Podstawowe pojęcia i definicje procesu katalitycznego:
Kataliza polega na zwiększeniu szybkości reakcji chemicznej i/lub skierowaniu reakcji na jedną z kilku możliwych termodynamicznie dróg prowadzących do różnych produktów, w obecności niewielkich ilości substancji zwanych katalizatorami.
Katalizator to substancja oddziaływująca chemicznie z substratami reakcji, tworzy przy tym nietrwałe produkty przejściowe. Katalizatory nie są jednak zużywane w trakcie reakcji i nie występują w ogólnym równaniu stechiometrycznym. Katalizator nie zmienia przy tym położenia równowagi chemicznej, wpływa jedynie na szybkość dochodzenia układu do tego stanu.
Miarą działania przyspieszającego daną reakcję chemiczną jest aktywność katalizatora, a działania ukierunkowującego - jego selektywność. Aktywność katalizatora to różnica między szybkościami reakcji w obecności katalizatora i bez niego. Aktywność katalizatora charakteryzuje nie samą substancję działającą jako katalizator, ale układ katalizator - reagenty.
Katalizatorami mogą być pierwiastki lub związki chemiczne, występujące w różnych stanach skupienia. Role katalizatorów w organizmach żywych pełnia enzymy. Spośród pierwiastków chemicznych najczęściej stosowanymi katalizatorami są metale grup VIII, VIA i VIIA, a spośród związków chemicznych tlenki i siarczki metali przejściowych, tlenki grup III i IV oraz kompleksy metaloorganiczne.
Katalizatory mogą być homogeniczne (tworzą jedna fazę z reagentami) lub heterogeniczne (reakcja zachodzi na granicy faz: katalizator - reagenty). Homogeniczne reakcje katalityczne zachodzą na drodze cyklicznej tj. po drodze składającej się z kilku następczych lub następczo - równoległych etapów, w wyniku, których jeden ze składników (katalizator), ulegający zużyciu w pierwszym etapie, jest regenerowany w ostatnim etapie. Pozostałe składniki mieszaniny reakcyjnej ulegają natomiast przemianie w produkty. W przypadku katalizy heterogenicznej katalizator jest zwykle ciałem stałym, reakcja przebiega zaś pomiędzy substancjami gazowymi (syntezy amoniaku, dwutlenku siarki), rzadziej ciekłymi (zmydlanie tłuszczów katalizowane tlenkami metali). w przypadku rozdrobnienia koloidalnego katalizatora mówimy o katalizie mikroniejednorodnej (mikroheterogenicznej) (koloidalna platyna, enzymy).
W reakcjach homogenicznych związkami przejściowymi mogą być obojętne cząsteczki, jony, rodniki i atomy.
Katalizę jednofazową można rozpatrywać w zależności od:
Rodzaju fazy (roztwory, faza gazowa)
Rodzaju reakcji (kwasowo - zasadowa, utleniająco - redukująca)
Mechanizmu katalizy (jonowy, rodnikowy, cząsteczkowy)
Energia aktywacji procesu katalitycznego
![]()
to różnica energii potencjalnej aktywnego kompleksu i nieoddziaływujących ze sobą substratów. ![]()
reakcji kontaktowej odpowiada różnicy energii chemisorbowanych na katalizatorze substratów i kompleksu aktywnego utworzonego przez substraty wraz z katalizatorem. Doświadczalnie przez badanie zależności szybkości reakcji od temperatury wyznaczamy tzw. pozorną ![]()
(różnica miedzy energią potencjalną substratów w fazie gazowej, czyli niezaadsorbowanych i powierzchniowego kompleksu aktywnego. Do wyznaczenia rzeczywistej ![]()
należy dodatkowo znać ciepło adsorpcji(zależy od stopnia obsadzenia powierzchni)
Kataliza jest zjawiskiem kinetycznym: katalizator zmienia szybkość reakcji lub jej kierunek, nie wpływając na położenie równowagi chemicznej.
Kompleks aktywny jest nietrwałym stanem przejściowym miedzy substratami i produktami reakcji. Schemat takiej reakcji jest następujący:![]()
. Aby mogła zajść reakcja cząsteczki substratów muszą przekroczyć barierę energetyczną równą różnicy energii kompleksu aktywnego i energii substratów, czyli muszą osiągnąć![]()
.
Stwierdzono, że głównym czynnikiem odpowiedzialnym za wzrost szybkości reakcji katalitycznej jest zmniejszenie entalpii tworzenia kompleksu aktywnego ![]()
, czyli wielkości związanej z ![]()
.
Wielość ![]()
tej samej reakcji zależy od rodzaju katalizatora, czyli mechanizm reakcji może być różny dla różnych katalizatorów i będzie również różnić się od mechanizmu reakcji nie katalizowanej. Dzięki obecności katalizatora reakcja może przebiegać nową drogą, prowadzącą przez kilka etapów, w których powstają produkty przejściowe substratów z katalizatorem. Można wyróżnić następujące etapy:
![]()
![]()
W pierwszym etapie o energii aktywacji ![]()
najpierw powstaje kompleks aktywny, który później tworzy się produkt przejściowy XZ. W drugim etapie reaguje utworzony produkt przejściowy z substratem Y pokonując barierę energetyczną ![]()
i przechodzi w produkt końcowy Z z odtworzeniem katalizatora. Jeżeli ![]()
i ![]()
są niższe od energii aktywacji reakcji nie katalizowanej ![]()
, to szybkość reakcji ulegnie zwiększeniu, jest ona przy tym określona wielkością najwyższej bariery energetycznej na drodze reakcji.
Może istnieć również sytuacja, gdzie oddziaływanie reagujących substancji z katalizatorem może polegać na wytworzeniu jedynie kompleksu aktywnego:![]()
. Powstanie wiązań miedzy atomami katalizatora i atomami substratów oraz związana z tym deformacja wiązań istniejących w wyjściowych substratach prowadzi do zmniejszenia energii tworzenia kompleksu aktywnego z udziałem atomów katalizatora w porównaniu z kompleksem aktywnym powstającym w reakcji nie katalizowanej, czyli do zmniejszenia energii aktywacji reakcji.
Kataliza homogeniczna (kataliza kwasowo - zasadowa, autokataliza, kataliza enzymatyczna)
Kataliza kwasowo - zasadowa: katalizatorem jest kwas lub zasada w sensie, Bronsteda, czyli substancja zdolna do odszczepienia albo przyłączenia protonu. (Obecność jonów ![]()
![]()
). Przykłady: inwersja sacharozy w roztworze wodnym katalizowana przez ![]()
, rozkład nitroamidu(![]()
) w roztworze wodnym (katalizowany przez ![]()
), dekarboksylacja ketokwasów w obecności jonów metali np.![]()
, alkilowanie związków aromatycznych, przy użyciu ![]()
, reakcja Friedla - Craftsa.
Kataliza w roztworach wodnych może polegać na działaniu tylko jonów![]()
lub tylko![]()
. Wówczas szybkość tej reakcji jest proporcjonalna do stężenia katalizującego jonu. Mechanizm działania katalizatorów związany jest z przenoszeniem protonów. Jeden z substratów przyłącza proton kwasowego katalizatora albo oddaje proton katalizatorowi zasadowemu. Powstają, zatem produkty przejściowe, aż w końcu produkty właściwe i następuje regeneracja katalizatora.
Do katalizatorów kwasowo zasadowych nalezą tlenki metali grup głównych: ![]()
![]()
![]()
i ich związki np. glinokrzemiany, charakteryzują się one obecnością na ich powierzchni centrów o charakterze kwasów i zasad Bronsteda i Lewisa. Centra te zdolne są do reagowania z cząsteczkami związków organicznych (węglowodorów) i generowania jonów organicznych np. karbokationów. Podstawowymi reakcjami są tutaj: izomeryzacja węglowodorów, kraking węglowodorów, alkilowanie, polimeryzacja, dehydratacja
Autokataliza: szybkość reakcji jest zwykle na początku mała i dopiero po upływie pewnego czasu tzw. okresu indukcji zaczyna wyraźnie rosnąć. Krzywa ma zwykle kształt litery S, oznacza to, że w pewnym momencie szybkość reakcji osiąga maksimum, któremu odpowiada punkt przegięcia krzywej, później szybkość maleje w związku z wyczerpaniem się substratów. Reakcje autokatalizy są reakcjami pierwszego rzędu. Przykład: hydroliza estrów w obecności kwasów.
Kataliza enzymatyczna: enzymami (fermentami) nazywamy substancje katalizujące w żywych organizmach wszelkie reakcje biochemiczne, składające się na przemianę materii. Z chemicznego punktu widzenia są to proste białka (pesyna - katalizująca hydrolityczne rozszczepienie wiązań peptydowych w białkach) lub białka złożone, w których oprócz części białkowej występuje jeszcze grupa prostetyczna tzw. koenzym, jeśli można ją odwracalnie odszczepić od białka, które wówczas nazywane jest apoenzymem. Enzymy są bardzo selektywne - często katalizują pewną reakcję tylko jednej odmiany enencjomerycznej danego związku. Jest to związane z istnieniem centrów aktywnych - fragmentów polipeptydowych o specyficznej konformacji i mogących dlatego tworzyć związki przejściowe jedynie z cząsteczka o określonej budowie.
Reakcje mogą zachodzić według ogólnego schematu reakcji katalitycznych lub w obecności inhibitora, który wiąże się z tymi samymi centrami aktywnymi enzymu co substrat może występować dodatkowo reakcja hamowania konkurencyjnego reakcji katalitycznej.
Działanie enzymów jest uzależnione od pH środowiska, które wpływa na równowagi kwasowo - zasadowe pomiędzy różnymi formami amfoterycznymi białek. Wraz ze wzrostem temperatury szybkość reakcji enzymatycznych rośnie, ale często w temp 20 - 30![]()
maleje gwałtownie, a ogrzanie do temp. 40 - 60![]()
powoduje nieodwracalną utrate aktywności enzymu (denaturacja białek).
Efektywność katalityczna enzymów jest bardzo duża.
Struktura powierzchni i centra aktywne
Według koncepcji centrów aktywnych jedynie część powierzchni, czyli tzw. miejsca lub centra aktywne bierze udział w reakcji katalitycznej, przy czym ułamek powierzchni, na którym zachodzi reakcja, zależy nie tylko od rodzaju katalizatora, ale dla danego katalizatora może zmieniać się w zależności od typu reakcji. Pozostała część powierzchni wykazuje znacznie mniejszą lub w ogóle nie katalizuje reakcji. Niejednorodność działania powierzchni katalizatora wynika z niejednorodności struktury powierzchni.
Przez centrum aktywne rozumie się pojedyncze atomy lub grupy atomów na powierzchni katalizatora, które tworzą wiązanie (wiązania)z cząsteczką reagującej substancji w procesie powstawania produktu przejściowego lub kompleksu aktywnego. Rolę centrów aktywnych mogą pełnić atomy o małych liczbach koordynacyjnych, na narożach lub krawędziach płaszczyzn krystalograficznych, nie wysycone w skutek zaburzenia periodyczności sieci kryształu na powierzchni. Centrami aktywnymi mogą być również defekty punktowe (obce atomy, wakancje w sieci), defekty liniowe (dyslokacje) czy defekty elektronowe.
Ze względu na charakter chemiczny centra aktywne podzielić można na centra:
- kwasowe i zasadowe (typu Bronsteda lub Lewisa)
- redoksowe:
* centra zawierające jony o zmiennej wartościowości, które w obecności reagentów ulegają utlenieniu lub redukcji
* centra zdolne do utworzenia kompleksów z przeniesieniem ładunku (charge transfer complexes) w obecności reagentów o charakterze akceptorów lub donorów elektronów
Czasami jedna substancja może na swojej powierzchni mieć centra aktywne o różnym charakterze chemicznym np. tlenki metali grup przejściowych (jony metalu o zmiennej wartościowości mogą działać jako centra redoksowe, bądź centra kwasowe Lewisa, jony ![]()
jako centra zasadowe Lewisa, a adsorbowana na powierzchni woda może pełnić rolę centrów kwasowych według Bronsteda. W zależności od typu reagentów i warunków reakcji „uruchamiane” są różne centra powierzchni katalizatora.
Centrum aktywne nie jest statyczną grupą atomów istniejącą na powierzchni niezależnie od przebiegającej reakcji. W wielu przypadkach centrum aktywne powstaje w pierwszym momencie kontaktu z substratami i za sprawą istniejących reagentów może dochodzić do modyfikacji powierzchni. Centrum aktywnym mogą być również przekształcone na atomach powierzchniowych katalizatora fragmenty cząsteczek substratów, związane w sposób trwały z powierzchnią.
Typy katalizatorów (metale, tlenki, siarczki metali przejściowych, katalizatory kwasowo - zasadowe)
Grupy katalizatorów:
Przewodniki elektronowe (metale, głównie przejściowe, i miedziowce) oraz niektóre substancje półprzewodzące (tlenki, rzadziej siarczki metali przejściowych i metali I i II grupy
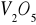
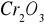
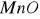
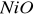
. Katalizują one reakcje utlenienia i redukcji głównie z udziałem gazowego tlenu i wodoru.Substancje o charakterze izolatorów - tlenki, krzemiany, często o charakterze żeli
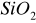
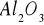
. Zazwyczaj aktywne są mieszaniny bądź połączenia takich tlenków tj. glinokrzemiany. Głównie katalizują one procesy uwadniania, odwadniania, krakowania, izomeryzacji, polimeryzacji węglowodorów.
Działanie katalizatorów metalicznych: chemisorpcja gazów związana jest z pobieraniem (lub oddawaniem) elektronów z najwyższego pasma energetycznego metalu. Aktywność metali przejściowych wynika z niecałkowitego wypełnione pasma elektronów d. Niezapełnione poziomy d mogą być zapełnione przez elektrony adsorbujących się cząsteczek, o czym świadczą stopniowe zmiany właściwości magnetycznych metalu zachodzące w miarę obsadzania jego powierzchni przez chemisorbowane cząsteczki. W działaniu katalitycznym istotna role odgrywa wymiana elektronów pomiędzy adsorbowanymi reagentami i metalem. Dodatkowa chemisorbowana cząsteczka na powierzchni katalizatora musi być odpowiednio zorientowana, wiążąc się niektórymi atomami czy grupami powierzchniowymi. Do tego wymagana jest pewna odpowiedniość geometryczna między strukturą cząsteczki a strukturą katalizującego metalu. Teoria „multipletu' mówi, że na powierzchni metalu muszą być rozmieszczone atomy tak, aby tworzyły zespoły umożliwiające właściwy sposób zaadsorbowania substratu.
Mówi się o istnieniu centrów aktywnych, których obecność świadczy o niejednorodności powierzchni katalizatorów. O istnieniu centrów aktywnych śwadczy fakt, że:
wraz ze wzrostem ułamka powierzchni zajętej przez adsorbent wzrasta
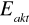
i maleje ciepło adsorpcjizjawisko zatrucia katalizatorów - pewne substancje silnie dezaktywują (niemetale V i VI grupy układu okresowego mające w cząsteczkach wolne pary elektronowe lub wiązania podwójne np.
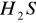
, tiofen,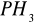
CO. Do dezaktywacji potrzebna jest znacznie mniejsza ilość trucizny niż wymagałoby tego pokrycie całej powierzchni katalizatora monomolekularną warstwą.
Działanie katalizatorów półprzewodników: związane jest z wymianą elektronów pomiędzy katalizatorem i reagentami w procesach chemisorpcji i reakcji na powierzchni. Tlenki półprzewodzące są półprzewodnikami typu n (nadmiarowymi ZnO, CdO, ![]()
![]()
) lub typu p (niedomiarowymi![]()
,![]()
,![]()
).
Półprzewodniki typu n zawdzięczają swoje właściwości nadmiarowi metalu, czyli niedomiarowi tlenu w stosunku do składu stechiometrycznego. Ich przewodnictwo elektronowe jest związane z istnieniem elektronów, których ładunek równoważy dodatni ładunek nadmiarowych międzywęzłowych kationów lub, które są związane z lukami anionowymi po tlenie. Przewodniki typu n są aktywniejsze w atmosferze redukującej, gdyż w tych warunkach rośnie stężenie wymienionych defektów donorowych (mogących dostarczyć elektrony do pasma przewodnictwa).
Półprzewodniki typu p mają natomiast niedomiar metalu w stosunku do tlenu. Jony o nienormalnie dużym ładunku dodatnim stanowią poziomy akceptorowe, których obecność umożliwia przewodnictwo dziurowe półprzewodnika. Lepiej, zatem adsorbowane są gazy utleniające i zwykle większa jest ich aktywność katalityczna w ich atmosferze.
Domieszki tlenków o kationach mających inną wartościowość niż kationy macierzystego tlenku silnie wpływają na stężenie poziomów donorowych czy akceptorowych, czyli również na przewodnictwo tlenku i jego właściwości katalityczne.
Aktywność katalityczna półprzewodników zależy silnie od temperatury, która wpływa na stężenie elektronów czy dziur. Dużą rolę odgrywają również czynniki geometryczne.
Podział katalizatorów:
Katalizatory o charakterze przewodników lub półprzewodników (metale, tlenki siarczki metali przejściowych), zwykle barwne, ich właściwości katalityczne zależą w dużym stopniu od obecności domieszek, struktury defektów, czyli od metody przygotowania katalizatora
Katalizatory o charakterze izolatorów (tlenki metali grup głównych np.
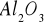
,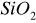
), ich własności nie zależą od dodatków innych składników a sposób ich przygotowania wpływa jedynie na właściwości fizyczne np. na wielkość powierzchni właściwej, porowatość struktury.
Inny sposób podziału:
Katalizatory reakcji homogenicznych - para elektronów wiązania kowalencyjnego między atomami A i B, ulegającego rozpadowi lub tworzeniu w czasie reakcji zostaje rozdzielona między oba atomy, które tworzyły wiązanie lub powstaje z elektronów pochodzących od obu atomów
Katalizatory reakcji heterogenicznych - para elektronów zostaje zlokalizowana przy jednym z atomów (rozpad wiązania) lub pochodzi od jednego tylko z atomów (tworzenie wiązania)
Oba w/w podziały pokrywają się:
Przewodniki i półprzewodniki są aktywne w reakcjach typu redoks (np. utlenienie, uwodornienie, odwodornienie), w których etap powstawania połączenia przejściowego jest reakcją typu homologicznego, a w tworzeniu wiązania cząsteczki zaadsorbowanej z katalizatorem bierze udział elektron katalizatora i elektron reagującej cząsteczki.
Izolatory są aktywne w reakcjach odwodnienia, krakingu, izomeryzacji węglowodorów, polimeryzacji, czyli w reakcjach z przeniesieniem elektronów (reakcje typu heterolitycznego). Na ich powierzchni istnieją centra o charakterze kwasów lub zasad typu Bronsteda lub Lewisa, a połączenia przejściowe mają charakter karbojonów.
Podział ten jednak nie jest zbyt ostry, gdyż wiele katalizatorów pierwszej grupy aktywnych jest także w reakcjach heterogenicznych.
Podział w/w nie obejmuje heterogenicznych katalizatorów, w których stopień rozdrobnienia fazy metalicznej lub tlenkowej na nośniku osiąga skale atomową czy cząsteczkową.
Podział:
Katalizatory na osnowie metali (metaliczne) - np. Pt, Pd, Ni
Katalizatory na osnowie tlenków metali przejściowych - np. Mo, V, Cr, Co
Katalizatory kwasowo - zasadowe(tlenki metali grup głównych, glinokrzemiany, amorficzne lub krystaliczne zeolity) - np. Al., Si, Mg, P
Kataliza na nośnikach
Pytanie nr. 11.
W celu maksymalnego rozwinięcia powierzchni katalizatora osadza się substancję czynna na porowatych nośnikach (ziemia okrzemkowa, azbest, naturalne glinokrzemiany, węgiel aktywny) utrzymują one katalizator w stanie dużego rozdrobnienia zabezpieczając jego ziarna przed spiekaniem się w wyższych temperaturach. Nośniki niekiedy mogą modyfikować działanie katalizatora. Do głównego katalizatora osadzonego na nośniku dodaje się często promotorów (nie stanowią katalizatorów danej reakcji, ale zwiększają znacznie aktywność fazy katalitycznie czynnej. Mogą to być promotory teksturalne (utrwalanie struktury katalizatora i zabezpieczanie przed rekrystalizacją, taki promotor nie zmienia![]()
osiąganej przy użyciu czystego katalizatora, ale wpływa na szybkość reakcji przez zwiększenie czynnika przedwykładniczego) lub, synergetyczne, czyli elektronowe (tworzą wraz z innymi składnikami masy kontaktowej nowe centra aktywne i obniżają![]()
)
Rola nośnika nie ogranicza się tylko do zwiększania stopnia dyspersji fazy metalicznej i jego stabilizowania w czasie reakcji. Nośnik może wpływać na zmiany aktywności, selektywności oraz rózne wielkości chemisorpcji. Oddziaływania metal - nośnik mogą polegać na:
Powstawaniu stopów lub związków międzymetalicznych miedzy nośnikiem a metalem
Przeniesieniu ładunku elektrycznego w celu wyrównania poziomu Fermiego obu faz, w wyniku, czego zmienia się stan elektronowy metalu
Ekspozycji różnych płaszczyzn krystalograficznych cząstek metalu
Oddziaływania między metalem a nośnikiem mogą być:
Słabe - metale naniesione na trudno redukowalne tlenki (
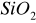
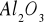
) lub węgiel aktywny
Średnie - małe cząstki metalu na zeolitach
Silne - metale na tlenkach łatwo redukujących się
Mechanizm adsorpcji na katalizatorach
Koniecznym warunkiem katalitycznego oddziaływania fazy stałej na reakcję zachodząca pomiędzy gazami jest chemisorpcja, co najmniej jednego ze substratów na powierzchni katalizatora. Na powierzchni tej następnie zachodzi reakcja> mogą w niej brac udział: wyłącznie substraty, chemisorbowane lub substrat chemisirbowany i inny, którego cząsteczki uderzają o powierzchnie katalizatora. Chemisorpcja z reguły związana jest z wymianą elektronów pomiędzy adsorbentem i adsorbatem, wytworzeniem między nimi silnych wiązań typu kowalencyjnego lub jonowego. Zaadsorbowana cząsteczka różni się od swobodnej rozkładem gęstości elektronów a często również deformacją szkieletu. Z związku, z czym adsorpcji towarzyszy niekiedy dysocjacja cząsteczek.
Adsorpcja jest procesem, w którym cząsteczki (cząstki) jednej substancji zostają związane na powierzchni innej substancji. Proces ten prowadzi do zwiększenia stężenia tych cząstek na granicy rozdziału obu faz. Kataliza heterogeniczna związana jest głównie z granicą faz gaz - ciało stałe. Zjawisko adsorpcji wynika z istnienia niezrównoważonych, niesymetrycznych sił na powierzchni adsorbenta lub tzw. niewysycenia powierzchni, która dzięki temu ma nadmiar energii swobodnej w stosunku do objętości fazy stałej.
Związek miedzy kataliza heterogeniczna a procesem adsorpcji wynika z samej definicji katalizy, która mówi, że w trakcie następuje czasowe związanie w sposób przejściowy reagujących substratów z katalizatorem.
Chemisorpcję substratów uważa się za wstępny etap właściwej reakcji na powierzchni, w czasie, którego następuje aktywacja reagujących cząsteczek tj. przejście ich w formy o większej reaktywności niż wyjściowe cząsteczki. Kompleks adsorpcyjny w następnych etapach ulega dalszym przekształceniom z wytworzeniem zaadsorbowanego produktu (ZS), który w ostatnim etapie reakcji ulega desorpcji do fazy gazowej. Dzięki procesowi adsorpcji mamy kilka etapów pośrednich, gdy adsorpcja ułatwia dany proces to ![]()
każdego z nich jest niższa niż ![]()
reakcji homogenicznej w zależności od wysokości bariery energetycznej etapów adsorpcji właściwej reakcji powierzchniowej i desorpcji każdy z tych etapów może determinować całkowitą szybkość heterogenicznej reakcji katalitycznej.
Według Langumira chemisorpcja jest niezbędnym warunkiem zajścia reakcji powierzchniowej
Reakcja jednocząsteczkowa - szybkość zależy od stężenia substratu na powierzchni
Reakcja dwucząsteczkowa
Reakcja miedzy X i Y w stanie zaadsorbowanym - szybkość proporcjonalna jest do iloczynu stężeń powierzchniowych
Jedna z reagujących substancji ulega adsorpcji - szybkość reakcji proporcjonalna jest do iloczynu stężenia powierzchniowego substancji zaadsorbowanej i ciśnienia substancji drugiej z fazy gazowej.
Ilość zaadsorbowanej substancji określona w pomiarach równowag czy kinetyki adsorpcji nie musi odpowiadać ilości, która bierze udział w powierzchniowej reakcji katalitycznej, spośród różnych form chemisorpcji tylko niektóre mogą pełnić rolę produktów przejściowych aktywnych w danej reakcji. Badania chemisorcji pozwalają na określenie mechanizmu reakcji heterogenicznej, określenie rodzaju i własności różnorodnych centrów powierzchniowych
Aspekty geometryczne, energetyczne, elektronowe zjawiska katalizy
Aspekt geometryczny - badania dotyczą tutaj wpływu: liczby atomów tworzących centrum aktywne, struktury geometrycznej (odległość, kąty miedzy atomami katalizatora tworzącymi centrum aktywne), struktury geometrycznej reagującej cząsteczki na mechanizm jej przekształceń, wzajemnego ułożenia cząsteczki i centrum aktywnego na powierzchni. Badano również wpływ rozmiarów (stopnia rozdrobnienia) cząstek metalu na nośniku na właściwości katalityczne. Na tej podstawie wyróżniono reakcje wrażliwe i niewrażliwe na ten parametr.
Teoria Balandina - założył, że w akcie elementarnym reakcji katalitycznej biorą udział tylko niektóre z atomów reagującej cząsteczki, miedzy, którymi zachodzi przegrupowanie wiązań chemicznych. Grupa atomów, która podczas reakcji jest przez określony czas w kontakcie z atomami powierzchni katalizatora to tzw. grupa indeksowa. Cząsteczka substratu jest zorientowana w stosunku do powierzchni katalizatora, a podstawniki grupy indeksowej nie będące w kontakcie z powierzchnią katalizatora nie wpływają w istotny sposób na przebieg reakcji. Czyli uwodornienie etylenu i uwodornienie nienasyconego kwasu tłuszczowego powinno zachodzić na tych samych katalizatorach.
Grupa atomów katalizatora w konfiguracji geometrycznej zapewniającej oddziaływanie z atomami indeksu to tzw. multiplet
Zgodnie z zasada odpowiedniości geometrycznej podczas reakcji katalitycznej atomy grupy indeksowej zachowując kąty miedzy wiązaniami nakładają się na atomy multipletu z wytworzeniem przejściowego kompleksu multipletowego, przy czym pomiędzy długościami wiązań w grupie indeksowej a odległościami atomów w multiplecie powinna istnieć zgodność. Dla każdej odległości międzyatomowej w różnych grupach indeksowych istnieje optymalna odległość między atomami zapewniająca przebieg reakcji z dużą szybkością przy minimalnej barierze energetycznej. Zbyt duże odległości to zbyt słabe oddziaływanie z atomami reagującej cząsteczki, zbyt małe odległości mogą być przyczyną odpychania między atomami powstającego produktu. Grupa indeksowa powinna wykazywać izomorfizm z powierzchnia katalizatora, jej atomy nadbudowują warstwę powierzchniową wchodząc w puste miejsce (wgłębienie) miedzy atomami na powierzchni
Współczesne badania geometrii zaadsorbowanych cząsteczek wskazują,
że w większości przypadków zaadsorbowane atomy zajmują takie miejsca na powierzchni w których są otoczone przez największą liczbę atomów adsorbenta.
długości wiązań między zaadsorbowanymi atomami a atomami adsorbenta są zbliżone do długości wiązań odpowiednich par atomów w związkach chemicznych.
Aktywność w reakcja z występowaniem wiązań C - H jest proporcjonalna do ilości schodków, a z C - C rośnie ze wzrostem stężenia uskoków (schodki i uskoki występują w monokryształach cząsteczki).
Aspekt energetyczny - czynnik energetyczny to innymi słowy zależność miedzy aktywnością katalityczna a wielkościami termodynamicznymi, charakteryzującymi energie wiązania między atomami reagującej cząsteczki i atomami na powierzchni katalizatora takimi jak np. ciepło tworzenia połączenia przejściowego lub ciepło adsorpcji reagentów. Aktywne katalizatory powinny charakteryzować się optymalna niezbyt dużą ani niezbyt małą energią wiązań w połączeniu przejściowym. Jeśli energia ta będzie zbyt mała połączenie przejściowe będzie mało trwałe i będzie tworzyć się w małych ilościach, co będzie prowadzić do niskiej aktywności. Natomiast, jeżeli połączenie przejściowe jest zbyt trwałe rozkład do produktów reakcji jest powolny. Ogólnie rzecz biorąc aktywność rośnie ze wzrostem energii wiązania cząsteczka - katalizator, jeśli powstanie tego wiązania jest etapem determinującym całkowitą szybkość reakcji, maleje zaś gdy w etapie determinującym szybkość następuje rozpad wiązania.
Aspekt elektronowy - rozważa się dwa podejścia:
Uwspólnione („kolektywne”) - oparte na teorii pasmowej ciała stałego, zakłada, że w tworzeniu wiązań uczestniczą uwspólnione elektrony z różnych poziomów energetycznych, rozszczepionych w krysztale na odpowiednie pasma. Szybkość elementarnego etapu reakcji katalitycznej zależeć będzie od stężenia elektronów lub dziur elektronowych w pasmach i energii elektronów w stosunku do energii Fermiego. Dla metali (nakładanie się pasm odpowiadających różnym poziomom energetycznym) poziom Fermiego odpowiada energii, poniżej której wszystkie poziomy są obsadzone, a powyżej której są puste. Dla półprzewodników poziom Fermiego leży w obszarze energii wzbudzonych, miedzy pasmem podstawowym a przewodnictwa. W obu przypadkach poziom Fermiego przedstawia potencjał elektrochemiczny elektronów w ciele stałym.
Lokalnym - rozpatruje się tutaj oddziaływania elektronowe między reagującą cząsteczką a wyizolowanym z sieci centrum aktywnym, którym może być pojedynczy atom, jon lub ich grupa.
Nie da się jednoznacznie rozstrzygnąć o roli własności uwspólnionych i lokalnych w katalizie, ich względny udział zależy od reakcji i katalizatora.
Preparatyka katalizatorów i ich regeneracja
Skład chemiczny i fazowy wnętrza i powierzchni oraz tekstura katalizatora zależą od metody jego otrzymania oraz od parametrów takich jak pH, temperatura, stężenie.
Z wyjątkiem nielicznych przypadków wykorzystania naturalnych surowców (np. Al.)większość katalizatorów otrzymuje się w wyniku reakcji chemicznych rozkładu lub syntezy z odpowiednich substancji wyjściowych: soli, wodorotlenków lub kompleksów (organicznych i nieorganicznych). Reakcje te zachodzą w fazie stałej (preparatyka „na sucho”) lub w roztworach (preparatyka „na mokro”). Preparatyka katalizatora obejmuje również procesy fizyczne: mieszanie, rozdział zawiesin, ogrzewanie, odparowanie, chłodzenie, krystalizacja, adsorpcja.
Etapy preparatyki katalizatora:
Wybór rodzaju i formy substancji wyjściowych
Procesy chemiczne lub fizykochemiczne prowadzące do otrzymania prekursora katalizatora albo jego masy aktywnej
Procesy formowania
Obróbka
Aktywacja katalizatora w określonej temp. i atmosferze
Strącanie osadów: polega na reakcji chemicznej w roztworze między związkiem chemicznym zawierającym aktywny składnik katalizatora a czynnikiem strącającym. Osad poddaje się starzeniu, oddziela od roztworu, suszy i praży. Tą metodą otrzymuje się wodorotlenki metali np. żelaza lub niklu, stanowiące prekursory katalizatorów tlenkowych lub metalicznych, wodorotlenek glinu, kwas glinokrzemowy. Czynnikiem strącającym są zwykle roztwory alkaliów lub amoniak, węglan sodu, potasu i amonu. Zaleta metod strąceniowych jest możliwość otrzymania mieszanych katalizatorów (głównie tlenkowych) w szerokim zakresie składników. Wada: trudność otrzymania dobrze zhomogenizowanych pod względem składu perkursorów katalizatorów mieszanych
Kompleksowanie: reakcja miedzy solami metali a kwasami i solami kwasów organicznych, w czasie, której powstają kompleksy o zmiennym składzie
Nanoszenie składników aktywnych z roztworów na nośnik: nośnik poddaje się działaniu roztworu zawierającego składnik aktywny. Nadmiar roztworu niezwiązany z nośnikiem usuwa się przez odsączenie, suszenie, odparowanie, a otrzymany prekursor poddaje się obróbce cieplnej. Oddziaływanie aktywnego składnika z nośnikiem:
Roztwór może ulegać adsorpcji na powierzchni nośnika z wytworzeniem kompleksów powierzchniowych
Składniki roztworu mogą ulegać wymianie jonowej z powierzchniowymi grupami(np. grupami hydroksylowymi) lub reakcji chemicznej
Siły oddziaływania mogą być słabe, a adsorpcja składnika aktywnego może mieć charakter adsorpcji fizycznej
W zależności od typu oddziaływania dzieli się metody nanoszenia z roztworu na: adsorpcyjne, jonowymienne i impregnacyjne.
Obróbka termiczna prekursora polega na jego prażeniu w wyniku, którego rozkłada się on z wytworzeniem właściwej fazy aktywnej katalizatora np. na rozkładzie prekursorów lub soli do odpowiednich tlenków albo kompleksów metali do czystych metali. Dobór temperatury zależy od rodzaju katalizatora i reakcji katalizatorów.
Katalizatory na nośnikach otrzymuje się w celu zwiększenia stosunku powierzchni aktywnej substancji do jej masy. Jako nośniki stosuje się:
Materiały naturalne występujące w przyrodzie np. ziemie okrzemkowe, gliny, pumeks
Syntetyczne nieorganiczne materiały np. tlenki glinu, krzem, magnez, węgiel aktywny
Polimery np. nylon
Charakteryzują się one zazwyczaj strukturą porowatą o średnicy porów od kilku do kilkunastu nanometrów. Rodzaj nośnika zależy od typu reakcji, katalizatora i warunków procesu katalitycznego. Np. ziemia okrzemkowa, pumeks w przypadku Ni (reakcja uwodornienia tłuszczów)
Glinokrzemiany i zeolity jako katalizatory (kwasowo - zasadowe)
Na powierzchni glinokrzemianów istnieją centra kwasowe wywołane zastąpieniem w sieci katalizatora jonu ![]()
przez ![]()
. Istnieją dwie prawdopodobne struktury: na powierzchni obecne są centra typu Lewisa, lub typu Bronsteda. Te oba rodzaje cent mogą współistnieć ze sobą. Katalizatory te zatruwane są zasadami i aktywowane kwasami.
Są to amorficzne katalizatory, charakteryzujące się nieuporządkowaną strukturą tetraedrów ![]()
![]()
połączonych wzajemnie mostkami tlenkowymi. Wprowadzenie atomów glinu w sieć ![]()
prowadzi do powstania centrów kwasowych zdolnych do generowania karbokationów. Każdy atom trójwartościowego glinu wprowadzony do sieci tetraedru krzemowego związany jest z czterema anionami tlenowymi tetraedr ma ładunek ujemny. Zatem dla zachowania elektroobojetności glinokrzemianów konieczna jest obecność kationów. Wprowadzone zostają, zatem protony i powierzchniowo tworzą się grupy OH (mocne centra kwasowe Bronsteda). Przy małych stężeniach![]()
liczba centrów kwasowych rośnie ze wzrostem stężenia atomów Al., przy stężeniach powyżej 20% mas. ![]()
liczba centrów typu Bronsteda maleje.
Różnorodność sposobów połączenia między sobą tetraedrów![]()
![]()
oraz zmienność składu amorficznych glinokrzemianów prowadzą do powstania różnych centrów na ich powierzchni.
Zeolity -charakteryzują się lepszymi własnościami katalitycznymi, wyższą aktywnością i lepszą selektywnością oraz mniejsza tendencją do zatruwania depozytem węglowym w porównaniu z amorficznymi glinokrzemianami. Zeolity to krystaliczne glinokrzemiany. Zbudowane z tetraedrów![]()
![]()
połączonych atomami tlenu w różny sposób prowadzący do uporządkowanej sieci. W sieci zeolitu obserwuje się rozwinięta strukturę porowatą, na którą składają się komory połączone kanałami o wymiarach molekularnych. Kationy podobnie jak u amorficznych glinokrzemianów kompensują ujemny ładunek. Aktywność katalizatorów zeolitowych przypisuje się większemu stężeniu centrów aktywnych w porównaniu z amorficznymi glinokrzemianami oraz większemu stężeniu węglowodorów w sąsiedztwie centrum aktywnego dzięki adsorpcji w mikroporach
1
Wyszukiwarka
Podobne podstrony:
Hydro Ściąga, semestr III, hydrologia, hydro-rożne materiały
GEOLOGIA SCIAGA I SEMESTR 2 PB, ochrona środowiska PB
sciąga semestr I, Technikum rolnicze, produkcja zwierzęca(2)
STANY PRZEDRAKOWE I STANY RZEKOMONOWOTWOROWE ściąga, Semestr II
sciąga 1 semestr (gotowe) mucha, Prywatne, Studia, Pedagogika
Bankowość, sciaga semestr2 bank
pedagogika opiekuńcza ściąga 1 semestr, College, Pedagogika, rok II, Pedagogika opiekuńcza
sciaga semestr II, geodezja, ROK II, Zarys budownictwa
ZABURZENIA ROGOWACENIA ściąga, Semestr II
sciaga1(1), semestr 6-7
CHOROBY ALERGICZNE SKÓRY ściąga, Semestr II
ZABURZENIA BARWNIKOWE ściąga, Semestr II
Finanse sciaga, Semestr 3, Ćwiczenia, Finanse
automaty sciaga, Semestr IV, Wspólne, Podstawy automatyki
EKOLOGIA - sciaga, semestr 2, Ekologia
metrologia sciaga, semestr 4, Metrologia, metrologia test
MATERIAŁOZNAWSTWO wykłady sciaga, V SEMESTR, Materiałoznawstwo
więcej podobnych podstron