I CHEMIA OGOLNA
1. Budowa atomu:
a) skład i struktura atomu,
b) konfiguracja elektronowa, promocje
elektronowe, bloki energetyczne, reguła
Hunda, teoria wzbudzenia,
c) elektroujemność, związek budowy atomu z
układem okresowym, teoria oktetu,
d) typy wiązań chemicznych,
e) hybrydyzacja,
f) VSEPR
2. Podstawy chemii:
a) podstawowe pojęcia,
b) stechiometria,
c) termochemia,
d) statyka i kinetyka reakcji chemicznych
3. Układy dyspersyjne
a) ogolna charakterystyka i podział układow
dyspersyjnych,
b) roztwory,
c) teorie kwasow i zasad,
d) ilościowy opis właściwości roztworow:
- stopień i stała dysocjacji,
- prawo rozcieńczeń Ostwalda,
- iloczyn jonowy wody,
- pH i pOH,
- iloczyn rozpuszczalności,
e) reakcje zachodzące w roztworach:
- miareczkowanie alkacymetryczne,
- amfoteryczność,
- zobojętnianie i hydroliza,
- wskaźniki,
- roztwory buforowe,
- związki kompleksowe,
f) koloidy
4. Elektrochemia
a) stopnie utlenienia pierwiastkow
b) reakcje redox w ujęciu elektronowym,
c) najwaŜniejsze utleniacze i reduktory,
d) związki chromu i manganu jako utleniacze,
e) szereg aktywności metali,
f) połogniwa,
g) ogniwa galwaniczne,
h) energetyka reakcji redox,
i) korozja metali i metody jej zwalczania,
j) elektroliza.
II CHEMIA ZWIĄZKOW NIEORGANICZNYCH
1. Systematyka związkow nieorganicznych:
a) tlenki,
b) wodorki,
c) wodorotlenki,
d) kwasy,
e) sole.
2. Litowce,
3. Berylowce,
4. Borowce,
5. Węglowce,
6. Azotowce,
7. Tlenowce,
8. Fluorowce,
9. Miedź,
10. Srebro,
11. Cynk,
12. Chrom,
13. Mangan,
14. śelazo.
CHEMIA OGÓLNA
Chemia
jako nauki o przemianach materii jest wiedza o jej budowie
BUDOWA ATOMU
SKŁAD I STRUKTURA ATOMU
Budowa atomu
Jądro atomowe
Protony
Neutrony
Chmura elektronowa
Elektrony
|
Nazwa |
Symbol |
Masa spoczynkowa |
Ładunek |
Trwałość
|
|
elektron |
e- |
9,109.10-31 kg ≈ 0 u |
- 1,602.10-19 C = - e |
trwały |
|
proton |
P+ |
1,673.10-27 kg ≈ 1 u |
+ 1,602.10-19 C = + e |
trwały |
|
neutron |
n0 |
1,675.10-27 kg ≈ 1 u |
0 |
T1/2=930s, n0 ---> p+ + e- + 00 ΰe
|
Pierwiastek
Zbiór atomów posiadających tę samą liczbę protonów w jądrze nazywamy pierwiastkiem.
Przy zapisie pierwiastka podajemy najczęściej najważniejsze informacje dotyczące jego składu ilościowego zgodnie ze schematem: AZX
Z to liczba atomowa (porządkowa) – jest ona równa liczbie protonów w jądrze, a tym samym ładunkowi jądra oraz liczbie elektronów otaczających jądro.
Z kolei A to liczba masowa, równa liczbie nukleonów tworzących jądro. Określenie ‘liczba masowa’ bierze się stąd, iż, jak łatwo zauważyć, nukleony są ok. 1836 razy cięższe niż elektron, dlatego w jądrze skupia się ponad 99% masy atomu.
Znając wielkości A i Z możemy określić wiele własności jądra.
Zakładając, że ma ono kształt kulisty, możemy jego promień wyrazić jako rj=r0 3√A, gdzie r0=1,23.10-15 m.
Liczba neutronów w jądrze jest różnicą całkowitej liczby nukleonów i liczby protonów, czyli A – Z.
Masa jądra jest mniejsza od sumy mas jego składników – różnicę tę określamy jako defekt masy i obliczamy następująco: Δm=Zmp+(A-Z)mn-mj, gdzie mp, mn, mj to odpowiednio masy spoczynkowe protonu i neutronu oraz rzeczywista masa jądra.
Nuklid
Wszystkie identyczne atomy tworzą nuklid. Pierwiastek może składać się z jednego rodzaju nuklidów lub z kilku, bowiem atomy tego samego pierwiastka mogą różnić się liczbą neutronów w jądrze.
Izotopy
Atomy o tym samym Z lecz różnym A nazywamy izotopami.
Przykładem jest wodór, składający się z trzech izotopów: protu 11H,deuteru 21H=D oraz trytu 31H=T.
Masę średniego składu izotopowego danego pierwiastka wyrażoną w unitach nazywamy masą atomową.
Izobary
Atomy o tym samym A lecz różnym Z nazywamy izobarami – mają one identyczną liczbę nukleonów, np. 3618 Ar i 3616S.
Izotony
Izotony to atomy o identycznej licznie neutronów, czyli o tej samej wartościróżnicy A – Z, np. 4020Ca i 3919K.
Promieniotwórczość
Zjawisko przekształcania się izotopów danego pierwiastka w izotop trwały lub nietrwały tego samego lub innego pierwiastka nazywamy promieniotwórczością.
Wyróżniamy kilka rodzajów rozpadu promieniotwórczego, z których najważniejsze to:
a) Rozpad α polega na wyrzuceniu z jądra cząstki 42 α2+, czyli jądra atomu helu. W wyniku tego liczba atomowa i masowa zmieniają się w następujący sposób: AZ X ---> A-4Z-2Y + 42 α
b) Rozpad β- polega na rozpadzie neutronu w jądrze na proton, elektron oraz antyneutrino elektronowe, zgodnie z równaniem: 10n ---> 11p + 0-1e- + 00ΰe – powstały elektron wyrzucany jest z jądra. Zmianę ilościową składu atomu ilustruje równanie: AZ X ---> AZ+1Y + 0-1 β- + 00ΰe
c) Rozpad β+ zachodzi w przyrodzie bardzo rzadko i w przeciwieństwie do pozostałych jest rozpadem sztucznym. Polega na rozpadzie protonu w jądrze na neutron, pozyton oraz neutrino elektronowe, zgodnie zrównaniem: 11p ---> 10n + 0+1e+ + 00ΰe Pozyton jest antycząstką w stosunku do elektronu – posiada taką samą masę i wartość bezwzględną ładunku, różni się natomiast jego znakiem. Po rozpadzie pozyton wyrzucany jest z jądra. Zmianę składu atomu przedstawia schemat: AZ X ---> AZ-1Y + 0+1 β+ + 00ΰe
d) Przemiana γ powiązana jest z wyrzutem wysokoenergetycznego fotonu z jądra. Najczęściej jest następstwem rozpadu α, po którym wzbudzone jądro przechodzi do stanu trwałego przez pozbycie się części energii właśnie w postaci fotonu.
Rozpadem promieniotwórczym rządzą określone prawa fizyczne.
Istotne
jest, iż rozpadowi w jednostce czasu nie ulega określona masa
materiału, lecz pewna jego część. Przykładowo jeżeli
promieniowanie pochodzi z 1 kg substancji promieniotwórczej, to
po czasie t rozpadnie się 0,5 kg, ale jeżeli weźmiemy 2 kg, to
po tym samym czasie rozpadnie się 1 kg! Wynika stąd, iż
szybkość rozpadu zależy wprost proporcjonalnie od liczby atomów
w próbce; po wprowadzeniu współczynnika proporcjonalności –
stałej rozpadu λ otrzymujemy:
− gdzie
τ=1/λ to średni czas życia jąder. Z prawa rozpadu wynika, iż
połowa próbki rozpadnie się po czasie:
gdzie
τ=1/λ to średni czas życia jąder. Z prawa rozpadu wynika, iż
połowa próbki rozpadnie się po czasie:
 zwanym
czasem połowicznego zaniku (okresem półtrwania).
zwanym
czasem połowicznego zaniku (okresem półtrwania).
Na podstawie tej wielkości dzieli się izotopy na trwałe i nietrwałe.
Izotop przyjmuje się za trwały, gdy jego okres półtrwania wynosi ponad miliard lat. Co ciekawe, niektóre pierwiastki mają ponad trzydzieści izotopów, jednak żaden z nich nie należy do trwałych.
teoria atomistyczna
Opiera się ona na założeniu, iż materia zbudowana jest z mikroskopijnych, niewidocznych gołym okiem cząstek, zwanych atomami. Poziom atomu przyjmowano przez długi czas za kres podzielności materii – zgodnie z tym założeniem każde ciało można dzielić tak długo, aż dojdzie się do cząstki juz dalej niepodzielnej. Współcześnie wiemy, iż budowa atomu nie jest jednorodna, lecz wyróżnić można w nim elementy składowe, dalej zresztą także podzielne. Nie zaprzecza to jednak doniosłości teorii atomistycznej ani jej znaczeniu, przeciwnie – stanowi raczej jej uzupełnienie.
Modele budowy atomu
Istnieje wiele modeli budowy atomu. Niektóre z nich mają już znaczenie wyłącznie historyczne, niektóre zaś są wciąż poprawiane i doskonalone.
Model rodzynkowy Thomsona
Jednym z pierwszych nowożytnych był tzw. model rodzynkowy, wprowadzony przez Josepha Thomsona w 1906r. Autor założył, iż atom składa się z fragmentów naładowanych ładunkiem elektrostatycznym ujemnym (elektronów), które znajdują się w dodatnio naładowanym ośrodku – niczym rodzynki w cieście. Całość atomu była dzięki temu elektrycznie obojętna. Choć zaletą było zaznaczenie istnienia ładunków dodatnich i ujemnych wewnątrz atomu, to ciężko wyobrazić sobie dlaczego nie przyciągną się one i nie wyrównają potencjałów do zera.
Doświadczenie Rutherforda
Niedługo później – w 1911r. – Ernest Rutherford przeprowadził doświadczenie obalające model Thomsona. Polegało ono na bombardowaniu naładowanymi cząstkami folii metalowej. Na „królika doświadczalnego” Rutherford wybrał złoto, gdyż można je walcować na bardzo cienkie płytki. Opracowując wyniki, stwierdził on, iż większość cząstek przeszła przez złotą folię bez zmiany kierunku, niewielka część uległa zakrzywieniu, a nawet odbiciu w stronę przeciwną. Wynikało stąd, iż atomy nie są „pełne”, jak to zakładał Thomson, lecz mają wewnątrz bardzo dużo wolnego miejsca. W ten sposób Thomson odkrył istnienie tworu zwanego jądrem atomowym.
Model Bohra
Problemem
do rozwiązania pozostawał opis ruchu elektronów wokół jądra.
Krążący elektron powinien bowiem stopniowo tracić energie
kinetyczną, a po jej wyczerpaniu zostać przyciągnięty przez
jądro. Rozwiązanie podał w 1913r. Niels Bohr w postaci teorii
orbit stacjonarnych. Zgodnie z nią elektron nie traci energii,
gdy porusza się po orbicie dozwolonej, czyli gdy moment pędu
elektronu jest całkowitą wielokrotnością stałej η=
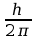 ,
gdzie h oznacza stałą Plancka (I postulat). Dodatkowo przejście
elektronu w wyższej orbity dozwolonej na niższą odpowiada
emisji fotonu o energii równej różnicy energii elektronu na
tych orbitach: Δ
,
gdzie h oznacza stałą Plancka (I postulat). Dodatkowo przejście
elektronu w wyższej orbity dozwolonej na niższą odpowiada
emisji fotonu o energii równej różnicy energii elektronu na
tych orbitach: Δ =En-Ek=hν (II postulat). Rozważania te pozwoliły na dokładny
opis ruchu elektronu, zgonie z determinizmem fizycznym.
=En-Ek=hν (II postulat). Rozważania te pozwoliły na dokładny
opis ruchu elektronu, zgonie z determinizmem fizycznym.
Teoria nieoznaczoności Heisenberga
Nowością natomiast okazała się udowodniona przez Wernera Heisenberga (1927r.) zasada nieoznaczoności, zgodnie z którą nie można jednocześnie i z dowolną dokładnością zmierzyć dwóch wielkości fizycznych kanonicznie sprzężonych tj. położenia i pędu, położenia kątowego i momentu pędu, energii i czasu itd. Dodatkowo iloczyn niepewności pary wielkości fizycznych kanonicznie sprzężonych jest nie mniejszy od stałej Plancka. Spostrzeżenie to ugodziło w przewidywalność ruchu elektronu i zasugerowało, iż brak dokładności nie jest konsekwencją niedoskonałości przyrządów czy technik, lecz wypływa z samej natury elektronu.
Hipoteza de Broglie
Kolejnych informacji dostarczyła hipoteza, jaką wysunął w 1924r. Ludwik de Broglie. Stwierdził on, iż z ruchem cząstki skojarzona jest fala materii (tzw. fala de Broglie’a) o określonej długości λ = h / p, gdzie h oznacza stałą Plancka, a p – pęd cząstki. Powyższy wzór można przedstawić w kilku alternatywnych formach, np. wiedząc, że pęd jest iloczynem masy i prędkości: λ = h / (mv). Z kolei energia ciała E = p2 / 2m, gdzie m to masa ciała, stąd p = √2mE, czyli λ = h / √2mE. Energia spoczywającej cząstki o ładunku q rozpędzonej przez różnicę potencjałów U wynosi E = e U, stąd λ = h / √2meU.
Równanie Schrodingera
Powiązanie
dotychczasowych zdobyczy fizyki pozwoliło Erwinowi
Schrodingerowi podać równanie ruchu elektronu (1926r.) w
postaci:
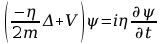 . Jego rozwiązaniami są funkcje falowe Ψ, opisujące orbitale
atomowe. Orbital atomowy jest fragmentem przestrzeni wokół
jądrowej, w której istnieje duże (≥90%) prawdopodobieństwo
znalezienia elektronu. Miarą tego prawdopodobieństwa jest
kwadrat modułu funkcji falowej |Ψ|2.
Na stan kwantowy elektronu składa się jego energia, zachowanie
w polu magnetycznym, orientacja wektora momentu pędu oraz spin,
czyli ruch wirowy wokół własnej osi. Wielkości te opisywane
są przy pomocy liczb kwantowych, wynikających z równania
falowego ruchu elektronu.
. Jego rozwiązaniami są funkcje falowe Ψ, opisujące orbitale
atomowe. Orbital atomowy jest fragmentem przestrzeni wokół
jądrowej, w której istnieje duże (≥90%) prawdopodobieństwo
znalezienia elektronu. Miarą tego prawdopodobieństwa jest
kwadrat modułu funkcji falowej |Ψ|2.
Na stan kwantowy elektronu składa się jego energia, zachowanie
w polu magnetycznym, orientacja wektora momentu pędu oraz spin,
czyli ruch wirowy wokół własnej osi. Wielkości te opisywane
są przy pomocy liczb kwantowych, wynikających z równania
falowego ruchu elektronu.
a) Głowna liczba kwantowa (n) kwantuje energię elektronu i decyduje o wielkości obszaru orbitalnego.
Związana jest z powłoką elektronową (poziomem energetycznym), czyli zbiorem elektronów o zbliżonych wartościach energii.
Elektron a n-tej powłoce posiada energię En=-E1/n2, gdzie E1 jest energią na powłoce pierwszej; przyjmuje się ją za wartość stałą dla danego atomu, np. dla atomu wodoru E1 = 13,6 eV.
Głowna liczba kwantowa przyjmuje wartości kolejnych liczb naturalnych: 1, 2, 3 itd. teoretycznie aż do nieskończoności (w praktyce do 7, gdyż atomy znanych pierwiastków mają od 1do 7 powłok elektronowych). Kolejne powłoki oznacza się wielkimi literami alfabetu:
|
n |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
nazwa powłoki |
K |
L |
M |
N |
O |
P |
Q |
b)Poboczna (orbitalna, azymutalna) liczba kwantowa (l) kwantuje orbitalny moment pędu elektronu i decyduje o kształcie obszaru orbitalnego.
Związana jest z podpowłoką elektronową (podpoziomem
energetycznym), czyli zbiorem elektronów o takich samych wartościach energii.
Liczba ta przyjmuje wartości od 0 do n - 1, gdzie n to główna liczba kwantowa. Stanom tym odpowiada położenie elektronu na określonych podpowłokach:
|
l |
0 |
1 |
2 |
3 |
4 |
|
nazwa podpowłoki |
s |
p |
d |
f |
g |
Danej wartości pobocznej liczby kwantowej odpowiada określony kształt orbitalu.
Orbital typu s ma kształt kulisty, co oznacza, iż prawdopodobieństwo napotkania elektronu w każdym kierunku, począwszy od jądra, jest jednakowe (rysunek lewy).
Orbital typu p ma kształt ósemki obrotowej, a położony jest na jednej z osi układu współrzędnych (x, y, z – m = -1, 0, 1), co oznacza, iż wzdłuż tej osi występuje największe prawdopodobieństwo napotkania elektronu (rysunek środkowy). Orbital typu d ma kształt dwóch ósemek obrotowych położonych różnie względem osi układu współrzędnych (np. rysunek prawy).
c) Magnetyczna liczba kwantowa (m) kwantuje rzut orbitalnego momentu pędu elektronu na wyróżniony kierunek w przestrzeni i decyduje o orientacji obszarów orbitalnych w polu magnetycznym.
Związana jest z poziomem orbitalnym.
Przyjmuje wartości od –l do l, gdzie l to poboczna liczba kwantowa, czyli razem 2n+1 różnych wartości. Dzięki temu warunkowi można określić, ile istnieje możliwych ustawień orbitalu, czyli ile orbitali mieści powłoka:
|
l=0 |
l=1 |
l=2 |
l=3 |
l=4
|
|
1 orbital s |
3 orbitale p |
5 orbitali d |
7 orbitali f |
9 orbitali g
|
Podpoziomy p, d i dalsze nazywamy zdegenerowanymi, gdyż występuje na nich więcej orbitali o tej samej energii, różniących się tylko orientacją przestrzenną.
d) Spinowa liczb kwantowa (s) kwantuje własny moment pędu elektronu. Ma ona wartość stałą, równa 1/2
e) Magnetyczna spinowa liczba kwantowa (ms) kwantuje rzut własnego momentu pędu elektronu na wyróżniony kierunek w przestrzeni. Przyjmuje ona dwie wartości: ms=1/2(tzw. spin dodatni) oraz ms=-1/2. (tzw. spin ujemny).
Stanem kwantowym elektronu nazywamy jego opis za pomocą czterech liczb kwantowych. Np. (3,1,1,1/2) oznacza powłokę M, podpowłokę 3p, orbital 3p oraz spin dodatni.
Elektrony objęte są zakazem Pauliego: nie istnieją w atomie elektrony mające takie same wartości wszystkich liczb kwantowych – muszą się różnić co najmniej jedną z nich. Wynika stąd, iż na jednym orbitalu mogą występować maksymalnie dwa elektrony o przeciwnych liczbach magnetycznych spinowych. Pojemności podpowłok wynoszą zatem:
s – 1 · 2 = 2 e-
p – 3 · 2 = 6 e-
d – 5 · 2 = 10 e-
f – 7 · 2 = 14 e-
Teoretyczną pojemność n-tej powłoki otrzymamy obliczając sumę: 2 + 6 + 10 + 14 + ... + 2 · (2m+1) = 2 ·[ 1 + 3 + 5 + 7 + 2m+1 ] = 2 · n2, gdzie n jest główną liczba kwantową – jest to tzw. wzór Bohra.
KONFIGURACJA ELEKTRONOWA, PROMOCJE ELEKTRONOWE, BLOKI ENERGETYCZNE, REGUŁA HUNDA, TEORIA WZBUDZENIA
Konfiguracja elektronowa
Konfiguracja elektronowa pierwiastka to rozmieszczenie jego elektronów na powłokach i podpowłokach atomu. Kolejność obsadzania elektronami poszczególnych stanów kwantowych w atomach wieloelektronowych określa schemat zwany „złotym deszczem” (rysunek obok).
Podczas zapisu konfiguracji elektronowej atomu lub jonu stosuje się schemat: numer powłoki – symbol podpowłoki – liczba elektronów na podpowłoce (w wykładniku symbolu). Dla uproszczenia zapisu, coraz dłuższego w przypadku dalszych pierwiastków, podaje się w nawiasie symbol poprzedzającego pierwiastek helowca oraz pozostałą część konfiguracji.
Ilustrują to poniższe przykłady:
20Ca = 1s2 2s2 2p6 3s2 2p6 4s2
25Mn2+ = [18Ar] 4s2 5d3
20Ca = [18Ar] 4s2
26Fe3+ = [18Ar] 3d5
26Fe = [18Ar] 4s2 3d6
16S2- = [10Ne] 3s2 3p6
82Pb = [54Xe] 4f14 5d10 6s2 2p2
20Ca2+ = [18Ar]
11Na+ = [10Ne]
13Al3+ = [10Ne]
Powłoka walencyjna
Powłoką walencyjną nazywamy powłokę o najwyższej w danym atomie wartości energii, wypełniającej się elektronami jako ostatnia.
Elektrony walencyjne
Znajdujące się na niej elektrony nazywamy walencyjnymi.
One właśnie biorą udział w reakcjach chemicznym i wiązaniach chemicznych, są ponadto odpowiedzialne za wartościowość pierwiastka.
Dla pierwiastków grup głównych układu okresowego liczba elektronów walencyjnych jest równa numerowi grupy.
Elektrony rdzenia
Pozostałe elektrony nazywamy elektronami rdzenia.
Te ostatnie wraz z jądrem atomu tworzą zrąb atomowy.
Promocje elektronowe
Promocja polega na tym, że konfiguracja elektronowa ulega zmianie ze względu na większą trwałość nowo powstałego układu. Najczęściej dotyczy ona jednego elektronu, który przeniesiony zostaje na wyższą podpowłokę powłoki niższej. Jest to konsekwencją faktu, iż kolejna powłoka zapełnia się szybciej niż dalsze podpowłoki powłoki niższej. Wynika to z przyjmowanych przez znajdujące się na nich elektrony wartości energii, a ilustruje to najlepiej „złoty deszcz”.
Typowymi układami wyjściowymi do promocji są: d4 s2 przekształcający się w d5 s1 oraz d9 s2 – w d10 s1. Wynika stąd, iż promocjom powinny ulegać pierwiastki grupy 6 i 11 i jest tak na prawdę. Przykładowo z położenia złota w układzie okresowym wynika konfiguracja: 79Au [54Xe] 4f14 5d9 6s2, jednak w rzeczywistości elektrony walencyjne są ułożone następująco: 4f14 5d10 6s1. Na uwagę zasługuje również pallad, gdzie konfiguracja 4d8 5s2 zmienia się w 4d10, czyli promocji ulegają aż dwa elektrony, pustosząc całkowicie podpoziom s. Oprócz wymienionych, promocji ulegają pierwiastki takie jak: 24Cr, 29Cu, 41Nb, 42Mo, 43Tc, 44Ru, 45Rh, 47Ag, 78Pt.
Bloki energetyczne
Ze względu na konfigurację elektronową w układzie okresowym wyróżnia się tzw. bloki energetyczne.
Forma długa (układ Wernera) grupuje pierwiastki według symbolu podpoziomu energetycznego. Dzięki znajomości konfiguracji w poszczególnych blokach można szybko ustalić budowę pierwiastka bez korzystania z „deszczu”.
|
s: ns1-2 |
d: (n-1)d1-10 ns2 |
|
p: ns2 np1-6 |
f: (n-2)f1-14 (n-1)d1 ns2 |
Zapis klatkowy
Ze względu na potrzebę czytelnego przedstawienia konfiguracji elektronów walencyjnych wprowadza się tzw. zapis klatkowy. Każda klatka oznacza jeden orbital będący w stanie pomieścić dwa elektrony o przeciwnie zorientowanych spinach. Zbiór odpowiedniej ilości klatek daje podpoziom walencyjny. Ilustruje to poniższy rysunek:
Reguła Hunda
W ustalaniu właściwej konfiguracji pomocna jest reguła Hunda, zwana też regułą maksymalnej różnorodności.
Składa się nań kilka zasad:
a) liczba niesparowanych elektronów na podpoziomie zdegenerowanym powinna być możliwie największa,
b) pary elektronowe tworzą się dopiero po zapełnieniu wszystkich orbitali danego podpoziomu przez elektrony niesparowane,
c) elektrony niesparowane danego podpoziomu mają ten sam kierunek spinu.
Pod wpływem rozmaitych czynników (zderzenie z elektronem, absorpcja fotonu, działanie termiczne) atomy mogą ulegać wzbudzeniu. Wówczas ich elektrony przechodzą na wyższe podpoziomy energetyczne, co związane jest ze zmianą konfiguracji oraz wzrostem wartościowości.
ELEKTROUJEMNOŚĆ, ZWIĄZEK BUDOWY ATOMU Z UKŁADEM OKRESOWYM, TEORIA OKTETU (właściwości pierwiastków - związek z budowa układu okresowego)
Elektroujemność
Elektroujemnością nazywamy zdolność pierwiastka do wiązania własnych i obcych elektronów walencyjnych.
Elektroujemność pierwiastków maleje w grupach a różnie w okresach wraz ze wzrostem liczby atomowej pierwiastka. Metale mają niską elektroujemność (są elektrododatnie).
Aktyność metali i niemetali
Aktywność metali rośnie z grupach i maleje w okresach wraz ze wzrostem liczby atomowej, ogólnie – wzrasta ze spadkiem elektroujemności.
Z kolei aktywność niemetali wzrasta ze wzrostem elektroujemności , maleje więc ona w grupach a rośnie w okresach wraz ze wzrostem liczby atomowej.
Efekt ekranowania jądra
Efektem ekranowania jądra nazywamy zjawisko osłabienia oddziaływania pomiędzy jądrem atomu a elektronami walencyjnymi, powstające wskutek obecności między nimi elektronów rdzenia.
Siła efektu rośnie wraz ze wzrostem numeru okresu – liczby powłok ekranujących.
Jest to jedna z przyczyn różnic elektroujemności pomiędzy pierwiastkami.
|
Związek budowy układu okresowego z właściwościami pierwiastków |
|
|
Wielkość |
Kierunek wzrostu |
|
elektroujemność |
|
|
energia jonizacji |
|
|
powinowactwo elektronowe |
|
|
promień atomu |
|
|
aktywność metali |
|
|
zasadowość tlenków i wodorków |
|
|
metaliczność |
|
|
aktywność niemetali |
|
|
kwasowość tlenków i wodorków |
|
|
niemetaliczność |
|
|
moc kwasów tlenowych |
|
|
moc kwasów beztlenowych |
|
|
maksymalna wartościowość względem tlenu |
→ |
|
maksymalna wartościowość względem wodoru |
→IV← |
Teoria oktetu
Każdy układ w przyrodzie dąży do możliwie najstabilniejszego stanu energetycznego. W przypadku atomu konsekwencje tego dążenia opisuje teoria oktetu – pierwiastki dążą do uzyskania trwałej, ośmioelektrodowej (ew. dwuelektronowej) powłoki zewnętrznej (tzw. oktet lub dublet elektronowy) – takiej, jaką mają gazy szlachetne. Mogą tego dokonać przez oddawanie, przyjmowanie lub uwspólnianie elektronów w wiązaniach chemicznych.
2He 1s2 K2 dublet
10Ne 1s2 2s2 2p6 K2 L8 oktet
18Ar 1s2 2s2 2p6 3s2 3p6 K2 L8 M8 oktet
a)Metale mają niską elektroujemność i liczbę elektronów walencyjnych, dlatego dążą do trwałości poprzez oddawanie elektronów, tworząc kationy. Np.:
11Na 1s2 2s2 2p6 3s1 ---> 11Na+ 1s2 2s2 2p6 (oktet) XNa =0,9
20Ca 1s2 2s2 2p6 3s2 3p6 4s2 ---> 20Ca2+ 1s2 2s2 2p6 3s2 3p6 4s2 (oktet) XCa =1,0
Metale grup pobocznych mogą tworzyć kationy poprzez oddawanie elektronów z podpoziomu s ostatniej powłoki oraz z podpoziomu d przedostatniej powłoki. Np.:
26Fe 1s2 2s2 2p6 3s2 3p6 3d6 4s2
26Fe2+ 1s2 2s2 2p6 3s2 3p6 3d6
26Fe3+ 1s2 2s2 2p6 3s2 3p6 3d5
b) Niemetale mają wysoką elektroujemność i liczbę elektronów walencyjnych, dlatego dążą do trwałości poprzez przyjmowanie elektronów, tworząc aniony. Np.:
17Cl 1s2 2s2 2p6 3s2 3p5 ---> 17Cl- 1s2 2s2 2p6 3s2 3p6 (oktet) XCl =3,0
8O 1s2 2s2 2p4 ---> 8O2- 1s2 2s2 2p6 (oktet) XO =3,5
TYPY WIĄZAŃ CHEMICZNYCH
a) Wiązanie jonowe tworzy się pomiędzy pierwiastkami skrajnie różniącymi się elektroujemnością (ΔX=|XA - XB|>1,7), czyli pomiędzy metalami i niemetalami.
Występuje najczęściej w tlenkach metali, wodorotlenkach i solach.
Najbardziej typowe wiązanie tworzy się pomiędzy pierwiastkami grup I i II a VI i VII.
Wiązanie jonowe polega na oddaniu elektronów walencyjnych przez np. metal i włączeniu ich do powłoki walencyjnej np. niemetalu. W ten sposób tworzą się kationy i aniony, które budują sieć krystaliczną związku jonowego.
Właściwości związków jonowych:
- trwałość sieci krystalicznej,
- stały stan skupienia i wysokie temperatury topnienia,
- przewodnictwo prądu elektrycznego po stopieniu lub rozpuszczeniu w wodzie.
Dla przykładu rozpatrzmy proces tworzenia chlorku potasu: Aby oderwać elektron od atomu potasu należy dostarczyć energii jonizacji E1. Przyłączeniu elektronu do atomu chloru towarzyszy wydzielenie energii powinowactwa elektronowego E2. Aby reakcja zaszła, należy do układu dostarczyć różnicę wartości tych energii, czyli deficyt energetyczny, gdyż E1 > E2. Warunkiem trwałości powstałego związku jest aby suma energii sieci krystalicznej oraz powinowactwa elektronowego była większa od energii jonizacji. Zazwyczaj energia sieci krystalicznej jest bardzo duża, co powoduje własności takie jak stały stan skupienia oraz wysokie temperatury topnienia. XK =0,9 XCl =3,0 ΔX=2,1>1,7 Nie istnieją cząsteczki chlorku potasu – tworzy on sieć krystaliczną. KCl jest jedynie wzorem empirycznym, wyrażającym stosunek liczby jonów w sieci.
Liczba koordynacyjna
O tym, jaka ilość jonów przeciwnego znaku otacza dany jon w sieci krystalicznej, decyduje tzw. liczba koordynacyjna. Wyznacza się ją na podstawie znajomości promienia kationu i anionu, co przedstawia poniższa tabela.
|
Stosunek promienia jonu metalu do niemetalu |
0,15-0,23 |
0,23-0,41 |
0,41-0,73 |
0,73-1,00 |
≥1,00
|
|
Liczba koordynacyjna jonu metalu |
2 |
4 |
6 |
8 |
12 |
wartościowść
Wartościowość jest liczbą wiązań, jakie może wytworzyć atom danego pierwiastka. Jest ona zawsze dodatnia i w grupach głównych równa jest numerowi grupy.
Stopień utlenienia pierwiastka
Stopień utlenienia pierwiastka to liczba ładunków elementarnych, które pojawiłby się na atomie danego pierwiastka, gdyby wszystkie wiązania w danym związku były czysto jonowe. W przeciwieństwie do wartościowości może przyjmować także wartości ujemne.
b) Wiązanie kowalencyjne powstaje pomiędzy atomami o takiej samej lub zbliżonej wartości elektroujemności (ΔX=0÷0,4).
Jego powstanie polega na uwspólnieniu elektronów walencyjnych brakujących do oktetu (ew.dubletu) z innymi atomami.
Jedno wiązanie kowalencyjne to jedna wspólna para elektronowa.
Najbardziej typowi wiązanie kowalencyjne występuje w cząsteczkach homoatomowych.
Właściwości związków kowalencyjnych:
- różne stany skupienia (chlor – gaz, brom – ciecz, siarka – ciało stałe),
- niskie temperatury topnienia i wrzenia – czasem lotność,
- rozpuszczalność w rozpuszczalnikach niepolarnych, brak w polarnych,
- brak przewodnictwa elektrycznego,
- reakcje z ich udziałem zachodzą przeważnie powoli i przy stosunkowo małej wydajności.
Przykłady związków o budowie kowalencyjnej:
• fosforowodór PH3:
• metan CH4:
• wodór H2:
Orbital
Orbital molekularny to funkcja opisująca stan elektronu w cząsteczce.
Orbital σ powstaje podczas czołowego nakładania się orbitali atomowych. Największe prawdopodobieństwo znalezienia wiążącej pary elektronowej występuje wzdłuż osi łączącej jądra atomów. Wiązanie σ jest wiązaniem kierunkowym, nadających cząsteczce określony kształt.
• tlen O2:
Orbital molekularny π powstaje podczas bocznego nakładania się orbitali atomowych p. Jest on zorientowany równolegle do osi wiązania σ. Składa się z dwóch jednakowych części, znajdujących się po obu stronach prostej przechodzącej przez jądra atomów tworzących wiązanie. Para elektronowa zajmująca obszar molekularny π tworzy wiązanie π. Nie występuje ono nigdy samodzielnie – pary elektronowe π wraz z parą elektronów σ tworzą wiązania wielokrotne, tj. podwójne lub potrójne.
c) Wiązanie kowalencyjne spolaryzowane (polarne = biegunowe) powstaje między atomami znacznie różniącymi się elektroujemnością.
Polega ono na uwspólnieniu elektronów walencyjnych, przy czym wiążąca para elektronowa znajduje się bliżej atomu o wyższej elektroujemności. Atom ten ładuje się ujemnie, a cała cząsteczka staje się dipolem.
Moment dipolowy wiązania
Miarą przesunięcia pary elektronowej jest moment dipolowy wiązania: μ = q l.
Naturalną jednostką jest kulombometr [C.m], ponieważ jednak niewygodnie jest posługiwać się tak małymi liczbami, przyjmuje się debje [D] (czyt. ‘debaj’). 1 D = 3,33.10-30 C.m.
Właściwości związków polarnych:
- rozpuszczalność w wodzie i w innych rozpuszczalnikach o budowie jonowej,
- dysocjacja jonowa w środowisku wodnym – możliwość przewodzenia prądu elektrycznego,
- niskie temperatury topnienia,
- w ciałach stałych tworzenie nietrwałych sieci krystalicznych – kryształy są miękkie i nietrwałe.
Przykłady związków o budowie polarnej:
• dwutlenek węgla CO2:
• woda H2O:
• amoniak NH3:
• czterochlorek węgla CCl4:
d) Wiązanie koordynacyjne (donorowo – akceptorowe) jest wiązaniem typu kowalencyjnego.
Polega na uwspolnieniu elektronów pochodzących od jednego atomu, zwanego donorem. Na schemacie zaznacza się je strzałką z grotem skierowanym do donora do akceptora.
Przykłady cząstek zawierających wiązanie koordynacyjne:
• kation amonowy NH4+:
• kation hydroniowy H3O+:
e) Wiązanie metaliczne (struktura metaliczna) występuje w kryształach metali lub ich stopów.
Atomy metali zostają przekształcone w elementy sieci krystalicznej: jądra atomowe obsadzają węzły sieci, zaś elektrony walencyjne tworzą chmurę elektronową, która scala całą sieć. Węzły są wierzchołkami komórek elementarnych. Dzięki bliskości węzłów, elektrony mogą swobodnie poruszać się we wnętrzu metalu, co jest warunkiem łatwego przewodzenia prądu elektrycznego.
Cechy metali wynikające z ich budowy:
- duża gęstość,
- wysokie temperatury topnienia i wrzenia,
- twardość, kowalność i ciągliwość,
- dobre przewodnictwo cieplne i elektryczne.
f) Wiązanie wodorowe polega na oddziaływaniu typu elektrostatycznego.
Występuje ono pomiędzy atomem o dużej elektroujemności a atomem wodoru związanym spolaryzowanym wiązaniem kowalencyjnym z innym silnie elektroujemnym atomem. Tym ostatnim jest najczęściej azot, tlen lub fluorowiec.
Wstępowanie wiązań wodorowych nie pozostaje bez wpływu na właściwości substancji. Najczęściej objawia się większą gęstością, lepkością i lotnością, czasem innym stanem skupienia. Różnice widać na przykładzie izomerycznych alkoholi i eterów – pomimo braku różnic w masie cząsteczkowej niższe alkohole są ciekłe, zaś etery – gazowe. Do ciekawych wniosków prowadzi również obserwacja stanu skupienia wodorków pierwiastków grupy 16 – wszystkie one są gazami wyjątkiem najlżejszego – wody. Ten pozorny paradoks tłumaczy duża różnica elektroujemności pomiędzy wodorem a tlenem, tak iż występujące pomiędzy cząsteczkami wody wiązania wodorowe utrzymują ją w stanie ciekłym (zjawisko asocjacji).
Przykłady substancji zawierających wiązania wodorowe:
- stały cyjanowodór (struktura liniowa): .....H-C≡N|.....H-C≡N|.....H-C≡N|.....H-C≡N|.....H-C≡N|.....
- stały fluorowodór (struktura zygzakowata):
- woda – asocjat (H2O)n:
HYBRYDYZACJA
Hybrydyzacja orbitali atomowych to metoda wyjaśniania geometrycznego kształtu cząsteczek.
Hybrydyzacja jest operacją matematyczną polegającą na uśrednieniu kształtu i energii orbitali walencyjnych atomu centralnego cząsteczki.
Wyróżnia się kilka podstawowych typów hybrydyzacji.
a) Hybrydyzacja tetraedryczna (sp3) polega na uśrednieniu energii i kształtu jednego orbitalu s i trzech orbitali p.
Orbitale zhybrydyzowane mają kształt zdeformowanej ósemki obrotowej, z której każda skierowana jest ku narożnikowi czworościanu foremnego. Atom węgla w stanie hybrydyzacji tetragonalnej otacza się wyłącznie wiązaniami pojedynczymi.
Przykłady związków z hybrydyzacją sp3:
• metan CH4:
Cząsteczka metanu ma kształt czworościanu foremnego – tetraedru. W każdym z naroży znajduje się taki sam element, stąd cząsteczka cechuje się regularnością i zerowym momentem dipolowym. Pomiędzy symetralnymi orbitali s-p (wiązaniami węgiel – wodór) występuje kąt 109o28’, będący kątem charakterystycznym tego typu hybrydyzacji.
• amoniak NH3:
W cząsteczce amoniaku występuje wolna para elektronowa. Fakt ten sprawia, iż budowa przestrzenna nie jest idealnie regularna, gdyż wolna para odpycha orbitale z większą siłą, niż one siebie nawzajem. Następuje lekkie wygięcie orbitali w stronę przeciwną, a kąt pomiędzy wiązaniami odbiega od wartości charakterystycznej – wynosi 107o.
• woda H2O:
W cząsteczce wody występują dwie wolne pary elektronowe. Z tego powodu wiązania ulegają odgięciu, a kąt pomiędzy nimi – zmniejszeniu (104o27’). Zgięta budowa cząsteczki wody pozwala na traktowanie ej jako dipola elektrostatycznego.
b) Hybrydyzacja trygonalna (sp2) polega na uśrednieniu energii i kształtu jednego orbitalu s i dwóch orbitali p.
Zhybrydyzowane orbitale skierowane są ku wierzchołkom trójkąta równobocznego. Kąt charakterystyczny hybrydyzacji wynosi zatem 120o.
Przykładem takiej cząsteczki jest fluorek boru BF3:
c) Hybrydyzacja digonalna polega na uśrednieniu energii i kształtu jednego orbitalu s i jednego p.
Atomy tworzące cząsteczkę są ułożone liniowo, a kat charakterystyczny hybrydyzacji wynosi 180o.
Przykładem takiej cząsteczki jest chlorek berylu BeCl2:
d) Istnieją również inne typy hybrydyzacji – pokażemy to na przykładzie sześciofluorku siarki SF6, którego cząsteczka ma kształt bipiramidy tetragonalnej.
VSEPR
VSEPR (ang. vallence shell electron pair repulsion – dosł. odpychanie się par elektronów powłok walencyjnych) jest metodą określania kształtu przestrzennego drobin chemicznych.
Może być stosowana pod kilkoma warunkami:
- drobina składa się wyłącznie z atomów grup głównych (bloki energetyczne s i p),
- drobina posiada tylko jeden atom centralny,
- podstawniki (ligandy) są atomami prostymi – nie mogą to być grupy złożone.
Etapy postępowania przy posługiwaniu się metodą VSEPR:
a) zapis cząstki w postaci EAn Hm, gdzie E to atom centralny, A – ligandy niewodorowe, H – ligandy wodorowe i odczytanie wartości n i m,
b) obliczenie całkowitej liczby elektronów walencyjnych w cząsteczce lub jonie (Lwal),
c) obliczenie liczby powstałych par elektronowych (Lpar = ½ Lwal),
d) obliczenie ilości własnych par elektronowych atomu centralnego (LwpE = Lpar - 4n - m),
e) obliczenie liczby przestrzennej drobiny (Lp = LwpE + n + m),
f) wykonanie rysunku odpowiednio do uzyskanej wartości liczby przestrzennej.
|
Wartość Lp |
Kształt cząsteczki
|
|
2 |
budowa liniowa |
|
3 |
trójkąt równoboczny |
|
4 |
tetraedr |
|
5 |
bipiramida trygonalna |
|
6 |
bipiramida tetragonalna |
|
7 |
bipiramida pentagonalna |
Ponadto zachodzą następujące relacje:
a) liczba wiązań σ łączących atom centralny z ligandami: Lσ = n + m, czyli Lp = LwpE + Lσ,
b) liczba wiązań π w drobinie (dla grup IV-VIII): Lπ = 4 - Lp; wartość ujemna oznacza brak wiązań π.
PODSTAWY CHEMII
PODSTAWOWE POJĘCIA
a) Zjawisko fizyczne to zjawisko, podczas którego następuje zmiana właściwości fizycznych substancji, np. stanu skupienia, gęstości, lepkości, sprężystości czy kształtu.
b) Zjawisko chemiczne (reakcja chemiczna) to zjawisko prowadzące do zmiany składu chemicznego substancji oraz ich właściwości fizycznych.
c) Atom to najmniejsza część pierwiastka zachowująca jeszcze jego właściwości chemiczne.
d) Cząsteczka to zespół co najmniej dwóch atomów (mogą być one takie same bądź różne). Cząsteczki dzielimy na homoatomowe (cząsteczki pierwiastków) oraz heteroatomowe (cząsteczki związków chemicznych).
e) Substancja chemiczna to rodzaj materii o ściśle określonych właściwościach fizycznych i chemicznych, posiadającej określoną masę. Substancje dzielimy na proste (pierwiastki) oraz złożone (związki chemiczne).
f) Pierwiastek chemiczny (pojęcie to wprowadził Robert Boyle) to najprostsza substancja, której nie da się już dalej rozłożyć metodami chemicznymi.
g) Symbol chemiczny pierwiastka to skrót łacińskiej nazwy pierwiastka (jedno- lub dwuliterowy).
h) Związek chemiczny to substancja składająca się z co najmniej dwóch pierwiastków, połączonych ze sobą w charakterystyczny, określony i uporządkowany sposób.
i) Wzór chemiczny to przedstawienie składu ilościowego i jakościowego związku chemicznego przy pomocy symboli chemicznych oraz indeksów stechiometrycznych.
j) Postulaty Johna Daltona (1804r.):
- Każdy pierwiastek chemiczny jest zbiorem małych cząstek zwanych atomami. Atomy tego samego pierwiastka mają identyczne właściwości chemiczne. (Postulat ten nie uwzględnia występowania izotopów.)
- Atomy różnych pierwiastków różnią się od siebie właściwościami fizycznymi i chemicznymi. Istnieje tyle rodzajów atomów o określonych właściwościach, ile jest rodzajów pierwiastków. (Także ten postulat nie uwzględnia występowania izotopów.)
- Atom danego pierwiastka nie może ulec przekształceniu w atom innego pierwiastka na drodze chemicznej. (Możliwe jest to natomiast na drodze fizycznej w sposób naturalny – przez rozpad promieniotwórczy bądź też w sposób sztuczny – przez fuzję jąder.)
- Łączenie się pierwiastków w związki chemiczne polega na łączeniu się ich atomów w większe zespoły zwane cząsteczkami.
- Związek chemiczny jest zbiorem cząsteczek. Wszystkie cząsteczki danego związku mają taki sam skład atomowy pierwiastków i identyczne właściwości chemiczne.
- Rozkład związku chemicznego na pierwiastki polega na rozpadzie cząsteczek tego związku na atomy poszczególnych pierwiastków. Może to być proces wieloetapowy.
- Atomy tego samego pierwiastka mogą również łączyć się ze sobą w cząsteczki.
k) Wartościowość pierwiastka w związku chemicznym to ilość wiązań chemicznych tworzonych przez atom tego pierwiastka w cząsteczce związku chemicznego.
l) Postulaty teorii wartościowości (J. Wislicenus, 1868r.):
- Pierwiastki w wstanie czystym nie posiadają wartościowości.
- Dany pierwiastek może mieć więcej wartościowości niż jedną.
- Wartościowość wodoru jest stała i wynosi I.
- Wartościowość tlenu jest również stała i wynosi II.
- Pierwiastki leżące w grupach nieparzystych mają na ogół wartościowości nieparzyste, zaś pierwiastki grup parzystych – wartościowości parzyste. („Na ogół”, ponieważ istnieją od tej reguły wyjątki, np. czterowartościowy atom chloru.)
m) Prawo stosunków stałych (prawo stałości składu, J. L. Proust, 1799r.): stosunek wagowy pierwiastków w danym związku chemicznym jest zawsze stały, charakterystyczny dla danego związku i nie zależy od sposobu otrzymywania tego związku.
n) Prawo stosunków wielokrotnych (J. Dalton): jeżeli dwa pierwiastki tworzą ze sobą kilka związków chemicznych, to ilości wagowe jednego pierwiastka, przypadające na tę samą ilość drugiego pierwiastka, pozostają do siebie w stosunku niewielkich liczb naturalnych.
o) Równanie chemiczne to zapis przebiegu reakcji chemicznej przy pomocy symboli i wzorów chemicznych. Każde równanie chemiczne musi spełniać prawo stałości składu oraz prawo zachowania masy.
p) Prawo zachowania masy (A. Lavoisser, M. Łomonosow): masa substratów biorących udział w reakcji jest równa masie produktów otrzymywanych w jej wyniku.
q) Reakcja syntezy to reakcja, podczas przebiegu której przynajmniej dwa substraty łączą się w jeden produkt o bardziej złożonej budowie.
r) Reakcja analizy to reakcja, podczas przebiegu której jeden złożony substrat rozpada się na kilka produktów o prostszej budowie.
s) Reakcja wymiany pojedynczej (prostej) to reakcja, podczas przebiegu której z pierwiastka i związku chemicznego powstaje inny pierwiastek i inny związek chemiczny.
t) Reakcja wymiany podwójnej to reakcja, podczas przebiegu której z dwóch związków chemicznych tworzą się dwa inne związki chemiczne o innych właściwościach.
STECHIOMETRIA
a) Międzynarodowa jednostka masy atomowej (1 unit) to masa odpowiadająca 1/12 masy izotopu węgla 12C. 1 u = 1,66.10-24 g 1 g = 6,022.1023 u
b) Masa atomowa to masa atomu pierwiastka wyrażona w unitach.
c) Masa cząsteczkowa to masa cząsteczki pierwiastka wyrażona w unitach. Oblicza się ją sumując masy poszczególnych atomów wchodzących w skład danego związku.
d) Mol to liczba cząstek równa ilości atomów węgla zawartych w 12 g jego izotopu 12C. Liczba ta nosi nazwę liczby Avogadra i wynosi: NA=6,022.1023 mol-1.
e) Masa molowa (M) to masa jednego mola cząstek. Masa molowa danej substancji równa się liczbowo jej masie atomowej lub cząsteczkowej, wyrażonej w g / mol.
f) Objętość molowa (VM) to objętość jednego mola cząstek. Objętość molowa gazu doskonałego jest stała i wynosi V0=22,4 dm3/mol. Najczęściej gazy rzeczywiste przybliża się do gazu doskonałego, stąd można przyjąć, że objętość mola dowolnego gazu wynosi V0.
g) Warunki normalne: T0=273,16K=0oC, p0=101325 Pa=1 atm.
h) Warunki standardowe: T0=292K=25oC, p0=101325 Pa=1 atm.
i) Prawo Avogadra (1811r.): jednakowe objętości różnych gazów zawierają w tych samych warunkach ciśnienia i temperatury jednakowe liczby cząsteczek tych gazów. W 22,4 dm3 dowolnego gazu znajduje się zatem, 6,022.1023 cząsteczek tego gazu.
j) Prawo Gay – Lussaca (1809r.): objętości reagujących ze sobą gazów i powstających w wyniku reakcji produktów gazowych mają się do siebie tak jak niewielkie liczby naturalne (w tych samych warunkach ciśnienia i temperatury).
k) W równaniu reakcji chemicznej często zestawia się ze sobą ilości reagujących substratów, jak również ilości powstałych w wyniku przemiany produktów. Porównywać możemy albo masy albo ilości moli (atomów lub cząsteczek) albo objętości wybranych reagentów. Dzieląc przez siebie odpowiednie masy otrzymujemy stosunek wagowy (masowy), odpowiednie ilości moli – stosunek molowy, zaś odpowiednie objętości – stosunek objętościowy. Zgodnie z matematyczną definicją równość dwóch stosunków nazywamy proporcją. Proporcje reagentów (proste bądź mieszane) wykorzystywane są w zadaniach w celu szybkiego obliczenia nieznanych wartości mas, ilości moli czy objętości.
TERMOCHEMIA
a) Układ chemiczny to ogół substancji chemicznych biorących udział w reakcji. Układy chemiczne można podzielić na otwarte, zamknięte oraz izolowane.
b) Układ otwarty to układ, który ma możliwość wymiany z otoczeniem zarówno masy jak i energii, np. otwarty cylinder.
c) Układ zamknięty to układ, który ma możliwość wymiany energii z otoczeniem, ale nie ma możliwości wymiany masy, np. zamknięty cylinder.
d) Układ izolowany to układ, który nie ma możliwości wymiany z otoczeniem ani masy ani energii. Układy izolowane istnieją wyłączenie hipotetycznie, gdyż nie znamy doskonałych izolatorów cieplnych. Wymianę energii z otoczeniem możne pominąć przy bardzo szybkim zachodzeniu procesu, np. w podczas przemiany adiabatycznej.
e) Energia wewnętrzna układu chemicznego (Ew, U) to całkowita ilość energii zawartej wewnątrz danego układu. Jej wartość jest niemożliwa do określenia, gdyż składa się na nią bardzo wiele rodzajów energii, z których pewne nie zostały jeszcze dokładnie poznane.
f) I zasada termodynamiki: zmiana energii wewnętrznej może odbywać się na sposób ciepła lub pracy objętościowej, z czego wynika zależność: ΔU= ±Q±Vobj.
g) Entalpia reakcji (H) to ciepło reakcji zachodzącej pod stałym ciśnieniem. Na podstawie I zasady termodynamiki zmiana entalpii wyraża się wzorem: ΔH=ΔU+pΔV (p=const), gdzie drugi składnik sumy określa wartość pracy objętościowej. Zmiana objętości, wynikająca ze zmiany liczby moli gazu o jeden, pod ciśnieniem normalnym, równoważna jest z wykonaniem pracy równej co do wartości: W=Vm p0 =0,0224m3/mol.101325N/m2=2271J/mol≈2,5kJ/mol.
h) Zmiana entalpii (entalpia reakcji) to różnica sumy entalpii tworzenia produktów oraz sumy entalpii tworzenia substratów; rozważając odpowiednie entalpie spalania składniki występują w odwrotnej kolejności. Każdy układ w przyrodzie dąży do zgromadzenia jak najmniejszej ilości energii, gdyż jest to sytuacja najstabilniejsza i najbardziej prawdopodobna, a więc dąży do zmniejszenia swojej entalpii. Pojęcie to tłumaczy zatem zachodzenie procesów egzoergicznych.
i) Entalpia standardowa (H0) to entalpia liczona w odniesieniu do jednego mola reagentu.
j) Reakcja egzoenergetyczna (egzoergiczna) to reakcja, podczas przebiegu której energia wydzielana jest do otoczenia. Entalpia takiego procesu jest ujemna. Reakcje tego typu są samorzutne – po zainicjowaniu zachodzą dalej samodzielnie.
k) Reakcja endoenergetyczna (endoergiczna) to reakcja, podczas przebiegu której energia jest pobierana z otoczenia, np. na sposób ciepła. Entalpia takiego procesu jest dodatnia. Reakcje tego typy są wymuszone, a do podtrzymania przebiegu wymagają stałego dopływu energii.
l) Entropia (S) jest miarą rozkładu energii w układzie chemicznym. Jest funkcję stanu, więc jej wartość zależy wyłącznie od początkowego i końcowego stanu układu. Jest wielkością addytywną, co oznacza, iż rozpatrywany proces można podzielić na kilka etapów i obliczyć entropie cząstkowe, które po dodaniu dadzą entropię całkowitą. W termodynamice fenomenologicznej entropia określana jest różniczką zupełną: dQ=TdS, stąd dS=dQ/T, czyli S=∫dQ/T + const, gdzie dQ oznacza nieskończenie małą ilość energii wymienionej na sposób ciepła w procesie odwracalnym, a T – temperaturę bezwzględną. Wynika stąd, iŜ w układach izolowanych, wobec braku wymiany ciepła z otoczeniem, entropia nie zmienia się. Podobnie każdy cykl odwracalny można podzielić na nieskończoną liczbę cząstkowych procesów adiabatycznych, zatem podczas przemiany odwracalnej entropia nie ulega zmianie. W termodynamice statystycznej przyjmuje się: S=k-lnW, gdzie k oznacza stałą Boltzmanna, zaś W – liczbę sposobów podziału energii w układzie, dla układów makroskopowych jest ona olbrzymia w porównaniu z liczbą Avogadra.
m) Entropia reakcji to różnica sumy entropii produktów oraz sumy entropii substratów. Każdy układ w przyrodzie dąży do stanu równowagi poprzez obniżenie energii wewnętrznej oraz zwiększenie entropii. Układy dążą zatem do zapewnienia możliwie największej różnorodności sposobów podziału energii między drobiny oraz poszczególne rodzaje form jej gromadzenia. Entropia rośnie ze wzrostem temperatury, o obniżaniem ciśnienia oraz przy rozpadzie wiązań chemicznych. Pojęcie entropii tłumaczy takie procesy jak zachodzenie przemian endoergicznych (czynnik entropowy przeważa nad energetycznym), zachodzenie reakcji w układach izolowanych oraz samorzutne mieszanie się gazów. Samorzutnie zachodzą wyłącznie procesy nieodwracalne, zwiększające entropię środowiska. Wszechświat dąży do stanu równowagi poprzez coraz większe nieuporządkowane (chaos) materii. Za jedyny prawdziwie izolowany układ można uznać jedynie cały Wszechświat (wobec braku otoczenia) – zachodzą więc w nim nieodwracalne procesy zwiększania entropii. Na tej podstawie można wnioskować, iż w odległej przyszłości procesy te doprowadzą do ustania wszelkiego ruchu i osiągnięcia przez Wszechświat stanu równowagi termodynamicznej (tzw. stan śmierci cieplnej Wszechświata). Teoria ta opisuje jednak zdarzenie bardzo mało prawdopodobne.
n) Entropia standardowa (S0) to entropia liczona w odniesieniu do jednego mola reagentu.
o) Entalpia swobodna (G) to funkcja stanu określająca samorzutność przebiegu oraz stan równowagi reakcji chemicznej. Jej zmianę podaje równania Gibbsa – Helmholtza postaci ΔG= ΔH-TΔS. Każdy układ dąży do zmniejszenia energii wewnętrznej (ΔH<0) oraz do wzrostu entropii (ΔS>0), co w efekcie daje ΔG<0 – stan taki oznacza reakcję samorzutną. Analogicznie ΔG=0 oznacza ustalenie się stanu równowagi w układzie, zaś ΔG<0 – zachodzenie reakcji wymuszonej. Entalpia swobodna reakcji to różnica sum entalpii swobodnych produktów oraz substratów.
p) Prawo Hessa: efekt cieplny reakcji nie zależy od drogi reakcji (pod warunkiem, że p,V=const), a praca objętościowa jest jedyną formą przemiany energii. Efekt cieplny reakcji zależy zatem wyłącznie od początkowego oraz końcowego stanu układu.
q) Prawo Lavoissier’a – Laplace’a: efekt cieplny reakcji odwrotnej do danej ma taką samą wartość jak reakcji danej, lecz przeciwny znak. Jest to swoista konsekwencja zasady zachowania energii.
STATYKA I KINETYKA REAKCJI CHEMICZNYCH
a) Reakcja nieodwracalna to reakcja, podczas której w środowisku powstają wyłącznie produkty. Przyjmuje się, że tego typu reakcje zachodzą do końca. Należą do tej grupy reakcje zobojętniania oraz te, w wyniku których wytrąca się nierozpuszczalny osad, wydziela się gaz lub powstaje słaby elektrolit.
b) Reakcja odwracalna to reakcja, podczas której gromadzące się produkty reagują ze sobą i odtwarzają substraty. W środowisku reakcji oprócz produktów występują nie przereagowane substraty. Reakcje tego typu nie zachodzą do końca. Należy tu np. dysocjacja słabych elektrolitów lub hydroliza soli. Większość reakcji należy właśnie do tej grupy.
c) Równowaga to ustalony stan np. reakcji chemicznej.
d) Równowaga statyczna to stan, w którym procesy chemiczne nie zachodzą, a stężenia reagentów nie zmieniają się w czasie.
e) Równowaga dynamiczna to stan, w którym zachodzą przemiany chemiczne, jednak tyle samo substratów przechodzi w produkty, co produktów w substraty. W stanie równowagi dynamicznej szybkość reakcji danej oraz reakcji do niej przeciwnej są sobie równe, a stężenia reagentów ustalają się wówczas na stałym poziomie.
f) Miarą szybkości reakcji jest szybkość pojawianie się produktów (i ubytku substratów) w środowisku, stąd v=dc/dt. Szybkość reakcji zależy proporcjonalnie od stężenia początkowego substratów, więc v=k.c0, gdzie k oznacza współczynnik proporcjonalności, zwaną stałą szybkości reakcji.
g) Reguła van’t Hoffa to empiryczna i orientacyjna reguła pozwalająca na określenie zmian szybkości reakcji pod wpływem zmian temperatury. W jej myśl szybkość reakcji wzrasta od dwóch do czterech razy przy podwyższeniu temperatury o 10oC. Liczba określająca, ile razy wzrasta szybkość reakcji, nazywana jest współczynnikiem temperaturowym: Θ=kT+10 /kT =kT /kT-10. Reguły tej nie stosuje się do reakcji wybuchowych i przebiegających w obecności stałych katalizatorów.
h) Dla większych ilości reagentów wyrażenia opisujące szybkość reakcji powiększają się o iloczyn odpowiednich stężeń.
A <=v1=> B, v1=k1[A],
A + A <=v1=> B, v1= k1[A][A]=k1[A]2,
aA <=v1=> B, v1=k1[A]a itd.
Ogólnie dla modelowej reakcji: aA + bB <=v1;v2=> cC + dD wyrażenia na stałe szybkości przyjmują postać: v1=k1[A]a[B]b, v2=k2[C]c[D]d. Ponieważ w stanie równowagi prędkości obu przeciwstawnych reakcji są równe, otrzymujemy: v1=v2 => k1[A]a[B]b=k2[C]c[D]d => k1/k2=[C]c[D]d/[A]a[B]b=const. Interpretacją otrzymanej zależności jest podaje prawo działania mas.
Prawo działania mas (prawo Guldberga – Waagego): w stanie równowagi chemicznej iloczyn stężeń (ciśnień) reagentów w wykładnikach potęgowych ich współczynników stechiometrycznych jest wielkością stałą dla danej reakcji i w określonej temperaturze i określaną stałą równowagi reakcji (stężeniową Kc lub ciśnieniową Kp – w praktyce najczęściej posługujemy się tą pierwszą). Pomiędzy stałymi równowagi tej samej reakcji zachodzi zależność: Kp=Kc(RT)Δn, gdzie R oznacza stałą gazową, T – temperaturę bezwzględną, zaś Δn – przyrost liczby moli produktów gazowych podczas reakcji. Stała stężeniowa i ciśnieniowa są więc sobie równe, gdy podczas przebiegu reakcji nie zmienia się liczba moli produktów gazowych. Stała równowagi jest wielkością chemiczną charakteryzującą liczbowo stan równowagi dynamicznej reakcji odwracalnej. Jej wartość nie zależy od stężeń reagentów, a jedynie od temperatury, zmieniając się w sposób zależny od efektu energetycznego reakcji.
j) Reguła przekory Le Chateliera – Browna: każdy układ w przyrodzie dąży do osiągnięcia stanu równowagi, a gdy na układ znajdujący się w stanie równowagi zadziała jakiś czynnik zewnętrzny, wówczas w układzie tym zachodzą zmiany zmniejszające skutki działania tego czynnika. Pod pojęciem czynników zewnętrznych rozumiemy zmianę temperatury, ciśnienia bądź stężenia jednego z reagentów. - wpływ temperatury na stan równowagi – stała równowagi zależy tylko i wyłącznie od temperatury:
• dla reakcji egzoergicznych (ΔH<0) maleje ze wzrostem temperatury,
• dla reakcji endoergicznych (ΔH>0) rośnie ze wzrostem temperatury, - wpływ ciśnienia na stan równowagi:
• dla reakcji, w których objętość produktów jest większa od objętości substratów, wzrost ciśnienia powoduje przesunięcie równowagi reakcji w lewo, zaś spadek ciśnienia – w prawo,
• dla reakcji, w których objętość produktów jest mniejsza od objętości substratów, wzrost ciśnienia powoduje przesunięcie równowagi reakcji w prawo, zaś spadek ciśnienia – w lewo,- wpływ zmiany stężenia jednego z reagentów: przesunięcie równowagi zmniejszenie stężenia zwiększenie stężenia substraty w lewo w prawo produkty w prawo w lewo
UKŁADY DYSPERSYJNE
OGOLNA CHARAKTERYSTYKA I PODZIAŁ UKŁADOW DYSPERSYJNYCH
Układ dyspersyjny (rozproszony) to układ złożony z co najmniej dwóch składników, zwanych fazami, z których jeden jest rozproszony w drugim. Fazy zmieszane są ze sobą w określonej (ogólnie jednak dowolnej) proporcji ilościowej, nie są połączone chemicznie, zachowują swoje indywidualne właściwości fizyczne i chemiczne, możne je rozdzielić metodami fizycznymi. Do tych ostatnich należą m. in.: dekantacja, sedymentacja, segregacja, sączenie, destylacja, ekstrakcja, adsorpcja, chromatografia, odparowanie, krystalizacja, wymrożenie itd.
Każdy układ dyspersyjny składa się z fazy rozpraszającej, czyli ośrodka dyspersyjnego oraz z fazy rozproszonej, czyli substancji zdyspergowanej. Zarówno jedną jak i drugą fazę mogą stanowić pierwiastki lub związki chemiczne we wszystkich stanach skupienia.
W zależności od stanu skupienia układy dyspersyjne dzielimy w następujący sposób:
|
rozproszona \ rozpraszająca |
gaz |
ciecz |
ciało stałe
|
|
gaz |
roztwory gazowe (np. powietrze) |
piany (np. woda sodowa) |
piany stałe (np. pumeks)
|
|
ciecz |
aerozole i mgły (np. deszcz) |
roztwory właściwe (np. ocet) i emulsje (np. mleko) |
(np. mokra gąbka)
|
|
ciało stałe |
dymy (np. kurz) |
roztwory właściwe, koloidy i zawiesiny |
stopy metali (np. brąz)
|
Układy dyspersyjne dzielimy na jednorodne (homogeniczne) oraz niejednorodne (heterogeniczne).
a) Układy homogeniczne charakteryzuje rozproszenie molekularne, a rozmiary cząsteczek rozproszonych są wielkości mniejszych od 1 nm. Nazywa się je roztworami właściwymi. Są one jednorodne fizycznie, tzn. każda próbka, pobrana z dowolnego miejsca układu, ma taki sam skład jakościowy i ilościowy.
b) Układy heterogeniczne cechuje rozproszenie koloidalne bądź makroskopowe. W pierwszy przypadku nazywamy je koloidami, a rozmiary cząstek rozproszonych zawierają się pomiędzy 1 nm a 200 nm. W drugim przypadku są to zawiesiny, gdzie rozmiary cząstek rozproszonych przewyższają 200 nm. Te ostatnie są zazwyczaj układami nietrwałymi, złożonymi z cząsteczek substancji stałej w cieczy lub gazie.
Określanie składu mieszanin:
a) procent wagowy i objętościowy:
pkw =mk /mm·100%, pk0 =Vk /Vm·100%, (indeks k dotyczy składnika numer k, zaś indeks m – mieszaniny),
b) przeliczanie procentu wagowego na procent objętościowy lub odwrotnie dla dwuskładnikowej mieszaniny gazowej:
p10 = p1w M2 100% (p1w M2 + p2w M1),
p1w = p10 M1 100% (p1w M11 + p2w M2),
ułamek wagowy, objętościowy i molowy:
ukw = mk /mm , Σuw =1,
uk0 = Vk /Vm , Σu0 =1,
ukmol = nk /nm , Σumol =1,
d) przeliczanie ułamków ilościowych dla mieszanin dwuskładnikowych:
|
|
umol |
u0 |
uw |
|
umol |
- |
u1mol
=
u2mol = 1 - u1mol |
u1mol
=
u2mol = 1 - u1mol |
|
u0 |
u1w
=
u2w = 1 - u1w |
- |
u1w
=
u2w = 1 - u1w |
|
uw |
u10
=
u20 = 1 - u10 |
u10
=
u20 = 1 - u10 |
- |




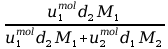
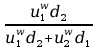
ROZTWORY
Roztworem nazywamy układ złożony z substancji rozpuszczonej (stałej, ciekłej bądź gazowej) w ciekłym rozpuszczalniku.
Roztwory podzielić można na właściwe (rzeczywiste, homogeniczne) oraz koloidalne (heterogeniczne).
Rozpuszczalność ciał stałych i cieczy silnie zależy od temperatury (zazwyczaj rośnie z jej wzrostem, gdyż proces rozpuszczania jest endoergiczny) i prawie nie zależy od ciśnienia (substancje te są praktycznie nieściśliwe). Na rozpuszczalność gazów widoczny wpływ mają oba wymienione czynniki – maleje ona ze wzrostem temperatury i rośnie ze wzrostem ciśnienia. W roztworach substancji stałych istnieje stan równowagi dynamicznej pomiędzy osadem a substancją rozpuszczoną, ilościowo opisywany przez iloczyn rozpuszczalności.
Ze względu na stopień nasycenia roztwór może być:
a) nasycony – kiedy po dalszym wprowadzaniu substancji rozpuszczonej stężenie nie ulega zmianie (nie da się dalej rozpuszczać danej substancji),
b) nienasycony – kiedy po wprowadzeniu dodatkowej ilości substancji rozpuszczonej stężenie wzrasta (można w nim dalej rozpuszczać daną substancję),
c) przesycony – o stężeniu większym od stężenia roztworu nasyconego w danej temperaturze. Może on powstać podczas obniżania temperatury roztworu nasyconego w warunkach utrudnionego powstawania zarodków krystalizacji. Wstrząśnięcie lub dodanie substancji rozpuszczonej powoduje gwałtowaną krystalizację jej nadmiaru i powstanie roztworu nasyconego.
Krystalizacja to proces tworzenia się i wzrostu kryształów z cieczy, roztworu bądź fazy gazowej.
Rozpuszczalność to maksymalna ilość gramów danej substancji, jaką w danych warunkach ciśnienia i temperatury można rozpuścić w 100g rozpuszczalnika, otrzymując roztwór nasycony.
Przy opisie właściwości roztworu największe znaczenie obok składu jakościowego ma podanie jego stężenia.
a) stężenie procentowe to ilość gramów substancji zawarta w 100g roztworu:
Cp
=
 · 100% =
· 100% =
 · 100%
· 100%
b) stężenie molowe to ilość moli substancji rozpuszczonej w 1dm3 rozpuszczalnika; jest wielkością zależną od temperatury:
Cmol
=
 =
=
 [
[
c) stężenie molalne – ilość moli substancji rozpuszczonej zawarta w 1kg roztworu; jest wielkością niezależną od temperatury:
Ca
=
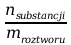 [
[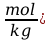
Przeliczanie stężeń (M – masa molowa substancji rozpuszczonej)
|
|
Cp |
cmol |
Ca |
|
Cp |
- |
Cp
=
|
Cp
=
|
|
cmol |
Cmol
=
|
- |
Cmol
=
|
|
Ca |
Ca
=
|
Ca
=
|
- |
 · 100%
· 100%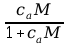 · 100%
· 100%
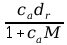
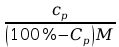

Reguła mieszania roztworów o znanych stężeniach procentowych:
 =
=
 (Cp2
> Cpx
> Cp1)
(Cp2
> Cpx
> Cp1)
Reguła mieszania roztworów o znanych stężeniach molowych:
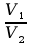 =
=
 (Cmol2
> Cmolx
> Cmol1
i
przy pominięciu kontrakcji – założeniu addytywności objętości)
(Cmol2
> Cmolx
> Cmol1
i
przy pominięciu kontrakcji – założeniu addytywności objętości)
Niektóre właściwości fizyczne roztworów:
a) Kontrakcja to efekt towarzyszący mieszaniu roztworów, polegający na otrzymaniu roztworu o objętości mniejszej od sumy objętości roztworów wyjściowych. Podobnie jak przy mieszaniu kamieni z piaskiem ten ostatni zajmuje wole przestrzenie pomiędzy większymi okruchami, tak samo zachowują się cząsteczki rozpuszczalnika i substancji rozpuszczonej. Przy mieszaniu roztworów wodnych o niewielkiej różnicy stężeń kontrakcja praktycznie nie występuje. Przy mieszaniu roztworu o dużym stężeniu z czystym rozpuszczalnikiem kontrakcja wynosi nie więcej niż kilka procent. Np. przy zmieszaniu 480cm3 wody z 20cm3 96% kwasu siarkowego (d=1,84g/cm3) otrzymujemy 495cm3, a nie przewidywane 500cm3; kontrakcja w tym przypadku wynosi: ΔV/V=5/500=1%. W typowych obliczeniach laboratoryjnych dotyczących stężeń molowych na ogół nie zachodzi potrzeba uwzględniania kontrakcji. W celu uzyskania bardzo dokładnego wyniku należy odpowiednie stężenia molowe zamienić na stężenia procentowe i zastosować regułę mieszania roztworów procentowych.
b) Zmiana temperatury topnienia i wrzenia roztworu w stosunku do czystego rozpuszczalnika: roztwory wrą w wyższych i zamarzają w niższych temperaturach niż występujące w nich rozpuszczalniki. Podwyższenie temperatury wrzenia, podobnie jak obniżenie temperatury topnienia, to wielkości koligatywne – charakterystyczne dla roztworu, a zależne wyłącznie od liczby niezależnych cząsteczek w danej objętości roztworu. Są one wprost proporcjonalne do stężenia molalnego roztworu i dla substancji nie dysocjującej wynoszą: ΔTk=Ek ma, ΔTw=Eb
.ma. Występujące w powyższych równaniach współczynniki
proporcjonalności noszą nazwy stałej krioskopowej oraz stałej ebulioskopowej. Wartości ich można
obliczyć z równań prawa Raoulta: Ek=RTk
2M/Lt, Eb=RTw
2M/Lp, gdzie R oznacza uniwersalną stałą gazową,
M – masę molową rozpuszczalnika, Tk, Tw – temperatury bezwzględne krzepnięcia i wrzenia
rozpuszczalnika, Lt, Lp – ciepła molowe topnienia i parowania rozpuszczalnika. Przykładowo dla wody
wartości te wynoszą: Ek=-1,87oC, Eb=0,51oC.
c) Ciśnienie osmotyczne: obecność w roztworze substancji rozpuszczonych powoduje powstawanie
dodatkowego ciśnienia, zwanego ciśnieniem osmotycznym. Aby roztwór pozostał w równowadze
osmotycznej z czystym rozpuszczalnikiem, należy nań wywrzeć nadwyżkę ciśnienia równą ciśnieniu
osmotycznemu. Przez równowagę osmotyczną rozumiemy, iż rozpuszczalnik, oddzielony od roztworu
przegrodą półprzepuszczalną, przenikliwą dla rozpuszczalnika a nieprzenikliwą dla substancji
rozpuszczonej, nie wnika osmotycznie do roztworu ani też nie jest z niego wyciskany nadwyżką ciśnienia w
roztworze. Zgodnie z prawem van’t Hoffa ciśnienie osmotyczne można interpretować jako ciśnienie równe
takiemu, jakie wywierałaby substancja rozpuszczona, jeśli znajdowałaby się w danej temperaturze w stanie
gazowym i zajmowała objętość równą objętości roztworu. Dla rozcieńczonych roztworów nieelektrolitów
można tą wartość wyrazić przekształcając równanie Clapeyrona w równanie van’t Hoffa:
cRT
V
nRT
pV = nRT ⇒ p = = ,
gdzie c oznacza stężenie molowe roztworu, R – uniwersalną stałą gazową, a T – temperaturę bezwzględną.
TEORIE KWASOW I ZASAD
Teorie kwasów i zasad są próbami wyjaśnienia zespołu cech wspólnych, charakterystycznych dla kwasów i
przeciwstawnego mu zespół cech zasad. Spośród kilkunastu zaproponowanych, największe uznanie zyskały:
teoria Arrheniusa (1887r.), teoria Bronsteda – Lowry’ego (1923r.) oraz teoria Lewisa.
Teoria Arrheniusa sformułowana została w postaci trzech postulatów.
Postulat I: Niektóre substancje, zwane elektrolitami, ulegają pod wpływem wody rozpadowi na dwa rodzaje
cząstek: jony dodatnie (kationy) oraz jony ujemne (aniony). Proces ten nazywamy dysocjacją jonową lub
elektrolityczną.
Z I postulatu wynika, iż wszystkie substancje można podzielić na dwie duże grupy:
- elektrolity, będące związkami o budowie jonowej bądź kowalencyjnej silnie spolaryzowanej (kwasy,
zasady, sole, wodorki kwasowe), których roztwory dobrze przewodzą prąd elektryczny dzięki obecności w
nich cząstek obdarzonych ładunkiem (jonów), które mogą się przemieszczać w polu elektrycznym,
- nieelektrolity, będące związkami o budowie kowalencyjnej wcale lub bardzo słabo spolaryzowanej
(cząsteczki pierwiastków oraz wiele związków organicznych), które nie dysocjują i nie przewodzą prądu.
Postulat II: Liczba wytworzonych podczas dysocjacji ładunków dodatnich musi być równa liczbie
wytworzonych ładunków ujemnych. Jest to konsekwencją zasady zachowania ładunku. Zgodnie z tym
założeniem przyjmuje się następujące definicje:
- kwasy to substancje, które w procesie dysocjacji odłączają katony wodoru oraz aniony reszt kwasowych,
ogólnie: HnR ---> nH+ + Rn-;
- zasady to substancje, ktore w procesie dysocjacji odłączają katony metali oraz aniony wodorotlenkowe,
ogólnie: M(OH)n ---> Mn+ + nOH-;
- sole to substancje, ktore w procesie dysocjacji odłączają katony metali oraz aniony reszt kwasowych,
ogólnie: MnRm ---> nMm+ + mRn-.
Postulat III: Niektóre elektrolity w swoich roztworach zawierają także niezdysocjowane cząstki – są to tzw.
elektrolity słabe.
Z III postulatu wynika, że wszystkie elektrolity można podzielić na:
- elektrolity mocne, całkowicie rozpadające się na jony, których roztwory nie zawierają niezdysocjowanych
cząstek; do grupy tej należą: wszystkie sole, kwasy: HNO3, H2SO4, HClO4, HCl, HBr, HI, wodorotlenki
litowców i berylowców za wyjątkiem berylu i magnezu,
- elektrolity słabe, rozpadające się na jony tylko częściowo, których roztwory zawierają duże ilości
niezdysocjowanych cząstek.
Teoria Bronsteda – Lowry’ego zwana jest także teorią protonową. W jej myśl kwasami nazywamy związki lub
jony zdolne do oddawania kationów wodoru, zaś zasadami – związki lub jony zdolne do ich przyjmowania.
Ponieważ kation wodoru to nic innego jak proton, toteż kwasy nazywamy donorami protonów
(protonodawcami), a zasady – akceptorami protonów (protonobiorcami). Każda reakcja kwasu z zasadą
prowadzi do wytworzenia nowej, sprzężonej pary kwas – zasada. Związki tworzące parę sprzężoną różnią się w
składzie cząsteczek co najwyżej o proton (kation wodoru). Im mocniejszy kwas, tym słabsza jest sprzężona z
nim zasada. Według teorii protonowej kwasy można podzielić na:
- kwasy cząsteczkowe (HCl, H2SO4),
- kwasy kationowe (NH4
+),
- kwasy anionowe (HSO4
-),
zaś zasady na:
- zasady cząsteczkowe (NH3),
- zasady anionowe (HSO4
-).
Teoria protonowa rozszerza zatem pojęcie kwasu i zasady o nowe grupy związków. Niektóre cząsteczki mogą
mieć ponadto charakter amfoteryczny, czyli wykazywać w zależności od warunków właściwości kwasowe bądź
zasadowe – np. HSO4
- może zarówno przyłączyć, jak i odszczepić kation wodoru.
Teoria Lewisa również poszerza pojęcie kwasów i zasad. Zgodnie z jej założeniami, kwasem jest związek lub
jon posiadający deficyt elektronowy – kwasami Lewisa są zatem zarówno wszystkie kationy metali, jak i
niektóre cząsteczki obojętne, np. BF3. Atom boru posiada w tym związku sześć elektronów walencyjnych, a
zgodnie z regułą oktetu dąży do uzyskania ośmiu – posiada zatem deficyt, gdyż 6<8. Analogicznie – zasadą jest
związek posiadający wolną parę elektronową, np. Cl- czy NH3. Dzięki swym właściwościom kwasy Lewisa
wykorzystywane są jako katalizatory w niektórych syntezach organicznych, np. w reakcjach Friedla – Craftsa.
ILOŚCIOWY OPIS WŁAŚCIWOŚCI ROZTWOROW
W celu ilościowego opisu procesu dysocjacji wprowadza się wielkości chemiczne: stopień oraz stałą dysocjacji.
a) Stopień dysocjacji (α) wyraża się stosunkiem ilości cząstek zdysocjowanych do ilości cząstek
wprowadzonych do roztworu: α=n/n0=c/c0. Np. dla reakcji AB <==> A+ + B-: α=[A+]/[AB]=[B-]/[AB]
(symbole w nawiasach kwadratowych oznaczają odpowiednie stężenia molowe). Dla elektrolitów
dysocjujących wielostopniowo wyróżnia się kolejne stopnie dysocjacji, przy czym α1>α2>α3 itd. Stopień
dysocjacji zależy od:
- rodzaju elektrolitu,
- temperatury – zasadniczo wzrasta z jej wzrostem,
- obecności innych substancji,
- stężenia substancji wprowadzonej
b) Stała dysocjacji (K), czyli stała równowagi reakcji dysocjacji, np. dla reakcji AB <==> A+ + B-:
K=[A+][B-]/[AB]. Dla elektrolitów dysocjujących wielostopniowo wyróżnia się kolejne stałe dysocjacji,
przy czym K1>K2>K3 itd. Stała dysocjacji zależy tylko i wyłącznie od temperatury.
Rozważmy następujący prosty przykład procesu dysocjacji:
równanie reakcji AB <==> A+ + B stężenie
początkowe c0 - -
stężenie reagujące c c c
stężenie równowagowe c0 - c c c
Powyższy związek to tzw. prawo rozcieńczeń Ostwalda. Można za jego pomocą wyznaczyć stopień dysocjacji:
Pierwsze rozwiązanie nie ma sensu chemicznego – stopień dysocjacji nie może przyjmować wartości ujemnych.
W przypadku, gdy α<5% lub gdy c0/K≥400, wówczas:
α ≈ ⇒ 1− α ≈ ⇒ Κ ≈ c α ⇒α = K .
Autodysocjacja wody to rozpad cząsteczki wody pod wpływem innej cząsteczki wody; sumarycznie proces ten
można przedstawić jako: H2O + H2O <==> H3O+ + OH-.
Eksperymentalnie wyznaczona stała dysocjacji powyższej reakcji wynosi: K=1,8.10-16. W wyrażeniu opisującym
stałą pojawia się stężenie molowe wody, przyjmowane za wartość stałą i obliczane następująco:
Powyższa wartość to tzw. iloczyn jonowy wody – jest ona zawsze stała dla wszystkich roztworów wodnych w
danej temperaturze. Pozwala ona na obliczenie stężenie H+ lub OH- w danym roztworze, gdy dana jest jedna z
tych wielkości. Wartości powyższych stężeń są bardzo małe, do rzędu czternastu miejsc po przecinku włącznie.
Dla uniknięcia stosowania niewygodnych liczb podaje się nie samą ich wartość, lecz potęgę, do jakiej należy
podnieść podstawę równą 10, aby tę wartość otrzymać. Ponieważ są to najczęściej liczby ujemne, poprzedza się
je znakiem minus. W ten sposób określa się pojęcie pH oraz pOH:
[ ] 10 log[ ] log[ ]
[ ] 10 log[ ] log[ ]
− − − −
+ − + +
= ⇒− = ⇒ = −
= ⇒− = ⇒ = −
OH pOH OH pOH OH
H pH H pH H
pOH
pH
.
Z powyższych definicji wynika następująca własność:
+ = −log[ + ]− log[ − ] = −log([ + ][ − ]) = −log10−14 =14 pH pOH H OH H OH .
Ze względu na wartości stężeń H+ i OH- wprowadza się pojęcia odczynu roztworu:
odczyn roztworu proporcje stężeń wartości stężeń pH i pOH
obojętny [H+]=[OH-] [H+]2=[OH-]2=10-14 => [H+]=[OH-]=10-7 mol/dm3 pH=pOH=7
kwasowy [H+]>[OH-] [H+]>10-7 mol/dm3 [OH-]<10-7 mol/dm3 pH<7 pOH>7
zasadowy [H+]<[OH-] [H+]<10-7 mol/dm3 [OH-]>10-7 mol/dm3 pH>7 pOH<7
Iloczyn rozpuszczalności to pojęcie wprowadzone dla roztworów nasyconych soli trudno rozpuszczalnych. Gdy
stężenie osadu przyjmuje się za stałe, wówczas iloczyn stałej dysocjacji i stężenia osadu także jest niezmienny, a
więc iloczyn stężeń jonów w wykładnikach potęgowych ich współczynników stechiometrycznych także będzie
stały i określany jako iloczyn rozpuszczalności (Ir). Jest to wielkość stała dla danej soli w danej temperaturze –
charakteryzuje ona sol pod względem rozpuszczalności: im niższa wartość Ir, tym słabiej rozpuszczalna jest dana
sol. Gdy do roztworu nasyconego dodaje się jonów trudno rozpuszczalnej soli, wówczas iloczyn stężeń jonów w
roztworze przekracza wartość Ir, co powoduje wytrącenie osadu; dodanie wody do wyjściowego roztworu
powoduje zmniejszenie stężenia i przejście części osadu w postaci jonów do roztworu. Jeżeli w roztworze
znajdują się jony dwóch soli o różnych Ir, to jako pierwsza wytrąci się z roztworu sol o mniejszej wartości Ir.
REAKCJE ZACHODZĄCE W ROZTWORACH
Miareczkowanie alkacymetryczne
Analiza chemiczne to zespół czynności zmierzających do ustalenia jakościowego i ilościowego składu próbki.
Jednym z jej działów jest analiza miareczkowa (objętościowa), polegająca na oznaczaniu składnika roztworu na
podstawie pomiaru objętości substancji zużytej w celu ilościowego przeprowadzenia reakcji z tym składnikiem
(titrantu). Titrant (roztwór mianowany) musi być roztworem o znanym z dużą dokładnością stężeniu
określonego składnika. Czynność w analizie miareczkowej, polegająca na dodawaniu z biurety titrantu do
roztworu substancji oznaczanej do momentu zakończenia reakcji (osiągnięcia punktu równoważnikowego)
nazywamy miareczkowaniem. Często w ramach analizy miareczkowej stosuje się alkacymetrię (miareczkowanie
alkacymetryczne), polegającą na oznaczaniu stężenia kwasu poprzez miareczkowanie zasadą (alkalimetria) bądź
zasady poprzez miareczkowanie kwasem (acydymetria). Punktem równoważnikowym (PR) nazywamy moment,
w którym oznaczany składnik przereagował ilościowo z titrantem. Określa się go za pomocą wskaźników lub
innymi metodami, np. elektrochemiczną. Roztwór znajdujący się w PR jest całkowicie zobojętniony, co nie
oznacza zawsze jednak odczynu obojętnego – sole hydrolizujące mogą mieć inny odczyn. Określa się również
punkt końcowy miareczkowania (PK), na ogół nieznacznie różniący się od PR.
Do ustalenia PR wykorzystuje się często metodę pomiaru przewodnictwa elektrycznego (η) roztworu
miareczkowanego. Przewodnictwo roztworu w PR ma wartość minimalną, gdyż obojętny roztwór w bardzo
małym stopniu przewodzi prąd elektryczny. Ilustrując zobojętnianie obserwujemy najpierw liniowy spadek
przewodnictwa, następnie linia wykresu ostro zaznacza PR, po czym przewodnictwo rośnie, gdyż przy dalszym
dodawaniu titrantu zaczyna on dysocjować, zwiększając liczbę jonów w roztworze. W przypadku wytrącania się
nierozpuszczalnego osadu przewodnictwo w PR spada do zera – np. przy miareczkowaniu roztworu kwasu
siarkowego wodorotlenkiem baru. Dla sformalizowania przewodnictwa elektrycznego roztworów wprowadza się
następujące wielkości chemiczne:
- przewodność elektrolityczną: χ=1/ς [m2/.] (ς=RS/l),
- przewodność molowa elektrolitu to stosunek przewodności elektrolityczne do stężenia molowego:
Λ=χ/c [m5/./mol]; jeżeli stężenie wyrażone jest w mol/dm3, wówczas Λ=1000χ/c.
Amfoteryczność to zdolność niektórych związków chemicznych, najczęściej tlenków i wodorotlenków, do
wykazywania zarówno właściwości kwasowych, jak i zasadowych. Związki takie nazywamy amfoterycznymi
lub amfoterami. Pierwiastki amfoteryczne leżą na przekątnej układu okresowego i mają elektroujemność
zbliżoną do wodoru (ok. 1,5-2). W wodorotlenkach amfoterycznych wiązania pomiędzy atomami wodoru i tlenu
oraz tlenu i metalu mają podobną moc. Tlenki i wodorotlenki amfoteryczne z reguły nie rozpuszczają się w
wodzie. W reakcjach z mocnymi kwasami zachowują się jak zasady, a z zasadami – jak kwasy.
Najważniejsze amfotery to związki następujących pierwiastków: Be+2, Al+3, V+4, V+5, Cr+3, Mn+4, Fe+2, Fe+3,
Co+2, Cu+1, Cu+2, Zn+2, Ga+3, Ge+4, As+3, Zr+4, Nb+5, Pd+4, Ag+1, Cd+2, In+3, Sn+2, Sn+4, Sb+3, Sb+5, Te+4, Hf+4,
Ta+5, Re+4, Ir+3, Pt+4, Au+3, Pb+2, Pb+4, Ce+4, U+6. Związki pierwiastków bloku d posiadających nie zapełnione
orbitale walencyjne są na ogoł barwne. Wśród związków organicznych charakter amfoteryczny wykazują przede
wszystkim aminokwasy z racji posiadania dwóch grup funkcyjnych – karboksylowej (kwasowej) oraz aminowej
(zasadowej).
Zobojętnianie to reakcja łączenia się kwasowych kationów wodorowych z zasadowymi anionami
wodorotlenkowymi w obojętne cząsteczki wody: H+ + OH- ---> H2O.
Hydroliza to reakcja odwrotna do zobojętniania – jest to reakcja jonów soli z cząsteczkami wody, w wyniku
której roztwory wodne soli mogą mieć odczyn różny od obojętnego.
Rodzaj soli Typ hydrolizy Odczyn środowiska
sol mocnego kwasu i słabej zasady hydroliza kwasowa (kationowa) kwasowy
sol słabego kwasu i mocnej zasady hydroliza anionowa (zasadowa) zasadowy
sol słabego kwasu i słabej zasady kwasowo-zasadowa (kationowo-anionowa) zbliżony do obojętnego
sol mocnego kwasu i mocnej zasady brak hydrolizy obojętny
Odczyn środowiska, w którym hydrolizuje sol słabego kwasu i słabej zasady, nie jest idealnie obojętny,
ponieważ stałe dysocjacji tworzących sol kwasu i zasady na ogół nieznacznie różnią się od siebie. O odczynie
roztworu decyduje zatem relacja KK i KZ.
Wskaźniki (indykatory) to najczęściej słabe kwasy lub zasady organiczne, występujące w dwóch formach
barwnych. Przykładami są: oranż metylowy (na kwasy), błękit tymolowy, fenoloftaleina (na zasady) oraz
wskaźnik uniwersalny, będący odpowiednio spreparowaną mieszaniną innych wskaźników.
Roztwór buforowy (bufor = zderzak) to roztwór mający zdolność przeciwdziałania zmianom pH zarówno przy
rozcieńczaniu, jak i dodawaniu do nich niewielkich ilości mocnych kwasów lub zasad. Roztworem buforowym
może być:
- roztwór słabego kwasu i jego soli z mocna zasadą,
- roztwór słabej zasady i jej soli z mocnym kwasem,
- roztwór dwóch soli kwasu wieloprotonowego.
Działanie buforujące przedstawione zostanie na przykładzie buforu octanowego (CH3COOH + CH3COONa).
protonodawca: CH3COOH <==> CH3COO- + H+ [CH3COOH]≈ckwasu
protonobiorca: CH3COONa <==> CH3COO- + Na+ [CH3COO-]≈csoli
a) protonodawca chroni bufor przed zmianami pH podczas dodawania do niego zasady, gdyż zobojętnia ją:
CH3COOH + OH- ---> CH3COO- + H2O
b) protonobiorca chroni bufor przed zmianami pH podczas dodawania do niego kwasu, gdyż zobojętnia go:
CH3COO- + H+ ---> CH3COOH
Dla buforu słabego kwasu można zapisać:
kwasu soli K K K
Analogicznie dla buforu słabej zasady:
zasady soli Z Z Z
W przypadku, gdy objętości kwasu (zasady) i soli są różne, w miejsce stosunków stężeń należy wstawić
odpowiednie stosunki molowe.
Związki kompleksowe to połączenia tworzące się jako trwalsze formy obecności cząsteczek lub jonów w
roztworach. Ich powstawanie jest przyczyną pogłębienia barwy roztworów. Kompleksy zbudowane są z
centralnie położonego atomu lub jonu oraz otaczających go ligandów. Kompleksy mogą zawierać jeden lub
więcej jonów (atomów) centralnych, zwanych rdzeniami, stąd wyróżnia się kompleksy jedno- oraz
wielordzeniowe. Ligandem może być cząsteczka lub jon posiadające wolną parę elektronową. Liczbę ligandów
otaczających atom centralny określa tzw. liczba koordynacyjna; zazwyczaj jest ona liczbą parzystą. Ligandy
podzielić moŜna na jednokleszczowe (monodentne – koordynujące jedną parę elektronową) oraz
wielokleszczowe (multidentne – koordynujące dwoma lub więcej parami elektronowymi). Ze względu na
ładunek, kompleksy moŜna podzielić na kationowe, anionowe oraz obojętne. Przy zapisie kompleksu w postaci
wzoru chemicznego kompleks ujmuje się w nawias kwadratowy.
Najczęściej występujące ligandy oraz skojarzone z nimi przedrostki:
Wzor Nazwa Wzor Nazwa
H2O akwa- S2O3
2- tiosiarczano-
NH3 amino- SCN- tiocyjaniano-
CO karbonylo- NO nitrozylo-
Cl- chloro- azotyno-
CH3COO- octano- nitro-
W celu ilościowego ujęcia mocy kompleksu się następujące wielkości chemiczne:
a) stała nietrwałości (rozpadu) kompleksu to stała rownowagi hipotetycznej reakcji ELn <==> En+ + nL-, czyli:
[ ]
[ ][ ]
EL
E L
K
n n
N
+ −
= ; im wyŜsza wartość stałej nietrwałości, tym dany kompleks jest mniej trwały;
b) stała trwałości (tworzenia kompleksu) to odwrotność stałej nietrwałości, a zarazem stała rownowagi reakcji
En+ + nL- <==> ELn, czyli:
n n
N
T E L
EL
K
K
[ ][ ]
1 [ ]
+ − = = ; im wyŜsza wartość stałej trwałości, tym trwalszy
jest dany związek kompleksowy.
KOLOIDY
Koloidy to układy dwu- lub wieloskładnikowe, heterogeniczne, w ktorych cząstki fazy rozproszonej mają
średnic rzędu 1-200nm. Cząstki koloidalne nie są widoczne gołym okiem ani pod mikroskopem, przechodzą
takŜe przez sączki filtracyjne. NajwaŜniejsze układy koloidowe to:
Created by Neevia Document Converter trial version http://www.neevia.com
25
Nazwa Stan skupienia faz Przykłady
zole ciało stałe w cieczy lub w gazie smog, dym w powietrzu, roztwor białka, skrobi, krochmal,
kleje pochodzenia roślinnego, budynie i kisiele
mgły ciecz w gazie aerozole mgła atmosferyczna
dymy ciało stałe w gazie dym z komina, kurz domowy
emulsje ciecz w cieczy mleko, majonez, masło, margaryna, kremy
piany ciekłe gaz w cieczy piana mydlana
stałe gaz w ciele stałym pumeks, styropian, materace piankowe
Koloidy moŜna podzielić na:
a) koloidy cząsteczkowe – fizycznie jednorodne, ktorych składnikami są makrocząsteczki, po wprowadzeniu
do wody zawsze otrzymujemy roztwor koloidowy, np. koloid białka, skrobi, celulozy, glikogenu, kauczuku;
b) koloidy fazowe – fizycznie niejednorodne, z nieciągłą powierzchnią podziału obu faz, na ktorej zachodzi
adsorpcja elektrolitu, warunkująca powstanie ładunku elektrycznego, np. zol złota (Au)x (tzw. purpura
Kasjusza), koloid srebra (Ag)y, koloid chlorku srebra (AgCl)z (x, y, z > 100).
Zasadniczo koloidy moŜna otrzymać na dwa sposoby:
a) metoda dyspersyjna polega na rozpraszaniu większych cząstek do rozmiarow koloidowych – do tego celu
wykorzystuje się specjalne urządzenia, zwane młynkami koloidowymi (dzielą się na termiczne, elektryczne
itd., w zaleŜności od rodzaju czynnika rozpraszającego),
b) metoda kondensacyjna i polimeryzacyjna polega na łączeniu atomow lub cząsteczek w większe zespoły o
rozmiarach odpowiadających cząsteczkom koloidowym (np. metoda strąceniowa).
Ze względu na stosunek do cząsteczek rozpuszczalnika koloidy dzielimy na:
a) liofilowe (gdy rozpuszczalnikiem jest woda – hydrofilowe), ktorych cząsteczki otaczają się zwartym
płaszczem cząsteczek rozpuszczalnika, dzięki czemu stają się bardziej odporne na działanie czynnikow
ścinających; są podatne na pęcznienie, tworzenia galaret i Ŝelow (np. koloid białka);
b) liofobowe (ew. hydrofobowe), w ktorych oddziaływanie z cząsteczkami rozpuszczalnika jest bardzo słabe,
przez co są one bardziej podatne na ścinanie od liofilowych (np. koloid srebra czy złota).
Właściwości roztworow koloidowych:
a) efekt Tyndalla – specyficzny sposob rozpraszania wiązki światła w charakterystyczny stoŜek Tyndalla,
b) barwa najczęściej biała; niektore koloidy posiadają barwę zaleŜną od rozmiarow cząstek koloidowych,
grubości warstwy, przez ktorą przechodzi światło oraz od sposobu obserwacji,
c) opalizowanie na świetle – obserwacja tęczowych refleksow,
d) zdolność do koagulacji, czyli łączenia się w większe zespoły, np.: koloid koagulat
koagulacja
peptyzacja
⇔ :
- koagulacja odwracalna: ( )
( 3 )
koloid białia wysolone bialko osad
r r soli NaNO
woda destylowana
−
⇔ ,
- koagulacja nieodwracalna (denaturacja): koloid białia zdenaturowane bialko
czynnik denaturujący
⇔ ;
do czynnikow denaturujących zaliczamy: wysoką temperaturę, jony metali cięŜkich, alkohol, kwasy, zasady,
formalinę,
e) tendencja do adsorpcji cząsteczek rozpuszczalnika na swojej powierzchni i wytwarzanie dzięki temu
ładunku elektrycznego o określonym znaku oraz otaczania się jonami o znaku przeciwnym na skutek
przyciągania elektrostatycznego.
ELEKTROCHEMIA
STOPNIE UTLENIENIA PIERWIASTKOW
Stopień utlenienia pierwiastka w związku o budowie jonowej jest rowny ładunkowi jonu, jaki tworzy atom tego
pierwiastka w związku. Stopień utlenienia pierwiastka w związku o budowie kowalencyjnej jest rowny liczbie
wszystkich ładunkow dodatnich lub ujemnych, ktore przypisalibyśmy atomowi pierwiastka, gdyby związek miał
budowę jonową.
Zasady ustalania stopnia utlenienia pierwiastka:
a) pierwiastki w stanie wolnym mają zawsze stopień utlenienia rowny zero,
Created by Neevia Document Converter trial version http://www.neevia.com
26
b) wodor przyjmuje z związkach chemicznych zawsze stopień utlenienia rowny +1, z wyjątkiem wodorkow
metali, gdzie posiada –1,
c) tlen przyjmuje w związkach chemicznych zawsze stopień utlenienia rowny –2, z wyjątkiem tlenku fluoru
(+2) oraz nadtlenkow (–1),
d) metale w związkach chemicznych mają zawsze dodatnie stopnie utlenienia, liczbowo rowne
wartościowości,
e) suma algebraiczna wszystkich stopni utlenienia w obojętnym związku chemicznym jest rowna zero, a w
jonie złoŜonym rowna jest ładunkowi tego jonu.
REAKCJE REDOX W UJĘCIU ELEKTRONOWYM
a) Utleniania (dezelektronacja) to reakcja oddawania elektronow i podwyŜszania stopnia utlenienia
pierwiastka.
b) Redukcja (elektronacja) to reakcja przyjmowania elektronow i obniŜania stopnia utlenienia pierwiastka.
c) Utleniacz (dezelektronator, elektronobiorca) to atom pierwiastka, ktory przyjmując elektrony powoduje
utlenienie atomu innego pierwiastka, a sam się redukuje. Typowe utleniacze chętnie przyjmują elektrony i
są „na minusie”.
d) Reduktor (elektronator, elektronodawca) to atom pierwiastka, ktory oddając elektrony powoduje redukcję
atomu innego pierwiastka, a sam się utlenia. Typowe reduktory chętnie oddają elektrony i są „na minusie”.
e) Prawo zachowania ładunku dla reakcji redox: liczba elektronow oddanych w procesie utleniania musi być
rowna liczbie elektronow przyjętych w procesie redukcji.
NAJWAśNIEJSZE UTLENIACZE I REDUKTORY
Utleniaczem moŜe być kaŜdy atom pierwiastka, ktory ma moŜliwość przyjęcia elektronow i obniŜenia swego
stopnia utlenienia. W związku z powyŜszym utleniaczami mogą być:
a) atomy niemetali w stanie wolnym (np. O2, Cl2, S8),
b) jony lub atomy metali występujących na swoich wyŜszych stopniach utlenienia (Fe3+, Sn4+, Pb4+, Cr6+,
Mn7+),
c) atomy niemetali występujące na swoich wyŜszych stopniach utlenienia (N5+, S6+, Cl7+).
Utleniaczami natomiast nie mogą być (są jednocześnie dobrymi reduktorami):
a) atomy metali w stanie wolnym (Mg0, Fe0),
b) atomy niemetali występujące na swoich najniŜszych stopniach utlenienia (Cl-, Br-, I-, F-, S2-, N3-).
ZWIĄZKI CHROMU I MANGANU JAKO UTLENIACZE
a) Chrom jest pierwiastkiem bloku d układu okresowego. Z połoŜenia wynika, iŜ powinien on mieć
następującą konfigurację: 25Cr: [18Ar]4s23d4, jednak w wyniku promocji elektrony walencyjne przyjmują
układ 4s15d5. Pozwala to atomowi chromu na przyjmowanie stopni utlenienia +2, +3 oraz +6. W tym
ostatnim przypadku tlenek chromu CrO3 wykazuje właściwości kwasowe. Chrom tworzy dwa głowne
kwasy tlenowe: chromowy (VI) K2CrO4 o barwie Ŝołtej oraz dwuchromowy (VI) K2Cr2O7 o barwie
pomarańczowej. W roztworze wodnym pomiędzy jonami chromianowymi (VI) i dwuchromianowymi (VI)
wytwarza się stan rownowagi opisany rownaniem: 2CrO4
- + H+ <==> Cr2O7
2- + OH-; wynika z niego, iŜ w
środowisku kwaśnym przewaŜa dwuchromian (VI), a w zasadowym – chromian (VI).
Badanie doświadczalne zdolności utleniających związkow chromu w zaleŜności od odczynu środowiska
moŜna przeprowadzić w następujący sposob. Do dwoch probowek z roztworem dwuchromianu (VI) potasu
dodaje się: do jednej kwasu siarkowego (VI), a do drugiego zasady potasowej, a następnie do obu wkrapla
się jodek potasu. Obserwujemy wydzielanie się pęcherzykow gazu (jod) w obu probowkach. Ponadto w
probowce z zasadą kolor roztworu zmienia się z pomarańczowego na zielony, co świadczy o pojawieniu się
jonow Cr3+ (zmiana o trzy stopnie utlenienia). W drugiej probowce zmiana następuje aŜ o cztery stopnie
utlenienia, dając jony Cr2+, łączące się w siarczan (VI) chromu (II), strącający się w postaci bezbarwnego
osadu. Z takiego przebiegu doświadczenia wnioskujemy, iŜ lepsze własności utleniające związku chromu
wykazują w środowisku kwaśnym, w obojętnym zapewne są one słabsze, a najsłabsze w zasadowym.
Chromiany znalazły rownieŜ zastosowanie w alkometrach, gdyŜ reagują z alkoholami i utleniają je do
związkow karbonylowych (aldehydow lub ketonow), np. izopropanol do acetonu:
3 CH3CH(OH)CH3 + K2Cr2O7 + 4H2SO4 ---> 3 CH3COCH3 + Cr2(SO4)3 + K2SO4 + 7H2O.
Wizualnym efektem zachodzenia reakcji jest zmiana barwy środowiska z pomarańczowego na zielony.
b) Mangan jest rownieŜ pierwiastkiem bloku energetycznego d. Posiada on konfigurację 25Mn: [18Ar]4s23d5, co
pozwala mu na przyjmowanie wartościowości od I do VII włącznie. Mangan bierze udział w roŜnych
reakcjach redox, np.:
Created by Neevia Document Converter trial version http://www.neevia.com
27
MnO2 + 4 HCl ---> MnCl2 + Cl2, + H2O,
MnO2 + H2O2 + H2SO4 ---> MnSO4 + O2 + 2 H2O,
2 KMnO4 + 16 HCl ---> 2MnCl2 + 2KCl + 5Cl2 + 8H2O.
Doświadczalne badanie zdolności utleniających związkow manganu w zaleŜności od odczynu środowiska
reakcji wykonuje się podobnie jak w przypadku związkow chromu. Do trzech probowek zawierających
roztwor manganianu (VII) potasu KMnO4 dodaje się: kwasu siarkowego (VI), wody destylowanej oraz
wodorotlenku potasu, a następnie – roztwor siarczanu (IV) sodu. W probowce pierwszej (kwas) fioletowy
roztwor odbarwia się do przezroczystego, w drugiej (woda) staje się czarno-brunatny, a w trzeciej (zasada) –
zielony. Tłumaczy się to zachodzeniem następujących reakcji:
I (kwaśne) MnO4
- ---H+---> Mn2+
2 KMnO4 + 5 Na2SO3 + 3 H2SO4 ---> 2 MnSO4 + 5 Na2SO4 + K2SO4 + 3 H2O,
II (obojętne) MnO4
- ---H2O---> MnO2↓
2 KMnO4 + 3 Na2SO3 + H2O ---> 3 Na2SO4 + 2 MnO2↓ + 2 KOH
III (zasadowe) MnO4
- ---OH----> MnO4
2-
2 KMnO4 + Na2SO3 + 2 KOH ---> Na2SO4 + 2 K2MnO4 + H2O.
Ostatnia reakcja wykazuje ogromną czułość – moŜna za jej pomocą wykryć obecność manganu w ilości 10-6
mg. Podobnie jak przy chromie, najsilniejsze własności utleniające nadmanganian potasu posiada w
środowisku kwaśnym, słabsze w obojętnym, najsłabsze zaś w środowisku zasadowym. Miarą mocy
utleniającej jest liczba elektronow wymienionych (tu: przyjętych) przez atomy magnezu rowna liczbie
utlenionych cząsteczek siarczanu (IV) sodu (odpowiednio: 5, 3, 1).
SZEREG AKTYWNOŚCI METALI
Szereg aktywności metali to ułoŜenie metali od najbardziej aktywnego do najmniej aktywnego. W szeregu tym
występuje rownieŜ wodor, ktory dzieli wszystkie metale na nieszlachetne (poprzedzające go w szeregu) i
szlachetne (występujące za nim w szeregu). Metale nieszlachetne wydzielają wodor z rozcieńczonych kwasow
nieorganicznych, podczas gdy metale szlachetne nie dają takiej reakcji. Dowolny metal występujący w szeregu
moŜe być reduktorem kationow wszystkich metali występujących za nim i nie moŜe być reduktorem kationow
metali występujących przed nim. Odwracając powyŜszą regułę – dowolny metal moŜe utlenić do kationu kaŜdy
inny metal występujący w szeregu przed nim i nie moŜe utlenić metalu występującego za nim. Wynika z tego, iŜ
najlepsze reduktory znajdują się na początku szeregu aktywności (przed nimi prawie nic juŜ nie ma), zaś
najlepsze utleniacze znajdują się na końcu szeregu (za nimi prawie nic juŜ nie ma).
Skrocony szereg aktywności ma następującą postać:
Li, K, Ba, Sr, Ca, Na, Mg, Be, Al, Mn, Zn, Cr, Fe, Cd, Co, Ni, Sn, Pb, H2, Bi, Sb, As, Cu, Ag, Hg, Pt, Au
POŁOGNIWA
a) definicja
Połogniwem (zwyczajowo – elektrodą) nazywamy układ złoŜony z co najmniej dwoch kontaktujących się
ze sobą faz: przewodnika metalicznego (I rodzaju, elektrody) zanurzonego w przewodniku jonowym (II
rodzaju). Liczba faz połogniwa moŜe być większa.
b) podział i przykłady
Klasyfikacja połogniw ma charakter umowny.
Ze względu na budowę (liczbę i rodzaj faz) rozroŜnia się kilka rodzajow połogniw.
- Połogniwo I rodzaju zbudowane jest z przewodnika metalicznego zanurzonego w roztworze elektrolitu.
Przykładem jest połogniwo miedziowe: Cu | Cu2+.
- Połogniwo II rodzaju zbudowane jest z przewodnika metalicznego, pokrytego solą trudno rozpuszczalną
tego metalu i zanurzonego w roztworze elektrolitu o anionie identycznym z anionem soli trudno
rozpuszczalnej. Obecne są dwie granice faz: roztwor – sol oraz sol – metal, na granicach ktorych ustala się
rownowaga. Przykładem jest połogniwo chlorkowo – srebrowe: Ag, AgCl | Cl-
(aq)
jako K(+): Ag+ + e- ---> Ag0, AgCl (rozpuszczanie) ---> Ag+ + Cljako
A(-): Ag0 ---> Ag+ + e-, Ag+ + Cl- ---> AgCl (osadzanie)
ogolnie: Ag + Cl- <==> AgCl + e-
StęŜenie roztworu zaleŜy od anionu, a dzięki stałemu stęŜeniu potencjał rownieŜ jest stabilny.
Połogniwem II rodzaju jest takŜe połogniwo kalomelowe: Hg(c), Hg2Cl2(s) | Cl-
(aq)
2 Hg + 2 Cl- <==> Hg2Cl2 + 2e-
- Połogniwo III rodzaju zbudowane jest z przewodnika metalicznego, pokrytego solą trudno rozpuszczalną
tego metalu, ktora z kolei jest pokryta drugą solą trudno rozpuszczalną o anionie wspolnym z pierwszą i
zanurzonego w roztworze elektrolitu zawierającego kation metalu występującego w drugiej z tych soli.
Created by Neevia Document Converter trial version http://www.neevia.com
28
Przykładem jest połogniwo ołowiowo – szczawianowo – wapniowe: Pb(s), PbC2O4(s), CaC2O4(s) | Ca2+
(aq).
Działanie jest analogiczne jak w przypadku połogniw II rodzaju.
Ze względu na typ reakcji elektrodowej wyroŜnia się:
- Połogniwo metaliczne (elektroda metaliczna) – kiedy faza metaliczna bierze udział w reakcji elektrodowej.
Zbudowane jest z przewodnika metalicznego zanurzonego w roztworze zawierającym jego jony. Zachodzi
na nim redukcja (elektronacja) lub utlenianie (dezelektronacja). Przykładem jest połogniwo cynkowe:
Zn | Zn2+.
- Połogniwo redox – kiedy faza metaliczna nie bierze udziału w reakcji elektrodowej, lecz jedynie spełnia rolę
przekaźnika elektronow. Do tego typu zaliczamy połogniwa gazowe – zbudowane z przewodnika
metalicznego zanurzonego w roztworze nasyconym gazem i zawierającym jony powstające w wyniku
reakcji elektrodowej tego gazu, np.: połogniwo wodorowe Pt, H2(g) | H+
(aq), chlorowe Pt, Cl(g) | Cl-
(aq),
tlenowe Pt, O2(g) | OH-
(aq).
c) odwracalność połogniw
Połogniwem odwracalnym nazywamy połogniwo znajdujące się w stanie rownowagi termodynamicznej.
Połogniwa I rodzaju są odwracalna względem kationu (reakcja potencjałotworcza: M0 <=> Mn+ + n e-), zaś
połogniwa II rodzaju – względem anionu (reakcja potencjałotworcza: Xn- <=> X + n e-).
d) potencjał połogniwa
RozwaŜmy granicę faz: przewodnik metaliczny – roztwor zawierający jego jony
(rysunek obok). Na granicy tej obecne są roŜnoimienne ładunki, występuje zatem skok
potencjału. Wartość bezwzględna potencjału połogniwa jest niemoŜliwa do
zmierzenia, podobnie jak całkowita energia wewnętrzna substancji, moŜna natomiast
mierzyć go metodą porownawczą – względem innego połogniwa o znanym
potencjale. Pomiaru tego dokonuje się względem normalnej (standardowej) elektrody
wodorowej, ktorej potencjał przyjęto umownie za rowny zeru w kaŜdej temperaturze.
NEW jest elektrodą gazową odwracalną względem kationu.
NEW: Pt, H2(g) | H+
(aq)
p=1013hPa, pH=0 => [H+]=1M
E H |H 0 V
0
2 + = (w kaŜdej temperaturze)
- pomiar dla cynku:
zestawienie ogniwa: A (-) Zn | Zn2+ || H+ | H2, Pt (+) K i odczytanie z woltomierza
wartości SEM
E SEM
SEM E
SEM E E
Zn Zn
Zn Zn
H H Zn Zn
= −
= −
= −
+
+
+ +
2
2
2
2
|
0
|
0
|
0
|
0
0
- pomiar dla miedzi:
zestawienie ogniwa: A (-) Pt, H2 | H+ || Cu2+ | Cu (+) K i odczytanie z woltomierza wartości SEM
E SEM
SEM E
SEM E E
Cu Cu
Cu Cu
Cu Cu H H
=
= −
= −
+
+
+ +
2
2
2
2
|
0
|
0
|
0
|
0
0
Potencjał standardowy (E0) to potencjał danej elektrody zanurzonej w 1-molowym roztworze kationow tego
metalu względem NEW. Potencjały standardowe elektrod metalicznych ułoŜone od najniŜszego do
najwyŜszego tworzą tzw. szereg napięciowy elektrod metalicznych – szereg elektrochemiczny. Kolejność
elektrod w szeregu napięciowym pokrywa się z kolejnością metali w szeregu aktywności.
e) rownanie Nernsta
Potencjał połogniwa zaleŜy od wielu czynnikow, m. in. od temperatury, stęŜenia, a w przypadku gazow –
takŜe od ciśnienia. Dla połogniwa odwracalnego zmiany potencjału ilustruje rownanie Nernsta postaci:
r
u
a
a
zF
RT
E = E0 ± ln , gdzie:
E – potencjał ogniwa w nowych warunkach,
E0 – potencjał standardowy połogniwa,
R – stała gazowa (R=8,31 J/mol/K),
F – stała Faradaya (F=86500 C),
z – liczba ładunkowa reakcji (liczba moli wymienionych elektronow w odniesieniu do jednego mola
reagenta),
Created by Neevia Document Converter trial version http://www.neevia.com
29
T – temperatura bezwzględna (w skali Kelvina),
au, ar – aktywności odpowiednio: formy utlenionej i zredukowanej.
Rownanie to dotyczy połogniw I i II rodzaju (dla połogniw gazowych dotyczy ciśnienia normalnego). Znak
występujący we wzorze zaleŜy od kierunku odwracalności procesu: + dla odwracalności względem kationu
(I rodzaj), – dla odwracalności względem anionu (II rodzaj). Aktywność obu form moŜna przedstawić jako
a=f.c, gdzie c oznacza stęŜenie reagentu, a f to wspołczynnik proporcjonalności, zwany wspołczynnikiem
aktywności: w roztworach rozcieńczonych jego wartość zbliŜona jest do jedności, podczas gdy w stęŜonych
przyjmuje wartość 0<f<1. Dla połogniw redox w miejscu stęŜenia pojawia się odpowiedni iloczyn stęŜeń w
wykładnikach wspołczynnikow stechiometrycznych (stała rownowagi).
Dla połogniwa metalicznego (odwracalnego względem kationu) w warunkach normalnych moŜna przyjąć
aktywność formy zredukowanej za jednostkową, a po wstawieniu odpowiednich stałych i wielkości
charakteryzujących warunki środowiska oraz zamianie logarytmu naturalnego na dziesiętny, wzor Nernsta
przyjmuje postać: u c
n
E E log
0 0,059 = + .
OGNIWA GALWANICZNE
Ogniwem nazywamy zestawienie dwoch połogniw. Aby popłynął prąd, musi wystąpić roŜnica potencjałow
miedzy elektrodami, a zatem musza one zawierać albo roŜne metale albo elektrolity muszą roŜnić się stęŜeniem
(tzw. ogniwo stęŜeniowe). Ogniwo metaliczne jest zestawieniem dwoch roŜnych metali połączonych
przewodnikiem i zanurzonych do roztworu elektrolitu. Pod wpływem roŜnicy potencjałow następuje
wytworzenie pola elektrycznego ( dx
E = −grad dV ) , ktorego siły powodują ruch elektronow od metalu
aktywniejszego (niŜszy potencjał) do metalu mniej aktywnego (wyŜszy potencjał). Oba połogniwa nazywamy
elektrodami: anoda to elektroda, na ktorej zachodzi utlenianie (w ogniwach ma ona znak –), zaś katoda to ta, na
ktorej zachodzi redukcja (w ogniwach ma ona znak +). Istnieje bardzo wiele rodzajow ogniw – tu podamy
jedynie kilka najpopularniejszych przykładow.
a) ogniwo Volty
Gdy do wodnego roztworu kwasu siarkowego (VI) zanurzymy dwie
płytki: cynkową oraz miedzianą, wowczas na pierwszej z nich zacznie
wydzielać się wodor zgodnie z rownaniem reakcji:
Zn + H2SO4 ---> ZnSO4 + H2,
natomiast miedź jest metalem połszlachetnym i nie daje podobnego
efektu (rysunek lewy). Układ ten nie wnosi nic ciekawego. JeŜeli
jednak obie płytki połączymy przewodnikiem, a w tak zmontowany
obwod włączymy Ŝarowkę, to zacznie ona świecić (rysunek prawy). W przewodniku odbywa się przepływ
elektronow wytworzonych w procesie utleniania na anodzie do katody, gdzie teraz redukowany jest wodor.
Ten ostatni wydziela się na płytce miedzianej w postaci pęcherzykow, ktore blokują przystęp nowych
kationow wodorowych, wobec czego praca ogniwa ustaje – zjawisko to nosi nazwę polaryzacji elektrody
(tu: katody). Dla ogniwa Volty moŜna zapisać:
A(-): Zn0 ---> Zn2+ + 2e- (utlenianie)
K(+): 2H+ + 2e- ---> H2↑ (redukcja)
sumarycznie: Zn + H2SO4 ---> ZnSO4 + H2↑
Zn + 2H+ + SO4
2- ---> Zn2+ + SO4
2- + H2↑
Zn + 2H+ ---> Zn2+ + H2↑
W roztworze powstaje siarczan (VI) cynku, zaś miedź spełnia wyłącznie rolę przewodnika. Ogniwo moŜna
przedstawić schematycznie: A (-) Zn | H2SO4 | Cu (+) K, gdzie kreski pionowe oznaczają granice faz.
b) Ogniwo Daniella, podobnie jak ogniwo Volty, jest ogniwem cynkowo – miedziowym,
posiada jednak tę zaletę, iŜ nie występuje w nim kłopotliwa polaryzacja katody.
Problem ten rozwiązano stosując zasadnicze zmiany w konstrukcji ogniwa. Ogniwo
Daniella składa się z dwoch oddzielnych połogniw – cynkowego i miedziowego –
połączonych przewodnikiem oraz kluczem elektrolitycznym (aby w obwodzie płynął
prąd, musi on być obwodem zamkniętym). Połogniwem cynkowym jest płytka
cynkowa zanurzona w roztworze zawierającym kationy cynkowe, podobnie
połogniwem miedziowym jest płytka miedziana zanurzona do roztworu z kationami miedziowymi. Kluczem
elektrolitycznym jest rurka szklana wypełniona ośrodkiem Ŝelującym (np. koloidowym roztworem
krzemionkowym), w ktorym rozpuszczony jest chlorek lub azotan (V) potasu.
schematycznie: A (-) Zn | ZnSO4 || CuSO4 | Cu (+) K, gdzie || oznacza klucz
zapis uproszczony: Zn | Zn2+ || Cu2+ | Cu
Created by Neevia Document Converter trial version http://www.neevia.com
30
A(-): Zn0 ---> Zn2+ + 2e-
K(+): Cu2+ + 2e- ---> Cu0
Zn + Cu2+ ---> Zn2+ + Cu
SEM V
SEM E E
Zn Cu
Zn Cu Cu Cu Zn Zn
0,34 ( 0,76) 1,1 /
/
0
/
0
/
2 2
= − − =
= + − +
c) Ogniwo Lenclanchego jest ogniwem nie podlegającym regeneracji. Generalnie zbudowane jest z dwoch
elektrod: cynkowej oraz węglowej, otoczonej workiem z dwutlenkiem manganu, zanurzonych w roztworze
chlorku amonu (salmiaku). W ogniwie „suchym” elektrodą ujemną jest cynkowa obudowa ogniwa. Te
ostatnie to popularne baterie R6 o SEM=1,5V.
ogniwo mokre: A (-) Zn | NH4Cl | MnO2, C (+) K
ogniwo suche: A (-) Zn | ZnCl2(aq), NH4Cl | MnO2, C (+) K
W ogniwie Lenclanchego zachodzi cały szereg reakcji chemicznych, warunkujących jego działanie. Na
anodzie zachodzi utlenianie metalicznego cynku (reakcja pierwotna):
A(-): Zn0 ---> Zn2+ + 2 e-.
Jednocześnie dysocjuje salmiak:
NH4Cl <==> NH4+ + Cl-,
a powstałe aniony chlorkowe łączą się z kationami cynkowymi (wtorna reakcja anodowa):
A(-): Zn2+ + 2 Cl- ---> ZnCl2.
Na katodzie zachodzi redukcja powstałych z dysocjacji kationow amonowych:
K(+): 2 NH4+ + 2 e- ---> H2 + 2 NH3.
Wydzielony amoniak kompleksuje z chlorkiem cynku, dając chlorek dwuaminocynku:
ZnCl2 + 2 NH3 ---> [Zn(NH3)2]Cl2,
zaś wodor łączy się z dwutlenkiem manganu, depolaryzując katodę:
2 MnO2 + H2 ---> 2 MnO(OH);
w wyniku reakcji tworzy się zasadowy tlenek manganu.
Sumaryczna reakcja ogniwa Lenclanchego ma postać:
2 Zn + 4 NH4Cl + 4 MnO2 ---> ZnCl2 + [Zn(NH3)2]Cl2 + 4 MnO(OH).
d) Ogniwo Clarka to ogniwo cynkowo – rtęciowe o następującym schemacie:
A (-) Zn(amalgamat) | ZnSO4 || HgSO4, Hg (+) K
A(-): Zn ---> Zn2+ + 2 e-
K(+): Hg2+ + 2 e- ---> Hg
Zn + Hg2+ ---> Zn2+ + Hg
SEM=1,433V
e) Ogniwo Westona jest ogniwem kadmowo – rtęciowym:
A (-) Cd(amalgamat 12%) | nas. 3 CdSO4
. 8 H2O || HgSO4, Hg (+) K
A(-): Cd ---> Cd2+ + 2 e-
K(+): Hg2+ + 2 e- ---> Hg
Cd + Hg2+ ---> Cd2+ + Hg
Ogniwo Westona określa się mianem ogniwa normalnego, gdyŜ utrzymuje ono przez długi czas stałą
wartość SEM=1,018V (w temperaturze 20oC). Ma ono zastosowanie w laboratoriach jako wzorzec napięcia.
f) Ogniwo Bunsena jest ogniwem cynkowo – azotowym:
A (-) Zn | ZnSO4(aq) (8-10%) || HNO3 (65-70%), C (+) K
SEM=1,88V
g) Ogniwo Greneta / Poggendorffa zbudowane jest z elektrody cynkowej oraz grafitowej, zanurzonych w
roztworze wodnym dwuchromianu (VI) potasu oraz kwasu siarkowego (VI):
A (-) Zn | K2Cr2O7, H2SO4 (aq) | C (+) K
SEM=2V
h) Ogniwo paliwowe – najprostszym przykładem jest ogniwo wodorowo – tlenowe,
w ktorym elektrolitem jest 50% roztwor wodorotlenku potasu, zaś elektrodami –
porowate płyty niklowe.
Created by Neevia Document Converter trial version http://www.neevia.com
31
A (-) Ni, H2 | KOH(aq) | O2, Ni (+) K
A(-): H2 + 2OH- ---> 2 H2O + 2 e-
K(+): . O2 + H2O + 2 e- ---> 2 OHH2
+ . O2 ---> H2O
W ogniwie następuje synteza wody z pierwiastkow. Ogniwa paliwowe są powszechnie stosowane – np. w
pojazdach kosmicznych.
i) Akumulator ołowiowy jest ogniwem regenerowalnym. Akumulator
samochodowy złoŜony jest z sześciu takich połogniw połączonych szeregowo.
A (-) Pb(ołow gąbczasty) | H2SO4(aq) | PbO2, Pb(płyta ołowiana pokryta tlenkiem) (+) K
H2SO4: C≈37%, d=1,27-1,29g/cm3
JeŜeli gęstość kwasu spadnie poniŜej wartości krytycznej: 1,19g/cm3, to ogniwa
nie będzie moŜna powtornie naładować.
A(-): Pb ---> Pb2+ + 2 e- (reakcja pierwotna)
Pb2+ + SO4
2- ---> PbSO4↓ (reakcja wtorna)
Pb + SO4
2- ---> PbSO4 + 2 e- (reakcja sumaryczna anody)
K(+): PbO2 + 4 H+ + 2 e- ---> Pb2+ + 2 H2O (reakcja pierwotna)
Pb2+ + SO4
2- ---> PbSO4↓ (reakcja wtorna)
PbO2 + SO4
2- + 4 H+ + 2 e- ---> PbSO4 + 2 H2O (reakcja sumaryczna katody)
Sumaryczne rownanie ilustrujące zasadę działania akumulatora ma postać:
Pb + PbO2 + 2 H2SO4 <====> 2 PbSO4 + 2 H2O
(rownanie przedstawia stan pracy; stan ładowania ilustruje rownanie czytane od prawej strony).
SEM=2,3V (1,8V oznacza wartość krytyczną)
SEMbaterii= 6 . SEM=6 . 2,2V=13,2V (napięcie akumulatora)
Trwałość akumulatorow ołowiowych (kwasowych) jest większa niŜ alkalicznych, chociaŜ wadą jest
stosunkowo duŜa masa. Z drugiej strony – w porownaniu z innymi częściami pojazdu akumulator nie
stanowi powaŜnego obciąŜenia.
j) Akumulator Edisona jest przykładem akumulatora zasadowego (alkalicznego). Anodę stanowi Ŝelazo, zaś
katodę mieszanina niklu i jego stałych związkow. Elektrolitem jest 10% roztwor wodny wodorotlenku
potasu.
A (-) Fe, Fe(OH)2 | KOH(aq) | Ni(OH)2, Ni(OH)3, Ni (+) K
A(-): Fe + 2 OH- ---> Fe(OH)2 + 2 e-
K(+): 2 Ni(OH)3 + 2 e- ---> 2 Ni(OH)2 + 2 OHFe
+ 2 Ni(OH)3 <===> 2 Ni(OH)2 + Fe(OH)2
Ze względu na relatywnie małą masę oraz większą trwałość w stanie rozładowania akumulator Edisona
znalazł szerokie zastosowanie w telekomunikacji, do zasilania przenośnej aparatury elektrycznej oraz
pojazdow elektrycznych.
k) Akumulator srebrowo – cynkowy jest rownieŜ przykładem ogniwa alkalicznego. Anodę stanowią związku
cynku, katodę – związku srebra, zaś elektrolitem jest wodorotlenek potasu.
A (-) Zn, ZnO | KOH(aq) | AgO, Ag2O, Ag (+) K
A(-): Zn + 2 OH- ---> ZnO + H2O + 2 e-
K(+): AgO + H2O + 2 e- ---> Ag + 2 OHZn
+ AgO <===> Ag + ZnO
Akumulator ten jest wprawdzie drogi, lecz stosowany jest wszędzie tam, gdzie wymagana jest duŜa
niezawodność aparatury, np. w samolotach i rakietach, do zasilania precyzyjnej aparatury pomiarowe, w
telewizji do silnego oświetlenia studia, a takŜe w lampach gorniczych.
l) Ogniwo rtęciowo – cynkowe znane jest rownieŜ pod nazwą ogniwa
pinezkowego. Jest ono powszechnie stosowane w zegarkach
elektronicznych oraz miniaturowych kalkulatorach.
A (-) Zn, ZnO, Zn(OH)2 | KOH(aq) | HgO, Hg (+) K
A(-): 2 Zn + 4 OH- ---> ZnO + Zn(OH)2 + H2O + 4 e-
K(+): 2 HgO + 2 H2O + 4 e- ---> 2 Hg + 4 OH-
2 Zn + 2 HgO + H2O ---> ZnO + Zn(OH)2 + 2 Hg
ENERGETYKA REAKCJI REDOX
a) Entalpia swobodna reakcji redox wyraŜa się wzorem ^G=-zF^E, gdzie z to liczba ładunkowa reakcji, F –
stała Faradaya, a ^E – siła elektromotoryczna ogniwa. JeŜeli SEM jest dodatnie, wowczas entalpia
swobodna reakcji jest ujemna, co gwarantuje samorzutność zachodzenia procesu. Aby uniknąć ujemnych
wartości SEM anodę ustawia się po lewej stronie, a katodę po prawej, gdyŜ zgodnie z konwencją
sztokholmską: ^E=Ep-El, gdzie Ep, El to potencjały odpowiednio prawego i lewego połogniwa.
Created by Neevia Document Converter trial version http://www.neevia.com
32
b) Przewidywanie kierunku przebiegu reakcji redox w oparciu o potencjały
standardowe połogniw – tzw. metoda zegara. Niech dane będą dwie reakcje
elektrodowe:
R1 <==> U1
n+ + n e- (E1),
R2 <==> U2
m+ + m e- (E2)
oraz niech E2>E1. Wowczas przemiany zachodzące w tak zestawionym
ogniwie ilustruje wykres – „zegar” (rysunek obok). W połogniwie o
niŜszym potencjale reakcja redox przebiega w stronę utleniania (połogniwo to staje się anodą), zaś w
połogniwie o wyŜszym potencjale reakcje redox przebiega w stronę redukcji (połogniwo to staje się katodą).
Przebieg reakcji sumarycznej moŜna zatem zilustrować następująco:
R1 ---> U1
n+ + n e- / . m
U2
m+ + m e- ---> R2 / . n
m R1 + n U2
m+ + nm e- ---> n R2 + m U1
n+ + nm em
R1 + n U2
m+ ---> n R2 + m U1
n+
Warto zwrocić uwagę, Ŝe liczba ładunkowa sumarycznej reakcji wynosi z=n.m.
c) Stała rownowagi reakcji redox. Kontynuując rozwaŜanie reakcji z poprzedniego podpunktu, moŜna wyrazić
potencjały połogniw następująco:
[ ]
[ ]
, ln
[ ]
[ ]
ln
2
0 2
2 2
1
0 1
1 1
R
U
mF
RT
E E
R
U
nF
RT
E E
n+ m+
= + = + .
Siła elektromotoryczna powstałego ogniwa to roŜnica potencjałow połogniw, czyli:
K
zF
RT
E
U R
U R
zF
RT
E
R
U
R
U
zF
RT
E
R
U
m
R
U
n
nmF
RT
E
R
U
nmF
mRT
R
U
nmF
nRT
E
R
U
nF
RT
R
U
mF
RT
E E
R
U
nF
RT
E
R
U
mF
RT
E E E E
n m n
m n m
m
n m
n
m n m n
m n m n
m n
ln
[ ] [ ]
[ ] [ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
[ ]
[ ]
ln
0
1
1 2
0 2 1
1
1
2
0 2
1
1
2
0 2
1
1
2
0 2
1
1
2
0 2
1
0
2
1
0 1
1
2
0 2
2 1 2
− _ =
= _ +
=
− + _ =
= _ + −
= − + − = _ + − =
=
+ −
_ = − = +
−
+
+
+ + + +
+ + + +
+ +
Gdy w środowisku reakcji ustali się stan rownowagi, wowczas ogniwo nie pracuje, a jego siła
elektromotoryczna wynosi zero.
RT
zF E
RT
zF E
K K
e
K
K
K e
RT
zF E
K E K
zF
RT
K
zF
RT
E E
2,3
0
0 0
0
0
2,3log 10
log
log
ln
0 ln 0 ln ln
_
_
= = ⇒ =
⇒ =
_
_ = ⇒ _ − = ⇒ = _ ⇒ =
Znając zatem roŜnicę potencjałow reakcji połowkowych oraz warunki środowiska, moŜemy określić stałą
rownowagi reakcji redox. Dodatkowo wprowadzając: zF^E0=-^G, otrzymujemy:
RT
G
RT
G
K e 102,3
−_ −_
= = .
Z ostatniego rownania wynika, iŜ:
- dla reakcji samorzutnej (^G<0) K>1,
- w stanie rownowagi (^G=0) K=1,
- dla reakcji wymuszonej (^G>0) K<1.
KOROZJA METALI I METODY JEJ ZWALCZANIA
Korozja jest procesem niszczenia metali na skutek wpływu czynnikow środowiska. Ze względu na przebieg
wyroŜniamy dwa typy korozji.
a) Korozja chemiczna zachodzi w środowisku suchym pod wpływem tlenu lub działania agresywnych gazow
przemysłowych. Prowadzi do powstania ochronnej warstewki tlenku (czasem takŜe wodorotlenku lub soli).
Ten typ korozji ma znaczenie pozytywne, gdyŜ pasywna warstwa chroni przed dalszą korozją. Np.
śniedzenie miedzi, czernienie srebra, pasywacja Ŝelaza, chromu, glinu.
Created by Neevia Document Converter trial version http://www.neevia.com
33
b) Korozja elektrochemiczna zachodzi w środowisku wilgotnym
pod wpływem tlenu atmosferycznego. Przyspiesza ją kwaśne
środowisko oraz obecność gazow przemysłowych, podczas gdy
zasadowość środowiska – hamuje. Istota korozji
elektrochemicznej polega na tworzeniu się tzw. mikroogniw
lokalnych, w ktorych Ŝelazo stanowi anodę. Ulegając
systematycznemu utlenianiu, przechodzi w postaci jonow z
metalu do środowiska i w ten sposob niszczeje.
c) W mikroogniwie zachodzą następujące reakcje:
A(-): Fe ---> Fe2+ + 2 e- (reakcja pierwotna)
Fe2+ + 2 OH- ---> Fe(OH)2↓ (biały) (reakcja wtorna)
4 Fe(OH)2 + 2 H2O + O2 ---> 4 Fe(OH)3↓ (brązowy) (reakcja wtorna)
K(+): . O2 + H2O + 2 e- ---> 2 OHEfektem
korozji jest powstanie rdzy: Fe2O3
. n H2O (n=1, 2, 3...), np. dla n=3 Fe2O3
. 3 H2O = 2 Fe(OH)3.
Metody walki z korozją:
a) wytwarzanie powłok ochronnych z farb, lakierow i tworzyw sztucznych,
b) wytwarzanie powłok ochronnych z innych metali
- Powłoki czynne wytwarzane są z metali o potencjale standardowym niŜszym
niŜ Ŝelaza, np. z cynku czy magnezu. Po uszkodzeniu nadal chronią one
przed korozją do czasu, gdy ulegną całkowitemu utlenienie. W powstałym
mikroogniwie Ŝelazo stanowi katodę, a więc nie niszczeje.
A(-): Zn ---> Zn2+ + 2 e- (reakcja pierwotna)
Zn2+ + 2 OH- ---> Zn(OH)2 (reakcja wtorna)
K(+): H2O + . O2 + 2 e- ---> 2 OH-
- Powłoki bierne wytwarzane są z metali o potencjale standardowym wyŜszym
niŜ Ŝelaza, np. z niklu, miedzi, srebra, złota. Po uszkodzeniu znacznie
przyspieszają one korozję, gdyŜ w powstałym mikroogniwie Ŝelazo stanowi
anodę i bardzo szybko ulega degradacji.
A(-): Fe ---> Fe2+ + 2 e- (reakcja pierwotna)
Fe2+ + 2 OH- ---> Fe(OH)2↓ (reakcja wtorna)
4 Fe(OH)2 + 2 H2O + O2 ---> 4 Fe(OH)3↓ (reakcja wtorna)
K(+): . O2 + H2O + 2 e- ---> 2 OHO
wyborze powłoki nie zawsze decyduje jej skuteczność, ale rownieŜ względy estetyczne – powłoki bierne
są bowiem o wiele ładniejsze od powłok czynnych.
c) osłabianie agresywności środowiska i / lub jego alkalizacja,
d) stosowanie ochrony protektorowej: protektor to metal aktywniejszy od Ŝelaza, przytwierdzany do
powierzchni wyrobu stalowego, np. kadłuba okrętu bądź rurociągu, działający jako anoda w powstałym
ogniwie; rolę protektora spełniają często płyty magnezowe,
e) stosowanie ochrony katodowej, czyli nadawanie niŜszego potencjału poprzez łączenie z ujemnym biegunem
ogniwa; metoda ta jest często praktykowana w odniesieniu do kotłow przemysłowych.
ELEKTROLIZA
a) istota procesu elektrolizy
RozwaŜmy procesy zachodzące w ogniwie
cynkowo – miedziowym. Zgodnie z
zasadami energetycznymi ^E>0 => ^G<0,
czyli procesy zachodzą samorzutnie w
przedstawionym kierunku. Zobaczmy co się
stanie, gdy do biegunow ogniwa przyłoŜymy
odwrotnie spolaryzowane napięcie o
wartości większej od siły elektromotorycznej
wytwarzanej prze to ogniwo. Obserwujemy
dokładne odwrocenie przebiegu samorzutnie
zachodzących reakcji redox. Zespoł
wymuszonych reakcji chemicznych
zachodzących na elektrodach i w elektrolitach towarzyszących przepływowi przezeń prądu stałego
nazywamy elektrolizą. Termin ten oznacza dosłownie „rozkład pod wpływem prądu”. Elektrolizę moŜna
przeprowadzać zarowno w stopionych elektrolitach (związkach o budowie jonowej – np. sole, tlenki metali i
wodorotlenki – ktore pod wpływem wysokiej temperatury ulegają termodysocjacji) jak i w roztworach
Created by Neevia Document Converter trial version http://www.neevia.com
34
wodnych elektrolitow (np. w roztworach kwasow, zasad i soli, gdzie pod wpływem wody zachodzi
dysocjacja elektrolityczna). Aparat, w ktorym przeprowadzamy elektrolizę, nazywamy elektrolizerem. W
przypadku zamiany ogniwa w elektrolizer anoda staje się katodą i odwrotnie, zmieniają się rownieŜ ich
znaki: anoda jest dodatnia, a katoda ujemna. Niezmiennym natomiast pozostaje fakt, iŜ na anodzie zachodzi
proces utleniania, a na katodzie – redukcji. Aby zaszła elektroliza, naleŜy do biegunow ogniwa przyłoŜyć
napięcie wyŜsze od jego SEM. Najmniejszą wartość zewnętrznego napięcia, przy ktorym obserwujemy
ciągłą reakcję elektrodową, nazywamy napięciem rozkładowym (Ur). Jest ono charakterystyczne dla danego
układu: przewodniki metaliczne – przewodnik jonowy. W praktyce przyjmuje się, iŜ Ur to napięcie
uzyskane przez ekstrapolację krzywej I=f(U) do I=0 (wykres). RoŜnicę pomiędzy
SEM ogniwa a Ur elektrolizera zeń uzyskanego nazywamy nadnapięciem
elektrolizy: η=Ur-E. RozroŜnia się nadnapięcie procesu katodowego (ηK) i
anodowego (ηA). Nadnapięcie zaleŜy od naboju powierzchni katody, gęstości
prądu zewnętrznego oraz od temperatury, co związane jest z energią i ruchliwością
jonow.
ZaleŜności pomiędzy przedstawionymi wielkościami ilustruje schemat:
pw
K = EK - ηK pw
A = EA - ηA
η = ηA + ηK
Ur = pw
A – pw
K = EA – EK + ηA + ηK
Ur = SEM + η
η > 0 => Ur > SEM
gdzie: pw – potencjały wydzielania, E – potencjały połogniw, η – nadnapięcie.
b) kolejność wydzielania produktow w elektrolizerze
Na anodzie zachodzi utlenianie reduktorow. Najlepsze reduktory mają najmniejszy potencjał standardowy,
dlatego na anodzie produkty wydzielają się w kolejności od najniŜszego potencjału począwszy.
Analogicznie na katodzie zachodzi redukcja utleniaczy. Najlepsze utleniacze mają największy potencjał
standardowy, dlatego na katodzie produkty wydzielają się w kolejności od najwyŜszego potencjału
począwszy. Bardzo często produktami elektrolizy są wodor i / lub tlen. Korzystając z rownania Nernsta
moŜna określić jaki odczyn środowiska sprzyja redukcji kationow wodorowych do wolnego wodoru, a jaki
utlenianiu anionow wodorotlenkowych do wolnego tlenu.
WODOR TLEN
K(-): 2 H2O + 2 e- ---> H2↑ + 2 OH- (alkalizacja) A(+): 2 H2O ---> O2↑ + 4 H+ + 4 e- (zakwaszenie)
pH=0 E = E0 = 0 V E = E0 - 0,059 . log 10-14 = 1,24 V pOH=14
pH=7 E = E0 + 0,059 . log 10-7 = -0,41 V E = E0 - 0,059 . log 10-7 = 0,82 V pOH=7
pH=14 E = E0 + 0,059 . log 10-14 = -0,83 V E = E0 = 0,41 V pOH=0
Wodor wydziela się na katodzie, zatem najkorzystniejszym jest potencjał najmniejszy, czyli redukcji
wodoru sprzyja środowisko zasadowe. Z kolei tlen wydziela się na anodzie, zatem najkorzystniejszy jest
potencjał największy, czyli utlenianiu tlenu sprzyja środowisko kwaśne. PowyŜsze wartości potencjałow
wydzielania wodoru i tlenu w zestawieniu z szeregiem napięciowym metali oraz z potencjałami normalnymi
reakcji redox informują nas o kolejności wydzielania się produktow.
Na anodzie utleniają się kolejno:
- aniony kwasow beztlenowych od najniŜszego potencjału (np. S2- ---> S0 + 2 e-),
- aniony wodorotlenkowe: 4 OH- ---> O2↑ + 2 H2O + 4 e-,
- woda: 2 H2O ---> O2↑ + 4 H+ + 4 e-.
Na katodzie redukują się kolejno:
- kationy metali od najwyŜszego potencjału (np. Cu2+ + 2 e- ---> Cu0),
- kationy wodorowe (gdy odczyn jest kwaśny): 2 H+ + 2 e- ---> H2↑,
- woda (gdy w roztworze znajdują się kationy metali aktywnych): 2 H2O + 2 e- ---> 2 OH- + H2↑.
c) produkty elektrolizy poszczegolnych grup związkow chemicznych – jakościowe ujęcie elektrolizy
- stopione elektrolity – substancje wyjściowe
• tlenek metalu ---> metal + tlen
Al2O3 ---^---> 2 Al3+ + 3 O2-
K(-): 2 Al3+ + 6 e- ---> 2 Al
A(+): 3 O2- ---> 3/2 O2↑ + 6 e-
Al2O3 ---> 2 Al + 3/2 O2
• wodorotlenek ---> metal + tlen + woda
NaOH ---^---> Na+ + OHK(-):
Na+ + e- ---> Na
A(+): OH- ---> . O2↑ + . H2O + e-
NaOH ---> Na + . O2↑ + . H2O
Created by Neevia Document Converter trial version http://www.neevia.com
35
• sol ---> metal + zredukowana reszta kwasowa
Fe2S3 ---^---> 2 Fe3+ + 3 S2-
K(-): 2 Fe3+ + 6 e- ---> 2 Fe
A(+): 3 S2- ---> 3 S + 6 e-
Fe2S3 ---> 2 Fe + 3 S
- roztwory wodne – według kolejności wynikającej z potencjałow; w przypadku identycznych jonow
pochodzących z roŜnych źrodeł wydzielają się one stamtąd, gdzie jest ich najwięcej
• zasada ---> wodor z wody + tlen z zasady
NaOH ---> Na+ + OHH2O
<==> H+ + OHK(-):
2 H+ + 2 e- ---> H2↑
A(+): 2 OH- ---> H2O + . O2↑ + 2 e-
H2O ---> H2↑ + . O2↑
Produkty wskazują na elektrolizę wody; w roztworze pozostaje zasada – środowisko ulega alkalizacji.
• kwas beztlenowy ---> wodor z kwasu + niemetal
HBr ---> H+ + Br-
K(-): 2 H+ + 2 e- ---> H2↑
A(+): 2 Br- ---> Br2↑ + 2 e-
2 HBr ---> H2↑ + Br2↑
• kwas tlenowy ---> wodor z kwasu + tlen z wody
H2SO4 ---> 2 H+ + SO4
2-
H2O <==> H+ + OHK(-):
2 H+ + 2 e- ---> H2↑
A(+): 2 OH- ---> . O2↑ + H2O + 2 e-
H2O ---> . O2↑ + H2↑
Produkty wskazują na elektrolizę wody; w roztworze pozostaje kwas – środowisko ulega zakwaszeniu.
• sol beztlenowa metalu lekkiego ---> wodor z wody + niemetal
KCl ---> K+ + Cl-
H2O <==> H+ + OHK(-):
2 H+ + 2 e- ---> H2↑
A(+): 2 Cl- ---> Cl2↑ + 2 e-
W roztworze pozostaje zasada – środowisko ulega alkalizacji.
• sol beztlenowa metalu cięŜkiego ---> metal + niemetal
CuS ---> Cu2+ + S2-
K(-): Cu2+ + 2 e- ---> Cu
A(+): S2- ---> S + 2 e-
CuS ---> Cu + S
• sol tlenowa metalu lekkiego ---> wodor z wody + tlen z wody
Li2SO4 ---> 2 Li+ + SO4
2-
H2O <==> H+ + OHK(-):
2 H+ + 2 e- ---> H2↑
A(+): 2 OH- ---> . O2↑ + H2O + 2 e-
H2O ---> . O2↑ + H2↑
Jest to właściwie elektroliza wody – jony soli spełniają jedynie rolę nośnikow ładunku elektrycznego.
• sol tlenowa metalu cięŜkiego ---> metal + tlen z wody
Cu(NO3)2 ---> Cu2+ + 2 NO3
-
H2O <==> H+ + OHK(-):
Cu2+ + 2 e- ---> Cu
A(+): 2 OH- ---> . O2↑ + H2O + 2 e-
W roztworze powstaje kwas azotowy – środowisko ulega zakwaszeniu.
d) prawa elektrolizy Faradaya – ilościowe ujęcie elektrolizy
RozwaŜmy następującą reakcję połowkową:
K(-): Un+ + n e- ---> R
1 cząstka n elektronow
mcz n . e
m q
Created by Neevia Document Converter trial version http://www.neevia.com
36
Z porownania otrzymujemy:
= ⋅
⋅
= ⋅ = ⋅ ⋅ =
⋅
⋅
⇒ =
⇒
⋅
⇒ =
⋅ ⋅
=
⋅
⇒
⋅
⇒ =
⋅
=
q I t
n F
M
k q k I t k
n F
M q
m
n F
q
M
m
n e N
q
m N
m
n e
q
m
m
q
n e
m
m
cz cz A A
cz
,
Pierwsze prawo elektrolizy stwierdza, iŜ masa wydzielonego produktu jest wprost proporcjonalna do
ładunku, jaki przepłynął przez elektrolizer, czyli iloczynowi wartości prądu stałego oraz czasu trwania
procesu. Wspołczynnik proporcjonalności k zwany jest rownowaŜnikiem elektrochemicznym; jest on
ilościowo rowny masie produktu wydzielonego w jednostkowym przedziale czasu w wyniku przepływu
prądu o jednostkowym natęŜeniu. Drugie prawo elektrolizy podaje wyraŜenie opisujące rownowaŜnik – jest
on wprost proporcjonalny do masy molowej produktu, a odwrotnie proporcjonalny do liczby ładunkowej
reakcji. Stała F zwana jest stałą Faradaya; liczbowo rowna jest ona iloczynowi liczby Avogadra i ładunku
elementarnego:
F = NA . e = 6,02 . 1023 mol-1 . 1,6 . 10-19 C ≈ 96 500 C / mol.
e) praktyczne zastosowanie elektrolizy:
- Otrzymywanie metali lekkich (sodu, wapnia, glinu) z ich stopionych elektrolitow. W przypadku glinu nie
moŜna stosować redukcji tlenku ze względu na zbyt kosztowny nakład energetyczny, dlatego stosuje się
termoelektrolizę. Surowcem wyjściowym jest boksyt: Al2O3 .
x H2O, do ktorego dodaje się topiku – kriolitu:
Na3AlF6.
Al2O3 ---^---> 2 Al3+ + 3 O2-
K(-): 2 Al3+ + 6 e- ---> 2 Al
A(+): 3 O2- ---> 3/2 O2↑ + 6 e-
Al2O3 ---> 2 Al + 3/2 O2
Rolę katody spełnia ciekły glin pozostały po poprzedniej
elektrolizie (nigdy nie spuszcza się go do końca), anodą zaś
szereg elektrod węglowych (grafitowych). Pod wpływem
wydzielającego się tlenu te ostatnie utleniają się do tlenku i dwutlenku węgla. Jest to zjawisko niepoŜądane,
lepiej jednak tracić grafit niŜ pozwalać na ponowne utlenianie glinu. Ostatecznymi produktami są zatem:
glin oraz tlenki węgla.
- pokrywanie metali mniej szlachetnych powłokami galwanicznymi z metali szlachetniejszych (np. cynk,
nikiel, miedź, srebro) – silniki, ramy rowerowe itp.
- rozdzielanie metali z mieszanin przy wykorzystaniu roŜnicy potencjałow
- produkcja przemysłowa wodorotlenku sodu
• metoda przeponowa – elektroliza roztworu chlorku sodu
NaCl ---> Na+ + Cl-
H2O ---> H+ + OHK(-):
2 H2O + 2 e- ---> H2↑ + 2 OHA(+):
2 Cl- ---> Cl2↑ + 2 e-
2 NaCl + 2 H2O ---> 2 NaOH + H2↑ + Cl2↑
Otrzymywany w ten sposob wodorotlenek jest pozostałością po elektrolizie, przez co jest silnie
zanieczyszczony.
• metoda rtęciowa
PoniewaŜ nadnapięcie wydzielania wodoru na katodzie rtęciowej jest bardzo wysokie (ηH = 1,2 - 1,3 V),
redukcji ulega sod. Tworzy on z rtęcią amalgamat, po czym jest wypłukiwany. Otrzymany w ten sposob
wodorotlenek jest zadowalająco czysty.
NaCl ---> Na+ + Cl-
K(-): 2 Na+ + 2 e- ---> Na
Hg + 2 Na ---> 2 Na|Hg
A(+): 2 Cl- ---> Cl2↑ + 2 e-
2 NaCl + Hg ---> 2 Na|Hg + Cl2↑
2 Na|Hg + 2 H2O ---> 2 NaOH + H2↑ + Hg
- elektrorafinacja miedzi – jeŜeli anoda wykonana jest z metalu, ktorego kationy znajdują się w roztworze, to
procesem anodowym jest utleniania metalu anody
K(-): Cu2+ + 2 e- ---> Cu
A(+): Cu ---> Cu2+ + 2 e-
Cu (zanieczyszczona z huty) ---elektrorafinacja---> Cu (czysta do uŜytku)
Created by Neevia Document Converter trial version http://www.neevia.com
37
CHEMIA ZWIĄZKÓW NIEORGANICZNYCH
SYSTEMATYKA ZWIĄZKOW NIEORGANICZNYCH
Związkami nieorganicznymi nazywamy generalnie związki chemiczne nie zawierające w swej budowie atomow
węgla. Wyjątkami od tej reguły są:
- węglany, czyli sole kwasu węglowego,
- węgliki, czyli połączenia o charakterze soli, w ktorym węgiel spełnia rolę anionu oraz
- inne związki węgla z roŜnymi pierwiastkami, np. dwusiarczek węgla CS2.
Związki nieorganiczne dzielimy na kilka grup; są to:
- tlenki (XnOm),
- wodorki (XHn lub HnX),
- kwasy (HnX lub HnR),
- wodorotlenki (Me(OH)n),
- sole (MenRm) oraz
- inne.
Do tych ostatnich zaliczmy:
- węgliki, azotki itp. (ogolnie związki o charakterze soli, w ktorych niemetaliczny pierwiastek niefluorowcowy
pełni rolę anionu),
- chlorki azotowcow, np. PCl5,
- związki międzyfluorowe: ClF3, BrF5, IF7,
- związki metali z niemetalami nie będące ani tlenkami ani kwasami,
- związki międzymetaliczne, np. CuZn oraz związki pomiędzy niemetalami, np. CS2.
TLENKI
Tlenkiem nazywamy związek chemiczny, w ktorym tlen połączony jest z innym pierwiastkiem. Wzor tlenku
zapisujemy jako XnOm, gdzie n i m to odpowiednie indeksy stechiometryczne. Kolejność w zapisie pierwiastkow
wynika z roŜnicy w elektroujemności – tlen posiada zawsze większą elektroujemność (3,5 w skali Paulinga) niŜ
pierwiastek, z ktorym się łączy, tworząc tlenek. Wyjątkiem jest tlenek fluoru, w ktorym fluor ma
elektroujemność wyŜszą niŜ tlen (4,0 w skali Paulinga), w związku z czym moŜna go zapisywać dwojako: F20
lub OF2. Ze względu na naturę pierwiastka wchodzącego w skład tlenku, związki te dzielimy na tlenki
metaliczne oraz niemetaliczne. Ze względu na charakter chemiczny tlenki podzielić moŜna na kwasowe,
zasadowe, amfoteryczne oraz obojętne. PrzynaleŜność tlenku do specyficznej grupy obydwu tych kategorii jest
ze sobą powiązana, o czy moŜna się przekonać analizując poniŜszą tabelę.
numer grupy: I A II A III A IV A V A VI A VII A
wartościowość
względem tlenu I II III IV, II V, III VI, IV VII, V, III, I
wzor ogolny
tlenku X2O XO X2O3 XO2 X2O5 XO3 X2O7
przykład tlenku
z 3. okresu Na2O MgO Al2O3 SiO2 P2O5 SO3 Cl2O7
nazwa tlenku tlenek sodu tlenek magnezu tlenek glinu
dwutlenek
krzemu / tlenek
krzemu (IV)
pięciotlenek
fosforu / tlenek
fosforu (V)
trojtlenek siarki
/ tlenek siarki
(VI)
siedmiotlenek
chloru / tlenek
chloru (VII)
charakter
wiązania (dX)
2,6
jonowy
2,3
jonowy
2,0
jonowy
1,7
kowalencyjny
polarny
1,4
kowalencyjny
polarny
1,0
kowalencyjny
polarny
0,5
kowalencyjny
polarny
charakter
chemiczny
zasadowy
słabo
zasadowy
amfoteryczny
słabo
kwasowy
kwasowy
silnie
kwasowy
bardzo silnie
kwasowy
<<---------------------------------------------------------------- --------------------------------------------------------------->>
wzrost jonowości i zasadowości wzrost kowalencyjności i zasadowości
Tlenki metali to ciała stałe o budowie jonowej, z wyjątkiem pierwiastkow I i II grupy nierozpuszczalne w
wodzie; zarowno rozpuszczalność, jak i charakter zasadowy rosną w obrębie grupie wraz ze wzrostem okresu.
W przypadku metali bloku d występujących na roŜnych wartościowościach tlenki niŜszych wartościowości są
nierozpuszczalne w wodzie (amfoteryczne), zaś tlenki na wyŜszych stopniach są rozpuszczalne w wodzie i dają
kwasy (np. H2CrO4, H2Cr2O7, KMnO4). Pierwiastki tego bloku bardzo często tworzą barwne związki (w tym
tlenki), co spowodowane jest pochłanianiem kwantow światła przez elektronu znajdujące się na częściowo
zapełnionej podpowłoce d (przy podpowłoce całkowicie zapełnionej bądź przy braku elektronow d jony proste
Created by Neevia Document Converter trial version http://www.neevia.com
38
są bezbarwne).
Tlenki niemetali to zazwyczaj gazy o budowie kowalencyjnej polarnej; w obrębie ich cząsteczek suma
momentow dipolowych wynosi przewaŜnie zero.
Tlenki kwasowe reagują z wodą dając kwasy oraz z zasadami, zobojętniając je, np.:
SO2 + H2O ---> H2SO3,
SO2 + 2NaOH ---> Na2SO3 + H2O.
Podobnie tlenki zasadowe reagują z wodą dając zasady oraz z kwasami, takŜe je zobojętniając, np.:
Na2O + H2O ---> 2NaOH,
Na2O + H2SO3 ---> Na2SO3 + H2O.
Tlenki obojętne to takie, ktore nie reagują ani z kwasami ani z zasadami, np.: CO, NO, N2O, SiO.
Tlenki amfoteryczne są tlenkami pierwiastkow o stosunkowo wysokiej elektroujemności (1,5 – 2), np.: Al2O3,
PbO, ZnO, Cr2O3, FeO, Fe2O3, CuO, Cu2O, Ag2O, Au2O3. Tlenki amfoteryczne nie rozpuszczają się w wodzie,
reagują z kwasami i bardzo mocnymi zasadami, dają przez to dwa rodzaje soli: kiedy pierwiastek z tlenku
spełnia w soli rolę kationu (klasyczna reakcja z kwasem) oraz kiedy ow pierwiastek wchodzi w skład reszty
kwasowej (reakcja z mocną zasadą). W reakcji z zasadami jon nie zmienia stopnia utlenienia) nie jest to reakcja
redox). Dla glinu i cynku reakcje wyglądają następująco:
Al2O3 + 6HCl ---> 2AlCl3 + 3H2O,
Al2O3 + 2NaOH + 3H2O ---> 2NaAl(OH)4 ---T---> 2NaAlO2 + 2H2O,
czterohydroksoglinian sodu
ZnO + 2HCl ---> ZnCl2 + H2O,
ZnO + 2NaOH + H2O ---> Na2Zn(OH)4 ---T---> Na2ZnO2 + 2H2O.
czterohydroksocynkan sodu
Tlenki moŜna otrzymywać roŜnorodnymi metodami. Najczęściej stosowanymi z nich są:
- spalanie pierwiastkow, np.:
S + O2 ---> SO2,
4Na + O2 ---> 2Na2O,
N2 + O2 ---> 2NO (w łuku elektrycznym – metoda Mościckiego),
- spalanie związkow chemicznych, np.:
ZnS + 3/2 O2 ---> ZnO + SO2, (sfaleryt – blenda cynkowa)
2NH3 + 5/2 O2 ---> 2NO + 3H2O,
2NH3 + 7/2 O2 ---> 2NO2 + 3H2O,
- rozkład związkow chemicznych (głownie soli i wodorotlenkow), np.:
CaCO3 ---T---> CaO + CO2,
NH4NO3 ---T---> N2O + 2H2O,
Cu(OH)2 ---T---> CuO + H2O,
- utlenianie niŜszych tlenkow do wyŜszych, np.:
2NO + O2 ---> 2NO2,
2SO2 + O2 ---V2O5+Pt/Pd--->2SO3
- redukcja wyŜszych tlenkow do niŜszych, np.:
Fe2O3 + C ---> 2FeO + CO,
Fe2O3 + CO ---> 2FeO + CO2.
WODORKI
numer grupy: I A II A III A IV A V A VI A VII A
wartościowość
względem
wodoru
I II III IV III II I
wzor ogolny
wodorku
XH XH2 XH3 XH4 XH3 H2X HX
przykład z
3. okresu
NaH MgH2 AlH3 SiH4 PH3 H2S HCl
nazwa
wodorku
wodorek
sodu
wodorek
magnezu
wodorek
glinu
krzemowodor fosforowodor siarkowodor chlorowodor
charakter
wiązania (dX)
1,2
spolaryzowany
0,9
spolaryzowany
0,6
spolaryzowany
0,3
kowalencyjny
0,0
kowalencyjny
0,4
spolaryzowany
0,9
spolaryzowany
charakter
chemiczny
zasadowy zasadowy słabo
zasadowy
obojętny obojętny słabo
kwasowy
kwasowy
Created by Neevia Document Converter trial version http://www.neevia.com
39
Wodorkami nazywamy dwuskładnikowe połączenia pierwiastkow z wodorem. Dzielimy je, podobnie jak tlenki,
według dwoch kategorii: na kwasowe, zasadowe i obojętne oraz na wodorki metali i niemetali. Pomiędzy
przynaleŜnością do obu grup istnieje ścisła zaleŜność.
Wodorki metali (umownie grupy I – V) to ciała stałe o małych gęstościach. Mają budowę polarną i charakter
zasadowy. Powstają w reakcjach wymuszonych, są bardzo aktywne chemicznie i łatwo się rozkładają z
wydzielenia ciepła. Reagują z wodą, w wyniku czego powstają zasady oraz gazowy wodor, np.:
NaH + H2O ---> NaOH + H2.
Wodorki niemetali (grupy VI i VII) to bezbarwne trujące gazy o charakterystycznych zapachach. Cechują się
roŜną rozpuszczalnością w wodzie, co wiąŜe się z charakterem chemicznych. Nierozpuszczalne i obojętne są:
CH4, BH3, PH3, SiH4, GeH4, natomiast rozpuszczalne są: NH3, H2S, H2Se, H2Te, HF, HCl, HBr, HI; oprocz
zasadowego amoniaku pozostałe są kwasowe. Charakter kwasowy, podobnie jak moc, rośnie w grupie i w
okresie (wzrost promienia jonowego osłabia wiązanie z wodorem, co ułatwia jego odłączanie i zwiększa stopień
dysocjacji). Rozpuszczalne wodorki reagują z wodą, w wyniku czego powstają kwasy beztlenowe (ew. zasada
amonowa: NH3 + H2O --> NH4OH).
Metody otrzymywania wodorkow:
- synteza z pierwiastkow, np.:
2Na + H2 ---> 2NaH,
H2 + Cl2 ---p,T--> 2HCl, \ specjalne
N2 + 3H2 ---kat--> 2NH3, / warunki
- z soli w reakcjach jonowych:
• amoniak w reakcji soli amonowej z roztworem mocnej zasady, np.:
(NH4)2SO4 + 2NaOH ---> 2NH4OH + Na2SO4 ---> 2NH3 + 2H2O + Na2SO4,
• wodorki kwasowe w reakcjach odpowiednich soli beztlenowych i mocnego kwasu, np.:
2NaCl + H2SO4 ---> 2HCl + Na2SO4.
WODOROTLENKI
1) Wodorotlenkami nazywamy połączenia metalu z jednowartościowymi grupami hydroksylowymi
(wodorotlenowymi). Zapisać je moŜna wzorem ogolnym M(OH)n, gdzie M to atom metalu, a –OH to grupa
wodorotlenowa. Wiązanie O-H jest wiązaniem kowalencyjnym silnie spolaryzowanym, zaś w wiązanie M-O jest
wiązaniem jonowym.
2) Wodorotlenki to krystaliczne ciała stałe o roŜnej barwie i rozpuszczalności w wodzie; dobrze rozpuszczalne
są wodorotlenki litowcow i Ba, słabo rozpuszczają się wodorotlenki Be, Mg, Ca i Sr, zaś pozostałe są
praktycznie nierozpuszczalne. Roztwory rozpuszczalnych wodorotlenkow nazywamy zasadami. Zasady barwią
indykatory (wskaźniki) chemiczne – fenoloftaleinę na malinowo, lakmus na niebiesko, wskaźnik uniwersalny na
niebiesko bądź zielono. Wodorotlenki litowcow są silnie higroskopijne, tzn. pochłaniają parę wodną z powietrza,
dlatego przechowuje się je w formie pastylek (mała powierzchnia kontaktowa); mają ponadto właściwości Ŝrące
i są śliskie w dotyku – dlatego rownieŜ śliskie jest mydło, gdyŜ podczas jego kontaktu z wodą wydziela się
zasada sodowa jako produkt hydrolizy. Zasady mocnych wodorotlenkow nazywamy ługami; niektore z nich
mają specyficzne nazwy zwyczajowe, np.: roztwor NaOH to soda kaustyczna, a KOH to potaŜ Ŝrący.
3) Wodorotlenki moŜna podzielić na zasadowe oraz amfoteryczne. Wodorotlenki zasadowe rozpuszczają się w
wodzie i reagują z kwasami, np.: NaOH, KOH, CaOH. Wiązanie M-O jest o wiele słabsze wobec O-H i przy
kontakcie z wodą ulega degradacji, co daje kation metalu i anion wodorotlenowy.
Wodorotlenki amfoteryczne nie rozpuszczają się w wodzie, reagują zarowno z kwasami, jak i z mocnymi
zasadami. Właściwości wodorotlenkow amfoterycznych tłumaczy się wysoką elektroujemnością wchodzącego
w ich skład pierwiastka, co prowadzi do upodobnienia siły wiązań M-O oraz O-H, w efekcie Ŝadne z nich nie
rozpada się i rozpuszczenie nie jest moŜliwe. Produkt reakcji zaleŜy dodatkowo od ilości uŜytych substratow.
Np.: Al(OH)3 + 3HCl --> AlCl3 + 3H2O,
Al(OH)3 + NaOH --> NaAlO2 + 2H2O,
Al(OH)3 + 3NaOH + --> Na3AlO3 + 3H2O.
4) Ze względu na zdolność do dysocjacji wodorotlenki dzielimy na mocne i słabe. Do pierwszej grupy zaliczmy
wodorotlenki litowcow, Ca, Ba, Sr, zaś do drugiej – Mg, Be i resztę.
5) Najpopularniejsze metody otrzymywania wodorotlenkow to:
- reakcja tlenkow litowcow i berylowcow z wodą, np.:
K2O + H2O --> 2KOH
CaO + H2O --> Ca(OH)2,
- reakcja bezpośrednio litowcow i berylowcow z wodą (Be i Mg wyłącznie w wyŜszych temperaturach), np.:
2Na + 2H2O --> 2NaOH + H2,
Ca + 2H2O --> Ca (OH)2 + H2,
Created by Neevia Document Converter trial version http://www.neevia.com
40
- reakcje strącania pomiędzy solami i zasadami, np.:
CuCl2 + 2KOH --> 2KCl + Cu(OH)2,
FeCl3 + 3NaOH --> 3NaCl + Fe(OH)3.
KWASY
1) Kwasy dzielimy na beztlenowe oraz tlenowe. Kwasami beztlenowymi nazywamy roztwory wodne wodorkow
kwasowych. Kwasy tlenowe to połączenia podobne w budowie chemicznej do zasad, składające się z
pierwiastka i jednej bądź więcej grupy –OH. Kwasy cechuje budowa kowalencyjna polarna. Istotną roŜnicą w
porownaniu do zasad jest moc wiązań M-O i O-H. W kwasach bowiem to pierwsze jest zdecydowanie
mocniejsze, w związku z czym przy kontakcie z wodą uwalnia się kation wodoru i reszta kwasowa.
Ze względu na ilość atomow wodoru w cząsteczce kwasy moŜna podzielić na jedno- (np. HNO3) oraz
wieloprotonowe, te ostatnie zaś na dwu- (H2SO4), trzy- (H3PO4), cztero- (H4SiO4), pięcio- (H5IO6),
sześcioprotonowe (H6TeO6) itd. Kwasy występują w stanie stałym (krzemowe, fosforowe, borowe) bądź
ciekłym (ogromna większość, np. HNO3). Kwasy cechują się roŜną rozpuszczalnością w wodzie – większość jest
rozpuszczalna (np. HCl), ale np. kwasy krzemowe są praktycznie nierozpuszczalne. Proces rozpuszczania
niektorych kwasow jest bardzo silnie egzotermiczny i moŜe doprowadzić do wrzenia rozpuszczalnika; tak dzieje
się dla kwasu siarkowego, dlatego teŜ podczas rozcieńczania naleŜy wkraplać go do wady, a nie wlewać doń
wody.
2) Kwasy beztlenowe otrzymuje się w reakcjach rozpuszczania kwasowych wodorkow niemetali w wodzie, np.:
HCl -------H2O------> H+ + Cl-,
HF -------H2O------> H+ + F-,
H2S -------H2O------> H+ + HS- -------H2O------> 2H+ + S+.
Z kolei kwasy tlenowe powstają w reakcjach bezwodnika kwasowego z wodą, np.:
SO2 + H2O ----> H2SO3,
SO3 + H2O ----> H2SO4,
N2O5 + H2O ----> 2HNO3.
Wyjątkowo kwasy krzemowe otrzymuje się w reakcjach strąceniowych mocnego kwasu z krzemianem:
2H+ + SiO3
2- ----> H2SiO3,
np.: H2SO4 + Na2SiO3 ----> H2SiO3 + Na2SO4.
3) Niektore kwasy (węglowy, siarkawy) rozkładają się pod wpływem temperatury na wodę oraz odpowiedni
bezwodnik:
H2CO3 ----T----> H2O + CO2,
H2SO3 ----T----> H2O + SO2.
4) Moc kwasow tlenowych zaleŜy od elektroujemności wchodzącego do nich niemetalu i wzrasta z jej wzrostem.
W związku z tym w okresach wzrasta na prawo, zaś w grupach malaje w doł. W przypadku kilku kwasow
tlenowych tego samego pierwiastka o roŜnych wartościowościach moc kwasu wzrasta ze wzrostem
wartościowości. Przy ogolnym porownywaniu mocy kwasow roŜnych pierwiastkow naleŜy brać pod uwagę ich
najwyŜsze wartościowości. Moc kwasow beztlenowych wzrasta w okresach ze wzrostem elektroujemności (na
prawo) i rośnie w grupach ze wzrostem promienia jonowego niemetalu (w doł).
Ogolnie kwasy moŜemy podzielić na mocne (dysocjujące w znacznym stopniu) oraz słabe (przewaga cząstek
niezdysocjowanych). NaleŜy zaznaczyć, iŜ tak określona moc kwasu nie pokrywa się zawsze ze skutkami
działania; przykładowo kwas fluorowodorowy jest kwasem słabym, jednak ze względu na duŜą agresywność jest
on jedynym kwasem uŜywanym do trawienia szkła.
5) Kwasy reagują z odczynnikami zasadowymi, tj. zasadami, tlenkami zasadowymi i amfoterycznymi, dając
sole. Reagują rownieŜ z metalami nieszlachetnymi (o ujemnych potencjałach standardowych) dając sole i wodor.
Nie reagują natomiast z metalami szlachetnymi (o dodatnich potencjałach standardowych). Np.:
2H3PO4 + 3Ca(OH)2 ---> Ca3(PO4)3 + 6H2O,
2H3PO4 + 3BaO ---> Ba3(PO4)3 + 3H2O,
3H2SO4 + Al2O3 ---> Al2(SO4)3 +3H2O,
Fe + H2SO4 ---> FeSO4 + H2,
Ag + H2SO4 --X--> nie zachodzi.
6) Istnieje grupa kwasow reagujących z tymi ostatnimi, jednak dzieje się to drogą reakcji redox. Ze względu na
rolę tych kwasow w takiej reakcji nazwano je kwasami utleniającymi. NaleŜą do nich: HNO3, stęŜony i gorący
H2SO4 oraz tlenowe kwasy chlorowe. Kwasy utleniające reagują z metalami szlachetnymi w ten sposob, Ŝe
utleniają pierwiastek na wyŜszy stopień utlenienia, po czym tworzą z nim sol. Jednocześnie część uŜytego do
reakcji kwasu redukuje się; ściślej mowiąc redukowany jest atom centralny występujący w kwasie, co powoduje
powstawanie obok wspomnianej soli rownieŜ cząsteczki tlenku zredukowanego atomu centralnego oraz wody.
W przypadku niektorych kwasow produkty zaleŜą dodatkowo od ich stęŜenia.
Created by Neevia Document Converter trial version http://www.neevia.com
41
Np.:
Cu + 4HNO3 -> Cu(NO3)2 + 2NO2 + 2H2O
(>15%) niebieski rudobrązowy
Cu + 8HNO3 -> 3Cu(NO3)2 + 2NO + H2O
(<15%) niebieski bezbarwny
Cu + HNO3 -X-> nie zachodzi
(<1%)
Cu + 2H2SO4 -> CuSO4 + SO2 + 2H2O
(stęŜony i gorący)
7) Do reakcji z metalami wybitnie szlachetnymi, złotem oraz platyną, zdolna jest wyłącznie mieszanina
stęŜonych kwasow – solnego oraz azotowego (V) – w stosunku objętościowym 3:1 (molowym 4:1). Reakcja ta
złoŜony przebieg: wpierw kwas solny reaguje z kwasem azotowym, wytwarzając chlorek nitrozylu, ktory
następnie atakuje metal szlachetny. Dla złota produktem reakcji jest kwas czterochlorozłotowy, co moŜna
zapisać następująco:
HNO3 + 3HCl -> NOCl + Cl2 + 2H2O,
Au + HCl + NOCl +Cl2 -> HAuCl4 + NO.
8) Natomiast kwasy utleniające nie reagują z niektorymi metalami nieszlachetnymi, np.: glinem, chromem czy
Ŝelazem na zimno. Przyczyną jest powstawanie na ich powierzchni ochronnej warstwy pasywnej tlenku, co
uniemoŜliwia dalszy przebieg reakcji. Proces powstawania takiej powłoki nazywamy pasywacją.
9) Kwasy utleniające mają takŜe moŜliwość utleniania niektorych niemetali, np. węgla, siarki czy fosforu.
Ilustrują to rownania:
3C + 4HNO3 -> 3CO2 + 4NO + 2H2O,
C + 2H2SO4 -> 2SO2 + CO2 + 2H2O,
3S + 4HNO3 -> 3SO2 + 4NO + 2H2O,
S + 2H2SO4 -> 3SO2 + 2H2O,
3P + 5HNO3 -> 3H3PO4 + 5NO.
10) W reakcjach kwasow utleniających z niektorymi metalami nieszlachetnymi będącymi silnymi reduktorami
nie obserwuje wydzielania wodoru. Pierwiastki te powodują znaczną redukcję atomu centralnego kwasu (nawet
o osiem stopni utlenienia!), co przyczynia się do powstawania innych produktow w środowisku reakcji, tj.
tlenkow bądź soli, np.:
4Zn + 10HNO3 -> 4Zn(NO3)2 + NH4NO3 + 4H2O,
4Mg + 10HNO3 -> 4Mg(NO3)2 + N2O + 5H2O.
11) StęŜone kwasu utleniające praktycznie nie dysocjują, poniewaŜ w niemal stuprocentowym roztworze prawie
nie występuje rozpuszczalnik (wowczas trudno taki układ nazwać roztworem). Ponadto w przypadku kwasu
siarkowego (VI) moŜna obserwować ciekawy paradoks stęŜeniowy; otoŜ moŜna w nim rozpuścić dodatkowo
duŜe ilości bezwodnika - SO3, przez co otrzymuje się tzw. dymiący kwas siarkowy bądź oleum o wzorze
umownym H2SO4
. SO3. Podczas dolewania do oleum wody zwiększamy objętość układu, natomiast nie
zmieniamy stęŜenia, poniewaŜ rozpuszczany bezwodnik wciąŜ daje nowe porcje kwasu i stęŜenie się nie
zmniejsza. Gdyby bezwzględnie przyjąć, iŜ dodawanie rozpuszczalnika zmniejsza stęŜenie roztworu, wowczas
okazałoby się, iŜ kwas siarkowy moŜe przyjmować wartości stęŜeń wyŜsze o stu.
12) Kwas azotowy (V) daje reakcję charakterystyczną z białkami. W wyniku nitrowania aminokwasow
aromatycznych powstają Ŝołte nitrozwiązki i całe środowisko przyjmuje barwę Ŝołtą. Reakcja ta nosi nazwę
reakcji ksantoproteinowej (ksantos – Ŝołty, proteina – białko). Po zalkalizowaniu środowiska przyjmuje ono
barwę pomarańczową. Skora ludzka po zetknięciu z kwasem azotowym rownieŜ przyjmuje zabarwienie Ŝołte.
SOLE
1) Solami nazywamy połączenia metali z resztami kwasowymi, skąd natychmiast wynika wzor ogolny MenRm.
W zaleŜności od typu kwasu, ktorego reszta wchodzi w skład soli, wyroŜniamy sole tlenowe oraz beztlenowe. W
przypadku tych pierwszych końcowkę kwasu –owy zamieniamy na –an i dopisujemy nazwę metalu w
dopełniaczu, natomiast w przypadku drugich zamieniamy końcowkę na –ek i postępujemy podobnie.
2) Sole są krystalicznymi ciałami stałymi o roŜnej barwie i rozpuszczalności w wodzie. Proces rozpuszczania
soli w wodzie jest endotermiczny, gdyŜ wiązania jonowe są tak silne, Ŝe energia pochłaniana do ich rozbicia nie
moŜe zostać pokryta przez energię uzyskaną podczas hydratacji powstałych jonow. W związku z powyŜszym
istnieje generalna prawidłowość, iŜ ze wzrostem temperatury rozpuszczalność soli rośnie, choć zaleŜności nie są
proporcjonalne, o czym świadczą zroŜnicowane krzywe rozpuszczalności. Choć większość soli jest bezbarwna,
wiele z nic ma określony kolor; zabarwienie moŜe pochodzić od kationu (barwa odkationowa) bądź o anionu
(barwa odanionowa). Przykłady: Cu2+ – niebieskie, Ni2+ – jasnozielone, Cr3+ - zielone, Fe2+ - brązowe, Fe3+ -
słomkowoŜołte; I- - Ŝołte, Br- - beŜowe, Se2- - roŜowe (szkło), MnO4
- - fioletowe, CrO4
2- - cytrynowo Ŝołte,
Cr2O7
2- - pomarańczowe. Metale nadające solom barwę leŜą w bloku energetycznym d, co zostało wytłumaczone
Created by Neevia Document Converter trial version http://www.neevia.com
42
przy okazji barwy tlenkow. Wskutek hydratacji jonow bądź tworzenia związkow koordynacyjnych barwa moŜe
ulec pogłębieniu.
3) - Sole są mocnymi elektrolitami, co oznacza, iŜ w roztworach prawie całkowicie występują jony.
- Uczestniczą w reakcjach wymiany w roztworach wodnych; wymiana moŜe być pojedyncza (typu metal + sol
lub sol beztlenowa + niemetal) bądź podwojna (typu sol + sol, sol + mocna zasada lub sol + mocny kwas).
- Sole pochodzące od słabych kwasow i / lub słabych zasad ulegają hydrolizie, ktory to proces szczegołowo
wytłumaczono w dziale Chemia ogolna.
– Wiele soli podatnych jest na rozkład pod wpływem czynnikow zewnętrznych (temperatury, światła), np.:
CaCO3 ---T---> CaO + CO2,
Na2SO3 ---T---> Na2O + SO2,
2KNO3 ---T---> 2KNO2 + O2,
2KClO3 ---T---> 2KCl + 3O2,
2KMnO4 ---T---> K2MnO4 + MnO2 + O2,
2AgCl ---hv---> 2Ag + Cl2,
2AgBr ---hv---> 2Ag + Br2.
4) Aby otrzymać sol naleŜy uŜyć odpowiednich odczynnikow, z ktorych jeden jest dawcą kationu, drugi zaś
dawcą anionu. W ogolnym przypadku daje to bardzo szeroki wachlarz moŜliwości, co ilustruje poniŜsza tabela:
Metody otrzymywania soli
niemetal tlenek niemetalu kwas sol
metal
2Na + Cl2 --> 2NaCl 2Na + 2CO2 -->
--> Na2CO3 + CO
Zn + 2HCl -->
--> ZnCl2 + H2
Fe + CuSO4 -->
--> FeSO4 + Cu
tlenek niemetalu
Na2O + Cl2 -->
--> NaCl + NaOCl
Na2O + CO2 -->
--> Na2CO3
CaO + 2HNO3 -->
--> Ca(NO3)2 + H2O
Na2O + 2NH4Cl -->
--> 2NaCl + 2NH3 + H2O
wodorotlenek
2NaOH + Cl2 -->
--> NaCl + NaOCl + H2O
Ca(OH)2 + CO2 -->
--> CaCO3 + H2O
NaOH + HCl -->
--> NaCl + H2O
Ca(OH)2 + 2NH4Cl -->
--> CaCl2 + 2NH4OH
sol
2KBr + Cl2 -->
--> 2KCl + Br2
3Na2CO3 + P2O5 -->
2Na3PO4 + 2HCl
CaCl2 + H2CO3 -->
--> 2HCl
AgNO3 + NaCl -->
--> AgCl + NaNO3
LITOWCE
Litowce są pierwszą grupą w układzie okresowym, co oznacza się I A (po staremu) lub 1 (według nowego
systemu zalecanego przez IUPAC). Posiadają konfigurację elektronową ns1
- posiadają zatem jeden elektron
walencyjny. Poza tym cechuje je mała energia jonizacji, co owocuje łatwym uleganiem wzbudzeniu i jonizacji.
Wzbudzone atomy pierwiastkow dają widmo w zakresie fal widzialnych, co jako tzw. proba płomienia słuŜy do
identyfikacji pierwiastkow; i tak: lit daje barwę karminową (czerwono - roŜową), sod – Ŝołtą (moŜna się o tym
przekonać rzucając szczyptę soli na palnik kuchenki gazowej), potas – liliową, rubid – fiołkowo-roŜową, a cez –
błękitną. Litowce są bardzo dobrymi przewodnikami elektryczności. Wszystkie mają nieduŜą gęstość, ktora
rośnie w grupie wraz ze wzrostem liczby atomowej (czyli lit jest najlŜejszym pierwiastkiem w przyrodzie). Są
metalami miękkimi, np. sod moŜna kroić noŜem. Niskie są rownieŜ temperatury topnienia i wrzenia, ktore
maleją w grupie w doł (wyjątkowo cez wrze w temperaturze wyŜszej niŜ rubid). Unikalną cechą litowcow jest
rozpuszczalność w ciekłym amoniaku.
Litowce z powodu higroskopijności i duŜej aktywności łatwo tworzą tlenki (X2O), a czasem rownieŜ nadtlenki
(XO - jeden atom tlenu więcej) i ponadtlenki (XO2 - dwa atomy tlenu więcej). Mowiąc o ponadtlenkach naleŜy
wymienić głownie nadtlenek potasu KO2 – pozostałe nie są tak powszechne. Tlenki litowcow wykazują
charakter silnie zasadowy. Nadtlenki i ponadtlenki są silnymi utleniaczami; te ostatnie mają charakter
rodnikowy, co oznacza, iŜ w cząsteczce ich obecny jest nie sparowany elektron, a sumaryczna liczba elektronow
jest nieparzysta. Spin nie sparowanego elektronu nadaje atomom właściwości magnetyczne – stąd wniosek, iŜ
ponadtlenki są paramagnetykami.
Sole litowcow najczęściej dobrze rozpuszczają się w wodzie. Nie mają one barwy odkationowej, łatwo ulegają
hydratacji, szczegolnie sodu i litu. Oznacza to, iŜ w ich cząsteczkach naleŜy spodziewać się obecności wody
krystalizacyjnej (np.: Na2SO4 . 10H2O - sol glauberska, Na2CO3 . 10H2O - soda krystaliczna czy Na2B4O7 .
10H2O - boraks). Sole litu rozpuszczają się stosunkowo najtrudniej – albo występują one w formie hydratow
albo są nierozpuszczalne. Częściej spotykane są sole sodu niŜ potasu. Ten ostatni wykorzystywany jest przez
rośliny, natomiast sod powoduje niszczenie roślin; wykorzystywano to niegdyś podczas wojny – posypując
ziemię solą wrog sprawiał, iŜ długo nie nadawała się ona do ponownej uprawy. W organizmach zwierzęcych sod
i potas mają niebanalne znaczenie w regulacji właściwości błon komorkowych; utrzymywanie jonow sodu
wewnątrz, a potasu – na zewnątrz komorki, warunkuje jej polaryzację, a przez to pośrednio jej moŜliwość do
przekazywania impulsow drugiej komorce.
Created by Neevia Document Converter trial version http://www.neevia.com
43
Ze względu na układ elektronow do grupy tej naleŜy wodor. Od pozostałych litowcow odroŜnia go wiele
właściwości – od stanu skupienia (gaz o gęstości 0,071 g/cm3) do elektroujemności (2,1, podczas gdy pozostałe:
0,7 – 1). Wodor jest pierwiastkiem reaktywnym: gwałtownie reaguje z fluorem, dając fluorowodor:
H2 + F2 ---> 2HF,
z tlenem tworzy mieszaninę wybuchową i po zapaleniu gwałtownie reaguje, dając wodę:
2H2 + O2 ---> 2H2O,
zaś pod działaniem promieni świetlnych reaguje z chlorem:
H2 + Cl2 ---> 2HCl.
Wodor moŜna otrzymać na wiele sposobow, m. in.:
a) z gazu wodnego:
C + H2O --1000oC--> CO + H2,
gaz wodny gaz syntezowy
b) w reakcji metanu z wodą:
CH4 + H2O ---Ni/1100oC---> CO + 3H2,
c) w reakcji metali nieszlachetnych z kwasami, np.:
Zn + 2HCl ---> ZnCl2 + H2,
d) poprzez elektrolizę wody:
2H2O -----> 2H2 + O2.
1) Do najwaŜniejszych związkow litowcow naleŜy wodorotlenek sodu NaOH. Jest to bardzo silna zasada, zwana
ługiem sodowym, atakująca nawet krzemionkę SiO2 (dlatego nie przechowuje się jej w szklanych butelkach).
Czysty NaOH jest białym, silnie higroskopijnym proszkiem, ktory w wyniku hydratacji „rozpływa się” na
powietrzu (aby temu zapobiec przechowuje się w pastylkach). MoŜna go otrzymać w reakcji kaustyfikacji:
Na2CO3 + Ca(OH)2 ---> 2NaOH + CaCO3.
Reaguje z dwutlenkiem węgla, dając wodorowęglan sodu, ktory ogrzany dalej daje sodę:
NaOH + CO2 ---> NaHCO3,
2NaHCO3 --T--> Na2CO3 + CO2 + H2O.
Znalazł zastosowanie w produkcji mydła, wypłukiwania celulozy drzewnej, fabrykacji barwnikow oraz środkow
czyszczących i piorących.
2) Wodorotlenek potasu KOH jest silniejsza zasadą od ługu sodowego. W podobny sposob reaguje z
dwutlenkiem węgla, dając w efekcie węglan potasu:
KOH + CO2 ---> KHCO3,
2KHCO3 --T--> K2CO3 + CO2 + H2O.
Jedyna roŜnica jest taka, iŜ wodorowęglan potasu łatwiej rozpuszcza się w wodzie od tworzącego zawiesinę
wodorowęglanu sodu. Jest on wykorzystywany do produkcji miękkich, szarych mydeł.
3) Sole sodowe i potasowe wykorzystywane są pospolicie w rolnictwie jako nawozy sztuczne. Najczęściej są to
azotany, znane pod nazwą saletr: NaNO3 – saletra chilijska, KNO3 – saletra indyjska.
4) Węglan sodu, zwany zwyczajowo sodą kalcynowaną, znajduje zastosowanie w produkcji szkła, mydła oraz
środkow zapobiegających zawilgoceniu (ze względu na właściwości higroskopijne). podobnie zresztą moŜna
uŜywać węglanu potasu. Na skalę przemysłową sodę otrzymuje się metodą Solvaya, ktora zostanie tu
szczegołowo opisana.
Surowcami jest sol kamienna NaCl oraz skała wapienna CaCO3. Proces technologiczny odbywa się dwoma
rownoległymi szlakami, ktore łączą się ze sobą we wspołzaleŜny cykl. Po oczyszczeniu sol kamienna
dostarczana jest do karbonizatora, ktory ma na celu przekształcenie chlorku sodu w jego wodorowęglan. Obecne
w karbonizatorze: woda i dwutlenek węgla reagują ze sobą, dając kwas węglowy, ktory łączy się z amoniakiem i
powstaje wodorowęglan amonu. Ostatni produkt wchodzi w reakcję wymiany podwojnej z chlorkiem sodu,
dając poŜądany wodorowęglan sodu oraz chlorek amonu (salmiak). Sumarycznie:
H2O + CO2 + NH3 +NaCl ----> NaHCO3 + NH4Cl.
Powstały wodorowęglan, znany po zwyczajową nazwą sody oczyszczonej, tworzy wpierw zawiesinę, po czym
wytrąca się w postaci delikatnego osadu. Połprodukt ten uŜywany jest jako środek spulchniający w przemyśle
piekarskim, a pospolicie stanowi głowny składnik proszku do pieczenia. Tak wytrącony osad podgrzewa się w
procesie kalcynacji, skutkiem czego wodorowęglan przekształca się w węglan:
2NaHCO3 ----T----> Na2CO3 + CO2 +H2O.
W ostatniej reakcji otrzymujemy końcowy produkt – sodę. Łatwo ulega ona uwodnieniu, tworząc hydrat znany
jako soda krystaliczna: Na2CO3 . 10H2O. Łatwość pochłaniania wody zadecydowała o zastosowaniu
osuszającym.
Rownolegle przebiega proces przetwarzania drugiego substratu, tj. węglanu wapnia. Związek ten poddaje się
działaniu wysokiej temperatury w procesie praŜenia, skutkiem czego otrzymuje się tlenek wapnia (wapno
palone):
CaCO3 ---1000oC---> CaO + CO2,
Created by Neevia Document Converter trial version http://www.neevia.com
44
ktore następnie wprowadza się do wody (gaszenie). Reakcja przebiega burzliwie, dając jako głowny produkt
wodorotlenek wapnia (wapno gaszone):
CaO + H2O ----> Ca(OH)2.
Wowczas następuje proces regeneracji, w ktorym powstałe wapno gaszone łączy się z salmiakiem jako
produktem karbonizacji, ktory nie wytrącił się z roztworu:
Ca(OH)2 + 2NH4Cl ----> CaCl2 + 2NH3 + H2O.
Powstały amoniak jest powtornie uŜywany w procesie karbonizacji, zaś chlorek wapnia jest zbędnym produktem
ubocznym.
Sumarycznie zapisany przebieg metody Solvaya przedstawia się następująco:
2NaCl + CaCO3 + H2O ---> Na2CO3 +CaCl2,
skąd wnioskować moŜna o jej substratach (sol kamienna i skała wapienna) i produktach (soda i chlorek wapnia).
Na uwagę zasługuje brak w rownaniu reakcji amoniaku; substrat ten spełnia w całym obiegu rolę jakby
katalizatora – reaguje z jednym z substratow, po czym jest odzyskiwany w procesie regeneracji. W praktyce
występuje potrzeba uzupełniania zapasow amoniaku ze względu na nie stuprocentową wydajność reakcji.
BERYLOWCE
Berylowce to druga grupa głowna w układzie okresowym – II A lub 2. Posiadają konfigurację elektronową ns2
-
dwa elektrony walencyjne na podpowłoce s. W wyniku wzbudzenia jeden z elektronow s przechodzi na
podpowłokę p; tak powstały układ ulega (teoretycznie!) hybrydyzacji digonalnej sp, co doprowadza do
powstania dwoch jednakowych orbitali zachowujących układ liniowy (znajdują się pod wzajemnym kątem
180o). Kształt liniowy posiadają zatem rownieŜ cząsteczki oparte w budowie na atomie berylowca. W
porownaniu do litowcow mają mniejszą objętość (większy ładunek jądra silniej przyciąga elektrony), większą
gęstość (co wynika z poprzedniego), wyŜsze temperatury topnienia i wrzenia oraz większą twardość. Promień
atomowy oraz związana z nim reaktywność rosną w grupie w doł, wraz ze wzrostem liczby atomowej. W
przyrodzie berylowce występują głownie w postaci soli, a zwłaszcza węglanow (CaCO3 - kamień wapienny,
kreda, kalcyt, aragonit, CaCO3 . MgCO3 – dolomit). Związki berylowcow rownieŜ wykazują tendencję do
tworzenia hydratow, ktora maleje w grupie w doł (np.: CuSO4 . 5H2O - kamień siny, CaSO4 . 2H2O – gips lub
alabaster, MgSO4 . 7H2O - sol gorzka, 3MgO . 4SiO2 . H2O – talk). Wszystkie berylowce posiadają ujemne
potencjały normalne, co stanowi o ich aktywności chemicznej. DuŜa aktywność wymusza stosowanie metody
elektrolizy ze stopionych soli podczas otrzymywania tych pierwiastkow. Berylowce są silnymi reduktorami,
przez co łatwo reagują z kwasami w wypierają z nich wodor. Począwszy od wapnia i idąc dalej w doł grupy,
związki berylowcow ulegają wzbudzeniu i emitują energię w postaci kwantow światła widzialnego. Oznacza to,
iŜ ich obecność badać moŜna testem płomieniowym, gdyŜ barwią płomień na określony kolor: wapń –
ceglastoczerwony (szybko znika), bar – jasnozielony, stront – purpurowy, rad – karminowy. W grupie tej
występuje izomorfizm (rownopostaciowość); np. BaSO4 i CaSO4 mają identyczny typ komorki elementarnej,
zbliŜone rozmiary komorek, taki sam typ wzoru chemicznego, podobną postać krystalograficzną i analogiczną
strukturę – warunkuje to zdolność do tworzenia kryształow mieszanych i naprzemienne narastanie kryształow
obu substancji
BERYL
Beryl jest najlŜejszy z całej grupy. W wilgotnym powietrzu pokrywa się warstwą tlenku. Nie reaguje z wodą ani
w temperaturze pokojowej, ani nawet po podgrzaniu. Podobnie jak tlenek, posiada właściwości amfoteryczne. W
kontakcie ze stęŜonym kwasem azotowym pasywuje, pokrywając się ochronna warstwą tlenku. Jest toksyczny
dla człowieka.
MAGNEZ
Magnez jest najmniej aktywny z całej grupy. Pozostawiony na powietrzu pokrywa się warstwą tlenku oraz
bardzo trudno rozpuszczalnego wodorotlenku, w małym stopniu wpływając na alkalizację środowiska. Reaguje z
wodą na gorąco, tj. w temperaturze >70oC. Silnie ogrzany (>760oC) zapala się oślepiającym płomieniem,
wydzielając duŜe ilości ciepła, co zadecydowało o wykorzystaniu magnezu jako materiału zapalnego do lamp
błyskowych, głownie podczas fotografowania nocą. W reakcji tej powstaje trudno rozpuszczalny tlenek
magnezu MgO, stosowany jako składnik materiałow ogniotrwałych. Twardość i odporność na niekorzystne
czynniki zadecydowały o technicznym zastosowaniu magnezu w budownictwie. Stosowany jest rownieŜ jako
protektor do pokrywania kadłubow okrętow i ochrony ich przed korozją. Jonow magnezowych Mg2+ nie moŜna
wykryć w obecności jonow amonowych NH4
+, ktore znacznie szybciej wiąŜą aniony wodorotlenowe.
WAPŃ
Wapń występuje w przyrodzie gownie wchodząc w skład soli – siarczanow oraz wapieni; te ostatnie jako
węglany najłatwiej wykryć działając kwasem, np. solnym:
Created by Neevia Document Converter trial version http://www.neevia.com
45
CaCO3 + 2HCl ---> CaCl2 + H2O + CO2;
reakcji towarzyszy silne pienienie roztworu spowodowane wydzielaniem pęcherzykow gazu.
Obecność jonow Ca2+ w roztworze moŜna stwierdzić dodając doń rozpuszczalnego szczawianu. Z roztworu
strąci się wowczas nierozpuszczalny biały osad szczawianu wapnia CaC2O4.
Z jonową budową węglanu wapnia związana jest jego budowa krystaliczna. Ze względu na sposob ułoŜenia
jonow w sieci krystalicznej wyroŜniamy dwie formy kryształow: kalcyt (forma podstawowa) oraz aragonit.
Drobnokrystaliczną odmianą kalcytu jest marmur. Czyste marmury są białe bądź przezroczyste (tzw. marmury
karryjskie z włoskiego kamieniołomu w Carras), natomiast drobne zanieczyszczenia nadają im określone barwy.
Kryształy kalcytu są dwojłomne, co oznacza, iŜ podające nań światło załamuje się pod dwoma roŜnymi kątami.
Powstają dzięki temu dwa promienie: zwyczajny, załamujący się zgodnie z prawem Snelliusa oraz
nadzwyczajny, nie spełniający powyŜszego warunku. Promienie te posiadają ponadto waŜną właściwość,
mianowicie oba są całkowicie spolaryzowane, a płaszczyzny polaryzacji są do siebie prostopadłe. Otrzymane
światło spolaryzowane słuŜyć moŜe do określania stęŜenia substancji optycznie czynnej w jej roztworze (np.
zawartości sacharozy w soku buraczanym).
Występujący w przyrodzie wapień poddaje się rozkładowi termicznemu w wapiennikach, gdzie zachodzi
reakcja:
CaCO3 ---1000oC---> CaO +CO2,
w ktorej powstaje aktywny i trudno rozpuszczalny tlenek wapnia, zwany wapnem palonym. Tak powstały
produkt zalewa się wodą:
CaO + H2O ----> Ca(OH)2.
Reakcja jest silnie egzotermiczne i prowadzi do powstania wapna gaszonego. Wodorotlenek wapnia jest słabo
rozpuszczalny w wodzie i tworzy tzw. mleko wapienne, jednak pomimo tego jest bardzo silną zasadą. W
laboratorium słuŜy np. do wykrywania dwutlenku węgla – wpierw pojawia się zmętnienie, zauwaŜa się strącenie
białego osadu, ktory następnie rozpuszcza się (wskutek przemiany w wodorowęglan, o czym poźniej):
Ca(OH)2 + CO2 ---> CaCO3 + H2O.
Reakcja ta znalazła zastosowanie w budownictwie: składniki zaprawy (wapno) pod wpływem atmosferycznego
dwutlenku węgla ulegają stwardnieniu i łączą elementy budowlane. Z powodu emisji do atmosfery
zwiększonych ilości dwutlenku węgla, skały wapienne oraz budowle są niszczone wskutek zachodzenia
następującego procesu:
2CaCO3 + CO2 + H2O <---> Ca(HCO3)2.
Kwasowość środowiska wpływa na przemianę węglanow w wodorowęglany charakteryzujące się o wiele
większą rozpuszczalnością w wodzie. Te ostatnie są po prostu wymywane – na tej samej zasadzie działają
procesy krasowe, jak i destrukcyjne skutki kwaśnych opadow (przykładowo zniszczenie rzeźb krakowskiej
Starowki). Wzrost temperatury sprzyja przebieganiu reakcji w drugą stronę – węglan wytrąca się, a lotne
składniki opuszczają środowisko.
Przy produkcji cukru zasadę wapniową wprowadza się do soku buraczanego celem oczyszczenia (defekacji).
Wiązaniu legają wszelkie zanieczyszczenia kwasowe, jak rownieŜ dwutlenek węgla (saturacja), ktore po
zobojętnieniu strącają się w postaci osadu, podczas gdy sacharoza pozostaje w roztworze.
Wapń wchodzi rownieŜ w skład gipsow. Miękki i łupliwy kamień gipsowy wydobywany w kamieniołomach to
nic innego jak uwodniony siarczan (VI) wapnia CaSO4 . 2H2O. W stosunkowo niewysokiej temperaturze (z
małym nakładem energetycznym) moŜna przeprowadzić dehydratację skały gipsowej:
2[CaSO4 . 2H2O] ---110oC---> 2CaSO4 . H2O + 3H2O,
i otrzymać popularny gips. Ten cechuje się łatwym wchłanianiem wody, ktoremu towarzyszy wzrost objętości
(powyŜsza reakcja zachodzi wowczas w drugą stronę). Ze względu na ostatnią właściwość odlewy gipsowe
rzeźb są niezwykle dokładne, gdyŜ w przeciwieństwie do gipsu beton kurczy się podczas twardnienia. Mosty
cementowe muszą być wylewane stopniowo wskutek dylatacji.
Drobnokrystaliczną odmianą gipsu jest alabaster (w przeciwieństwie do pierwszego jest bezwodny). ZbliŜona do
marmuru struktura pozwoliła na zastosowanie go do celow dekoracyjnych juŜ w czasach sumeryjskich.
Szczegolnie trudno rozpuszczalnymi solami są fluorki ze względu na duŜe roŜnice elektroujemności i energie
wiązań. Sprawia to, iŜ fluorek wapnia CaF2 buduje szkliwo zębowe – element skrajnie nierozpuszczalny.
BAR
Bar jest najaktywniejszym berylowcem - posiada aktywność porownywalną z potasem, dzięki czemu z łatwością
wypiera wodor z wody. Podczas otrzymywania baru z jego tlenku stosuje się metodę aluminotermiczną.
Siarczany berylowcow są solami trudno rozpuszczalnymi, a rozpuszczalność maleje w grupie w doł:
siarczan (VI) baru jest nierozpuszczalny nawet w stęŜonych kwasach, co pozwoliło na zastosowanie go w
diagnostyce medycznej. Pacjent zjada papkę zawierającą BaSO4, po czym tworzy się zdjęcie rentgenowskie
jamy brzusznej. Ze względu na duŜą liczbę masową bar pochłania promieniowanie, co uwidocznione jest na
zdjęciu jako białe plamy. Analizując wynik specjalista moŜe stwierdzić chorobę wrzodową lub inne schorzenia.
Reakcja, w ktorej strąca się biały osad nierozpuszczalny w kwasach, jest reakcją charakterystyczną na jony Ba2+.
Created by Neevia Document Converter trial version http://www.neevia.com
46
Przy okazji berylowcow omowimy pojęcie twardości wody. W naturalnej wodzie zawsze znajdują się pewne
ilości jonow wapniowych i magnezowych. W przypadku wod mineralnych jest to zjawisko poŜądane, poniewaŜ
występujące w formie słabo wysyconych i rozpuszczalnych wodorowęglanow makroelementy są dobrze
przyswajalne przez organizm ludzki (nawet 20 – 40%). Natomiast obecność takich jonow w wodzie uŜywanej do
prania, zwana twardością wody, jest wysoce niewskazane, gdyŜ tworzą one z mydłami nierozpuszczalne sole. W
praniu pojawia się kłaczkowaty osad a tkanina staje się sztywna. TakŜe włosy umyte szarym mydłem będą
matowe (w celu uzyskania połysku moŜna dodać kwasku cytrynowego). W zaleŜności od czynnika
powodującego twardość moŜna podzielić ją na:
a) twardość przemijającą (węglanową), spowodowaną obecnością w wodzie wodorowęglanow wapnia i
magnezu. Podczas gotowania wody pod wpływem temperatury powstaje lotny z nich dwutlenek węgla oraz
nierozpuszczalne węglany, strącające się w postaci kamienia kotłowego, np.:
MgCO3 + H2O ---> Mg(OH)2 + CO2.
Powstający kamień kotłowy, będący mieszaniną węglanu wapnia i magnezu, osadza się na elementach
grzewczych i powoduje ich nadmierne ogrzewanie – łatwiej się wtedy topią i niszczą, nie mowiąc juŜ o
dodatkowym zuŜyciu energii.
Twardość przemijającą moŜna usunąć dodając do wody określone substancje, a następnie odsączając powstały
osad, np.:
Ca(OH)2 + Ca(HCO3)2 ---> 2CaCO3 + 2H2O,
2Ca(OH)2 + Mg(HCO3)2 ---> 2CaCO3 + Mg(OH)2 + 2H2O
Na2CO3 + Ca(HCO3)2 ---> CaCO3 + 2NaHCO3.
Usuwaniu twardości słuŜą rownieŜ jonity. Są to specjalne substancje, ktore dzięki posiadanemu na powierzchni
ładunkowi elektrycznemu osadzają na niej niepoŜądane jony – w tym przypadku Ca2+ i Mg2+.
b) twardość nieprzemijającą (siarczanową), spowodowaną obecnością w wodzie siarczanow wapnia i magnezu.
W usuwaniu twardości pomocne są detergenty, czyli syntetyczne środki piorące. Oprocz zmniejszania napięcia
powierzchniowego roztworu posiadają bardzo często naturalny charakter zasadowy.
Twardość wody jest wielkością mierzalną. Określa się ją zawartością soli utwardzających w przeliczeniu na CaO
i oznacza w stopniach. 1o niemiecki odpowiada zawartości 10 mg CaO w 1 dm3 wody. 1o niemiecki = 1,25o
angielskiego = 1,79o francuskiego. Wodę o twardości 1-7o niemieckich uznaje się za wodę miękką, zaś 7-14o –
za wodę twardą.
BOROWCE
Borowce to trzecia grupa głowna w układzie okresowym – III A lub 13. Posiadają konfigurację elektronową
ns2np1
- zatem trzy elektrony walencyjne rozmieszczone na podpoziomach s i p.. W wyniku wzbudzenia jeden z
elektronow s przechodzi na podpowłokę p; tak powstały układ ulega (teoretycznie!) hybrydyzacji trygonalnej
sp2, co doprowadza do powstania trzech jednakowych orbitali zachowujących układ trojkątny (orbitale
skierowane w wierzchołki trojkąta rownobocznego znajdują się pod wzajemnym kątem 120o). Cząsteczki oparte
w budowie na atomie borowca posiadają zatem budowę płaską, trojkątną. Borowce mają mniejsza aktywność
chemiczną w porownaniu z pierwiastkami grupy I i II. WiąŜąc się z innymi pierwiastkami trzema wiązaniami
zapewniają sobie układ sześciu elektronow walencyjnych. Jest to ilość mniejsza od potrzebnego do uzyskania
trwałości oktetu - wobec deficytu elektronowego zgodnie z teorią Lewisa związki takie są kwasami. Dzięki tej
właściwości związki borowcow z fluorowcami, a zwłaszcza halogenki glinu (AlCl3, AlBr3, AlF3), pełnią rolę
katalizatorow w reakcjach syntez organicznych, m. in. w reakcjach Friedla – Craftsa, ktorych mechanizm
zostanie omowiony poźniej. Borowce łatwo tworzą tlenki, mogą nawet odbierać tlen związany z innymi
pierwiastkami, przez co redukują środowisko.
BOR
Pierwszym pierwiastkiem w grupie, od ktorego zresztą bierze się jej nazwa, jest bor. Jest to pierwiastek
połmetaliczny, występujący w związkach z wartościowością III. W przyrodzie występuje on w wielu formach.
Czysty bor występuje w dwoch postaciach alotropowych: forma krystaliczna jest bardzo twarda, odporna na
działanie tlenu stęŜonego kwasu azotowego, zaś forma bezpostaciowa jest miękka i łatwo utlenia się na
powietrzu w podwyŜszonej temperaturze. W warunkach takich bor moŜe rownieŜ redukować parę wodną,
dwutlenek węgla oraz krzemionkę. Identyfikacja boru opiera się na analizie widmowej i teście płomieniowym:
wprowadzony w ogień bor daje barwę zieloną, jednak nie tak jaskrawą jak bar.
Naturalnym związkiem o duŜej zawartości boru jest boraks, czyli uwodniony czteroboran sodu Na2B4O7 .
10H2O. Stopiony boraks rozpuszcza tlenki metali, dając charakterystycznie zabarwione szkliwo, tzw. perłę
boraksową. PoniewaŜ jej kolor zaleŜy od uŜytego pierwiastka, moŜe być wykorzystywana do identyfikacji
pierwiastkow przy dostępie tlenu (jakościowa analiza chemiczna). I tak: Mn2O7 stopiony z boraksem ma kolor
fioletowy, CaO – ciemnoniebieski lub granatowy, zaś CrO3 – zielony. Rozpuszczanie tlenkow na powierzchni
Created by Neevia Document Converter trial version http://www.neevia.com
47
metali pozwoliło uŜyć boraksu jako środka odrdzewiającego. Perły boraksowej uŜywa się ponadto przy
produkcji płytek ceramicznych, gdyŜ zmniejsza to wymaganą temperaturę. Znajduje takŜe zastosowanie jako
topik przy lutowaniu. Dodawany jest do gorących emalii, gdyŜ ze względu na duŜy wspołczynnik załamania
światła nadaje im połysk. UŜywany jest do produkcji szkła Ŝaroodpornego, w połączeniu z ołowiem stanowi
składnik szkła kryształowego, zaś stopiony z berylem – składnik szkła uŜywanego w mikrofalowkach. Boraks
reaguje z nadtlenkiem wodoru, dając nadtlenoboran o silnych właściwościach wybielających. Oprocz wybielania
tkanin ten ostatni słuŜy do barwienia jedwabiu oraz drogich wyrobow skorzanych.
Bor występujący w postaci tlenku B2O3 występuje w towarzystwie krzemianow i glinokrzemianow. Tlenek boru
jest ciałem stałym, rozpuszczającym się w wodzie ze nieznacznym wydzieleniem ciepła (jest to proces
niewymuszony). Reakcja prowadzi do powstania kwasu ortoborowego:
B2O3 + 3H2O ---> 2H3BO3.
Związek ten ma właściwości antyseptyczne, dlatego jest wykorzystywany jako środek dezynfekcyjny w postaci
silnie rozcieńczonego roztworu pod nazwą wody borowej. Podczas ogrzewania kwas ortoborowy traci jedną
cząsteczkę wody i przechodzi w kwas metaborowy:
H3BO3 ---T---> HBO2 + H2O.
Kwasy borowe posiadają słaby charakter kwasowy.
Bor tworzy z niektorymi pierwiastkami nietrwałe borki, np. borek magnezu Mg3B2. Są to na ogoł związki
nietrwałe, łatwo rozkładające się pod działaniem wilgoci i kwasow. Wyjątkowo węglik boru CB4 posiada
ogromną twardość, co zadecydowało o zastosowaniu go do produkcji materiałow ściernych. W naturalnych
złoŜach jest zanieczyszczony grafitem, obok ktorego występuje. Czysty węglik boru, dzięki duŜej zdolności
pochłaniania neutronow, uŜywany był do budowy reaktorow jądrowych oraz schronow atomowych.
W połączeniu w wodorem bor tworzy borowodory. Najprostszym byłby BH3 – byłby, gdyŜ naturalnie związek
taki nie występuje. Spotykany jest jego dimer B2H6 charakteryzujący się deficytem elektronowym, co ułatwia
reakcję ze związkami posiadającymi wolną parę elektronową (np. z amoniakiem). Dimery i polimery
borowodoru aŜ do B6H10 włącznie są gazami o bardzo nieprzyjemnym zapachu, wydzielającymi się podczas
gnicia substancji organicznych.
GLIN
Rownie waŜnym pierwiastkiem w grupie borowcow jak sam bor jest glin. Pierwiastek ten występuje pospolicie
w skorupie ziemskiej w postaci glinokrzemianow, np. skalenia czy miki. Praktycznie wykorzystywana jest
glinka porcelanowa, czyli kaolin (uwodniony krzemian glinu – [Al2(OH)4][Si2O5] – w zapisie tlenkowym:
Al2O3 . 2SiO2 . 2H2O), z ktorej wypala się cegły. Następstwem ruchow geologicznych jest występowanie w
kaolinie zanieczyszczeń, np. czerwonych związkow Ŝelaza Fe2O3, co nadaje cegłom kolor czerwony. Tlenek
glinu Al2O3 występuje w dwoch alotropowych odmianach: krystalicznej, jako korund, minerał bardzo twardy i
wykorzystywany w produkcji materiałow ściernych oraz ziemistej, takŜe uŜywanej w materiałach szlifierskich.
Następstwem jonowej budowy tlenku glinu jest wysoka temperatura topnienia. Siła wiązań sprawia, iŜ tlenku
glinu nie redukują typowe reduktory jak węgiel czy dwutlenek węgla. Cechuje go ponadto wysoki wspołczynnik
załamania światła. Chemicznie czysty minerał jest bezbarwny, natomiast zanieczyszczenia pierwiastkami bloku
d nadają mu piękną barwę. Powstają w ten sposob kamienie szlachetne: ciemnoczerwony rubin przy
domieszkowaniu jonami Cr3+ oraz niebieski szafir przy domieszkowaniu jonami Ti3+. Związki glinu wykazują
charakter amfoteryczny, stąd mowi się o kwasach: ortoglinowym H3AlO3 oraz metaglinowym HAlO2. Te
ostatnie występują jednak wyłącznie w postaci anionow, wchodząc w skład odpowiednich soli. Związki glinu
często biorą udział w tworzeniu połączeń koordynacyjnych z liczbą koordynacyjną zaleŜną od stopnia utlenienia
– odpowiednio 4 dla –1 oraz 6 dla –3.
WaŜnym z punktu widzenia praktyki związkiem glinu jest kriolit Na3AlF6, ktory dzięki obniŜaniu temperatury
topnienia znalazł zastosowanie w elektrolitycznym otrzymywaniu glinu na skalę przemysłową. Innym istotnym
związkiem jest chlorek glinu AlCl3, ciało stałe dobrze rozpuszczalne w wodzie. Swe znaczenie zawdzięcza
istnieniu deficytu elektronowego, dzięki czego moŜe być jako kwas wykorzystywany w syntezie organicznej, np.
jako katalizator przy bromowaniu benzenu. Wodorek litowo – glinowy Li[AlH4] lub LiH . AlH3 posiada bardzo
silne właściwości redukujące, gdyŜ występujący w nim wodor -1 dąŜy do utlenienia się na stopień 0 bądź +1.
Zadecydowało to o jego wykorzystaniu w chemii organicznej w reakcjach hydrogenacji, czyli uwodornienia.
Niektore metale jednowartościowe (potas, sod, rubid, cez) oraz kation amonowy tworzą z metalami
dwuwartościowymi (Al, Cr, Fe, Co, Ga, In, Ti, V) tzw. sole podwojne typu MIMIII(SO4)2 . 12H2O, co moŜna
przedstawić alternatywnym wzorem MI
2SO4 . MIII
2(SO4)3 . 24 H2O. Sole te, zwane ałunami, tworzą dorodna
barwne kryształy. Ałun chromowo – sodowy stosowany jest w garbarstwie do wyprawiania skor, podobnie jak
ałun glinowo – potasowy, ktory znalazł teŜ zastosowanie w farbiarstwie , jako wypełniacz przy produkcji
papieru oraz do tamowania krwawień.
Created by Neevia Document Converter trial version http://www.neevia.com
48
WĘGLOWCE
Węglowce to czwarta grupa głowna w układzie okresowym – IV A lub 14. Posiadają konfigurację elektronową
ns2np2
- zatem cztery elektrony walencyjne rozmieszczone po rowno na podpoziomach s i p. W wyniku
wzbudzenia jeden z elektronow s przechodzi na podpowłokę p; tak powstały układ ulega hybrydyzacji
tetragonalnej sp3, co doprowadza do powstania czterech jednakowych orbitali skierowanych w naroŜa
czworościanu foremnego; shybrydyzowane orbitale znajdują się pod wzajemnym kątem 109o. Cząsteczki oparte
w budowie na atomie węglowca posiadają zatem budowę czworościanu.
WĘGIEL
Pierwiastkiem rozpoczynającym grupę jest węgiel. Występuje on w kilku odmianach alotropowych: diament,
grafit oraz fulleren. Istotne roŜnice pomiędzy dwoma pierwszymi wynikają z występującego w obrębie ich
cząsteczek typu hybrydyzacji. W diamencie występuje hybrydyzacja sp3, co oznacza, iŜ atomy są ułoŜone blisko
siebie i kaŜdy połączony jest z czterema innymi obecnymi dookoła. Powoduje to relatywnie duŜą gęstość i
twardość – jest to najtwardszy minerał w przyrodzie. Brak swobodnych elektronow sprawia, iŜ diament jest
izolatorem. O wysokim wspołczynniku załamania światła w diamencie przekonać się moŜna obserwując barwne
refleksy powstające podczas przejścia światła przez brylant. Z kolei w graficie
występuje hybrydyzacja sp2, co oznacza, iŜ kaŜdy atom połączony jest płasko z
trzema dookoła. Grafit ma zatem budowę warstwową, a poszczegolne
płaszczyzny atomow mogą się względem siebie przesuwać. Z tego powodu
grafit jest miękki i ma mniejszą gęstość od grafitu. Czwarty elektron, nie
związany z Ŝadnym innym atomem, jest wolny i moŜe być nośnikiem prądu
elektrycznego – dlatego teŜ grafit posiada doskonałe właściwości przewodzące.
Odmianą grafitu jest węgiel aktywny; cechuje go bardzo duŜa powierzchnia
wchłaniania i sorpcji, dzięki czemu stosowany jest do zatrzymywania
szkodliwych substancji. Podawany jako lekarstwo zatrzymuje biegunkę, zaś
stosowany w maskach gazowych oczyszcza zatrute powietrze.
Fullereny to specyficzna odmiana węgla; naleŜą do nowych, intensywnie badanych materiałow. Najprostszy
fulleren - C60 jest zamkniętą klatką zbudowaną z 60 atomow węgla w kształcie piłki noŜnej, złoŜoną z 12
pięciokątow i 20 sześciokątow foremnych (patrz rysunek). MoŜna go otrzymać przez laserowe odparowanie grafitu
w atmosferze helu. Fullereny znalazły juŜ zastosowanie jako doskonałe smary. Z tych wielkich cząsteczek
C60 moŜna zbudować kryształ molekularny, ktory nazywa się fullerytem i ktory jest połprzewodnikiem o prostej
przerwie energetycznej rownej 1,9 eV. Istnieją takŜe sferyczne obiekty z wielu wspołśrodkowych fullerenow
(tzw. struktury cebulowe): C60, C240, C540, C960, ktore nazywane są hiperfullerenami. Ponadto moŜna
domieszkować fullereny „zapakowując" obce atomy w C60 i tworzyć tzw. fullerydki: np. fullerydek z metalem
alkalicznym K3C60. Wykazuje on nadprzewodnictwo wysokotemperaturowe z temperaturą przejścia 33 K.
Metodą wyładowania łukowego moŜna tworzyć rurki fullerenowe (tzw. nanorurki kwantowe) - wspołosiowe
cylindry odległe o 0,34 nm i długości ok. l μm, ktore są bardzo trwałymi włoknami, stosowanymi jako
światłowody dla promieniowania gamma. Interesująca właściwość „samozasklepiania" rurek fullerenowych daje
ich duŜą odporność na uszkodzenia. MoŜna wreszcie „patroszyć" fullereny; np. dodając do sześciokątow
siedmiokąty, wewnętrzną stronę odwracać na zewnątrz. Dokonując „mariaŜu topologii z węglem" moŜna
prowadzić geometryczne zabawy budowania roŜnych obiektow przestrzennych z atomow węgla otrzymując
nowe struktury o nieoczekiwanych właściwościach.
Ogromną rolę w przyrodzie i gospodarce odgrywają związki węgla. Związki organiczne, ze względu na ich
mnogość, omowione będą osobno. Wśrod nieorganicznych związkow węgla waŜną rolę pełnią tlenki.
1) Najprostszym tlenkiem węgla jest CO, znany jako czad. Jest to bezbarwny i bezwonny gaz, nieco lŜejszy od
powietrza, bardzo słabo rozpuszczalny w wodzie i nie reagujący z nią. Na uwagę zasługuje budowa cząsteczki:
otoŜ po dwa niesparowane elektrony z tlenu i węgla tworzą dwa wiązania koordynacyjne spolaryzowane, na obu
atomach pozostają wolne pary, a dodatkowo jedna para z atomu tlenu tworzy trzecie, koordynacyjne wiązanie z
atomem węgla. Cząsteczka wykazuje słaby moment dipolowy. Taka sama budowa strukturalna, identyczna masa
molowa oraz liczba elektronow walencyjnych upodabnia cząsteczkę CO do N2 – mowimy, iŜ są izosteryczne.
Dzięki dysponowaniu wolnymi parami w wyŜszych temperaturach tworzy lotne związki z metalami, zwane
karbonylkami, np.: Ni(CO)4, Fe(CO)5. Wiązanie jest potrojne, czyli silnie nienasycone. Dzięki temu tlenek
węgla bardzo łatwo wchodzi w reakcje, jest gazem palnym, a w wyŜszych temperaturach moŜe odbierać tlen
innym związkom. Redukcyjne właściwości sprzyjają zastosowaniu go przy otrzymywaniu metali. W procesie
wielkopiecowym najpierw jest on otrzymywany w wyniku przepuszczanie strumienia powietrza przez
rozŜarzony koks (reakcja Braundarda):
C + O2 ---> CO2, CO2 + C --->2CO,
Created by Neevia Document Converter trial version http://www.neevia.com
49
a następnie wchodzi w reakcję z rudą:
Fe2O3 + 3CO ---T---> 3CO2 + 2Fe.
Podobnie przebiega proces otrzymywania cynku; wpierw ruda siarczkowa (sfaleryt lub wurtzyt) jest spalana do
tlenku:
2ZnS + 3O2 --->2ZnO + 2SO2,
ktory następnie poddawany jest redukcji:
ZnO + CO ---> Zn + CO2.
W odpowiednio wysokiej temperaturze tlenek węgla odtlenia wodę:
CO + H2O <---1100oC---> CO2 + H2.
W przedstawionym rownaniem układzie ustala się stan rownowagi. Jest to przykład reakcji konwersji, o ktorej
mowimy, gdy jeden gaz lub mieszanina przekształca się w drugą (w powyŜszej reakcji ze względu na
temperaturę wszystkie substraty występują w stanie gazowym).
Czad tworzy się podczas połspalania związkow organicznych, jak rownieŜ przy kontakcie metanu z wodą:
CH4 + H2O ---T---> CO + 3H2.
MoŜna go takŜe otrzymać działając stęŜonym kwasem siarkowym na kwas mrowkowy. Ten pierwszy odbiera
drugiemu cząsteczkę wody, w wyniku czego pozostaje tlenek węgla:
HCOOH ---st. H2SO4 ---> H2O (natychmiast pochłaniana przez higroskopijny kwas) + CO.
MoŜe on wchodzić w reakcję z zasadą sodową, dając mrowczan sodu:
CO + NaOH ---T, p---> HCOONa.
W wyniku reakcji koksu z wodą w wysokiej temperaturze powstaje mieszanina wodoru i tlenku węgla, zwana
gazem syntezowym (generatorowym, wodnym):
C + H2O ---1000oC---> H2 + CO.
Odpowiednia proporcja składnikow oraz właściwe warunki termiczno – ciśnieniowe pozwalają na otrzymanie z
gazu syntezowego roŜnorodnych związkow organicznych.
Reaguje rownieŜ z siarką, dając ostatecznie dwusiarczek węgla:
2CO + 2S ---> 2COS (nietrwały) ---> CO2 + CS2.
Z chlorem tworzy fosgen, ktory wykorzystywany był jako gaz bojowy:
CO + Cl2 ---> COCl2.
Fosgen ma jednak rownieŜ bardziej cywilizowane zastosowania – przykładowo w reakcji z amoniakiem daje
mocznik, wykorzystywany w produkcji tworzyw sztucznych (np. Ŝywic):
COCl2 + 2NH3 ----> NH2CONH2 + 2HCl.
Czad jest gazem silnie trującym. Śmiertelne jest dla człowieka juŜ stęŜenie 0,3% w powietrzu. Czad łączy się z
hemoglobiną krwi 250 razy trwalej niŜ czyni to tlen, co blokuje funkcję transportującą tego związku, gdyŜ
powstała karboksyhemoglobina HbCO nie przenosi tlenu.
2) Najbardziej znanym jest dwutlenek węgla, biorący udział w procesach biologicznych. Powstaje przez pełne
spalanie związkow organicznych (przy bogatym dostępie tlenu). Otrzymać go moŜna przez rozkład węglanow
metodą termiczną bądź przez działanie nań mocnym kwasem np.:
CaCO3 ---1000oC---> CaO + CO2,
CaCO3 + 2HCl ---> CaCl2 + H2O + CO2.
Ostatnia reakcja moŜe być wykorzystywana do identyfikacji węglanow, gdyŜ towarzyszy jej charakterystyczne
silne pienienie roztworu spowodowane wydzielaniem pęcherzykow gazu.
Dwutlenek węgla jest bezbarwnym i bezwonnym gazem, cięŜszym od powietrza. Przez krotki czas moŜna go
przechowywać w naczyniach otwartych. Nie jest gazem palnym ani nie podtrzymuje palenia. Jest związkiem
niepolarnym. W obrębie jego cząsteczki zachodzi hybrydyzacja digonalna sp. Cząsteczka CO2 ma budowę
liniową, a sumaryczny moment dipolowy wynosi zero. Rozpuszcza się w wodzie, przy czym rozpuszczalność
maleje ze wzrostem temperatury i rośnie ze wzrostem ciśnienia. Przykładowo – w butelce wody i w małym
stopniu reaguje z nią, dając kwas węglowy:
H2O + CO2 <---> H2CO3.
Reakcja jest silnie odwracalna, powstały produkt jest bardzo nietrwały i gdy stęŜenie jego przekroczy setną
część procenta, rozkłada się. Kwas węglowy jest kwasem dwuprotonowym, przechodzi więc dysocjację
dwustopniową i moŜe tworzyć dwa rodzaje soli: obojętne węglany oraz kwaśne wodorowęglany. Wodorosole
rozpuszczają się lepiej od soli obojętnych, o czym mowa była w temacie poświęconym berylowcom. Węglany
litowcow (nie dotyczy litu i amonu) są dobrze rozpuszczalne, podobnie wodorowęglany litowcow, trochę gorzej
w przypadku berylowcow. Dwutlenek węgla moŜna skroplić pod zwiększonym ciśnieniem oraz zestalić w
temperaturze –78,5oC – stosunkowo wysokiej w porownaniu z innymi naturalnymi gazami. Zestalony znany jest
pod nazwą suchego lodu; sublimuje on, a wrzucony do wody unosi jej cząsteczki ponad powierzchnię, co daje
efekt „dymu”. Obecnie wykorzystuje się go w produkcji środkow chłodzących. W małych ilościach dwutlenek
węgla nie wywiera szkodliwego działania na organizm ludzki, jednak stęŜenie jego nie moŜe być całkowicie
dowolne. Za dopuszczalne stęŜenie, tzw. wskaźnik Pettenkofera, przyjmuje się 0,1%.
Created by Neevia Document Converter trial version http://www.neevia.com
50
Substancje o bardzo duŜym powinowactwie do tlenu mogą go zredukować, np.:
Mg + CO2 ---> 2MgO + C.
Dwutlenek węgla wykorzystywany jest w analizie jakościowej. Wpuszczony do roztworu zasady wapiennej
powoduje zmętnienie i wytrącenie białego osadu węglanu wapnia:
Ca(OH)2 + CO2 ---> CaCO3 + H2O.
Przy nieustannym dostarczaniu nowych porcji zaczyna się reakcja w wodą:
H2O + CO2 <---> H2CO3.
Powstający kwas węglowy reaguje z węglanem i przekształca go w bardziej rozpuszczalny wodorowęglan:
2CaCO3 + H2CO3 <---> Ca(HCO3)2.
Skutek jest taki, Ŝe powstały osad ulega rozpuszczeniu; taki cykl przemian pozwala jednoznacznie stwierdzić
obecność w środowisku wymienionych powyŜej substancji.
Dwutlenku węgla uŜywa się ponadto do produkcji sody metodą Solvaya, produkcji wod gazowanych,
oczyszczania soku buraczanego w procesie saturacji, wiązania zanieczyszczeń zasadowych oraz do wypełniania
gaśnic.
3) Dwa opisane powyŜej tlenki wymieniane są zawsze gdy mowa o związkach węgla. Ale oprocz nich istnieją
rownieŜ inne tlenki. Podczas ogrzewania związku organicznego jakim jest kwas malonowy HOOC-CH2-COOH
w obecności pięciotlenku fosforu otrzymuje się jego bezwodnik:
HOOC-CH2-COOH<------>C3O2 + 2H2O.
MoŜna mowić zatem o kolejnym tlenku węgla o wzorze C3O2. Zaproponować moŜna dla niego liniowy wzor
cząsteczki, w ktorym wszystkie wiązania są podwojne: O=C=C=C=O. W przedstawionej reakcji związek fosforu
ze względu na duŜą higroskopijność pełni rolę osuszacza.
Naukowo dowiedziono rownieŜ istnienia kolejnych tlenkow węgla o wzorach sumarycznych C6O9 oraz C12O9,
przy czym dla tego ostatniego zaproponowano budowę cykliczną.
4) W wyniku trzykrotnego chlorowania metanu otrzymuje się trichlorometan CHCl3, znany pod zwyczajową
nazwą chloroform. Wykorzystywany był do narkozy podczas zabiegow medycznych. W wyniku całkowitego
chlorowania metanu otrzymuje się waŜnym technicznie związek - czterochlorek węgla CCl4. Związek ten jest
cieczą wielokrotnie cięŜszą od wody. Choć trujący dla człowieka, znalazł zastosowanie dzięki skutecznemu
rozpuszczaniu tłuszczy.
5) Węgiel wchodzi w skład reszt kwasowych kwasow: cyjanowodorowego, piorunowego oraz rodanowego.
Kwas cyjanowodorowy HCN występuje w dwoch odmianach mezomerycznych, pomiędzy ktorymi ustala się
stan rownowagi dynamicznej: HCN <=>HNC. Posiada charakterystyczny zapach gorzkich migdałow i jest
bardzo silną trucizną: 50 mg czystego HCN powoduje śmierć w przeciągu kilku sekund. Jego sole, cyjanki, są
rownieŜ bardzo silnymi truciznami o krotkim czasie efektu śmiertelnego. Najgroźniejsze są sole rozpuszczalne w
wodzie, a więc cyjanki litowcow i berylowcow, np. cyjanek potasu KCN. Jony cyjankowe biorą udział w
tworzeniu związkow kompleksowych rozpuszczalnych w wodzie, np.:
AgCN + CN- ---> Ag(CN)2
-.
(osad) (rozpuszczalny)
W cyjankach rozpuszczają się metale szlachetne, takie jak srebro czy złoto, stąd w wielu rejonach świata
wykorzystuje się je do wypłukiwania tych metali ze zmielonych skał. śelazocyjanek potasu K4[Fe(CN)6] znany
jest jako błękit pruski i słuŜył niegdyś do barwienia tkanin na niebiesko. Jego hydrat K4[Fe(CN)6] . 3H2O tworzy
Ŝołtawe kryształy uŜywane do cementacji stali.
Kwas piorunowy (fulminowy) HOCN jest nietrwały i istnieje tylko w roztworach wodnych. Jest aktywny
chemicznie, dając reakcje przyłączania i polimeryzacji. Istotną rolę spełniają jego sole – pioruniany. Piorunian
rtęci Hg(OCN)2 to biała substancja stała wybuchająca przy uderzeniu. Właściwości te stanowią o jej
zastosowaniu w spłonkach jako detonator. Otrzymuje się ją jako produkt reakcji azotanu (V) rtęci z etanolem.
Kwas rodanowy (tiocyjanowy) HSCN jest mocnym, nietrwałym kwasem beztlenowym. Jest bezbarwną cieczą
mieszającą się z wodą. Otrzymuje się go przez gotowanie siarki w wodnym roztworze cyjankow. Zastosowanie
mają jego sole, głownie w analizie chemicznej, gdyŜ jony rodankowe tworzą barwne kompleksy z niektorymi
kationami, np. z Cu2+ szafirowy a z Fe3+ krwistoczerwony.
KRZEM
Krzem połmetalem występującym w dwoch odmianach alotropowych. Pierwsza - metaliczna ma budowę
krystaliczną. Cechuje się wyŜszą od grafitu twardością i potrafi rysować szkło. Jest przewodnikiem, a
przewodnictwo to rośnie ze wzrostem temperatury; przypomnijmy, iŜ dla metali obowiązuje odwrotna zaleŜność
– wraz ze wzrostem temperatury rośnie ich opor. Na zimno krzem jest bierny chemicznie, natomiast w wyŜszych
temperaturach łączy się z tlenem, fluorem i azotem (tworzy azotki). W łuku elektrycznym reaguje z wodorem
tworząc wodorokrzemiany (silany). Na gorąco rozpuszcza się w wielu metalach, a z niektorymi z nich tworzy
krzemki o teoretycznym charakterze kwasowym, np. krzemek magnezu Mg2Si. Niewielkimi ilościami krzemu
domieszkuje się stale o podwyŜszonych właściwościach spręŜystych. Druga – bezpostaciowa odmiana posiada
większą powierzchnię czynną, czemu zawdzięcza szybsze wchodzenie w reakcje chemiczne. Rozpuszcza się w
Created by Neevia Document Converter trial version http://www.neevia.com
51
bardzo mocnych zasadach, tworząc krzemiany z wydzieleniem wodoru:
Si + 2OH- + H2O ---> SiO3
2- + 2H2.
W przyrodzie krzem występuje ponadto w postaci związanej. Przykładem jest węglik krzemu SiC, znany jako
karborund; minerał ten jest bardzo twardy, dzięki czemu uŜywany jest jako materiał do produkcji wyrobow
szlifierskich.
1) Wśrod związkow krzemu waŜne miejsce zajmują jego połączenia z wodorem. Wspomniano juŜ wcześniej, iŜ
reagując z wodorem w łuku elektrycznym, krzem daje silany. MoŜna mowić tu o całym szeregu
homologicznym, podobnie jak przy związkach organicznych: SiH4, Si2H6, Si3H8, Si4H10 itd. Przedstawione
związki są gazami o zroŜnicowanej gęstości. Są bardzo nietrwałe, zapalają się na powietrzu i ulegają działaniu
zasad:
SiH4 + 2OH- + H2O ----> SiO3
2- +4H2.
Mogą polimeryzować dając łatwopalne polisilany (SiH2)n. Gdy atom wodoru jest w nich zastępowany przez
grupy alkilowe, wowczas mowimy o alkilosilanach.
2) Krzem tworzy związki rownieŜ z fluorowcami. NajwaŜniejszym w tej grupie jest fluorek krzemu SiF4,
pozostałe nie odgrywają istotnej roli. Fluorek krzemu powstaje podczas trawienia szkła z wykorzystaniem kwasu
fluorowodorowego:
SiO2 + 4HF <==> 2H2O + SiF4.
Z powodu trujących właściwości tego ostatniego coraz częściej proces ten przeprowadza się za pomocą reakcji
alternatywnej:
SiO2 + 2CaF2 + 2H2SO4 ---> 2CaSO4 + 2H2O + SiF4.
Fluorek krzemu reaguje rownieŜ z samym kwasem fluorowodorowym:
2 SiF4 + 4HF ---> 2H2[SiF6].
Produkt ostatniej reakcji to kwas fluorokrzemowy; posiada relatywnie dosyć silne właściwości kwasowe i
dobrze rozpuszcza się w wodzie.
3) Bardzo powszechną formą występowania krzemu w przyrodzie jest krzemionka, czyli tlenek krzemu SiO2.
Jest to krystaliczne ciało stałe o bardzo uporządkowanej strukturze, w ktorej występują wyłącznie wiązania
pojedyncze, a kaŜdy atom tlenu związany jest z dwoma atomami krzemu. Konstrukcja oparta jest na
czworościanach o duŜym stopniu upakowania, co gwarantuje duŜą gęstość i twardość. Największe ilości
krzemionki znajdują się w piasku. W przyrodzie występuje w postaci amorficznej, skrytokrystalicznej, a
zabarwiony przez zanieczyszczenia zyskuje barwę (np. kamienie – opal, agat, krwawnik). Czysto krystaliczna
odmiana nosi nazwę kwarcu, a dokładnie kryształu gorskiego. Ze względu na czynność optyczną wyroŜniamy
kwarc prawo- oraz lewoskrętny. Zabarwiony tlenkami metali daje kamienie połszlachetne: fioletowy ametyst,
czarny morion czy Ŝołty cytryn. Kwarc jest kryształem dwojłomnym, co oznacza iŜ posiada zdolność
podwojnego załamania światła spolaryzowanego. Na tym zjawisku oparta jest budowa nikoli, czyli przyrządy
mierzące zawartość cukrow w ich roztworach. Stopiona krzemionka zastyga na bezpostaciowe szkło kwarcowe,
przepuszczające promieniowaniu ultrafioletowe. Lampy kwarcowe wykorzystywane są zarowno w szpitalach do
celow dezynfekcyjnych, jak i w solariach do opalania. Bezpostaciowa odmiana krzemionki to tzw. ziemia
okrzemkowa. Jest to materiał wybitnie porowaty o duŜych zdolnościach sorpcyjnych, uŜywany do produkcji
dynamitu przez wysycenie go nitrogliceryną. Ze względu na swe właściwości wchłania ona trzykrotnie więcej
materiału wybuchowego niŜ sama waŜy.
Krzemionki uŜywa się do produkcji szkła. Pomimo odporności na działanie wielu chemikaliow wchodzi w
reakcję podczas stapiania z wodorotlenkiem sodu:
SiO2 + 2NaOH ---> Na2SiO3 + H2O;
w rzeczywistości uŜywa się do tego celu sody:
Na2CO3 + SiO2 ---> Na2SiO3 + CO2.
Oczywiście moŜna stosować odpowiednie sole potasu. W wyniku powyŜszych reakcji otrzymuje się
rozpuszczalne w wodzie krzemiany, tzw. szkło wodne. Następnie w celu stabilizacji przeprowadza się podobny
szereg reakcji dla węglanu wapnia. Powstały metakrzemian wapnia nie jest juŜ rozpuszczalny w wodzie, przez
co zwiększa odporność i wytrzymałość nowo powstałego szkła. Gotowy wyrob jest zatem stopioną mieszaniną
krzemianu sodu (ew. potasu), krzemianu wapnia i krzemionki. W zaleŜności od uŜytych składnikow cechują go
określone właściwości. I tak: szkło potasowo – wapniowe charakteryzuje się duŜym połyskiem i
wspołczynnikiem załamania światła, przez co stosowane jest do wyrobu soczewek. z kolei szkło sodowe jest
tańsze, bardziej wraŜliwe na zmiany temperatury i słabiej załamuje światło. Szkło dekoracyjne z dodatkiem
ołowiu posiada piękny połysk; wykonanych z niego „kryształow” nie moŜna jednak uŜywać do podawania
produktow spoŜywczych, gdyŜ łatwo pękają pod wpływem ciepła. Typowe szkło ołowiowe stosowane jest w
optyce do wyrobu soczewek okularowych i mikroskopowych. W naczyniach szklanych nie wolno
przechowywać wodorotlenku sodu, ktory powoduje nadŜerki i przecieki; z tego powodu pastylki wodorotlenku
trzyma się w pojemnikach z tworzyw sztucznych.
Krzemian sodu moŜe reagować z kwasami, np. z kwasem solnym:
Na2SiO3 + 2HCl ---> H2SiO3 + 2NaCl.
Created by Neevia Document Converter trial version http://www.neevia.com
52
Powstały kwas metakrzemowy posiada właściwości kwasowe słabsze od (bardzo słabego przecieŜ) kwasu
węglowego, ponadto słabo rozpuszcza się w wodzie i strąca się w postaci białego galaretowatego osadu.
Wytwarza się faza koloidowa SiO2 . H2O. Dodatek wody powoduje przejście kwasu metakrzemowego w kwas
ortokrzemowy:
H2SiO3 + H2O ---> H2SiO4,
ktory rownieŜ jest kwasem bardzo słabym i źle rozpuszczalnym w wodzie, strącając się w postaci
galaretowatego osadu.
CYNA
Cyna jest metalem występującym na dwoch stopniach utlenienia: +2 i +4, w związku z czym tworzy dwa tlenki:
zasadowy SnO i amfoteryczny SnO2. Pierwszy, dąŜąc do stopnia utlenienia +4 wykazuje właściwości
redukcyjne. Drugi jako amfoter ulega reakcji z mocnymi zasadami:
SnO2 + 2NaOH ---> Na2SnO3 + H2O.
W roztworze powstaje oczywiście forma bogatsza w cząsteczki wody, sześciohydroksocynian sodu
Na2[Sn(OH)6], ktory następnie przechodzi w powyŜszy cynian sodu.
Cyna jest odporna na czynniki atmosferyczne, nie ulega działaniu słabych kwasow ani słabych zasad. UŜywana
jest do lutowania, jak rownieŜ do produkcji blachy białej, z ktorej powstają puszki do konserw. Jest plastyczna i
moŜe być rozwałkowywania na cienkie paski. Powstała folia cynowa słuŜy do opakowania wyrobow
cukierniczych. Prawdziwość tego moŜna łatwo zbadać, gdyŜ dobrej jakości cyna dzięki ziarnistej budowie
chrzęści w charakterystyczny sposob, zaś folia aluminiowa nie daje takiego efektu. Podobnie sprawdzić moŜna
czystość cienkich profili cynowych, gdyŜ zanieczyszczenia rownieŜ pozbawiają cynę efektu szelestu.
OŁOW
Metal ten występuje w przyrodzie w postaci związanej jako roŜnorodne minerały, np.: PbO – glejta, PbS –
galena, PbCO3 – cerusyt, PbSO4 – anglezyt, Pb3(PO4)2 – piromorfit. Jest to metal cięŜki, pozostawiony na
powietrzu pokrywa się warstwą zasadowego węglanu ołowiu PbO . Pb(OH)2 . PbCO3, co zabezpiecza go przed
korozją. W obecności stęŜonych kwasow utleniających (siarkowego, azotowego) ulega pasywacji, tworząc
nierozpuszczalne sole, np. PbSO4. Jest giętki, ciągliwy i kowalny; posiada niską temperaturę topnienia, dzięki
czemu wykorzystuje się go do wykonywania odlewow. Dla organizmow Ŝywych jest silnie trujący, kumulując
się głownie w mozgu. Dzięki duŜej liczbie masowej uŜywany jest do zatrzymywania promieniowania –
przykładowo przed działaniem promieniowania gamma chroni dopiero gruba warstwa ołowiu. Przypomnijmy
tylko, iŜ zanik natęŜenia promieniowania w warstwie absorbującej ma charakter wykładniczy: I=I0e-kx, gdzie x to
grubość płytki, a k – wspołczynnik proporcjonalności. Tworzy bardzo trwałe związki na +2 stopniu utlenienia,
natomiast te na +4 wykazują pewien charakter amfoteryczny. Bardzo łatwo łączy się z siarką, co znalazło
zastosowanie przy identyfikacji białek (choć siarka występuje w trzech tylko aminokwasach – cystynie, cysteinie
i metioninie – to zawiera ją praktycznie kaŜde naturalne białko). Podczas ogrzewania białka z octanem ołowiu
(CH3COO)2Pb wydziela się powoli czarny osad siarczku ołowiu PbS, co jest charakterystyczne dla produktow
zawierających siarkę.
AZOTOWCE
Azotowce to piąta grupa głowna w układzie okresowym – V A lub 15. Posiadają konfigurację elektronową
ns2np3
- zatem pięć elektronow walencyjnych rozmieszczonych na podpoziomach s i p. W grupie tej występują
zarowno niemetale (azot, fosfor), połmetale (arsen, antymon) jak i metale (bizmut). Azotowce niemetaliczne
występują na wyŜszych stopniach utlenienia, zaś metaliczne na niŜszych.
AZOT
Pierwiastkiem rozpoczynającym grupę jest azot. Jest to gaz stanowiący głowny składnik (78%) powietrza i nieco
od niego lŜejszy. Spotykamy z się z nim najczęściej w postaci związanej, np. saletr: NaNO3 – saletra chilijska,
KNO3 – saletra indyjska, Ca(NO3)2 – saletra norweska, NH4NO3 – saletra amonowa (dawniej pospolity środek
słuŜący do konserwacji mięsa). Wchodzi w skład aminokwasow, a przez to rownieŜ peptydow i białek. Na skalę
przemysłową otrzymywany jest przez frakcjonowaną destylację powietrza w temperaturze –210oC. W
laboratorium otrzymać go moŜna przez rozkład termiczny wybuchowego azotanu (III) amonu:
NH4NO3 ---T---> N2 + 2H2O.
Ciekły azot jest powszechnie wykorzystywany do szeroko rozumianego chłodzenia: od transplantowanych
organow medycznych do procesow przemysłowych. W cząsteczce azotu występują trzy wiązania kowalencyjne,
co nadaje jej ogromną trwałość i energię jonizacji (947 kJ/mol). Z tego powodu temperaturze pokojowej jest
bierny, zaś w temperaturach wyŜszych tworzy z wieloma metalami (Ca, Mg, Si) związki zwane azotkami. Łączy
się z wodorem dając amoniak oraz w wysokiej temperaturze z tlenem dając tlenki. Zapach świeŜości
występujący w powietrzu po burzy to efekt powstawania tlenkow azotu w łuku elektrycznym piorunu.
Created by Neevia Document Converter trial version http://www.neevia.com
53
1) Amoniak NH3 powstaje w wyniku reakcji azotu z wodorem:
N2 + 3H2 ---(war)--> 2NH3.
Aby taka reakcja zaszła, wymagane są odpowiednie warunki: ze względu na zmniejszanie objętości stosuje się
wysokie ciśnienie, ze względu na endotermiczność – wysoką temperaturę (reguła przekory). Oprocz tego dodaje
się odpowiednich katalizatorow: najbardziej optymalnymi są osm, iryd bądź platyna, moŜna rownieŜ stosować
tańszą, ale zdecydowanie mniej wydajną mieszaninę Ŝelaza, tlenku glinu i wodorotlenku potasu.
Amoniak jest lŜejszym od powietrza gazem o intensywnym zapachu. Łatwo się skrapla, a dzięki pochłanianiu
znacznych ilości ciepła znalazł zastosowanie w chłodziarkach. W stanie skroplonym przewodzi prąd (ale bardzo
słabo) wskutek autodysocjacji na kation amonowy i anion amidowy:
2NH3 ---> NH4+ + NH2-.
MoŜe rozpuszczać metale i z niektorymi z nich tworzyć amidki, np.: amidek sodu NaNH2, wapnia Ca(NH2)2 czy
glinu Al(NH2)3. W powietrzu spala się z wydzieleniem azotu:
4NH3 + 3O2 ---> 2N2 + 6H2O.
Spalany w tlenie wobec katalizatora daje mieszaninę tlenkow azotu:
4NH3 + 5O2 ---> 6H2O + 4NO (reakcja głowna),
4NH3 + 7O2 ---> 6H2O + 4NO2 (reakcja uboczna).
W obrębie cząsteczki występują wiązania kowalencyjne. Azot jest w stanie hybrydyzacji tetragonalnej sp3,
dzięki czemu cząsteczka ma budowę zgiętą. W trzech naroŜach czworościanu foremnego znajdują się jądra
wodoru, zaś w czwartym – wolna para elektronowa. Ta cecha budowy dalece wpływa na właściwości amoniaku;
są to przede wszystkim: słaby charakter zasadowy oraz zdolność do tworzenia związkow kompleksowych.
Omowimy teraz kolejno obie te właściwości.
W wodzie amoniak łączy się z kationami wodorowymi dzięki przekazaniu im swej wolnej pary:
NH3 + H+ ---> NH4+,
co jako reakcja z wodą przedstawia się następująco:
NH3 + H2O <==> NH4OH <==> NH4+ + OH-.
Powstaje więc związek o charakterze zasadowym – zasada amonowa. Jej właściwości zasadowe są słabe, ale
bardzo łatwo reaguje z wieloma kwasami. Wszystkie sole amonowe przy ogrzewaniu bądź działaniu
mocniejszymi zasadami wydzielają wonny amoniak, co słuŜy identyfikacji tych soli. Przykładowo jeśli w
nachylonej do poziomu rurce ogrzewać będziemy biały proszek salmiaku NH4Cl, a na obu końcach umieścimy
zwilŜone papierki lakmusowe, to dolny zabarwi się na czerwono, a gorny na niebiesko. Mechanizm
doświadczenia jest następujący: w wyniku rozkładu termicznego soli amonowej wydzielają się lotne gazy –
amoniak oraz chlorowodor:
NH4Cl ---T---> NH3 + HCl.
Amoniak jako lŜejszy od powietrza unosi się w gorę, gdzie rozpuszcza się w wodzie i daje aniony
wodorotlenowe barwiące papierek na niebiesko:
NH3 + H2O <==> NH4OH <==> NH4+ + OH-.
Z kolei chlorowodor jako cięŜszy od powietrza spływa w doł rurki, rozpuszcza się w wodzie i daje kationy
wodorowe, barwiące papierek na czerwono.
HCl + H2O ---> H3O+ + Cl-.
Amoniak tworzy wiele związkow kompleksowych z nierozpuszczalnymi bądź trudno rozpuszczalnymi solami
(np. halogenkami), w ktore to połączenia wchodzi z liczbą koordynacyjną 2, np.:
AgCl + 2NH3 ---> [Ag(NH3)2]Cl,
AgBr + 2NH3 ---> [Ag(NH3)2]Br.
Istotne jest, iŜ powstałe związki kompleksowe są juŜ rozpuszczalne. Natomiast nie jest w stanie rozpuścić w
podobny sposob jodku srebra AgI, ktorego wiązanie jest zbyt silne. Z jonami pierwiastkow bloku d (miedź,
mangan, Ŝelazo, nikiel, kobalt) tworzy kationy kompleksowe o charakterystycznych barwach: Cu2+ - niebieski,
Fe2+ - zołty, Fe3+ - brunatny, Ni2+ - Ŝołty, Co2+ - niebieski. MoŜna to wytłumaczyć dwustopniowo: pierwiastki
bloku d posiadające niesparowane elektrony d powodują rozpraszanie się na nich światła, zaś dodatek amoniaku
wprowadzającego wolne pary elektronowe znacznie ten efekt pogłębia. Przykładowo jon miedzi Cu2+ otacza się
w wodzie czterema cząsteczkami wody (na kaŜdej występują dwie pary elektronowe), tworząc kation
tetraakwamiedzi (II), zaś po dodaniu amoniaku otacza się czterema jego cząsteczkami, dając kation
tetraamionomiedzi.
Amoniak słuŜy do produkcji mocznika:
COCl2 + 2NH3 ----> NH2CONH2 + 2HCl
oraz kwasu azotowego i nawozow sztucznych:
Created by Neevia Document Converter trial version http://www.neevia.com
54
Pochodnymi amoniaku są: hydrazyna NH2NH2 oraz hydroksyloamina NH2OH, o ktorych zastosowaniach
powiemy poźniej.
2) WaŜne znaczenie mają tlenki azotu. Tlenkiem o najniŜszej wartościowości jest podtlenek azotu N2O.
Otrzymać go moŜna przez termiczny rozkład azotanu (V) amonu:
NH4NO3 ---T---> N2O + 2H2O.
W temperaturze pokojowej jest to bezbarwny gaz o przyjemnym, słodkawym zapachu. Wdychany działa na
organizm ludzki oszałamiająco, dlatego stosowano go jako gaz rozweselający, np. do krotkotrwałego
znieczulenia przy zabiegach dentystycznych. Tlenek azotu (I) posiada parzystą liczbę elektronow, z ktorych
wszystkie są sparowane, co przyczynia się zapewne do stosunkowo duŜej bierności chemicznej w zwykłej
temperaturze.
3) Tlenek azotu (II) powstaje podczas reakcji tlenu z azotem przy dostarczeniu duŜych ilości energii. MoŜe to się
odbywać na sposob katalityczny (termiczny):
N2 + O2 ---1500oC---> 2NO,
bądź w łuku elektrycznym, np. podczas wyładowań atmosferycznych lub w specjalnym reaktorze (metoda
Mościckiego). Tlenek azotu wydziela się rownieŜ podczas reakcji niezbyt stęŜonego (1-15%) kwasu azotowego
z metalami szlachetnymi, np.:
Cu + 8HNO3 ---> 3Cu(NO3)2 + 2NO + H2O.
W warunkach normalnych jest to bezbarwny gaz nieco cięŜszy od powietrza, bardzo słabo rozpuszczalny w
wodzie. Pierwiastki o duŜym powinowactwie do tlenu (jak magnez czy glin) po zakończeniu reakcji spalania w
powietrzu mogą palić się dalej w atmosferze tlenku azotu. Posiada on nieparzystą liczbę elektronow (11),
dlatego wykazuje właściwości paramagnetyczne i charakter rodnikowy. W dąŜeniu do uzyskania sparowanych
elektronow reaguje egzotermicznie z powietrzem:
2NO + O2 ---> 2NO2.
Efektem ostatniej reakcji jest czerwonobrunatny, cięŜszy od powietrza dwutlenek azotu. Omawiany tlenek
tworzy połączenia kompleksowe z fluorowcami – nitrozyle, np. chlorek nitrozylu NOCl czy bromek nitrozylu
NOBr. Dla analizy jakościowej istotna jest reakcja tworzenia kompleksu z jonami Ŝelaza Fe2+:
FeSO4 + NO ---> Fe(NO)SO4.
Powstały siarczan nitrozyloŜelaza (II) tworzy w środowisku reakcji ciemnobrunatną obrączkę.
4) Tlenek azotu (III) N2O3 jest cieczą o barwie niebieskawej. Trwałość zapewnia temperatura poniŜej –10oC, w
wyŜszych bowiem ulega dysproporcjonowaniu na tlenki azotu (II) i (IV). Jest bezwodnikiem kwasu
azotowego (III):
N2O3 + H2O ---> 2HNO2,
ktory rownieŜ jest związkiem nietrwałym i dysproporcjonuje:
3HNO2 ---> HNO3 + 2NO + H2O.
Nieco bardziej trwałe są przynajmniej jego sole – azotany (III). Ze względu na pośredni stopień utlenienia moŜe
pełnić zarowno funkcję utleniacza, jak i reduktora.
5) Tlenek azotu (IV) NO2 jest czerwonobrunatnym, cięŜszym od powietrza i silnie trującym gazem. Powstaje
przez utlenianie tlenku azotu (II). Ze względu na nieparzystą liczbę elektronow walencyjnych samorzutnie i
egzotermicznie dimeryzuje:
2NO2 <==> N2O4.
Powstały dimer jest produktem bezbarwnym. Przesunięcie rownowagi reakcji w ktorąś stronę jest odpowiedzią
na zmianę temperatury – w niŜszych występuje raczej N2O4, zaś w wyŜszych NO2. Dwutlenek azotu łatwo
rownieŜ ulega dysproporcjonowaniu:
2NO + H2O ---> HNO2 + HNO3.
Powstaje w reakcjach kwasu azotowego (V) z metalami o dodatnich potencjałach normalnych, np.:
Cu + 4HNO3 ---> Cu(NO3)2 + 2NO2 + 2H2O.
Jest silnym utleniaczem; wiele pierwiastkow o duŜym powinowactwie do tlenu świetnie pali się w jego
atmosferze.
6) Pięciotlenek azotu N2O5 jest ciałem stałym. Bezwodnik kwasu azotowego (V), jest związkiem nietrwałym i
juŜ w temperaturze pokojowej rozkłada się z wydzieleniem tlenu:
2N2O5 ---> 2N2O4 + O2,
przez co staje się niebezpieczny w przechowywaniu.
7) Kwas azotowy (V) HNO3 jest bardzo waŜnym produktem przemysłowym. Na skalę przemysłową otrzymuje
się go poprzez utlenianie amoniaku. Miesza się z wodą w kaŜdym stosunku. Jest związkiem nietrwałym, a
przechowywany na świetle brunatnieje wskutek wydzielania się tlenku azotu (IV):
2HNO3 ---> 2NO2 + .O2 + H2O.
Bardzo rozcieńczony (<1%) kwas azotowy zachowuje się jak klasyczny kwas: przewodzi prąd elektryczny,
reaguje tylko z metalami o ujemnych potencjałach standardowych i wydziela w takich reakcjach wodor. StęŜony
natomiast nie dysocjuje, dlatego nie przewodzi prądu i zachowuje się jak typowy środek utleniający. Jest bardzo
silnym utleniaczem, utlenia niemetale w stanie czystym (fosfor, siarkę, węgiel) do ich tlenkow i kwasow z
Created by Neevia Document Converter trial version http://www.neevia.com
55
wydzieleniem tlenku azotu(II):
3P + 5HNO3 -> 3H3PO4 + 5NO,
3S + 4HNO3 -> 3SO2 + 4NO + 2H2O,
3C + 4HNO3 -> 3CO2 + 4NO + 2H2O.
Działaniu stęŜonego kwasu azotowego ulegają metale wybitnie szlachetne, np. iryd, platyna czy złoto. Procesy te
są jednak bardziej złoŜone. Do reakcji z metalami wybitnie szlachetnymi zdolna jest wyłącznie mieszanina
stęŜonych kwasow – solnego oraz azotowego (V) – w stosunku objętościowym 3:1 (molowym 4:1). Reakcja ta
przebieg dwuetapowy: wpierw kwas solny reaguje z kwasem azotowym, wytwarzając chlorek nitrozylu, ktory
następnie atakuje metal szlachetny. Dla złota produktem reakcji jest kwas czterochlorozłotowy, co moŜna
zapisać następująco:
HNO3 + 3HCl -> NOCl + Cl2 + 2H2O,
Au + HCl + NOCl +Cl2 -> HAuCl4 + NO.
Metale o niŜszych potencjałach, będące silnymi reduktorami mogą go zredukować nawet o 8 stopni utlenienia:
4Zn + 10HNO3 -> 4Zn(NO3)2 + NH4NO3 + 4H2O,
4Mg + 10HNO3 -> 4Mg(NO3)2 + N2O + 5H2O.
Niektore metale (Ŝelazo, chrom, glin) pokrywaj się w jego obecności pasywną warstwą tlenku; jest to czasami
zjawisko poŜądane – przykładowo moŜna dzięki temu przewozić stęŜony kwas azotowy w stalowych cysternach.
Kwas azotowy reaguje z dymiącym kwasem siarkowym (oleum, roztwor SO3 w H2SO4), odszczepiając kation
nitroniowy:
2H2SO4 + HNO3 ---> 2HSO4
- + NO+ + H3O+,
ktory bardzo agresywnie atakuje inne związki, np. benzen czy toluen. Z tego powodu do nitrowania
(wprowadzania grupy nitrowej do) związkow chemicznych nie stosuje się czystego kwasu azotowego, lecz tzw.
mieszaninę nitrującą, złoŜonej z HNO3 i H2SO4. W reakcjach tych powstają Ŝołte nitrozwiązki, często cechujące
się właściwościami wybuchowymi, np. sym-trinitrotoluen (TNT) znany powszechnie pod nazwą trotylu.
Obecność kwasu azotowego wykrywa się na podstawie reakcji z siarczanem (VI) Ŝelaza (II) FeSO4, w ktorej to
wytwarza się wspomniana juŜ wcześniej ciemnobrunatna obrączka siarczanu (VI) nitrozyloŜelaza (II).
Jego sole – azotany(V) – są nośnikami duŜych ilości tlenu, dlatego znalazły zastosowanie jako składniki
materiałow wybuchowych. Zmieszane z substancjami łatwopalnymi ulegają samozapłonowi; przykładowo
azotanu amonu nie naleŜy mieszać z węglem drzewnym, trocinami, papierem ani nie rozbijać metalowymi
przedmiotami.
8) Rzadko wymieniany jest związek zwany kwasem azotowodorowym HN3 (H-N=N=>N|). Istotne znaczenie
mają jedynie jego sole, azydki. Są bardzo nietrwałe i pobudzenie mechaniczne wyzwala w nich wybuchowe
reakcje rozkładu. Z tego powodu wiele azydkow stanowi głowny składnik detonatorow, np.: azydek ołowiu
Pb(N3)2, azydek rtęci Hg2(N3)2 czy azydek srebra AgN3.
FOSFOR
Następnym w grupie azotowcow pierwiastkiem jest fosfor. W przyrodzie występuje on w postaci związanej -
pięciotlenku fosforu P2O5, fosforanow (np. jednym z głownych składnikow kości jest fosforan wapnia) bądź w
postaci czystej – jednej z kilku odmian alotropowych.
a) Fosfor biały jest bezbarwną substancją stałą, bardzo miękką i plastyczną. Nie rozpuszcza się w wodzie ani w
alkoholach, za to rozpuszcza się w PCl3, SCl2 i CS2. Posiada niską temperaturę topnienia (44oC) i wrzenia
(280oC). Występuje w postaci tetraedrycznych cząsteczek P4. Jest bardzo aktywny chemicznie: w atmosferze
czystego chloru samorzutnie się zapala, łatwo reaguje z bromem i siarką. Rozdrobniony lub podgrzany do 60oC
zapala się na powietrzu:
4P + 5O2 ---> P4O10 <==> 2P2O5.
Naświetlony świeci w ciemności, stąd wyrabia się z niego elementy świecące w zegarkach nocnych. Jest bardzo
silną trucizną, dawka śmiertelna czystego fosforu białego wynosi 0,1 g. Z powodu zagroŜenia samozapłonem i
wybuchem przechowywany jest pod wodą.
b) Fosfor czerwony moŜna otrzymać z odmiany białej przez powolne ogrzewanie bez dostępu powietrza. W
powyŜszych warunkach tetraedry polimeryzują, wytwarzając długą, liniową cząsteczkę Pn (n>>1). Z roŜnicy w
strukturze wynikają roŜnice we właściwościach: forma czerwona jest trwalsza, nietrująca, mało aktywna
chemicznie, nierozpuszczalna w chlorkach i siarczkach, posiada wyŜsze temperatury zmian fazy, zapala się w
temperaturze powyŜej 240oC. Wykorzystywany jest do produkcji zapałek: głowka i draska wykonane są z
mieszaniny fosforu czerwonego i chloranu, będącego nośnikiem tlenu.
c) Pozostałe odmiany alotropowe – fosfor czarny i fioletowy – nie są szczegolnie wykorzystywane.
1) W połączeniu z wodorem fosfor tworzy fosforowodor PH3. W przeciwieństwie do podobnego w budowie
amoniaku nie posiada on właściwości zasadowych. Jest związkiem silnie trującym, o przykrym zapachu
zgniłych ryb.
2) Fosfor tworzy kilka tlenkow: P2O3, P2O5 oraz nietrwały P2O4. Dwa pierwsze z nich są bezwodnikami
kwasowymi. Pięciotlenek fosforu P2O5 jest białą substancją stałą o konsystencji sypkiej; łatwo sublimuje i jest
Created by Neevia Document Converter trial version http://www.neevia.com
56
bardzo higroskopijny, dzięki czemu moŜe być wykorzystywany jako środek osuszający substancji o charakterze
obojętnym lub kwasowy.
3) Fosfor tworzy dwa szeregi kwasow – w jednym występuje na stopniu utlenienia +3 , a w drugim na +5.
Powstanie konkretnego kwasu uzaleŜnione jest od ilości dostępnej w środowisku reakcji wody:
P2O3 + H2O ---> 2HPO2 (kwas metafosforowy (III)),
P2O3 + 2H2O ---> H4P2O5 (kwas pirofosforowy (III) = difosforowy (III)),
P2O3 + 3H2O ---> 2H3PO3 (kwas ortofosforowy (III)),
P2O5 + H2O ---> 2HPO3 (kwas metafosforowy (V)),
P2O5 + 2H2O ---> H4P2O7 (kwas pirofosforowy (V) = difosforowy (V)),
P2O5 + 3H2O ---> 2H3PO4 (kwas ortofosforowy (V)).
Interesującym z pewnego względu jest kwas
ortofosforowy (III). Jako kwas trzyprotonowy powinien
tworzyć on trzy rodzaje soli, a tworzy tylko dwie.
MoŜna stwierdzić, iŜ dysocjuje on dwustopniowo, a
ostateczny anion ma postać HPO3
2- (patrz rysunek).
Dzieje się tak, poniewaŜ jeden z atomow wodoru
połączony jest bezpośrednio z atomem fosforu i nie ma przez to moŜliwości dysocjacji.
Kwas ortofosforowy (V) tworzy trzy szeregi soli: sole obojętne, wodorosole oraz diwodorosole. Te dwa ostatnie
rozpuszczają się w wodzie lepiej niŜ sole obojętne. Z tego powodu nawozy fosforowe mają postać diwodorosoli,
np. superfosforat Ca(H2PO4)2. Wśrod soli obojętnych rozpuszczalne są ortofosforany litowcow ( z wyjątkiem
litu) oraz amonu, pozostałe są nierozpuszczalne. Fosforany i polifosforany stosowane były jako składniki
środkow piorących i zmiękczających. Zostały jednak wycofane ze względu na skutki, jakie wywoływały ścieki
fosforowe, m. in. nadmierny rozwoj roślin, obrastanie zbiornikow wodnych i kadłubow statkow oraz deficyt
tlenowy i śmierć zwierząt wodnych.
4) Połączenia fosforu z fluorowcami, np. PCl5, stosowane są w przemyśle chemicznym do chlorowania
aldehydow.
ARSEN
Arsen jest połmetalem występującym rownieŜ w odmianach alotropowych: arsen metaliczny As4 oraz arsen
Ŝołty. Jest to pierwiastek o aktywności mniejszej od fosforu. Wszystkie jego związki są silnie trujące.
1) Arsenowodor AsH3 posiada zapach zbliŜony do czosnku. Jest silną trucizną. Podczas ogrzewania rozkłada się
na pierwiastki, co w kryminalistyce funkcjonuje jako proba lustra arsenowego (wykrywanie otrucia związkami
arsenu juŜ w niewielkich stęŜeniach):
2AsH3 ---T---> 2As + 3H2.
2) Tlenek arsenu (III) As2O3, znany jako arszenik, jest rownieŜ silną trucizną. Bardzo źle rozpuszcza się w
wodzie, z ktorą tworzy kwas arsenowy (III):
As2O3 + 3H2O ---> 2H3AsO3.
Powstały produkt jest bardzo słabym kwasem, wykazującym pewien charakter amfoteryczny.
ANTYMON
Antymon jest pierwiastkiem połmetalicznym o właściwościach zbliŜonych do arsenu. Występuje w odmianach
alotropowych. Nie ulega działaniu nawet stęŜonych kwasow, jedynie na gorąco roztwarza się w wodzie
krolewskiej – jest zatem metalem szlachetnym. Antymonowodor SbH3 posiada właściwości trujące
porownywalne z arsenowodorem. RownieŜ podobnie jak on rozkłada się w wyŜszych temperaturach na
pierwiastki, dając lustro antymonowe (proba na antymon juŜ w małych stęŜeniach).
BIZMUT
Bizmut jest jedynym w grupie azotowcow pierwiastkiem o właściwościach metalicznych. Wprowadzony do
stopu znacznie zmniejsza jego temperaturę topnienia.
TLENOWCE
Tlenowce to szosta grupa głowna w układzie okresowym – VI A lub 16. Posiadają konfigurację elektronową
ns2np4
- zatem sześć elektronow walencyjnych rozmieszczonych na podpoziomach s i p. W stanie podstawowym
atom tlenowca posiada dwie pary elektronowe – jedną s i jedną p – oraz dwa niesparowane elektrony walencyjne
p, ktore mogą brać udział w wiązaniach. Ogranicza to wartościowość do II – taka sytuacja występuje w
przypadku tlenu. JeŜeli tlenowiec dysponuje wolnym orbitalem walencyjnym d (okres trzeci i poniŜsze), to w
wyniku wzbudzenia mogą nań przemieszczać się elektrony z par s i p. Rozszerza to zakres wartościowości o IV
(wzbudzenie pojedyncze) i VI (wzbudzenie podwojne). Oznacza to rownieŜ, iŜ mogą pojawić się rzadko
Created by Neevia Document Converter trial version http://www.neevia.com
57
wymieniane typy hybrydyzacji, np. sp3d2 w sześciofluorku siarki SF6. W grupie tej występują zarowno niemetale
(tlen, siarka, selen), połmetale (tellur) jak i metale (polon).
TLEN
Pierwiastkiem rozpoczynającym grupę jest tlen. W warunkach normalnych jest to gaz o gęstości 1,43 g/cm3, a
zatem nieco cięŜszy od powietrza. Stanowi około jedną piątą (21%) składu powietrza. Występuje w dwoch
odmianach alotropowych: zwykły tlen (O2) oraz ozon (O3), ktorego największe ilości występują w gornej
warstwie atmosfery zwanej ozonosferą. Występuje pomiędzy nimi stan rownowagi:
2O3 <==> 3O2.
Jest pierwiastkiem bardzo aktywnym (przy czym ozon jest aktywniejszy), łączy się praktycznie z wszystkimi
pierwiastkami i daje tlenki. Występuje na –2 stopniu utlenienia, wyjątkowo w nadtlenkach na –1 ze względu na
istnienie ugrupowania -O-O- oraz w tlenku fluoru OF2 na +2 ze względu na wyŜszą elektroujemność fluoru. W
przeciwieństwie do pozostałych tlenowcow nie przyjmuje wyŜszych stopni utlenienia, co zostało wyjaśnione
wcześniej.
Tlen występuje w postaci trzech izotopow: 16O, 17O oraz 18O. Ostatni izotop jest promieniotworczy, dlatego
wykorzystywany jest do badania mechanizmow reakcji metodą tzw. tlenu znaczonego. Przykłady zastosowania
tej metody podane będą poźniej, np. przy reakcjach estryfikacji.
SIARKA
Siarka jest Ŝołtym ciałem stałym występującym w kilku odmianach alotropowych. W kaŜdej z nich występują
cząsteczki siarki S8, roŜniące się jedynie konfiguracją przestrzenną, a ściślej typem sieci krystalicznej.
a) Odmiana alfa (rombowa) jest trwała w temperaturze do 95,6oC. Ma barwę Ŝołtą, nie rozpuszcza się w wodzie,
bardzo słabo zaś w substancjach organicznych. Ogrzana powyŜej punktu trwałości topi się i przechodzi w formę
beta.
b) Odmiana beta (jednoskośna) w zwykłej temperaturze jest nieaktywna chemicznie, ulega działaniu fluorowcow
z wyjątkiem jodu. W wysokich temperaturach łączy się niemal z wszystkimi pierwiastkami, z wyjątkiem azotu,
złota, platynowcow i helowcow. W powietrzu spala się do dwutlenku siarki SO2. Jako pierwiastek elektroujemny
(X=2,5) łatwo reaguje z metalami tworząc siarczki; jest to przyczyną czernienia wyrobow z miedzi i srebra.
PowyŜsze reakcje są egzotermiczne i wymagają jedynie inicjacji. Z wodorem siarka łączy się tylko w wysokich
temperaturach, ze względu na małą roŜnicę elektroujemności. Siarka ma wiele zastosowań. W lecznictwie
wykorzystuje się ją w postaci maści siarkowej do leczenia chorob skory, gdyŜ niszczy drobnoustroje trądzikowe.
UŜywana jest do konserwacji tanich wyrobow alkoholowych (np. win, na ktorych etykiecie widnieje
dopuszczalna zawartość dwutlenku siarki), rownieŜ sokow i przecierow owocowych (jagodzianki po przetarciu
poddaje się działaniu dwutlenku siarki). Przy wulkanizacji w wysokiej temperaturze dodana siarka tworzy
mostki siarczkowe pomiędzy łańcuchami polimerow, np. kauczuku czy gumy, przez co zmniejsza płynność tych
wyrobow. Siaka wykorzystywana jest ponadto do produkcji barwnikow, prochu strzelniczego oraz w przemyśle
chemicznym do produkcji kwasy siarkowego (VI).
1) Jak powiedziano, w wyŜszych temperaturach siarka łączy się z wodorem, dając siarkowodor H2S. Jest
bezbarwny gaz o bardzo przykrej woni zgniłych jaj, silnie trujący o działaniu duszącym z powodu redukcji
oksyhemoglobiny. W warunkach laboratoryjnych otrzymać go moŜna działając na siarczek metalu kwasem
solnym, np.:
Na2S + 2HCl ---> H2S + NaCl.
Siarkowodor jest wraŜliwy na wysokie temperatury i rozkłada się wowczas na pierwiastki:
H2S ---T---> H2 + S.
Jest gazem palnym, przy czym produkt zaleŜy od dostępności tlenu w środowisku:
2H2S + 3O2 ---> 2H2O + 2SO2,
H2S + .O2 ---> H2O + S.
Wykazuje dosyć silne właściwości redukcyjne, gdyŜ potencjał reakcji S2- -->S0 wynosi –0,51V. Ponadto dobrze
rozpuszcza się w wodzie i dysocjuje dwustopniowo:
H2S <==> H+ + HS- <==> 2H+ + S2-.
Jest słabym kwasem, moŜe tworzyć dwa szeregi soli: obojętne siarczki oraz kwaśne wodorosiarczki. Siarczki są
bardzo trudno rozpuszczalne w wodzie i mają ciemne (czarne bądź brunatne) zabarwienie; wyjątek stanowią
siarczki litowcow i amonu. Siarczki litowcow mogą rozpuścić w sobie znaczne ilości siarki, a w wyniku
zachodzącej reakcji tworzą wielosiarczki. Siarczki rozpuszczalne w wodzie hydrolizują zasadowo, np. (NH4)2S
jest solą słabego kwasu i słabej zasady, lecz ostatecznie jego wodny roztwor ma odczyn zasadowy, gdyŜ zasada
amonowa dysocjuje lepiej niŜ siarkowodor. Niektore siarczki wskutek hydrolizy nie istnieją w roztworach
wodnych, np. siarczek magnezu MgS:
MgS ---H2O---> Mg2+ + S2-,
2H2O <==> 2H+ + 2OH-,
Created by Neevia Document Converter trial version http://www.neevia.com
58
Mg2+ + 2OH- --> Mg(OH)2,
S2- + 2H+ ---> H2S.
Produkty hydrolizy to ulatniający się gaz (H2S) oraz strącający się osad (Mg(OH)2). Ze względu na słabą
rozpuszczalność wiele siarczkow metali to rudy tych metali, np.: Cu2S – chalkozyn, CuS – kowelin, CuS . FeS –
chalkopiryt, PbS – galena ołowiowa, FeS2 – piryt lub markosyt, HgS – cynober, Ag2S – argentyt, ZnS – sfaleryt
lub wurtzyt, MoS2 – molibdenit. Istnieje generalna prawidłowość, iŜ wodorosiarczki rozpuszczają się w wodzie
lepiej od siarczkow obojętnych.
2) Tlenek siarki SO2 jest związkiem trwałym. Występuje w przyrodzie jako składnik wyziewow wulkanicznych.
W warunkach domowych otrzymać go moŜna przez spalanie zapałek, o czym świadczy charakterystyczny
zapach. W przemyśle powstaje podczas utleniania siarczkow (proces technologiczny polega na przeprowadzeniu
siarczku w tlenek, a następnie jest redukcji koksem) np.:
4FeS2 + 11O2 --> 2Fe2O3 + 8SO2.
Tlenek siarki powstaje rownieŜ jako produkt reakcji stęŜonego kwasu siarkowego z metalami o dodatnim
potencjale normalnym, np.:
Cu + 2H2SO4 -> CuSO4 + SO2 + 2H2O.
Wydziela się, gdy działamy stęŜonym kwasem siarkowym na niemetale, np.:
S + 2H2SO4 -> 3SO2 + 2H2O,
C + 2H2SO4 -> 2SO2 + CO2 + 2H2O.
Tlenek siarki jest gazem bezbarwnym o ostrym zapachu, dwukrotnie cięŜszym od powietrza. Jest szkodliwy
zarowno dla człowieka, jak i dla mikroorganizmow. Cząsteczka ma budowę zgiętą ( |O=S=>O| ), posiada
niezerowy moment dipolowy, a w związku z tym budowę polarną. Łatwo się skrapla; w ciekłym dwutlenku
siarki rozpuszcza się wiele soli, np. PCl3. Jest pochłaniany przez wodę i jako bezwodnik tworzy niezbyt mocny
kwas siarkowa (IV):
SO2 + H2O ---> H2SO3.
Dzięki pośredniemu stopniowi utlenienia siarczany (IV) mogą być utleniaczami i reduktorami. Rozpuszczalne w
wodzie siarczany (IV) stosowane były jako środki bielące, np. Na2SO3 utleniając się redukuje niepoŜądane
związki barwne w tkaninie, zaś zredukowane produkty są łatwo rozpuszczalne i mogą zostać usunięte przez
pocieranie. Preparaty siarkowe miały przewagę nad wybielaczami chlorowymi dzięki miłemu zapachowi.
3) Trojtlenek siarki SO3 otrzymywany jest katalitycznie przez utlenianie dwutlenku w aparatach kontaktowych:
SO2 + .O2 ---(war)---> SO3.
Ze względu na zmniejszanie objętości podczas przebiegu reakcji stosuje się wysokie ciśnienie, a ze względu na
endotermiczność – temperaturę 400oC (reguła przekory). Dodatkowo obecny jest zazwyczaj katalizator
wanadowy, ktorym najczęściej jest pięciotlenek wanadu V2O5. W typowych warunkach trojtlenek siarki jest
ciałem stałym, sublimującym w biały dym. UŜywany jest głownie do produkcji kwasu siarkowego (VI), moŜe
się w nim rozpuszczać i tworzyć tzw. dymiący kwas siarkowy (oleum). Reakcja z wodą jest tak egzotermiczna,
iŜ powstający wrzący kwas mogłby rozprysnąć się i stanowić zagroŜenie, dlatego teŜ SO3 uŜywane jest w
nadmiarze. Pełniąc rolę inhibitora sprawia, iŜ reakcja jest w pewien sposob kontrolowana.
4) Kwas siarkowy (VI) rozpuszcza się w wodzie w kaŜdym stosunku. Tworzenie wiązań wodorowych i
samorzutna hydratacja są procesami bardzo egzotermicznymi, stąd rozpuszczaniu towarzyszy wydzielenie
duŜych ilości ciepła. Występuje rownieŜ zjawisko kontrakcji – objętość mieszaniny maleje. Dzięki ogromnemu
powinowactwu do wody oleum uŜywane jest do osuszania gazow, a nawet odbiera wodę substancjom, w ktorych
jest ona związana chemicznie, np.:
C2H5OH ---st. H2SO4/120-130oC---> C2H4 + H2O (pochłaniana),
2C2H5OH ---st. H2SO4---> C2H5OC2H5 + H2O (pochłaniana),
eter dietylowy
CnH2nOn = Cn(H2O)n ---st. H2SO4---> nC + nH2O (pochłaniana).
Ostatnia reakcja ilustruje zwęglanie cukrow przez stęŜony kwas siarkowy; moŜe to być zarowno cukier domowy,
jak i na drewno (celuloza). Skora poparzona kwasem siarkowym pokrywa się czarnymi strupami. StęŜony kwas
siarkowy jest silnym utleniaczem: utlenia niemetale (węgiel, siarkę, fosfor), a na gorąco rownieŜ metale
szlachetne. Metale takie jak Ŝelazo, glin czy chrom pasywują w jego obecności i pokrywają się ochronną
warstwą tlenkow. Rozcieńczony kwas siarkowy w temperaturze pokojowej na ma właściwości utleniających i
zachowuję się jak zwykły kwas dwuzasadowy. Tworzy dwa szeregi kwasow – obojętne siarczany (VI) i kwaśne
wodorosiarczany (VI). Większość tych pierwszych jest dobrze rozpuszczalna w wodzie, wyjątkiem są siarczany
wapniowcow, np. BaSO4 lub innych – PbSO4.
5) Gdyby w cząsteczce kwasu siarkowego (VI)
jeden z atomow tlenu wymienić na atom siarki
na stopniu utlenienia –2, to otrzymalibyśmy
kwas tiosiarkowy H2S2O3. Jest on związkiem
nietrwałym i w środowisku kwaśnym powoli
się rozkłada:
Created by Neevia Document Converter trial version http://www.neevia.com
59
H2S2O3 ---H+---> S + SO2 + H2O.
Jego sole, tiosiarczany, są dobrze rozpuszczalne w wodzie i przede wszystkim – trwałe. Anion tiosiarczanowy
S2O3
2- posiada duŜo (10) par elektronowych, dzięki czemu moŜe być donorem par w połączeniach
koordynacyjnych, ktore z łatwością tworzy. Przykładowo w fotografii tiosiarczan sodu stosowany jest jako
utrwalacz – wypłukuje i usuwa z negatywu i pozytywu nie rozłoŜone halogenki srebra:
AgBr + 2Na2S2O3 ---> Na3[Ag(S2O3)2] + NaBr.
osad rozpuszczalny
Powstałe związki: tiosrebrzan sodu i bromek sodu mogą być łatwo wypłukane. Tiosiarczan sodu
wykorzystywany jest w przemyśle papierniczym i włokienniczym do usuwania nadmiaru chloru uŜytego do
bielenia, stąd nazwa techniczna: antychlor. W reakcji aniony tiosiarczanowe działają redukująco na chlor, ktory
zjonizowany tworzy NaCl, podczas gdy sam tiosiarczan utlenia się do siarczanu (VI):
Na2S2O3 + 4Cl2 + 10NaOH --> 2Na2SO4 +8NaCl + H2O.
6) Niektore związki siarki warte są uwagi ze względu na udział w reakcjach substytucji w chemii organicznej; są
to np.: dwuchlorek siarki SCl2 i jego dimer S2Cl4 oraz chlorotlenek siarki SO2Cl2.
7) Jak juŜ wcześniej wspomniano, siarka uŜywana była przez wiele wiekow do produkcji prochu strzelniczego.
W najprostszym przypadku naleŜy zmieszać siarkę (zapalnik) z materiałem palnym (węgiel drzewny) w
obecności utleniacza (np. saletry indyjskiej) w proporcjach 1:1,5:7,5. Po zainicjowaniu przebiega reakcja:
S + 3C + 2KNO3 --> 3CO2 + N2 + K2S.
Dwa pierwsze produkty są gazami, trzeci natomiast – ciałem stałym, dlatego teŜ pisany proch spala się z
wydzieleniem dymu. Głownym składnikiem prochu bezdymnego jest triazotan (V) celulozy, opisany dokładniej
w dziale poświęconym chemii organicznej. Jako zapalniki inicjujące reakcję uŜywane są np. piorunian rtęci
Hg(OCN)2 bądź azydek ołowiu Pb(N3)2.
8) W przyrodzie siarka wspołtworzy wiele naturalnych związkow dezynfekujących. Na przykład podczas
krojenia cebuli uwalnia się z niej propantiol C3H6SO, ktory reagując z wodą znajdującą się na powierzchni oka
daje kwas siarkowy, będący przyczyną łzawienia. Z tego teŜ powodu krojenie cebuli pod wodą nie powoduje
męczącego „płaczu”. Znanym warzywem o działaniu dezynfekującym jest czosnek; zawiera on szereg związkow
siarkowych o działaniu odkaŜającym, co ilustruje poniŜszy schemat:
.
SELEN
Selen jest pierwiastkiem rozpoczynającym w grupie tlenowcow przejście niemetali w metale. Występuje w
odmianach alotropowych. W związkach przyjmuje roŜne stopnie utlenienia, jednak najtrwalsze są związki w
ktorych ma +4. Nie przewodzi prądu elektrycznego – jest izolatorem. Natomiast pod wpływem kwantow
promieniowania świetlnego generuje impulsy elektryczne, dzięki czemu moŜe być stosowany jako zamiennik
impulsow świetlnych na sygnały w postaci prądu. Ze względu na tą ostatnią właściwość znalazł zastosowanie w
urządzeniach alarmowych, fotokomorkach, telewizorach i urządzeniach kserograficznych. Selen jest
mikroelementem, niezbędnym w śladowych ilościach do prawidłowego funkcjonowania organizmu ludzkiego
(erytrocyty). Dodatek selenu barwi szkło na kolor roŜowy lub czerwono – rubinowy, przez co neutralizuje
zieloną barwę pochodzącą od zanieczyszczeń krzemionki związkami Ŝelaza. W połączeniu z wodorem selen
tworzy selenowodor H2Se, ktory wprowadzony do wody wykazuje mocniejsze właściwości kwasowe niŜ
siarkowodor. Tworzy dwa szeregi soli: selenki i wodoroselenki.
Tellur i polon tworzą związki o typowej wartościowości +4. Porownując selen, tellur i polon pod względem
elektrycznym stwierdzamy, iŜ potencjał normalny maleje w grupie wraz ze wzrostem liczby atomowej.
FLUOROWCE
Fluorowce to siodma grupa głowna w układzie okresowym – VII A lub 17. Posiadają konfigurację elektronową
ns2np5
- zatem siedem elektronow walencyjnych rozmieszczonych na podpoziomach s i p. W stanie
podstawowym atom fluorowca posiada trzy pary elektronowe – jedną s i dwie p – oraz jeden niesparowany
elektron walencyjny p, ktory moŜe brać udział w wiązaniach. Ogranicza to wartościowość do I – taka sytuacja
występuje w przypadku fluoru. JeŜeli fluorowiec dysponuje wolnym orbitalem walencyjnym d (okres trzeci i
poniŜsze), to w wyniku wzbudzenia mogą nań przemieszczać się elektrony z par s i p. Rozszerza to zakres
Created by Neevia Document Converter trial version http://www.neevia.com
60
wartościowości tak, Ŝe pozostałe fluorowce przyjmują wartościowości I, III, V, i VII. Oznacza to rownieŜ, iŜ
mogą pojawić się niestandardowe typy hybrydyzacji, np. sp3d3 w siedmiofluorku jodu IF7. W grupie tej
dominują niemetale; połmetaliczny astat występuje w warunkach naturalnych w śladowych ilościach 10-24% i
aktualnie nie ma zastosowania. Fluorowce cechuje zroŜnicowany stan skupienia: fluor i chlor to gazy o
cząsteczkach dwuatomowych, brom jest cieczą a jod – łatwo sublimujących ciałem stałym. Elektroujemność
maleje w grupie wraz ze wzrostem liczby atomowej. Fluorowce tworzą wodorki kwasowe, ktore wprowadzone
do wody dają kwasy beztlenowe. Tak powstałe roztwory mają stosunkowo silne właściwości kwasowe, przy
czym moc kwasu wzrasta w grupie wraz ze wzrostem liczby atomowej. Tłumaczyć to naleŜy wzrastaniem
promienia jonowego, a tym samym odległości pomiędzy jądrami fluorowca i wodoru – to z kolei znacznie
osłabia działającą między nimi siłę; w efekcie łatwiej wodor moŜe być odszczepiany łatwiej, a ze wzrostem
stopnia dysocjacji rośnie pH oraz moc kwasu. KaŜdy fluorowiec wypiera z soli sąsiada leŜącego poniŜej siebie w
grupie (fluor wypiera wszystkie pozostałe, chlor wypiera brom i jod, brom wypiera jod, a jod moŜe być
wyłącznie wypierany przez inne fluorowce ze swoich soli).
FLUOR
Pierwiastkiem rozpoczynającym grupę jest fluor. W warunkach normalnych jest gazem o barwie Ŝołtozielonej o
intensywnym, nieco chlorowym zapachu. Atakuje drogi oddechowe człowieka. Ma najwyŜszą elektroujemność
w układzie okresowym (X=4,0), jest w związku z tym najaktywniejszym pierwiastkiem. Bezpośrednio nie łączy
się jedynie z tlenem, chlorem, azotem i helowcami. W wysokiej temperaturze działaniu chloru ulegają: diament,
złoto i platynowce. Bardzo energicznie reaguje z wodorem nawet w ujemnych temperaturach i bez udziału
światła. Odbiera wodor innym związkom chemicznym, np. wodzie.
W wyniku reakcji fluoru z wodorem powstaje fluorowodor:
F2 + H2 ---> 2HF.
MoŜna go rownieŜ otrzymać przez działanie stęŜonym kwasem siarkowym (VI) na fluorek wapnia:
CaF2 + H2SO4 ---> CaSO4 + H2F2.
Ze względu na bardzo duŜą roŜnicę elektroujemności cząsteczki fluorowodoru wytwarzają bardzo silne wiązania
wodorowe, skutkiem czego wytwarza się struktura liniowa: .
Skutek jest taki, iŜ forma dimeru H2F2 jest trwalsza od formy monomeru. Czasami tworzone są długie łańcuchy
polimeru (HF)n o budowie liniowej. Kwas fluorowodorowy w stosunkowo małym stopniu ulega dysocjacji i jest
kwasem słabym, choć agresywnym. Jego sole – fluorki – cechują się zroŜnicowaną rozpuszczalnością w wodzie:
rozpuszczalne są fluorki litowcow i srebra, natomiast reszta jest nierozpuszczalna. Wybitnym przykładem złej
rozpuszczalności fluorkow jest fluorek wapnia CaF2, wchodzący w skład szkliwa zębowego. Fluorowodor
niszczy substancje organiczne, przez co stosowany jest jako środek dezynfekcyjny i bakteriobojczy. Wykazuje
odporność na działanie czynnikow utleniających. UŜywany jest do trawienia szkła:
Na2SiO3 + 6HF ---> 2NaF + SiF4 + 3H2O,
CaSiO3 + 6HF ---> CaF2 + SiF4 + 3H2O.
Powstały w powyŜszych reakcjach fluorek krzemu reaguje rownieŜ z samym kwasem fluorowodorowym:
2 SiF4 + 4HF ---> 2H2[SiF6].
Produkt ostatniej reakcji to kwas fluorokrzemowy; jest bardzo mocnym kwasem i dobrze rozpuszcza się w
wodzie.
CHLOR
Chlor występuje w przyrodzie w postaci minerałow, np.: NaCl – halit, KCl – sylwin. W warunkach
laboratoryjnych moŜna go otrzymać przez elektrolizę chlorkow bądź odpowiednią reakcją, np. działanie kwasem
solnym na braunsztyn:
4HCl + MnO2 ---> MnCl2 + Cl2 + 2H2O.
W warunkach normalnych chlor jest Ŝołtozielonym gazem o duszącej woni. Nazwa jego wzięła się od barwy,
gdyŜ gr. chloros oznacza zielony. Chlor działa draŜniąco na błony śluzowe człowieka, podczas pierwszej wojny
światowej uŜywany był jako gaz bojowy. Jest wielokrotnie cięŜszy od powietrza, dzięki czemu „pełznie” po
podłoŜu. Rozpuszcza się w wodzie i wchodzi z nią w reakcję dysproporcjonowania:
H2O + Cl2 ---> HCl + HClO.
Jest pierwiastkiem bardzo czynnym chemicznie i silnym utleniaczem, a bezpośrednie reakcje z chlorem mogą
przebiegać bardzo gwałtownie. Jest mniej aktywny od fluoru, gdyŜ reakcja z wodorem nie przebiega w
ciemności:
H2 + Cl2 ---> 2HCl.
Natomiast przy dostępie światła reakcja przebiega bardzo gwałtownie, gdyŜ powstałe pod wpływem światła
rodniki natychmiast dąŜą do sparowania elektronow walencyjnych. Działa utleniająco na substancje organiczne,
stosowany jest do bielenia tkanin i papieru. Nadmiaru jego moŜna się pozbyć za pomocą „antychloru”, czyli
tiosiarczanu sodowego:
Na2S2O3 + 4Cl2 + 10NaOH --> 2Na2SO4 +8NaCl + H2O.
Created by Neevia Document Converter trial version http://www.neevia.com
61
1) W wyniku łączenia się chloru z wodorem otrzymujemy chlorowodor HCl. Nie dysponując powyŜszymi
odczynnikami moŜna go otrzymać np. działając mocnym kwasem na chlorek:
2NaCl + H2SO4 ---> Na2SO4 + 2HCl,
albo przez hydrolizę fluorku fosforu (III):
PCl3 + 3H2O ---> H3PO3 + 3HCl.
Chlorowodor jest gazem bezbarwnym, cięŜszym od powietrza. Rozpuszcza się w wodzie i dysocjuje, dzięki
czemu roztwor przewodzi prąd elektryczny. Łączy się z wodą egzotermicznie w wyniku automatycznej
hydratacji. Jest mocnym kwasem , zdysocjowanym niemal stuprocentowo. Jest związkiem trwałym, a rozkład na
pierwiastki stanowi problem. Wchodzi w skład wody krolewskiej wraz z kwasem azotowym (V). Kwas
chlorowodorowy jest kwasem jednozasadowym, dlatego tworzy tylko jeden szereg soli obojętnych. Większość
chlorkow jest dobrze rozpuszczalna w wodzie, z wyjątkiem AgCl, PbCl2 oraz Hg2Cl2. Pierwszy z nich, chlorek
srebra AgCl, wchodzi w postaci kompleksu z amoniakiem i cyjanowodorem w skład materiałow światłoczułych
na kliszach fotograficznych.
2) Chlor tworzy cały szereg tlenkow, co spowodowane jest mnogością stopni utlenienia. Są to: Cl2O, Cl2O3,
ClO2, Cl2O5, Cl2O6 (dimer ClO3) oraz Cl2O7. Jak widać, chlor tworzy związki zarowno na parzystych, jak i na
nieparzystych stopniach utlenienia. Tlenki chloru są związkami nietrwałymi, są nośnikami tlenu i mają silne
właściwości utleniające. W reakcjach z wodą tworzą cały szereg tlenowych kwasow chlorowych:
3) HClO (kwas podchlorowy, kwas chlorowy (I)) jest związkiem nietrwałym, występuje wyłącznie w duŜym
rozcieńczeniu. Powstaje w wyniku reakcji dysproporcjonowania chloru w wodzie:
H2O + Cl2 ---> HCl + HClO.
Jego sole, podchloryny, uŜywane były jako środki bielące.
4) HClO2 (kwas chlorowy (III)) jest nietrwałym związkiem, słabym kwasem, ale za to silnym utleniaczem.
5) HClO3 (kwas chlorowy (V)) jest w porownaniu z poprzednimi trwały. RownieŜ posiada właściwości
utleniające
6) HClO4 (kwas chlorowy (VII)) jest najtrwalszym i najmocniejszym kwasem z grupy. Jest bardzo silnym
utleniaczem, a aniony chloranowe (VII) rozkładają się wybuchowo.
Moc kwasow chlorowych rośnie wraz ze wzrostem stopnia utlenienia, czyli kwas chlorowy (VII) jest mocniejszy
od chlorowego (V), ten z kolei jest mocniejszy od chlorowego (III), zaś chlorowy (I) jest najsłabszy.
BROM
W warunkach normalnych brom jest ciemnobrunatną cieczą. Słabo rozpuszcza się w wodzie, a powstała woda
bromowa ma kolor jasnobrązowy; o jej znaczeniu dla wykrywania nienasyconego charakteru związkow
organicznych powiemy poźniej. Brom rozpuszcza się dobrze w rozpuszczalnikach organicznych. Brom działa
niszcząco na substancje organiczne (choć wolniej niŜ chlor) i szkodliwie na organizmy Ŝywe; pary bromu mają
silnie duszący zapach.
1) Podczas łączenia się bromu z wodorem na świetle powstaje bromowodor. Jest to gaz cięŜszy od powietrza, a
mocnym zapachu. Rozpuszcza się w wodzie i daje kwas bromowodorowy, silniejszy od chlorowodorowego.
Bromki są związkami barwnymi, o kolorze jasnoŜołtym bądź jasnobeŜowym.
2) Bromek srebra AgBr o barwie jasnoŜołtej stanowi element masy światłoczułej na kliszach fotograficznych.
3) Podobnie jak chlor, rownieŜ brom tworzy kwasy tlenowe: kwas bromowy (I) HBrO i kwas bromowy (III)
HBrO3. Są to związki nietrwałe, o charakterze utleniającym słabszym od kwasow chlorowych. Ze względu na
rozkład przechowuje się je w formie 50% roztworow.
JOD
W warunkach normalnych jod jest ciałem stałym, występującym w postaci szarych metalicznych łusek. Bardzo
łatwo sublimuje, a jego pary są fiołkowe. Słabo rozpuszcza się w wodzie, rozpuszczalność tą podnosi obecność
anionow jodkowych, gdyŜ tworzone są wowczas jony trojjodkowe:
J2 + J- ---> J3
-.
Roztwor przyjmuje wowczas barwę ciemnobrunatną. Jod stosunkowo dobrze rozpuszcza się w
rozpuszczalnikach organicznych, np. 3% roztwor jodu z dodatkiem jodku potasu KI w etanolu to jodyna,
uŜywana niegdyś powszechnie do dezynfekcji ran. Spośrod fluorowcow jest najmniej aktywny, posiada
największy potencjał normalny. Ze skrobią tworzy barwę ciemnogranatową, przez co stosowany jest do jej
wykrywania, np. margarynę poddaje się probie z jodem, czy nie zawiera nawet niewielkich ilości skrobi. W
analizie chemicznej stosowany jest podczas jodometrii, opartej na reakcjach redox, np.
Cr2O7
2- + 6J- + 14H+ ---> 2Cr3+ + 3J2 + 3H2O.
1) Przez syntezę jodu z wodorem przy uŜyciu nakładu energetycznego bądź przez reakcję jodkow z mocnymi
kwasami moŜna otrzymać jodowodor. Jest to gaz cięŜszy od powietrza o przenikliwym zapachu. Rozpuszcza się
w wodzie i daje kwas jodowodorowy. Powstałe w roztworze jony jodkowe I- są czynnikiem redukującym. Jodki
są na ogoł solami rozpuszczalnymi, jednak np. AgI, PbI2 i Hg2I2 to związki bardzo trudno rozpuszczalne.
Dodając do roztworu amoniaku nie moŜna rozpuścić jodkow przez ukompleksowienie, tak jak to wychodzi z
Created by Neevia Document Converter trial version http://www.neevia.com
62
nierozpuszczalnymi chlorkami. Wiele jodkow to substancje barwne: AgI jest Ŝołty, HgI2 – ceglastoczerwony,
zaś Hg2I2 – szarozielony.
2) Podobnie jak poprzednicy, jod tworzy kwasy tlenowe. Kwas jodowy (I) HIO jest kwasem nietrwałym i
słabszym od sąsiadow.
3) Kwas jodowy (V) HJO3 jest dosyć silnym utleniaczem i relatywnie trwałym związkiem, gdyŜ moŜna go
wydzielić z roztworu w postaci kryształow.
MIEDŹ
Miedź jest metalem połoŜonym w grupie I B lub 11 i w 4 okresie. Rozpoczyna grupę pierwiastkow zwanych
miedziowcami, do ktorej naleŜy rownieŜ srebro i złoto. Znajduje się w grupie pobocznej, w bloku
energetycznym d. Powinna posiadać konfigurację nd9(n-1)s2, jednak w wyniku promocji wszystkie miedziowce
zyskują konfigurację nd10(n-1)s1. Atom miedzi posiada zatem jeden niesparowany elektron, co wskazywałoby na
jej jednowartościowość. Istnieje teoretyczna moŜliwość wzbudzenia atomu i przeniesienia elektronow na orbital
p; dzięki temu miedź moŜe być dwu-, a w rzadkich przypadkach nawet trojwartościowa. Elektroujemność tego
pierwiastka wynosi X=1,9 w skali Paulinga, przez co niewiele roŜni się od wodoru. NaleŜałoby spodziewać się
amfoteryczności i rzeczywiście ona występuje. Na tym przykładzie potwierdza się prawidłowość, iŜ pierwiastki
amfoteryczne leŜą na umownej przekątnej układu okresowego. W atomie miedzi odległości pomiędzy jądrem a
elektronami walencyjnymi nie są zbyt duŜe. Miedź jest bardzo dobrym przewodnikiem ciepła i elektryczności –
posiada drugi w kolejności najmniejszy opor właściwy, dzięki czemu stosuje się ją w przemyśle
elektrotechnicznym do produkcji przewodow (najmniejszy opor właściwy posiada srebro, ktorego rzadkość i
wysoka cena wyklucza podobne zastosowanie). Jest bardzo ciągliwa i kowalna, co umoŜliwia obrobkę
plastyczną. Wprowadzona w płomień palnika, zabarwia go na kolor zielony.
Cechuje się bardzo niewielką aktywnością chemiczną. Podobnie zachowuje się reszta miedziowcow, przy czym
szlachetność rośnie w doł – świadczą o tym dodatnie i rosnące wartości potencjałow normalnych. Miedź reaguje
z silnymi kwasami utleniającymi, doprowadzając do redukcji atomu centralnego, np.:
Cu + 4HNO3 -> Cu(NO3)2 + 2NO2 + 2H2O
(>15%) niebieski rudobrązowy
Cu + 8HNO3 -> 3Cu(NO3)2 + 2NO + H2O
(<15%) niebieski bezbarwny
Cu + HNO3 -X-> nie zachodzi
(<1%)
Cu + 2H2SO4 -> CuSO4 + SO2 + 2H2O
(stęŜony i gorący).
Natomiast Ŝaden z miedziowcow nie reaguje z kwasami beztlenowymi. Miedź tworzy związki, w ktorych
występuje na stopniu utlenienia +1 oraz +2, przy czym te drugie są trwalsze. Ze względu na amfoteryczny
charakter tlenki i wodorotlenki miedzi są trudno rozpuszczalne. Wodorotlenek miedzi (I) jest silniejszą zasadą
niŜ miedzi (II).
W przyrodzie miedź wstępuje w postaci minerałow siarczkowych oraz, w małych ilościach, tlenowych, np.:
Cu2O – kupryt, CuO – melakonit, Cu2S – chalkozyn, CuS – kowelin, CuS . FeS (CuFeS2) – chalkopiryt,
Cu(OH)2 . CuCO3 - malachit, CuSO4 . 5H2O - kamień siny. W procesie przetwarzania rud miedzi wpierw
siarczki przeprowadza się w tlenki, np.:
Cu2S + 2O2 ---> 2CuO + SO2,
a następnie redukuje koksem:
CuO + C ---> Cu + CO.
Wyroby miedziane pokrywają się warstwą czerwonego tlenku miedzi (I) Cu2O, dlatego mają charakterystyczny
odcień. W wyniku łączenia się miedzi z siarką, występującą w niewielkich ilościach w powietrzu, przyjmują one
barwę czarno-brunatną. Nie ulega działaniu suchego tlenu, natomiast w wilgotnym powietrzu pokrywa się
zasadowym węglanem miedzi Cu(OH)2 . CuCO3 = [Cu(OH)]2CO3, czyli patyną – z tego powodu miedziane
dachy z czasem zielenieją. Bezwodne sole miedzi są bezbarwne, natomiast uwodnione mają kolor niebieski lub
niebieskozielony. Solidarnie z resztą związkow miedzi, sole ulegają działaniu kwasow utleniających. Miedź ma
szerokie zastosowanie: wymieniono juŜ produkcję przewodow elektrycznych, poza tym blachy miedziowe
uŜywane są w roŜnych konstrukcjach, wyrabia się z niej kotły parowe, aparaty destylacyjne, kryje się nią dachy.
Miedź wchodzi w skład licznych stopow, np. z cyną tworzy brązy, z cynkiem – mosiądze, z glinem – brąz
aluminiowy (brązal); stop uŜywany do bicia monet to połączenie miedzi i niklu oraz glinu i cynku.
Tlenek miedzi (I) występuje w przyrodzie jako minerał kupryt. Tlenek ma barwę ceglastoczerwoną. Powstaje w
wyniku proby Trommera, stosowanej do wykrywania grupy aldehydowej – zawarty w odczynniku Fehlinga
niebieski wodorotlenek miedzi (II) redukuje się do czerwonego tlenku miedzi (I), a aldehyd utlenia się do kwasu
karboksylowego. Dzięki tworzeniu połączeń kompleksowych nierozpuszczalne związki miedzi rozpuszczają się
Created by Neevia Document Converter trial version http://www.neevia.com
63
w nadmiarze amoniaku, natomiast ze stęŜonym kwasem solnym łączą się koordynacyjnie. Szereg soli kwasow
beztlenowych miedzi (I) jest trwały (np.: CuCl, CuCN, CuSCN), natomiast sole kwasow tlenowych w
roztworach wodnych dysproporcjonują z wydzieleniem czystej miedzi oraz soli miedzi (II).
Tlenek miedzi (II) ma barwę czarną, nie rozpuszcza się w wodzie ze względu na amfoteryczny charakter. Jest
utleniaczem i redukuje inne substancje. Bezwodne sole są białe, zaś hydraty przyjmują barwę niebieską bądź
zielonkawą (np. CuSO4 . 5H2O). Większość soli miedzi jest rozpuszczalna w wodzie – CuCl2, CuBr, CuI2 itd.
Sole miedzi moŜna rozpuszczać w stęŜonym kwasie solnym, roztworze amoniaku czy mocnych wodorotlenkach.
Miedź w postaci kationow Cu2+ jest mikroelementem, stanowi składnik enzymow oddechowych u wielu grup
zwierząt oraz wchodzi w skład enzymow. W nadmiernych ilościach jej sole wywierają szkodliwe działanie na
organizmy Ŝywe za względu na denaturację białek – szkodliwe jony moŜna wykryć a pomocą amoniaku
(tworzenie kolorowych kompleksow – patrz tekst o amoniaku).
SREBRO
Srebro występuje w samorodkach, lecz procentowy udział molowy metalu jest niewielki. Czyste srebro
wydobywa się przez ługowania amoniakiem, amalgamację rtęcią bądź metodą elektrolityczną. Jest to metal o
barwie srebrzystej, bardzo kowalny i ciągliwy, świetny przewodnik ciepła i elektryczności, dzięki czemu
uŜywany jest do wyrobu specjalistycznych obwodow elektrycznych. Jest nieaktywny chemicznie i odporny na
działanie tlenu cząsteczkowego, natomiast z ozonem reaguje juŜ w temperaturze pokojowej. Reakcje srebra z
fluorowcami zachodzą bardzo powoli. Natomiast łatwo następuje łączenie z siarką z wydzieleniem
nierozpuszczalnego czarnego siarczku srebra AgS – jest to przyczyną czernienia wyrobow srebrnych, np.
sztućcow. Ze względu na duŜy dodatni potencjał srebro naleŜy do silnych utleniaczy i reaguje jedynie z silnymi
kwasami utleniającymi. Trwałością odznaczają się związki srebra za +1 stopniu utlenienia. Srebro tworzy
połączenia kompleksowe z amoniakiem i cyjanowodorem, w ktorych występuje z liczbą koordynacyjną 2.
Czyste srebro jest miękkie, dlatego zazwyczaj do uŜytku wprowadza się jego stopy z miedzią. Srebro tworzy
stopy z wieloma metalami, z wyjątkiem Ŝelaza, manganu i chromu. Stopy z duŜa zawartością srebra uŜywane są
do produkcji biŜuterii; zawartość srebra w wyrobie oznacza się przez podanie proby: I – 916, II – 875, III – 800
(ułamki masowe w promilach). Dla porownania stopy złota stosowane w dentystyce maja proby: I – 750, II –
575, III – 573. Tradycyjnie odsetek złota w wyrobie podaje się w karatach: 24 karaty odpowiadają 100%, zaś
zazwyczaj uŜywa się złota 14-karatowego, co odpowiada 58,3%. Karat to rownieŜ uŜywana w jubilerstwie
jednostka masy odpowiadająca 2,5g. Srebro moŜna odroŜnić od złota poprzez reakcję z kwasem azotowym (V) –
w przypadku srebra powstanie niebieska plama. Srebro jest metalem szlachetnym szkodliwym dla organizmow
Ŝywych (np. lapis AgNO3 zwany jest kamieniem piekielnym). Ze względu na bakteriobojcze działanie jonow
Ag+ srebro stanowi składnik środkow dezynfekcyjnych. Ze względu na połysk było wykorzystywane do
produkcji luster.
Wiele związkow srebra jest barwnych.
1) Tlenek srebra (I) Ag2O jest czerwonobrunatnym proszkiem. Rozpuszcza się w wodzie i w wyniku hydrolizy
nadaje jej odczyn zasadowy.
2) Azotan AgNO3 i chloran (V) AgClO3 srebra są nośnikami tlenu, zaś fluorek AgF ma właściwości
wybuchowe.
3) Tiosiarczan srebra Ag2S2O3 w reakcji z wodą i mocniejszym kwasem daje trudno rozpuszczalny czarny osad
siarczku srebra oraz kwas siarkowy (VI),np.:
2Ag2S2O3 + 2HCl + 2H2O ---> Ag2S + 2AgCl + 2H2SO4.
4) Węglan Ag2CO3 i fosforan (V) Ag3PO4 srebra maja barwę Ŝołtą, a chromian (VI) Ag2CrO4 – ciemnobrunatną.
CYNK
Cynk jest metalem połoŜonym w grupie II B lub 12 i w 4 okresie. Ze względu na rozpoczynania grupy, nazwano
ją cynkowcami (cynk, kadm, rtęć). Cynk posiada konfigurację elektronow walencyjnych 3d104s2 i występuje
wyłącznie na +2 stopniu utlenienia. Posiada niski potencjał standardowy i jest bardzo silnym reduktorem:
4Zn + 10HNO3 -> 4Zn(NO3)2 + NH4NO3 + 4H2O.
W powyŜszej reakcji cynk powoduje zmianę stopnia utlenienia azotu z +5 na –3, a więc o 8 jednostek, a moŜna
to powiedzieć o niewielu reakcjach. Jest metalem amfoterycznym, a produkt reakcji z kwasami zaleŜy od
warunkow środowiska reakcji:
Zn + 4HNO3 -> Zn(NO3)2 + 2NO2 + 2H2O,
Zn + 8HNO3 -> 3Zn(NO3)2 + 2NO + H2O.
Created by Neevia Document Converter trial version http://www.neevia.com
64
Reaguje z mocnymi zasadami, np.:
ZnO + 2NaOH + H2O ---> Na2Zn(OH)4 ---T---> Na2ZnO2 + 2H2O;
czterohydroksocynkan sodu
[Zn(OH)4]2- ----2H
2
O---> [ZnO2]2-.
MoŜe tworzyć zasadowe hydroksysole oraz sole obojętne. Sole cynku w roztworach wodnych wskutek hydrolizy
kationowej wykazują odczyn kwasowy, np. ZnCl2.Do trudno rozpuszczalnych soli cynku zaliczmy: ZnS, ZnCO3,
Zn(PO4)3 i ZnF2. Wodorotlenek Zn(OH)2 rownieŜ posiada charakter amfoteryczny – nie rozpuszcza się w
wodzie, wytrącając się w formie galaretowatego osadu. Rozpuszcza się w nadmiarze mocnych zasad, słabo zaś
w amoniaku, z ktorym tworzy połączenie kompleksowe [Zn(NH3)4]2+.
Cynk występuje w przyrodzie w postaci minerałow, np.: ZnS – sfaleryt lub wurtzyt, ZnCO3 – galman. Wszystkie
rudy doprowadza się do formy tlenkowej, ktorą następnie redukuje się koksem lub tlenkiem węgla. Tak powstały
cynk poddaje się elektrolitycznej rafinacji (elektrorafinacji). Czysty metal ma niebieskawy połysk, jest ciągliwy i
kowalny oraz łatwo topliwy (ok. 400oC). Ogrzany do 100oC staje się kruchy. Stosowany jest do produkcji blachy
cynkowej oraz do cynkowania blach stalowych – jest to ochrona przeciwkorozyjna czynna (aktywna), gdyŜ w
razie uszkodzenia w powstałym ogniwie galwanicznym cynk utlenia się aŜ do całkowitego rozpuszczenia. Cynk
jest rownieŜ składnikiem wielu stopow: z miedzią tworzy mosiądze, z miedzią i srebrem – tzw. nowe srebro,
uŜywane do produkcji biŜuterii i sztućcow, stop miedzi z dodatkiem do 10% cynku daje tombak imitujący złoto,
stopiony z glinem daje znal.
1) Tlenek glinu ZnO jest jedną z form występowania tego pierwiastka. Czysty tlenek uŜywany jest jako biała
farba o duŜych zdolnościach kryjących, znana pod nazwą bieli cynkowej. Cynk jako mikroelement niezbędny
jest człowiekowi w procesie regeneracji naskorka; dzięki dodatkowym właściwościom antyseptycznym idealnie
nadaje się do produkcji lekow naskornych, tzw. maści cynkowej. W kosmetyce uŜywany jest jako składnik
pudrow. W przemyśle chemicznym katalizuje reakcję syntezy metanolu z gazu syntezowego:
CO + H2 ---ZnO---> CH3OH.
2) Siarczek cynku ZnS to występująca jako minerał blenda cynkowa. W przeciwieństwie do większości
siarczkow, ktorych barwa jest czarna lub brązowa, siarczek cynku jest biały. Zmieszany z siarczanem (VI) baru
daje białą farbę znaną jako litopan.
3) Siarczan (VI) cynku ZnSO4 jest solą dobrze rozpuszczalną w wodzie. Roztwor ZnSO4 słuŜy do impregnacji
drewna – zmniejsza się przez to jego palność, a dzięki antyseptycznym właściwościom jonow Zn2+ zabezpiecza
je przed rozkładem.
CHROM
Chrom połoŜony jest w grupie VI B lub 6 i w 4 okresie. Rozpoczyna grupę pierwiastkow zwanych
chromowcami, do ktorej naleŜy rownieŜ molibden i wanad. Znajduje się w grupie pobocznej, w bloku
energetycznym d. Z połoŜenia w układzie okresowym wynika konfiguracja nd4(n-1)s2, jednak w wyniku
promocji wszystkie chrom i molibden zyskują konfigurację nd5(n-1)s2 - atom chromu posiada zatem jeden
niesparowany elektron. Ogolnie chromowce są metalami bardzo trwałym i trudno topliwymi, wykazują
odporność na czynniki atmosferyczne i silne środki utleniające. Ulegają pasywacji poddane działaniu stęŜonych
kwasow, a nie wskutek ujemnego potencjału normalnego. Chrom występuje na kilku stopniach utlenienia (+2,
+3, +6), z czego wynikają roŜnice we właściwościach. Ze względu na duŜą liczbę elektronow walencyjnych
związki chromu są barwne: Cr2+ jest niebieskie w roztworze wodnym, Cr3+ - niebieskie, CrO4
2- - Ŝołte, a Cr2O7
2-
- pomarańczowe.
1) Tlenek chromu (III) Cr2O3 ma barwę zieloną. Jest związkiem nierozpuszczalnym w wodzie, wykazującym
charakter amfoteryczny. Tworzy połączenia koordynacyjne, w ktore wchodzi z liczbą koordynacyjną +6, np.:
Na3[Cr(OH)6] ----3H
2
O---> Na3CrO3.
W wyniku słabej hydratacji dole chromu przyjmują czasem barwę fioletową.
2) Tlenek chromu (VI) CrO3 posiada sam w sobie barwę czerwoną. Jest substancją bardzo higroskopijną.
Reaguje z wodą, dając kwas chromowy (VI):
CrO3 + H2O ---> H2CrO4.
Roztwor przyjmuje wowczas barwę od Ŝołtopomarańczowej do czerwonej, gdyŜ wytworzone jony
chromianowe (VI) bardzo łatwo ulegają dimeryzacji do jonow dwuchromianowych (VI); w roztworze wytwarza
się stan rownowagi dynamicznej pomiędzy dwiema formami jonow:
2CrO4
2- + H+ <====> Cr2O7
2- + OH-.
Ŝołty pomarańczowoczerwony
3) Chromiany (VI) i dwuchromiany (VI) to silne utleniacze, redukujące się podczas reakcji do zielonych Cr3+.
Dwuchromiany utleniają: S(+4)->S(+6), S(-2)->S(0), a w środowisku silnie kwaśnym: Cl(-1)->Cl(0), Br(-1)-
>Br(0) oraz I(-1)->I(0). Dwuchromiany rozpuszczają się w wodzie lepiej od chromianow. Rozpuszczalne są
chromiany litowcow, amonu, magnezu, wapnia i strontu.
Created by Neevia Document Converter trial version http://www.neevia.com
65
MANGAN
Mangan połoŜony jest w grupie VII B lub 7 i w 4 okresie. Rozpoczyna grupę pierwiastkow zwanych
manganowcami, do ktorej naleŜy rownieŜ technet i ren. Znajduje się w grupie pobocznej, w bloku
energetycznym d. Posiada konfigurację elektronow walencyjnych 4s23d5 oraz wolny do zagospodarowania
orbital p. Pozwala to manganowi na przyjmowanie wartościowości od II do VII, przy czym najtrwalsze są
związki o wartościowości II. Mangan jest metalem aktywnych chemicznie, łatwo reaguje z kwasami i
rozpuszcza się w nich. Ulega działaniu chlorowcow juŜ w zwykłej temperaturze, dając halogenki manganu
(MnCl2, MnBr2). Łączy się chętnie z tlenem i daje wiele tlenkow: MnO (zasadowy), Mn2O3 (amfoteryczny),
Mn2O7 (kwasowy).
1) Tlenek manganu (II) MnO jest w warunkach pokojowych szarozielonym proszkiem, bardzo słabo
rozpuszczalnym w wodzie. Związek ten łatwo się utlenia, podobnie jak Mn(OH)2 utlenia się na powietrzu i
powstaje wodorotlenek manganu (IV) MnO(OH)2. Jon Mn2+ tworzy połączenia kompleksowe, w ktore wchodzi
z liczbą koordynacyjną 6, np. jon sześcioamionomanganowy (II) [Mn(NH3)6]2+. W roztworze wodnym jon Mn2+
jest prawie bezbarwny, moŜe lekko roŜowy, dlatego trwały zanik zabarwienia utleniających związkow manganu
oznacza koniec reakcji rodox.
2) Związki manganu na +3 stopniu utlenienia są nietrwałe i szybko dysproporcjonują.
3) Tlenek manganu (IV) MnO2 jest czarnym proszkiem. Jest nierozpuszczalny w wodzie i wykazuje charakter
amfoteryczny. Ze względu na pośredni stopień utlenienia moŜe być zarowno utleniaczem jak i reduktorem.
Związki manganu na +4 stopniu utlenienia są trwałe w środowisku zasadowym.
4) Manganiany (VI) są trwałe w środowisku zasadowym.
5) Tlenek manganu (VII) Mn2O7 to w warunkach normalnych oleista ciecz. Związek ten jest tlenkiem
kwasowym. Istniejący teoretycznie kwas manganowy (VII) znany jest tylko w rozcieńczonych roztworach. Jon
manganowy (VII) jest w roztworze wodnym fioletowy. Manganiany (VII) to silne utleniacze, utleniające m. in.:
S(+4)->S(+6) i S(-2)->S(0). Procesy redox zaleŜą od pH środowiska: najsilniejsze właściwości utleniające
osiąga się w środowisku kwasowym (mangan przechodzi ze stopnia utlenienia +7 na +2, a fioletowy roztwor
staje się bezbarwny), słabsze w obojętnym (+7 na +4, powstaje czarny brunatny osad MnO2), zaś najsłabsze w
zasadowym (+7 na +6, roztwor staje się trawiastozielony). Manganiany są przenośnikami tlenu i mogą utleniać
wiele substancji, zaś wymieszane z materiałami palnymi (papier) powodują samozapłon.
śELAZO
W układzie okresowym Ŝelazo jest pierwiastkiem grupy VIII B lub 8, w okresie 4. Posiada konfigurację
elektronow walencyjnych 3d64s2. Tworzy związki na +2 i +3 stopniu utlenienia. Przykładem tego są tlenki:
Ŝelaza (II) FeO, nieco zasadowy oraz Ŝelaza (III) Fe2O3, bardziej amfoteryczny. Rzadko spotykane i nietrwałe
są związki Ŝelaza na stopniu +6 – są to Ŝelaziany o pozornie kwasowym charakterze. Rozpuszczone sole Ŝelaza
(II) hydrolizują kationowo i są prawie bezbarwne, bardziej stęŜone mają zabarwienie zielonkawoŜołte. Z kolei
sole Ŝelaza (III) rozpuszczone w wodzie wskutek hydrolizy tworzą po pewnym czasie czerwonobrunatny osad.
W wyniku reakcji Ŝelaza z kwasami powstają sole Ŝelaza (II), co wynika z szeregu napięciowego (roŜnica
potencjałow wodoru i Ŝelaza (III) jest niewystarczająca do wydzielenia wodoru, a potrzeba 0,20); pozostawiony
na powietrzu roztwor powoli brunatnieje wskutek przemiany jonow Fe2+ w Fe3+. Wobec stęŜonych kwasow
utleniających Ŝelazo pasywuje, czyli pokrywa się ochronną warstwą tlenku; czasami jest to zjawisko poŜądane –
pozwala to przykładowo na przewoz stęŜonego kwasu siarkowego (VI) w stalowych cysternach. śelazo
wystawione na działanie czynnikow atmosferycznych lub zanurzone w wodzie koroduje. Korozji sprzyja
nasycenie środowiska reakcji tlenem, rozpuszczenie w nim soli oraz zakwaszenie, podczas gdy środowisko
zasadowe hamuje postęp korozji. śelazo, nikiel i kobalt posiadają właściwości ferromagnetyczne, jednak w
przypadku Ŝelaza są one największe.
W przyrodzie Ŝelazo występuje w postaci rud, np.: Fe2O3 – hematyt, FeO . Fe2O3 – magnetyt, FeS2 – piryt lub
markosyt, FeCO3 – syderyt, Fe(OH)3 – limonit. Rudę Ŝelazową poddaje się obrobce w wielkim piecu: w wyniku
redukcji dwutlenku węgla rozŜarzonym koksem powstaje tlenek węgla:
C + CO2 --> 2CO,
ktory stopniowo redukuje Ŝelazo – wpierw do tlenkow:
3Fe2O3 + CO --> 2Fe3O4 + CO2,
Fe3O4 + CO --> 3FeO + CO2,
a następnie do czystego metalu:
FeO + CO --> Fe + CO2.
W procesie wielkopiecowym następuje wytapianie surowki, ktorą następnie wzbogaca się i powstaje stal. Istotny
fakt, to iŜ czyste Ŝelazo jest kruche i raczej nie nadaje się do praktycznego zastosowania. Dodatek 0,5 - 1,5%
węgla tak dalece zmienia właściwości stali, szczegolnej jej wytrzymałość mechaniczną, twardość i kruchość, iŜ
moŜe być wszechstronnie wykorzystywana w konstrukcjach technicznych. Dalsze dodawanie węgla do 2,5 - 3%
powoduje powstanie kruchego Ŝeliwa.
Domieszki hematytu w naturalnej krzemionce powodują, iŜ najtańsze szkło jest Ŝołtawe bądź zielonkawe (tzw.
szkło luxver, np. w starych przychodniach). Neutralizację tego koloru uzyskuje się przez dodanie selenu,
barwiącego szkło na roŜowo lub czerwono – rubinowo. Wytworzone domieszkami barwy znoszą się i szkło staje
się przezroczyste.
Wyszukiwarka
Podobne podstrony:
Organizacja [Automatycznie zapisany]
kinetyka, studia, ochrona środowiska UJ, chemia ogólna i nieorganiczna, wyrównawcze
instrukcja - HYDROLIZA SOLI, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, laboratorium
pHmetr-instrukcja obsługi, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, laboratorium
kationy, Polibuda, II semestr, fizyka, FIZA, lab, Chemia laborki, chemia ogolna nie organiczna
Cząsteczka (VB), CHEMIA, semestr 1, chemia ogólna, wykłady
Ćw.1 Wybrane reakcje chemiczne przebiegające w roztworach wodnych ćwiczenie 1, Chemia ogólna i żywno
pato 1 10 (Automatycznie zapisany)
Chemia ogolna wyklady 5 6 2012 Nieznany
CHEMIA OGÓLNA
Chemia ogolna zagadnienia na zaliczenie wykla
Jasiorski, chemia ogólna, Opracowane zagadninia na kolowium
Wykład 3. Reakcje chemiczne, chemia, CHEMIA OGÓLNA -Walkowiak- (WPC 1002w) DOC
instrukcja - CHEMIA ORGANICZNA II, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, laboratori
rownowagi1, studia, ochrona środowiska UJ, chemia ogólna i nieorganiczna, wyrównawcze
przemiany jadrowe, Nauka, CHEMIA, Szkoła, Chemia ogólna
download, chemia ogólna
więcej podobnych podstron