
CHEMIA 1

Termochemia
◦
Równanie stanu gazu doskonałego i rzeczywistego
◦
Podstawy termodynamiki
◦
Prawo Hessa
Wykład 11

Równanie stanu gazu
doskonałego
i rzeczywistego
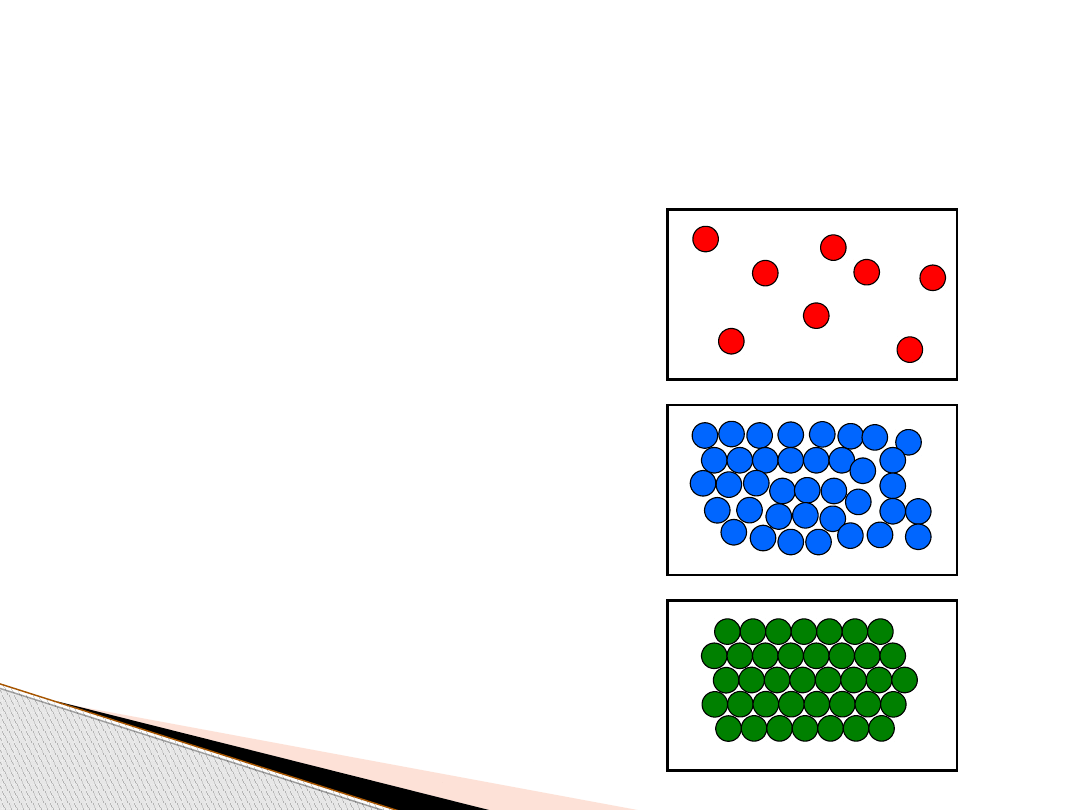
Stany skupienia materii –
przypomnienie
1. Gaz (wypełnia całkowicie
objętość naczynia w którym
się znajduje)
2. Ciecz (przybiera kształt
naczynia w którym się
znajduje)
3. Ciało stałe (zachowuje swój
własny kształt)
2-wymiarowy model
uporządkowania w skali atomowej
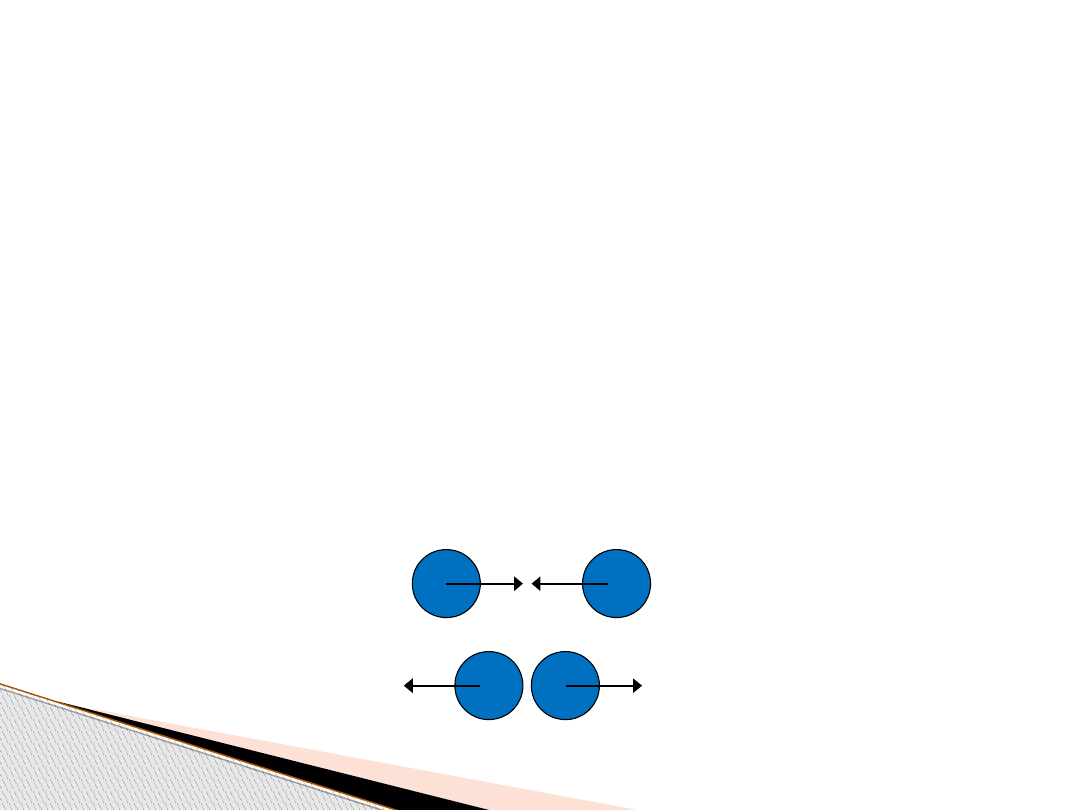
Gazy doskonałe – założenia teorii
1. Cząsteczki gazu zachowują się jak jednorodne kule, doskonale
sprężyste
2. Objętość własna, którą zajmują może być zaniedbana
3. Cząsteczki znajdują się w ciągłym ruchu postępowym,
zderzając się miedzy sobą doskonale sprężyście
4. Pomiędzy zderzeniami cząstki poruszają się ruchem
prostoliniowym
5. Generalnie, ruch cząsteczek nie ma uprzywilejowanego
kierunku
zderzenie doskonale sprężyste

Gazy doskonałe – parametry
Stan gazowy można określić parametrami:
p
– ciśnienie
jednostki: Pa (paskal), atm (atmosfera fizyczna), 1 Pa = 1 N/1
m
2
,
1 atm = 101325 Pa = 1013 hPa ~ 0,1 MPa
T
– temperatura
jednostki: K (kelwin),
o
C (stopień Celsjusza),
0
o
C = 273,15 K
V
– objętość
jednostki: m
3
, cm
3
, litr,
1 m
3
= 1000 litrów, 1 litr = 1000 cm
3
= 1 dm
3
n
– liczba moli
1mol zawiera N
A
cząsteczek
N
A
– liczba Avogadra = 6,022·10
23
mol
-1

Gazy doskonałe – historia
1662r – prawo Boyle’a:
p·V = const
w danej temperaturze
1802r – prawo Gay-Lussaca:
V = V
0
(1 + a·T)
dla danego ciśnienia (T w stopniach Celsjusza a
V
0
to objętość w temp. 0
o
C, a oznacza współczynnik
rozszerzalności cieplnej)
jeśli oznaczymy a = 1/T
0
to V = V
0
(1+T/T
0
), T
0
+ T daje
temperaturę w Kelwinach, więc V = V
0
T/T
0
, w efekcie
otrzymujemy:
V/T = const
1811r – prawo Avogadra:
Przy tym samym ciśnieniu i w tej samej temperaturze jednakowe
objętości różnych gazów zawierają taką samą liczbę cząsteczek.
Równanie stanu gazu doskonałego
(Clapeyron, 1834)
T
R
n
V
p
ciśnienie
[Pa]
objętość
[m
3
]
ilość moli
[mol]
stała gazowa
8,314 [J·mol
-1
·K
-1
]
temperatura
[K]
Gaz doskonały (izotermy, T = const)
0,0
0,2
0,4
0,6
0,8
1,0
0,0
5,0x10
4
1,0x10
5
1,5x10
5
2,0x10
5
Przykładowe izotermy gazu doskonałego (298K)
ci
śn
ie
ni
e
[P
a]
objętość [m
3
]
1 mol
5 moli
10 moli
p
1
V
1
= p
2
V
2
= const
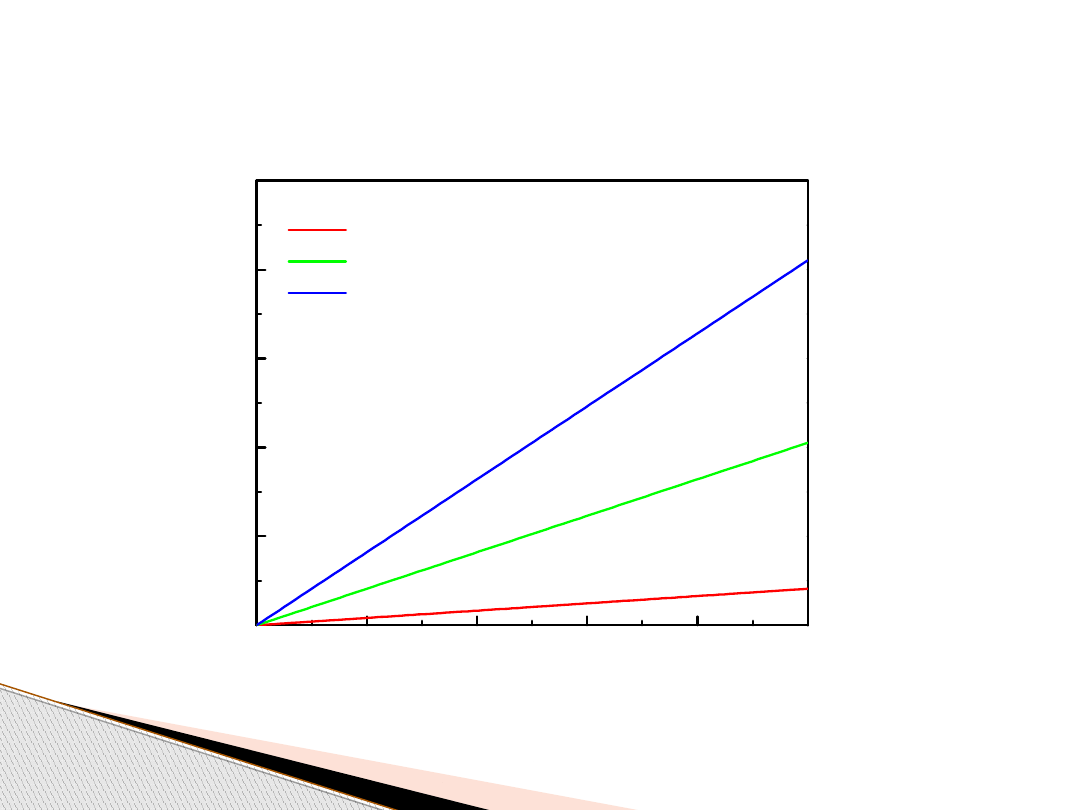
Gaz doskonały (izobary, p = const)
0
200
400
600
800
1000
0,0
0,2
0,4
0,6
0,8
1,0
Przykładowe izobary gazu doskonałego (p = 1013hPa = 1atm)
ob
ję
to
ść
[m
3
]
temperatura [K]
1 mol
5 moli
10 moli
V
1
/T
1
= V
2
/T
2
= const
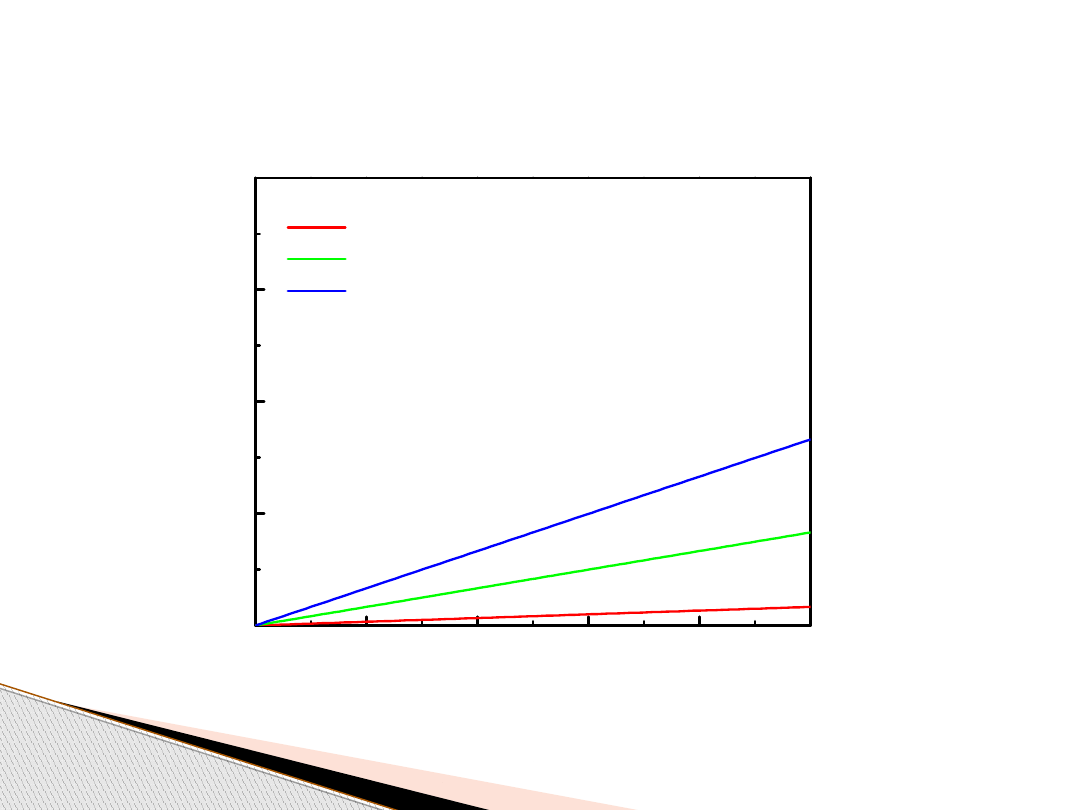
Gaz doskonały (izochory, V = const)
0
200
400
600
800
1000
0,0
5,0x10
4
1,0x10
5
1,5x10
5
2,0x10
5
Przykładowe izochory gazu doskonałego (V = 1m
3
)
ci
śn
ie
ni
e
[P
a]
temperatura [K]
1 mol
5 moli
10 moli
p
1
/T
1
= p
2
/T
2
= const

Gazy doskonałe – przykładowe
zadanie
Obliczyć objętość 1 mola gazu doskonałego w temperaturze 0
o
C
znajdującego się pod ciśnieniem 1 atm:
p·V = n·R·T
V = n·R·T/p
V = 1 mol · 8,314 J·mol
-1
·K
-1
· 273,15 K / 101300 Pa
V = 22,4 dm
3
= 0,0224 m
3
Co oznacza, że w warunkach normalnych (1 atm, 0
o
C) 1 mol gazu
doskonałego zajmuje objętość 22,4 dm
3
.

Gazy doskonałe – elementy
kinetycznej teorii gazów
Załóżmy, że gaz doskonały znajduje się w sześciennym
pojemniku o boku
a
.
Ciśnienie
p
wywierane na ściankę to całkowita zmiana pędu
cząsteczek zderzających się z daną ścianką liczona dla czasu 1
sekundy oraz przypadająca na jednostkową powierzchnię.
Pęd cząstki w kierunku osi
x
wynosi
m·u
x
gdzie
m
to masa
cząstki a
u
x
to składowa x-owa prędkości cząstki.
Zmiana pędu po zderzeniu doskonale sprężystym =
2m·u
x
.
Ilość zderzeń w ciągu sekundy z daną ścianką wynosi
u
x
/2a
.
Zmiana pędu podczas jednej sekundy dla 1 cząstki =
2m·u
x
·u
x
/2a
=
m·ū
x
2
/a
gdzie
ū
x
2
to średni kwadrat prędkości składowej x-owej.

Gazy doskonałe – elementy
kinetycznej teorii gazów
Wobec tego dla
N
cząstek wywierane ciśnienie na ściankę
wynosi:
bo składowe dla różnych kierunków są sobie równe, a
prawdopodobieństwo ruchu wzdłuż jednej osi wynosi 1/3, pole
ścianki wynosi
a
2
.
Dla jednego mola cząsteczek ciśnienie wynosi:
Wiedząc, że
p·V = n·R·T
i
n = 1
:
2
2
a
a
u
m
N
3
1
p
V
u
m
N
3
1
p
2
A
2
A
u
m
N
3
1
T
R

Gazy doskonałe – elementy
kinetycznej teorii gazów
Energia kinetyczna dana jest wzorem
E = 0,5m·ū
x
2
, a więc:
k
B
= 1,381·10
-23
J·K
-1
to jedna z głównych stałych fizycznych
(stała Boltzmanna).
Podstawowy wniosek:
temperatura jest miarą energii
kinetycznej cząsteczek
.
Na każdy z kierunków w przestrzeni (stopień swobody) dla
jednej cząsteczki (jednoatomowego) gazu doskonałego
E =
0,5k
B
·T
, a dla jednego mola:
E = 0,5R·T
T
k
2
3
2N
T
3R
E
E
N
3
2
T
R
B
A
A
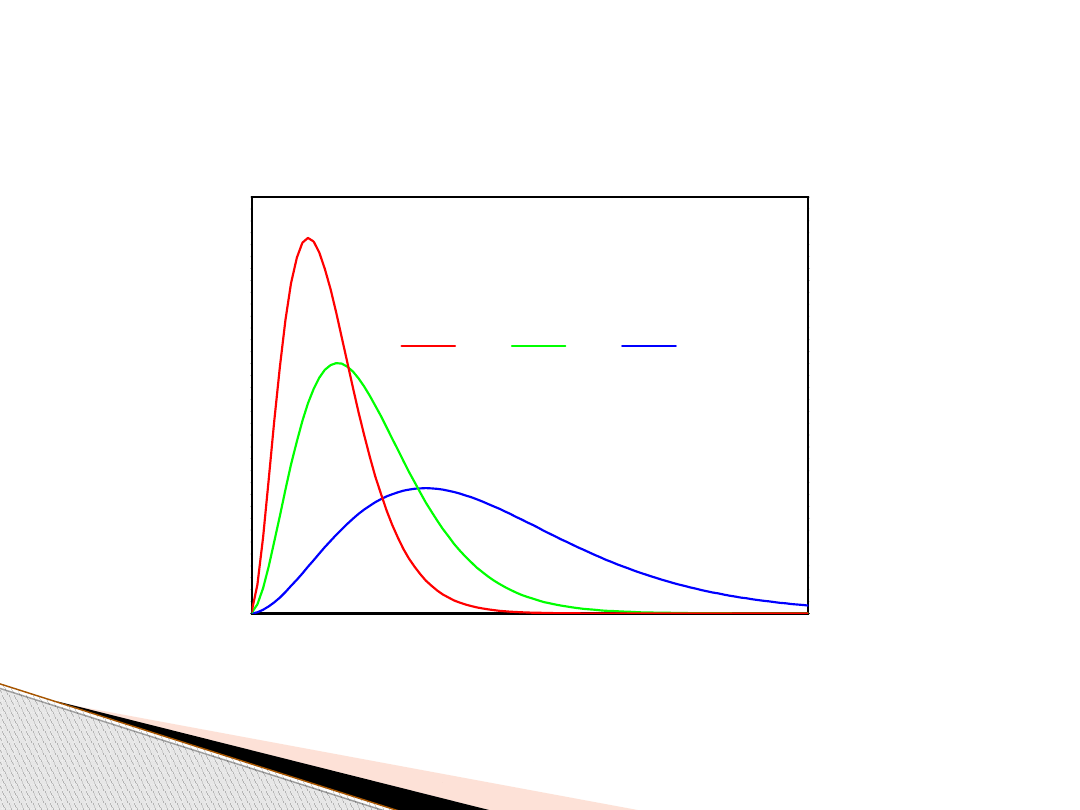
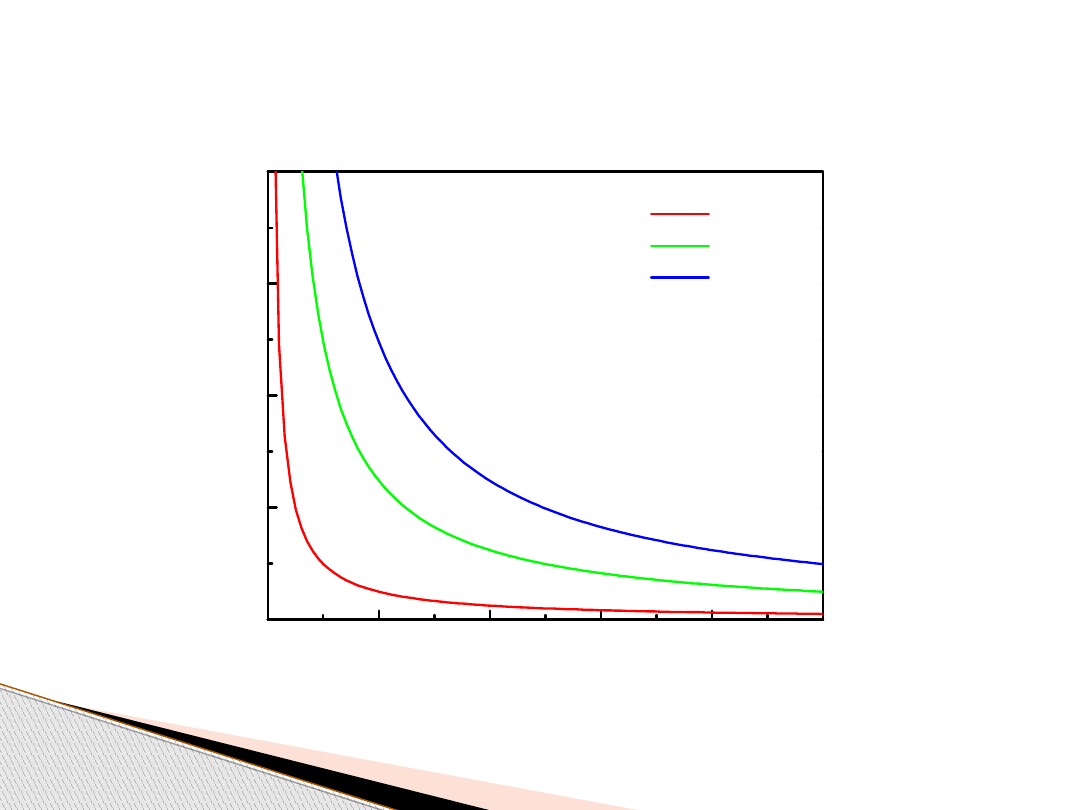
Gazy doskonałe – elementy
kinetycznej teorii gazów
Rozkłady prędkości cząsteczek gazu
dla różnych temperatur (na postawie rozkładu Boltzmanna)
ilo
ść
c
zą
st
ec
ze
k
o
da
ne
j p
rę
dk
oś
ci
prędkość cząsteczek gazu
T
1
<
T
2
<
T
3

Gazy rzeczywiste
Obserwowane parametry mikroskopowe:
1. Średnia prędkość cząsteczek
100-1000 m·s
-1
2. Średnia droga pomiędzy zderzeniami
100-1000 Å
(1 Å = 10
-10
m)
3. Częstość zderzeń
10
10
s
-1
Różnice pomiędzy gazami rzeczywistymi, a opisem prostej teorii
gazów doskonałych wynikają głównie z:
• wpływu oddziaływań międzycząsteczkowych w gazie,
• niezaniedbywalnej objętości własnej cząsteczek gazu.

Gazy rzeczywiste
Odstępstwa będą tym większe im niższa temperatura (małe
k
B
·T) oraz im wyższe ciśnienie (mniejsze odległości między
cząsteczkami).
Poprawione przez van der Waalsa równanie stanu dla gazów ma
postać:
gdzie:
a
– stała uwzględniająca oddziaływanie pomiędzy cząsteczkami
(m
3
·Pa·mol
-2
),
b
– stała uwzględniająca rozmiar cząsteczek (m
3
·mol
-1
).
T
R
n
b
n
V
V
a
n
p
2
2

Podstawy termodynamiki i
prawo Hessa

Termodynamika – wstęp i definicje
Układ
– to wydzielona (umownie) przestrzeń wraz ze
znajdującymi się tam substancjami, w której będzie zachodził
interesujący nas proces.
Parametry stanu
– to zmienne określające stan
(termodynamiczny) układu.
•
Parametry ekstensywne
– to parametry, które zależą od
wielkości układu np. objętość układu
V
, masa układu
m
,
energia
E
.
•
Parametry intensywne
– ich wielkość nie zależy od wielkości
układu np. temperatura
T
, ciśnienie
p
.
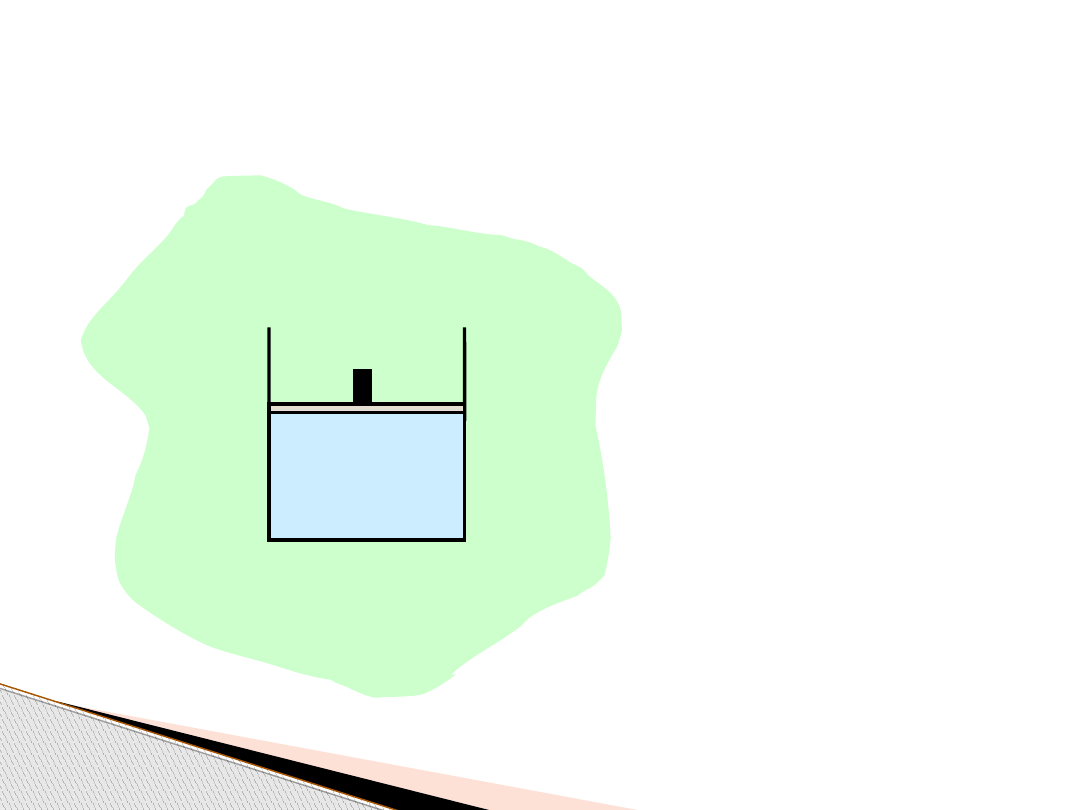
Termodynamika – wstęp i definicje
układ
V, p, T, n
otoczenie układu
Układ izolowany
– nie ma wymiany
masy ani energii (ciepła) z otoczeniem
Układ zamknięty
– nie ma wymiany
masy z otoczeniem
Układ otwarty
– jest wymiana
masy z otoczeniem

Termodynamika – I zasada
W układzie izolowanym energia wewnętrzna jest zachowana. U
= const. ΔU = 0.
Energia wewnętrzna jest sumą wszystkich rodzajów energii
układu: np. energii kinetycznej, oscylacji i rotacji cząstek
układu, energii elektronów, wiązań chemicznych, oddziaływań
międzycząsteczkowych, itd...
Zmiana energii wewnętrznej układu zamkniętego może nastąpić
na sposób ciepła albo na sposób pracy. DU = Q + W.
Q
– ciepło,
W
– praca. Q > 0 – do układu przepływa energia na
sposób ciepła, Q < 0 – układ traci energię na sposób ciepła. W >
0 – do układu przepływa energia na sposób pracy, W < 0 – układ
traci energię na sposób pracy.
Energia wewnętrzna jest funkcją stanu.
Jej zmiany zależą
jedynie od stanu początkowego i końcowego (przed i po dla
dowolnego procesu w układzie), nie zależą natomiast od drogi
tego procesu.
DU = U
2
– U
1
(w ogólnym przypadku Q i W
funkcjami stanu nie są!).
Dla tzw. procesu elementarnego (dowolnego, nieskończenie
małego):
dU = Q
el
+ W
el
(U posiada różniczkę zupełną, Q i W nie).
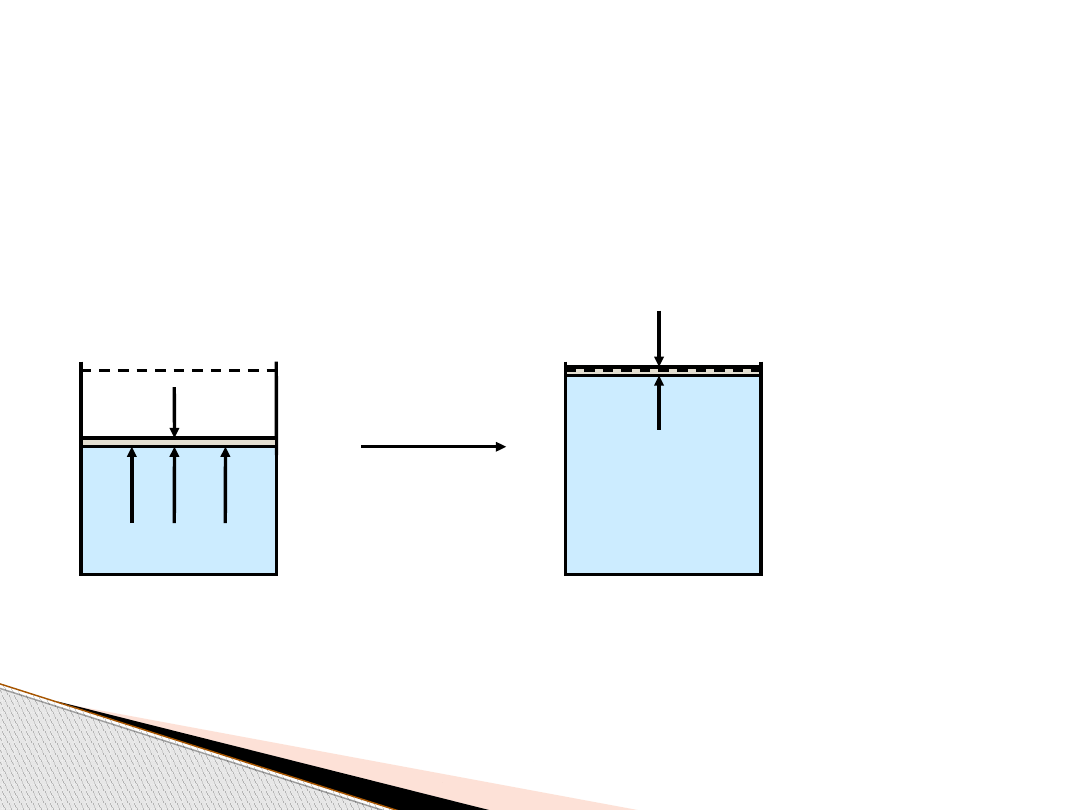
P
rocesy odwracalne i nieodwracalne
Nieodwracalne rozprężanie gazu
: ciśnienie zewnętrzne
jest stałe,
ciśnienie wewnętrzne maleje z początkowej wartości do
wartości ciśnienia
zewnętrznego
p
z
= const
p
w
p
z
V
1
V
2
p
w
p
z
V
2
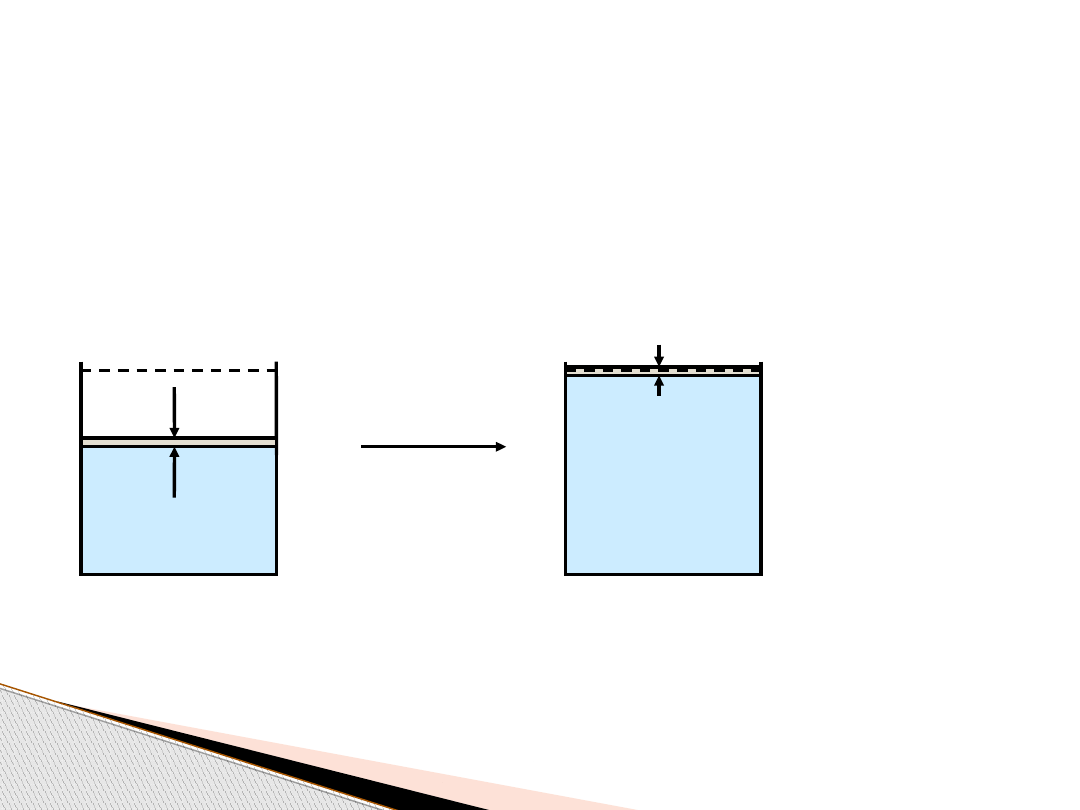
P
rocesy odwracalne i nieodwracalne
Odwracalne rozprężanie gazu
: ciśnienie zewnętrzne w
każdym momencie
równoważy ciśnienie wewnętrzne, oba ciśnienia
równocześnie maleją.
p
z
= p
w
p
w
p
z
V
1
V
2
p
w
p
z
V
2
W procesach odwracalnych przemiana następuje poprzez kolejne stany
równowagi.
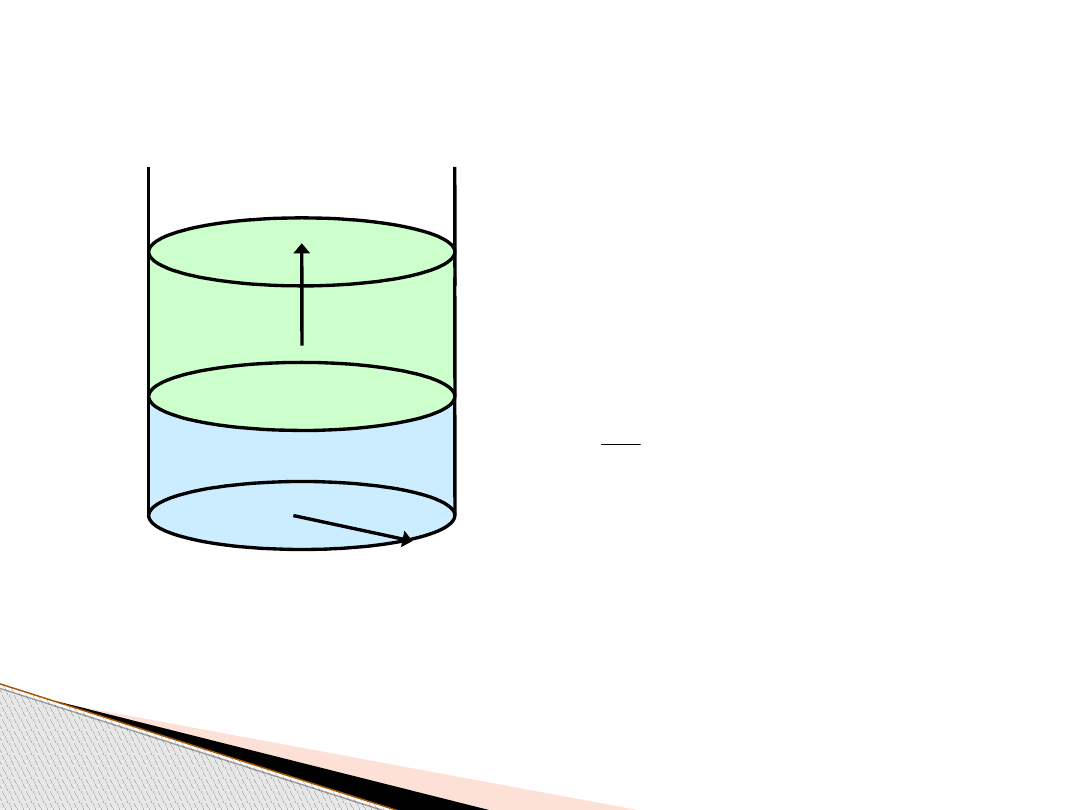
Praca objętościowa gazu – W
r
V
1
V
2
f
p
z
< p
w
, p
z
= const
f – siła działająca na tłok
pr
2
– pole powierzchni tłoka
V
p
Δl
πr
p
W
Δl
πr
V
V
ΔV
πr
f
p
Δl
f
W
2
2
1
2
2
Jeśli układ traci energię (wykonuje pracę lub oddaje
ciepło) to znak pracy
i ciepła jest ujemny:
W
el
= –p·dV
. A więc:
dU = Q
el
–
p·dV

P
rocesy odwracalne i nieodwracalne
Nieodwracalne rozprężanie gazu doskonałego (p
z
< p
w
, p
z
=
const, T = const):
Odwracalne rozprężanie gazu doskonałego (p
z
= p
w
i maleją w
trakcie procesu,
T = const):
2
1
2
1
V
V
1
2
z
z
V
V
el
V
p
)
V
(V
p
dV
p
W
W
2
1
2
1
V
V
V
V
1
2
el
el
V
V
ln
T
R
n
V
dV
T
R
n
W
W
dV
V
T
R
n
dV
p
W
T
R
n
V
p

I zasada termodynamiki dla procesów
izochorycznych
ΔU = Q + W
dU = Q
el
– p·dV
w procesach izochorycznych V = const, z czego wynika że dV =
0, a więc:
dU = Q
el
ΔU = U
2
– U
1
= Q
V
Ciepło reakcji (chemicznej) przebiegającej w stałej objętości jest
równe różnicy energii wewnętrznej w stanie końcowym i
początkowym – czyli Q
V
jest funkcją stanu.

I zasada termodynamiki dla procesów
izobarycznych
Wprowadźmy tzw. entalpię
H
jako:
H = U + p·V
dH = dU + p·dV +V·dp
dU = Q
el
– p·dV
dH = Q
el
+ V·dp
co jest równoważne z I zasadą termodynamiki dla procesu
izobarycznego.
Entalpia jest funkcją stanu.
Oznacza to, że jej zmiany zależą
jedynie od stanu początkowego i końcowego.
Dla procesu izobarycznego p = const co daje, że dp = 0 czyli:
dH = Q
el

Prawo Hessa
DH = H
2
– H
1
= Q
p
Ciepło reakcji (chemicznej) przebiegającej przy stałym ciśnieniu
jest równe różnicy entalpii w stanie końcowym i początkowym –
czyli Q
p
jest funkcją stanu.
Prawo Hessa:
Ciepło reakcji chemicznej (w warunkach izobarycznych lub
izochorycznych) nie zależy od drogi (reakcji), a jedynie od stanu
początkowego i końcowego reakcji. Czyli ciepło w takich
warunkach jest funkcją stanu.
Dla dowolnej reakcji chemicznej:
substraty ↔ produkty
w stanie
początkowym mamy substraty, a w końcowym produkty.
W
warunkach
izochorycznych
(
lub izobarycznych
) jeśli od sumy
energii wewnętrznych
(
entalpi
i) produktów odjąć sumę
energii
wewnętrznych
(
entalpii
) substratów otrzymamy wyrażenie na
ciepło reakcji
:
i
i
)
(substraty
i
i
(produkty)
i
i
V
ΔU
n
ΔU
n
Q
ΔU
i
i
)
(substraty
i
i
(produkty)
i
i
p
ΔH
n
ΔH
n
Q
ΔH
Document Outline
- Slide 1
- Wykład 11
- Slide 3
- Stany skupienia materii – przypomnienie
- Gazy doskonałe – założenia teorii
- Gazy doskonałe – parametry
- Gazy doskonałe – historia
- Równanie stanu gazu doskonałego (Clapeyron, 1834)
- Gaz doskonały (izotermy, T = const)
- Gaz doskonały (izobary, p = const)
- Gaz doskonały (izochory, V = const)
- Gazy doskonałe – przykładowe zadanie
- Gazy doskonałe – elementy kinetycznej teorii gazów
- Gazy doskonałe – elementy kinetycznej teorii gazów
- Gazy doskonałe – elementy kinetycznej teorii gazów
- Gazy doskonałe – elementy kinetycznej teorii gazów
- Gazy rzeczywiste
- Gazy rzeczywiste
- Slide 19
- Termodynamika – wstęp i definicje
- Termodynamika – wstęp i definicje
- Termodynamika – I zasada
- Procesy odwracalne i nieodwracalne
- Procesy odwracalne i nieodwracalne
- Praca objętościowa gazu – W
- Procesy odwracalne i nieodwracalne
- I zasada termodynamiki dla procesów izochorycznych
- I zasada termodynamiki dla procesów izobarycznych
- Prawo Hessa
Wyszukiwarka
Podobne podstrony:
Wykład Chemia kwantowa 11
Chemia fizyczna wykład 11
chemia analityczna wyklad 11 i 12
wykladChK-11, Chemia UŁ, teoretyczna wykład
11.jony, AGH różne, chemia wykłady
wyklad 11, Studia, Chemia Ogólna, Chemia Ogólna - notatki z wykładów, wykłady
Chemia budowlana Wykład 11
Wykład Chemia kwantowa 11
Chemia fizyczna wykład 11
chemia analityczna wyklad 11 i 12
wyklad 11
WYKŁAD 11 SPS 2 regulatory 0
wyklad 11 toksyczno niemetali
BUD OG wykład 11 3 Geosyntetyki
Psychometria 2009, Wykład 11, Inwentarz MMPI
Chemia wyklad I i II (konfiguracja wiÄ…zania Pauling hybrydyzacja wiazania pi i sigma)
więcej podobnych podstron