
HEMOSTAZA
Mgr Katarzyna Lewandowska
Zakład Diagnostyki Laboratoryjnej
i Immunologii Klinicznej Wieku Rozwojowego
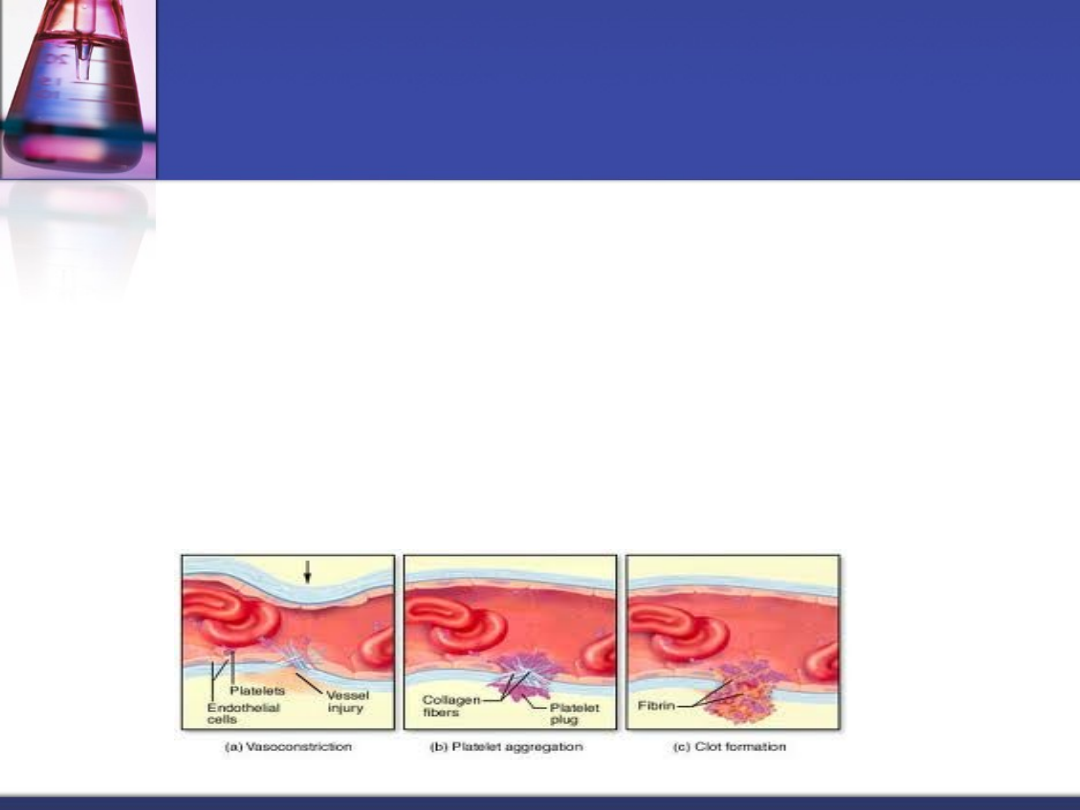
FUNKCJA
HEMOSTAZY
Zapewnienie płynności krążącej krwi
Utrzymanie szczelności łożyska
naczyniowego
Hamowanie krwawienia po przerwaniu
ciągłości ściany naczyń krwionośnych
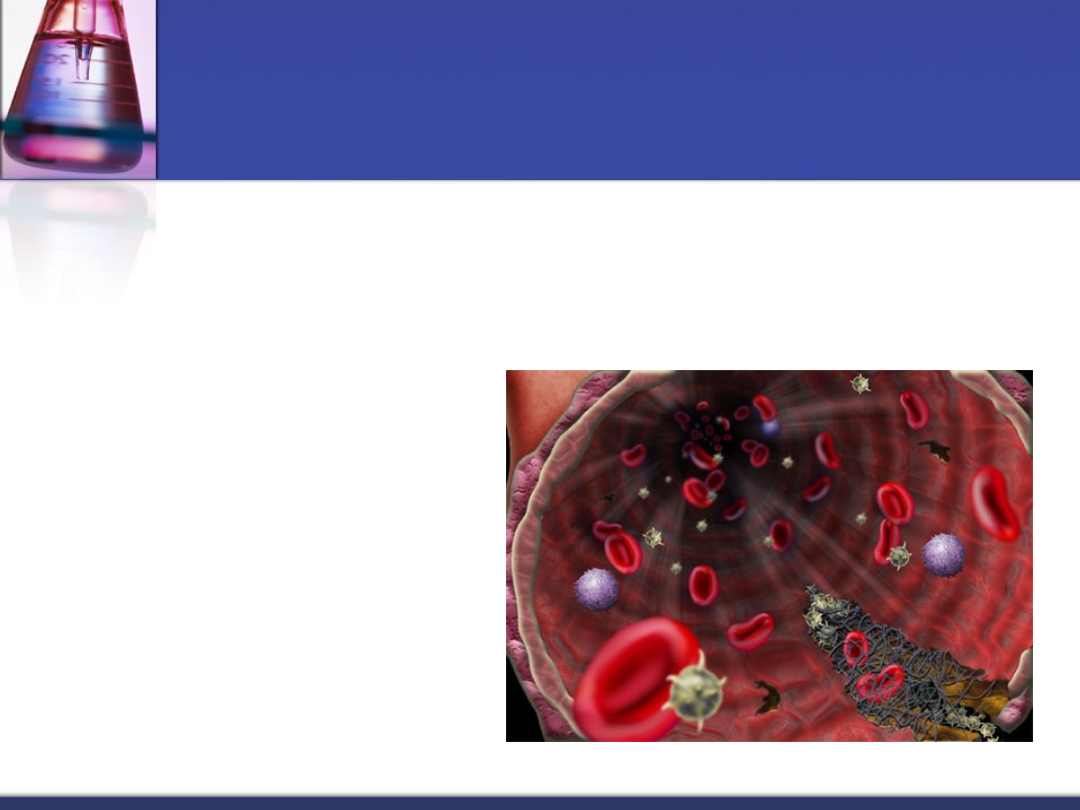
Główne elementy
hemostazy
Ściana naczyń krwionośnych
Płytki krwi
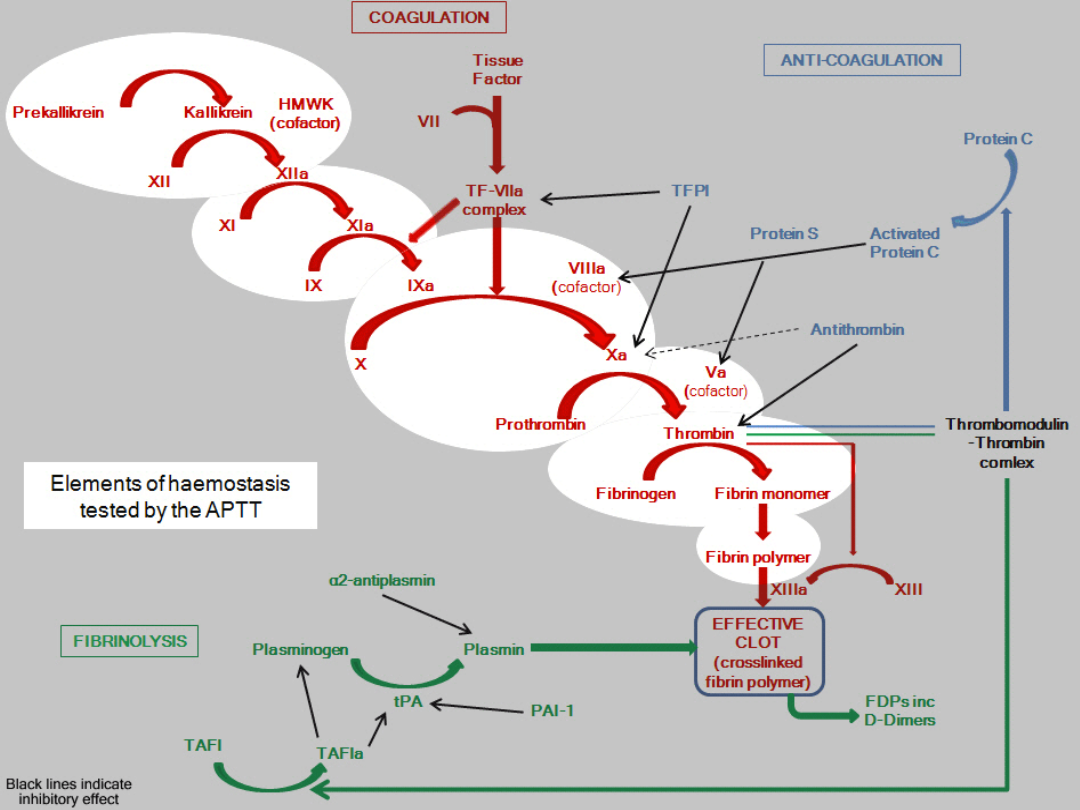
Układ krzepnięcia
Układ fibrynolizy
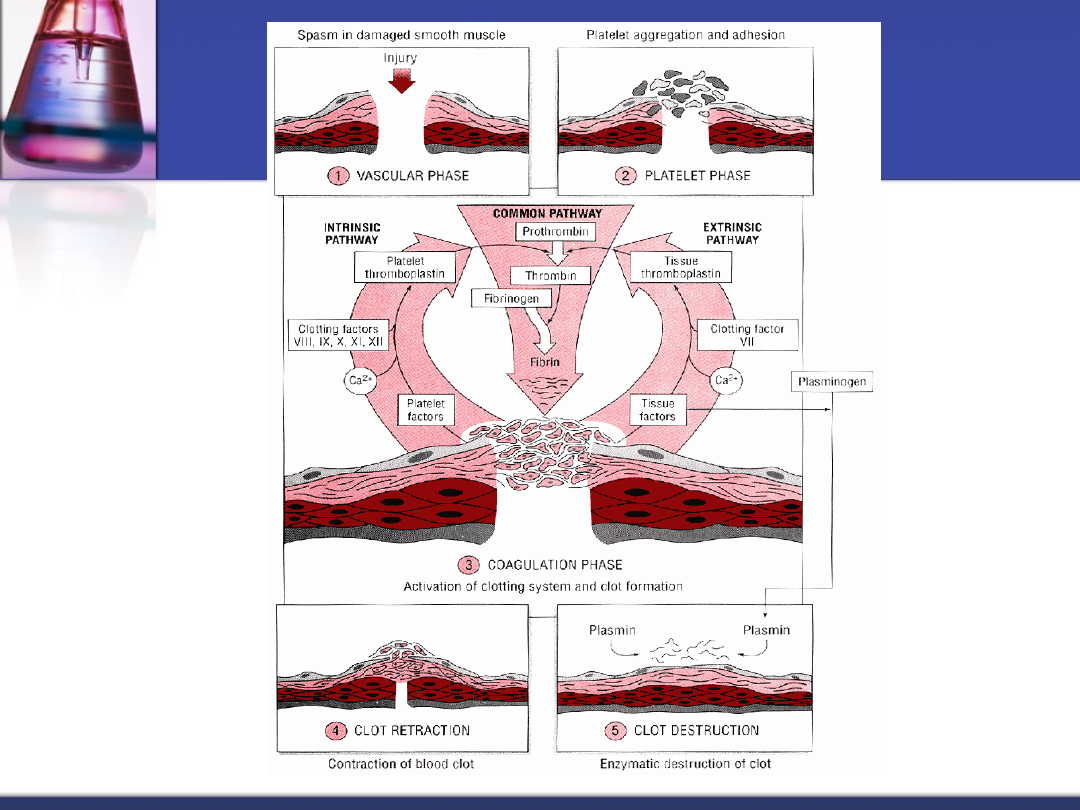
czynniki
krzepnięcia
fosfolipidy
Ca ++
AT III
białko C
białko S
+
_
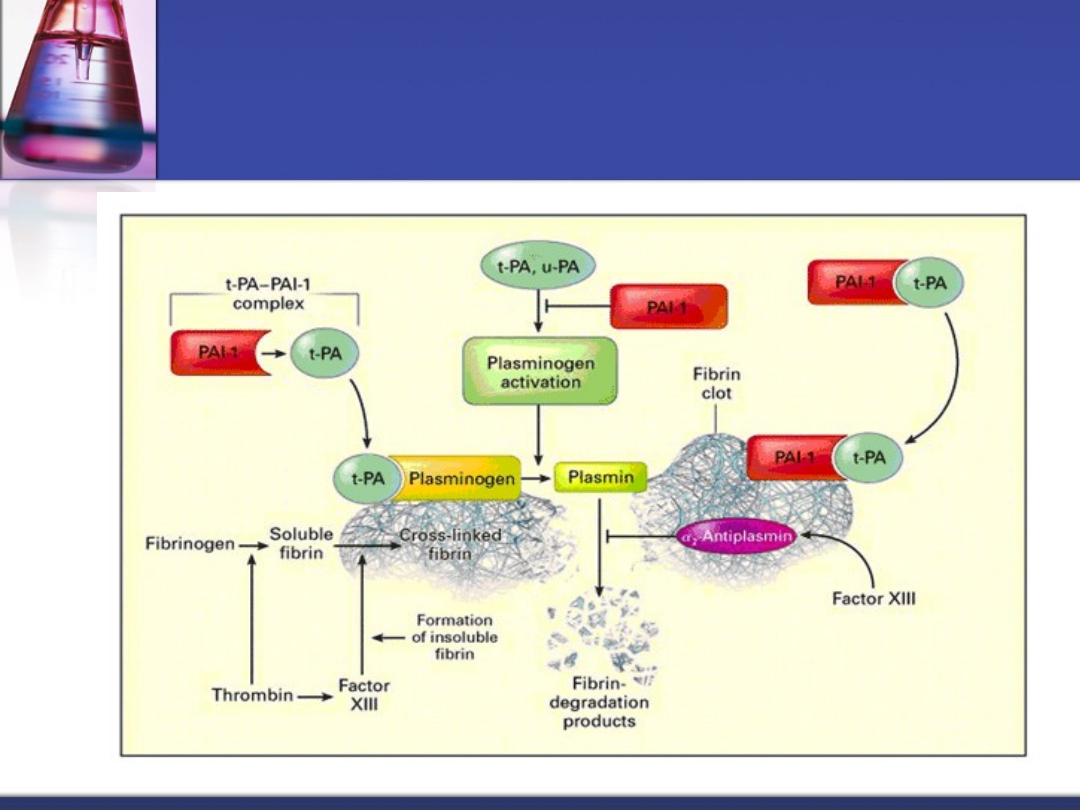
tP
A
uP
A
PAI-1
antyplazmina
+
_
fibrynoliza
plazmina
FIBRYNA
Równowaga w układzie
krzepnięcia
krzepnięcie
trombina
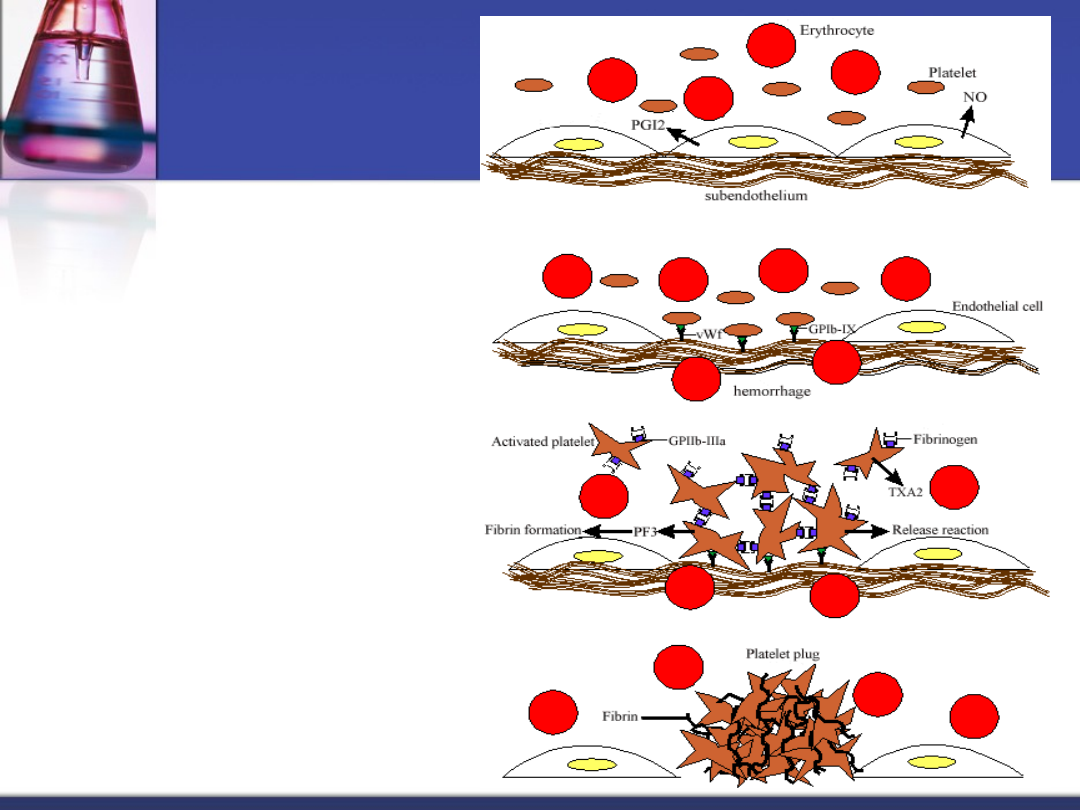
Udział ściany
naczyń
krwionośnych w
hemostazie

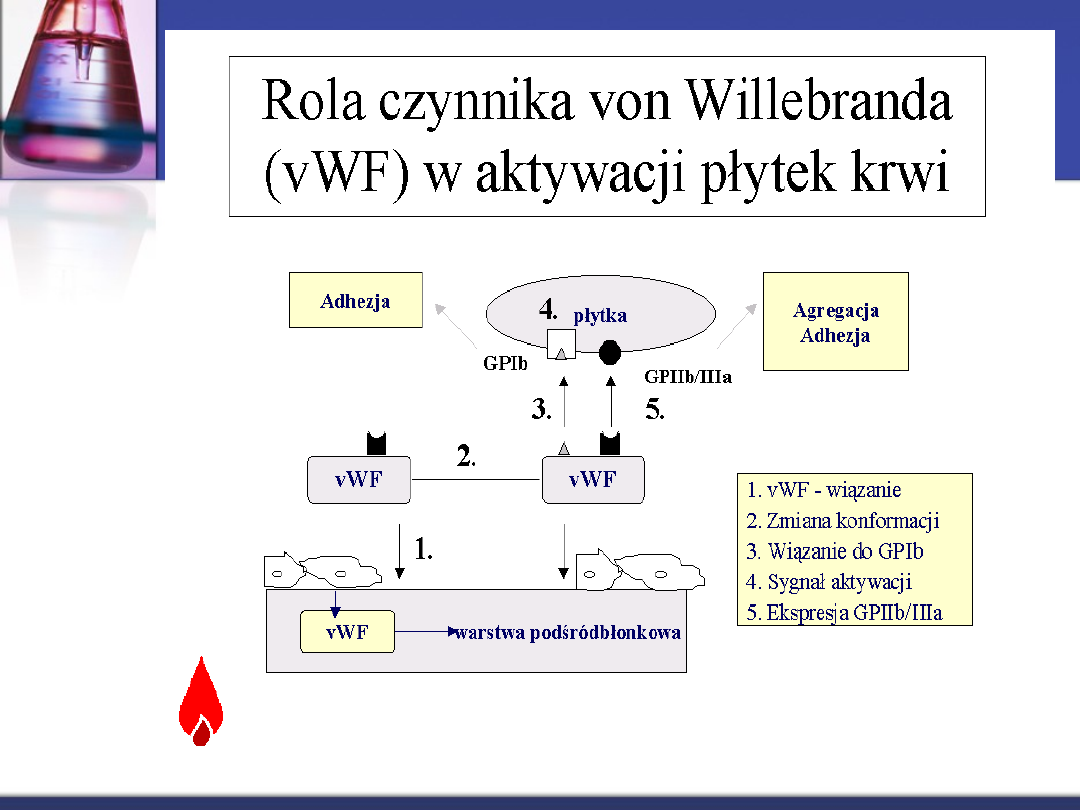
Płytki krwi

PŁYTKI KRWI =
PŁYTKI KRWI =
TROMBOCYTY – PLT
TROMBOCYTY – PLT
* powstają w szpiku w procesie trombopoezy z
megakariocytów przy udziale
trombopoetyny, Il-3, 11
* bezjądrzaste elementy morfotyczne krwi
* czas przeżycia płytek – 8-12 dni (rozpad w ukł. s-ś
śledziony i wątroby)
wartości prawidłowe PLT
PLT = 150 – 400 x 10
3
/ul
(x 10
9
/l)
poziom hemostatyczny płytek (najmniejsza liczba płytek
warunkująca
czynność hemostatyczną płytek)
PLT = 35-40 x 10
3
/ul
poziom zagrażający życiu
PLT poniżej
10 x 10
3
/ul
małopłytkowość
PLT poniżej
100 x 10
3
/ul
nadpłytkowość
PLT powyżej
600 x 10
3
/ul

Czynniki krzepnięcia
Czynnik tkankowy (TF)
Jest białkiem błonowym znajdującym się na powierzchni
komórek, które w warunkach fizjologicznych nie mają kontaktu
z krwią. Najobficiej występuje on na powierzchni komórek
nabłonkowych (w tym także na śródbłonku), fibroblastów
ściany naczyń krwionośnych i na powierzchni aktywowanych
monocytów.

Czynniki zależne od witaminy K
(czynniki zespołu protrombiny)
Czynnik X
Czynnik X = cz. Stuarta-Prowera
Czynnik IX
Czynnik IX = cz. Christmasa =
cz. przeciwhemofilowy –B
Czynnik VII
Czynnik VII = Prokonwertyna = cz.
Stabilny
Czynnik II
Czynnik II = Protrombina

Czynniki zależne od witaminy K
(czynniki zespołu protrombiny)
Czynnik VII
Jest jednołańcuchową glikoproteiną syntetyzowaną w wątrobie.
Podwyższone stężenie czynnika VII w osoczu może towarzyszyć
nadkrzepliwości,
chorobie
niedokrwiennej
serca,
udarom
niedokrwiennym mózgu oraz koreluje ze śmiertelnością z powodu
choroby wieńcowej i zawału mięśnia sercowego.
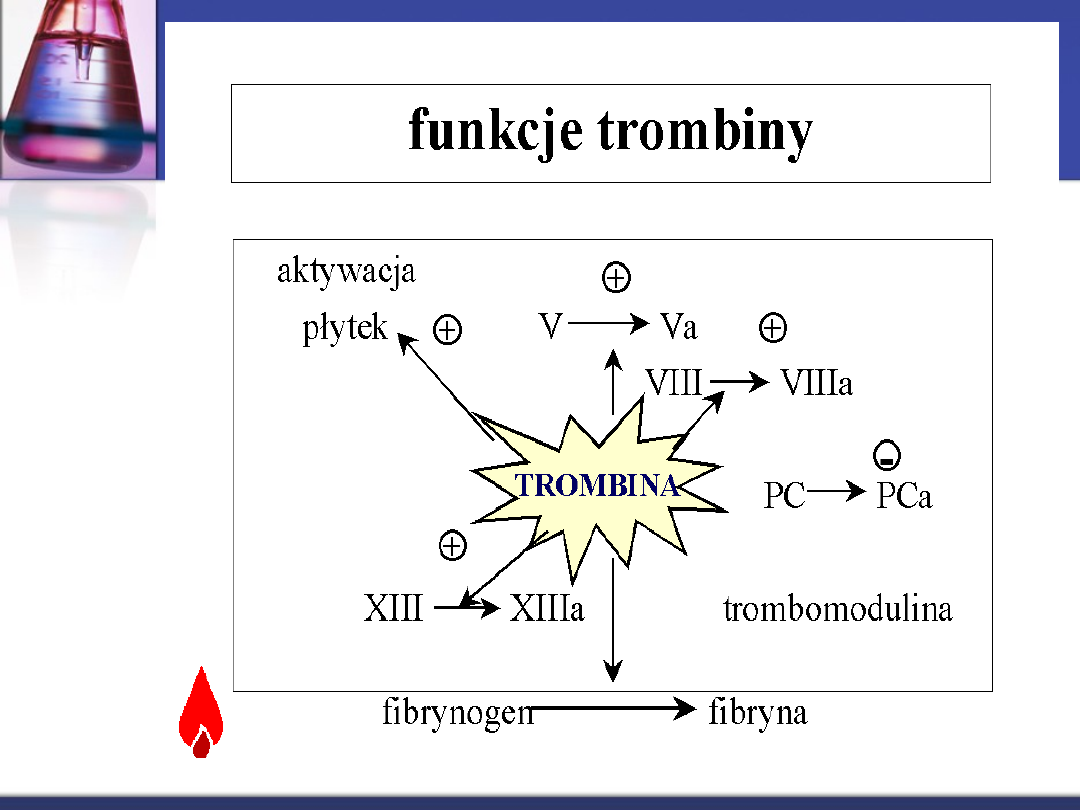
Protrombina (czynnik II)
Proteaza syntetyzowana w wątrobie. Przekształcana jest przez
czynnik Xa do trombiny. Podstawowa funkcja trombiny to
odszczepianie fibrynopeptydów A i B z fibrynogenu , tworzenie
włóknika, aktywacja płytek krwi, czynnika XI, V, VIII, XIII.
Antykoagulacyjna aktywność trombiny jest związana z tworzeniem
kompleksów z trombomoduliną.

Czynniki zależne od
witaminy K (czynniki
zespołu protrombiny)
Czynnik IX
Czynnik IXa wchodzi w skład kompleksu tenazy, uczynniającego
czynnik X.
Defekt genu wywołuje hemofilie B.
Hemofilia B występuje 7 razy rzadziej niż hemofilia A.
Czynnik X
Czynnik Xa wchodzi w skład protrombinazy – kompleksu
enzymatycznego przekształcającego protrombinę w trombinę.
Posiada zdolność przekształcania czynnika V w Va.
U homozygot skaza jest jawna, u heterozygot obserwuje się niewielką
skłonność do krwawień. Występuje bardzo rzadko.

CZYNNIKI
CZYNNIKI
KONTAKTU
KONTAKTU
Białka kofaktorowe , produkowane w wątrobie, niezbędne do aktywacji
krzepnięcia w kontakcie z ujemnie naładowanymi powierzchniami:
włókna kolagenu, kaolin, szkło
Czynnik XII
Czynnik XII
= cz. kontaktu = cz. Hagemana
Prekalikreina
Prekalikreina
= Kalikreinogen = cz. Fletschera – proenzym
kalikreiny,
- aktywator cz. XII i plazminogenu
Wielkocząsteczkowy
Wielkocząsteczkowy
kininogen
kininogen = cz. Fitzgeralda = HMWK – kofaktor
aktywacji cz. XII, XI i kalikreinogenu
Czynnik XI
Czynnik XI = cz. Rosenthala = cz. przeciwhemofilowy- C

CZ
CZ
YNNIKI WRAŻLIWE NA
YNNIKI WRAŻLIWE NA
TROMBINĘ
TROMBINĘ
Czynniki V i VIII są najbardziej labilnymi czynnikami i szybko
ulegają degradacji w próbce krwi przechowywanej w
temperaturze pokojowej lub w podgrzanym osoczu.
C
C
zynnik XIII
zynnik XIII =
czynnik stabilizujący fibrynę,
transglutaminaza osoczowa
C
C
zynnik VIII
zynnik VIII =
czynnik
przeciwhemofilowy A,
globulina antyhemofilowa
C
C
zynnik V
zynnik V
=
proakceleryna
C
C
zynnik I
zynnik I = fibrynogen

Czynnik V i VIII
Czynnik V
Białko produkowane w 80% w wątrobie i komórkach śródbłonka,
źródłem pozostałych 20% są ziarnistości alfa płytek krwi.
Syntetyzowany w postaci nieaktywnej, do jego aktywacji dochodzi
na drodze ograniczonej proteolizy przez trombinę i czynnik Xa.
Pełni rolę prokoagulacyjną oraz działa jako aktywator inaktywacji
czynnika VIIIa przez aktywne białko C. Czynnik V jest kojarzony z
trombofilią.
Czynnik VIII
Synteza czynnika VIII prawdopodobnie nie zachodzi w wątrobie
(śródbłonek naczyń płucnych?). W osoczu czynnik VIII występuje w
kompleksie z czynnikiem von Willebranda, którego niedobór
przekłada się na obniżone stężenie czynnika VIII. Należy do białek
ostrej fazy, jego stężenie wzrasta w stanach zapalnych. Niedobór
czynnika VIII jest ściśle związany z hemofilią A. Wysoka aktywność
prokoagulacyjna czynnika VIII koreluje z zachorowalnością na
choroby sercowo-naczyniowe.
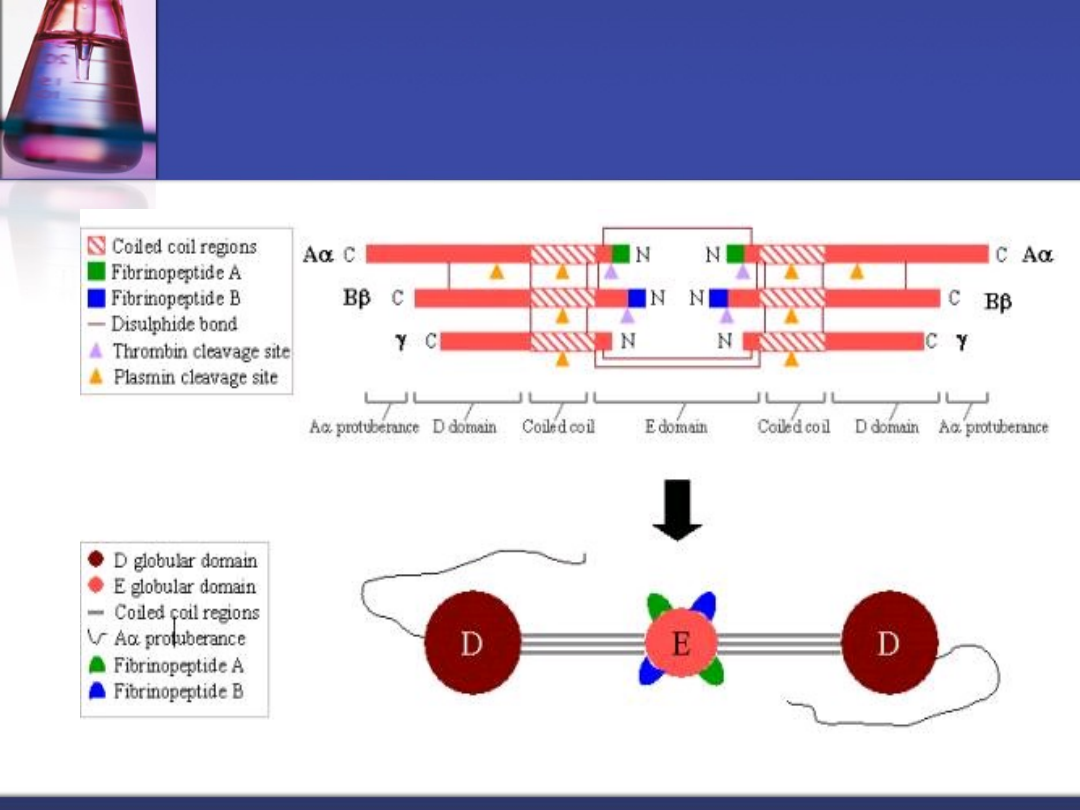
Fibrynogen ( czynnik
I )
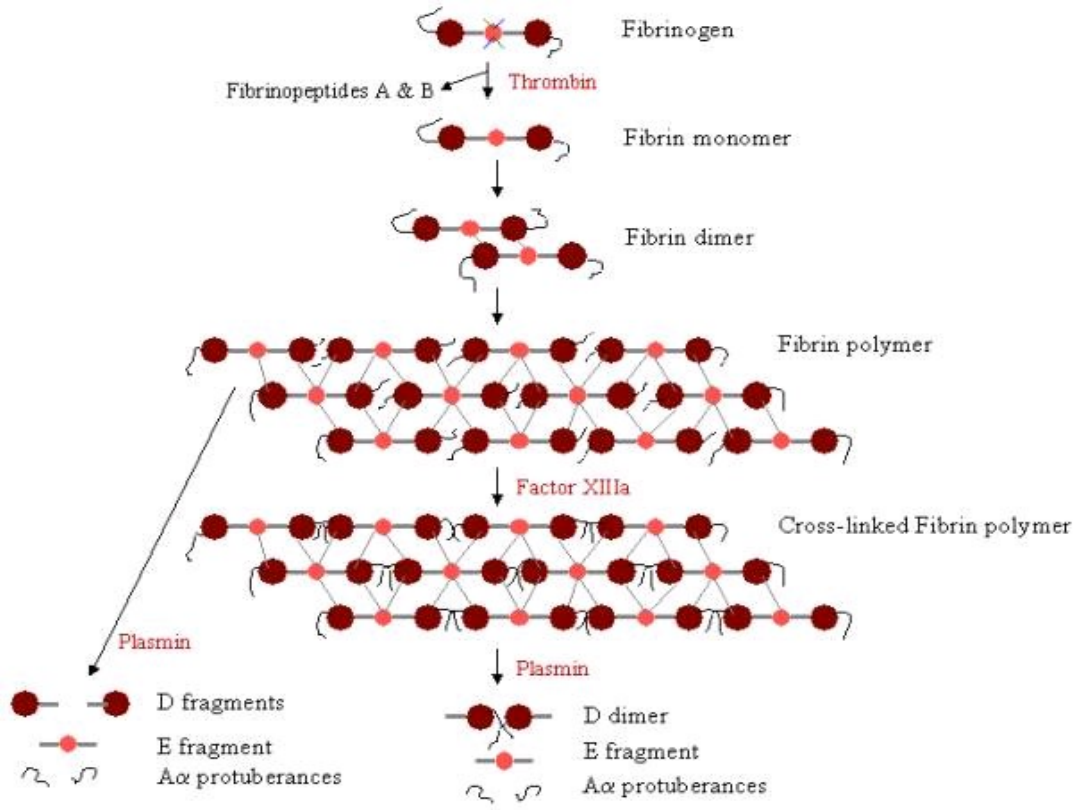

FDP ( produkty
degradacji
fibrynogenu/fibryny)
Plazmina działając na fibrynogen doprowadza do powstania
produktów degradacji fibrynogenu (FDP), które dzieli się na
1.Wczesne – fragment Bᵝ 1-42, Bᵝ 1-118, X i Y
2. Późne – fragmenty D i E
Plazmina degradując nieusieciowaną fibrynę doprowadza do
powstania takich samych FDP, jednak zamiast peptydów Bᵝ 1-42
powstają peptydy Bᵝ 15-42, gdyż 14 pierwszych aminokwasów
odszczepionych jest wcześniej przez trombinę.
FDP posiadają właściwości antykoagulacyjne, hamują funkcję płytek,
polimeryzację monomerów fibryny, stymulują syntezę fibrynogenu.
Zwiększają przepuszczalność naczyń włosowatych, działają
immunosupresyjnie i cytotoksycznie.
Wzrost stężenia FDP:
↑
Zespół DIC, zespoły zakrzepowo-zatorowe, po operacjach, stany zapalne,
nowotwory, choroby wątroby, nerek, doustne środki antykoncepcyjne,
ciąża, leczenie trombolityczne, pierwotna fibrynogenoliza.

D- dimer
D-dimery to produkty rozpadu fibryny.
D-dimery są bezpośrednimi markerami fibrynolizy
(produkcji plazminy) oraz pośrednim wskaźnikiem
procesu koagulacji (produkcji trombiny). Okres
półtrwania D-dimerów we krwi wynosi 8 godzin.
Wzrost stężenia D-dimerów
Zespół DIC, zespoły zakrzepowo-zatorowe, po
operacjach, stany zapalne, nowotwory, choroby
wątroby, nerek, doustne środki antykoncepcyjne,
ciąża, leczenie trombolityczne, wtórna fibrynoliza.

WSKAZANIA KLINICZNE DO
OZNACZENIA POZIOMU D-
DIMERÓW
WYKLUCZENIE ZAKRZEPICY ŻYLNEJ LUB
TĘTNICZEJ
WYKLUCZENIE ZATOROWOŚCI PŁUCNEJ
ZESPOŁY WYKRZEPIANIA
WEWNĄTRZNACZYNIOWEGO (DIC)

D-DIMER W OSOCZU TO
NAJBARDZIEJ PRZYDATNY MARKER
LABORATORYJNY
DO BADAŃ PRZESIEWOWYCH U OSÓB Z
PODEJRZENIEM
ZAKRZEPICY ŻYLNEJ LUB ZATOROWOŚCI
PŁUCNEJ
SZCZEGÓLNIE W WARUNKACH
AMBULATORYJNYCH LUB W IZBIE PRZYJĘĆ

D - dimery
D- dimery posiadają wysoką wartość
predykcyjną ujemną - niski poziom
pozwala z dużym
prawdopodobieństwem wykluczyć
patologie, natomiast podwyższony
poziom nie jest pewnym dowodem ich
występowania (niska wartość
predykcyjna dodatnia).

Naturalne inhibitory
krzepnięcia
Utrzymanie płynności krwi krążącej oraz zapobieganie nadmiernemu narastaniu czopu
ostatecznego
Najważniejsze inhibitory: Antytrombina III – AT III
Białko C (PC)
Białko S (PS) jako kofaktor białka C
Inhibitor zewnątrzpochodnego szlaku krzepnięcia –TFPI
ANTYTROMBINA III
ANTYTROMBINA III
- synteza w wątrobie, w mniejszym stopniu w śródbłonku naczyń oraz w
megakariocytach
- należy do rodziny serpin inaktywujących proteazy serynowe poprzez ich wiązanie w
kompleks stechiometryczny w stos. 1:1
- kofaktorem AT III jest heparyna, która tworząc z nią kompleks przyśpiesza reakcję
inaktywacji cz. krzepnięcia 1000 x.
AT III inaktywuje : trombinę, XIIa , XIa, IXa, Xa ( ostatnio uważa się, że także VIIa)
Naturalne inhibitory
krzepnięcia
BIAŁKO C
BIAŁKO C
- synteza formy nieaktywnej w wątrobie przy udziale witaminy K
- aktywacja białka C uruchamiana jest przez pojawienie się trombiny we
krwi
trombina
wiąże się w kompleks z
Trombomodulina
Białko C
APC + PS
inaktywacja czynnika Va i VIIIa
Niedobór białka C : DIC, leczenie L-asparaginazą, doustnymi antykoagulantam
TFPI
TFPI
- inhibitor zewnątrzpochodnego szlaku
- inhibitor zewnątrzpochodnego szlaku
krzepnięcia
krzepnięcia
- białko występujące w osoczu w formie związanej z Lp
- w obecności cz. Xa wiąże i inaktywuje kompleks TF – VII a

Zasady pobierania
krwi do badań układu
krzepnięcia
RANO, NA CZCZO ( lub lekki posiłek beztłuszczowy)
W WARUNKACH SPOKOJU, BEZ STRESU
KREW ŻYLNA NA 3,2% CYTRYNIAN SODU
(1 cz. cytrynianu - 9 cz. krwi )
BEZ STAZY lub UCISK ok. 30 sek
OSTRE, DOŚĆ GRUBE IGŁY
KREW PO ODRZUCENIU PIERWSZYCH 2-3 ML
PLASTIKOWE PROBÓWKI
BARDZO DELIKATNE MIESZANIE KRWI TUŻ PO POBRANIU
(3-4 krotne odwrócenie probówki do góry dnem - nie wolno spienić! )
NIEZWŁOCZNIE ODWIROWAĆ PRÓBKĘ KRWI
(10 min – 3 tys. obrotów/min)
BADANIA WYKONAĆ W CIĄGU MAX 2 GODZIN OD POBRANIA

Aktywność fizyczna – aktywacja fibrynolizy, ↑ stężenia t-PA, PAP,D-
dimerów, vWF, cz.VIII
Stres – przedłużający się stres psychiczny nie powoduje zmian w
zakresie układu fibrynolitycznego, prowadzi jednak do znamiennego
obniżenia cz. V, VIII i IX.
Zmienność dobowa – zdolność płytek do agregacji jest największa w
godzinach porannych. Aktywność czynników VIIa, VIIIa i stężenia
białek C i S są wyższe rano niż wieczorem. Fibrynoliza jest mniej
aktywna rano niż wieczorem, wyższe stężenia PAI-1 i niższe stężenia
t-PA w godzinach rannych.
Wpływ żeńskich hormonów płciowych – fibrynogen osiąga
najwyższe wartości w fazie lutealnej, najniższe w okresie miesiączki.
Faza folikularna – rośnie VII i spada poziom białka C. Czynnik VIII i vW
są najniższe w fazie folikularnej i najwyższe w fazie lutealnej.

Badania w diagnostyce zaburzeń
układu krzepnięcia
NACZYNIOWE
NACZYNIOWE
Czas krwawienia metodą Ivy
Testy specjalistyczne – test oporności kapilarowej
- biopsja skóry
PŁYTKOWE
PŁYTKOWE
Czas krwawienia - BT
Liczba płytek krwi –PLT
Rozmaz krwi obwodowej
Badania czynnościowe:
Adhezja i agregacja płytek
Reakcja uwalniania
Ocena zawartości ziarnistości płytek
Czas zużycia protrombiny
OSOCZOWE
OSOCZOWE
Czas kaolinowo-kefalinowy – aPTT – (k-k) -czas częściowej tromboplastyny po aktywacji Czas
protrombinowy – PT
Czas trombinowy – TT
Czas rekalcynacji – CT
Fibrynogen
Poszczególne czynniki krzepnięcia: VIII, VII, XIII
Krążące antykoagulanty
Inhibitory krzepnięcia:
Antytrombina III –AT III
Białko C, białko S

Badania układu
hemostazy
Metody ogólne czynnościowe,
oceniające funkcję naczyń i płytek krwi
Metody koagulometryczne, oceniające
globalnie sprawność krzepnięcia
osoczowego
Metody biochemiczne chromogenne
Metody immunologiczne
Metody diagnostyki molekularnej

Czas od chwili uszkodzenia naczynia do samoistnego ustania
krwawienia
• jest jednoznaczny z czasem trwania hemostazy pierwotnej
• jest miarą: czasu utworzenia czopu płytkowego
skurczu naczyń
adhezji i agregacji płytek do śródbłonka naczyń
• próba czynnościowa płytek krwi zależna częściowo od stanu naczyń, nie
zależy od
czynników krzepnięcia
• metody oznaczania :
Metoda Duke’a
Nakłucie skóry opuszki palca na głębokość 2-3 mm i pomiar czasu do
momentu zaprzestania wypływu krwi
(metoda obarczona
błędem)
Norma - do 5 min
Czas
krwawienia

Czas krwawienia
Metoda Ivy z modyfikacją Mielke – met. zalecana
Metoda Ivy z modyfikacją Mielke – met. zalecana
Standaryzacja pomiaru – pomiar czasu wypływu krwi od momentu dokonania nacięcia na
skórze przedramienia o dł. 5 mm i głębokości ok. 0,5 mm (zestawy np. Simplate) przy
ciśnieniu
40 mmHg (ciśn. nadmuchane po założeniu opaski ciśnieniomierza) do braku śladu krwi na
przykładanej do nacięcia bibule filtracyjnej.
Norma – 2 – 10 min
Nie powinien przekraczać 7 min
CZAS KRWAWIENIA ↑
małopłytkowość
trombastenia Glanzmana (wrodzone zaburzenia agregacji płytek)
choroba von Willebranda
niektóre trombocytopatie
skazy krwotoczne z hipofibrynogenemią
po aspirynie ( blokada aktywności cyklooksygenazy płytkowej)

CZAS REKALCYNACJI OSOCZA - CK
CZAS REKALCYNACJI OSOCZA - CK
czas krzepnięcia po dodaniu do osocza nadmiaru jonów Ca
2+
w probówce szklanej
ocenia aktywność układu wewnątrzpochodnego
Norma CT: 100 – 210 sek
próba mało czuła, czas wydłuża się dopiero po zmniejszeniu aktywności jakiegoś czynnika
poniżej 3 – 5% normy ( skazy krwotoczne występują już przy wartościach 10 – 20 %
normy)
CZAS KRZEPNIĘCIA WG. METODY LEE – WHITE’A (CZAS L-W)
CZAS KRZEPNIĘCIA WG. METODY LEE – WHITE’A (CZAS L-W)
pomiar czasu od pobrania pełnej krwi do jej skrzepnięcia w szklanej probówce w temp.
37 C
określa sprawność układu wewnątrzpochodnego
(próba mało czuła, czas wydłuża się dopiero po zmniejszeniu aktywności jakiegoś
czynnika
poniżej 1 – 3 % normy)
Norma czasu L-W : 8 – 12 min
Czas krzepnięcia
niedobór cz. układu wewnątrzpochodnego (XII, XI, IX, VIII)
niedobór cz. drogi wspólnej ( X, V, II, I)
obecność inhibitorów czynników krzepnięcia np. podczas leczenia heparyną

APTT

PT
złożony niedobór cz. VII, V, X, II
przewlekłe ch. miąższu wątroby
awitaminoza K
hypo- dysfibrogenemia
obecność inhibitorów krzepnięcia – heparyna, FDP
ch. krwotoczne noworodków (niedobór wit. K, niedorozwój wątroby)
rozsiane wykrzepianie wewnątrznaczyniowe (DIC)
krążące antykoagulanty u chorych z toczniem rumieniowatym
inne :białaczki, mocznica, leczenie salicylanami
leczenie antykoagulantami doustnymi
PT
zakrzepica
stany nadkrzepliwości m.in. ciężarnych i okołoporodowej
zwiększona aktywność cz. VII
niewłaściwe przechowywanie próbki (powyżej 2 h w +4 ?C)

PT – ocena zewnątrzpochodnego
PT – ocena zewnątrzpochodnego
układu krzepnięcia
układu krzepnięcia
CZAS
PROTROMBINOWY
Czas (sekundy)
11 – 13 sek
12 – 16 sek
WSKAŹNIK
PROTROMBINOWY
PT prawidłowy x 100
PT pacjenta
80 – 120 %
WSPÓŁCZYNNIK
PROTROMBINOWY - R
PT pacjenta
PT prawidłowy
0,85 – 1,15
INR – międzynarodowy
współczynnik
znormalizowany
R
ISI
ISI – międzynarodowy
indeks
czułości
0,9 – 1,25

Czas trombinowy
TT – ocenia końcowy etap wspólnej drogi krzepnięcia. Czas
trombinowy jest miarą przejścia fibrynogenu w fibrynę. Zależy on od
stężenia fibrynogenu, obecności nieprawidłowego fibrynogenu,
aktywności antytrombin oraz procesów polimeryzacji i stabilizacji
fibryny.
↑ TT
:
Hipofibrynogenemia
Dysfibrynogenemia
Obecność immunologicznych inhibitorów trombiny
Zaburzenia polimeryzacji fibryny
Leczenie heparyną niefrakcjonowaną
Wartości prawidłowe 16-21 sek.

Czas batroksobinowy (BT),
reptylazowy (RT) lub
ankrodowy
Jest to czas krzepnięcia osocza po aktywacji
trombinopodobnym enzymem – reptylazą lub ankrodem
wyizolowanym z jadu węża. Reptylaza odszczepia od
fibrynogenu fibrynopeptyd A.
Podobnie jak czas trombinowy, czas reptylazowy jest miarą
przejścia fibrynogenu w fibrynę. Na pomiar czasu
reptylazowego nie mają wpływu heparyna, hirudyna i
immunologiczne antytrombiny, dlatego w trakcie leczenia
heparyną czas reptylazowy pozostaje niezmieniony.
W dysfibrynogenemi czas reptylazowy jest bardziej
przedłużony niż TT, w obecności FDP czy D-dimerów – mniej.
Wartości prawidłowe: 16-22
sek.

Fibrynogen
Glikoproteina syntetyzowana w wątrobie, niezbędna w procesie krzepnięcia do
tworzenia czopu płytkowego (warunkuje agregację płytek) i ostatecznego
(tworzenie siatki fibrynowej).
Białko ostrej fazy, którego stęż. rośnie w początkowym okresie infekcji (wpływ na
wzrost OB)
niezależny czynnik ryzyka choroby wieńcowej, zawału m. sercowego, udaru mózgu
(bierze udział w transporcie cholesterolu i tworzeniu k. piankowatych, powoduje
rozrost
mięśni gładkich – zw. miażdżycorodny)
wzrost między 3-5 dniem w zawale, stabilizacja w ciągu 20 dni
im wyższy fbg, tym gorsze rokowanie w zawale
Norma - 2,0 – 5,0 g/l 200-500 mg/dl
Metody oznaczania fibrynogenu:
metoda chronometryczna Claussa
(ścisła korelacja między trombinowym czasem krzepnięcia i stężeniem fibrynogenu w
osoczu)
metoda kolorymetryczna z odczynnikiem Folina i Ciocalteu

Czas lizy skrzepu
euglobulin
próba ogólna układu fibrynolitycznego
czas upłynnienia skrzepu euglobulin osocza wytrąconych pod wpływem
roztworu o niskiej sile jonowej, pH-5 i temp. 4C, a następnie
rozpuszczonych w buforze i wykrzepionych CaCl
2
mierzy się czas od momentu powstania skrzepu do chwili rozpuszczenia w
temp. 37C
euglobulinowa frakcja białkowa zawiera : plazminogen
plazmina
aktywatory plazminogenu
fibrynogen
cz. krzepnięcia: m.in. VIII,
XII, XIII
brak inhibitorów plazminogenu np. antyplazmin (które pozostają w
supernatancie)
Norma - 120 – 240 min (2 – 4 godz. )

ELT
czas lizy euglobulin
czas lizy euglobulin
III trymestr ciąży
okres pooperacyjny
choroby zatorowo-zakrzepowe
zawał serca i miażdżyca
cukrzyca
nadciśnienie
czas lizy euglobulin
czas lizy euglobulin
skazy krwotoczne przebiegające z hiperfibrynolizą
zaawansowana choroba nowotworowa
marskość wątroby
ostre stany septyczne
po przetoczeniu krwi obcej grupowo
przy znacznym skróceniu czasu lizy euglobulin przed operacją
podaje się
leki hamujące aktywność t-PA

Algorytm
postępowania
APTT ↑
PT - N, TT - N , BT - N niedobór cz. XII, XI, IX, VIII ( hemofilia lub ch. vW), PK
obecność krążącego antykoagulantu
próba korekcji APTT
prawidłowa brak korekcji
niedobór czynników krążący antykoagulant (anty-VIII lub LA)
oznaczyć poziom poj. czynników miano
APTT↑ PT ↑
TT – N, BT -N niedobór cz. II, V, X, VII (choroby wątroby)
złożony niedobór cz. zależnych od witaminy K (znaczny)
zaburzenia po masywnych przetoczeniach
próba korekcji APTT oznaczyć poziom cz. II, V, VII, X
APTT ↑ PT ↑ TT ↑
BT – N hypo- i dysfibrynogenemie
DIC
ch. wątroby oznaczyć stęż. fibrynogenu
aktywacja fibrynolizy FDP
wpływ heparyny czas reptylazowy
APTT – N
PT↑
TT – N, BT – N niedobór cz. VII
rozpoczęcie leczenia antykoagulantami doustnymi
APTT- ↑ / N
, PT – N, TT- N,
BT↑
ch. von Willebranda
APTT – N, PT – N, TT- N,
BT↑
zaburzenia płytkowo-naczyniowe

Podział skaz
krwotocznych
NACZYNIOWE
NACZYNIOWE
Wrodzone:
Wrodzone:
· wrodzona naczyniakowatość krwotoczna (ch. Rendu-Oslera)
· plamice we wrodzonych zaburzeniach tkanki łącznej np. zespół Marfana
· samoistna hemosyderoza płuc
Nabyte:
Nabyte:
· zespół Schönleina-Henocha
· plamice w przebiegu zakażeń
· plamice w przebiegu amyloidozy
· plamica starcza, ortostatyczna, mechaniczna, polekowe, z zapaleniem naczyń
włosowatych,
szkorbut
- rozpoznanie : przedmiotowe badanie lekarskie + wywiad rodzinny
wyniki testów z zakresu krzepnięcia krwi i hemostazy - norma

Podział skaz
krwotocznych
PŁYTKOWE
PŁYTKOWE
Zmiany ilościowe:
A.
Małopłytkowości - Trombocytopenie
Małopłytkowości - Trombocytopenie
a) zmniejszone wytwarzanie płytek
wrodzone:
* wrodzona hipoplazja szpiku – zespół Fanconiego
nabyte :
* nk aplastyczna
* aplazja megakariocytowa
* nacieczenie szpiku kostnego (ALL, chłoniaki, gruźlica)
* zwłóknienie szpiku
* leki mielosupresyjne, zw. chemiczne, promienie jonizujące
* nk megaloblastyczne, z niedoboru żelaza, nocna napadowa hemoglobinuria
* zakażenia wirusowe, niewydolność nerek
b) nadmierne niszczenie płytek
wrodzone:
* samoistna autoimmunologiczna plamica małopłytkowa
nabyte: immunologiczne: * małopłytkowość poprzetoczeniowa, polekowa
* autoimmunologiczna nk hemolityczna
* toczeń rumieniowaty układowy
* wstrząs anafilaktyczny

Podział skaz
krwotocznych
nieimmunologiczne: * DIC
* zakażenia
* zespół hemolityczno- mocznicowy
* zakrzepowa plamica małopłytkowa
c) nieprawidłowe rozmieszczenie płytek w ustroju
* hipersplenizm
d) utrata płytek: * krążenie pozaustrojowe, krwotoki
B.
B.
Nadpłytkowości
Nadpłytkowości
pierwotne: * samoistna,
* zespóły mieloproliferacyjne
wtórne: * stany zapalne (gruźlica, sarkoidoza, RKZ, wrzodziejące zap. jelita
grubego)
* ch. nowotworowe
* po splenektomii i innych zabiegach pooperacyjnych
* po krwotokach
* w niedoborze żelaza
* polekowa ( np. winkrystyna)
* powysiłkowa (trwająca ok. 15-30 min)

Zmiany jakościowe (zaburzona czynność płytek)- TROMBOCYTOPATIE:
Zmiany jakościowe (zaburzona czynność płytek)- TROMBOCYTOPATIE:
Wrodzone
a)
a) anomalie błony płytkowej:
* zespół Bernarda-Souliera (defekt kompleksu GP Ib/IX/V- płytkowy rec. dla cz.vW
zaburzona adhezja płytek krwi
* trombastenia Glanzmanna ( defekt kompleksu GP IIb/IIIa – rec. dla fbg)
zaburzona agregacja płytek krwi
* defekt receptora kolagenu ( defekt GP Ia/IIa) - łagodna skaza krwotoczna
* zespół Scotta (zaburzenia prokoagulacyjnej czynności płytek)
b) zaburzenia sekrecji ziarnistości płytkowych
B. Nabyte :
* wpływ leków
* ch. krwi (ALL, MDS, zespoły mieloproliferacyjne),
* DIC
* inne choroby np. mocznica, krążenie pozaustrojowe, przewlekłe ch. wątroby

Podział skaz
krwotocznych
OSOCZOWE
OSOCZOWE
Wrodzone
Wrodzone:
* Ch. von Willebranda
* Hemofilia A
*Hemofilia B
* A-, hipo-, dysfibrynogenemia
* Niedobór pojedynczego cz. II, V, VII, X, XI, XII, XIII (występują rzadko)
Nabyte:
Nabyte:
a) niedobór witaminy K
upośledzenie wytwarzania wit. K ( ch. krwotoczna noworodków)
upośledzenie wchłaniania wit.K ( kamica, nowotwór, zespół złego
wchłaniania)
upośledzone wykorzystanie wit. K (doustne antykoagulanty-
pochodne dihydroksykumaryny)
b) zaburzenia krzepnięcia z powodu chorób wątroby

Trombofilia -
nadkrzepliwość
Wrodzona lub nabyta skłonność do zakrzepów
przyczyny zaburzeń nabytych:
obecność p/c antyfosfolipidowych (Lupus antykoagulant- LA)
nadpłytkowość
czerwienica prawdziwa
zwiększona aktywność inhibitorów fibrynolizy
przyczyny wrodzonej trombofilii ( wyst. poniżej 40 r.ż., bez czynników ryzyka):
oporność na aktywne białko C (APC-R)
– 12 –64 % częstość występowania
niedobór białka C
- 5 –8 %
niedobór białka S
- 5 – 8 %
niedobór AT III
- 2 – 4 %
APC-R
zaburzenie związane z mutacją punktową w obrębie cz. V typu Leiden,
zmieniony cz. V ma aktywność prokoagulacyjną, natomiast nie ulega proteolizie
pod
wpływem APC

Hemofilia- skaza
krwotoczna osoczowa
Hemofilia to wrodzone zaburzenie krzepnięcia krwi związane
chromosomem X (dziedziczenie recesywne sprzężone z płcią -
chorują chłopcy, kobiety są nosicielami, chorują przy spadku
poziomu czynnika VIII poniżej 40 %):
hemofilia A – wrodzony niedobór cz. VIII
hemofilia B – wrodzony niedobór cz. IX
hemofilia C – wrodzony niedobór cz. XI
H
H
EMOFILIA A
EMOFILIA A
-
- wrodzony niedobór cz. VIII, stopień ciężkości
choroby
zależy od aktywności cz. VIII

Hemofilia
Postać
hemofilii
Aktywność cz. VIII
w surowicy
Objawy
ciężka
poniżej 1 %
częste nawracające krwawienia do
stawów (zniekształcenia), do
mięśni i narządów wewnętrznych
umiarkowana
1 – 5 %
krwawienia samoistne (rzadko)
Ciężkie krwawienia po ekstrakcji
zębów,
zabiegach chirurgicznych, krwiaki
pourazowe
łagodna
5 – 15 %
Krwawienia po zabiegach, urazach
subhemofilia
15 – 30 %
bezobjawowo

Choroba von
Willebranda
Jest najczęstszą skazą wrodzoną (1-2% w populacji ogólnej)
Występuje u obu płci, dziedziczy się autosomalnie dominująco lub recesywnie.
Czas krwawienia (defekt hemostazy pierwotnej) – cecha różnicująca chorobę von
Willebranda z hemofilią.
Wyróżnia się 3 typy choroby:
Typ I – łagodny ilościowy niedobór vWF
Typ II – jakościowy defekt vWF
Podtyp 2A – nadmierna proteoliza i defekt uwalniania vWF (brak dużych
multimerów vWF)
Podtyp 2B – wzrost powinowactwa vWF do GP Ib (małopłytkowość)
Podtyp 2M – spadek powinowactwa vWF do GP Ib
Podtyp 2N – spadek powinowactwa multimerów vWF do FVIII
Typ III – ciężki ilościowy niedobór vWF, wtórny niedobór FVIII
Czynnik vWF jest syntetyzowany w megakariocytach i śródbłonku.
Niższy poziom czynnika vWF obserwuje się u osób z grupą krwi O

Monitorowanie
leczenia heparyną
Heparyna niefrakcjonowana przyspiesza
inhibitorowe działanie AT III w stosunku
do trombiny 1000-krotnie, a czynnika X
– 4000 razy. Kompleks AT III – heparyna
inaktywuje także czynniki IX a, XI a, XII
a i czynnik VII w kompleksie z
czynnikiem tkankowym. Heparyna
unieczynnia także trombinę.

Monitorowanie
leczenia
p
obieranie krwi: bezpośrednio przed podaniem heparyny
po 4-5 godz – w przypadku wlewu ciągłego
po 4 godz - przy podaniu podskórnym
terapeutyczne stęż. heparyny
–
1,5 - 2,5 x wzrost APTT w porównaniu z APTT przed leczeniem
– 1,5 – 3 x wzrost CT w porównaniu z CT przed leczeniem
Objawy uboczne UFH : małopłytkowość na poczatku leczenia, utajony wrzód żołądka lub
dwunastnicy, skaza krwotoczna, osteoporoza (po okresie 3 miesięcy)
(hirudyna -wyciąg ze ślinianek pijawek lekarskich -działa bezpośrednio na AT III
ANTYKOAGULANTY
ANTYKOAGULANTY
DOUSTNE
DOUSTNE
hamują cykl przemian witaminy K hamowanie produkcji cz. wit. K zależnych (II, VII, IX, X)
hamują produkcję białka C i S
monitorowanie leczenia poprzez pomiar PT terapeutyczne stęż. leku
pobieranie krwi: bezpośrednio przed podaniem
INR – 2,0 – 3,0
w 3, 5, 7 dniu leczenia,
INR – 2,5-3,5 (w ciężkich przypadkach)
co tydzień w pierwszym m-cu leczenia
co 2-3 tyg. w II i III m-c
co 4-6 tyg w okresie póżniejszym
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
Wyszukiwarka
Podobne podstrony:
HEMOSTAZA ratownictwo 2
FIZJOLOGIA UKLADU BIALOKRWINKOWEGO I HEMOSTAZA
Diagnostyka Laboratoryjna hemostaza
pytania z hemostazy, umb rok 3, rok III, materiały, patofizjo, III kolo, hemostaza
3.Skazy krwotoczne, Farmacja, Farmakologia(1), Hemostaza, Układ krwionośny
ZABURZENIA HEMOSTAZY, Wykłady
Hemostaza, Biochemia
LEKI WPLYWAJACE NA KRZEPNIECIE I HEMOSTAZE, 000-Nasze Zdrowko, Leki i Witaminy
zakrzepica, Farmacja, Farmakologia(1), Hemostaza, Układ krwionośny
hemostaza2
Patofizjologia mechanizmów hemostazy
hemost, patologia
wyklad hemostaza
Hemostaza 2009 BAZA
Zaburzenia hemostazy
UKŁAD HEMOSTAZY CZ II
BADANIA UKŁADU HEMOSTATYCZNEGO
więcej podobnych podstron