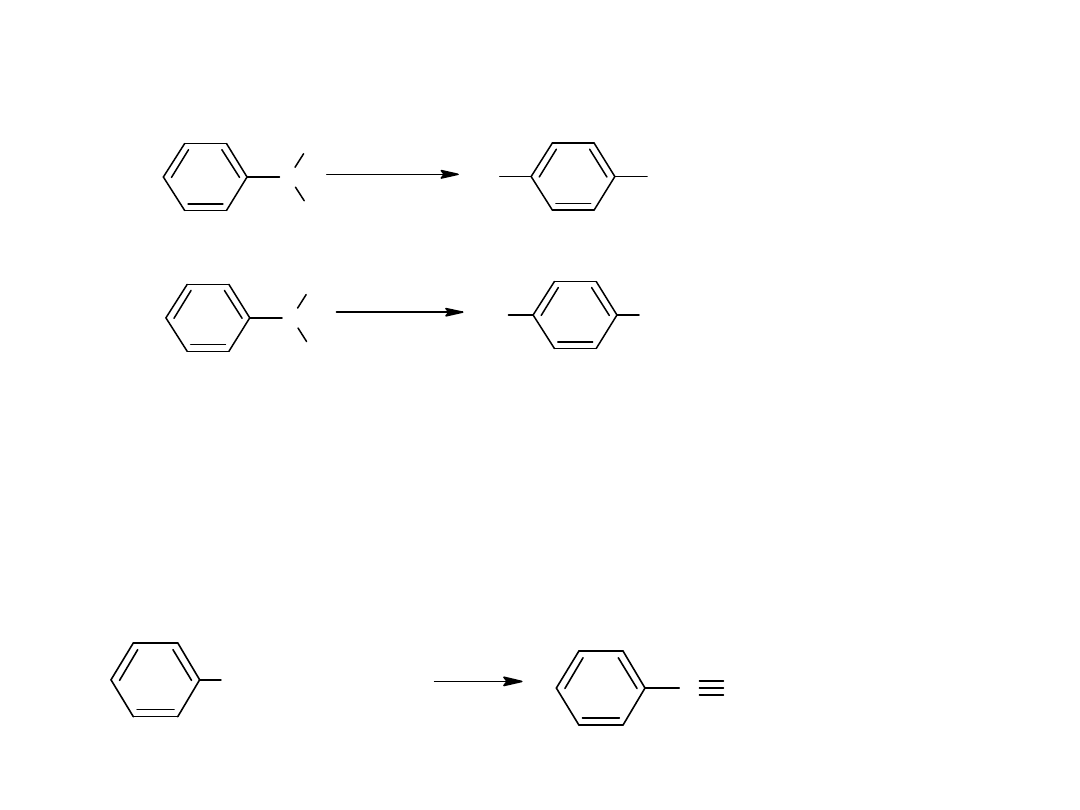
5. Wewnątrzcząsteczkowe przegrupowanie pochodnych aniliny
Izomeryzacja zachodzi dla wszystkich pochodnych aniliny, w których 1
atom H grupy aminowej podstawiony jest inną grupą
N
H
OH
o g r z
O
H
NH
2
p -hydroksyanilina
N -fenylohydroksyloamina
N
Br
COCH
3
o g r z
Br
NHCOCH
3
o -bromoacetamid
Gdy położenie para- jest zajęte, przegrupowanie może zajść w położenie
orto.
6. Diazowanie.
Bardzo ważną reakcją I
o
amin aromatycznych jest reakcja diazowania.
Aminy 1
o
w roztworze kwasu azotowego (III) ulegają przemianie w sól
diazoniową.
Reakcja została odkryta przez Griess’a
NH
2
+
NaNO
2
+ HX
< 1 0 �C
N
N
+
: X
-
+ NaX +H
2
O
s�l diazoniowa
Natychmiast po otrzymaniu roztworu soli diazoniowej, przeprowadza się
następną reakcję, ponieważ związki te ulegają rozkładowi.
Związki diazoniowe (dobrze rozpuszczalne w wodzie) odznaczają się
szczególnie dużą reaktywnością, dzięki której znalazły szerokie
zastosowanie w syntezie organicznej. W szeregu aromatycznym, sole
diazoniowe odgrywają podobną rolę, jeśli chodzi o różnorodność
zastosowań w syntezie, jak chlorowcopochodne w szeregu alifatycznym.
Budowa kationu diazoniowego była przedmiotem długich badań, z powodu
skomplikowanych oddziaływań dodatnio naładowanej grupy N
2
z
pierścieniem aromatycznym:
N
+
N
C
H
+
N
+
N
-
CH
+
N
+
N
-
CH
+
N
+
N
-
Sole diazoniowe ulegają licznym reakcjom, np:
1. reakcje wymiany, w których od soli odrywa się cząsteczka, a na jej
miejsce przyłącza się do pierścienia inny atom lub grupa
2. reakcje sprzęgania, w których atomy N pozostają w cząsteczce
3. redukcja
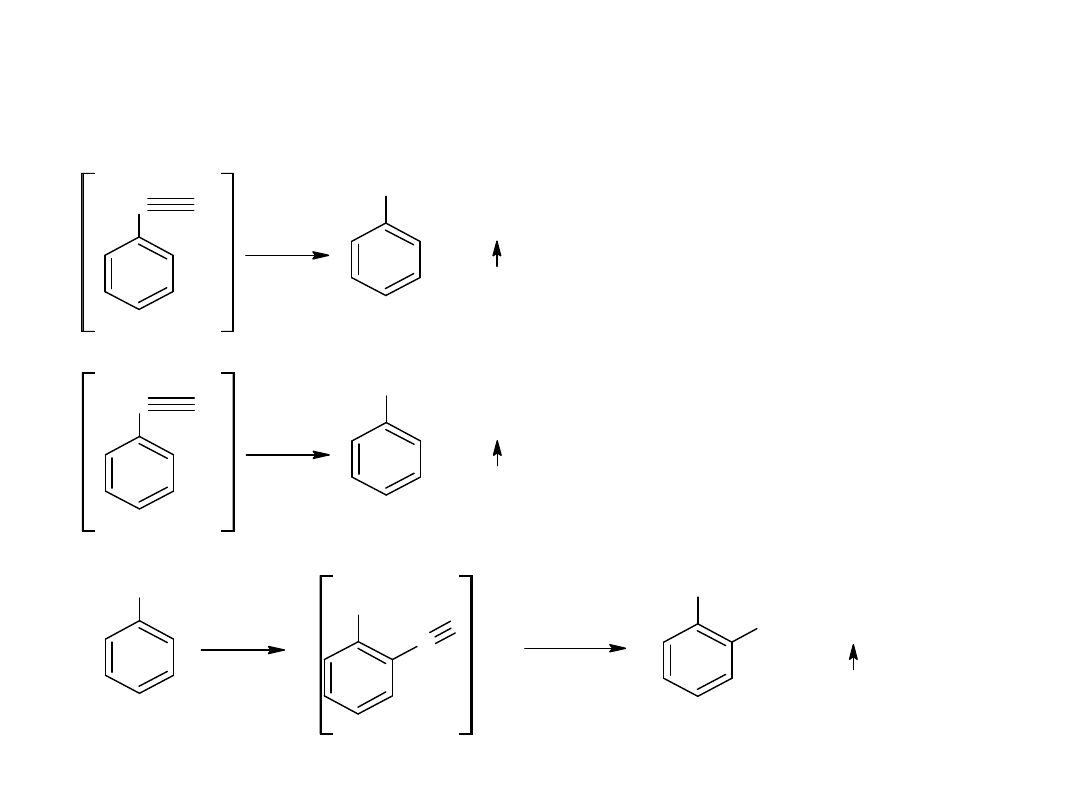
Ad 1.
Redukcja Sandmeyera.
Jest to reakcja wymiany grupy diazoniowej na chlor, brom lub grupę
cyjanową w obecności soli miedziowych
N
N
+
C u C l
Cl
+
N
2
chlorobenzen
N
N
+
Br
+
N
2
bromotoluen
C u B r
CH
3
N a N O
2
, H C l
N
N
CH
3
+
Cl
-
C u C N
chlorek o -toluenodiazoniowy
CH
3
CN
N
2
+
o -tolunitryl
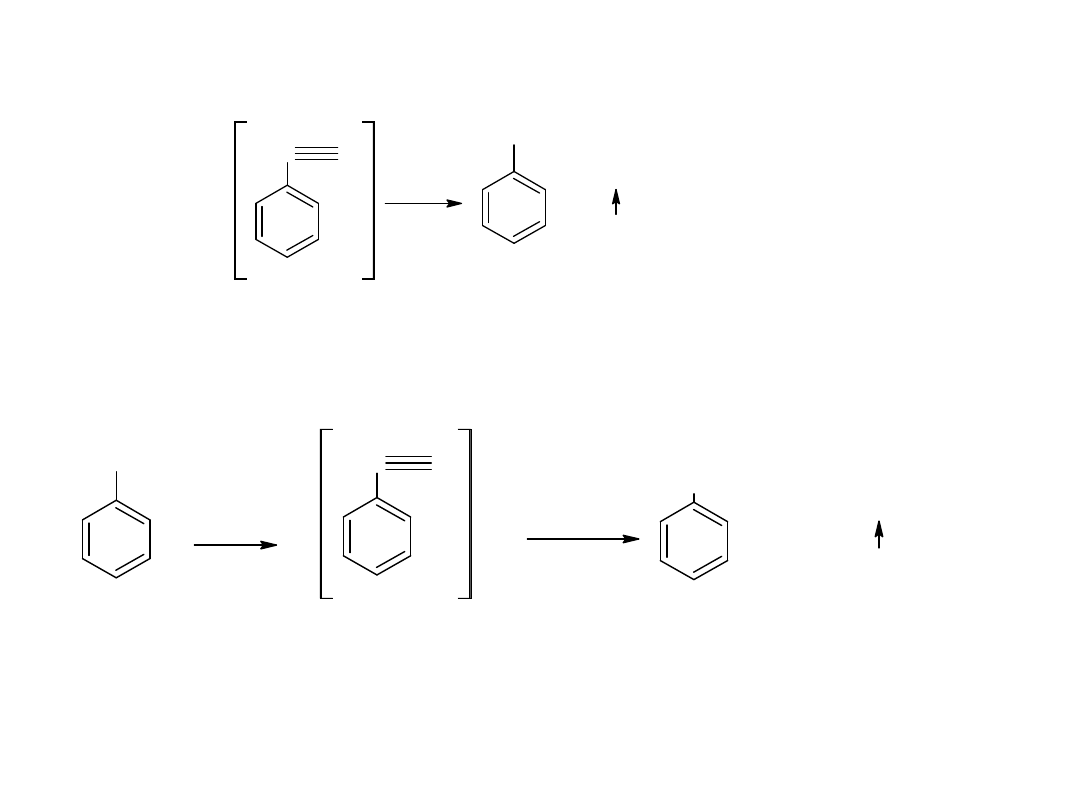
reakcja Gattermanna
Jest reakcją wymiany grupy diazoniowej na jod, w obecności
sproszkowanej miedzi.
N
N
+
I
+
N
2
K I
reakcja Schiemanna
Jest to reakcja wymiany grupy diazoniowej na fluor, polegająca na
termicznym rozkladzie suchych fluoroboranów diazoniowych
N
N
+
NH
2
+
N
2
N a N O
2
, H B F
4
BF
-
4
1 5 0 �C
F
+
BF
3
Jest to niebezpieczna reakcja, ponieważ sole diazoniowe, w stanie
suchym a zwłaszcza po podgrzaniu, są wybuchowe.
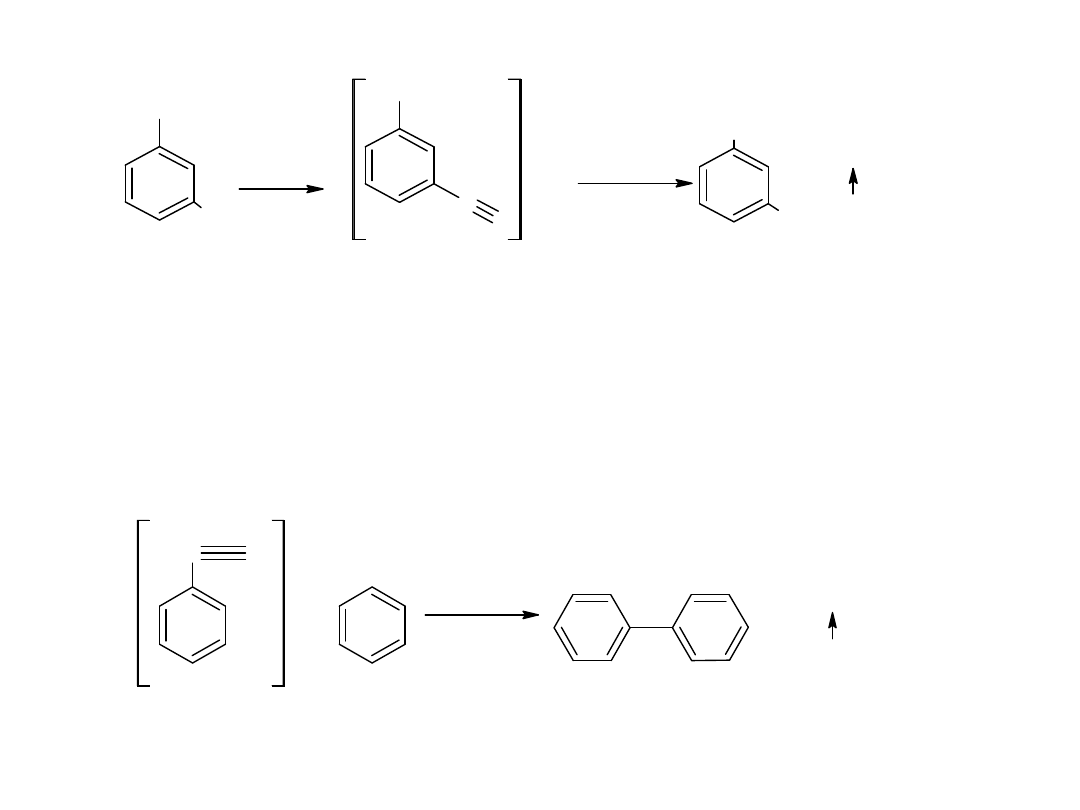
Reakcja powstawania fenoli
NO
2
N
+
N
+
NO
2
NH
2
N
2
Cl
-
NO
2
OH
+
N a N O
2
, H C l
H
2
O
1 0 0 �C
m -nitrofenol
Wymiana na grupę –SH zachodzi w sposób analogiczny.
Redukcja Gomberga i Bachmanna.
W reakcji pomiędzy benzenem lub innym węglowodorem aromatycznym
z solą diazoniową powstaje pochodna bifenylu.
N
N
+
+
bifenyl
N
2
+
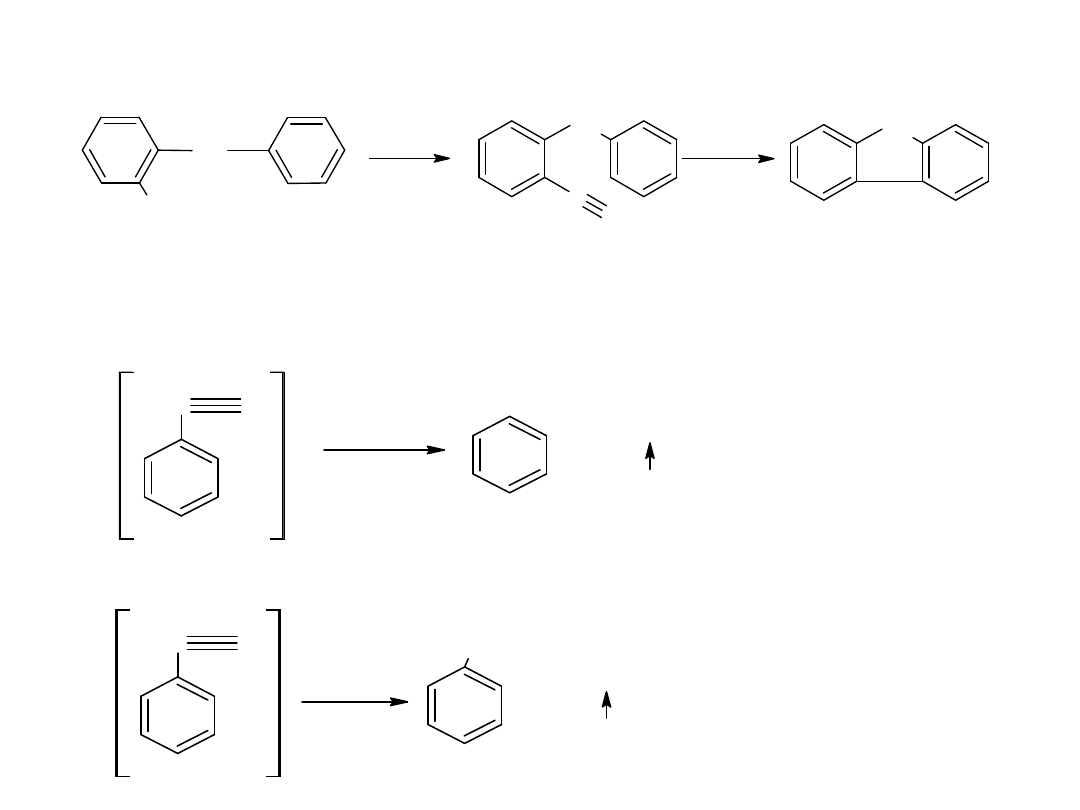
Psorr wykazał, że istnieje również możliwość otrzymania węglowodorów o
pierścieniach skondensowanych.
CH
2
NH
2
difenylometan
N a N O
2
, H C l
CH
2
N
+
N
CH
2
fluoren
Wymiana na wodór lub grupę alkoksylową zachodzi w obecności
niewielkich ilości soli miedzi, podczas ogrzewania soli diazoniowych z
alkoholami lub H
3
PO
4
N
N
N
N
lub
+
+
C
2
H
5
O H
H
3
P O
4
C
2
H
5
O H
OC
2
H
5
N
2
+
N
2
+
fenetol (eter fenylowoetylowy)
Ad.2
W środowisku zasadowym kationy diazoniowe przekształcają się w
aniony kwasów diazowych, które można wyodrębnić z roztworu w postaci
soli z metalami
N
N
Cl
-
+
N
N
O
-
K
+
+
N
N
O
-
K
+
anion kw cis(syn)diazowegoanion kw trans(anty)diazowego
Skłonność związków diazoniowych do reakcji z odczynnikami nukleofilowymi
prowadząca do soli kwasów diazowych, stała się przesłanką do prób
przeprowadzenia reakcji związków diazoniowych ze związkami
aromatycznymi.
Okazało się, że reakcje, znane jako reakcje sprzęgania zachodzą bardzo
łatwo w niskiej temperaturze(0
o
C), ale tylko ze związkami aromatycznymi
posiadającymi silnie aktywujące grupy, tj. z fenolami i aminami. Sprzęganie
jest przykładem reakcji substytucji elektrofilowej, w której pierścień ulega
atakowi soli diazoniowej. Substytucja zachodzi najczęściej w położenie para-
w stosunku do grupy aktywującej pierścień.
Produkty sprzęgania noszą nazwę związków azowych. Związki azowe
stanowią grupę związków o wyraźnym zabarwieniu.
Ze względu na barwę lub jej zmianę w zależności od pH, związki azowe są
powszechnie stosowane jako barwniki oraz wskaźniki kwasowo-zasadowe.
N
+
N
+
Cl
-
OH
fenol
N a O H
N
N
OH
p -hydroksyazobenzen
zw azowy (zolty)
Gdy polozenie para jest zajete, sprzeganie zachodzi w pozycji orto
-
.
N
+
N
+
Cl
-
C
H
3
OH
+
N a O H
N
N
O
H
CH
3
zw azowy
benzoazo -p -krezol
N
+
N
+
Cl
-
N
CH
3
CH
3
+
N
N
N
CH
3
CH
3
benzenoazodimetyloanilina
(zolcien maslowa)
SO
3
H
NH
2
N a N O
2
, H C l
SO
3
-
N
+
N
+
N
CH
3
CH
3
kw sulfakilowy
N,N -dimetyloanilina
sol wew po diazowaniu
N
N
N
CH
3
CH
3
SO
3
H
p -sulfonobenzeno -azodimetyloanilina (heliantyna,oranz metylowy)
N
H
2
COOH
kw antranilowy
N a N O
2
, H C l
N
+
N
COO
-
sol wew po diazowaniu
+
N
CH
3
CH
3
N
N
N
CH
3
CH
3
COOH
o -karboksybenzeno -azodimetyloaminobenzen
(czerwien metylowa)
NH
2
N
H
2
(benzydyna)
+
NH
2
SO
3
H
NH
2
SO
3
H
N
N
SO
3
H
NH
2
[(czerwien kongo)
W wyniku sprzęgania z aminami 1
o
powstają najpierw związki diazowe,
które po ogrzaniu z solami amin aromatycznych, ulegają przekształceniu
w związki aminoazowe.
N
+
N
+
Cl
-
N
H
2
+
diazoaminobenzen
C
6
H
5
N H
2
* H C l
N
N
NH
N
N
NH
N
N
NH
2
p -aminoazobenzen (zolcien anilinowa)
Ad.3
Pod wpływem
, grupę diazoniową można zredukować do
grupy hydrazynowej. Metoda jest wykorzystywana do otrzymywania
aromatycznych pochodnych hydrazyny
NaHSO
3
lub SnCl
2
N
+
N
+
Cl
-
N a H S O
3
+ H
2
O
NH
NH
2
(*HCl)
chlorowodorek fenylohydrazyny
Występowanie i znaczenie amin
W przyrodzie występuje olbrzymia różnorodność związków aminowych,
od najprostszej metyloaminy do złożonych amin zwanych alkoloidami,
znanych ze swego działania biologicznego.
Aminy są licznie reprezentowane wśród syntetycznych leków, jakimi
dysponuje współczesna terapia, np. acetanilid noszący nazwę
antyfebryny, trujący alkaloid atropina służący w małych dawkach do
rozszerzania źrenicy w trakcie leczenia oczu, fenacetyna i anestezyna –
środki przeciwbólowe stanowiące składniki gazów anestetycznych.
Nie można zapominać o szkodliwości większości amin. Wiele z nich ma
silne właściwości narkotyczne, w większych dawkach działają paraliżująco
na układ nerwowy np. morfina, strychnina, muskaryna (składnik
muchomorów), meskalina (wyodrębniona z kaktusa).
N
N
CH
3
nikotyna
OH
3
C
OH
3
C
OH
3
C
CH
2
CH
2
NH
2
meskalina
N
C
H
3
COOCH
3
COOCH
3
kokaina
Kodeina, która jest eterem metylowym morfiny. Jest ordynowana przez
lekarzy jako lek przeciwkaszlowy i przeciwbólowy. Heroina, inna bliska
pochodna morfiny, nie występuje w postaci naturalnej, ale jest
otrzymywana na drodze diacetylowania morfiny
O
H
O
H
H
O
H
H
N
CH
3
morfina
OH
3
C
O
H
H
OH
H
N CH
3
kodeina
OH
3
C
O
H
H
OCH
3
C
H
N CH
3
O
O
heroina
Morfina i jej pochodne są wyjątkowo użytecznymi substancjami
farmakologicznymi, pomimo to stwarzają one poważny problem społeczny
ze względu na ich właściwości wywołania uzależnienia narkotycznego.
Wiele wysiłku włożono więc, w rozpoznawanie sposobu działania morfiny i
opracowanie zmodyfikowanych analogów morfiny, które zachowują jej
działania analegtyczne, ale nie powodują uzależnienia fizycznego.
Meperydyna (Demerol), szeroko stosowany analgetyk, oraz Metadon,
substancja stosowana w leczeniu uzależnienia heroinowego, to dwa
związki, które spełniają „regułę morfinową”
C
C
C
C
C
N
C
C
C
6
H
5
CH
3
O
N
CH
3
CH
3
metadon
CO
2
H
5
C
2
N
CH
3
meperydyna
Produkowane masowo przez przemysł organiczny aminy aromatyczne są
bardzo szkodliwe dla zdrowia. Są to związki toksyczne, w większości
rakotwórcze. Począwszy od aniliny wywołują one nie gojące podrażnienia
skóry; wchłaniane przez skórę mogą doprowadzić do zatrucia organizmu.
Do najbardziej niebezpiecznych należą naftyloaminy i benzydyna.
Naftyloaminy otrzymuje się w procesie redukcji nitronaftalenów, natomiast
benzydynę w wyniku przegrupowania benzydynowego związków
hydrazowych.
NH
NH
o g r z
N
H
2
NH
2
hydrazobenzen
benzydyna
Największym zagrożeniem dla zdrowia są nitrozoaminy, stanowiące
produkty niecałkowitego utleniania amin 2º. Są to związki silnie
rakotwórcze
(CH
3
)
2
NH
u t l
1 0 �C
(CH
3
)N -NO
N,N -dimetylo -N -nitrozoamina
dimetyloamina
Obecność związków nitrozowych została wykryta w konserwowanych
produktach spożywczych.
Przykładem aminy 3º, mającym duże znaczenie, jest tzw. iperyt azotowy
i jego pochodne. Związek ten wywołuje silne podrażnienia skóry
wchodząc w reakcję z białkiem i niszcząc je (hamuje podział komórek).
Pochodne iperytu stosowane są jako składniki leków
antynowotworowych.
N
H
CH
2
CH
2
Cl
CH
2
CH
2
Cl
i
R
CH
2
CH
2
OH
CH
2
CH
2
OH
iperyt azotowy (gaz bojowy)
N -alkilo,N,N -dietanoloamina
R
NH
2
( C H
2
)
2
O
R
CH
2
CH
2
OH
CH
2
CH
2
OH
S O C l
2
R
CH
2
CH
2
Cl
CH
2
CH
2
Cl
Ad.III
Nitryle i izonitryle (cyjanki i izocyjanki) są pochodnymi cyjanowodoru.
Cyjanowodór istnieje tylko w postaci H-CΞN, natomiast jego pochodne
tworzą 2 odmiany tautometryczne
R
C
N
R
N
NH
nitryl (cyjanek)
izonitryl (izocyjanek)
W chemii organicznej ważną rolę odgrywają nitryle, związki o bardzo
reaktywnej grupie funkcyjnej.
Izonitryle mają mniejsze znaczenie.
Reaktywnym miejscem w grupie nitrylowej jest atom C grupy -CN; ponadto
wykazują one reaktywność przy atomie C
C
C
N
C
N
miejsca aktywne w nitrylach
W związkach aromatycznych, grupa -CN wywiera wpływ dezaktywujący
na pierścień i kieruje podstawniki elektrofilowe w położenie –meta.
Metody otrzymywania nitryli i izonitryli.
1.
z chlorowcopochodnych z cyjankami metali
R-X + NaCN
R-CN cyjanek
R-X + AgCN
R-NC izocyjanek
Różnica w produktach reakcji jest wynikiem różnic budowy cyjanków
metali. Cyjanek sodowy jest zdysocjowany, a zatem nukleofilem
atakującym chlorowcopochodną jest anion CN
-
.
Cyjanek srebrowy (nierozpuszczalny) o budowie kowalencyjnej
uniemożliwia reakcję przy atomie C.
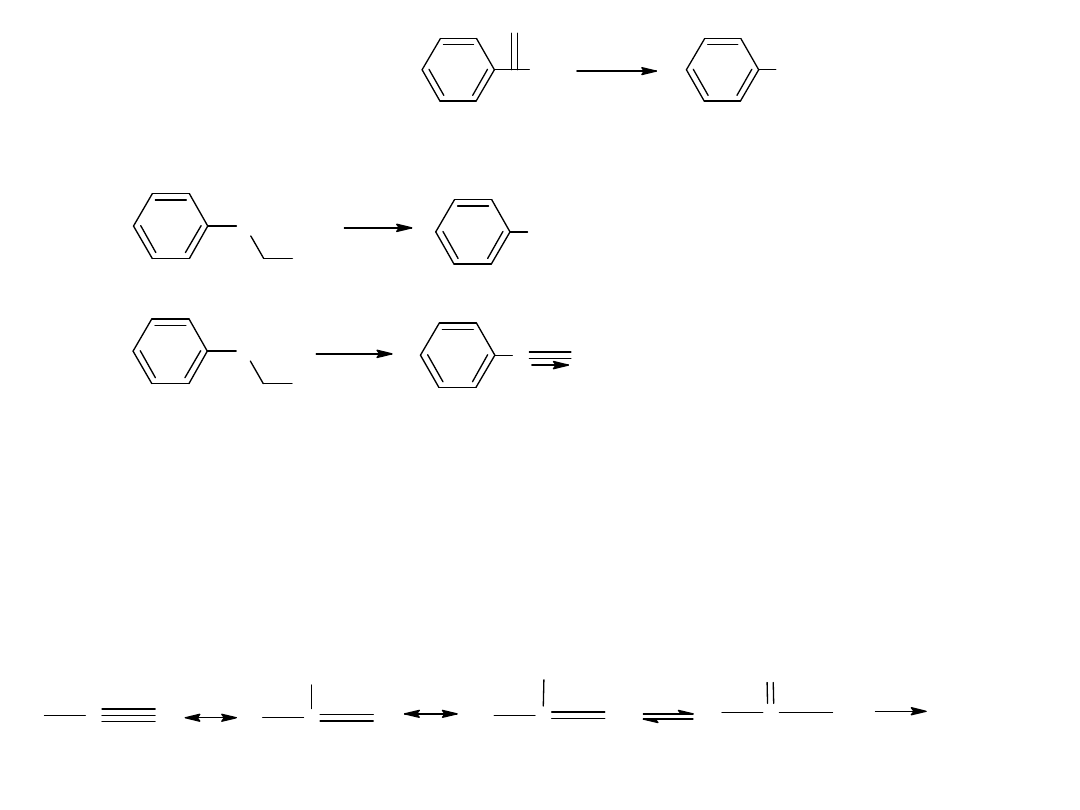
2.
z amidów kwasowych
O
NH
2
CN
S O C l
2
, p i r y d y n a
8 0 �C
benzonitryl
benzoamid
3.
z amin 1
o
przez odwodnienie
CH
2
NH
2
CH
2
NH
2
benzyloamina
N i O
2
- 2 H
2
N a O H , C H C l
3
CN
benzonitryl
N
CH
2
Właściwości chemiczne nitryli
1. hydroliza do kwasów karboksylowych.
Nitryle ulegają hydrolizie do kwasów zarówno w środowisku kwaśnym jak
i zasadowym. Produktem przejściowymi są w tej hydrolizie amidy
kwasowe, ale reakcji nie udaje się zatrzymać na etapie amidu.
W hydrolizie zasadowej nukleofilem atakującym atom C grupy CN jest
OH-.
R
C
N
O H
-
R
C
N
-
OH
H
+
OH
R
C
NH
O
R
C
NH
2
H
2
O
R -COOH

W hydrolizie kwasowej, najpierw proton przyłącza się do atomu N grupy
-CN a potem następuje atak nukleofilowy cząsteczki wody na atom C
R
C
N
R
C
N
+
H
OH
2
+
R
C
NH
O
H
R
C
NH
H
2
O
R -COOH
H
+
H
2
O
- H
+
O
R
C
NH
2
2. redukcja do amin 1
o
R-CN
N a ( a l k o h o l )
l u b L iA lH
4
lu b n ik ie l R a n e y a
R
2
CH
2
NH
2
3. reakcja Rittera.
Nitryle z alkoholami 3º tworzą N-acylowe pochodne amin
R
C
R
R
OH
+
R -CN
H
2
S O
4
, H
2
O
R
C
R
R
NH
C
R
O
Jest to rzadka reakcja, w której grupa aminowa łączy się z 3º
atomem C
Reakcje zachodzące przy węglu
4. alkilowanie w położenie
R -CH
2
-CN
R - X
C
H
3
C
H
3
CH CN
5. reakcje kondensacyjne – zachodzą tylko dla nitryli posiadających H
przy węglu . Są to reakcje analogiczne do kondensacji aldolowej
Ad. IV
Cyjaniany i izocyjaniany
Kwas cyjanowy istnieje tylko w postaci zawierającej H przy atomie N, a
więc w postaci kwasu izocyjanowego
N
C
OH
N
H
2
CH O
kw cyjanowy
kw izocyjanowy (tantomeria kw cyjanowego)
Trwałe są pochodne organiczne kwasu izocyjanowego. Kwas cyjanowy
otrzymuje się z kwasu cyjanurowego a ten z mocznika
N
H
2
N
H
2
O
3
- 3 N H
3
NH
N
H
N
H
O
O
O
3HNCO
mocznik
kw cyjanurowy
kw izocyjanowy
Izocyjaniany są związkami
o dużej reaktywności
względem odczynników
nukleofilowych. Cyjaniany
nie mają większego
znaczenia
Właściwości chemiczne izocyjanianów
1. hydroliza do amin.
Produktem przejściowym, w reakcji są nietrwałe kwasy karbaminowe,
ulegające samorzutnej dekarboksylacji
R
N
C
O
H O H
R
NH
COOH
kw karbaminowy
- C O
2
R
NH
2
amina
2. z alkoholami tworzą karbaminiany tzw. uretany
R
N
C
O
R
NH C
O
OR
R O H
(karbaminian,uretan)
3. z NH
3
i aminami tworzą pochodne mocznika
R
N
C
O
R
NH C
O
NH
R
(pochodna dialkilowa mocznika)
R N H
2
Ważną pochodną kwasu cyjanowego, z punktu widzenia praktycznego,
jest melamina, czyli cyjanamid, służąca do wyrobu tworzyw melaminowo-
fenolowych
N
C
NH
2
3
cyjanamid
N
C
C
N
N
C
NH
2
NH
2
N
H
2

Organiczne związki fosforu, arsenu, antymonu i bizmutu
W celu uzupełnienia wiadomości o organicznych związkach azotu, zostaną
scharakteryzowane związki organiczne pozostałych pierwiastków grupy V.
Im pierwiastek grupy V jest położony w wyższym okresie układu
okresowego, tym wykazuje bardziej metaliczny charakter.
Azot ma wyraźnie niemetaliczny charakter, o czym świadczy jego trwały
związek z wodorem – amoniak i jego nietrwałe tlenki.
Różnice zasadowości między NH
3
i analogicznymi związkami pozostałych
pierwiastków są następujące:
NH
3
>PH
3
>AsH
3
>SbH
3
c h a ra k te r z a s a d o w o �c i
Podobnie w przypadku związków analogicznych do amin.
a)
Aminy – zasadowe
b)
Fosfiny – słabo zasadowe.
c)
Arsyny – obojętne
a) Związki fosfoorganiczne
Fosfor tworzy połączenia analogiczne do amin, noszące nazwę – fosfiny.
Fosfiny wywodzą się z fosforiaku –PH
3
.
Istnieją fosfiny 1, 2, 3 i 4
o
.
Wykazują mniej zasadowy charakter od amin. Utleniają się bardzo łatwo:
1 i 2
o
do odpowiednich kwasów fosfonowych a 3
o
do tlenków trialkilofosfin
R -PH
2
u t l
OH
P
R
OH
O kw alkilofosforowy
Ist
IIst
R
2
-PH
u t l
OH
P
R
O
R kw alkilofosfinowy
IIIst
R
3
-P
u t l
R
P
R
O
R kw trialkilofosfinowy
Fosfiny są związkami o bardzo
nieprzyjemnych zapachach. Są trujące
F
P
OHC(H
3
C)
2
O
CH
3
sarin (ester izopropylowy kwasu fluorometylofosfinowego)
b) Związki arsenoorganiczne.
Arsen tworzy arsyny, będące pochodnymi arsenowodoru. – AsH
3
.
Istnieją arsyny 1, 2, 3 i 4
o
.
W powietrzu, utleniają się samorzutnie, najczęściej są samozapalne.
Otrzymuje się je z kakodylu.
Kakodylem nazywane są wszystkie związki arsenoorganiczne zawierające
ugrupowania (CH
3
)
2
As-
Kakodyl został odkryty przez Cadeta, w czasie destylacji arszeniku z
octanem potasu.
As
O
O
As
O
arszenik
+
4CH
3
COOK
- C O
2
CH
3
CH
3
As
O
As
C
H
3
CH
3
tlenek kakodylu
Wolny kakodyl powstaje w reakcji chlorku kakodylu z cynkiem
CH
3
As
CH
3
C
H
3
O
As
CH
3
tlenek kakodylu
+
2
As Cl
C
H
3
C
H
3
2
Z n
- Z n C l
2
chlorek kakodylu
As
CH
3
C
H
3
dimer

c) Organiczne związki antymonu
Antymon tworzy tylko stybiny 3 i 4
o
.
Wykazują charakter metaliczny.
Wobec kwasów zachowują się jak metale, wypierając wodór
R
3
Sb + 2 HCl
C
H
3
Sb
Cl
Cl
+ H
2
d) Organiczne związki bizmutu.
Bizmut tworzy co prawda, połączenia typu R
3
Bi o nazwie bizmutyny, ale
nie są to związki o charakterze zasadowym.
Pod wpływem kwasów rozkładają się:
(CH
3
)
3
bI + 3HCl
BiCl
3
+ 3CH
4
Bizmutyny, częściej traktuje się jako metaliczne pochodne
węglowodorów, tj.
C
H
3
H
C
H
3
H
C
H
3
H
+
Bi
(CH
3
)
3
Bi
trimetylobizmut
Bizmutyny stanowią pomost pomiędzy związkami czysto organicznymi
a związkami metaloorganicznymi

Związki metaloorganiczne
Związkami metaloorganicznymi określa się te substancje, w których atom
metalu jest bezpośrednio połączony z atomem C.
Spośród znanych obecnie związków metaloorganicznych do
najważniejszych należą pochodne: litu, sodu, potasu, magnezu, glinu,
cynku, kadmu, rtęci, cyny i ołowiu.
Nazwy ich tworzy się, traktując grupy organiczne jako podstawniki przy
atomach metalu. (Wyjątek stanowią pochodne acetylenu zwane
acetylenkami).
Można je uważać za związki pochodne alkanów lub bardziej
skomplikowanych połączeń, w których atom H został zastąpiony metalem
C
H
3
H
C
H
3
H
+
Zn
CH
3
CH
3
Zn
cynkodimetyl lub dimetylocynk
CH
3
CH
2
CH
2
CH
2
-H + Li
CH
3
CH
2
CH
2
CH
2
Li
n
-
butylolit lub litobutyl
CH
3
CH
2
MgBr bromek etylomagnezowy
C
6
H
5
HgCl chlorek fenylorteciowy
Br -CH
2
-COOC
2
H
5
+ Zn ------> Br -Zn -CH
2
-COOC
2
H
5
ester etylowy kw
a - bromooctowego (r. Reformackiego))

Wszystkie związki metaloorganiczne odznaczają się dużą reaktywnością
chemiczną, przy czym najbardziej reaktywnym miejscem cząsteczki jest
wiązanie węgiel-metal. Przyjmuje się, że wiązanie to jest typu jonowego
lub jest to wiązanie atomowe spolaryzowane.
Wyjątkowo reaktywne są połączenia metaloorganiczne: litu, sodu, potasu,
magnezu, glinu i cynku. Związków tych nie udaje się wyodrębnić w stanie
czystym. Istnieją tylko jako roztwory, produkty lub substraty, pewnych
reakcji chemicznych.
Na powietrzu łatwo się utleniają, są samozapalne!
Każdy metal, tworzący związek metaloorganiczny, jest mniej
elektroujemnym pierwiastkiem od węgla i dlatego wiązanie między
atomem C i metalem jest silnie polarne
R -M
d
-
d
+
Związki metaloorganiczne mają ogromne zastosowanie, głównie dzięki
jednej wspólnej właściwości: mogą służyć jako źródło atomów C.
Przyjmuje się, że reakcja wymiany metalu jest reakcją podstawiania
elekrofilowego S
E2
przy atomie C, ponieważ w stanie przejściowym z
atomem C połączona jest zarówno grupa odchodząca jak i przyłączony
odczynnik elektrofilowy:
C
M
+
X
+
C
X
Md
+
d
-
C
X
+
M
+
schemat podstawienia elektrofilowego SE
2
Wśród związków metaloorganicznych, najbardziej znane i
rozpowszechniane są związki magnezoorganiczne, zwane związkami
Grignarda (1900r.)
Związki Grignarda otrzymuje się w reakcji halogenków z metalem w
obecności suchego eteru. Większość z nich jest wybuchowa!
Znane są 2 typy połączeń magnezoorganicznych
R
Mg
R
1
R
Hg
R
+
Mg
R
Mg
R
+
Hg
2
R -MgX
RX + Mg
s u c h y
e t e r
RMgX
Połączenia typu 1 otrzymuje się w reakcji wymiany dialkilortęci z
magnezem, podobnie jak w przypadku otrzymywania dialkilosodu.
Reakcja halogenku alkilu z Mg lub Na typu 2 zawsze prowadzi do bardziej
złożonych, symetrycznych alkilów. (synteza Wurtza).
CH
3
J 2Na
CH
3
J CH
3
Na
CH
3
Na
CH
3
-CH
3
2CH
3
J 2Mg
C
H
3
Mg
C
H
3
+
MgJ
2CH
3
J
C
H
3
Mg
C
H
3
2CH
3
-CH
3
+
+
+
+
Podstawienie elektrofilowe przy atomie C w związkach Grignarda
CH
3
MgJ + HOH
CH
4
+ MgOHJ
CH
3
MgJ + HO -R
CH
4
+
OR
Mg
I
RMgX + HNH
2
R -H
Mg
NH
2
X
+
RMgX + R -NH
2
R -H
+
Mg
NHR
X
RMgX
+
R
1
R
1
O
R
1
R
1
R
OMgX
H O H
R
1
R
1
R
OH
+
Mg
OH
X
alkohol 3st
RMgX
+
R
1
H
O
R
1
H
R
OMgX
H O H
R
1
H
R
OH
+
Mg
OH
X
alkohol 2st
RMgX + HSH
R -SH (tiol) +
RMgX + CO
2
R -COOMgX
H
+
R -COOH kwas
3(C
6
H
5
)MgBr + PCl
3
P(C
6
H
5
)
3
+
Mg
Cl
Br
3
fosfina
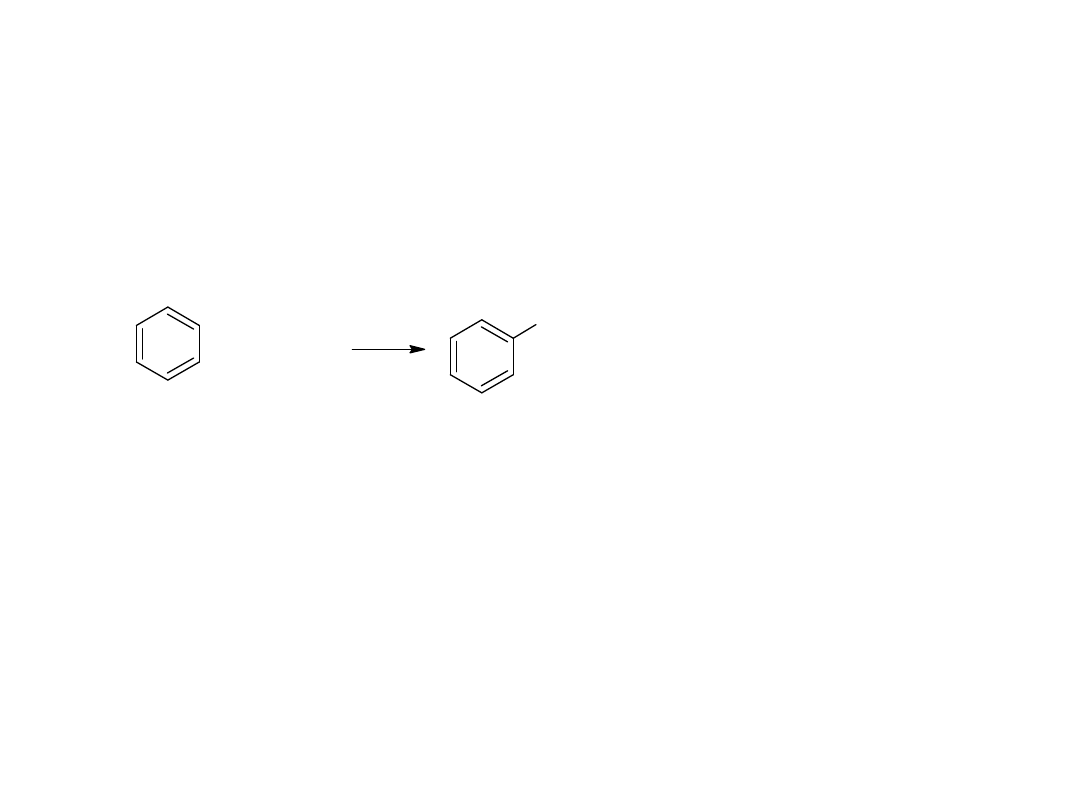
Drugą grupą związków metaloorganicznych są związki rtęcioorganiczne. Są one
mniej reaktywne od związków Grignarda, ale za to nie utleniają się na
powietrzu (są bezpieczne) i są odporne na działanie wody. Wiązanie C-Hg jest
jednak bardzo reaktywne i dlatego służy między innymi do otrzymywania
innych związków metaloorganicznych. Ze względu na bardzo łatwy sposób
powstawania tych związków, są one bardzo niebezpieczne dla zdrowia. Mogą
się kumulować np. w rybach, gdyż środowisko wodne nie przeszkadza w
tworzeniu tego typu połączeń i ich pochodnych.
Dość często wykorzystuje się w syntezie organicznej organiczne związki talu
+
(CF
3
COO)
3
Tl
Tl(OOCCF
3
)
2
+
CF
3
COOH
90%
Praktyczne znaczenie reakcji talowania w syntezie organicznej wynika z kilku
powodów:
1. grupa Tl(OOCCF
3
)
2
może być łatwo zastąpiona innymi podstawnikami
2. talowanie podstawionych pochodnych benzenu prowadzi do powstawania
prawie wyłącznie tylko jednego izomeru, a nie mieszaniny wynikającej z
wpływu skierowującego podstawników. Fluorowce i grupy alkilowe
skierowują grupę Tl(OOCCF
3
)
2
w pozycję para, a niektóre podstawniki
zawierające tlen skierowują wyłącznie w położenie orto, nawet jeśli jest to
niezgodne z ich normalnym wpływem skierowującym.
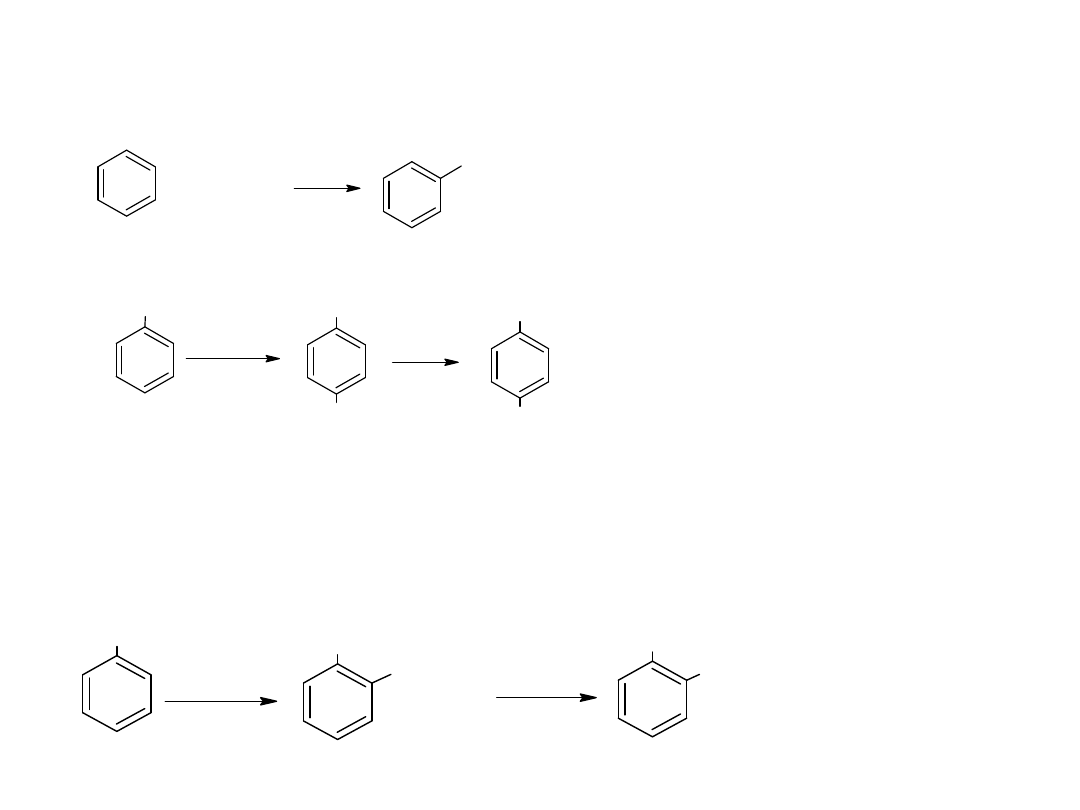
Grupę
można szczególnie łatwo zastąpić atomem jodu przez
proste ogrzewanie z wodnym roztworem KI. Jest to w tej chwili najprostsza
metoda otrzymywania dość trudno dostępnych aromatycznych
pochodnych jodu
Tl(OOCCF
3
)
2
+
(CF
3
COO)
3
Tl
Tl(OOCCF
3
)
2
+
CF
3
COOH
90%
C(CH
3
)
3
( C F
3
C O O )
3
T l
Tl(OOCCF
3
)
2
C(CH
3
)
3
K I , H
2
O
I
C(CH
3
)
3
93%
Do innych grup, które można wprowadzić na miejsce wiązania węgiel –
tal, należą OH, CN, SH, SCN, NH
2
.
Przykładem przebiegu reakcji niezgodnego z wpływem skierowującym
podstawnika jest talowanie kwasu benzoesowego.
COOH
( C F
3
C O O )
3
T l
COOH
Tl(OOCCF
3
)
2
K I
COOH
I
kwas o -jodobenzoesowy
Ferrocen
Jednym z najbardziej osobliwych związków metaloorganicznych jest
dicyklopentadienylożelazo (ferrocen), najważniejszy przedstawiciel grupy
związków zwanych metalocenami. Ferrocen można łatwo otrzymać
różnymi sposobami, np. w reakcji sodowej pochodnej cyklopentadienu z
chlorkiem żelazawym
2C
5
H
5
Na + FeCl
2
(C
5
H
5
)
2
Fe + 2NaCl
Atom żelaza w ferrocenie jest połączony z pierścieniami
cyklopentadienowymi za pośrednictwem elektronów ¶, które w łącznej
liczbie 12 uzupełniają wypełnianie poziomów 3d i 4s żelaza do trwałej
konfiguracji 18 elektronów. Dzięki temu ferrocen jest wyjątkowo trwałym
związkiem o wyraźnie zaznaczonych właściwościach aromatycznych. W
cząsteczce ferrocenu atom żelaza znajduje się między ułożonymi
równolegle, jeden nad drugim, pierścieniami cyklopentadienowymi. Inne
metaloceny są zbudowane podobnie
Fe
budowa ferrocenu

Organiczne związki siarki
Spośród organicznych związków siarki najważniejsze to:
I
Tioalkohole (merkaptany, tiole)
II
Tioetery (sulfidy, siarczki alkilowe)
III
Kwasy sulfonowe
Ad. I
Tioalkohole są to związki zawierające grupę -SH. Są one pochodnymi H
2
S
i jako takie mają bardziej kwaśny charakter od alkoholi, których
pierwowzorem jest H
2
O. Ich kwaśny charakter ujawnia się w zdolności do
tworzenia soli, noszących nazwę merkaptydów.
Metody otrzymywania tioalkoholi
RX
+
KSH
RSH
+
KJ
C
2
H
5
J
KSH
+
wodorosiarczek potasowy
C
2
H
5
SH
+
KJ
merkaptan etylowy
ROH
+
S
H
2
300�C
ThO
2
RSH
+
O
H
2
C
H
3
OH
+
S
H
2
-H
2
O
CH
3
SH
merkaptan metylu
Właściwości merkaptanów
Merkaptany bardzo łatwo utleniają się do disiarczków (disulfidów)
2C
2
H
5
SH
H
2
O
2
C
2
H
5
- S - S - C
2
H
5
disiarczek etylu
Pod wpływem silniejszych utleniaczy merkaptany ulegają przekształceniu
w kwasy sulfonowe
2C
2
H
5
SH
+
6MnO
4
+
18H
+
C
H
3
S
O
O
OH
+
6Mn
2
+
+
9H
2
O
Taki proces redox jest podstawą wielu reakcji zachodzących w
organizmach żywych, w których białka zawierające grupę –SH podlegają
procesom utleniania i redukcji.
W obecności zasad tworzą sole (merkaptydy)
C
2
H
5
SH
+
NaOH
-H
2
O
C
2
H
5
SNa
etylomerkaptyd sodowy
2C
2
H
5
SH
+
HgO
-H
2
O
(C
2
H
5
S)Hg
merkaptyd rteci
Merkaptany są oleistymi cieczami o odrażającym zapachu, nawet w
stanie wielkiego rozcieńczenia.
C
4
H
9
SH – merkaptan butylu jest głównym składnikiem płynu
wydzielanego przez skunksa.
Poddane redukcji w obecności silnych reduktorów, rozpadają się do
odpowiednich węglowodorów i H
2
S. Reakcja jest bardzo przydatna, w
procesie oczyszczania zasiarczonej ropy naftowej
RSH
red. H
2
S
H
2
+
RH
Ad. II
Tioetery można także traktować jako pochodne H
2
S
S
H
H
S
R
R
(tioeter, sulfid, siarczek)
W stanie czystym mają dość przyjemny zapach, np. disulfid allilu
CH
2
=CH-CH
2
-S-S-CH
2
-CH=CH
2
znajduje się w olejku eterycznym czosnku.
Metody otrzymywania tioeterów
1. W reakcjach K
2
S z chlorowcopochodnymi alkilowymi
2C
2
H
5
Cl
+
K
2
S
-2KCl
C
2
H
5
- S - C
2
H
5
siarczek dietylu
(sulfid dietylowy)
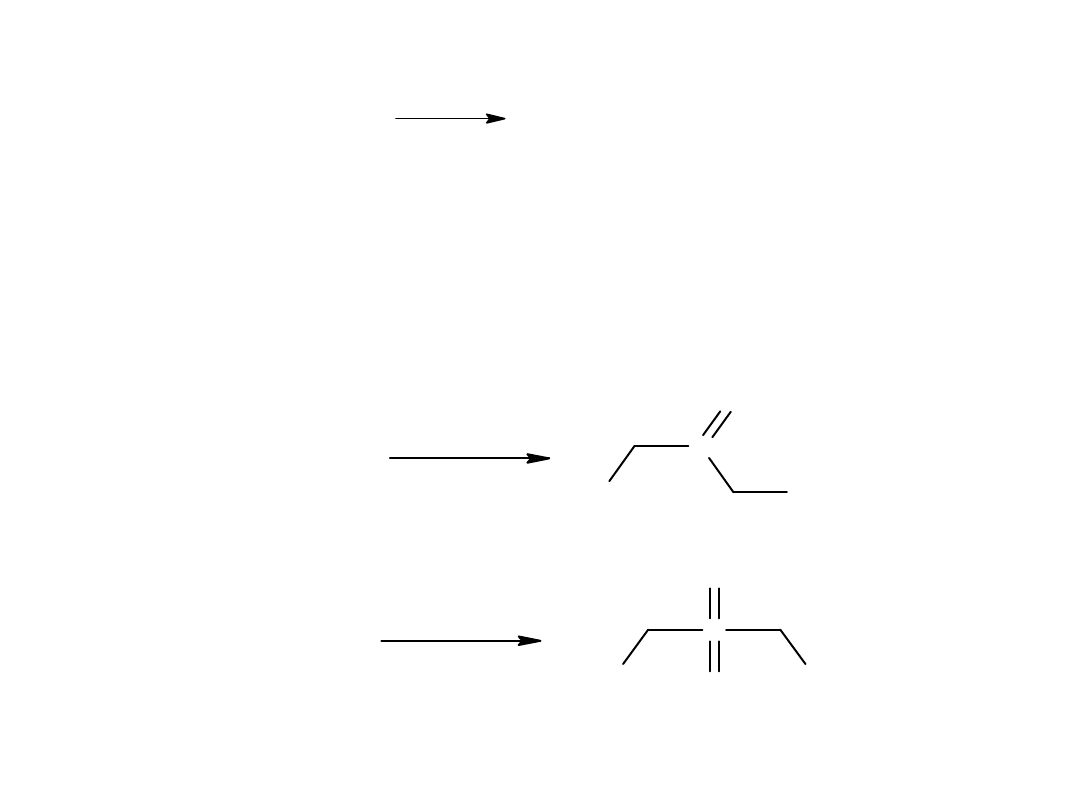
2. W reakcji Williamsona, z merkaptydów z chloropochodnymi
alkilowymi
C
2
H
5
SNa
+
JCH
3
etylomerkaptyd sodowy
-NaJ
C
2
H
5
- S - CH
3
sulfid metylowo-etylowy
tioeter metylowo-etylowy
Właściwości tioeterów
Tioetery można utleniać w zależności od środka utleniającego do
sulfotlenków lub do sulfonów
C
2
H
5
- S - C
2
H
5
H
2
O
2
C
H
3
S
O
CH
3
sulfotlenek dietylu
C
2
H
5
- S - C
2
H
5
KMnO
4
C
H
3
S
O
CH
3
O
dietylosulfan
Tioetery mają bardzo słabo zasadowy charakter, ujawniający się w
reakcjach z halogenkami alkilowymi. W wyniku reakcji tworzą się sole
sulfoniowe (analogiczne do soli amoniowych).
S
R
R
+
RX
S
+
R
R
R
X
-
s�l sulfoniowa
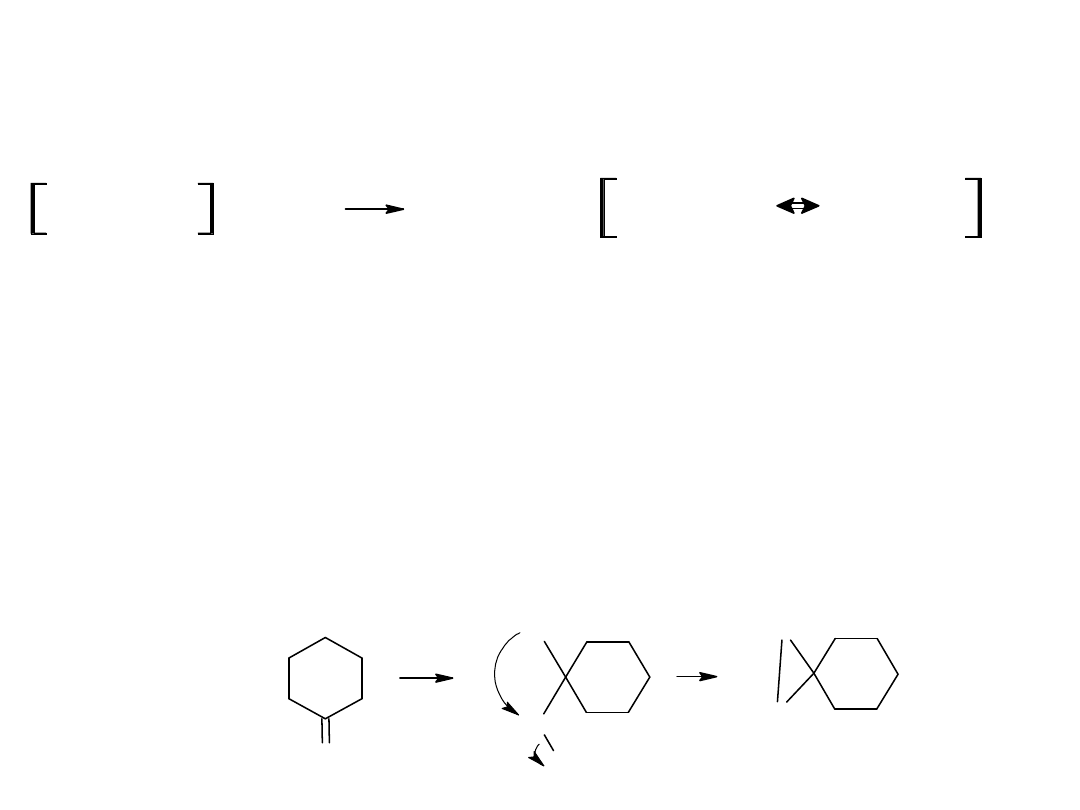
Oderwanie atomu wodoru w pozycji przekształca sole sulfoniowe w
osobliwe związki, zawierające ujemnie naładowany atom węgla, połączony
bezpośrednio z dodatnio naładowanym atomem siarki.
(CH
3
)
2
S
+
- CH
3
I
-
+
NaH
NaI
H
2
+
+
(CH
3
)
2
S
+
- CH
2
-
(CH
3
)
2
S=CH
2
dwie struktury graniczne ylidu siarkowego
Związki tego typu, w których ujemny atom węgla sąsiaduje z dodatnim
heteroatomem, a jednocześnie mogą utworzyć wiązanie podwójne
węgiel-heteroatom, są nazywane ylidami.
Ylidy są z reguły mało trwałymi związkami, ale można je łatwo otrzymać
i wykorzystywać jako reaktywne czynniki w syntezie organicznej, pod
warunkiem, że natychmiast po otrzymaniu zostaną użyte do dalszych
reakcji.
Przykładem zastosowania ylidów siarkowych są reakcje, w których
ylidowy atom węgla przyłącza się do wiązań podwójnych C=O, C=C.
(CH
3
)
2
S
+
- CH
2
-
+
O
O
-
H
2
C
S
+
(CH
3
)
2
O
H
2
C
+
(CH
3
)
2
S
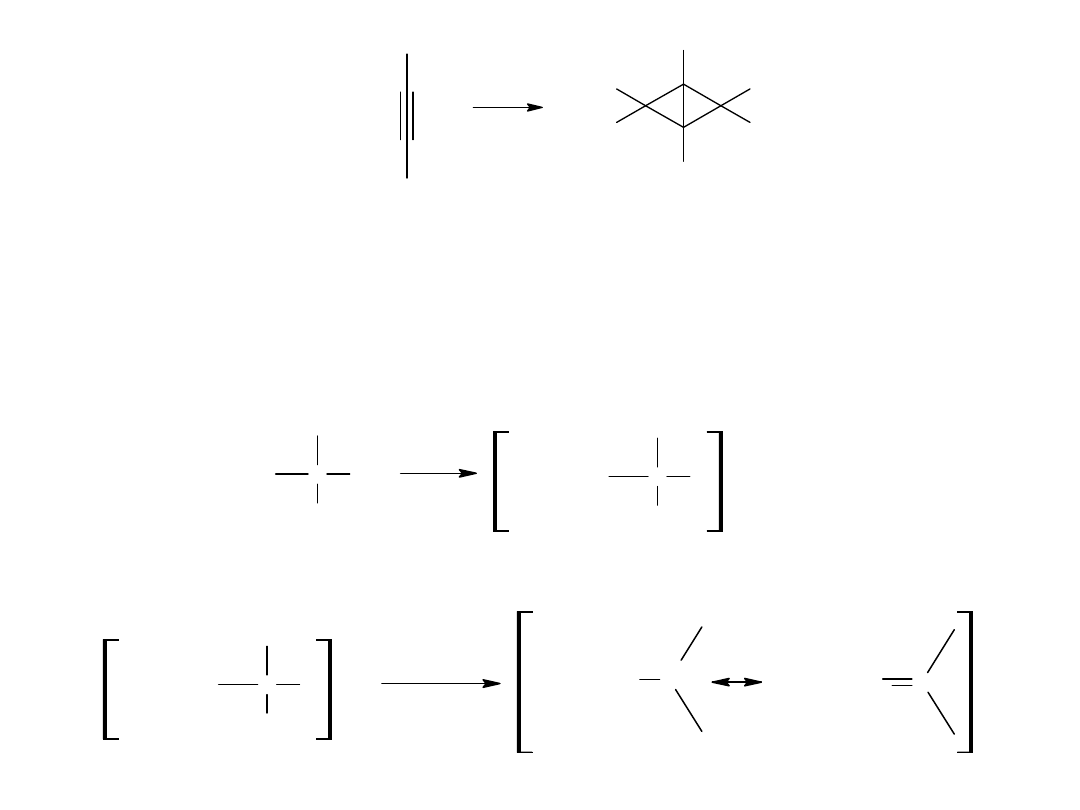
2(C
6
H
5
)
2
S
+
- C
-
(CH
3
)
2
+
COOCH
3
COOCH
3
COOCH
3
C
H
3
C
H
3
CH
3
CH
3
COOCH
3
Ylidy fosforowe
Ylidy siarkowe, aczkolwiek ich znaczenie w chemii organicznej jest
pokaźne, nie znajdują tak szerokiego zastosowania, jak odkryte w roku
1954 przez Wittiga ylidy fosforowe. Ogólna metoda syntezy tych ylidów
polega na reakcji trifenylofosfiny z fluorowcopochodnymi alkilowymi, po
czym na otrzymaną w ten sposób czwartorzędową sól fosfoniową działa
się silną zasadą. Wytworzony ylid bez wydzielania z mieszaniny
reakcyjnej poddaje się dalszym reakcjom
(C
6
H
5
)
3
P
+
C
X
H
trifenylofosfina
halogenopochodna
C
P
+
(H
5
C
6
)
3
X
H
X
-
czwartorzedowa s�l fosfinowa
C
P
+
(H
5
C
6
)
3
X
H
X
-
butylolit
P
+
(H
5
C
6
)
3
C
-
P(H
5
C
6
)
3
C
struktury graniczne ylidu fosforowego

Najprostszym przykładem ylidu fosforowego jest
trifenylometylenofosforan, ale przez zastosowanie fluorowcopochodnych
o różnej budowie możliwe jest otrzymywanie ylidów fosforowych z
różnymi podstawnikami przy ylidowym atomie węgla.
(C
6
H
5
)
3
P
+
CH
3
I
[(C
6
H
5
)
3
P-CH
3
]I
butylolit
(C
6
H
5
)
3
P - CH
2
trifenylometylenofosforan
(C
5
H
5
)
3
P
+
Br - CH
2
- CO
2
C
2
H
5
[(C
6
H
5
)
3
P - CH
2
- CO
2
C
2
H
5
]Br
butylolit
(C
6
H
5
)
3
P - CH - CO
2
C
2
H
5
trifenylokarboetoksymetylenofosforan
Ylidy fosforowe ulegają rozmaitym reakcjom, wśród których największe
znaczenie ma synteza związków nienasyconych, przebiegająca wg
schematu.
C
O
+
C
P(C
6
H
5
)
3
C
H
3
C
C
H
3
C
CH
3
CH
3
+
P(C
6
H
5
)
3
O
aldehyd lub keton
ylid fosforowy
tlenek trifenylofosfiny
Ad. III
Alifatyczne kwasy sulfonowe nie mają większego znaczenia, natomiast
aromatyczne kwasy sulfonowe i wszelkie możliwe ich pochodne znalazły
liczne zastosowanie. Są powszechnie produkowane przez przemysł
farmaceutyczny jako tzw. sulfonamidy (sulfamidy), leki o działaniu
antybakteryjnym.
Aromatyczne kwasy sulfonowe można traktować jako arylowe pochodne
kwasu siarkowego
H
SO
3
H
SO
3
H
Kwasy sulfonowe, podobnie jak kwas siarkowy, są silnymi kwasami. Z
metalami tworzą sole, zwane sulfonianami. Sole wapnia i baru są lepiej
rozpuszczalne w wodzie od soli metali alkaicznych.
Kwasy sulfonowe powstają w reakcji sulfonowania odpowiednich
substratów aromatycznych. Środkiem sulfonującym jest kwas siarkowy.
Wprowadzona do pierścienia aromatycznego grupa sulfonowa -SO
3
H jest
podstawnikiem II rodzaju, dezaktywującym pierścień, co oznacza, że
atak elektrofilowy innej grupy będzie kierowany w położenie –meta.
Otrzymywanie kwasów sulfonowych
1.
Sulfonowanie stęż. H
2
SO
4
H
2
SO
4
SO
3
H
kwas benzenosulfonowy
2.
Sulfonowanie oleum
HO
3
S
oleum
HO
3
S
SO
3
H
kwas meta-benzenosulfonowy
3.
Chlorosulfonowanie kwasem chlorosulfonowym – HOSO
2
Cl
W wyniku chlorosulfonowania otrzymuje się sacharynę i chloraminę T.
CH
3
HSO
2
Cl
CH
3
SO
2
Cl
CH
3
SO
2
Cl
i
p-sulfochlorek (sta�y)
o-sulfochlorek (ciek�y)
Z o-sulfachlorku otrzymuje się sacharynę
CH
3
SO
2
Cl
NH
3
CH
3
SO
2
NH
2
KMnO
4
COOH
SO
2
NH
2
NH
S
O
O
O
sacharyna
(imid kwasu o-sulfonobenzoesowego)
Sacharyna była I syntetycznym słodzikiem, odkrytym w 1879.
Sacharyna jest kilkaset razy bardziej słodka od sacharozy.
NH
S
O
O
O
saccharin
NHSO
3
Na
cyclamate
W 1937 zsyntezowano następny syntetyczny słodzik – cyklamat.
W 1969 r. udowodniono, że cyklamat wywołuje raka u zwierząt
doświadczalnych. Rok później został wycofany z użycia.
W 1977 r. odkryto kancerogenne działanie sacharyny. Pojawienie się raka
u szczurów skarmianych sacharyną spowodowało zakaz jej stosowania.
W latach 1980 popularnym środkiem słodzącym był aspartam. Aspartam
nadawał się do słodzenia jedynie napojów chłodzących, gdyż w
podwyższonych temperaturach tracił słodki smak. Szybko stwierdzono, że
spożywanie aspartamu wywołuje problemy medyczne, zwłaszcza u osób
cierpiących na fenyloketonurię.
Obecnie stosuje się sorbitol – cukier prosty, którego główną zaletą jest to,
że nie powoduje próchnicy zębów, gdyż jego rozkład w jamie ustnej ma
inny przebieg niż sacharozy.
O
N
H
2
O
OH
NH
O
O
CH
3
C
6
H
5
aspartam
CH
2
OH
H
OH
OH
H
H
OH
H
OH
CH
2
OH
sorbitol
Z p-sulfochlorku otrzymuje się chloraminę T – środek antyseptyczny.
CH
3
SO
2
Cl
NH
3
CH
3
SO
2
NH
2
NaOH
NaCl
CH
3
O
2
S
N
Na
Cl
chloramina T
(s�l sodowa chloramidu
kw. p-toluenosulfonowego)
Wielkie znaczenie w tej grupie związków ma kwas sulfanilowy, powstający
z aniliny
NH
2
H
2
SO
4
-H
2
O
NH - SO
3
H
anilina
kwas fenyloaminobenzoesowy
izomeryzacja
200�C
NH
2
SO
3
H
kwas sulfanilowy
(p-aminobenzenosulfonowy)
Kwas sulfanilowy z metalami nie tworzy soli, natomiast tworzy tzw. sól
wewnętrzną (jest jonem dwubiegunowym).
N
H
2
SO
3
H
H
3
N
SO
3
Znaczenie praktyczne kwasów sulfonowych
Amid kwasu sulfanilowego i niektóre jego podstawione pochodne mają
duże znaczenie w medycynie jako leki sulfamidowe. Sulfanilamid (amid
kwasu sulfanilowego) ma działanie przeciwbakteryjne.
Do chwili obecnej otrzymano i zbadano aktywność przeciwbakteryjną
ponad 6000 sulfamidów, ale tylko kilkanaście znajduje zastosowanie w
medycynie.
Działanie sulfanilamidu jest przykładem działania biologicznego opartego
na analogii strukturalnej. Sulfanilamid wykazuje podobieństwo
strukturalne do kwasu
p-aminobenzoesowego i może blokować miejsca aktywne białek
przeznaczone dla tego kwasu. Sulfanilamid hamuje więc rozwój bakterii,
zbudowanych z takiej konfiguracji białek, które nie odróżniają kwasu p-
aminobenzoesowego (powstającego w jednym etapów biosyntezy) od
sulfanilamidu.
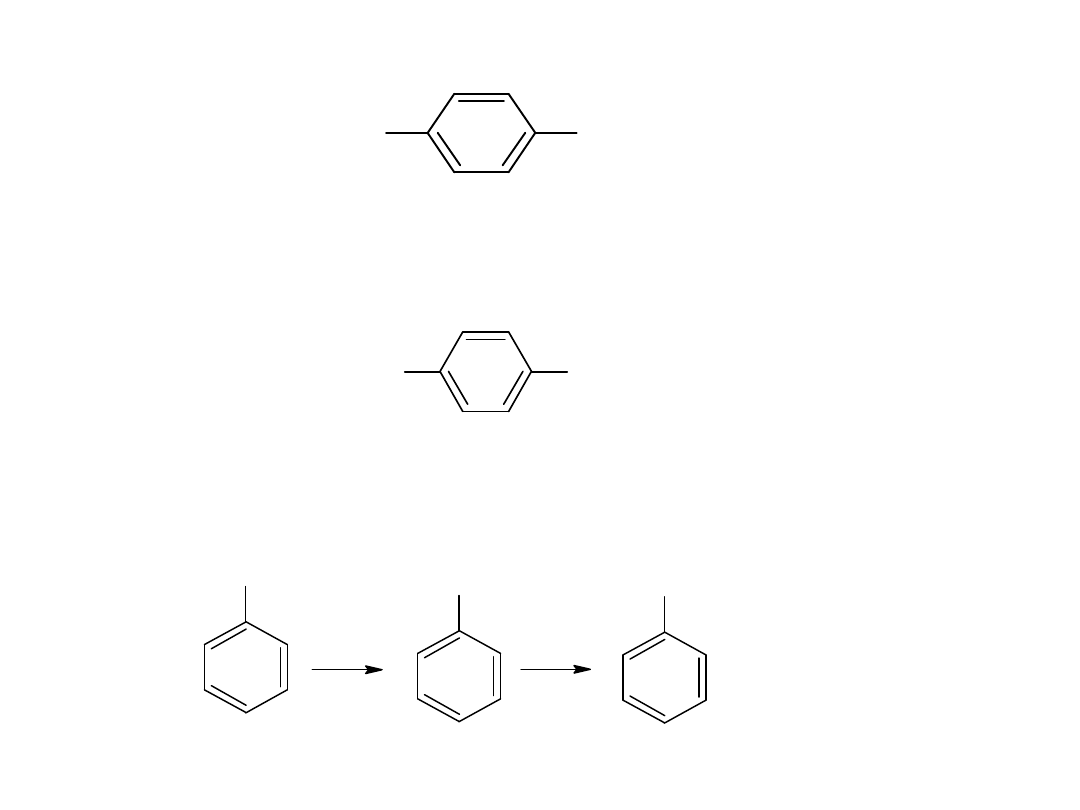
Bakterie wykorzystują kwas p-aminobenzoesowy do biosyntezy kwasu
foliowego, niezbędnego do ich rozwoju.
N
H
2
CO
2
H
kwas p-aminobenzoesowy
Sulfanilamid strukturalnie przypomina kwas p-aminobenzoesowy
N
H
2
SO
2
NH
2
sulfanilamid
Sulfamidy otrzymuje się z kwasów sulfonowych, a właściwie z ich
chlorków
SO
3
H
PCl
5
SO
2
Cl
NH
3
SO
2
NH
2
kwas
benzenosulfonowy
chlorek kw.
amid kw. benzenosulfonowego
Sulfanilamidy otrzymuje się najczęściej aniliny przez sulfonowanie,
stosując acetylowanie grupy aminowej, w celu zabezpieczenia przed
ubocznymi reakcjami pomiędzy grupą –NH
2
jednej cząsteczki z –SO
3
Cl
drugiej
NH
2
CH
3
COOH
anilina
NHCOCH
3
acetanilid
H
2
SO
4
NHCOCH
3
SO
3
H
kwas p-acetamidobenzenosulfonowy
PCl
5
NHCOCH
3
SO
2
Cl
chlorek p-acetamidobenzenosulfonowy
NHCOCH
3
SO
2
Cl
chlorek
NH
3
NHCOCH
3
SO
2
NH
2
amid
HOH
rozc. HCl
NH
2
SO
2
NH
2
sulfanilamid
W 1930 roku po raz pierwszy zaobserwowano antybakteryjne działanie
barwnika azowego, będącego pochodną sulfanilamidu, o nazwie Prontosil
SO
2
NH
2
NH
2
NaNO
2
HCl
SO
2
NH
2
N
2
Cl
SO
2
NH
2
N
2
Cl
+
NH
2
NH
2
m-fenylenodiamina
N
N
NH
2
H
2
N
SO
2
NH
2
Obecnie stosuje się dziesiątki leków sulfonowych
N
H
2
SO
2
NH
sulfapridyna
N
H
2
SO
2
N
NH
2
NH
2
sulfaguanidyna
N
H
2
S
SO
2
N
N
OCH
3
OCH
3
madroksim
Synteza sulfatiazolu
O
Cl
+
N
H
2
S
NH
2
N
S
NH
2
aldehyd chlorooctowy
tiomocznik
2-aminotiazol
N
S
NH
2
+
H
3
N
SO
3
N
S
NH
SO
2
NH
2
Właściwości kwasów sulfonowych
I
Reakcje w obrębie grupy sulfonowej
1.
Z metalami tworzą sole
SO
3
H
NaOH
SO
3
Na
benzenosulfonian sodu
2.
Dają analogiczne pochodne jak kwasy karboksylowe. Nie dają
się tylko bezpośrednio estryfikować, w związku z czym, amidy i estry
otrzymuje się z chlorków kwasów sulfonowych
SO
3
H
PCl
5
SO
2
Cl
benzenosulfochlorek
SO
2
Cl
NH
3
SO
2
NH
2
benzenosulfoamid
SO
2
Cl
C
2
H
5
ONa
SO
2
OC
2
H
5
ester etylowy kwasu benzenosulfonowego
3.
Chlorki kwasów sulfonowych ulegają reakcjom Friedla-Craftsa
SO
2
Cl
+
AlCl
3
SO
2
difenylosulfan
II
Reakcje z rozerwaniem wiązania C-S
1.
Hydroliza rozcieńczonym H
2
SO
4
SO
3
H
HOH
rozc. H
2
SO
4
150-160�C
+
H
2
SO
4
2.
Otrzymywanie fenoli przez stapianie sulfonianów sodu z NaOH
SO
3
Na
NaOH
300�C
ONa
+
NaHSO
3
fenolan sodu
3.
Otrzymywanie nitryli (reakcja nie ma znaczenia praktycznego)
SO
3
Na
KCN
200�C
CN
K
2
SO
4
+
cyjanek fenylu
4.
Otrzymywanie aromatycznych merkaptanów
SO
3
Na
NaHS
SH
merkaptan fenylu
SH
KMnO
4
SO
3
H
Reakcje utleniania aromatycznych markaptanów świadczą o
bezpośrednim wiązaniu S z pierścieniem.

Aromatyczne związki heterocykliczne
Związki heterocykliczne zawierają jako podstawę budowy pierścień,
którego jeden lub więcej członów stanowi heteroatom, zazwyczaj O, S lub
N. Znane są również układy heterocykliczne zawierające bor, krzem, fosfor,
arsen, selen. Istnieje ogromna różnorodność związków heterocyklicznych
zwłaszcza, jeśli wziąć pod uwagę te z nich, które zawierają więcej niż jeden
heteroatom.
Do chwili obecnej poznano kilkanaście tysięcy podstawowych układów
pierścieniowych zawierających heteroatomy.
Przedmiotem wykładu będą układy najważniejsze i najczęściej spotykane,
czyli heterocykle o pierścieniach pięcio- i sześcioczłonowych, zawierające
atomy O, S i N.
W nazewnictwie pierścieni heterocyklicznych obowiązują dwa systemy.
I – opiera się na nazwach zwyczajowych
II – opracowany przez IUAPC, polegający na tym, że za podstawę nazwy
uważany jest nasycony lub aromatyczny węglowodór, a obecność
heteroatomu oznacza się przez umieszczenie przedrostka oksa (tlen), aza
(azot), tia (siarka).
Pozycję heteroatomu, określa się takim numerem, aby był jak najmniejszy.
Jeśli w pierścieniu znajdują się dwa lub więcej heteroatomów tego samego
rodzaju np. N numerację zaczyna się od nasyconego atomu N.
N
H
azol (pirol)
N
N
H
1, 2-diazol (pirazol)
O
N
S
oksol (furan)
S
tiol (tiofen)
1, 2-tiazol (tiazol)
O
oksan
O
O
1, 4-diokan
I. Związki heterocykliczne o pierścieniach pięcioczłonowych
Najprostszymi związkami heterocyklicznymi
pięcioczłonowymi zawierającymi
1 heteroatom są: pirol, furan i
tiofen. Każdy z omawianych związków
wykazuje charakter aromatyczny,
który jest wynikiem budowy.
Każdy atom pierścienia pirolu, zarówno C jak i N połączony jest z 3
innymi atomami za 3 wiązań σ. Wiązania te są utworzone przez trzy
orbitale sp2 leżące w tej samej płaszczyźnie. Po oddaniu po jednym
elektronie do każdego wiązania σ, każdy atom C ma jeszcze po 1
elektronie, natomiast atom N ma jeszcze 2 elektrony. Elektrony te
zajmują orbitale p.
Na skutek bocznego nałożenia orbitali p powstają 2 chmury elektronów
¶, powyżej i poniżej płaszczyzny pierścienia. Każda z tych chmur zawiera
po 6 elektronów, czyli sekstet elektronowy.
Delokalizacja elektronów ¶ powoduje stabilizację pierścienia.
N
H
azol (pirol)
O
oksol (furan)
S
tiol (tiofen)

Dodatkowa para elektronów N, która jest odpowiedzialna za zasadowość
wykazywaną zazwyczaj przez związki azotu, wchodzi w skład chmury
elektronów ¶
i nie może być wykorzystana do wiązania kwasów. Efektem tego jest
bardzo słabo zasadowy charakter pirolu.
Z tej samej przyczyny, pierścień pirolu charakteryzuje duża gęstość
elektronowa powodująca, że jest on bardzo reaktywny w reakcjach
substytucji elektrofilowej, nawet takich jak nitrozowanie i sprzęganie z
solami diazoniowymi.
Strukturę pirolu najlepiej przedstawia wzór, z którego jasno wynika, że N
oddaje elektrony do pierścienia.
Analogicznie przedstawia się struktury furanu i tiofenu
N
H
lub
N
H
O
lub
O
S
lub
S
Cząsteczki furanu i tiofenu mają wolną parę elektronową na orbitalu sp2.
Podobnie jak atom N w pirolu, atomy O i S dostarczają po 2 elektrony do
chmury elektronów ¶, w wyniku czego furan i tiofen zachowują się jak
bardzo reaktywne pochodne benzenu.
Metody otrzymywania pirolu, furanu i tiofenu.
Pirol
Pirol został odkryty przez Rungego, w produktach suchej destylacji olejku
kostnego. Syntetycznie pirol otrzymuje się wieloma metodami
1.
Z bursztynianiu amonu przez destylację i redukcję powstałego
imidu
COONH
4
COONH
4
bursztynian amonu
ogrz.
O
N
H
O
imid kwasu bursztynowego
H
2
/Zn (py�)
N
H
pirol
2.
Z acetylenu i amoniaku podczas przepuszczania przez
rozżarzone rury
C
H
CH
+
NH
3
+
C
H
CH
ogrz.
N
H
pirol
+
H
2
Furan
Furan znajduje się w produktach drewna drzew iglastych. Furan
otrzymuje się z furfuralu, który powstaje podczas działania kwasami
mineralnymi na łuski owsa, ryżu lub kaczany kukurydzy.
Furfural powstaje z zawartych w nich pentoz.
O
H
O
H
OH
OH
O
H
pentoza
cyklizacja
-3H
2
O
O
O
H
furfural
kat. tlenkowy
para wodna
400�C
O
furan
Tiofen
Na skale przemysłową tiofen otrzymuje się z butanu i siarki, dawniej
stosowano ogrzewanie acetylenu z siarką
1.
Z butanu z siarką
C
H
3
CH
3
+
S
560�C
S
+
H
2
S
2.
Z bursztynianu sodu przez ogrzewanie z P
2
S
3
COONa COONa
bursztynian sodu
P
2
S
3
ogrz.
S
tiofen
Reakcje podstawienia elektrofilowego w pierścieniach pirolu,
furanu i tiofenu
Ze względu na to, że pierścienie aromatyczne pirolu, furanu i tiofenu są
wzbogacone w elektrony podstawienie elektrofilowe w pierścieniu
zachodzi bardzo łatwo.
W reakcjach podstawienia elektrofilowego związki te wykazują
reaktywność podobną do fenoli i amin. Uprzywilejowanym miejscem
przyłączenia odczynnika elektrofilowego są pozycje sąsiadujące z
heteroatomem, a więc 2 i 5.
Mechanizm podstawienia elektrofilowego w pierścieniach pirolu, furanu i
tiofenu jest dokładnie taki sam jak w pierścieniu benzenu i polega na
przyłączeniu odczynnika elektrofilowego i następującej po tym eliminacji
protonu.
Warto zwrócić uwagę na łagodne warunki, w których zachodzą reakcje
podstawienia elektrofilowego, np.
N
H
+
C
H
3
Br
N
H
CH
3
2-propylopirol
O
+
(CH
3
CO)
2
O
bezwodnik octowy
eter dietylowy
BF
3
0�C
O
CO
CH
3
2-acetylofuran
S
+
HNO
3
(CH
3
CO)
2
O
S
NO
2
2-nitrotiofen
O
+
SO
3
pirydyna
20�C
O
SO
3
H
kwas 2-furanosulfonowy
SO
3
O
SO
3
H
HO
3
S
kwas 2, 3-furanodisulfonowy
O
+
Br Br
dioksan
25�C
O
Br
2-bromofuran
N
H
+
C
6
H
5
N
N Cl
0�C
N
H
N=N-C
6
H
5
2-(fenyloazo)pirol
O
+
HNO
2
O
NO
2
2-nitrozofuran
Pochodne pirolu, furanu i tiofenu
W wyniku katalitycznego uwodornienia (Ni) reakcje Sabatiera, każdy ze
związków traci charakter aromatyczny a zyskuje właściwości odpowiednich
związków cyklicznych.
N
H
H
2
, Ni, 200-250�C
N
H
pirolidyna (amina)
O
H
2
, Ni, 50�C
O
tetrahydrofuran (eter)
S
H
2
, Ni, 50�C
S
tetrahydrotiofen (tioeter)
Tetrahydrotiofen otrzymywany jest innymi metodami, gdyż obecność
siarki w jego cząsteczce zatruwa katalizator
Najważniejszą pochodną furanu jest fural (furfural), związek ulegający
reakcji Cannizzaro
O
CHO
fural
OH
O
CH
2
OH
+
O
COOH
alkohol alfa-furynowy
kwas piro�luzowy
Pod wpływem KCN fural ulega kondensacji do furoiny:
O
CHO
KCN
O
OH
O
O
furoina
(zwi�zek analogiczny do benzoiny)
Do najważniejszych pochodnych pirolu należą związki będące jego
homologami, a mianowicie, hemopirol i kryptopirol, stanowiące produkty
degradacji barwnika krwi – hemoglobiny (hemopirol) i zielonego barwnika
roślin – chlorofilu (kryptopirol)
N
H
H
3
C
H
3
C
CH
2
CH
3
hemopirol
N
H
H
3
C
CH
2
CH
3
CH
3
kryptopirol
Związek złożony z 2 skondensowanych pierścieni: pirolowego i
benzenowego o nazwie benzopirol (indol) jest bardzo rozpowszechnioną
pochodną pirolu. Indol jest produktem gnicia substancji białkowych,
wchodzi w skład niektórych hormonów (ludzkich), oraz np. hormonu
wzrostu roślin.
Indol ulega substytucji elektrofilowej w pozycji 3.
3-metyloindol (skatol) ma zapach kału.
N
H
indol
N
H
CH
3
skatol
Pochodną indolu jest indygotyna, podstawowy składnik indyga (błękitu
indygowego) barwnika znanego już w starożytności, obecnie
stosowanego do barwienia jeansów.
Indygotyna jest ciemnoniebieskim proszkiem, przesublimowana ma
postać miedzianoczerwonych igiełek. Nie rozpuszcza się w wodzie,
eterze, alkoholu, rozcieńczonych kwasach i zasadach, natomiast
rozpuszcza się na gorąco w anilinie i terpentynie. Stąd pochodzi trwałość
barwienia.
Ponieważ indygotyna nie rozpuszcza się w wodzie, aby umożliwić
barwienie tkanin, redukuje się ją do tzw. bieli indygowej, rozpuszczalnej
w wodzie i dopiero nasyca tkaninę. Roztwór natychmiast utlenia się
wprost na tkaninie tlenem z powietrza i regeneruje błękitny kolor.
Analogiczną pochodną tiofenu jak indol dla pirolu, jest tionaften. Z
tionaftenu otrzymuje się barwnik o nazwie czerwień tioindygowa.
S
tionaften
II. Związki heterocykliczne o pierścieniach sześcioczłonowych
Z aromatycznych związków o pierścieniach sześcioczłonowych
najważniejszym jest pirydyna.
N
alfa
beta
gamma
pirydyna
Konfiguracja elektronowa atomu N w cząsteczce pirydyny, różni się
znacznie od konfiguracji N w cząsteczce pirolu. Tak samo, jak w
cząsteczce pirolu, atom N i wszystkie atomy C są w stanie hybrydyzacji
sp2. Każdy z atomów C dostarcza 1 elektron do chmury elektronowej ¶
(razem 5). Atom N tworzy 2 wiązania, natomiast trzeci orbital sp2
atomu N zawiera po prostu wolną parę elektronową, która nie wchodzi
w skład chmury elektronów ¶. Ta wolna para elektronowa może wiązać
kwasy.
W wyniku takiej struktury pierścień pirydyny, jest zubożony w elektrony
i w związku z tym z trudnością ulega podstawieniu elektrofilowemu.
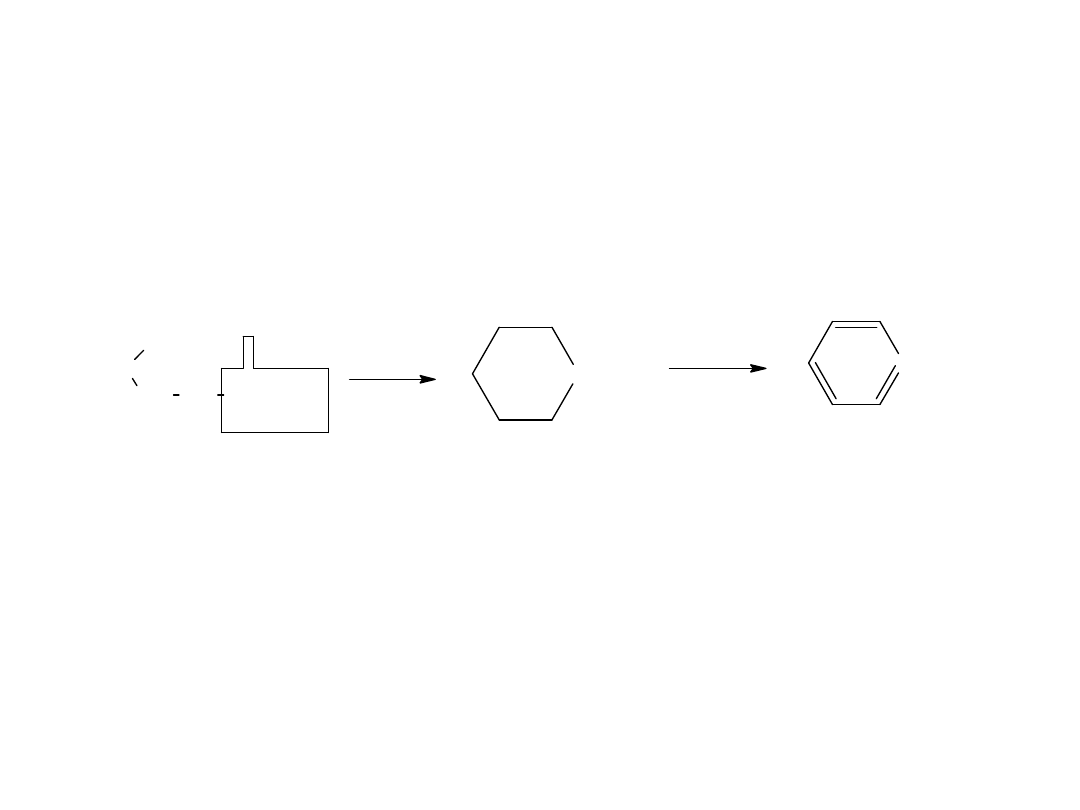
Otrzymywanie pirydyny
Głównym źródłem otrzymywania pirydyny oraz jej metylowych
pochodnych zwanych pikolinami jest smoła pogazowa z węgla
kamiennego. Pirydyna została odkryta przez Andersena w oleju kostnym.
Wyizolowano ją również z licznych alkaloidów np. koniiny. Syntetycznie
otrzymano ją kilkoma metodami. Najprostszy sposób polega na suchej
destylacji chlorowodorku pentametylenodiaminy, czyli kadaweryny, i
utlenianiu powstałej przy tym piperydyny
C
H
2
CH
2
CH
2
CH
2
CH
2
NH
2
* HCl
NH
2
* HCl
ogrz.
-NH
4
Cl
NH * HCl
chlorowodorek kadaweryny
chlorowodorek piperydyny
utl.
N
pirydyna
Reakcje pirydyny
1.
Reakcje substytucji elektrofilowej
Pirydyna, w swych reakcjach substytucji elektrofilowej przypomina silnie
zdezaktywowane pochodne benzenu. Tylko w drastycznych warunkach
ulega nitrowaniu, sulfonowaniu i chlorowcowaniu.
Nie ulega natomiast reakcjom Friedla-Craftsa.
Substytucja w cząstce pirydyny zachodzi głównie w pozycji 3 ().
N
KNO
3
, H
2
SO
4
300�C
N
O
2
N
3-nitropirydyna
N
H
2
SO
4
350�C
N
HO
3
S
kwas 3-pirydynosulfonowy
N
Br
2
300�C
N
Br
Br
2
N
Br
Br
3-bromopirydyna
3, 5-bromopirydyna
2.
Reakcja substytucji nukleofilowej
Ze względu na to, że pirydyna wykazuje słabe właściwości aromatyczne
(ma osłabiony pierścień), możliwe są w jej przypadku reakcje substytucji
nukleofilowej, za pomocą amidku sodowego lub fenylolitu.
Atak nukleofilowy ma miejsce w pozycjach 2 i 4 ( i ).
a)
reakcja Cziczibabina – aminowanie za pomocą amidu sodowego
N
NaNH
2
100�C
N
NH
2
2-aminopirydyna
+
H
2
b)
reakcja z fenylolitem
C
6
H
5
Li
100�C
N
C
6
H
5
2-fenylopirydyna
N
3.
Utlenianie i redukcja
W wyniku katalitycznej redukcji pirydyny, powstaje piperydyna będąca
alifatyczną aminą 2º.
N
H
2
, Pt, HCl
25�C
NH
Pirydyna utlenia się z trudnością, do N-tlenku pirydyny
N
kwas nadbenzoesowy
N
O
N-tlenek pirydyny
Natomiast pikoliny utleniają się z łatwością do odpowiednich kwasów:
N
COOH
kwas nikotynowy
N
COOH
kwas pikolinowy
N
CH
3
kwas izonikotynowy
Kwasy te mają charakter amfoteryczny, w roztworach występują jako
sole wewnętrzne.
Kwas nikotynowy, niacyna, jest otrzymywany na szerszą skalę przez
przemysł farmaceutyczny jako witamina PP.
Dietylowy amid kwasu nikotynowego
tzw. koramina, jest znany jako
popularny lek pobudzający akcję
serca, kardiamid.
N
N
O
C
2
H
5
C
2
H
5
koramina
Przedstawicielem tej grupy związków
(sześcioczłonowe z 1 heteroatomem) jest
także piran, związek zawierający O w
pierścieniu.
O
piran
Piran nie ma większego znaczenia, natomiast jego pochodna benzo--
piran znana jest jako kumaryna.
O
kumaryna
O
O
i
Kumaryna jest substancją zapachową występującą między innymi w
marzannie wonnej.
Dikumarol jest pochodną kumaryny, stosowaną jako trucizna na szczury.
Jest to związek o silnie toksycznych właściwościach.
III. Związki heterocykliczne o pierścieniach skondensowanych
Najważniejszą pochodną pirydyny jest chinolina, zawierająca pierścień
benzenowy, skondensowany z pierścieniem pirydynowym
N
1
2
3
4
5
6
7
8
a
b
chinolina
Chinolina ma budowę przypominającą naftalen i w związku z tym należy
oczekiwać podobnych izomerów. Istnieje 7 jednopodstawionych
pochodnych chinoliny. Właściwości chinoliny są wynikiem jej budowy,
tzn. połączenia czysto aromatycznego pierścienia benzenowego z
pierścieniem pirydyny, posiadającym słabo aromatyczny charakter.
Utlenianie chinoliny, prowadzące do kwasu chinolinowego, świadczy o
tym, że pierścień benzenowy skondensowany jest z pierścieniem
pirydynowym w położeniach 2 i 3.
N
1
2
3
4
5
6
7
8
utl.
N
COOH
COOH
kwas chinolinowy
Metody otrzymywania chinoliny
1.
Synteza Skraupa – z aniliny z gliceryną, stężonym H
2
SO
4
i
utleniaczem
NH
2
+
OH
OH
OH
anilina
gliceryna
C
6
H
5
NO
2
+
H
2
SO
4
N
chinolina
+
C
6
H
5
NH
2
Synteza Skraupa biegnie przez kilka etapów:
- dehydratacja gliceryny do akroleiny (aldehydu akrylowego)
OH
OH
OH
gliceryna
H
2
SO
4
-2H
2
O
C
H
2
H
O
akroelina
- addycja nukleofilowa aniliny do akroleiny
NH
2
+
H
O
CH
2
akroleina
NH
O
aldehyd beta-(fenyloamino)-propionowy
(akroleinoanilina)
- cyklizacja (elektrofilowy atak karbonylowego atomy C aldehydu na
pierścień aromatyczny)
NH
H
O
H
2
SO
4
NH
H
OH
H
2
SO
4
-H
2
O
N
H
1, 2-dihydrochinolina
- utlenianie 1,2-dihydrochinoliny za pomocą nitrobenzenu
N
H
C
6
H
5
NO
2
-H
2
O
N
C
6
H
5
NH
2
+
chinolina
+
1, 2-dihydrochinolina
2.
Metoda Friedlandera – kondensacja aldehydu o-
aminobenzoesowego z aldehydem octowym w środowisku zasadowym
NH
2
H
O
aldehyd o-aminobenzoesowy
+
CH
3
H
O
aldehyd octowy
N
chinolina
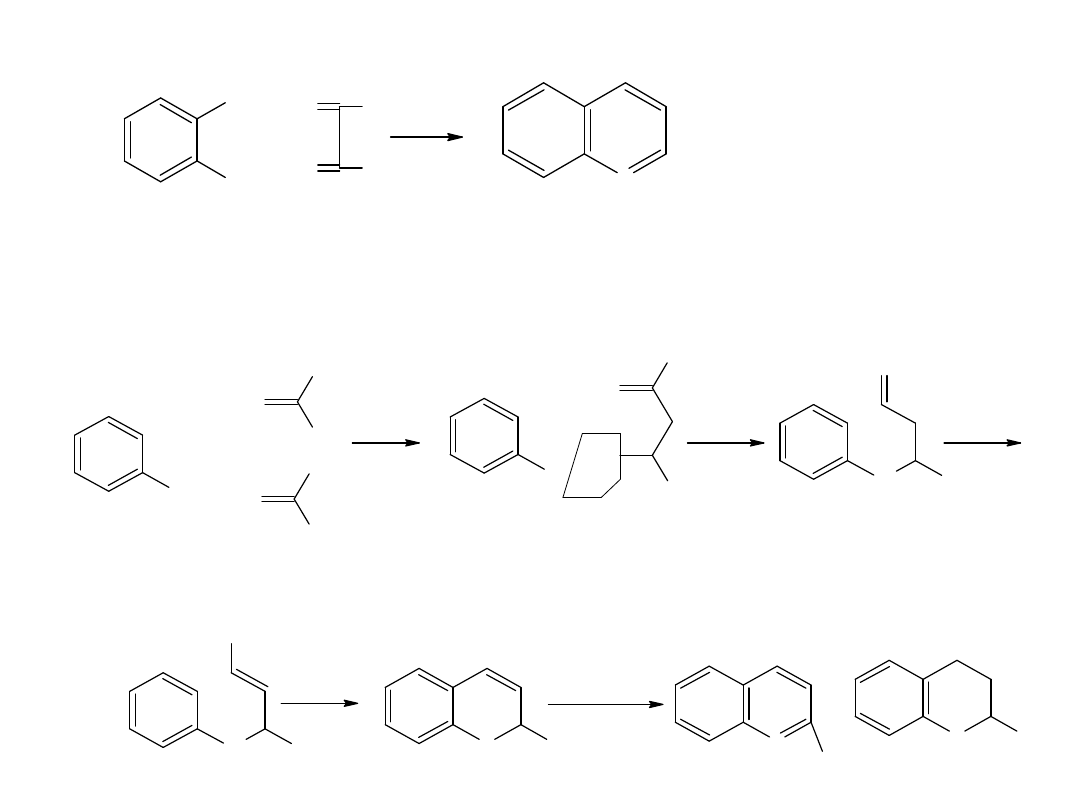
3.
Kondensacja o-toluidyny z glioksalem
NH
2
CH
3
o-toluidyna
+
O
O
H
H
NaOH
N
chinolina
glioksal
4.
Metoda Döbnera i Millera
W wyniku tej reakcji można np. otrzymać chinaldyny (metylochinoliny)
przez kondensację aniliny z aldehydem octowym w obecności
stężonego HCN.
NH
2
O
CH
3
H
O
CH
3
H
+
anilina
2 cz. aldehydu octowego
HCl
NH
2
O
H
CH
3
O
H
dimer aldehydu
HCl
-H
2
O
NH
CH
3
O
HCl
NH
CH
3
OH
posta� enolowa
HCl
N
H
CH
3
dysmutacja
N
CH
3
+
N
H
CH
3
chinaldyna
tetrahydrochinolina
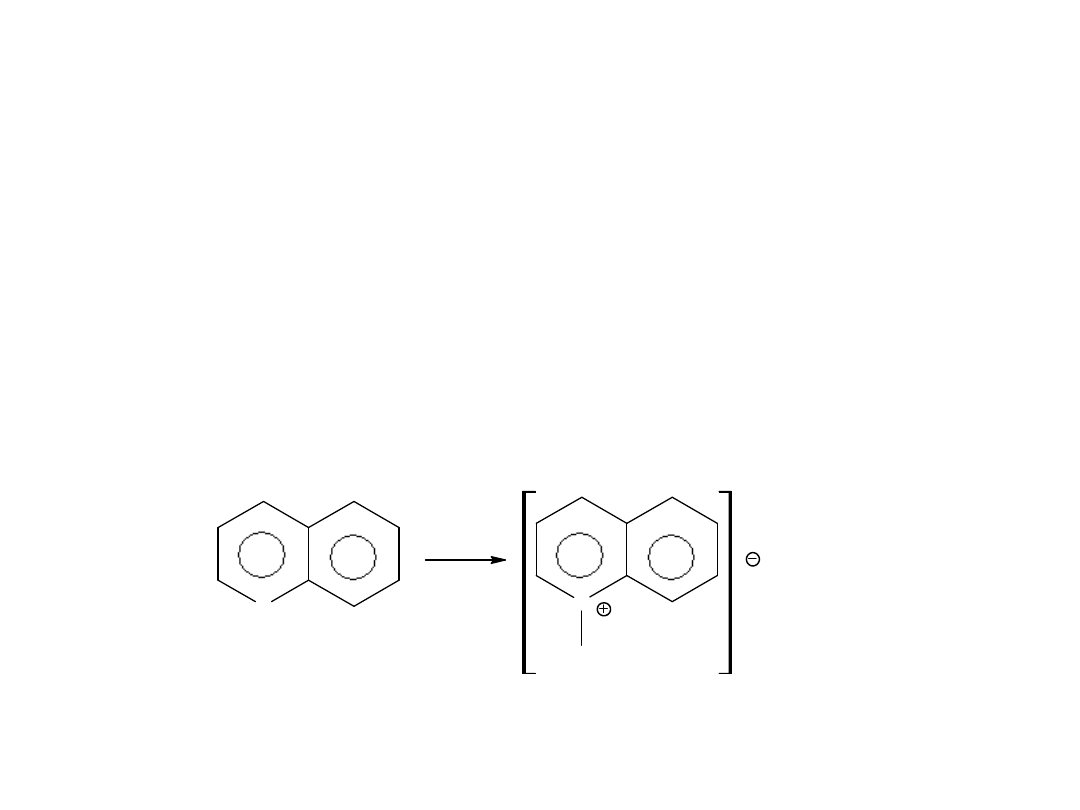
Chinolina występuje obok pirydyny w smole pogazowej i olejku kostnym.
Powstaje również przy degradacji wielu alkaloidów np. chininy i
cynchoniny występującej w korze pewnych drzew w Ameryce
Południowej.
Reakcje chinoliny
Chinolina w przeciwieństwie do pirydyny nie rozpuszcza się w wodzie.
1.
Jest znacznie słabszą zasadą od pirydyny, co jest wynikiem jej
budowy chemicznej. Z mocnymi kwasami chinolina tworzy sole
chinoliniowe, łączy się również z halogenkami alkilowymi dając także sole
chinoliniowe.
N
CH
3
J
N
CH
3
J
jodek N-metylochinoliny
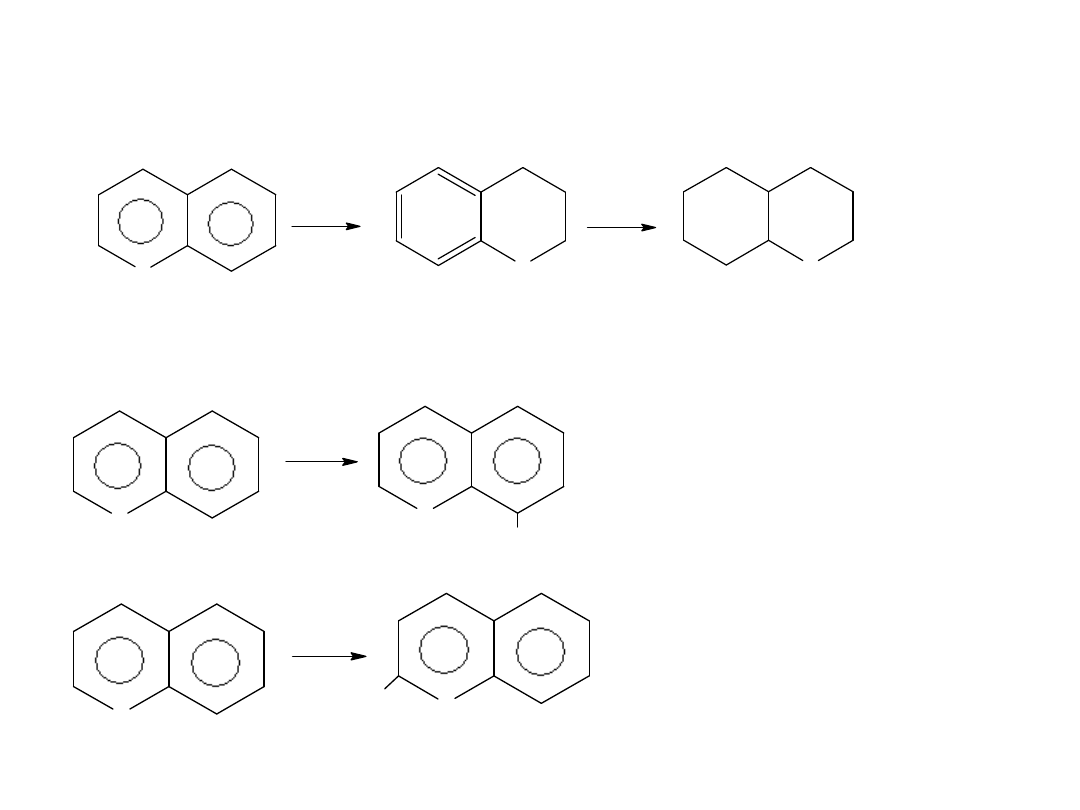
2.
Hydrogenizacja chinoliny przebiega stopniowo, najpierw tworzy
się tetrahydrochinolina, ciecz zawierająca uwodorniony pierścień
pirydynowy. Później, hydrogenizacji ulega pierścień benzenowy, w skutek
czego tworzy się dekahydrochinolina, substancja stała o właściwościach
mocnej zasady.
N
H
2
N
H
chinolina
tetrahydrochinolina
H
2
N
H
dekahydrochinolina
3.
W przypadku nitrowania i sulfonowania chinoliny, podstawniki
wchodzą do pierścienia benzenowego.
N
HNO
3
N
NO
2
Z sulfonowych pochodnych
przez stapianie z alkaliami
otrzymuje się
hydroksypochodne.
N
NaNH
2
N
N
H
2
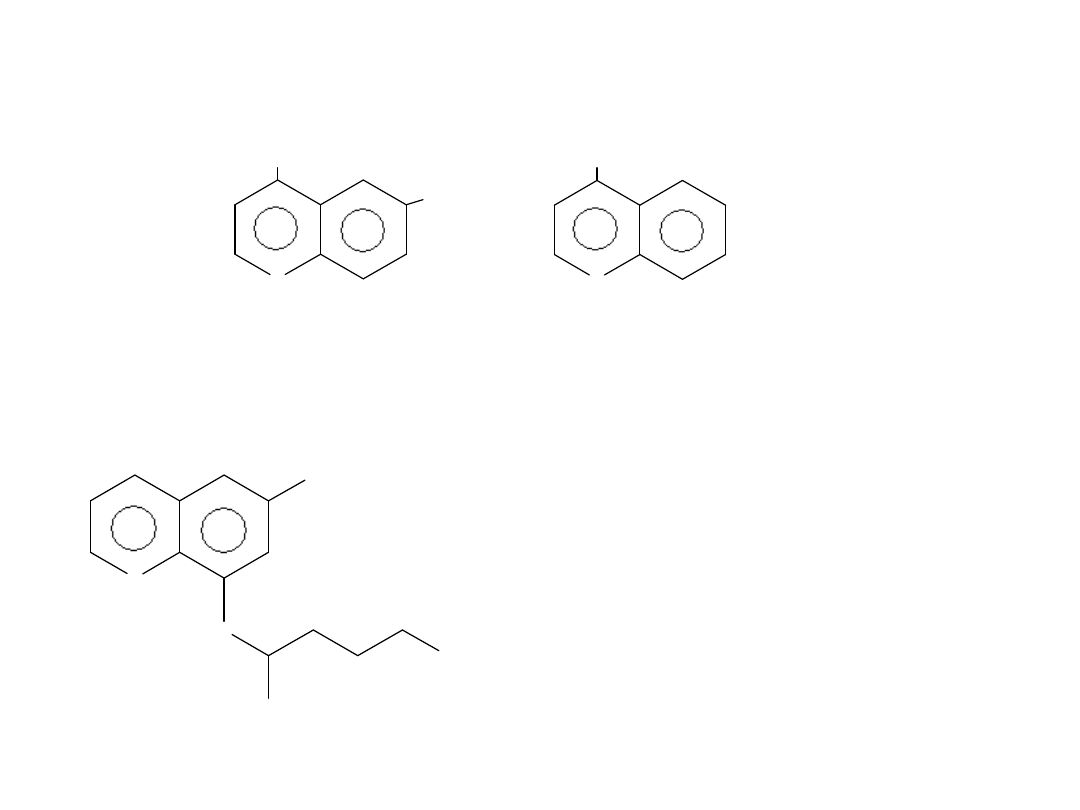
4.
Kwasy karboksylowe chinoliny tworzą się podczas utleniania
odpowiednich metylopochodnych. Powstają one również w wyniku
utleniania niektórych alkaloidów, np. chinina utleniana daje kwas
chininowy, cynchonina zaś – kwas cynchoninowy.
N
COOH
OCH
3
kwas chininowy
N
COOH
kwas cynchoninowy
Plazmochina jest chlorowodorkiem 6-metoksy-8-(4’-dietyloamino-1’-
metylo)-butylo-aminochinoliny
N
N
H
CH
3
N(C
2
H
5
)
2
OCH
3
plazmochina
Znajduje ona zastosowanie jako
środek przeciwmalaryczny,
zabijający jedną z form
rozwojowych pierwotniaka malarii
tzw. gamety.
Związkiem izomerycznym do chinoliny jest izochinolina
N
1
2
3
4
5
6
7
8
izochinolina
Izochinolina oprócz licznych alkaloidów, występuje w smole pogazowej.
Jest nieco silniejszą zasadą od chinoliny.
Utlenianie izochinoliny prowadzi do kwasu ftalowego, lub do kwasu
cynchomerowego, w zależności od warunków.
Izochinolinę otrzymuje się metodą Bischlera-Napieralskiego, w wyniku
kondensacji -fenyloetyloaminy z aldehydem mrówkowym i utleniania
powstałej 1,2,3,4-tetrahydroizochinoliny
N
izochinolina
utl.
COOH
COOH
N
COOH
COOH
lub
kwas ftalowy
kwas cynchomerowy
NH
2
+
HCHO
beta-fenylortyloamina
aldehyd mr�wkowy
cyklizacja
P
2
O
5
NH
tetrahydroizochinolina
utlenianie
Pd
N
izochinolina
Głównym alkaloidem kurary, trucizny stosowanej przez Indian, jest -
tubokuraryna, zawierająca w swojej cząsteczce dwa pierścienie
izochinolinowe, połączone ze sobą przez dwa pierścienie benzenowe.
IV. Związki heterocykliczne pięcioczłonowe zawierające 2
heteroatomy
Pirazol (1,2-diazol)
N
N
H
pirazol
Pirazol jest słabą zasadą, którą można otrzymać z acetylenu i
diazometanu w zimnym roztworze eterowym.
CH
CH
+
CH
2
N
N
acetylen
diazometan
N
N
N
H
N
pirazol
Pochodne pirazolu dają się nitrować, chlorowcować, sulfonować w
pozycji 5, wykazują, więc charakter aromatyczny.
Pod wpływem wodoru redukują się do pirazolin lub pirazolidyn.
N
H
N
pirazolina
N
H
NH
pirazolityna
Pirazoliny znajdują zastosowanie
w medycynie jako leki
przeciwgorączkowe i
przeciwbólowe.
Ketopochodne pirazoliny noszą
nazwę pirazolonu.
NH
HC
HC
N
H
O
Najważniejsze leki z tej grupy to antypiryna i piramidon.
N
N
O
CH
3
CH
3
C
6
H
5
antypiryna
N
N
CH
3
CH
3
C
6
H
5
O
N
CH
3
C
H
3
piramidon
Imidazol
Imidazol jest związkiem izomerycznym do pirazolu
N
N
H
imidazol (glioksalina)
Imidazol i jego pochodne są to mocne
zasady występujące mi. wśród produktów
hydrolizy substancji białkowych
Pierścień imidazolowy jest nietrwały, w przyrodzie występuje jako
składnik budowy wielu związków np. alkaloidów i puryn. Imidazol zwany
glioksaliną, można syntetycznie otrzymać w wyniku kondensacji
glioksalu z amoniakiem i formaldehydem.
O
H
H
O
glioksal (dialdehyd)
+
NH
3
NH
3
+
O
H
H
formaldehyd
-H
2
O
N
N
N
N
imidazol
Pochodne imidazolu znajdują również zastosowanie jako środku lecznicze
(choroby układy nerwowego).
Tiazol
Pierścień tiazolowy zawiera jako heteroatomy azot i siarkę, oddzielone od
siebie atomem C.
N
S
tiazol
Właściwości chemiczne i fizyczne (nawet zapach) tiazolu są bardzo
podobne do pirydyny.
Tiazol powstaje podczas ogrzewania tioformamidu z aldehydem
chlorooctowym.
O
H
Cl
+
N
H
2
S
aldehyd chlorooctowy
tioformamid
ogrz.
O
H
Cl
+
N
H
S
H
N
S
tiazol
Pochodne tiazolu są bardzo ważnymi lekami, z grupy antybiotyków.
Najważniejsze z nich to sulfatiazol i penicyliny – antybiotyki działające
przeciw bakteriom Gram-dodatnim.
Sulfatiazol powstaje z 2-aminotiazolu z kwasem sulfanilowym.
N
S
NH
S
O
O
NH
2
sulfatiazol
( 2-tiazoliloamid kwasu sulfanilowego)
Penicyliny są pochodnymi tetrahydrotiazolu
S
N
HOOC
C
H
3
C
H
3
O
NH
R
O
penicyliny
W tzw. penicylinie krystalicznej R=CH
2
-C
6
H
5
V. Związki heterocykliczne sześcioczłonowe zawierające 2
heteroatomy
Z biologicznego punktu widzenia najważniejszymi układami pierścieni
heterocyklicznych są pirymidyna i puryna. Pirymidyna zawiera
sześcioczłonowy pierścień z dwoma atomami azotu, natomiast puryna
ma cztery atomu azotu w strukturze dwóch pierścieni
skondensowanych. Oba te
związki heterocykliczne są
podstawowymi składnikami
kwasów nukleinowych,
niezwykle ważnej klasy
biocząsteczek
N
N
1
2
3
4
5
6
pirymidyna
N
N
N
N
H
1
2
3
4
5
6
7
8
9
puryna
Pirymidyna ulega reakcjom substytucji elektrofilowej w pozycji 5.
W kwasie dezoksyrybonukleinowym DNA występują cztery różne,
podstawowe zasady heterocykliczne. Dwie z nich to podstawione puryny
(adenina i guanina), a dwie są podstawionymi pirymidynami (cytozyna i
tymina). Adenina, guanina i cytozyna występują również w RNA, ale tymina
jest w RNA zastąpiona inną zasadą pirymidynową zwaną uracylem.
N
N
N
N
H
N
H
2
adenina (A) DNA RNA
N
N
N
N
H
O
H
NH
2
guanina (G) DNA RNA
N
H
N
NH
2
O
cytozyna (C) DNA RNA
N
H
NH
O
O
C
H
3
tymina (T) DNA
N
H
NH
O
O
uracyl (U) RNA
Kwasy nukleinowe i nukleotydy
Kwasy nukleinowe, kwas dezoksyrybonukleinowy (DNA) i rybonukleinowy
(RNA), są chemicznymi nośnikami informacji genetycznej w komórce. W
komórkowym DNA zakodowana jest cała informacja, która decyduje o
naturze komórki, kontroluje jej wzrost i podział oraz kieruje biosyntezą
enzymów i innych białek potrzebnych do funkcjonowania komórki.
Składnikiem cukrowym w RNA jest ryboza, a element cukrowy obecny w
DNA to 2’-deoksyryboza (przedrostek 2’-deoksy- oznacza, że brakuje
atomu tlenu w pozycji 2’ rybozy. Liczby ze znakiem „’„ odnoszą się do
cukrowego fragmentu nukleotydu, a liczby bez tego znaku oznaczają
pozycję pierścienia zasady heterocyklicznej).
O
HOH
2
C
OH
HO
OH
ryboza
O
HOH
2
C
OH
HO
2-deoksyryboza

Zarówno w DNA, jak i w RNA zasada heterocykliczna jest związana z
pozycją C1’ pierścienia cukrowego, a kwas fosforowy związany jest
wiązaniem fosfoestrowym z pozycją C5’ cukru.
Chociaż chemicznie są podobne, DNA i RNA różnią się rozmiarami i pełnią
różne funkcje w komórce. Cząsteczki DNA są olbrzymie. Ich ciężary
cząsteczkowe sięgają 150 miliardów, a długość może wynosić do 12 cm i
znajdują się one głównie w jądrach komórkowych. W przeciwieństwie do
nich cząsteczki RNA są o wiele mniejsze (masa cząsteczkowa zaledwie
35 000) i znajdują się głównie poza jądrem komórkowym.
Cząsteczki DNA można, przyrównać do nici skręconej z dwóch
pojedynczych nitek.
Ich budowę oddaje słynny model „podwójnej spirali” zaproponowany dla
DNA przez Watsona i Cricka w przełomowym dla biochemii i genetyki roku
1953. Według tego modelu wewnątrz podwójnej spirali znajdują się i
stykają ze sobą heterocykliczne pierścienie zasad purynowych i
pirymidynowych, a na zewnątrz, niejako na powierzchni nitek, leżą
łańcuchy cukrowo-fosforowe.
Oddziaływanie między heterocyklicznymi zasadami polega na wiązaniach
wodorowych.
Tak więc, względy strukturalne natury raczej subtelnej decydują o tym, że
w DNA tylko pary A-T i C-G mogą leżeć naprzeciwko siebie w podwójnej
spirali, Są to tzw. zasady komplementarne czyli uzupełniające się.
N
N
H
H
N
O
H
O
N
N
N
H
N
C
C
C1 dezoksyrybozy
C1 dezoksyrybozy
108 nm
N
H
3
C
O
C
O
H
H
N
N
N
H
N
C
111 nm
tymina
adenina
Wiązania wodorowe między
komplementarnymi parami
zasad C-G i A-T.
AIDS
The drug most used to treat AIDS patients in the Unitek States is
zidovudine, also known as azidothymine or AZT. AZT does not kill the
AIDS wirus but rather intereferes with its ability to reproduce. Thus AZT
controls an AIDS indefction but does not cure it.
O
HOH
2
C
N
3
N
NH
H
3
C
O
O
Zidovudine (AZT)
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Slide 99
- Slide 100
- Slide 101
- Slide 102
- Slide 103
- Slide 104
- Slide 105
- Slide 106
- Slide 107
- Slide 108
- Slide 109
- Slide 110
- Slide 111
- Slide 112
- Slide 113
- Slide 114
- Slide 115
- Slide 116
- Slide 117
- Slide 118
- Slide 119
- Slide 120
- Slide 121
- Slide 122
- Slide 123
- Slide 124
- Slide 125
- Slide 126
- Slide 127
- Slide 128
- Slide 129
- Slide 130
- Slide 131
- Slide 132
- Slide 133
- Slide 134
- Slide 135
- Slide 136
- Slide 137
- Slide 138
- Slide 139
- Slide 140
- Slide 141
- Slide 142
- Slide 143
- Slide 144
- Slide 145
- Slide 146
- Slide 147
- Slide 148
- Slide 149
- Slide 150
- Slide 151
- Slide 152
- Slide 153
- Slide 154
- Slide 155
- Slide 156
- Slide 157
- Slide 158
- Slide 159
- Slide 160
- Slide 161
- Slide 162
- Slide 163
- Slide 164
- Slide 165
- Slide 166
- Slide 167
- Slide 168
- Slide 169
- Slide 170
- Slide 171
- Slide 172
- Slide 173
- Slide 174
- Slide 175
- Slide 176
- Slide 177
- Slide 178
- Slide 179
- Slide 180
- Slide 181
- Slide 182
- Slide 183
- Slide 184
- Slide 185
- Slide 186
- Slide 187
- Slide 188
- Slide 189
- Slide 190
- Slide 191
- Slide 192
- Slide 193
- Slide 194
- Slide 195
- Slide 196
- Slide 197
- Slide 198
- Slide 199
- Slide 200
- Slide 201
- Slide 202
- Slide 203
- Slide 204
Wyszukiwarka
Podobne podstrony:
Chemia organiczna czesc I poprawiona
chemia organiczna wykład 6
Wykład 9 CHEMIA ORGANICZNA
Chemia Organiczna 4
Chemia organiczna IV
CHEMIA- CHEMIA ORGANICZNA, CHEMIA
bromoacetanilid, Studia, Sprawozdania, Chemia organiczna
Przykladowy egzamin chemia organiczna - ICiP - 2010-zima. , Egzamin
I POPRAWKA EGZAMINU Z CHEMII ORGANICZNEJ, Technologia chemiczna, Chemia organiczna, 4 semestr, organ
chemia organiczna 2003 cała PDF(1)
więcej podobnych podstron