
A.18. LICZBOWO ŚREDNIA MASA CZĄSTECZKOWA I ROZKŁAD MASY
CZĄSTECZKOWEJ POLIMERÓW
a) METODA
Niniejsza metoda chromatografii żelowej jest odpowiednikiem metody OECD TG 118 (1996). Podstawowe
zasady i dalsze informacje techniczne podano w piśmiennictwie
1)
.
1.1. Wstęp
Ze względu na bardzo różne właściwości polimerów, nie jest możliwe opisanie jednej metody, która dokładnie
podawałaby warunki rozdziału i oceny oraz obejmowała wszystkie możliwości i szczególne przypadki
występujące podczas rozdziału polimerów. Zwłaszcza w przypadku złożonych układów polimerycznych często
nie można stosować chromatografii żelowej (GPC). Masę cząsteczkową można wówczas oznaczać za pomocą
innych metod („Inne metody oznaczania średniej masy cząsteczkowej (M
n
) polimerów”) podając równocześnie
wszystkie informacje i uzasadnienie wybranej metody.
Opisana metoda jest zgodna z normą DIN 55672 1). W normie tej zawarte są dokładne informacje o wykonaniu
doświadczeń i ocenie wyników. Wszelkie zmiany dotyczące modyfikacji warunków doświadczenia należy
uzasadnić. Można stosować inne wzorce, jeżeli są certyfikowane. W opisanej metodzie w celu kalibracji stosuje
się próbki polistyrenu o znanej polidyspersyjności. Jednak dla pewnych polimerów może być konieczna
modyfikacja tej metody, na przykład dla polimerów rozpuszczalnych w wodzie i polimerów o długich,
rozgałęzionych łańcuchach.
1.2. Definicje i jednostki
Liczbowo średnią masę cząsteczkową M
n
i wagowo średnią masę cząsteczkową M
w
oznacza się stosując
następujące równania:
∑
∑
=
=
=
n
1
i
i
i
1
i
n
M
/
H
H
M
n
i
∑
∑
=
=
×
=
n
1
i
i
1
i
i
w
H
M
H
M
n
i
gdzie:
H
i
–
wielkość sygnału detektora od linii podstawowej dla objętości retencji V
i
M
i
– masa
cząsteczkowa frakcji polimeru dla objętości retencji V
i
n –
liczba
składników polimeru
M
w
/M
n
– szerokość rozkładu masy cząsteczkowej, będąca miarą dyspersyjności układu
1.3. Materiały kontrolne
Ze względu na to, że GPC jest metodą względną, należy wykonać kalibrację. Zwykle stosuje się w tym celu
wzorce polistyrenowe o budowie liniowej i znanych średnich masach cząsteczkowych M
n
i M
w
oraz o znanym
wąskim rozkładzie masy cząsteczkowej. Do oznaczania masy cząsteczkowej nieznanej próbki stosuje się krzywą
kalibracyjną tylko wtedy, jeżeli warunki rozdziału tej próbki i wzorców są identyczne.
Oznaczona zależność pomiędzy masą cząsteczkową i objętością elucji jest prawdziwa jedynie dla określonych
warunków danego doświadczenia. Warunki te obejmują przede wszystkim temperaturę, rozpuszczalnik (lub
mieszaninę rozpuszczalników), warunki chromatograficzne i kolumnę dzielącą lub układ kolumn.
Masy cząsteczkowe próbki oznaczone w ten sposób są wartościami względnymi i opisuje się je jako „masy
cząsteczkowe równoważne masom polistyrenu”. Oznacza to, że zależnie od różnic strukturalnych i chemicznych
1)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
- 101 -

pomiędzy próbką i wzorcami, masy cząsteczkowe mogą różnić się od wartości absolutnych w większym lub
mniejszym stopniu. Gdy stosuje się inne wzorce, na przykład glikol polietylenowy, tlenek polietylenu,
poli(metakrylan metylu), kwas poliakrylowy, należy podać uzasadnienie.
1.4. Zasada metody badań
Stosując GPC można oznaczyć zarówno rozkład masy cząsteczkowej próbki jak i średnie masy cząsteczkowe
(M
n
, M
w
). GPC jest szczególnym rodzajem chromatografii cieczowej, w której próbka jest rozdzielana
w zależności od objętości hydrodynamicznych poszczególnych składników
Rozdział następuje w miarę jak próbka przechodzi przez kolumnę wypełnioną materiałem porowatym, zwykle
żelem organicznym. Małe cząstki mogą penetrować pory, podczas gdy duże cząstki są wykluczane. Droga
dużych cząstek jest przez to krótsza i one wymywają się w pierwszej kolejności. Cząstki średniej wielkości
penetrują niektóre z porów i wymywają się później. Najmniejsze cząstki, o średnim promieniu
hydrodynamicznym mniejszym niż pory żelu, mogą penetrować wszystkie pory. One wymywają się jako
ostatnie.
W idealnym przypadku o rozdziale decyduje wyłącznie wielkość cząstek, ale w praktyce trudno jest zapobiec co
najmniej niektórym interferencyjnym efektom absorpcyjnym. Niejednolite wypełnienie kolumny i martwe
objętości mogą utrudnić rozdział
2)
.
Detekcję prowadzi się wykorzystując na przykład współczynnik załamania lub absorpcję w UV i uzyskuje się
krzywą rozkładu. Jednakże w celu przypisania krzywej rzeczywistych wartości mas cząsteczkowych, konieczna
jest kalibracja kolumny przez przepuszczenie przez nią polimerów o znanej masie cząsteczkowej i dość
podobnej budowie, na przykład różnych wzorców polistyrenowych. Powstaje typowa krzywa Gaussa, niekiedy
zniekształcona przez niewielkie ogonowanie po stronie małych mas cząsteczkowych. Oś pionowa wskazuje ilość
(wagowo) wymywanych cząstek o różnej masie cząsteczkowej, a oś pozioma logarytm masy cząsteczkowej.
1.5. Kryteria jakościowe
Powtarzalność (względne odchylenie standardowe: RSD) objętości elucji powinna być lepsza niż 0,3%. Jeżeli to
kryterium nie jest spełnione, należy wymaganą powtarzalność analizy zapewnić przez korygowanie za pomocą
wzorca wewnętrznego, a chromatogram oceniać w zależności od czasu 2). Polidyspersyjność zależy od mas
cząsteczkowych wzorców. W przypadku wzorców polistyrenowych typowymi wartościami są:
M
p
<2000
M
w
/M
n
<1,20,
2000
≤ M
p
≤10
6
M
w
/M
n
<1,05,
M
p
>10
6
M
w
/M
n
<1,20,
M
p
- masa cząsteczkowa wzorca w maksimum piku.
1.6. Opis metody badań
1.6.1. Przygotowanie roztworów wzorcowych polistyrenu
Wzorce polistyrenowe rozpuszcza się przez mieszanie ostrożnie w wybranym eluencie. Podczas przygotowania
roztworów należy stosować się do zaleceń producenta.
Stężenia wybranych wzorców zależą od różnych czynników, na przykład wstrzykiwanej objętości, lepkości
roztworu i czułości detektora analitycznego. Maksymalną objętość wstrzykiwaną należy dostosować do długości
kolumny, aby zapobiec przeładowaniu. Typowe objętości wstrzykiwane w celu rozdziałów analitycznych
z zastosowaniem GPC z kolumną 30 cm
×7,8 mm mieszczą się na ogół pomiędzy zawartościami 40 i 100 µl.
Można stosować większe objętości, ale nie powinny one przekraczać 250
µl. Przed kalibracją kolumny należy
oznaczyć optymalny stosunek pomiędzy wstrzykiwaną objętością i stężeniem.
1)
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds. (1979). Modern Size Exclusion Liquid Chromatography, J.
Wiley and Sons.
2)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
- 102 -

1.6.2. Przygotowanie roztworu próbki
Do przygotowania roztworów próbek stosuje się takie same wymagania. Próbkę rozpuszcza się ostrożnie
wstrząsając w odpowiednim rozpuszczalniku, na przykład w tetrahydrofuranie (THF). W żadnym wypadku nie
należy jej rozpuszczać stosując łaźnię ultradźwiękową. W razie potrzeby roztwór próbki oczyszcza się za
pomocą membrany filtracyjnej o wielkości porów pomiędzy 0,2 i 2
µm.
W sprawozdaniu należy uwzględnić obecność nierozpuszczonych cząstek, ponieważ mogą one pochodzić
z substancji o dużym ciężarze cząsteczkowym. W celu oznaczenia zawartości procentowej rozpuszczonych
cząstek należy zastosować odpowiednią metodę. Roztwór należy wykorzystać w ciągu 24 godzin.
1.6.3. Aparatura
- pojemnik na rozpuszczalnik,
- urządzenie do odgazowania (gdy to wskazane),
- pompa,
- wytłumiacz pulsacji (gdy to wskazane),
- układ dozujący,
- kolumny chromatograficzne,
- detektor,
- przepływomierz (gdy to wskazane),
- urządzenie rejestrujące i przetwarzające dane,
- naczynie na odpady.
Należy zapewnić, aby układ GPC był obojętny w stosunku do stosowanych rozpuszczalników (na przykład
kapilary stalowe do THF).
1.6.4. Układ dozujący i układ zasilania rozpuszczalnikiem
Na kolumnę, w ściśle zdefiniowanej strefie, wprowadza się ręcznie lub za pomocą automatycznego dozownika
określoną objętość próbki. Pociągając lub wciskając zbyt szybko tłok strzykawki podczas operacji manualnych
można spowodować zmiany w
obserwowanym rozkładzie mas cząsteczkowych. Układ zasilania
rozpuszczalnikiem powinien być, w miarę możliwości, bezpulsacyjny, dzięki zainstalowaniu wytłumiacza
pulsacji. Szybkość przepływu powinna być rzędu 1 ml/min.
1.6.5. Kolumna
Polimer (w zależności od próbki) ocenia się stosując jedną albo kilka kolumn połączonych kolejno. W handlu
dostępne są liczne materiały porowate do kolumn, które mają określone właściwości (np wielkość porów,
granice wykluczania). Wybór żelu do rozdzielania lub długości kolumny zależą zarówno od właściwości próbki
(objętości hydrodynamicznych, rozkładu masy cząsteczkowej) jak i od określonych warunków rozdziału, takich
jak rozpuszczalnik, temperatura i szybkość przepływu
1.6.6. Półki teoretyczne
Kolumnę lub zestaw kolumn stosowany do rozdziału należy scharakteryzować za pomocą liczby półek
teoretycznych. Obejmuje to, w przypadku THF jako rozpuszczalnika wymywającego, wprowadzenie na
kolumnę o znanej długości roztworu etylobenzenu lub innej odpowiedniej rozpuszczonej substancji niepolarnej.
1)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
2)
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds. (1979). Modern Size Exclusion Liquid Chromatography, J.
Wiley and Sons.
3)
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight
Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography – GPC). American
Society for Testing and Materials, Philadelphia, Pennsylvania.
- 103 -

Liczba półek teoretycznych obliczana jest z następujących równań:
2
1/2
e
)
W
V
5,54(
N
=
lub
2
e
)
W
V
16(
N
=
gdzie:
N –
liczba
półek teoretycznych,
V –
objętość elucji w maksimum piku,
W –
szerokość podstawy piku,
W
1/2
– szerokość piku w połowie wysokości.
1.6.7. Skuteczność rozdziału
Oprócz liczby półek teoretycznych, która decyduje o szerokości pasma, rolę odgrywa również skuteczność
rozdziału, określana przez nachylenie krzywej kalibracyjnej. Skuteczność rozdziału kolumny otrzymuje się
z następującej zależności:
⎥
⎦
⎤
⎢
⎣
⎡
≥
−
2
3
)
e(10M
M
e,
cm
cm
0
,
6
kolumny
przekroju
i
powierzchn
pole
V
V
x
x
gdzie:
V
e,Mx
– objętość elucji dla polistyrenu o masie cząsteczkowej M
x
,
V
e,(10Mx)
– objętość elucji dla polistyrenu o 10 razy większej masie cząsteczkowej,
Rozdział układu definiuje się na ogół następująco:
)
M
/
M
(
log
1
W
W
V
V
2
R
1
2
10
2
1
e2
e1
1,2
×
+
−
×
=
gdzie:
V
e1
, V
e2
– objętości elucji dwóch wzorców polistyrenowych w maksimum piku,
W
1
, W
2
– szerokości podstaw pików,
M
1
, M
2
– masy
cząsteczkowe w maksimum piku (powinny różnić się 10 razy).
Wartość R dla układu kolumn powinna być większa niż 1,7
1.6.8. Rozpuszczalniki
Wszystkie rozpuszczalniki powinny mieć wysoką czystość (stosuje się THF o czystości 99,5%). Pojemnik na
rozpuszczalnik (gdy to konieczne w atmosferze gazu obojętnego) powinien być odpowiednio duży w celu
kalibracji kolumny i licznych analiz próbki. Rozpuszczalnik należy odgazować za pomocą pompy, zanim
zostanie podany na kolumnę.
1.6.9. Kontrola temperatury
Temperatura krytycznych składników układu (pętli dozującej, kolumn, detektora i przewodów) powinna być
stała i odpowiednia dla danego rozpuszczalnika.
1)
ASTM D 5296-92 (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight
Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for
Testing and Materials, Philadelphia, Pennsylvania.
- 104 -

1.6.10. Detektor
Celem detektora jest ilościowa rejestracja stężenia próbki wymytej z kolumny. W celu zapobieżenia
niepożądanemu poszerzeniu pików należy stosować jak najmniejszą objętość kuwety celki detektora. Nie
powinna być ona większa niż 10
µl, z wyjątkiem detektorów lepkościowych i do światła rozproszonego. W celu
detekcji stosuje się na ogół refraktometrię różnicową. Jednak, o ile wymagają tego szczególne właściwości
próbki lub rozpuszczalnika wymywającego, można stosować również inne rodzaje detektorów, np UV/VIS, IR,
detektory lepkościowe itd.
2. WYNIKI BADAŃ
W zakresieszczegółowych kryteriów oceny, jak również wymagań dotyczących zbierania i przetwarzania
danych, należy odwołać się do normy DIN
Dla każdej próbki należy przeprowadzić dwa niezależne doświadczenia i analizować je indywidualnie.
Dla każdego pomiaru należy podać wartość M
n
, M
w
, M
w
/M
n
i M
p
i wyraźnie wykazać, że są one wartościami
względnymi, równoważnymi masom cząsteczkowym stosowanego wzorca.
Po oznaczeniu objętości retencji lub czasów retencji (w miarę możliwości skorygowanych z zastosowaniem
wzorca wewnętrznego) wykreśla się wartość log M
p
(M
p
- maksimum piku wzorca do kalibracji) względem
jednej z tych wielkości. Konieczne są co najmniej dwa punkty kalibracyjne na dziesięć mas cząsteczkowych
i wymaga się co najmniej pięciu punktów pomiarowych dla pełnej krzywej, która powinna obejmować
szacowaną masę cząsteczkową próbki. Punkt końcowy krzywej kalibracyjnej dla małych mas cząsteczkowych
określa się za pomocą n-heksylobenzenu lub innej odpowiedniej niepolarnej substancji. Masy cząsteczkowe
średnie liczbowo i średnie wagowo oznacza się na ogół za pomocą elektronicznego przetworzenia danych
w oparciu o wzory z punktu 1.2. W przypadku przekształceń manualnych należy oprzeć się na normie ASTM D
3536-91
Krzywa rozkładu powinna mieć postać tabeli lub rysunku (częstotliwość różnicowa lub suma zawartości
procentowych względem log M).
Przy przedstawianiu graficznym dziesięciu masom cząsteczkowym powinno odpowiadać około 4
cm
szerokości, a maksymalny pik powinien mieć około 8 cm wysokości.
W przypadku całkowych krzywych rozkładu różnica rzędnych pomiędzy 0 i 100% powinna wynosić około
10 cm.
3. SPRAWOZDANIE
Sprawozdanie powinno zawierać, z uwzględnieniem zakresu przeprowadzonych badań, następujące informacje:
1) Badana substancja:
a) dostępne informacje dotyczące badanej substancji (tożsamość, dodatki, zanieczyszczenia),
b) - opis postępowania z próbką, spostrzeżenia, problemy;
2) Oprzyrządowanie:
a) pojemnik na eluent, gaz obojętny, odgazowanie eluenta, skład eluenta, zanieczyszczenia,
b) pompa, wytłumiacz pulsacji, układ dozujący,
c) kolumny rozdzielcze (producent, wszystkie informacje dotyczące charakterystyki kolumn, takie jak
wielkość porów, rodzaj materiału rozdzielnego itd., liczba, długość i kolejność stosowanych kolumn),
d) liczba półek teoretycznych kolumny (lub kombinacji), skuteczność rozdziału (rozdzielczość układu),
e) informacje na temat symetrii pików,
f) temperatura kolumny, sposób kontroli temperatury,
g) detektor (zasada pomiaru, objętość kuwety),
h) przepływomierz, o ile jest stosowany (producent, zasada pomiaru),
i) układ rejestrujący i przetwarzający dane (sprzęt komputerowy i oprogramowanie);
3) Kalibracja układu:
a) szczegółowy opis metody stosowanej do sporządzania krzywej kalibracyjnej,
1)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
2)
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight
Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography – GPC). American
Society for Testing and Materials, Philadelphia, Pennsylvania.
- 105 -

b) informacje na temat kryteriów jakościowych tej metody (np współczynnika korelacji, sumy
kwadratów błędów, itd.),
c) informacje dotyczące wszelkich ekstrapolacji, założeń i przybliżeń dokonanych podczas procedury
doświadczalnej oraz oceny i przetwarzania danych,
d) wszelkie pomiary zastosowane w celu konstruowania krzywej kalibracyjnej należy zestawić w tabeli
zawierającej następujące informacje dotyczące każdego punktu kalibracji:
- nazwa próbki,
- producent próbki,
- wartości charakteryzujące wzorce M
p
, M
n
, M
w
i M
w
/M
n
, dostarczane przez producenta lub
pochodzące z kolejnych pomiarów, wraz ze szczegółami odnośnie metody oznaczenia,
- dozowana objętość i stężenie dozowanej próbki,
- wartość M
p
stosowana do kalibracji,
- objętość elucji lub skorygowany czas retencji mierzony w maksimum piku,
- M
p
obliczona dla maksimum piku,
- błąd procentowy obliczonej wartości M
p
i wartości kalibracji;
4) Ocena:
a) ocena na podstawie czasu: metody stosowane do zapewnienia wymaganej powtarzalności (metoda
korygowania, wzorzec wewnętrzny, itd.),
b) informacje o tym czy ocenę przeprowadzono na podstawie objętości elucji czy czasu retencji,
c) informacje dotyczące granic oceny, jeżeli pik nie jest w pełni analizowany,
d) opis metod wygładzania danych, jeżeli były stosowane,
e) procedury przygotowania i wstępnej obróbki próbki,
f) obecność nierozpuszczonych cząstek, jeżeli występują,
g) objętość dozowana (
µl) i stężenie dozowanej próbki (mg/ml),
h) spostrzeżenia dotyczące wpływów, które prowadzą do odchyleń od idealnego profilu GPC,
i) szczegółowy opis wszelkich modyfikacji procedur badawczych,
j) szczegóły dotyczące zakresów błędów,
k) wszelkie inne informacje i spostrzeżenia istotne dla interpretacji wyników.
- 106 -

INNE METODY OZNACZANIA LICZBOWO ŚREDNIEJ MASY CZĄSTECZKOWEJ (M
n
)
POLIMERÓW
Chromatografia żelowa (GPC) jest preferowaną metodą oznaczania M
n
zwłaszcza, gdy dostępny jest zestaw
wzorców, których budowa jest porównywalna z budową polimerów. Jednak, gdy występują trudności praktyczne
ze stosowaniem GPC lub zachodzi obawa, że dana substancja nie spełni (co wymaga potwierdzenia) ustalonych
kryteriów M
n
, dostępne są metody alternatywne, takie jak:
Wykorzystanie właściwości koligatywnych
Ebuliometria/kriometria
Dokonuje się pomiaru podwyższenia temperatury wrzenia (ebuliometria) albo obniżenia temperatury krzepnięcia
(kriometria) rozpuszczalnika, gdy dodany jest polimer. Metoda polega na tym, że wpływ rozpuszczonego
polimeru na temperaturę wrzenia/krzepnięcia zależy od masy cząsteczkowej polimeru
.
Możliwość stosowania: M
n
<20000.
Obniżenie prężności par
Dokonuje się pomiaru prężności pary wybranej cieczy przed i po dodaniu znanych ilości polimeru
1), 2)
.
Możliwość stosowania: M
n
<20000 (teoretycznie; w praktyce bardziej ograniczona).
Osmometria membranowa
Podstawą tej metody jest zasada osmozy, to jest na naturalnej tendencji cząstek rozpuszczalnika do
przechodzenia przez błonę półprzepuszczalną z roztworu rozcieńczonego do stężonego w celu osiągnięcia
równowagi. Podczas badania roztwór rozcieńczony ma stężenie zero, natomiast roztwór stężony zawiera
polimer. Przechodzenie rozpuszczalnika przez błonę powoduje różnicę ciśnienia, zależną od stężenia i masy
cząsteczkowej polimeru 1),
.
Możliwość stosowania: M
n
pomiędzy 20000-200000.
Osmometria parowa
Polega na porównaniu szybkości parowania aerozolu czystego rozpuszczalnika z co najmniej trzema aerozolami
zawierającymi polimer o różnych stężeniach
1), ), )
.
Możliwość stosowania: M
n
<20000.
Analiza grup końcowych
Stosowanie tej metody wymaga wiedzy zarówno na temat budowy polimeru, jak i charakteru grup końcowych
w łańcuchu (które powinny dać się odróżnić od głównego łańcucha za pomocą, na przykład, NMR lub
miareczkowania/derywatyzacji). Oznaczenie stężenia cząsteczkowego końcowych grup obecnych w polimerze,
może prowadzić do wartości mas cząsteczkowych
Możliwość stosowania, M
n
do 50000 (ze zmniejszającą się niezawodnością).
1)
Billmeyer, F.W.Jr., (1984). Textbook of Polymer Science, 3
rd
Edn., John Wiley, New York.
2)
Glover, C.A., (1975). Absolute Colligative Property Methods. Chapter 4. In: Polymer Molecular Weights, part
I P.E.Slade, Jr.ed., Marcel Dekker, New York.
3)
ASTM D 3750-79, (1979). Standard Practice for Determination of Number-Average Molecular Weight of
Polymers by Membrane Osmometry. American Society for Testing and Materials, Philadelphia, Pensylvania.
4)
Coll, H. (1989). Membrane Osmometry. In: Determination of Molecular Weight, A.R.Cooper ed., J.Wiley and
Sons, pp. 25 to 52.
5)
ASTM 3592-77, (1977). Standard Recommended Practice for Determination of Molecular Weight by Vapour
Pressure, American Society for Testing and Materials, Philadelphia, Pennsylvania.
6)
Morris, C.E.M., (1989). Vapour pressure osmometry. In: Determination of molecular weight, A.R. Cooper ed.,
John Wiley and sons.
7)
Schröder, E., Müller, G., and Arndt, K-F., (1989). Polymer Characterisation, Carl Hanser Verlag, Munich.
8)
Garmon, R.G., (1975). End-Group Determinations, Chapter 3. In: Polymer molecular weights, Part I, P.E.
Slade, Jr. ed. Marcel Dekker, New York.
9)
Amiya, S., et al. (1990). Pure and Applied Chemistry, 62, 2139-2146.
- 107 -

A.19. ZAWARTOŚĆ POLIMERÓW O MAŁEJ MASIE CZĄSTECZKOWEJ
1. METODA
Niniejsza metoda chromatografii żelowej jest odpowiednikiem metody OECD TG 119 (1996). Podstawowe
zasady i dalsze informacje techniczne podano w piśmiennictwie.
1.1. Wstęp
Ze względu na bardzo różne właściwości polimerów, nie jest możliwe opisanie jednej metody, która dokładnie
podawałaby warunki rozdziału i oceny oraz obejmowała wszystkie możliwości i szczególne przypadki
występujące podczas rozdziału polimerów. Zwłaszcza w przypadku złożonych układów polimerycznych często
nie można stosować chromatografii żelowej (GPC). Masę cząsteczkową można wówczas oznaczać za pomocą
innych metod („Wskazówki dotyczące korygowania zawartości cząstek o małej masie cząsteczkowej ze względu
na obecność nierozpuszczalnego polimeru”) podając równocześnie wszystkie informacje i uzasadnienie
wybranej metody.
Opisana metoda jest zgodna z normą DIN 55672
. W normie tej zawarte są dokładne informacje o wykonaniu
doświadczeń i ocenie wyników. Wszelkie zmiany dotyczące modyfikacji warunków doświadczenia należy
uzasadnić. Można stosować inne wzorce, jeżeli są certyfikowane. W opisanej metodzie w celu kalibracji stosuje
się próbki polistyrenu o znanej polidyspersyjności. Jednak dla pewnych polimerów może być konieczna
modyfikacja tej metody, na przykład dla polimerów rozpuszczalnych w wodzie i polimerów o długich,
rozgałęzionych łańcuchach.
1.2. Definicje i jednostki
Małą masę cząsteczkową definiuje się jako masę cząsteczkową poniżej 1000 daltonów.
Liczbowo średnią masę cząsteczkową M
n
i wagowo średnią masę cząsteczkową M
w
oznacza się stosując
następujące równania:
∑
∑
=
=
=
n
1
i
i
i
1
i
n
M
/
H
H
M
n
i
∑
∑
=
=
×
=
n
1
i
i
1
i
i
w
H
M
H
M
n
i
gdzie:
H
i
– wielkość sygnału detektora od linii podstawowej dla objętości retencji V
i
M
i
– masa
cząsteczkowa frakcji polimeru dla objętości retencji V
i
n –
liczba
składników polimeru
M
w
/M
n
– szerokość rozkładu masy cząsteczkowej, będąca miarą dyspersyjności układu
1.3. Materiały kontrolne
Ze względu na to, że GPC jest metodą względną, należy wykonać kalibrację. Zwykle stosuje się w tym celu
wzorce polistyrenowe o budowie liniowej i znanych średnich masach cząsteczkowych M
n
i M
w
oraz o znanym
wąskim rozkładzie masy cząsteczkowej. Do oznaczania masy cząsteczkowej nieznanej próbki stosuje się krzywą
kalibracyjną tylko wtedy, jeżeli warunki rozdziału tej próbki i wzorców są identyczne.
Oznaczona zależność pomiędzy masą cząsteczkową i objętością elucji jest prawdziwa jedynie dla określonych
warunków danego doświadczenia. Warunki te obejmują przede wszystkim temperaturę, rozpuszczalnik (lub
mieszaninę rozpuszczalników), warunki chromatograficzne i kolumnę dzielącą lub układ kolumn.
Masy cząsteczkowe próbki oznaczone w ten sposób są wartościami względnymi i opisuje się je jako „masy
cząsteczkowe równoważne masom polistyrenu”. Oznacza to, że zależnie od różnic strukturalnych i chemicznych
1)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
- 108 -

pomiędzy próbką i wzorcami, masy cząsteczkowe mogą różnić się od wartości absolutnych w większym lub
mniejszym stopniu. Gdy stosuje się inne wzorce, na przykład glikol polietylenowy, tlenek polietylenu,
poli(metakrylan metylu), kwas poliakrylowy, należy podać uzasadnienie.
1.4. Zasada metody badań
Stosując GPC można oznaczyć zarówno rozkład masy cząsteczkowej próbki jak i średnie masy cząsteczkowe
(M
n
, M
w
). GPC jest szczególnym rodzajem chromatografii cieczowej, w której próbka jest rozdzielana
w zależności od objętości hydrodynamicznych poszczególnych składników
Rozdział następuje w miarę jak próbka przechodzi przez kolumnę wypełnioną materiałem porowatym, zwykle
żelem organicznym. Małe cząstki mogą penetrować pory, podczas gdy duże cząstki są wykluczane. Droga
dużych cząstek jest przez to krótsza i one wymywają się w pierwszej kolejności. Cząstki średniej wielkości
penetrują niektóre z
porów i
wymywają się później. Najmniejsze cząstki, o
średnim promieniu
hydrodynamicznym mniejszym niż pory żelu, mogą penetrować wszystkie pory. One wymywają się jako
ostatnie.
W idealnym przypadku o rozdziale decyduje wyłącznie wielkość cząstek, ale w praktyce trudno jest zapobiec co
najmniej niektórym interferencyjnym efektom absorpcyjnym. Niejednolite wypełnienie kolumny i martwe
objętości mogą utrudnić rozdział
1)
.
Detekcję prowadzi się wykorzystując na przykład współczynnik załamania lub absorpcję w UV i uzyskuje się
krzywą rozkładu. Jednakże w celu przypisania krzywej rzeczywistych wartości mas cząsteczkowych, konieczna
jest kalibracja kolumny przez przepuszczenie przez nią polimerów o znanej masie cząsteczkowej i dość
podobnej budowie, na przykład różnych wzorców polistyrenowych. Powstaje typowa krzywa Gaussa, niekiedy
zniekształcona przez niewielkie ogonowanie po stronie małych mas cząsteczkowych. Oś pionowa wskazuje ilość
(wagowo) wymywanych cząstek o różnej masie cząsteczkowej, a oś pozioma logarytm masy cząsteczkowej.
Z
tej krzywej określa się zawartość substancji o małej masie cząsteczkowej. Obliczenia mogą być dokładne
tylko wówczas, jeżeli substancje o małych masach cząsteczkowych odpowiadają równoważnie na jednostkę
masy polimeru jako całości.
1.5. Kryteria jakościowe
Powtarzalność (względne odchylenie standardowe: RSD) objętości elucji powinna być lepsza niż 0,3%. Jeżeli to
kryterium nie jest spełnione należy wymaganą powtarzalność analizy zapewnić przez korygowanie za pomocą
wzorca wewnętrznego, a chromatogram oceniać w zależności od czasu
. Polidyspersyjność zależy od mas
cząsteczkowych wzorców. W przypadku wzorców polistyrenowych typowymi wartościami są:
M
p
<2000
M
w
/M
n
<1,20
2000
≤ M
p
≤10
6
M
w
/M
n
<1,05
M
p
>10
6
M
w
/M
n
<1,20
M
p
- masa cząsteczkowa wzorca w maksimum piku
1.6. Opis metody badań
1.6.1. Przygotowanie roztworów wzorcowych polistyrenu
Wzorce polistyrenowe rozpuszcza się przez ostrożne mieszanie w wybranym eluencie. Podczas przygotowania
roztworów należy stosować się do zaleceń producenta.
Stężenia wybranych wzorców zależą od różnych czynników, na przykład wstrzykiwanej objętości, lepkości
roztworu i czułości detektora analitycznego. Maksymalną objętość wstrzykiwaną należy dostosować do długości
kolumny, aby zapobiec przeładowaniu. Typowe objętości wstrzykiwane w celu rozdziałów analitycznych
z zastosowaniem GPC z kolumną 30 cm
×7,8 mm mieszczące się na ogół pomiędzy zawartościami 40 i 100 µl.
Można stosować większe objętości, ale nie powinny one przekraczać 250
µl. Przed kalibracją kolumny należy
oznaczyć optymalny stosunek pomiędzy wstrzykiwaną objętością i stężeniem.
1)
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds. (1979). Modern Size Exclusion Liquid Chromatography,
J. Wiley and Sons.
2)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
- 109 -

1.6.2. Przygotowanie roztworu próbki
Do przygotowania roztworów próbek stosuje się takie same wymagania. Próbkę rozpuszcza się ostrożnie
wstrząsając w odpowiednim rozpuszczalniku, na przykład w tetrahydrofuranie (THF). W żadnym wypadku nie
należy jej rozpuszczać stosując łaźnię ultradźwiękową. W razie potrzeby roztwór próbki oczyszcza się za
pomocą membrany filtracyjnej o wielkości porów pomiędzy 0,2 i 2
µm.
W sprawozdaniu należy uwzględnić obecność nierozpuszczonych cząstek, ponieważ mogą one pochodzić
z substancji o dużym ciężarze cząsteczkowym. W celu oznaczenia zawartości procentowej rozpuszczonych
cząstek należy zastosować odpowiednią metodę. Roztwór należy wykorzystać w ciągu 24 godzin.
1.6.3. Korekta ze względu na zawartość zanieczyszczeń i dodatków
Zwykle konieczna jest korekta ze względu na zawartość cząstek o M<1000, spowodowana udziałem obecnych
związków niepolarnych (np zanieczyszczeń i/lub dodatków), chyba że mierzona zawartość wynosi <1%. Osiąga
się to przez bezpośrednią analizę roztworu polimeru lub GPC eluatu.
W przypadkach gdy eluat po przejściu przez kolumnę jest zbyt rozcieńczony do dalszej analizy, należy go
zatężyć. Może być konieczne odparowanie eluatu do sucha i ponowne rozpuszczenie go. Stężenie eluatu należy
zmieniać w warunkach, które zapewnią, że nie nastąpią zmiany w eluacie. Traktowanie eluatu po etapie GPC
zależy od metody analitycznej stosowanej w celu oznaczania ilościowego.
1.6.4. Aparatura
- pojemnik na rozpuszczalnik,
- urządzenie do odgazowania (gdy to wskazane),
- pompa,
- wytłumiacz pulsacji (gdy to wskazane),
- układ dozujący,
- kolumny chromatograficzne,
- detektor,
- przepływomierz (gdy to wskazane),
- urządzenie rejestrujące i przetwarzające dane,
- naczynie na odpady.
Należy zapewnić, aby układ GPC był obojętny w stosunku do stosowanych rozpuszczalników (na przykład
kapilary stalowe do THF).
1.6.5. Układ dozujący i układ zasilania rozpuszczalnikiem
Na kolumnę, w ściśle zdefiniowanej strefie, wprowadza się ręcznie lub za pomocą automatycznego dozownika
określoną objętość próbki. Pociągając lub wciskając zbyt szybko tłok strzykawki podczas operacji manualnych
można spowodować zmiany w
obserwowanym rozkładzie mas cząsteczkowych. Układ zasilania
rozpuszczalnikiem powinien być, w miarę możliwości, bezpulsacyjny, dzięki zainstalowaniu wytłumiacza
pulsacji. Szybkość przepływu powinna być rzędu 1 ml/min.
1.6.6. Kolumna
Polimer (w zależności od próbki) ocenia się stosując jedną albo kilka kolumn połączonych kolejno. W handlu
dostępne są liczne materiały porowate do kolumn, które mają określone właściwości (np wielkość porów,
granice wykluczania). Wybór żelu do rozdzielania lub długości kolumny zależą zarówno od właściwości próbki
(objętości hydrodynamicznych, rozkładu masy cząsteczkowej) jak i od określonych warunków rozdziału, takich
jak rozpuszczalnik, temperatura i szybkość przepływu
1)
DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
2)
Yau, W.W., Kirkland, J.J., and Bly, D.D. eds. (1979). Modern Size Exclusion Liquid Chromatography,
J. Wiley and Sons.
3)
ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight
Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography – GPC). American
Society for Testing and Materials, Philadelphia, Pennsylvania.
- 110 -
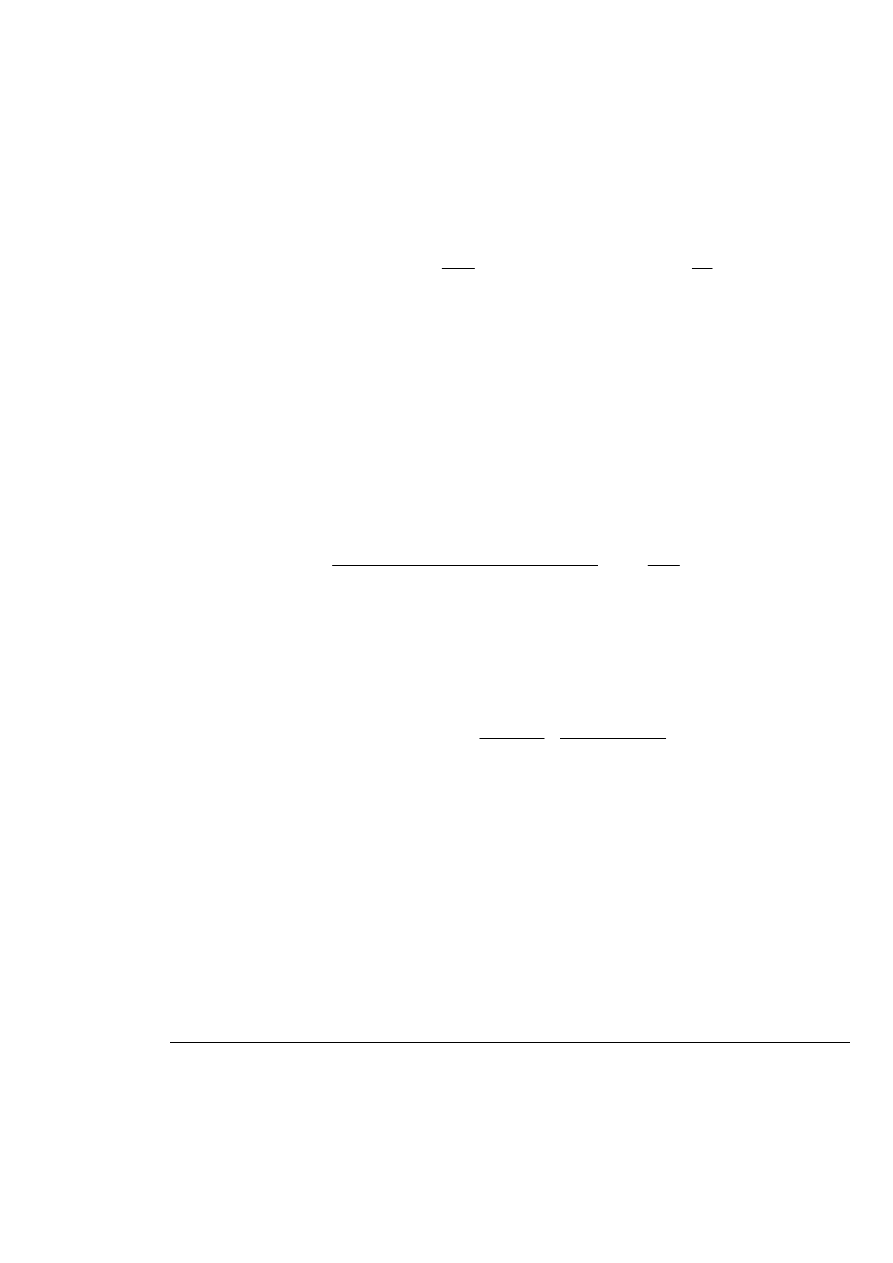
1.6.7. Półki teoretyczne
Kolumnę lub zestaw kolumn stosowany do rozdziału należy scharakteryzować za pomocą liczby półek
teoretycznych. Obejmuje to w przypadku THF jako rozpuszczalnika wymywającego, wprowadzenie na kolumnę
o znanej długości roztworu etylobenzenu lub innej odpowiedniej rozpuszczonej substancji niepolarnej. Liczba
półek teoretycznych obliczana jest z następujących równań:
2
1/2
e
)
W
V
5,54(
N
=
lub
2
e
)
W
V
16(
N
=
gdzie:
N –
liczba
półek teoretycznych,
V
e
– objętość elucji w maksimum piku,
W –
szerokość podstawy piku,
W
1/2
– szerokość piku w połowie wysokości.
1.6.8. Skuteczność rozdziału
Oprócz liczby półek teoretycznych, która decyduje o szerokości pasma, rolę odgrywa również skuteczność
rozdziału, określana przez nachylenie krzywej kalibracyjnej. Skuteczność rozdziału kolumny otrzymuje się
z następującej zależności:
⎥
⎦
⎤
⎢
⎣
⎡
≥
−
2
3
)
e(10M
M
e,
cm
cm
0
,
6
kolumny
przekroju
i
powierzchn
pole
V
V
x
x
gdzie:
V
e,Mx
– objętość elucji dla polistyrenu o masie cząsteczkowej M
x
,
V
e,(10Mx)
– objętość elucji dla polistyrenu o 10 razy większej masie cząsteczkowej.
Rozdział układu definiuje się na ogół następująco:
)
M
/
M
(
log
1
W
W
V
V
2
R
1
2
10
2
1
e2
e1
1,2
×
+
−
×
=
gdzie:
V
e1
, V
e2
– objętości elucji dwóch wzorców polistyrenowych w maksimum piku,
W
1
, W
2
– szerokości podstaw pików,
M
1
, M
2
– masy
cząsteczkowe w maksimum piku (powinny różnić się 10 razy).
Wartość R dla układu kolumn powinna być większa niż 1,7
1.6.9. Rozpuszczalniki
Wszystkie rozpuszczalniki powinny mieć wysoką czystość (stosuje się THF o czystości 99,5%). Pojemnik na
rozpuszczalnik (gdy to konieczne w atmosferze gazu obojętnego) powinien być odpowiednio duży w celu
kalibracji kolumny i licznych analiz próbki. Rozpuszczalnik należy odgazować za pomocą pompy, zanim
zostanie podany na kolumnę.
1)
ASTM D 5296-92 (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight
Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for
Testing and Materials, Philadelphia, Pennsylvania.
- 111 -

1.6.10. Kontrola temperatury
Temperatura krytycznych składników układu (pętli dozującej, kolumn, detektora i przewodów) powinna być
stała i odpowiednia dla danego rozpuszczalnika.
1.6.11. Detektor
Celem detektora jest ilościowa rejestracja stężenia próbki wymytej z kolumny. W celu zapobieżenia
niepożądanemu poszerzeniu pików należy stosować jak najmniejszą objętość kuwety celki detektora. Nie
powinna być ona większa niż 10
µl, z wyjątkiem detektorów lepkościowych i do światła rozproszonego. W celu
detekcji stosuje się na ogół refraktometrię różnicową. Jednak, o ile wymagają tego szczególne właściwości
próbki lub rozpuszczalnika wymywającego, można stosować również inne rodzaje detektorów, np UV/VIS, IR,
detektory lepkościowe itd.
2. WYNIKI BADAŃ
W celu szczegółowej oceny kryteriów jak również wymagań dotyczących zbierania i przetwarzania danych
należy odwołać się do normy DIN
Dla każdej próbki należy przeprowadzić dwa niezależne doświadczenia i analizować je indywidualnie. Ważne
jest, aby we wszystkich przypadkach wykonać oznaczenia dla ślepych prób, traktowanych w tych samych
warunkach, co próbka.
Należy również wyraźnie wykazać, że mierzone wartości są wartościami względnymi równoważnymi masom
cząsteczkowym stosowanego wzorca.
Po oznaczeniu objętości retencji lub czasów retencji (możliwie skorygowanych z zastosowaniem wzorca
wewnętrznego), wykreśla się wartość log M
p
(M
p
- maksimum piku wzorca do kalibracji) względem jednej
z tych wielkości. Konieczne są co najmniej dwa punkty kalibracyjne na dziesięć mas cząsteczkowych i wymaga
się co najmniej pięciu punktów pomiarowych dla pełnej krzywej, która powinna obejmować szacowaną masę
cząsteczkową próbki. Punkt końcowy krzywej kalibracyjnej dla małych mas cząsteczkowych określa się za
pomocą n-heksylobenzenu lub innej odpowiedniej niepolarnej substancji. Masy cząsteczkowe średnie liczbowo
i średnie wagowo oznacza się na ogół za pomocą elektronicznego przetworzenia danych. W przypadku
przekształceń manualnych należy oprzeć się na normie ASTM D 3536-91
.
Jeżeli na kolumnie pozostaje jakiś nierozpuszczalny polimer, jego masa cząsteczkowa jest prawdopodobnie
większa niż masy we frakcji rozpuszczalnej, i jeżeli się go nie uwzględni, będzie wpływał on na podwyższenie
zawartości małych mas cząsteczkowych. Wskazówki, dotyczące korygowania zawartości niskich mas
cząsteczkowych w przypadku nierozpuszczalnych polimerów podano w rozdziale „Wskazówki dotyczące
korygowania zawartości cząstek o małej masie cząsteczkowej ze względu na obecność nierozpuszczalnego
polimeru”.
Krzywa rozkładu powinna mieć postać tabeli lub rysunku (częstotliwość różnicowa lub suma zawartości
procentowych względem log M).
Przy przedstawianiu graficznym dziesięciu masom cząsteczkowym powinno odpowiadać około 4 cm szerokości,
a maksymalny pik powinien mieć około 8 cm wysokości.
W przypadku całkowych krzywych rozkładu różnica rzędnych pomiędzy 0 i 100% powinna wynosić
około 10 cm.
3. SPRAWOZDANIE
Sprawozdanie powinno zawierać, z uwzględnieniem zakresu przeprowadzonych badań, następujące informacje:
1) Badana substancja:
c) dostępne informacje dotyczące badanej substancji (tożsamość, dodatki, zanieczyszczenia),
d) - opis postępowania z próbką, spostrzeżenia, problemy;
1) DIN 55672 (1995). Gelpermeationschromatographie (GPC) mit Tetrahydrohuran (THF) als Elutionsmittel,
Teil 1.
2) ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight
Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography – GPC). American
Society for Testing and Materials, Philadelphia, Pennsylvania.
- 112 -

2) Oprzyrządowanie:
j) pojemnik na eluent, gaz obojętny, odgazowanie eluenta, skład eluenta, zanieczyszczenia,
k) pompa, wytłumiacz pulsacji, układ dozujący,
l) kolumny rozdzielcze (producent, wszystkie informacje dotyczące charakterystyki kolumn, takie jak
wielkość porów, rodzaj materiału rozdzielnego itd., liczba, długość i kolejność stosowanych kolumn),
m) liczba półek teoretycznych kolumny (lub kombinacji), skuteczność rozdziału (rozdzielczość układu),
n) informacje na temat symetrii pików,
o) temperatura kolumny, sposób kontroli temperatury,
p) detektor (zasada pomiaru, objętość kuwety),
q) przepływomierz, o ile jest stosowany (producent, zasada pomiaru),
r) układ rejestrujący i przetwarzający dane (sprzęt komputerowy i oprogramowanie);
3) Kalibracja układu:
e) szczegółowy opis metody stosowanej do sporządzania krzywej kalibracyjnej,
f) informacje na temat kryteriów jakościowych tej metody (np współczynnika korelacji, sumy
kwadratów błędów, itd.),
g) informacje dotyczące wszelkich ekstrapolacji, założeń i przybliżeń dokonanych podczas procedury
doświadczalnej oraz oceny i przetwarzania danych,
h) wszelkie pomiary zastosowane w celu konstruowania krzywej kalibracyjnej należy zestawić w tabeli
zawierającej następujące informacje dotyczące każdego punktu kalibracji:
- nazwa próbki,
- producent próbki,
- wartości charakteryzujące wzorce M
p
, M
n
, M
w
i M
w
/M
n
, dostarczane przez producenta lub
pochodzące z kolejnych pomiarów, wraz ze szczegółami odnośnie metody oznaczenia,
- dozowana objętość i stężenie dozowanej próbki,
- wartość M
p
stosowana do kalibracji,
- objętość elucji lub skorygowany czas retencji mierzony w maksimum piku,
- M
p
obliczona dla maksimum piku,
- błąd procentowy obliczonej wartości M
p
i wartości kalibracji;
4) Informacje dotyczące zawartości polimerów o małej masie cząsteczkowej:
a) opis metod stosowanych podczas analizy i sposób w jaki prowadzono doświadczenia,
b) informacje dotyczące zawartości procentowej cząstek o małej masie cząsteczkowej (w/w) w stosunku
do całej próbki,
c) informacje na temat zanieczyszczeń, dodatków i innych niepolarnych cząstek w procentach wagowych
w stosunku do całej próbki;
5) Ocena:
a) ocena na podstawie czasu: metody stosowane do zapewnienia wymaganej powtarzalności (metoda
korygowania, wzorzec wewnętrzny, itd.),
b) informacje o tym czy ocenę przeprowadzono na podstawie objętości elucji czy czasu retencji,
c) informacje dotyczące granic oceny, jeżeli pik nie jest w pełni analizowany,
d) opis metod wygładzania danych, jeżeli były stosowane,
e) procedury przygotowania i wstępnej obróbki próbki,
f) obecność nierozpuszczonych cząstek, jeżeli występują,
g) objętość dozowana (
µl) i stężenie dozowanej próbki (mg/ml),
h) spostrzeżenia dotyczące wpływów, które prowadzą do odchyleń od idealnego profilu GPC,
i) szczegółowy opis wszelkich modyfikacji procedur badawczych,
j) szczegóły dotyczące zakresów błędów,
k) wszelkie inne informacje i spostrzeżenia istotne dla interpretacji wyników.
- 113 -

WSKAZÓWKI DOTYCZĄCE KORYGOWANIA ZAWARTOŚCI CZĄSTEK
O MAŁEJ MASIE CZĄSTECZKOWEJ ZE WZGLĘDU NA OBECNOŚĆ
NIEROZPUSZCZALNEGO POLIMERU
Gdy w próbce obecny jest nierozpuszczalny polimer, powoduje to ubytek masy podczas analizy GPC.
Nierozpuszczalny polimer jest nieodwracalnie zatrzymywany na kolumnie lub w trakcie sączenia próbki,
podczas gdy część rozpuszczalna próbki przechodzi przez kolumnę. W przypadku, gdy możliwe jest zmierzenie
lub oszacowanie przyrostu współczynnika załamania polimeru (dn/dc), można oszacować ubytek masy próbki na
kolumnie. W takim przypadku dokonuje się korekty stosując zewnętrzną kalibrację z materiałami odniesienia
o znanym stężeniu i dn/dc w celu kalibracji wskazań refraktometru. W powyższym przykładzie stosuje się
wzorzec poli(metaktrylanu metylu) (pMMA).
Podczas kalibracji zewnętrznej w celu analizy polimerów akrylowych analizuje się metodą GPC wzorzec
pMMA o znanym stężeniu w tetrahydrofuranie, a uzyskane dane wykorzystuje się do określenia stałej
refraktometru zgodnie z równaniem:
K = R/(C
×V×dn/dc)
gdzie:
K –
stała refraktometru (w mikrowoltosekundach/ml)
R –
wartość sygnału dla wzorcowego pMMA (w mikrowoltosekundach/ml)
C –
stężenie wzorcowego pMMA (w mg/ml)
V –
objętość wstrzykiwaną (w ml)
dn/dc – przyrost
współczynnika załamania dla pMMA w tetrahydrofuranie (w ml/mg)
Dla wzorcowego pMMA typowe są następujące dane:
R = 2937891
C = 1,07 mg/ml
V = 0,1 ml
dn/dc = 9
× 10
-5
ml/mg
Uzyskaną wartość K, 3,05
× 10
11
, wykorzystuje się następnie do obliczenia teoretycznej wartości sygnału
detektora, gdy 100% wstrzykniętego polimeru wymywa się przez detektor.
- 114 -

A.20. ROZPUSZCZANIE/EKSTRAKCJA POLIMERÓW W WODZIE
1. METODA
Niniejsza metoda jest odpowiednikiem poprawionej wersji metody OECD TG 120 (1997). Dalsze informacje
techniczne podano w piśmiennictwie 1).
1.1. Wstęp
Dla niektórych polimerów, takich jak polimery emulsyjne, mogą być konieczne wstępne prace przygotowawcze
zanim będzie można zastosować podaną tu metodę. Metoda ta nie nadaje się dla polimerów ciekłych i dla
polimerów, które reagują z wodą w warunkach badania.
Gdy metoda ta okaże się niepraktyczna albo nie jest możliwa, rozpuszczanie/ekstrakcję można badać za pomocą
innych metod. W takich przypadkach należy podać dokładne szczegóły i uzasadnienie dla stosowanej metody.
1.2. Substancje kontrolne
Brak.
1.3. Zasada metody badań
Rozpuszczanie/ekstrakcję polimerów w środowisku wodnym oznacza się stosując metodę z kolbą (Metoda:
„Rozpuszczalność w wodzie”) z modyfikacjami opisanymi poniżej.
1.4. Kryteria jakościowe
Brak.
1.5. Opis metody badań
1.5.1. Aparatura
Metoda wymaga stosowania następującej aparatury:
- urządzenie kruszące, np młyn do wytwarzania cząstek o znanej wielkości,
- wytrząsarka z możliwością kontroli temperatury,
- układ filtrów membranowych,
- odpowiednie wyposażenie analityczne,
- sita znormalizowane.
1.5.2. Przygotowanie próbki
Reprezentatywną próbkę należy najpierw zredukować do cząstek o wielkości pomiędzy 0,125 a 0,25 mm
stosując odpowiednie sita. Ze względu na stabilność próbki lub podczas procesu rozdrabniania może być
wymagane chłodzenie. Substancje o charakterze gumowatym można rozdrabniać w temperaturze ciekłego
azotu
Jeżeli nie można uzyskać frakcji o wymaganej wielkości cząstek, należy podjąć działania w celu maksymalnego
zmniejszenia wymiaru cząstek i opisać wynik. Konieczne jest podanie w
sprawozdaniu sposobu
przechowywania rozdrobnionej próbki przed badaniem.
1.5.3. Sposób postępowania
Trzy próbki badanej substancji o masie 10 g odważa się do trzech kolb zamykanych szklanym korkiem i do
każdej z nich dodaje się 1000 ml wody. Jeżeli operacje z ilością 10 g polimeru okażą się niewygodne, należy
zastosować inną najwyższą ilość, którą można użyć i odpowiednio dobrać objętość wody.
Kolby zamyka się szczelnie i wytrząsa w 20
°C. Należy zastosować urządzenie do wytrząsania lub mieszania,
mogące pracować w stałej temperaturze.
1)
DIN 53733 (1976) Zerkleinerung von Kunststofferzeugnissen für Prüfzwecke.
- 115 -

Po okresie 24 godzin zawartość każdej kolby odwirowuje się lub sączy i oznacza się stężenie polimeru
w klarownej fazie wodnej za pomocą odpowiedniej metody analitycznej. Jeżeli nie są dostępne odpowiednie
metody analityczne dla fazy wodnej, zdolność do całkowitego rozpuszczania/ekstrakcji można ocenić na
podstawie suchej masy pozostałości z sączenia lub osadu z odwirowania.
Na ogół konieczne jest ilościowe określenie różnicy pomiędzy zanieczyszczeniami i dodatkami z jednej strony,
a cząstkami o niskiej masie cząsteczkowej z drugiej. W przypadku oznaczania grawimetrycznego konieczne jest
również wykonanie ślepej próby bez użycia badanej substancji w celu określenia pozostałości wynikłych ze
stosowanej procedury doświadczalnej.
Rozpuszczanie/ekstrakcję polimerów w wodzie w 37
°C przy wartościach pH=2 i pH=9 można oznaczać w ten
sam sposób, co opisany dla prowadzenia doświadczenia w 20
°C. Wartości pH można uzyskać przez dodawanie
odpowiednich roztworów buforowych lub odpowiednich kwasów, zasad, takich jak: kwas solny, kwas octowy,
wodorotlenek sodu lub potasu albo NH
3
o czystości analitycznej.
W zależności od stosowanej metody analitycznej należy przeprowadzić jedno lub dwa badania. Gdy dla
składnika polimeru istnieją wystarczająco specyficzne metody bezpośredniej analizy fazy wodnej, powinno
wystarczyć jedno opisane powyżej badanie. Jednak, gdy takie metody nie są dostępne i oznaczanie
rozpuszczania/ekstrakcji polimeru ogranicza się do analizy pośredniej za pomocą oznaczania jedynie całkowitej
zawartości węgla organicznego (CWO) w ekstrakcie wodnym, należy przeprowadzić badanie dodatkowe. To
dodatkowe badanie należy również przeprowadzić trzykrotnie, stosując 10 razy mniejsze próbki polimeru i takie
same ilości wody jak użyte w pierwszym badaniu.
1.5.4. Analiza
1.5.4.1. Badanie prowadzone z jedną wielkością próbki
Mogą być znane metody bezpośredniej analizy składników polimerów w fazie wodnej. Można również
rozważyć pośrednią analizę rozpuszczonych/ekstrahowanych składników polimerów za pomocą oznaczania
ogólnej zawartości części rozpuszczonych i
korygowania na zawartość określonych składników
niepolimerycznych.
Analiza fazy wodnej na ogólną zawartość substancji polimerycznych możliwa jest za pomocą wystarczająco
czułej metody, np:
- z zastosowaniem rozkładu do CO
2
za pomocą nadsiarczanu albo dichromianu, a następnie oznaczania
metodą IR lub za pomocą analizy chemicznej,
- atomowej spektrometrii absorpcyjnej (AAS) albo równoważenia emisji plazmy wzbudzonej indukcyjnie
(ICP) w przypadku silikonów lub polimerów zawierających metal,
- absorpcji w UV lub spektrofluorymetrii w przypadku polimerów arylowych,
- LC-MS dla próbek polimerów o niskiej masie cząsteczkowej,
- przez odparowanie do sucha wodnego ekstraktu pod obniżonym ciśnieniem i spektroskopową (IR, UV itp.),
za pomocą AAS/ICP analizę pozostałości.
Jeżeli nie można zastosować analizy fazy wodnej jako takiej, należy ekstrahować ekstrakt wodny nie
mieszającym się z wodą organicznym rozpuszczalnikiem, np chlorowanym węglowodorem. Następnie
odparowuje się rozpuszczalnik, a pozostałość analizuje jak wyżej na zawartość polimeru. Wszelkie składniki tej
pozostałości zidentyfikowane jako zanieczyszczenia lub dodatki odejmuje się w celu oznaczenia stopnia
rozpuszczenia/ekstrakcji polimeru jako takiego.
Gdy obecne są stosunkowo duże ilości takich substancji, może być konieczne poddanie pozostałości na przykład
analizie HPLC lub GC w celu odróżnienia obecnych zanieczyszczeń od monomeru i pochodnych monomeru tak,
aby można było oznaczyć prawdziwą zawartość tych ostatnich.
W niektórych przypadkach może wystarczyć proste odparowanie do sucha rozpuszczalnika organicznego
i zważenie suchej pozostałości.
1.5.4.2. Badanie prowadzone z dwiema różnymi wielkościami próbek
Wszystkie wodne ekstrakty analizuje się pod kątem CWO.
Wykonuje się oznaczenie grawimetryczne nierozpuszczonej/niewyekstrahowanej części próbki. Jeżeli po
odwirowaniu lub przesączeniu zawartości każdej kolby na ściankach naczynia pozostają pozostałości polimeru,
kolbę należy płukać przesączem, dopóki pozostałości nie przestaną być widoczne. Następnie przesącz znów
sączy się lub odwirowuje. Pozostałości na sączku lub w probówce wirówkowej suszy się w 40
°C pod
obniżonym ciśnieniem i waży. Kontynuuje się suszenie, aż do osiągnięcia stałej masy.
- 116 -

2. WYNIKI BADAŃ
2.1. Badanie przeprowadzone z jedną wielkością próbki
Należy podać wyniki dla każdej z trzech kolb oraz wartości średnie i wyrazić je w jednostkach masy na objętość
roztworu (zwykle mg/ml) lub masy na masę próbki polimeru (zwykle mg/g). Ponadto należy również podać
utratę masy próbki (obliczoną jako masa rozpuszczonej substancji podzielona przez masę początkowej próbki).
Należy obliczyć względne odchylenia standardowe (RSD). Należy podać oddzielne dane dla całej substancji
(polimeru + głównych dodatków itd.) oraz dla samego polimeru (to jest po odjęciu udziału tych dodatków).
2.2. Badanie prowadzone z dwiema różnymi wielkościami próbki
Należy podać pojedyncze wartości CWO ekstraktów wodnych z dwóch trzykrotnie prowadzonych badań
i wartość średnią dla każdego doświadczenia w jednostkach masy na objętość roztworu (zwykle mgC/l), jak
również w jednostkach masy na masę początkowej próbki (zwykle mgC/g).
Jeżeli nie ma różnicy pomiędzy wynikami dla wysokiej i niskiej wartości stosunku próbka/woda, może to
wskazywać, że wszystkie dające się ekstrahować składniki rzeczywiście zostały wyekstrahowane. W takim
przypadku analiza bezpośrednia nie jest na ogół konieczna.
Poszczególne masy pozostałości należy podawać i wyrażać jako zawartość procentową początkowych mas
próbek. Należy obliczyć średnie dla doświadczeń. Różnice pomiędzy 100 i znalezionymi zawartościami
procentowymi oznaczają zawartości procentowe rozpuszczalnej i dającej się wyekstrahować substancji
w pierwotnej próbce.
3. SPRAWOZDANIE
Sprawozdanie powinno zawierać, z uwzględnieniem zakresu przeprowadzonych badań, następujące informacje:
1) Badana substancja:
a) dostępne informacje na temat badanej substancji (tożsamość, dodatki, zanieczyszczenia, zawartość
substancji o niskich ciężarach cząsteczkowych);
2) warunki doświadczenia:
a) opis stosowanych procedur i warunków doświadczenia,
b) opis metod analizy i detekcji;
3) wyniki:
a) wyniki rozpuszczania/ekstrakcji w mg/l; poszczególne i średnie wartości uzyskane z badań ekstrakcji,
w różnych roztworach, dane dotyczące zawartości polimeru, zanieczyszczeń, dodatków, itd.,
b) wyniki rozpuszczania/ekstrakcji w mg/g polimeru,
c) wartości TOC ekstraktów wodnych, masy substancji rozpuszczonej i obliczone zawartości procentowe,
o ile zostały zmierzone,
d) pH każdej próbki,
e) informacje na temat wyników ślepej próby,
f) gdy to konieczne, odnośniki na temat nietrwałości chemicznej badanej substancji, zarówno podczas
badania jak i analizy,
g) wszelkie informacje istotne do interpretacji wyników.
- 117 -
Wyszukiwarka
Podobne podstrony:
WYKŁAD 2 ŚREDNIA MASA CZĄSTECZKOWA
06 Średnia Masa Cząsteczkowa Polimeru, wiskozymetria (alkohol)
zadania dodatkowe - masa cząsteczkowa, …………………&hell
Chemia Fizyczna masa czasteczkowa id 112216
kartk. - masa czasteczkowa i prawo stałości składu, chemia, kartkówki
07 Masa cząsteczkowa polimerówid 6896 ppt
sprawozdanie1 masa cząsteczkowa (ostwald) (2)
polimery fizykochemia masa czasteczkowa
06 Średnia Masa Cząsteczkowa Polimeru, wiskozymetria (glikol), Marek Mokrzycki IM sem
Masa atomowa i cząsteczkowa, NAUKA, chemia, lab
MASA ATOMOWA I CZĄSTECZKOWA PIERWISTKA
Ważne informacje o budowie cząsteczek można uzyskać?dając wzajemne oddziaływanie materii obdarzonej
Cząsteczkowa budowa materii
Obliczanie masy cząsteczkowej
WYKúAD 4 MASA» J CH cd
WYKúAD 2 TECHNIKI MASA»U
więcej podobnych podstron