Hemostaza to kompleks dynamicznych układów utrzymujących krążącą krew w stanie płynnym. Czynność tych układów prowadzi do hamowania krwawienia i naprawy ściany naczynia po zranieniu. Procesy hemostazy przebiegają z jednoczesnym udziałem:
• Ściany naczynia (hemostaza naczyniowa)
• Krwinek płytkowych (hemostaza płytkowa) i innych elementów morfotycznych krwi
• Białek osocza (hemostaza osoczowa)
Hemostaza naczyniowa : wszystkie warstwy ściany naczynia uczestniczą w utrzymaniu krążącej krwi w ścianie płynnym i w hamowaniu krwawienia po uszkodzeniu naczynia. Najbardziej aktywny metabolicznie z wszystkich warstw ściany naczynia jest śródbłonek.
Śródbłonek naczyniowy (endotelium) jest nieadhezyjną błoną, do której w warunkach fizjologicznych nie przywierają elementy upostaciowane krwi. Łączna masa komórek śródbłonka naczyniowego wynosi ok. 1500 g. Tworzą one narząd odpowiedzialny za kontrole przepływu krwi poprzez wydzielanie substancji działających rozszerzające lub zwężające na naczynia krwionośne, a także regulujących ich przebudowe i rozrost oraz utrzymanie równowagi pomiędzy krzepnięciem a fibrynoliza. Laminarną cześć błon śródbłonków pokrywa glikokaliks , w którego glikozaminoglikanach 80% stanowi siarczan heparanu. Nadaje to ujemny ładunek powierzchni ściany naczynia co zapobiega przyleganiu krwinek do śródbłonka.
Głównymi składnikami błon podstawnych i podśródbłonka są : kolagen, retikulina, elastyna oraz proteoglikany. Obecne są również fibronektyna i trombospondyna oraz czynnik von Willebranda.
Obok komórek śródbłonka syntetyzują je komórki mięśni gładkich , warstwy środkowej i fibroblasty przydanki. Wszystkie komórki ściany naczynia zawierają trombcplastynę tkankową. Warstwa środkowa i przydanka warunkują właściwości mechaniczne ściany i stanowią barierę chroniącą przed utratą krwi.
Za regulacje czynności komórek śródbłonka odpowiedzialne są : bradykinina, serotonina. angiotensyny, aminy katecholowe, histamina, ADP, ATP poprzez swoiste dla siebie receptory obecne na powierzchni tych komórek.
Proteoglikany błonowe wzmagają lipolityczną aktywność osocza a siarczany dermatanu i heparanu w kompleksie z antytrombiną III i kofaktorem II heparyny unieczynniają trombinę, czynnik Xa oraz inne osoczowe proteinazy. Sródbłonkowa proteinaza - neksyna- w ścianie naczyń wiąże oraz unieczynia trombinę i inne enzymy.
Błonowa glikoproteina - trombomodulina - stanowi w śródbłonku receptor trombiny, w połączeniu z którym enzym traci powinowactwo do fibrynogenu i aktywuje białko C co stanowi naturalny system antykoagulacyjny.
Badania przeprowadzone w latach 80-tych rzuciły nowe światło na rolę śródbłonka, którego komórki tworzą aktywna metabolicznie tkankę. Stwierdzono wówczas, że pod wpływem acetylocholiny naczynia z nieuszkodzonym śródbłonkiem ulegają rozkurczowi a z uszkodzonym - kurczą się. Zwróciło to uwagę na fakt, że śródbłonek naczyniowy może być ważnym narządem parakrynnym, uwalniającym czynniki naczyniorozszerząjące i naczyniokurczące.
Do najważniejszych wazodylatatorów należą tlenek azotu /NO = EDRF ang. endothelium derived relaxing factor/ prostacykilina /PGI1/ oraz śródbłonkowy czynnik hiperpolaryzujący /EDHF ang. endothelium derived hyperpolarizing factor/.
Głównymi czynnikami obkurczającymi naczynia czyli wazokonstryktorami są endoteliny, prostaglandyna PGH2 oraz angiotensyna II
Z czynników naczyniorozszerzajacych szczególne zainteresowanie od czasów doniesień Monkady z 1987r. budzi tlenek azotu. Jest on produkowany w komórkach śródbłonka z argininy przy udziale syntaz tlenku azotu:
a) śródbłonkowej,
b) neuronalnej (te dwie syntetazy określa się mianem konstytutywnej)
c) indukowalnej (makrofagowej).
Izoenzym śródbłonkowy znajduje się w śródbłonku naczyń, w płytkach krwi, w sercu, izoenzym neuronalny w neuronach ośrodkowch i obwodowych, przy czym obie formy są stale obecnymi składnikami komórek. Do ekspresji formy indukowalnej dochodzi po pobudzeniu makrofagów przez produkty procesu zapalnego - bakteryjne egzo- lub endotoksyny, cytokiny, TNF, interleukinę 1. Ekspresja tego izoenzymu może mieć miejsce w komórkach mięśni gładkich naczyń, serca, jelit. Wytwarza ona dużo większe ilości tlenku azotu niż forma konstytutywna.
Forma konstytutywna jest zależna od kompleksu wapń - kalmodulina. Powstanie tego kompleksu może być indukowane przez substancję P, endoteliny, bradykininę, acetylocholinę czy ADP. Mechanizm jego działania związany jest ze stymulacja cyklazy guanylowej, co zwiększa stężenie cGMP, który rozszerza naczynia, hamuje adhezję i agregację płytek krwi oraz działa antyproliferacyjnie.
Forma indukowalna jest niezależna od jonów wapnia. Tlenek azotu syntetyzowany przez syntazę indukowalną także aktywuje cyklazę guanylową ale jego obecność w wysokich stężeniach powoduje inaktywację enzymów zawierających metale grup przejściowych. Podkreślić należy, ze tlenek azotu nie wymaga receptora, bezpośrednio bowiem penetruje do wnętrza komórek. Tlenek azotu jest bardzo nietrwały, jego półokres trwania wynosi zaledwie 3 - 50 sek. Jest on unieczyniany przez anionorodnik ponadtlenkowy O2, który natychmiast utlenia NO do NO2.Tlenek azotu syntetyzowany w sposób ciągły w śródbłonku naczyń zapewnia utrzymanie stałego napięcia ścian tętnic i tętniczek. Wytwarzany w naczyniach płucnych zapewnia utrzymanie odpowiedniego przepływu krwi przez płuca. Działa przeciwagregacyjnie, wykazując synergizm z działaniem prostacyklin. Wydaje się być jednym z neurotransmiterów w neuronach nieadrenergicznych i niecholinergicznych odgrywających istotną rolę w rozszerzaniu niektórych naczyń (ciał jamistych), rozszerzaniu zwieracza Odiego, oskrzeli.
Innym czynnikiem naczyniorozszerzającym jest czynnik hiperpolaryzujacy mięśnie gładkie. Spotyka się różne opinie co do istoty tego czynnika. Są doniesienia identyfikujące go jako nadtlenek wodoru. Powoduje on otwieranie ATP-zależnych kanałów potasowych w mięśniach gładkich, ucieczkę potasu z komórki i w następstwie hiperpolaryzację mięśnia oraz jego rozkurcz. (są to metabolity kwasu arachidonowego, powstające przy udziale cytochromu P-450 kw. epoksyeikozatrienowe).
Kolejnym, istotnym czynnikiem naczyniorozszerzającym są prostacykliny. Głównym źródłem prostacyklin w naczyniach są komórki śródbłonka, lecz syntetyzowane mogą być także w obrębie mięśni gładkich naczyń. Prostacyklina powstaje z kwasu arachidowego, znajdującego się w fosfolipidach błonowych. Uwalniany przez fosfolipaze A2 kwas arachidonowy jest utleniany przez cyklooksygenazę, a powstające cyklicznie nadtlenki /PGH2/ są przekształcanie przez syntetazę PGI2 w prostacyklinę, przeciwdziałającą agregacji płytek oraz rozszerzająca naczynia. Prostacykliny hamują akumulacje cholesterolu w makrofagach i hamują powstawanie komórek piankowatych. PGI2 posiada okres półtrwania krótszy od. jednego cyklu krążeniowego. Mechanizm jej działania polega na aktywacji cyklazy adenylowej powodującej wzrost stężenia cAMP w mięśniówce naczyń i płytkach krwi.
Do czynników śródbłonkowych działających kurcząco należy tromboksan A2 /TXA2/. Powstaje on z PGH2 wytwarzanych w płytkach krwi, gdzie przekształcane są w tromboksan A2 zwężający naczynia i zwiększający agregację płytek krwi, przez co jest on fizjologicznym przeciwstawieniem PGI2. Płytki krwi intensywniej wytwarzają TXA2, aniżeli śródbłonek produkuje PGI:, stąd inhibitory cyklooksygenazy (np. aspiryna, indometacyna) hamują agregację płytek krwi. Obok tromboksanu czynnikami kurczącymi naczynia są endoteliny /TT/ reprezentujące rodzinę polipeptydów złożonych z 21 aminokwasów. W zależności od zmiennej sekwencji aminokwasów wyróżnia się trzy formy endotelin, wykazujące różnice w aktywności biologicznej. Oprócz czynności wazokonstrykcyjnej, co głównie dotyczy izoform ET1 i ET3 endoteliny wywierają dodatni efekt inotropowy i chronotropowy na serce.
Działanie endotelin odbywa się poprzez swoisty receptor w mięśniówce gładkiej naczyń krwionośnych. Wyróżnia się dwa rodzaje receptorów dla endotelin : ETA - charakterystyczne dla ET1 oraz ETβ - z którymi wiążą się wszystkie endoteliny. Uważa się, że endoteliny wywierają efekt kurczący naczynia poprzez aktywację kanałów wapniowych. Stwierdzono także, że ET1 poprzez receptory ETβ może spowodować wzrost poziomu prostaglandyn i tlenku azotu i na tej drodze wywierać efekt naczyniorozszerzający. Dzieje się to w przypadku niskich stężeń ET1, co powoduje aktywacje kanału potasowego zależnego od jonów wapnia. Na tej podstawie wysunięto hipotezę, że endoteliny odpowiedzialne są za długotrwale zmiany napięcia naczyń prawdopodobnie zarówno przez obkurczanie jak i rozszerzanie naczyń.
Czynnikami, które wykazują istotny wpływ naczyniorozszerzający, a także współdziałają w przemianie kwasu arachidonowego oraz poprawiają metabolizm energetyczny są kininy.
W 1928 roku Frey i Wede odkryli związek, który określili jako ciepłochwiejną substancje, niepoddającą się dializie, a znajdującą się w moczu (93, 94). Podanie jej powodowało długotrwałe obniżenie ciśnienia. Stwierdzili, że jest ona uwalniana w czasie pracy mięśni. Kolejne badania dowiodły, że jest ona uwalniana przez trzustkę i ten fakt pozwolił na nadanie nazwy temu związkowi - od greckiej nazwy trzustki - kalikreina. Kalikreiny są proteazami, które uwalniają peptydy zwane kininami. Znane są dwie kalikreiny - kalikreina osoczowa która krąży we krwi w formie nieaktywnej oraz kalikreina tkankowa , która jest zlokalizowana na powierzchni błon tych komórek, w których zachodzi transkomórkowy transport elektrolitów. Kalikreina osoczowa jest zasadową glikoproteiną o m. c. ok 100 kD. Syntetyzowana jest w wątrobie w postaci nieaktywnej prekalikreiny. Nieaktywna prekalikreina przekształcana jest w formę aktywną przez aktywatory, które są proteolitycznymi fragmentami aktywnej postaci XII czynnika krzepnięcia (czynnika Hagemana). Rolę katalizatora w procesie aktywacji czynnika Hagemana odgrywa plazmina i na zasadzie dodatniego sprzężenia zwrotnego sama kalikreina. Po uaktywnieniu kalikreina osoczowa działa na kininogen wysokocząsteczkowy o masie 100 - 200 k D tworząc bradykininę.
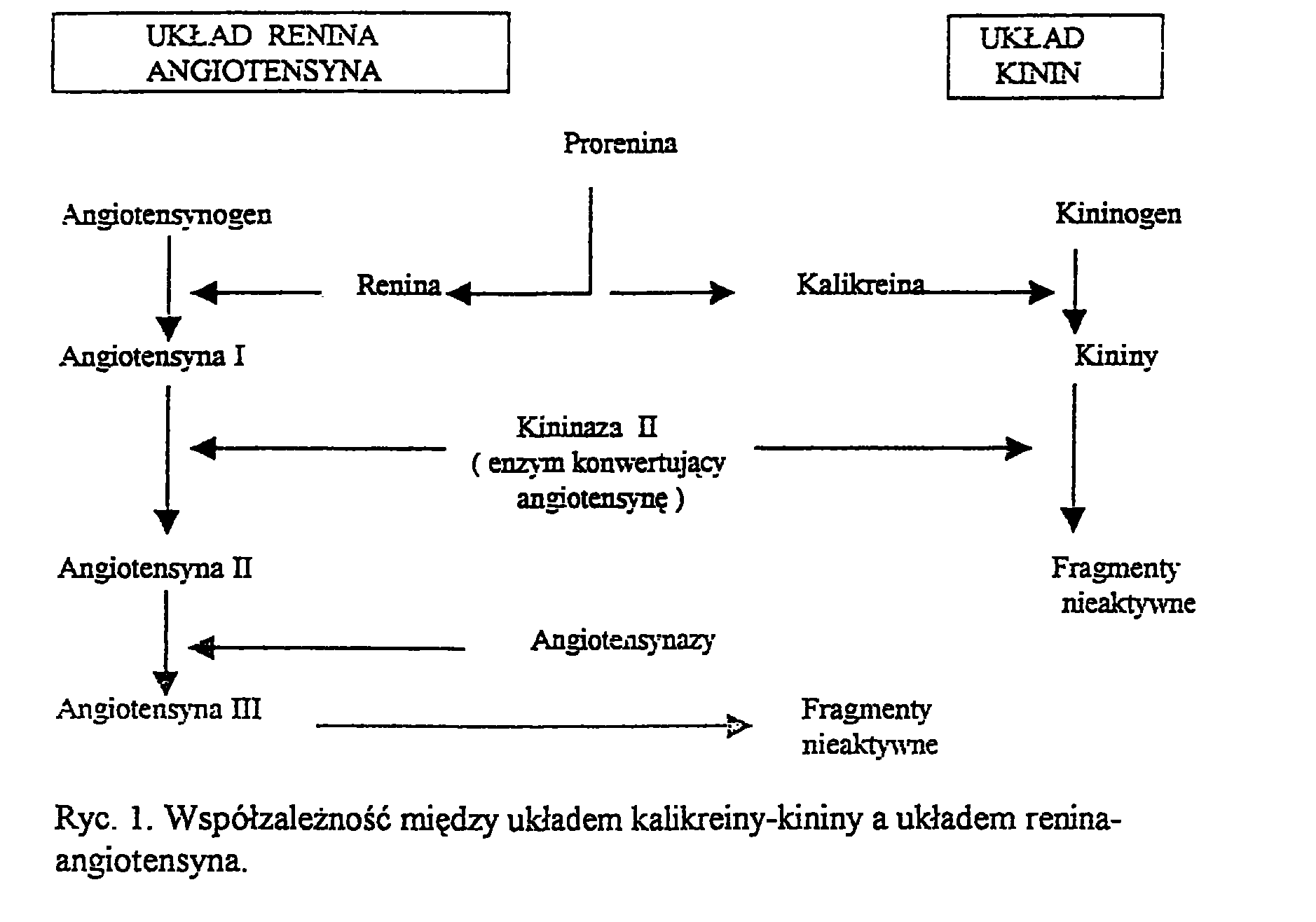
Kalikreina tkankowa jest kwaśną glikoproteiną o m .c. między 2,7 a 4,3kD działającą na kininogen wysokocząsteczkowy i kininogen niskocząsteczkowy, wytwarzając lizylobradykininę zwaną również kalidyną. Lizobradykinina może być przekształcana w bradykininę przez aminopeptydazę.
Obydwa peptydy rozkładane są na nieczynne fragmenty przez kininazę I syntetyzowaną w wątrobie i płucach, która odłącza arginine z C-końcowego odcinka bradykininy. Dodatkowo dipeptydylokarboksypeptydaza (kininaza II) unieczynnia bradykinine i lizylobradykinine odczepiając dipeptyd Phe-Arg z C-końcowego odcinka bradykininy.
Kininaza II występuje w śródbłonku naczyń, szczególnie w płucach i nerkach i jest tym samym enzymem co enzym konwertujący angiotensynę, odłączającym dipeptyd His-Leu z C-końcowego odcinka angiotensyny I i przekształcając ją w angiotensynę II. (Rycina 1.)
Hemostaza płytkowa
Płytki krwi są bezjądrzastymi, dyskowatymi komórkami produkowanymi w szpiku kostnym w procesach rnegakariocytopoezy i trombocytopoezy. Produkcja płytek krwi regulowana jest przy udziale trombopoetyny TPO oraz Mk-CSF (megakariocytic coiony-stimulating factor). Ten ostatni reguluje różnicowanie oraz podziały macierzystych komórek hemopoetycznych , prekursorów megakariocytów. Poza szpikiem dojrzale megakariocyty mogą uwalniać płytki krwi w płucach, we krwi, śledzionie i w wątrobie. Prawidłowy poziom płytek we krwi wynosi 150-400 x 109 /l. Procesy hemostazy mogą przebiegać bez zaburzeń przy poziomie płytek we krwi 35-40 x l09 /l. Jest to tzw. poziom hemostatyczny płytek krwi warunkujący prawidłową czynność hemostatyczną płytek. Prawidłowy czas przeżycia płytek wynosi 8-12 dni.
Powierzchnię płytki stanowi dwuwarstwa fosfolipidów błony plazmatycznej płytki pokryta glikokaliksem. Glikokaliks zawiera kwasy sialowe, które nadają powierzchni płytki ujemny ładunek, który zapobiega wzajemnemu zlepianiu się płytek oraz przyleganiu ich do innych komórek krwi i do powierzchni śródbłonka. Glikoproteiny obecne w dwuwarstwie fosfolipidów decydują o własnościach receptorowych i antygenowych błony plazmatycznej, a w połączeniu z białkami szkieletu uczestniczą w procesach zmian kształtu płytek. Płytkowe glikoproteiny błonowe określa się symbolem GP (glycoprotein) dodając oznaczenia cyframi rzymskimi i mała literę alfabetu łacińskiego.
Czynność niektórych glikoprotein błon płytek
Glikoproteina (GP) |
Czynność |
GP Ia |
receptor kolagenu |
GP Ib |
receptor czynnika Y.Willebranda i trombiny |
GP Ic |
nie ustalona |
GP IIa |
nie ustalona |
GP IIb |
receptor fibrynogenu i czynnika Y.Willebranda, płytkowy antygen P1A1 |
GP IIIa |
|
GP V |
Substrat trombiny |
GP IX |
Czynna w kompleksie z GP Ib |
Krwinki płytkowe posiadają ziarnistości α i β a także wgłębienia błony plazmatycznej tworzące otwarte kanały, stanowiące łączność miedzy wnętrzem komórki a środowiskiem.
Składniki błony plazmatycznej wiążą lub adsorbują białka osocza .W tejże błonie znajdują się enzymatyczne układy transportowe oraz enzymy uczestniczące w metabolizmie nukleotydów. Kontakt krwinki płytkowej z powierzchnią aktywującą oraz rozpuszczalne induktory i proteinazy prowadzi do udostępnienia receptorów błonowych. Odsłonięte, udostępnione fosfolipidy błonowe uczestniczą w likrywacji osoczowego układu krzepnięcia jako czynnik płytkowy trzeci (PF 3).
Składniki płytkowe biorące udział w osoczowej fazie hemostazy noszą skrótową nazwę PF (plateled factor) z dodaną cyfrą arabską.
Krwinka płytkowa zawiera, gęsty układ kanałów i aparat Golgiego. W błonach tych struktur znajdują się enzymy uczestniczące w przemianach kwasu arachidonowego:
- fosfolipazy,
- cyklcoksygenaza,
- lipoksygenaza,
- svntaza tromboksanu a także jony wapnia.
W cytoplazmie obok białek szkieletu aktyny i miozyny, znajduje się fibrynogen , czynnik XIII, fosfolipaza C, płytkowe enzymy proteolityczne, niektóre inhibitory proteinaz oraz metaboliczna pula nukleotydów.
Krwinki płytkowe charakteryzują się bardzo intensywnym metabolizmem . Zachodzi w nich synteza glikogenu, lipidów i w niewielkim stopniu synteza białek.
Krwinki płytkowe są jednym z podstawowych czynników odpowiedzialnych za utrzymanie integralności ściany naczynia. Ujemny ładunek powierzchni śródbłonka pozwala na zapobieganie przyleganiu płytek .
Uwalniane z komórek śródbłonka prostacyklina i tlenek azotu są antagonistami procesów przylegania i agregacji płytek. Uszkodzenie ściany naczynia udostępnia płytkom składniki ściany naczyniowej.
Pierwotny proces przylegania przebiega z udziałem
- struktur ściany naczyniowej,
- czynników fizycznych ,
- białek osocza oraz
- receptorów płytkowych i czynników aktywujących płytki.
Obecne w ścianie naczynia lub pochodzące z krwi multimery czynnika von Willebranda (vWf) w kompleksie z włóknami kolagenu wiążą się z GP Ib powierzchni płytek.
Spolimeryzowany kolagen bezpośrednio łączy się z GP la.
Zlokalizowane w centralnej pozycji strumienia krwi erytrocyty przesuwają płytki na obwód a uszkodzone w miejscu zranienia erytrocyty są źródłem ADP, który jest induktorem adhezji i agregacji płytek oraz skurczu ściany naczynia. W kompleksie z fibronektyną kolagen łączy się z receptorami płytek.
Ziarnistości płytek i uwalniane z nich substancje.
Ziarnistości |
α |
β |
Lizosomy |
Uwalniane |
Albumina |
ADP |
β-heksozoaminidaza |
Substancje |
Osoczowe czynniki krzepnięcia, fibrynogen, V, XIII, vWf, HMW kininogen |
ATP, GDP, Ca2+ |
β-glukuronidaza β-galaktozydaza β-arabinozydaza |
|
Czynniki płytkowe PF2, PF4 |
Mg2+ |
Arylosulfataza |
|
β-tromboglobulina (βTG) |
K+ |
Katepsyny |
|
Płytkowy cz. wzrostowy (PDGF) |
HPO42+ |
Kolagenazy |
|
Transformujący cz. wzrostowy β (TGF β). |
Serotonina |
|
|
Inhibitory proteinaz osoczowych: C1-INH, α2-AP, α1-P1, α2-M, białko S |
|
|
|
Inhibitor aktywatora plazminogenu (PAI-1) |
|
|
|
Cz. chemotaktyczne, przeciwbakteryjne, zwiększające przepuszczalność naczyń, immunoglobuliny, białka anionowe i kationowe, trombospondyna, fibronektyna |
|
|
Agonistami płytek są: tromboksan ADP, PAF, czynnik vWf, kolagen, trombina, kwas arachidonowy, serotonina, PGG2, PGH2, wazopresyna, norepinefryna, epimefryna, kompleksy immunologiczne, wirusy i bakterie.
Agonista po połączeniu z receptorem, z udziałem kompleksu białek G aktywuje fosfolipazę C, która inicjuje powstanie diacyloglicerolu oraz inozytolotrifosforanu.
Efektem tych przemian jest uwalnianie jonów wapnia z depozytów błonowych do cytoplazmy. Przy udziale ADP i norepinefryny wapń napływa do cytoplazmy ze środowiska. Jony wapnia aktywują kinazę białkową C, fosfolipazę A2 oraz białka kurczliwe płytki. Fosfolipaza A2 aktywuje kaskadę przemian kwasu arachidonowego, głównie torem cyklooksygenazy.
Powstające endogenne cykliczne nadtlenki PGG2 i PGH2 są prekursorami antagonistów płytkowych (PGI2, PGE1, PGH2) oraz głównego ich agonisty - tromboksanu A2.
Tromboksan jest najsilniejszym aktywatorem krwinek płytkowych, wywołuje również skurcz mięśni gładkich naczyń.
Przy udziale fosfolipazy A2 powstaje w płytkach PAF.
Aktywacja płytek powoduie ich:
- rozpościeranie na powierzchni podsrodbicuka.
- przyleganie.
- centralizacje ich ziarnistości i
- narastanie zlepu płytkowego - zwanego pierwotnym czopem hemostatycznym.
Antagonistą tromboksanu jest prostacyklina powstająca w nieuszkodzonych komórkach śródbłonka z płytkowych endonadtlenków przy udziale syntetazy prostacykliny.
Prostacyklina z udziałem kompleksu białek G i GTP powoduje przemieszczenie jonów wapnia z cytozolu do depozytów błonowych czego konsekwencja jest hamowanie:
- czynności białek kurczliwych ,
- aktywności fosfolipaz i cyklooksygenazy.
Efektem działania prostacykliny jest:
- hamowanie przylegania płytek do śródbłonka (działanie antyadhezyjne)
- hamowanie zlepiania się płytek (działanie antyagregacyjne)
Podobne działanie wykazuje produkowany przez komórki śródbłonka tlenek azotu.
Wynikiem aktywacji enzymów działających na składniki błony plazmatycznej jest jej destabilizacja, ułatwienie uwalniania składników ziarnistości płytek.
Efektem tych procesów jest udostępnienie PF3 do łączenie się z osoczowymi czynnikami krzepnięcia i jonami wapnia. ADP z beta ziarnistości razem z jonami wapnia indukuje receptorową czynność kompleksu GP IIb - IIIa, który wiąże głównie fibrynogen. Poprzez te receptory z płytkami wiążą się również czynnik vW, fibronektyna, kolagen, trombospondyna, wirusy i bakterie.
Udostępnienie tromboplastyny tkankowej oraz kontakt kompleksu czynników XII, HMW kininogenu, prokalikreiny i czynnika XI ze składnikami podśródbłonka w uszkodzonym naczyniu , przy dostępnym PF3 prowadzą do generacji trombiny.
Początkowo proces ten zachodzi na obwodzie zlepu płytkowego. Stabilizowana fibryna wzmacnia zlepy płytkowe i razem z białkami kurczliwymi prowadzi do kurczenia się skrzepu (retrakcji).
Trombina pobudzając napływające płytki uczestniczy w narastaniu zakrzepu.
Uwalniane z ziarnistości PF4, β-tromboglobulina i trombospondyna wiążą heparynę , siarczany heparanu i dermatanu, zapobiegając aktywacji antytrombiny III i kofaktora II heparyny. Jednocześnie uwalniane z płytek inhibitory proteinaz ograniczają aktywność osoczowych i komórkowych enzymów proteolitycznych.
Uwalniane z ziarnistości enzymy: kolagenaza, elastaza powodują degradacje białek osocza w tym czynników krzepnięcia.
Jednocześnie przy udziale trombiny dochodzi do ograniczenia procesów krzepnięcia i działania profibrynolitycznego w miejscu, gdzie śródbłonek jest nieuszkodzony.
Zaburzenia hemostazy płytkowej.
Skazy płytkowe mogą przebiegać jako:
- małopłytkowości (trombocytopenie) .
- nadpłytkowości (trombocytemie)
a przy prawidłowej liczbie płytek mogą być spowodowane
• pierwotnym lub wtórnym zaburzeniem metabolizmu płytek lub też
• zmianą strukturalną składników krwinek płytkowych (trombastenie, trombopatie)
Skazy płytkowe są przyczyną występowania
- wybroczyn o różnym nasileniu,
- samoistnych zewnętrznych lub wewnętrznych krwawień
- przedłużenia krwawień po zranieniu
W badaniach laboratoryjnych stwierdza się :
- przedłużenie standardowego czasu krwawienia
- zmienioną liczbę płytek, zaburzenie procesu adhezji bądź agregacji płytek
Mechanizm osoczowej fazy krzepnięcia
Proces ten wymaga serii kaskadowych reakcji zachodzących pod wpływem proteazy serynowej, polegających na proteolitycznym przekształceniu nieaktywnych prekursorów osoczowych białek do białek aktywnych. Proteoliza cząsteczki prekursora bez zmiany jej masy , z uwolnieniem peptydu aktywacyjnego , bądź z odłączeniem podjednostki prowadzi do odsłonięcia centrum aktywnego enzymu.
Osoczowe czynniki krzepnięcia
Czynnik |
niektóre synonimy |
I II III IV V VII VIII vWf IX X XI XII XII Prekalikreina HMW kininogen |
fibrynogen protrombina tromboplastyna tkankowa jony Ca2+ proakceleryna prokonwertna antyhemofilowa globulina A, przeciwhemofilowy czynnik A czynnik von Willebranda czynnik Christmas, czynnik przeciwhemofilowy B czynnik Stuart czynnik przeciwhemofilowy C czynnik Hageman, czynnik kontaktu czynnik stabilizujący fibryne, stabilizator włóknika czynnik Fletcher czynnik Fitzgerald |
Osoczowe czynniki krzepnięcia, poza III i IV są glikoproteinami i można je podzielić na trzy grupy:
- czynniki kontaktu cz. XII, XI prokalikreina osoczowa, wielkocząsteczkowy kininogen biorą udział w aktywacji krzepnięcia przez kontakt z powierzchniami o ujemnym ładunku (np. szkło, kwasy tłuszczowe, fosfolipidy), syntetyzowane są w hepatocytach.
- czynniki rodziny protrombiny: II, VII, IX, X , syntetyzowane w hepatocytach z udziałem witaminy K
- czynniki rodziny fibrynogenu: fibrynogen, czynniki: V, VIII, XIII. Są one substratami trombiny, plazminy i niektórych proteinaz komórkowych, zaś czynniki V i VIII funkcjonują jako kofaktory w procesie aktywacji czynnika X i protrombiny. Synteza zachodzi w hepatocytach, megakariocytach a także w komórkach śródbłonka i fagocytach.
Czynnik III (tromboplastyna tkankowa) jest kompleksem glikoproteiny z fosfolipidami zlokalizowanymi w błonach komórek większości narządów i wszystkich warstw ściany naczynia. Szczególnie dużo tromboplastyny występuje w mózgu, płucach, łożysku i nerkach. Obecna jest również w erytrocytach i leukocytach, nie występuje w płytkach krwi
Jony wapnia uczestniczą w:
- asocjacji czynników II, VII, IX i X z tromboplastyną tkankową, bądź z fosfolipidami błon komórkowych.
- asocjacji trombiny z czynnikiem XIII, XIII a i monomerami fibryny
- oddziaływania między czynnikami XIa i IX
Krzepnięcie krwi jest złożonym procesem fizykochemicznym, w wyniku którego rozpuszczalne białko osocza, fibrynogen zostaje przekształcony w nierozpuszczalna fibrynę. Aktywacja osoczowych czynników krzepnięcia zachodzi w kontakcie z krwią z błoną podstawna śródbłonków i podśródbłonkiem, z tromboplastyną pobudzonych lub uszkodzonych komórek krwi, ściany naczynia i tkanek.
Prowadzą do tego urazy mechaniczne, termiczne lub chemiczne narządów.
Inicjacja krzepnięcia może także nastąpić w kontakcie krwi z powierzchnią sztucznych zastawek i protez naczyniowych.
W miejscu zranienia aktywacja osoczowych czynników krzepnięcia w kontakcie z kolagenem i i tromboplastyna tkankowa przebiega łącznie z przyleganiem i aktywacją krwinek płytkowych. Rozmiar i jakość hemostatycznego czopu płytkowo-fibrynowego uwarunkowane procesami aktywacji protrombiny i aktywnością trombiny.
Reakcje prowadzące do aktywacji trombiny porządkuje się w dwa układy krzepnięcia zewnątrzpochodny i wewnatrzpochodny.
Aktywne enzymy uczynniają szereg cząsteczek proenzymu następnego etapu w jednym lub jednocześnie w obu układach. Stanowi to zwielokrotnieniu i przyspieszeniu procesów krzepnięcia, co w 1964 roku Macfarane nazwał " kaskadową", zaś Davie i Ratnoff "wodospadową" aktywacją krzepnięcia.
W zewnątrzpochodnym układzie krzepnięcia
- skrzep powstaje po 15 sekundach
- aktywacje tego układu inicjuje kompleks tromboplastyny tkankowej z czynnikiem VII i jonami Ca2+, w którym zmiana konformacji cząsteczki prowadzi do utworzenia czynnika VIIa.
Enzymatycznymi aktywatorami czynnika VII trombina oraz czynniki IXa. Xa, XIIa i kalikreina. W kompleksie tworzonym przez czynnik VIIa, Ca2+, fosfolipidy błonowe i czynniki X lub IX powstają formy aktywne tych czynników. Czynnik VIIa jest zatem alternatywnym aktywatorem układu wewnątrzpochodnego, zaś aktywne enzymy tego układu, czynnik XIIa. IXa i kalikreina aktywatorami czynnika VII. Czynnik Xa i trombina w kompleksach z Ca2+ i fosfolipidami również aktywują czynnik IX.
W układzie wewnątrzpochodnym
- skrzep powstaje po upływie 8 minut.
- aktywację zaś krzepnięcia inicjują kompleksy czynnika XII z kolagenem, bądź z innymi składnikami podśródbłonka lub sztuczna powierzchnią, z którymi wiąże się również wielkocząsteczkowy kininogen z przyłączoną prokalikreiną, lub czynnikiem XI.
Kininogen wielkocząsteczkowy we krwi znajduje się w kompleksach z prokalikreiną lub czynnikiem XI. Wczesna aktywacja proenzymu Hageman w kontakcie z powierzchnią polega na zmianie konformacji cząsteczki, z odsłonięciem centrum aktywnego. Pobudzone komórki śródbłonków, bądź niektóre leukocyty są źródłem enzymatycznych aktywatorów czynnika kontaktu. Czynniki XIIa, o masie cząsteczki 80kd, aktywuje prokalikreinę, czynniki XI, VII i plazminogen. Kalikreina, plazmina i czynnik XIa, poprzez proteolizę cząsteczki czynnika XII, tworzą formę XIIa, a następnie formę o masie cząsteczki 28 kd, czynnik XIIa, który aktywuje głównie prokalikreinę. W kompleksie powierzchniowym kalikreina jest najbardziej wydajnym, enzymatycznym aktywatorem czynnika XII. W łączności z powierzchnią aktywującą pozostaje czynnik XIa, który bez udziału fosfolipidów, w obecnościCa2+ aktywuje czynnik IX.. Kalikreina może uwalniać się do krwi, a w osoczu jest jednym z enzymatycznych aktywatorów proenzymu Hageman. Cząsteczki czynnika IXa łącznie z czynnikiem VIIIa, fosfolipidami i jonami Ca2+ formują kompleks aktywujący czynnik X. Alternatywne sposoby aktywacji czynnika IX sprawiają, iż niedobory czynników kompleksu kontaktu nie są przyczyną skazy krwotocznej. Produkt aktywacji obu układów - czynnik Xa pozostaje związany z fosfolipidami błonowymi poprzez jony Ca2+, co chroni go przed dostępem inhibitorów oraz zwiększa stężenie w miejscu aktywacji. W tym kompleksie, w obecności czynnika Va, czynnik Xa dokonuje wydajnej konwersji protrombiny do trombiny.
Trombina zaś poprzez proteolizę cząsteczek protrombiny powoduje powstawanie aktywnego enzymu, bądź nieczynnych produktów.
Niefizjologicznymi aktywatorami protrombiny są trypsyna, papaina i stafylokoagulaza. Trombina nie zawiera reszt kwasu gamma-karboksyglutaminowego, opuszcza zatem kompleks aktywujący, a jej miejsce zajmują kolejne cząsteczki protrombiny.
Trombina posiada kluczowe znaczenie w procesach hemostazy.
- bierze udział w wielu reakcjach osoczowego procesu krzepnięcia,
- wpływa na hemostatyczna funkcję krwinek płytkowych i śródbłonków naczyń,
- jako agonista płytkowy powoduje narastanie czopu hemostatycznego,
- aktywuje udostępnianie fosfolipidów błonowych /PF3/,
- pobudza uwalnianie składników ziarnistości i agregacji,
- trombina jest również aktywatorem czynników V i VIII
- w kompleksie zaś z trombomoduliną śródbłonków traci powinowactwo do fibrynogenu i innych czynników krzepnięcia, a staje się aktywatorem białka C.
Zawarty w osoczu lub uwolniony z alfa ziarnistości czynnik V, czynniki płytkowe i inhibitory proteinaz regulują nasilenie osoczowego krzepnięcia i aktywacji fibrynolizy.
Białko C w kompleksach z fosfolipidami błonowymi, głównie z fosfolipidami błonowymi krwinek płytkowych, jonami Ca2+ i kofaktorem białkiem S powoduje degradacje czynników Va i VIIIa oraz inaktywuje PAI-1. Ogranicza zatem proces krzepnięcia, a osłania aktywacje plazminogenu. Obok tego układu barierę przeciwzakrzepową stanowią osoczowe inhibitory proteinaz głównie antytrombina III i kofaktor II heparyny, reaktywne w obecności heparyny i heparynopodobnych proteoglikanów. Ponadto fibryna adsorbuje i przejściowo unieczynnia trombine.
Trombina pełni czynność koagulacyjną zasadniczo poprzez ograniczoną proteolizę fibrynogenu, który jest jej głównym fizjologicznym substratem.
Cząsteczkę fibrynogenu tworzą dwie identyczne podjednostki. Każda z nich składa się z trzech łańcuchów polipeptydowych. Poszczególne łańcuchy i podjednostki są związane miedzy sobą mostkami dwusiarczkowymi. Trombina odszczepia z N-końcowych regionów łańcuchów po jednym 16-aminokwasowyrn fibrynopeptydzie A oraz po jednym aminokwasowym fibrynopeptydzie B z tegoż łańcucha B. Fibrynopeptyd A pobudza uwalnianie tPA, a w dużych stężeniach jest inhibitorem trombiny. Fibrynopeptyd B aktywuje migrację leukocytów oraz powoduje wzrost napięcia mięśni gładkich, szczególnie w ścianach naczyń żył płucnych. Cząsteczki fibrynogenu pozbawione fibrynopeptydów noszą nazwę monomerów fibryny. Asocjujące monomery fibryny, z udziałem wiązań wodorowych tworzą labilny skrzep fibryny, który ulega depolimeryzacji w roztworach związków rozrywających te wiązania, takich jak: kwas jednochlorooctowy, mocznik i inne.
W obrębie czopu hemostatycznego trombina z udziałem Ca2+ aktywuje osoczowy i płytkowy czynnik XIII . Czynnik XIIIa katalizuje tworzenie walencyjnych wiązań krzyżowych glutamino - lizynowych między łańcuchami alfa lub włókien fibryny. W ten sposób powstaje stabilizowana fibryna zwana również desmofibryną, a skrzep staje się nierozpuszczalny w roztworach związków zrywających wiązania wodorowe i oporny na odkształcanie mechaniczne.
Czynnik XIIIa wprowadza również kowalencyjne wiązania krzyżowe między fibryna i alfa2 antyplazminą. Wbudowanie tego inhibitora do sieci fibrynowej ochrania ją przed działaniem plazminy. Przy braku czynnika XIII lub antyplazminy ułatwiona fibrynoliza i labilność skrzepu mogą prowadzić do skazy krwotocznej.
Substratami czynnika XIIIa są również fibronektyna, trombospondyna i inne białka osocza oraz białka błonowe. Tworzenie kowalencyjnych wiązań krzyżowych miedzy fibronektyną, trombospondyną i fibryną z białkami błon komórkowych ułatwia gojenie ran.
Do badań osoczowej fazy krzepnięcia zalicza się :
- Czas protrombinowy (PT - oceniający czynniki II, V, VII, X )
- Czas częściowej tromboplastyny (PTT - oceniający czynniki II, VI, VIII, IX. XII, prekalikreinę i kininogen)
- Czas trornbinowy (TT - oceniający konwersję fibrynogenu we włóknik)
- Ilościowe oznaczenie fibrynogenu
Wrodzone i nabyte zaburzenia krzepnięcia występują stosunkowo często. Do urodzonych zaburzeń krzepnięcia zalicza się chorobę von Willebranda i hemofilie podczas gdy choroby wątroby, skazy ze zużycia w przebiegu urazu, wstrząsu lub infekcji należą co często spotykanych skaz nabytych.
Układy fibrynolityczne
W warunkach fizjologicznych tendencje do krzepnięcia krwi są równoważone przez fibrynolizę czyli reakcje ograniczające powstawanie skrzepu wewnątrz naczyń krwionośnych i powodujące rozpad skrzepu. który się utworzył.
Reakcje te obejmują:
- inaktywację niektórych aktywnych czynników krzepnięcia z krążenia przez wątrobę
- wzajemne oddziaływanie pomiędzy tromboksanem A i prostacykliną
Wzajemne oddziaływania miedzy tromboksanem A2 i prostacykliną powodują powstawanie skrzepu w ścianie uszkodzonego naczynia krwionośnego a jednocześnie zapobiega utworzeniu skrzepu wewnątrz naczynia co zapobiega zamknięciu jego światła.
Mechanizmy, dzięki którym aktywność prokoagulacyjna i zapoczątkowane procesy naprawcze zostają ograniczone do miejsca urazu są ważnymi regulatorami prawidłowej hemostazy. Wymienić należy tu dwa podstawowe układy będące jej regulatorami.
Układ inhibitorów składających się głównie z krążących i zawartych w śródbłonku inhibitorów proteinaz, oraz układ fibrynolityczny odpowiedzialny za proteolityczne rozpuszczenie skrzepu włóknikowego. Do inhibitorów białkowych zalicza się: antytrombinę III z szeroką swoistością w stosunku do prokoagulacyjnych proteaz serynowych i alfa-2 makroglobulinę.
Dwa antykoagulanty proteazy serynowej - białko C i białko S prowadzą sprzężoną zwrotnie kontrolę krzepnięcia przez proteolizę czynnika V i czynnika VIII. Niedobory antytrombiny III, białka S mogą prowadzić do patologicznej zakrzepicy.
Fibrynolizą steruje enzym zwany plazminą, który powstaje z nieczynnego krążącego plazminogenu w wyniku uwolnionych ze śródbłonka jego aktywatorów. Plazminogen i jego aktywatory wiążą się z tworzącym się skrzepem włóknikowym, zapewniając mechanizm miejscowego rozpuszczania skrzepimy od wewnątrz.. Naturalnie występujący osoczowy inhibitor plazminy, inhibitor alfa-2-plazminy, odgrywa ważną fizjologiczną rolę, ponieważ jego niedobór prowadzi do niekontrolowanego działania plazminy i skazy krwotocznej.
Plazmina jest bardzo aktywnym składnikiem układu fibryno litycznego. Rozpuszcza ona fibrynę i fibrynogen oraz uwalnia produkty degradacji fibrynogenu.
Plazminą powstaje z nieaktywnego prekursora jakim jest plazminogen.
Aktywatory plazminogenu odgrywają rolę w inicjacji fibrynolizv. Najbardziej znane to tPA (tkankowy aktywator plazminogenu), urokinaza, streptokinaza.
Komórki śródbłonka syntetyzują tPA (tkankowy aktywator plazminogenu) i zwiększają jego uwalnianie pod wpływem:
- fibryny, trombiny, plazminy
- acetylocholiny, noradrenaliny, histaminy. bradykininy
- PGE1, PGE2, PGI1, PAF,
- estrogenów
- stężenie wzrasta we krwi po wysiłku, w stanach niedotlenienia tkanek,
Urokinaza jest produkowana przez śródbłonek kanalików nerkowych.
Streptokinaza, zewnątrzpochodny aktywator plazminogenu, produkt paciorkowców hemolizujących. Są one wykorzystywane w leczeniu wczesnych zawałów mięśnia serca ze względu na własności rozpuszczania skrzepów.
Najważniejszym, fizjologicznym aktywatorem plazminogenu jest tPA.
TPA jest obecnie wytwarzany przy pomocy techniki rekombinacji DNA i wykorzystywany w klinice. Wykazuje on własności rozpuszczania skrzepu w tętnicach wieńcowych jeżeli jest zastosowany bezpośrednio po wystąpieniu zawału mięśnia serca.
TPA, osoczowe proaktywatory plazminogenu i plazminogen wiążą się z fibryną w zakrzepie lub z powierzchnia komórek, co chroni je przed inaktywacją a poprzez zagęszczenie ułatwia powstawanie plazminy.
Śródbłonek naczyń krwionośnych odgrywa istotna role w zapobieganiu i rozprzestrzenianiu się skrzepu na nieuszkodzone naczynia krwionośne.
Wszystkie komórki śródbłonka wytwarzają trombomodulinę z wyjątkiem mikrokrażenia mózgowego.
Trombomodulina jest białkiem wiążącym trombinę. Trombomodulina zamienia trombinę w aktywator białka C.
Białko C jest naturalnie występującym białkowym antykoagulantem inaktywującym:
- inhibitor tkankowego aktywatora plazminogenu
- czynnik V i VIII zwiększa ono powstawanie plazminy
Antytrombina III jest inhibitorem proteazy krążącej we krwi, wiąże się proteazą serynową w procesie koagulacji i blokuje jej aktywność jako czynnika krzepnięcia. Wiązanie antytrombiny III z proteazą serynową jest ułatwione przez heparynę. Heparyna hamuje aktywność czynników : IX, X, XI, XII.
Degradacja fibryny w czopie hemostatycznym składa się na przywracanie sprawności ściany naczynia i utrzymanie krwi w stanie płynnym.
Produkty pochodzące z degradacji fibryny i fibrynogenu wykazują szereg własności biologicznych:
- hamują trombinę,
- blokują tromboplastynę tkankowa
- hamują adhezję
- agregacje płytek
drobnocząsteczkowe produkty:
- hamują agregacje płytek,
- aktywują migracje leukocytów
- pobudzają uwalnianie IL-1 przez monocyty
- pobudzają biosyntezę fibrynogenu w hepatocytach
- hamują czynność limfocytów
- w układzie sercowo - naczyniowym wykazują aktywność bradykininy oraz potencjalizują działanie bradykininy, histaminy, angiotensyny, amin katecholowych i serotoniny
Laboratoryjnymi wykładnikami aktywacji układu fibrynolitycznego jest obecność we krwi wolnej plazminy. Ze względu na trudności w oznaczeniu obecności wolnej plazminy oceny aktywacji układu fibrynolitycznego można dokonać pośrednio przy użyciu oznaczenia:
- obniżenia stężenia fibrynogenu w osoczu
- stężenia plazminogenu
- wydłużenia czasu trombinowego (TT) lub czasu częściowej tromboplastyny (PTT)
Leczenie przeciwkrzepliwe
Heparyna zmniejsza krzepliwość krwi poprzez aktywację głównego inhibitora układu krzepnięcia - antytrombiny III (AT III). Tworzy z AT III kompleks unieczynniajacy trombinę i aktywny czynnik Xa. W mniejszym stopniu wpływa także na inne aktywne postaci czynników krzepnięcia będące proteazami serynowymi (IXa, XIa, XIIa). Heparyna katalizuje równocześnie działanie antytrombinowe drugiego białkowego kofaktora osoczowego zwanego kofaktorem heparyny II (heparin cofactor II, HC II). To działanie wymaga bardzo wysokich stężeń heparyny i nie zależy od powinowactwa do AT III. Heparyna wiąże się też z komórkami śródbłonka makrofagami i komórkami siateczki wywierając wpływ przeciwkrzepliwy. Poprzez wiązanie się z płytkami krwi hamuje czynność płytek i przyczynia się do występowania powikłań krwotocznych. Działanie to jest zależne od masy cząsteczkowej heparyny - im wyższa masa cząsteczkowa tym silniejsze działanie na płytki.
Heparyny drobnoczasteczkowe.
Heparyny drobnoczasteczkowe (LMWH) to wg European Pharmacopeia Commission sole heterogennych siarczanów glikozaminoglikanów o średniej masie cząsteczkowej mniejszej niż 8kD z których 60% cząsteczek posiada masę cząsteczkową mniejszą niż 8 kilodaltonów. Rozpiętość mas cząsteczkowych łańcuchów polisacharydowych występujących w preparatach handlowych LMWH jest duża i wynosi 1,8-12 kD. Są one związkami uzyskanymi z heparyny standardowej (unfractionated heparin- UFH ) o średnim ciężarze cząsteczkowym ok. 15 kD.
Wykazują, one działanie przeciwzakrzepowe dzięki:
- uwalnianiu z komórek śródbłonka TFPI (tissue factor pathway inhibitor)
- łączeniu swoich łańcuchów pentasacharydowych z antytrombiną III
- przyspieszeniu inaktywacji aktywnych czynników krzepnięcia IIa, IXa, Xa.
- reakcji z kofaktorem II heparyny oraz płytkowym czynnikiem 4 (PF4)
- wzmaganiu uwalniania śródbłonkowego tkankowego aktywatora plazminogenu (tPa)
- modulacji aktywnego białka C i inhibitora aktywatora plazminogenu (PAI -1)
- nasileniu ekspresji komórkowej P-selektyny
Za stosowaniem heparyn drobnocząsteczkowych przemawiają następujące fakty:
- dłuższy okres półtrwania w porównaniu do UFH
- zdecydowanie wyższa niż UFH biodostępność (powyżej 90%) po podaniu podskórnym
- brak konieczności kontroli działania LMWH w codziennej praktyce klinicznej (pod warunkiem podawania ich w dawkach dostosowanych do masy ciała lub profilaktycznych)
- zmniejszone występowanie trombocytopenii poheparynowej (Heparin Induced Thrombocytopenia, HIT) przy zastosowaniu LMWH
Trombocytopenie po heparynowe (HIT) dzielimy na dwa typy:
Typ I HIT - łagodne powikłanie rozwijające się najczęściej w pierwszych dniach stosowania heparyny (do 5 dni), trwa od 2, do kilku dni, ustępuje samoistnie, bez żadnych następstw, charakteryzuje się obniżeniem ilości płytek krwi do wartości nie mniejszej jednak niż 100 000/ml.
Typ II HIT - ciężkie powikłanie rozwijające się miedzy 5 a 14 dniem leczenia, charakteryzuje się znacznym obniżeniem liczby płytek (poniżej 100 000/ml), wywołane przez immunoglobuliny klasy G przeciwko kompleksowi heparyna - PF4, często dochodzi też do powikłań zakrzepowo-zatorowych w układzie żylnym i tętniczym oraz zaburzeń w funkcji nadnerczy, śmiertelność może sięgać 30% chorych.
W praktyce lekarskiej do dyspozycji mamy kilkanaście LMWH, min:
- sól sodowa ardeparyny (ardeparin)
- sól sodowa certpoaryny (certoparin)
- sól sodowa dalteparyny (dalteparin)
- sól sodowa enoksaparyny (enoxaparin)
- sól sodową nadroparyny (nadroparin)
- sól sodową rewiparyny (reviparin)
W Polsce zastosowanie znalazły : dalteparyna (Fragmin), enoksaparyna (Clexan) oraz nadroparyna (Fraxiparyna) różniące się masą cząsteczkowa a także aktywnością anty Xa.
Na podstawie licznych badań zebranych przez Tomkowskiego wynika, że podanie identycznej dawki mierzonej aktywnością anty Xa, różnych heparyn drobnocząsteczkowych wiąże się z różnym nasileniem działania przeciwzakrzepowego i może powodować odmienne efekty kliniczne. Wiąże się z tym spostrzeżeniem brak możliwości wymiennego stosowania LMHW.
Interesujące spostrzeżenia zawarł Hajduk opracowując metaanalizy wielu prób klinicznych obejmujących kilka tysięcy chorych z zakrzepicą żył głębokich kończyn dolnych. W badaniach wzięto pod uwagę: częstość nawrotów żchzz, powikłania krwotoczne, śmiertelność oraz liczbę niepowodzeń leczenia zżg. Stwierdzono znamienną statystycznie różnicę na korzyść LMWH w odniesieniu do ograniczenia częstości zżg a także zmniejszenie częstości nawrotów żchzz, poważnych krwawień i ogólnej śmiertelności.
Stwierdzono również, że heparyny drobno cząsteczkowe w dawkach dostosowanych do masy ciała oraz bez kontroli laboratoryjnej są skuteczniejsze i bezpieczniejsze w leczeniu zżg kończyn dolnych od heparyny standardowej podawanej dożylnie, w dawkach dostosowanych do APTT.
Wykład z fizjologii - Hemostaza
- 2 -
www.stomka.prv.pl
Wyszukiwarka
Podobne podstrony:
ZABURZENIA HEMOSTAZY, Wykłady
2. PATOFIZJOLOGIA - ZABURZENIA HEMOSTAZY, Wykłady
ZABURZENIA HEMOSTAZY, Wykłady
Napęd Elektryczny wykład
wykład5
Psychologia wykład 1 Stres i radzenie sobie z nim zjazd B
Wykład 04
geriatria p pokarmowy wyklad materialy
ostre stany w alergologii wyklad 2003
WYKŁAD VII
Wykład 1, WPŁYW ŻYWIENIA NA ZDROWIE W RÓŻNYCH ETAPACH ŻYCIA CZŁOWIEKA
Zaburzenia nerwicowe wyklad
Szkol Wykład do Or
Strategie marketingowe prezentacje wykład
Wykład 6 2009 Użytkowanie obiektu
wyklad2
wykład 3
więcej podobnych podstron