Co to jest STS ?
STS mapping - mapowanie miejsc znakowanych sekwencją; rodzaj mapowania fizycznego; wykorzystujemy w nim komplet pociętych fragmentów dużego genomu (np. ludzkiego) wstawionych w np. sztuczne chromosomy drożdżowe YAC; fragmenty te muszą zawierać specyficzne sekwencje - każda z sekwencji występuje tylko raz w całym genomie, ale w każdym fragmencie występują przynajmniej dwie różne sekwencje specyficzne; tworzymy specjalnie wyznakowane sondy, komplementarne do odpowiednich sekwencji (każda sonda wyznakowana inaczej, w zależności od tego do której sekwencji jest komplementarna); jeśli ta sama sonda przyłączy się do sekwencji w 2 różnych fragmentach genomu (a do drugiej sekwencji przyłączą się w obu przypadkach inne sondy), to oznacza, że fragmenty te zachodzą na siebie; wykorzystując te obserwacje można uporządkować bibliotekę fragmentów genomu i ułożyć mapę STS.
Określ liczbę możliwych struktur pentapeptydu, jeśli wykres Ramachandrana wygląda następująco:
Będą to trzy struktury:
- lewy górny obszar - β kartka
- lewy dolny obszar - prawoskrętna α helisa
- prawy obszar - lewoskrętna α helisa
Oblicz odległość między dwoma punktami na drzewie metodą UPGMA (tu odpowiedni schemat drzewa).
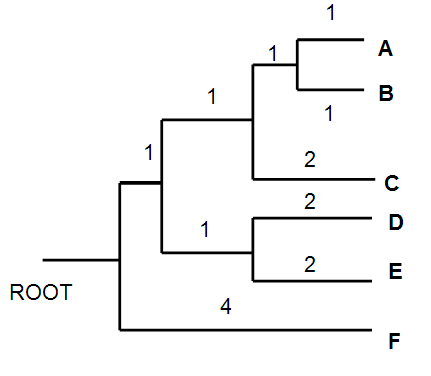
np. tu odległość między B i E wynosi:
1+1+1+1+2=6
Albo odległość między AB i C wynosi:
1+1+2=4
Co oznacza węzeł na drzewie pokrewieństwa?
Węzeł - reprezentuje jednostkę taksonomiczną (populację, organizm, gen), może przedstawiać współcześnie istniejący takson lub jego przodka.
Grupy jakich aminokwasów mogą tworzyć wiązania wodorowe?
Wiązania wodorowe są utworzone przez wszystkie grupy CO i NH aminokwasowego łańcucha głównego z wyjątkiem aminokwasów skrajnych (np. na końcu helisy α)
Dlaczego opuszczamy obszary o małym dopasowaniu?
Opuszczamy je, ponieważ chcemy uzyskać optymalne dopasowanie. Porównujemy każdą parę znaków 2 sekwencji uwzględniając wszelkie dopasowania, niedopasowania i przerwy, po czym wybierana jest tylko optymalna ścieżka (optymalne dopasowanie)
Wady metody ab initio
- wymagają ogromnych mocy obliczeniowych (trzeba wykonać ogromną ilość obliczeń)
- za pomocą tej metody można dziś obliczać tylko proste związki, bo te większe i bardziej skomplikowane wymagają zbyt wielu obliczeń
Ograniczenia i wady NMR
- ograniczenia wielkości białek - metoda ta nie może być wykorzystywana do pomiarów białek o masach przekraczających 30kD
- kosztowne znakowanie izotopowe
- wymaga dużej ilości materiału oraz długotrwałych pomiarów
- mniejsza dokładność niż w krystalografii
Które aminokwasy są najbardziej mutabilne w metodzie PAM?
Najbardziej mutabilne są te, które mają najwyższe prawdopodobieństwo substytucji przez inny aminokwas - najwyższe wartości w okienkach macierzy (wartości prawdopodobieństw nie mogą leżeć na diagonali)
Jak rozpoznać nieuporządkowane obszary metodą krystalograficzną?
W metodzie krystalograficznej na regiony nieuporządkowane wskazuje brak współrzędnych (koordynatów) na danym obszarze.
Narysuj wiązania wodorowe w niżej wymienionych wzorach strukturalnych.
Wyznacz konsensus dla pewnych sekwencji:
AAAAAAA
ABBBACA
ABCBADA
ABDACBA
100%-75%-25%-50%-75%-25%-100%
LCRy - tu nie wiem
Co to jest proteom, transkryptom?
proteom - zespół wszystkich białek zawartych w organizmie
transkryptom - zespół wszystkich transkryptów (sekwencji mRNA) zawartych w organizmie
Definicja genu.
GEN - podstawowa jednostka dziedziczenia; sekwencja genomowa (DNA lub RNA) bezpośrednio kodująca fukcjonalne produkty w postaci RNA lub białek
Jaki rodzaj mapy przedstawiono (genetyczna/fizyczna)?
Wiadomo
Aj = Pe/Po, uzupełnić: a)jeżeli Aij<0 to ………. jest większe b) jeżeli Aij>0 to……….. jest mniejsze c) jeżeli Aij=0 to………….. jest takie samo d) jeżeli Aij + 1 to logarytm 10 x większy
Tu chyba jest błąd i powinno być Po/Pe. dla Aij = Po/Pe:
jeżeli Aij<0 to prawdopodobieństwo substytucji jest mniejsze
jeżeli Aij>0 to prawdopodobieństwo substytucji jest mniejsze
jeżeli Aij=0 to prawdopodobieństwo substytucje jest takie samo
jeżeli Aij +1 to prawdopodobieństwo substytucji jest 10 razy większe
Które genomy nie zostały w pełni zsekwencjonowane: Canis lupus, Arabitopsis thaliana, E. coli K12, Saccharomyces pombe, Bos taurus?
Canis lapus, Bos Taurus
Wypisz 3 metody tworzenia drzew filogenetycznych, podaj tylko pełne nazwy
- Metoda maksymalnej parsymonii MP
- Metoda maksymalnej wiarygodności ML
- Metody oparte na odległościach - metody dystansowe (p-dystans)
Który program wybrać przy przeszukiwaniu bazy SwissProt mając daną sekwencję X aminokwasową w poszukiwaniu homologów?
blastp - białko na białko
blastn - sekw nukleotydowa na sekw nukleotydową
blastx - sekw nukleotydowa na białko
tblastn - białko na transkrypt
tblastx - transkrypt na transkrypt
Jak najlepiej przedstawić wiązanie atom-ligand (metody do wyboru: all-atom, powierzchniowo, strukturą wstążki)?
Chyba all-atom
Jakiego programu użyć do przewidywania homologów struktur białkowych?
SwissModel
Co to jest OTU?
OTU - operacyjna jednostka taksonomiczna; dla poszczególnych OTU analizujemy zależności filogenetyczne, np. metodą UPGMA czy NJ
Ograniczenia metody krystalografii w tworzeniu struktur trzeciorzędowych białek, wady krystalografii
- trudność rozwiązania struktury białka posiadającego dynamiczną pętlę
- konieczność posiadania kryształu białka o wysokiej jakości, którego „wyhodowanie” jest procesem długotrwałym i często wymaga dużej ilości materiału
Długość wiązania N-C, C-C w białkach
N-C - 1,32Å
C-C - 1,51Å
Co to jest STRs? a ) mikrosatelity b) minisatelity c) powtorzenia tandemowe d) markery genetyczne
Co to jest RFLP - marker genetyczny, miejsce cięcia enzymów restrykcyjnych
E value = 10 - co to znaczy?
Oznacza to, że oczekuje się 10 dopasowań z punktacją równą przynajmniej S.
Możliwość dopasowania dwóch sekwencji z istotna wartością S oznacza wskazówka że kodowane białka mają podobną strukturę i funkcje, oraz że mogą być homologami.
5.STS to : mapowanie miejsc znakowanych sekwencją , jedna z metod mapowania fizycznego
14.do metod fizycznego mapowania genów nie należy:
mapowanie restrykcyjne
fish
mapowanie miejsc znaczonych sekw.
Krzyżówka trójpunktowa
7.Skrót EST oznacza
znaczniki sekwencji ulegających ekspresji
expressed segment tags
expressed segment flags
żadne
8.E = 10 oznacza że:
p-stwo zaobserwowania podobieństwa równego zaobserwowanej wartości S między dwiema losowymi sekwencjami =10
oczekiwana liczba losowych dopasowań o wartości S wyliczonych dla znalezionego alighmentu =10
znaleziono 10 dopasowań o takiej samej wartości
wszystkie prawidłowe
13.wymień przynajmniej 2 zastosowania wielokrotnego alighmentu.
1. Do utworzenia drzew filogenetycznych
2.
10. Porównując odległe ewolucyjnie sekwencje aminokwasów powinniśmy oprzeć system wartościowania alighmentu na:
macierzach PAM 250 i BLOSUM 45
PAM 1 i BLOSUM 62
PAM 120 i BLOSUM 80
Unikać macierzy PAM i BLOSUM
16. program t blast x służy do:
przeszukiwania bazy nukleotydowej sekwencji DNA
-`'-`'-`'-`'-`'-`'-`'-`'- białkowej sekwencji aminokwasowych
-`-`'-`'-`'-`'-`'-`'-`'-`'nukleotydowej (tłumaczonej w ??? )sekwencją nukleotydową (także tłumaczoną na aa)
tworzenia macierzy PSSM
7.Algorytm programu Basic Local Aligsment Search Tool opiera się na Tworzenie listy słów znaków o zadanej długości i określenie sąsiadujących (podobnych) słów - neighorhood words (dla sekwencji aminokwasowych).
10.Podaj metody tworzenia drzew filogen.,które się nie opierają na macierzach odległości MP (metoda maksymalnej parsymonii) i ML (metoda maksymalnej wiarygodności)
14.Odczynnikiem do mapowania mogą być?
18.Możliwość dopasowania 2 sekwencji z istotną wartością,coś tam? - programowanie dynamiczne (?)
17. Algorytm tworzenia alighmentu opiera się na..... Czy punktacja dopasowania jest znacząco większa od punktacji oczekiwanej dla dopasowania losowych sekwencji o tej samej długości i składzie?
1.Tworzenie metodą Monte Carlo losowych(-ej) sekwencji (o tej samej długości i składzie co rzeczywiste).
2.Przyrównanie losowych(-ej) sekwencji (powtórzenie np. 100-1000 razy) przy tych samych parametrach dopasowania.
3.Określenie rozkładu punktacji, średniej i odchylenia standardowego (SD).
4.Wyliczenie Z-score
11. metody odległościowego tworzenia drzew filogenetycznych to:
NJ, MP, ML
Maxymalnej parsymonii, NJ, , ML
UPMGA, NJ, Fitch- Margolish
Wszystkie prawidłowe
11.E value 0.005 oznacza,że?
oczekiwana (wg rozkładu) liczba dopasowań z punktacją równą przynajmniej S wynosi 0,005
6.W bazie Uni Gene można znaleźć sekwencje trankrypcyjne , które pochodzą z tego samego genu lub pseudogenu, oraz podobieństwa białek, ekspresji genów oraz umiejscowienie w genomie.
12.Do porównania 2 sekwencji nukleotydowych używane sąmacierz punktowa, programowanie dynamiczne oraz Metody słów ( z wykorzystaniem programów FASTA i BLAST)
15.Macierz PSSM charakteryzuje się:
przypisaniem różnych wart. Score różnym w zależności od ich pozycji w sekwencji
przypisaniu tych samych wartości score różnym aa niezależnie od ich pozycji w sekwencji
stałą liczbą wieszy i kolumn
3.Wymień przykłady 3 zasadniczych grup metod przewidywania struktury białek
Przewidywanie struktur drugorzędowych: metoda Chou-Fassmana (statystyczna), metoda GOR, metoda zachodzących okien (zasadnicze metody) oprócz tego metoda najbliższego sąsiada
Definicja wiązania wodorowego: Wiązanie wodorowe - rodzaj stosunkowo słabego wiązania chemicznego polegającego głównie na przyciąganiu elektrostatycznym między atomem wodoru i atomem elektroujemnym zawierającym wolne pary elektronowe. Klasyczne wiązanie wodorowe powstaje, gdy atom wodoru jest połączony wiązaniem kowalencyjnym z innym atomem o dużej elektroujemności (np. tlenem) i w ten sposób uzyskuje nadmiar ładunku dodatniego. W wyniku tego oddziaływania pierwotne, kowalencyjne wiązanie wodór - inny atom ulega częściowemu osłabieniu, powstaje zaś nowe, stosunkowo słabe wiązanie między wodorem i innym atomem (akceptorem wiązania wodorowego).
Przykłady transformacji posttranslacyjnych to proteoliza (cięcie łańcucha białkowego), acetylacja (modyfikacje końców), fosforylacja (modyfikacje łańcuchów bocznych)
1.Regiony nieuporządkowane w białkach występują:
wyłącznie w wysokiej temp, denaturacja
tam gdzie nie ma alfa helis i beta kartek
w w ielu białkach tam gdzie jest to pożądane ze względu na specyficzne oddziaływania międzycząsteczkowe
2.Skrót CalfaRMSD oznacza.....(root mean square deviation) średnie odchylenie atomowe, najczęściej Calfa
BIOINFORMATYKA - egzamin 2008, I termin
Wyliczyć score dla alignmentu z uwzględnieniem maskowania LCR
Ograniczenia metody krystalografii w tworzeniu struktur trzeciorzędowych białek, wady krystalografii
Wady i ograniczenia tworzenia obrazów trójwymiarowych białek za pomocą metody magnetycznego rezonansu jądrowego - NMR
Co to jest gen
Długość wiązania N-C, C-C w białkach
Dorysować wiązania wodorowe w strukturach beta- kartka
Wybrać aminokwasy, których łańcuchy boczne mogą być akceptorami/donorami wiązań wodorowych
Podać pełne nazwy 3 głównych metod odległościowych konstruowania drzew filogenetycznych
Obliczyć odległość OTU w drzewku skonstruowanym metodą UPMGA
Genom którego zwierzęcia nie jest w pełni zsekwencjonowany Odp. Canis lupus/ Bos taurus
Wykres Ramachandrana
Którą metoda najlepiej przedstawia wiązanie receptor - ligand? a ) wstążka b) all atom c) powierzchniowa d)……
Który program wybrać przy przeszukiwaniu bazy Swiss Prot mając daną sekwencję aminokwasową, szukamy homologow? a ) blast p b) tblastn c)…… d)……..
Co to jest STRs? a ) mikrosatelity b) minisatelity c) powtorzenia…….. d) markery genetyczne
Istotna wartość S mówi o…….
Co to jest RFLP (odp. marker genetyczny, miejsce cięcia enzymów restrykcyjnych)
Wybrać aminokwasy o największej/ najmniejszej zmienności - macierz BLOSUM62/65
Co to jest transkryptom/ proteosom
Rysunek mapy chromosomu - rozpoznać czy fizyczna, czy genetyczna
E value = 10 - co to znaczy
Jak rozpoznać nieuporządkowane obszary w krystalografii rentgenowskiej
Konsensus ????
Aj = Pe/Po, uzupełnić: a)jeżeli Aij<0 to ………. jest większe b) jeżeli Aij>0 to……….. jest mniejsze c) jeżeli Aij=0 to………….. jest takie samo d) jeżeli Aij + 1 to logarytm 10 x większy (??????????)
1.Regiony nieuporządkowane w białkach występują:
wyłącznie w wysokiej temp, denaturacja
tam gdzie nie ma alfa helis i beta kartek
w w ielu białkach tam gdzie jest to pożądane ze względu na specyficzne oddziaływania międzycząsteczkowe
2.Skrót CalfaRMSD oznacza.....
3. wymień wady 2 najczęściej stosowanych metod wyznaczania struktur makrocząsteczek biologicznych
4. wymień nazwy 3 zasadniczych metod przewidywania struktur białek
5. zredukowane reprezentacje (?)struktury białek stosuje się przede wszystkim aby:
zmniejszyć wymiar przestrzeni konformacyjnej białek
uprościć obraz białkowy dla celów ilustrowania oddziaływań makromolekularnych
przyspieszyć poszukiwanie przestrzeni konformacyjnej białek
6.wylicz score dla alighmentu, zastosuj pełne maskowanie dla elementów o małej złożoności (LCR) - podany alighment
7.Skrót EST oznacza
znaczniki sekwencji ulegających ekspresji
expressed segment tags
expressed segment flags
żadne
8.E = 10 oznacza że:
p-stwo zaobserwowania podobieństwa równego zaobserwowanej wartości S między dwiema losowymi sekwencjami =10
oczekiwana liczba losowych dopasowań o wartości S wyliczonych dla znalezionego alighmentu =10
znaleziono 10 dopasowań o takiej samej wartości
wszystkie prawidłowe
9.Stwórz wyrażenie regularne na podstawie wielokrotnego dopasowania 5 sekwencji
10. Porównując odległe ewolucyjnie sekwencje aminokwasów powinniśmy oprzeć system wartościowania alighmentu na:
macierzach PAM 250 i BLOSUM 45
PAM 1 i BLOSUM 62
PAM 120 i BLOSUM 80
Unikać macierzy PAM i BLOSUM
11. metody odległościowego tworzenia drzew filogenetycznych to:
NJ, MP, ML
Maxymalnej parsymonii, NJ, , ML
UPMGA, NJ, Fitch- Margolish
Wszystkie prawidłowe
12. do ukorzenienia drzewa filogenetycznego stworzonego metoda MP dla grupy homologów pochodzących z Mus muscarus, ......różne łacińskie nazwy....moglibysmy użyć sekwencji ortologa pochodzącego z: i tu były różne łacińskie nazwy
13.wymień przynajmniej 2 zastosowania wielokrotnego alighmentu.
14.do metod fizycznego mapowania genów nie należy:
mapowanie restrykcyjne
fish
mapowanie miejsc znaczonych sekw.
Krzyżówka trójpunktowa
15.Macierz PSSM charakteryzuje się:
przypisaniem różnych wart. Score różnym w zależności od ich pozycji w sekwencji
przypisaniu tych samych wartości score różnym aa niezależnie od ich pozycji w sekwencji
stałą liczbą wieszy i kolumn
16. program t blast x służy do:
przeszukiwania bazy nukleotydowej sekwencji DNA
-`'-`'-`'-`'-`'-`'-`'-`'- białkowej sekwencji aminokwasowych
-`-`'-`'-`'-`'-`'-`'-`'-`'nukleotydowej (tłumaczonej w ??? )sekwencją nukleotydową (także tłumaczoną na aa)
tworzenia macierzy PSSM
17. Algorytm tworzenia alighmentu opiera się na.....
18.określ konsensus dla dopasowania 5 sekwencji
19. podaj definicję wiązań wodorowych
20. podaj 3 przykłady transformacji potranslacyjnych w białku
21. typowy eksperyment mikromacierzowej ekspresji genów polega na:
jednoczesnym pomiarze poziomu transkryptów większości genów w badanej próbie
pmiarze liczby kopii genów ulegających ekspresji w badanym genomie
jednoczesnym pomiarze długości genów w badanej próbie
1.Zrekombinowane reprezentanty struktury białek stosuje się przede wszystkim:?
2.Definicja wiązania wodorowego
3.Wymień przykłady 3 zasadniczych grup metod przewidywania struktury białek
4.Typowy eksperyment mikromacierzowy ekspresji genów polega na: ?
5.STS to : ?
6.W bazie Uni Gene można znależć:
7.Algorytm programu Basic Local Aligsment Search Tool opiera się na ?
8.Program tBlast opiera się na ?
9.Wymień 2 zastosowania wielokrotnego aligsmentu
10.Podaj metody tworzenia drzew filogen.,które się nie opierają na macierzach odległości
11.E value 0.005 oznacza,że?
12.Do porównania 2 sekwencji nukleotydowych używane są?
13.PSI-Blast to?
14.Odczynnikiem do mapowania mogą być?
15.3 typowe modyfikacje posttranskrypcyjne
16.Regiony nieuporządkowane w białkach wyst?
17.CαRMSD oznacza:
18.Możliwość dopasowania 2 sekwencji z istotną wartością,coś tam?
co to gen
narysowac na rys wiazania wodorowe
modyfokacje potranskrypcyjne
co to OTU
odleglosc C-C-1.51 A
N-C- 1.32(tak dla wiedzy)
obliczyc odleglosci miedzy dwoma OTU na drzewku filogen. metoda UPGMA
jakie struktury na wykresie ramachandrana
dlaczego opuszczamy opszar w malym dopasowaniu
ograniczenia i wady metody NMR
wady i ograniczenia - Krystalograficznej <chodzila o tą ale nazwabyla
jakos bardziej rozbudowana.. trzebaby w wykladach
sprawdzic;P>wyznaczyc konsensus
ile u eukariotow zajmuje transkrypto,w calym genomie
jaka mape przedstawiono genetyczna czy fizyczna
to zadanie z Aj=Pe/Po
co to rflp
Wyszukiwarka
Podobne podstrony:
giełda 14 bioinf z odpowiedziami
TEST zalicz mikroskopia czescETI z odpowiedz
obowiazki i odpowiedzialnosc nauczyciela
025 odpowiedzialnosc cywilnaid 4009 ppt
Czynniki warunkuj ce wybor metod nauczenia odpowiednich dla
odpowiedzialnosc
Charakterystyka odpowiedzi immunologicznej typu GALT faza indukcji
odpowiedzi
Odpowiedzialność cywilna
odpowiedź6 2
cw 16 odpowiedzi do pytan id 1 Nieznany
form3 odpowiedż na pozew
podstawy robotyki odpowiedzi
Odpowiedzi Przykladowy arkusz PP Fizyka (2)
Benjamin06 odpowiedzi
więcej podobnych podstron