
1
semestr VI (studia I stopnia)
wersja: 10. lutego 2010
Metody badań materiałów – laboratorium
________________________________________________________________________________
ćwiczenie nr 5:
Oznaczanie wielkości cząstek w dyspersjach metodą DLS oraz
oznacza
nie ciężarów cząsteczkowych polimerów metodą GPC
prowa
dzący:
dr inż. Ireneusz Wielgus
________________________________________________________________________________
Oznaczanie ciężarów cząsteczkowych metodą chromatografii żelowej
Wprowadzenie
Polimery naturalne i otrzymywane przez człowieka, poza specyficznymi wyjątkami, są zawsze mie-
szaniną homologów o różnej długości łańcucha, a zatem różniących się ciężarem cząsteczkowym.
Dlatego w ich przypadku podaje się średnie wartości ciężaru cząsteczkowego, liczone według róż-
nych kryteriów: M
n
, M
w
, M
z
, M
z+1
. Są to ważne parametry, gdyż wiążą się z właściwościami użyt-
kowymi i przetwórczymi tworzywa sztucznego (wytrzymałość mechaniczna i termiczna, twardość,
wrażliwość na działanie rozpuszczalników, temperatura płynięcia w maszynie przetwórczej).
Do oznaczenia
średniej wartości ciężaru cząsteczkowego można korzystać z różnych metod pomia-
rowych
, jednak wyznaczenie rozkładu masy (czyli podanie udziału procentowego łańcuchów o kon-
kretnej długości w badanej próbce) jest w większości przypadków bądź niemożliwe, bądź szczegól-
nie pracochłonne. Chromatografia żelowa, zwana też chromatografią filtracji żelowej (GPC – Gel
Permeation Chromatography
), albo chromatografią wykluczania (SEC – Size Exclusion Chromato-
graphy) jest w tym
przypadku skuteczną i względnie prostą techniką analityczną.
Zasada chromatografii żelowej
Wype
łnienie kolumny GPC składa się z porowatych ziaren, tworzących przestrzenie między-
ziarnowe, przez które p
łynie ciecz, oraz z ogromnej liczby otwartych mikroporów (kanalików)
wewn
ątrz ziaren wypełnienia. W przestrzeni mikroporów ciecz praktycznie nie płynie a cząstki
polimeru dostają się tam spontanicznie drogą dyfuzji, której prędkość uzależniona jest od ich
średnicy hydrodynamicznej, związanej z wielkością cząsteczki i średnim ciężarem cząsteczkowym.
Po wprowadzeniu roztworu
próbki na kolumnę zaczyna się proces jej wymywania za pomocą stru-
mienia odpowiednio dobranego rozpuszczalnika (eluentu). Najwi
ększe cząsteczki, o bardzo dużych
promieniach hydrodynamicznych (i bardzo wysokich masach molekularnych), nie s
ą w stanie
wnikn
ąć do żadnych porów wewnątrz wypełnienia kolumny i przepływają szybko przez przestrzeń
w której p
łynie eluent, tzn. przestrzeń międzyziarnową. Mówimy wówczas, że takie molekuły są
wykluczane z kolumny. Nie mo
żna opisać rozkładu ich ciężarów cząsteczkowych, gdyż nie uległy
one rozdziałowi (opuszczają kolumnę jako mieszanina).
Nie mo
żna także w warunkach chromatografii żelowej scharakteryzować rozkładu masy cząstecz-
kowej najmniejszych cz
ąsteczek, które wnikają do wszystkich porów wypełnienia kolumny i
opuszczają ją razem w mieszaninie jako jeden sygnał na elugramie. Do ich rozdzielania trzeba
zastosowa
ć wypełnienie o mniejszych średnicach porów.
Substancje o po
średnich rozmiarach, a więc o pośrednich masach cząsteczkowych i takichże warto-
ściach współczynnika dyfuzji, wnikają do porów na głębokość odpowiadająca ich rozmiarom, a
potem wskutek ruchów Browna opuszczają ziarna wypełnienia i są unoszone przez eluent w kie-
runku wylotu kolumny. Proces wnikania i opuszczania kanalików żelu powtarza się wielokrotnie,
st
ąd sygnały retencji przyjmują wartości pomiędzy pikami dla cząstek wykluczanych a bardzo
2
małych. W konsekwencji uzyskuje się określoną zależność funkcyjną między wartością logarytmu
masy molekularnej rozdzielanych cz
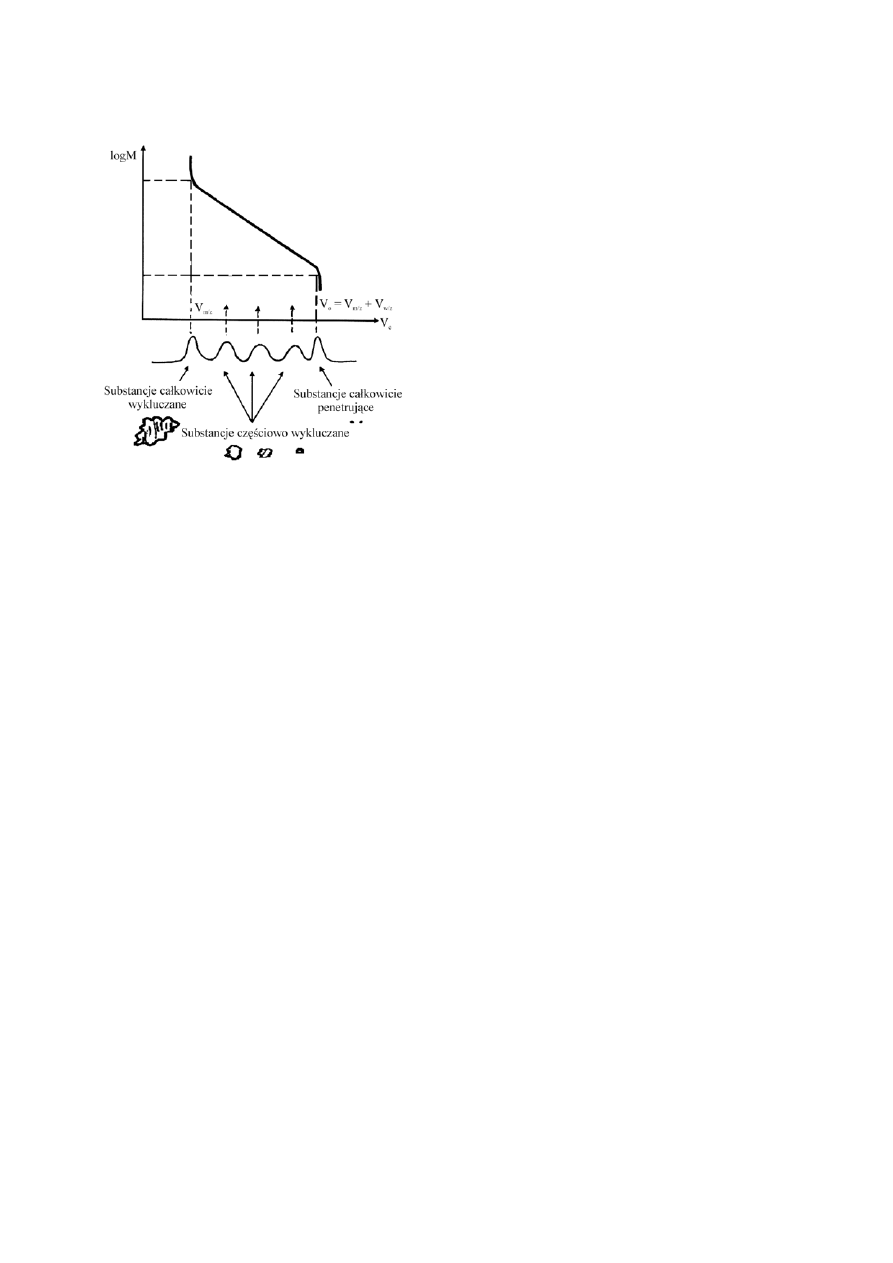
ąsteczek i objętością ich elucji z kolumny (Rys. 1.)
Rys. 1.
Zależność logarytmu masy cząsteczkowej od objętości elucji (log M
w
= f(V
e
) podczas
badania rozkładu masy cząsteczkowej mieszaniny metodą chromatografii żelowej.
Należy podkreślić w tym miejscu, że separacja cząstek na kolumnie żelowej jest procesem czysto
mechanicznym, podobnym do działania sita. W warunkach chromatografii żelowej dąży się do
wyeliminowania jakichkolwiek oddzia
ływań sorpcyjnych, mających kluczowe znaczenie w chro-
matografii cieczowej i gazowej (oddziaływania dipolowe, różnice polarności, tworzenie wiązań
wodorowych) mi
ędzy powierzchnią wypełnienia kolumny i cząsteczkami rozdzielanych substancji.
W przeciwnym razie
oddziaływania konkurencyjne wobec prostego mechanizmu wykluczania będą
prowadzić do pogorszenia selektywności kolumny, a w skrajnych przypadkach do całkowitego
zafałszowania wyników – cząstki silniej wiążące się z wypełnieniem będą opuszczać kolumnę
później, niezależnie od wielkości.
Kolumn
ę żelową dobiera się w ten sposób, aby rozkład wielkości porów był dostosowany do roz-
k
ładu wartości hydrodynamicznych średnic cząsteczek rozdzielanych substancji, a więc do rozkładu
masy cz
ąsteczkowej badanej próbki. Wypełnienia kolumn do chromatografii żelowej przygotowuje
si
ę w ten sposób, że rozkład średnic porów mieści się w określonym wąskim zakresie i jest podany
w
świadectwie jakości kolumny. W praktyce częste jest łączenie kolumn o różnych zakresach roz-
k
ładu wielkości porów. Wskazane jest zachowanie następującej kolejności: dozownik – kolumna o
najwi
ększych porach – kolumna o pośrednich porach – kolumna o najmniejszych porach – detektor.
Alternatyw
ą jest zastosowanie kolumny wypełnionej mieszaniną ziaren o różnych zakresach wiel-
ko
ści porów (tzw. kolumny typu „mix” lub „mixed bed”). Stosowanie mieszanych wypełnień jest
mniej korzystne ze wzgl
ędu na poszerzenie i rozmycie sygnałów w detektorze, lecz ze względu na
ni
ższy koszt kolumny mix bywają jednak często stosowane dla wstępnego oszacowania zakresu mas
cz
ąsteczkowych.
Faza stacjonarna w chromatografii żelowej
Rodzaje faz stacjonarnych w chromatografii żelowej (ze względu na oddziaływanie z eluentem):
1) Pęczniejące – (wymagają przygotowania – spęcznienia). Zmiana wartości przepływu (i w kon-
sekwencji ciśnienia w kolumnie) bardzo silnie wpływa na opory przepływu i zmienia warunki
wymywania.

3
2) Twarde –
nieściśliwe do pewnej wartości ciśnienia. Są to zazwyczaj silnie usieciowane kopoli-
mery, albo wypełnienia zawierające żel krzemionkowy, szkło porowate, tlenek cyrkonu lub inne
trwałe sorbenty. Ze względu na silne oddziaływania polarne wypełnienia te mają dezaktywowaną
powierzchnię, np. w procesie silanizacji (reakcja ze związkami krzemoorganicznymi blokuje silnie
polarne grupy –
OH i =O na powierzchni wypełnienia).
Wypełnieniem najczęściej wykorzystywanym do rozdzielania nisko- i średniopolarnych polimerów
jest kopolimer styrenu i diwinylobenzenu, przestrzennie usieciowany, o kulistych ziarnach wielko-
ści 5 mikrometrów, będący praktycznie sorbentem twardym.
W przypadku stosowania chromatografii żelowej do rozdzielania biopolimerów wykorzystuje się
wypełnienia o znacznie większej polarności, np. żele dekstranowe, agarozowe, akrylowe. Ze wzglę-
du na silną skłonność do adsorpcji na powierzchni żelu poza jej dezaktywacją (np. przy użyciu
metylosilanu) dodatkowo w charakterze fazy ruchomej stosuje się wodne roztwory buforów z
dodatkiem EDTA, mocznika, etanolu itp.
Eluenty w chromatografii żelowej
Chromatografia żelowa jest wykonywana w dwóch podstawowych układach separacyjnych:
1) W warunkach niewodnych, z zastosowaniem takich eluentów, jak: THF, dioksan, czterochloro-
etylen, chlorobenzen, dichlorobenzen, toluen, ksylen i tym podobne. Jako faza stacjonarna stoso-
wane s
ą odporne na działanie organiki kopolimery styrenu i diwinylobenzenu, albo poliestry. Jest to
układ do badań rozkładu masy cząsteczkowej polimerów nisko i średnio polarnych, rozpuszczal-
nych w rozpuszczalnikach niepolarnych (np. polistyrenu w toluenie), a
także lipidów (tłuszczow-
ców), fosfolipidów, wosków itp. niepolarnych substancji pochodzenia naturalnego.
2) W roztworach wodnych, albo z zastosowaniem silnie polarnych niewodnych eluentów, takich
jak dimetyloformamid, metanol, acetonitryl i ich mieszani
ny z wodą i z wodnymi roztworami soli,
kwasów i zasad, z zastosowaniem wypełnień kolumn wykonanych z polarnych materiałów, takich
jak polidekstrany i inne policukry, poli
węglany, szkła porowate, silanizowany żel krzemionkowy.
Takie układy stosowane są do rozdzielania, a także do charakteryzowania rozkładu ciężaru czą-
steczkowego polimerów polarnych (np. poli(glikol etylenowy) w wodzie), a
zwłaszcza biopolime-
rów (policukry, białka, nukleotydy).
Warunkiem stosowania chromatografii do oznaczania rozkładu ciężarów cząsteczkowych, poza
oczywistym warunkiem całkowitej rozpuszczalności próbki w eluencie, jest taka siła elucyjna fazy
ruchomej, aby eliminowała ona adsorpcję cząsteczek polimeru na powierzchni wypełnienia ko-
lumny. Dla przypomnienia w chromatografii
cieczowej LC i HPLC siłą napędową procesu
rozdzielania jest właśnie wykorzystanie różnicy powinowactwa składników badanej substancji do
fazy ruchomej i stacjonarnej wywołana różnicami polarności. W przypadku średnio polarnych
polimerów syntetycznych w char
akterze eluentu stosuje się najczęściej tetrahydrofuran (THF),
będący dobrym rozpuszczalnikiem wielu polimerów i jednocześnie substancją dobrze minima-
lizującą adsorpcję nawet do żelu krzemionkowego a także oddziaływania hydrofobowe. W nie-
których przypadk
ach stosuje się chloroform, który wymaga specjalnej uwagi ze względu na
działanie korodujące (wobec czynników utleniających niszczy stal nierdzewną).
Polimery niepolarne są na ogół trudno rozpuszczalne. Do ich rozpuszczania i elucji wykorzystuje
się rozpuszczalniki takie jak toluen, ksylen, dekalina i podwyższoną temperaturę.
W chromatografii żelowej biopolimerów w roli eluentu stosuje się roztwór buforowy będący
dobrym rozpuszczalnikiem polimeru, a jednocześnie zapewniający minimalne oddziaływania
sorpcyjne. Niekiedy dodaje się acetonitryl CH
3
CN, lub alkohol izopropylowy, aby ograniczyć
oddziaływania hydrofobowe z powierzchnią fazy stacjonarnej.

4
Problemy i
zakłócenia w czasie pracy
Jeśli polimer rozpuszcza się tylko na gorąco, np. polietylen w ksylenie, niezbędne jest termostato-
wanie kolumny, zaworu dozującego i, o ile to możliwe, detektora. Należy zwrócić uwagę aby rów-
nież przewody łączące miały odpowiednia izolację termiczną.
Inne problemy i zak
łócenia, które trzeba brać pod uwagę:
– Mo
żliwość oddziaływań sorpcyjnych rozdzielanych polimerów.
–
Pęcznienie i kurczenie się ziaren wypełnień kolumn do chromatografii żelowej pod wpływem
ró
żnych eluentów. Pochopna zmiana eluentu w kolumnach do chromatografii żelowej może trwale
zniszczy
ć kolumnę!
– Okre
ślony stopień ściśliwości żelów (nawet twardych) – ograniczona odporność na ciśnienie.
– Wolna dyfuzja makromoleku
ł i konieczność ograniczenia prędkości przepływu eluentu w celu
niedopuszczenia do nadmiernego rozmycia pików.
– Przeszkadzanie sobie nawzajem przez du
że molekuły penetrujące wnętrze porów i zatykanie nie-
których porów przez cz
ąsteczki makropolimerów.
– Mo
żliwość polikondensacji makromolekuł w warunkach podwyższonego ciśnienia i wzrost cię-
żaru cząsteczkowego próbki w trakcie pomiaru lub wręcz „zaklejenie” kolumny przez powstający
polimer.
W przypadku stosowania kolumn do chromatografii
żelowej należy zachować ostrożność przy wy-
mianie eluentu na inny. Przede wszystkim trzeba przestrzega
ć ściśle zaleceń producenta kolumny.
Gdy dane takie s
ą niedostępne, należy przestrzegać ogólnej reguły, aby w przypadku kolumn prze-
znaczonych do rozdzielania nisko i
średnio polarnych polimerów syntetycznych nie zastosować
cieczy polarnych, takich jak woda, metanol, etanol, izopropanol, acetonitryl. W przeciwnym razie
mo
że dojść do nieodwracalnego skurczenia się ziaren wypełnienia kolumny i w konsekwencji
utraty zarówno jej selektywno
ści, jak i sprawności. Podobnie ma się sprawa z hydrofilowymi że-
lami mi
ękkimi i półtwardymi, których spęcznienia dokonuje się z użyciem roztworów wodnych. W
takim przypadku zastosowanie heksanu czy toluenu mo
że zniszczyć porowatą strukturę ziaren wy-
pe
łnienia, a aceton, albo THF mogą spowodować nadmierne spęcznienie sorbentu, a nawet go roz-
pu
ścić – zawartość kolumny wypłynie do detektora (i przy okazji trwale go uszkodzi !!!
Detektory wykorzystywane w chromatografii żelowej
W chromatografii żelowej stosuje się przede wszystkim detektory, których sygnał jest proporcjo-
nalny (choćby w przybliżeniu) nie tylko do stężenia badanych składników eluatu, ale i do masy
cząsteczkowej. Są to detektor refraktometryczny (RID) – najczęściej wykorzystywany, reaguje na
zmianę współczynnika załamania światła roztworu zawierającego rozpuszczoną substancję w sto-
sunku do czystego rozpuszczalnika,
detektor laserowy światła rozproszonego (LLSD – Laser Light
Scattering Detector), spektrometr mas do pracy w układzie HPLC-MS, detektor pomiaru lepkości.
Natomiast unika się stosowania detektora UV, gdyż – pomijając brak proporcjonalności sygnału od
ciężaru cząsteczkowego – większość polimerów nie absorbuje światła w tym zakresie.
Ostatnio w coraz większym zakresie znajduje zastosowanie detektor mierzący intensywność rozpro-
szenia przez cząstki badanej substancji wiązki światła laserowego pod różnymi kątami równocześ-
nie. Na tej podstawie można bezpośrednio obliczyć rozkład ciężarów cząsteczkowych polimeru bez
stosowania kalibracji za pomocą wzorców. Jednoczesne użycie połączonych szeregowo detektorów
RID, pomiaru lepkości oraz LLSD też umożliwia uniknięcie stosowania polimerów kalibracyjnych.
Sposób ten jest jednak kosztowny.

5
Kalibracja
Chromatograf żelowy podaje zawartość składnika o konkretnej wielkości promienia hydrodynami-
cznego w funkcji czasu wymywania a w praktyce w funkcji
objętości eluatu. Aby poznać odpo-
wiadającą mu wartość ciężaru cząsteczkowego konieczne jest wykonanie kalibracji, czyli zmierze-
nie czasów retencji kilku wzorców o znanym ciężarze cząsteczkowym i odpowiednie przeliczenie
skali.
Czyni się przy tym założenie, że ciężary cząsteczkowe związków chemicznych określonego
typu (tzn. o bardzo podobnej budowie chemicznej) s
ą proporcjonalne do ich promieni hydrodyna-
micznych
i szybkości retencji z kolumny (w odpowiedniej potędze). Kalibracja jest tym bardziej
wiarygodna, im bardziej cz
ąsteczki badanego polimeru i polimeru, który został wykorzystany do
kalibracji, zbli
żone są budową i kształtem (budowa liniowa, cykliczna, rozgałęziona). Teoretycznie
więc, aby zmierzyć rozkład ciężarów np. poliamidu-6,6, należy wykonać kalibrację stosując wzorce
z poliamidu, i to najlepiej PA-
66. W praktyce jednak handlowo dostępne są tylko nieliczne poli-
mery, a samodzielne przygotowanie wzorca np. metodą frakcjonowania przez wytrącanie nieroz-
puszczalnikiem jest niesłychanie żmudne i nie zawsze wykonalne.
W badaniach polimerów nisko i
średnio polarnych do kalibracji wykorzystywane są zazwyczaj
wzorce polistyrenowe, dla których stosunek maksymalnej i minimalnej
masy cząsteczkowej subs-
tancji M
max
/ M
min
zawiera się w granicach od 1,05 do 1,20. Z wzorca polistyrenowego korzysta się
również w przypadku, gdy nie dysponujemy odpowiednim polimerem. W takim przypadku ko-
nieczne jest zaznaczenie w opisie metody pomiaru „
ciężar wyznaczono w oparciu o wzorce PS”, zaś
wyznaczone wartości ciężarów nie muszą odpowiadać rzeczywistości – służą do porównań z
innymi próbkami
. Wzorzec polistyrenowy jest więc w pewnym sensie uniwersalny.
W zależności od możliwości i wymagań jakościowych stosuje się następujące metody prowadzenia
kalibracji:
– Kalibracja z wykorzystaniem serii odpowiednio dobranych wzorców o w
ąskich frakcjach masy
cz
ąsteczkowej. Jest to metoda najdokładniejsza.
– Kalibracja z wykorzystaniem jednej próbki polimeru polidyspersyjnego, ale o znanym rozk
ładzie
masy molekularnej.
Rozkład ten wyznacza się według metody z punktu poprzedniego albo np.
przez frakcjonowanie.
– Kalibracja uniwersalna – okre
śla się zależność:
log([
η] M
w
) = f (V
e
)
gdzie:
[
η] – jest ekstrapolowan
ą do zerowego stężenia lepkością dynamiczną wzorca masy molekularnej o
średniej masie cząsteczkowej M (tzw. graniczna liczba lepkościowa),
M
w
–
wagowo średni ciężar cząsteczkowy,
V
e
–
objętość elucji mierzona do maksimum albo do środka ciężkości piku.
Zasto
sowanie chromatografii żelowej
Główny zakres zastosowań chromatografii żelowej to rozdzielanie substancji różniących się cięża-
rem cząsteczkowym (i związaną z nim wielkością cząsteczek), a w pierwszej kolejności różnego
typu polimerów, a na mniejszą skalę smarów, olejów, kosmetyków. Poza badaniem składników
podstawowych (głównych) chromatografia żelowa wykorzystywana jest także do oznaczania za-
wartości dodatków modyfikujących i uszlachetniających, a zatem do oznaczania zawartości plasty-
fikatorów, antyutleniaczy, konserwantów, barwników. S
łuży wówczas do wyodrębnienia skład-
ników próbki, które ró
żnią się zasadniczo masą molową od pozostałych, np. plastyfikatora w
polimerze,
zagęszczacza w oleju smarowym, modyfikatora polimerowego w masie bitumicznej (np.
asfalcie drogowym) itp.

6
Inn
ą grupą zastosowań chromatografii żelowej jest przygotowanie próbek do oznaczania nisko-
cz
ąsteczkowych zanieczyszczeń w żywności, produktach naftowych i w środowisku. Zasada
post
ępowania polega na wydzieleniu frakcji o niskich wartościach ciężaru cząsteczkowego i pod-
daniu wyizolowanej frakcji analizie z zastosowaniem innego rodzaju uk
ładu chromatograficznego,
najcz
ęściej w układzie odwróconych faz. Można w ten sposób przygotować próbki do oznaczania
wielopier
ścieniowych węglowodorów aromatycznych, pestycydów, toksycznych związków metalo-
organicznych (tetraetyloołów) i podobnych im zanieczyszczeń w glebie, żywności (mięsie, warzy-
wach i owocach, zbożach, mleku i przetworach), w materiale biologicznym pochodzenia roś-
linnego,
zwierzęcego i ludzkiego (analiza toksykologiczna i kryminalistyka) a także w olejach
technicznych, asfaltach
(związki siarki i azotu, tzw. trucizny katalizatora).
Chromatografię żelową wykorzystuje się powszechnie do wstępnego frakcjonowania badanych
materia
łów w badaniach biochemicznych, mikrobiologicznych i w biotechnologii, a także w prepa-
ratyce, np. przy pozyskiwaniu substancji biologicznie czynnych w medycynie, chemii kosmetyków,
czy do odsalania roztworów peptydów i bia
łek, otrzymanych po zastosowaniu wysalania frakcyj-
nego, albo str
ącania w punkcie izoelektrycznym.
Cel ćwiczenia
Celem ćwiczenia jest zapoznanie z działaniem zestawu do chromatografii żelowej, oraz
scharakteryzowanie próbki polimeru (technicznego polistyrenu) metodą GPC.
Literatura
D. Berek, M. Dressler, M. Kubin, K. Marcinka „Chromatog
rafia żelowa” PWN Warszawa 1989.
Pytania dla PT Studentów
Wymień angielskie akronimy używane w literaturze na określenie chromatografii żelowej.
Opisz zasadę działania kolumny do chromatografii żelowej. Wskaż na istotną cechę odróżniającą
chromatografię żelową i chromatografię cieczową.
W jaki sposób można rozdzielić na kolumnie żelowej mieszaninę związków o bardzo dużym
rozrzucie ciężarów cząsteczkowych.
Wymień przykład eluentu i materiału wypełnienia do prowadzenia rozdziału: a) polistyrenu, b)
mieszaniny peptydów.
Dysponujesz wzorcami M
cz
z polistyrenu. Czy można użyć ich do kalibracji kolumny, w której
oznaczany będzie rozgałęziony poli(tlenek etylenu) / poli(kwas akrylowy)?
Dlaczego wypełniacze tlenkowe (np. SiO
2
)
stosowane w chromatografii żelowej traktuje się przed
użyciem silanami?
Czy można przyśpieszyć cykl pracy zwiększając kilkakrotnie przepływ w czasie rozdzielania
substancji białkowych na żelu dekstranowym?
Wymień rodzaje detektorów najczęściej używane w zestawach do chromatografii żelowej.

7
Oznaczanie wielkości cząstek metodą dynamicznego rozproszenia
światła DLS
Wprowadzenie
Cząsteczki związków chemicznych tego samego rodzaju w pewnych warunkach mogą łączyć się
tworząc większe struktury zwane ogólnie cząstkami. W roztworach zasocjowane cząsteczki
osiągając pewien rozmiar (stopień asocjacji) mogą tworzyć niejednorodną mieszaninę dwufazową,
w której wyróżniamy cząstki rozproszone (zawieszone) i fazę rozpraszającą (ciągłą). Jeżeli
rozdrobnienie (czyli dyspersja) substancji rozproszonej jest odpowiednio duże, to fizycznie
mieszanina sprawia wrażenie substancji jednorodnej, jednak nie jest to wymieszanie na poziomie
pojedynczych cząsteczek. Według IUPAC układ dyspersyjny jest układem koloidalnym, gdy
rozmiary cząstek fazy rozproszonej albo rozmiary nieciągłości układu koloidalnego są w zakresie
od 1 nm do 1 µm przynajmniej w jednym kierunku.
W koloidach stopień dyspersji (stosunek
powierzchni fazy rozproszonej do objętości tej fazy) wynosi od 10
5
do 10
7
cm
-1
– wówczas
wielkość cząstek fazy zawieszonej (zdyspergowanej) sprawia, że ważne są zarówno oddziaływania
pomiędzy nią i fazą dyspergującą, jak i oddziaływania wewnątrz obu faz.
Aglomeracja cząsteczek lub cząstek w cieczach jest spowodowana oddziaływaniami wynikającymi
z ich struktury i właściwości oraz czynnikami zewnętrznymi. Należą do nich m.in.:
–
budowa cząsteczek i wynikająca z niej polarność, zdolność do tworzenia wiązań wodorowych,
koordynacyjnych czy jonowych,
–
rozwinięcie powierzchni cząstek (cząstki o dużej powierzchni właściwej dążą samorzutnie do jej
zredukowania),
–
polarność fazy rozpraszającej,
–
kwasowość (pH) w przypadku wodnej fazy rozpraszającej,
– temperatura,
–
stężenie substancji w fazie rozpraszającej,
–
możliwość wzajemnych oddziaływań pomiędzy fazą rozpraszającą a rozproszoną.
Jeżeli z jakiegoś powodu proces aglomeracji nie kończy się na utworzeniu koloidalnego układu
cząstek (trwałej dyspersji) to następuje proces koagulacji fazy rozproszonej koloidu. Wynikiem
tego procesu może być zjawisko żelowania, tworzenia się past i materiałów stałych, sedymentacji
lub pokrywania powierzchni mieszaniny warstwą fazy rozproszonej. Istnieje koagulacja odwracalna
i nieodwracalna, a także spontaniczna i wymuszona.
Z punktu widzenia chemii i technologii polimerów procesy aglomeracji są ważne między innymi w
odniesieniu do proszkowych napełniaczy tworzyw sztucznych. Napełniaczami takimi są zazwyczaj
nieorganiczne lub hybrydowe (organiczno-
nieorganiczne) związki (tlenki, sole) metali, które dodaje
się do tworzyw w celu zmiany ich właściwości mechanicznych, reologicznych (przetwórstwo),
obniżenia palności czy obniżenia ceny. Otrzymanie tworzywa o dobrych właściwościach jest
uzależnione między innymi od stopnia dyspersji cząstek napełniacza, czyli od ich rozmiaru. Jedną z
najnowszych grup napełniaczy są nanonapełniacze, czyli proszki, których cząstki charakteryzują się
tym, że przynajmniej jeden z ich rozmiarów mieści się w zakresie 1 – 100 nm. Jednakże po
wprowadzeniu ich do tworzywa lub żywicy mogą one aglomerować (separacja faz). Określenie
wielkości cząstek napełniaczy zarówno w dyspersjach rozpuszczalnikowych czy też w
nieusiecio
wanych żywicach oraz w gotowych kompozytach (utwardzona żywica, przetworzony
termoplast) ma bardzo duże znaczenie z punktu widzenia potencjalnego i praktycznego
zastosowania napełniaczy.
Rozmiary cząstek są istotnym parametrem także materiałów biologicznych, w medycynie,
przemyśle spożywczym, farb i lakierów, poligrafii i innych.
8
Dynamiczne rozpraszanie światła – DLS (Dynamic Light Scattering)
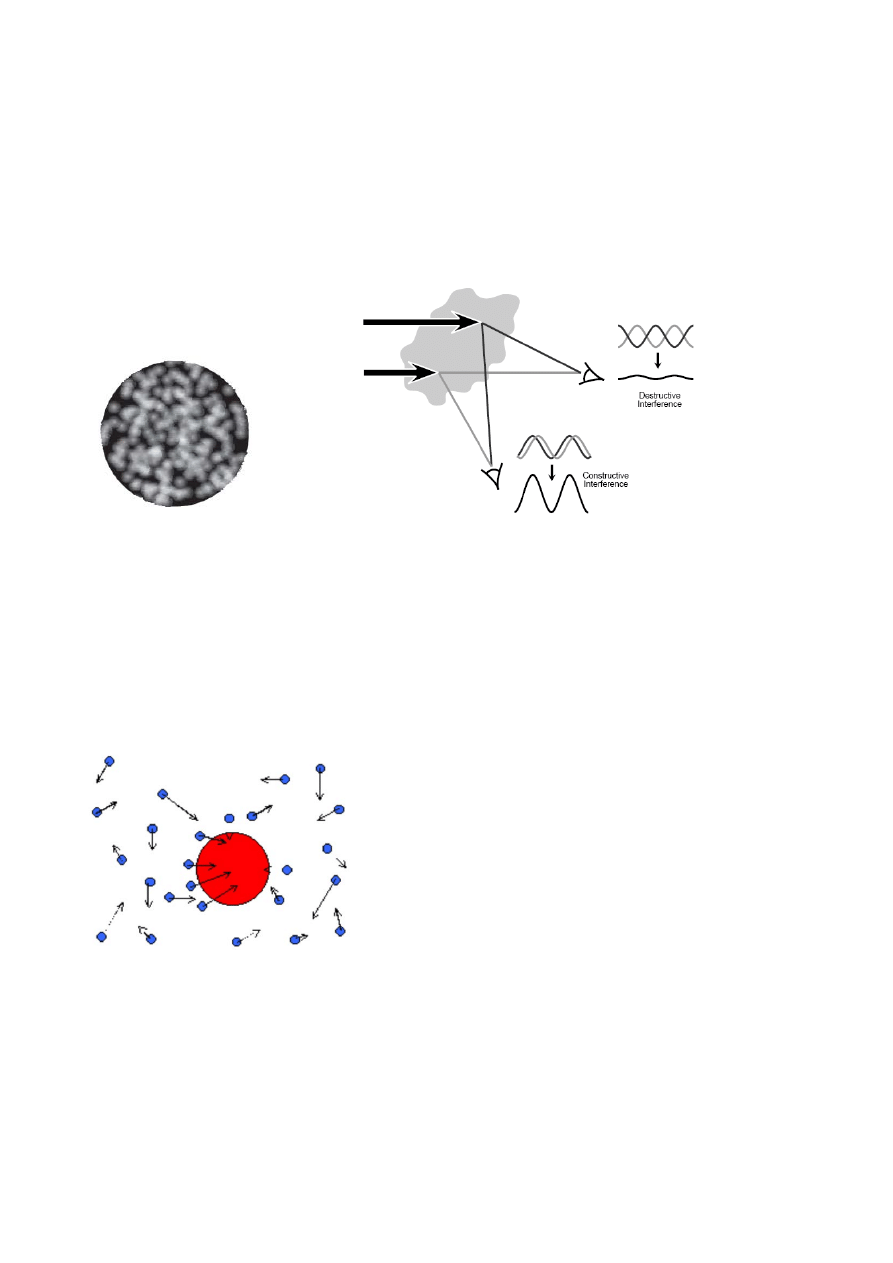
Jeżeli na małą cząstkę pada wiązka światła (np. laserowego), następuje rozproszenie tej wiązki we
wszystkich kierun
kach. Gdy w odpowiednio bliskiej odległości od rozważanej cząstki umieszczony
jest ekran (detektor), to zostanie on oświetlony przez światło rozproszone. Zastąpienie pojedynczej
cząstki tysiącami nieruchomych cząstek spowodowałoby pojawienie się na ekranie wzoru z
złożonego wielu jasnych i ciemnych plamek (Rys. 1a). Jasne plamki pochodzą od nakładających się
fal rozproszonego światła, które są w tej samej fazie i wzmacniają się w wyniku interferencji,
podczas gdy ciemne obszary powstają wtedy, gdy nakładające się fale są w fazie przeciwnej i
ulegają wygaszeniu (Rys. 1b).
a)
b)
Rys. 1.
Obraz interferencyjny światła rozproszonego na dyspersji i zasada jego powstawania.
W przedstawionym przykładzie założono, że cząstki nie poruszają się, dlatego obraz na ekranie jest
statyczny. W rzeczywistości cząstki zdyspergowane w cieczy są w ciągłym ruchu (ruchy Browna).
Zjawisko to zostało odkryte przez brytyjskiego biologa Roberta Browna, który obserwując przez
mikroskop pyłki kwiatowe w zawiesinie wodnej dostrzegł, iż znajdują się one w nieustannym,
chaotycznym ruchu, a nie –
jak sądził – stoją w miejscu. Ruchy Browna definiuje się jako
przypadkowe ruchy małych cząstek w cieczy, spowodowane bombardowaniem ich przez cząsteczki
cieczy (fazy rozpraszającej) –Rys. 2.
Rys. 2. Ruchy Browna.
To bombardowanie drobiny otaczającymi cząsteczkami jest statystycznie biorąc takie samo z
każdej strony. Jednak jeśli rozpatrywana cząstka jest wystarczająco mała, to zdarza się, że liczba
cząsteczek zderzających się z nią z jednej strony będzie w jakimś momencie inna (większa lub
mniejsza) od cząsteczek uderzających z przeciwnej strony. W efekcie drobina dostaje co jakiś czas
silniejszy impuls w stronę wyznaczoną przez uderzenia większej (w danej chwili) grupy cząsteczek
(mówimy w takiej sytuacji o tzw. fluktuacji
). Ważną cechą ruchów Browna jest fakt, że większe
9
cząstki poruszają się wolniej a mniejsze szybciej. Zależność pomiędzy wielkością cząstek a
szybkością ich ruch Browna jest opisana równaniem Stokesa-Einsteina:
D
kT
R
h
πη
6
=
gdzie: R
h
–
promień hydrodynamiczny cząstki, k – stała Boltzmanna, T – temperatura (K), η -
lepkość rozpuszczalnika a D – współczynnik dyfuzji.
Jako że cząstki są w ciągłym ruchu, plamisty wzór na ekranie również będzie sprawiał wrażenie
ruchu. Wzmacniające i wygaszające nakładanie się fal światła rozproszonego od poruszających się
cząstek będzie powodowało, że ciemne i jasne obszary będą zmniejszały i zwiększały swoją
intensywność w czasie. Takie zjawisko nazywa się fluktuacją światła rozproszonego. Szybkość
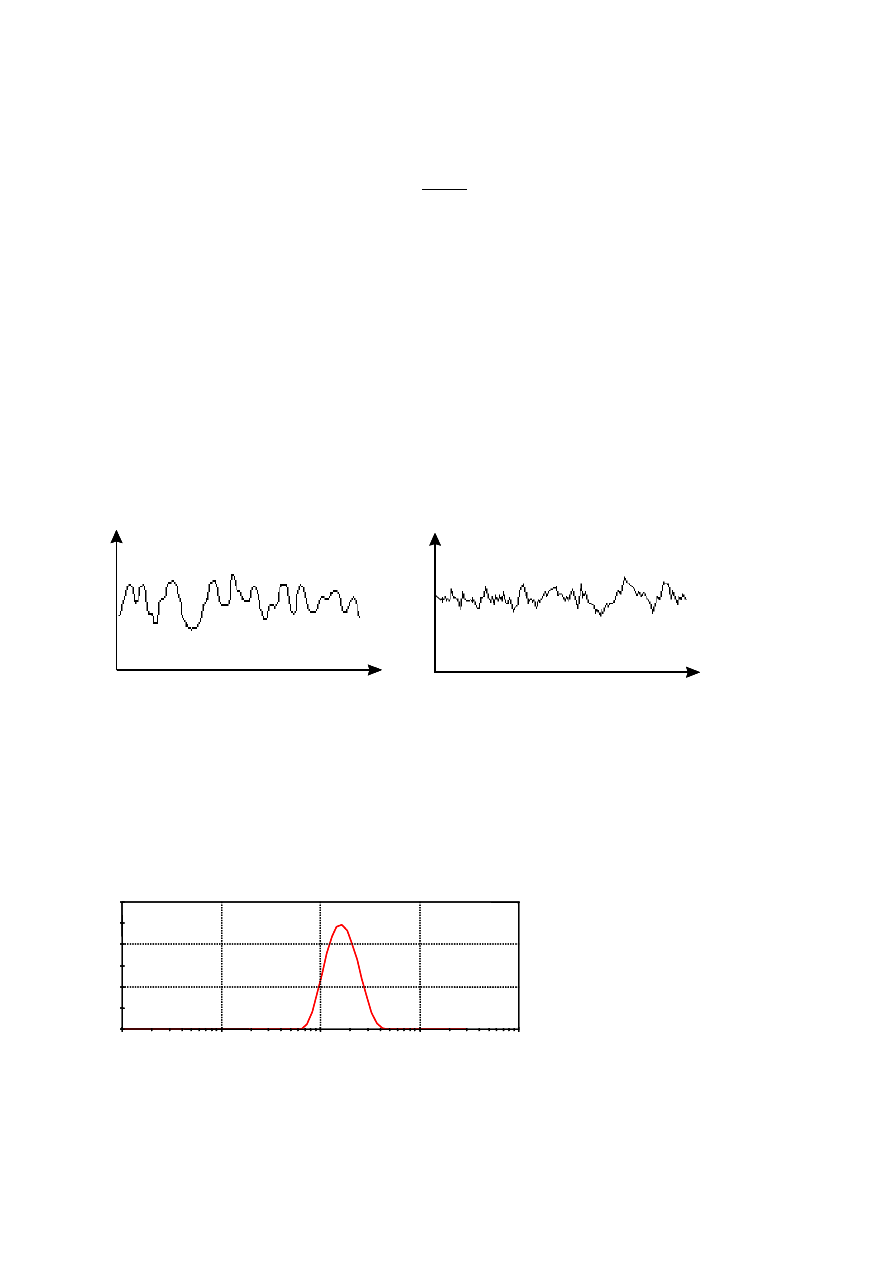
zmian intensywności światła rozproszonego zależy od wielkości cząstek i dlatego może posłużyć do
obliczenia ich rozmiaru. Metoda ta nosi nazw
ę dynamicznego rozproszenia światła (ang. Dynamic
Light Scattering, DLS).
Dla dużych cząstek sygnał zmienia się wolniej, dla mniejszych szybciej,
jednak ogólnie rzecz biorąc są to zjawiska zachodzące z dużą prędkością – skala czasu wyrażona
jest w mikro- a nawet w nanosekundach (Rys. 3).
du
że cząstki
czas
in
ten
syw
n
ość
du
że cząstki
czas
in
ten
syw
n
ość
du
że cząstki
czas
in
ten
syw
n
ość
ma
łe cząstki
czas
in
ten
syw
n
ość
ma
łe cząstki
czas
in
ten
syw
n
ość
Rys. 3.
Fluktuacje intensywności światła rozproszonego w zależności od wymiaru
geometrycznego cząstek rozpraszających.
W wyniku odpowiednich przekształceń i zastosowania odpowiednich algorytmów matematycznych
zarejest
rowane przez detektor zmiany intensywności światła rozproszonego zostają przekształcone
w wykres rozkładu wielkości cząstek (czyli udziału procentowego cząstek w zależności od ich
rozmiaru). Przykładowy (typowy) wykres przedstawia Rys. 4.
0
5.
10.
15.
1.
10.
100.
1000.
1.e+4
In
te
n
s
y
w
n
ość
(%
)
Średnica (nm)
Rozk
ład wielkości po intensywności
0
5.
10.
15.
1.
10.
100.
1000.
1.e+4
In
te
n
s
y
w
n
ość
(%
)
Średnica (nm)
Rozk
ład wielkości po intensywności
Rys. 4.
Rozkład wielkości cząstek w próbce (przykład).
W metodzie DLS bezpośredni pomiar dotyczy intensywności światła rozproszonego i jest to
podstawowy wykres prezentowany jako rezultat (size by intensity). Jednak przy analizie wyników
10
istotniejsze od porównania inten
sywności rozproszenia dwu próbek może być określenie objętości
fazy rozpraszającej, lub liczby cząstek rozpraszających w obu próbkach. Przez odpowiednie
przeliczenia, wykorzystujące teorię Mie, oprogramowanie sterujące aparatem udostępnia również
wykres size by volume i size by number
. Dla właściwej interpretacji wyników niezbędne jest
zrozumienie różnic między nimi.
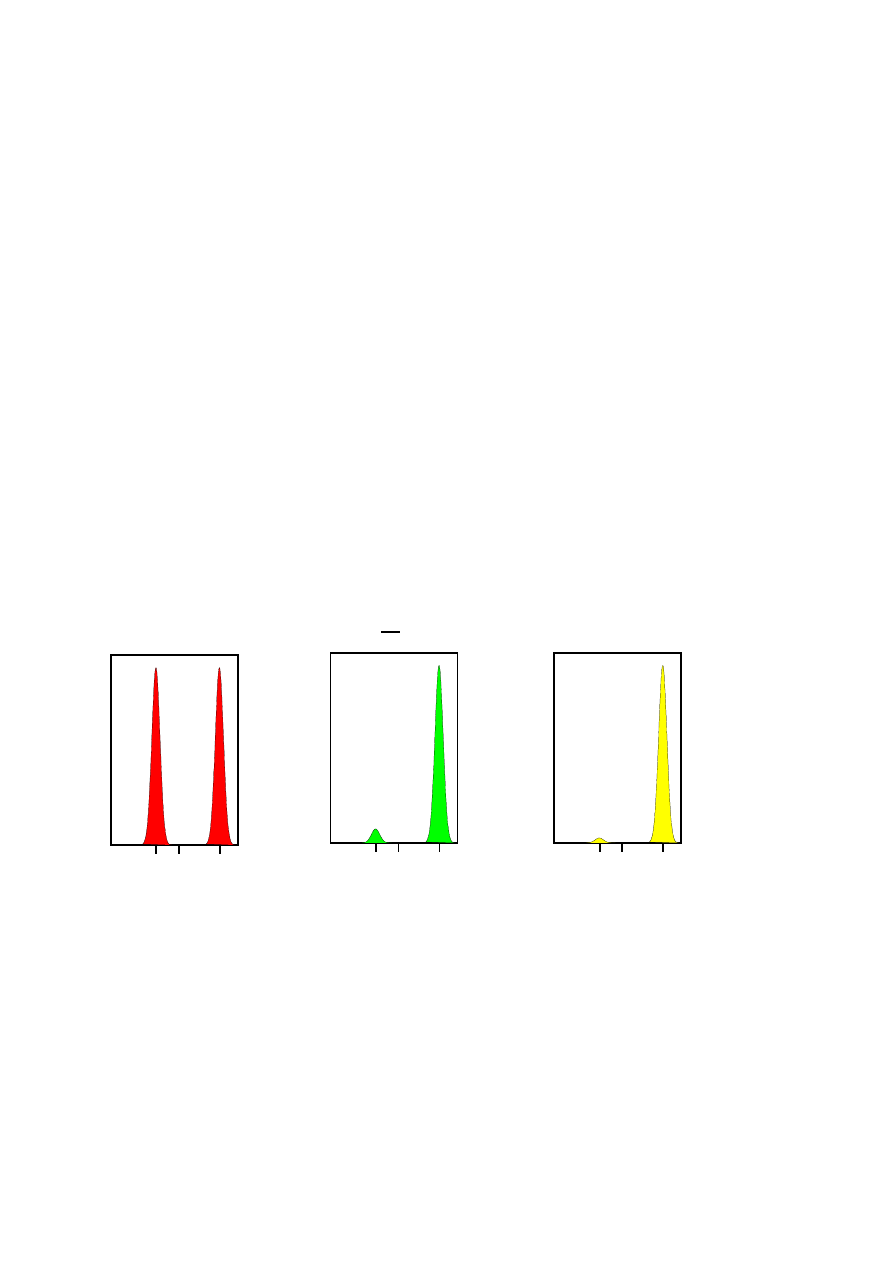
Rozważmy próbkę, w której znajdują się jednakowe liczby cząstek o średnicy 5 nm i o średnicy 50
nm. Na Rys. 5. na pierwszym od lewej wykresie
przedstawiony jest wynik pomiaru rozkładu
wielkości cząstek liczonego względem liczby tych cząstek. Jak należało się spodziewać przy
odpowiednich wartościach średnicy (na osi odciętych) występują dwa piki tej samej wielkości
(stosunek pików 5 nm i 50 nm wy
nosi 1:1), ponieważ zgodnie z założeniem w próbce jest ta sama
liczba cząstek obu rozmiarów. Na środkowym wykresie pokazano wyniki dla rozkładu liczonego po
objętości cząstek. Powierzchnia piku dla cząstek o średnicy 50 nm jest 1000 razy większa od tej dla
cząstek o mniejszej średnicy (stosunek 1:1000). Wynika to z tego, że objętość cząstek o średnicy 50
nm jest 1000 razy większa od łącznej objętości cząstek o średnicy 5 nm (objętość kuli jest równa V
d
= 4/3
πr
3
; V
5nm
= 65,45 nm
3
, V
50nm
= 65 450 nm
3
). W przypadku trzeciego wykresu, na którym
przedstawiono rozkład wielkości cząstek liczonych po intensywności światła rozproszonego,
stosunek powierzchni piku cząstek 50 nm jest 1 000 000 razy większy od piku dla 5 nm (stosunek
1:1 000
000). Dzieje się tak dlatego, że większe cząstki rozpraszają znacznie więcej światła niż
małe – według przybliżenia Rayleigha intensywność rozproszenia światła jest proporcjonalna do
średnicy cząstek rozpraszających w szóstej potędze.
Może się więc zdarzyć się tak, że rozkład wielkości cząstek o małej średnicy może być niemalże
niewidoczny na wykresie, jeżeli w dyspersji występują również cząstki o dużo większej średnicy.
Dlatego zawsze należy sprawdzać rozkłady po objętości i liczbie cząstek.
ILO
ŚĆ
4
3
OBJ
ĘTOŚĆ
=
πr
3
INTENSYWNO
ŚĆ
= d
6
Średnica (nm)
W
zg
lę
dny
%
5
10
50
1
1
5
10
50
1
1000
5
10
50
1
1,000,000
W
zg
lę
dny
%
W
zg
lę
dny
%
Średnica (nm)
Średnica (nm)
Rys. 5.
Wpływ wybranej metody obliczania na prezentację wyników DLS (opis w tekście).
Stabilność dyspersji – potencjał Zeta
Ważnym parametrem charakteryzującym dyspersję cząstek w cieczy jest jej stabilność, to znaczy
zdolność do utrzymywania cząstek w formie dyspersji koloidalnej. Niestabilne dyspersje ulegają
procesom sedymentacji czy też koagulacji w czasie, w wyniku czego po pewnym okresie może
okazać się, że badana próbka składa się z dwóch warstw: osadu i cieczy nad nim. Każdy z Państwa
widział zapewne starą farbę dyspersyjną – kiedyś nazywano je „emulsyjnymi”, która rozwarstwiła
się – uległa sedymentacji, albo „zwarzyła się” – uległa nieodwracalnej koagulacji.
Do liczbowego określenia stabilności dyspersji stosuje się tzw. potencjał Zeta. Jest to parametr
wyznaczany z pomiarów elektroforetycznych –
ruchliwości cząstek w polu elektrycznym. Prędkość
11
cząstek między elektrodami mierzy się w aparacie Malvern przy wykorzystaniu efektu Dopplera
(LDV – Laser Doppler Velocimetry) w specjalnych kuwetach z zainstalowanymi elektrodami.
Przy
jmuje się, że dyspersja jest stabilna wtedy, gdy wartość bezwzględna potencjału Zeta jest
większa od 30 mV (Zeta > +30 mV lub Zeta < –30 mV). Największy wpływ na potencjał Zeta ma
warto
ść pH badanej próbki. Wraz ze zmianą pH dodatni potencjał Zeta może zmniejszać swoją
wartość do zera a następnie osiągać wartości ujemne. Wartość pH przy której potencjał Zeta osiąga
zero i następuje zmiana jego znaku nazywa się punktem izoelektrycznym.
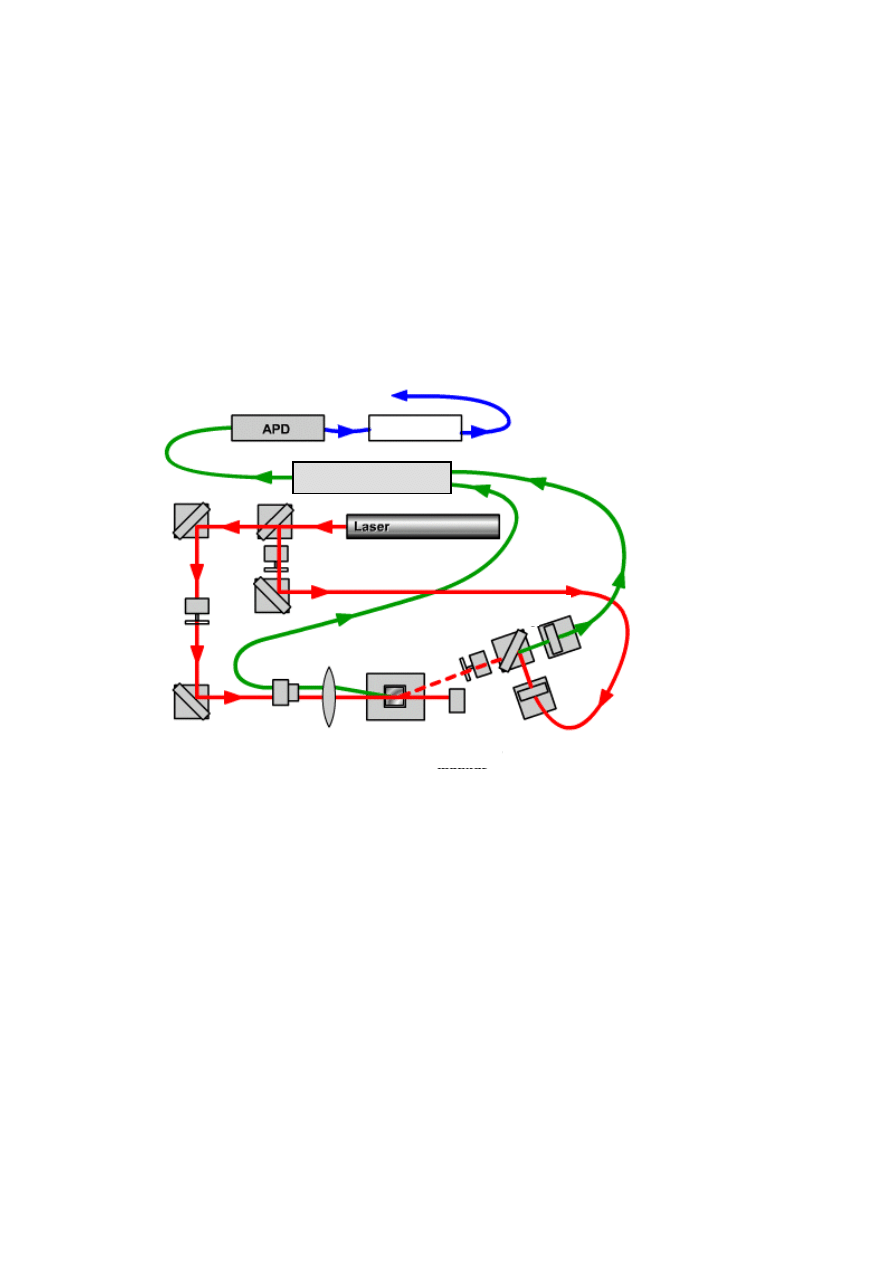
Budowa aparatu Zetasizer Nano ZS
Na Rys. 6. przedstawiono schemat analiza
tora wielkości cząstek Nano ZS firmy Malvern
Instruments
. Urządzenie zbudowane jest z lasera, układu pomiarowego wielkości cząstek i układu
pomiarowego potencjału Zeta, komory pomiarowej, korelatora i komputera z odpowiednim
oprogramowaniem.
Wi
ązka odniesienia
(Zeta)
Komora
pomiarowa
Sprz
ęganie wiązek
T
łumik sygnału
(rozmiar)
Korelator
T
łumik sygnału
(Zeta)
Zespó
ł
soczewek
Uk
ład kompensacyjny
(Zeta)
Detektor
mocy lasera
Komputer z oprogramowaniem
Wi
ązka odniesienia
(Zeta)
Komora
pomiarowa
Sprz
ęganie wiązek
T
łumik sygnału
(rozmiar)
Korelator
T
łumik sygnału
(Zeta)
Zespó
ł
soczewek
Uk
ład kompensacyjny
(Zeta)
Detektor
mocy lasera
Komputer z oprogramowaniem
Rys. 6. Anal
izator wielkości cząstek Malvern Zetasizer Nano ZS (schemat budowy).
Wykonanie
ćwiczenia
Z wykorzystaniem analizatora wielkości cząstek Malvern Zetasizer Nano ZS oznaczyć wielkość
cząstek hybrydowego napełniacza w postaci wodnej dyspersji lub innej dyspersji dostarczonej przez
prowadzącego. Przed pomiarem właściwym sprawdzić poprawność działania urządzenia mierząc
wzorcowy lateks polistyrenowy.
Document Outline
- Metody badań materiałów – laboratorium
- Oznaczanie ciężarów cząsteczkowych metodą chromatografii żelowej
- Wprowadzenie
- Zasada chromatografii żelowej
- Faza stacjonarna w chromatografii żelowej
- Eluenty w chromatografii żelowej
- Problemy i zakłócenia w czasie pracy
- Detektory wykorzystywane w chromatografii żelowej
- Kalibracja
- Zastosowanie chromatografii żelowej
- Cel ćwiczenia
- Literatura
- Pytania dla PT Studentów
- Oznaczanie wielkości cząstek metodą dynamicznego rozproszenia światła DLS
Wyszukiwarka
Podobne podstrony:
Oznaczenie aktywnosci czastek p Nieznany
kuran,Metrologia wielkosci geom Nieznany
oznaczanie ciezarow czasteczkowych polimerow
3 Oznaczanie kwasowooci wymienn Nieznany (2)
oznaczenia 2 id 343343 Nieznany
oznaczenia substancji niebezpie Nieznany
7 Oznaczanie wegla organicznego Nieznany (2)
ćwiczenie 5 kształt i wielkość cząstek MO
metodyka oznaczania glukozy Che Nieznany
Oznaczanie granicy plastycznosc Nieznany
Lechowski Pojęcia i oznaczenia wielkości przyjętych w metodzie modelowania charakterów ludzkich M M
2 Energia jako wielkosc bazowa Nieznany (2)
8 Oznaczanie utlenialnosci id 4 Nieznany (2)
REKOLEKCJE WIELKOPOSTNE w szkol Nieznany
Cw 2 Oznaczenie gestosci grunt Nieznany
CWICZENIE wielkosc partii (1) i Nieznany
2wyklad 5 Calka oznaczona id 60 Nieznany (2)
więcej podobnych podstron