3. Ilościowy opis podziału składników mieszaniny w układzie
chromatograficznym. Warunki rozdziału składników miesza-
niny
Są dwie koncepcje termodynamicznego opisu retencji składników mieszaniny w układzie chromatograficznym uwzględniające zjawisko podziału składników między dwie fazy układu chromatograficznego lub zjawisko adsorpcji
faza ruchoma
składnik podział składnika
mieszaniny zgodnie z K
faza stacjonarna
Ilościowo rozdział każdego ze składników mieszaniny pomiędzy fazę stacjonarną a ruchomą opisuje stała podziału K:
K = cS / cM
(1)
cS - stężenie składnika w fazie stacjonarnej
cM - stężenia składnika w fazie ruchomej
RT ln K = 2.3 RT lgK = 2.3 RT lg cS/cM = W
Stężenie substancji w fazie ruchomej wyraża się w [mol/l-1]. Stężenie substancji w fazie stacjonarnej odnosi się albo do jej objętościVS [mol/l-1]lub do jej masy AS [mol/kg-1].
Składniki mieszaniny wymywane są z układu chromatograficznego zgodnie z ich współczynnikami podziału:
Jeśli KA > KB to tA > tB
większy współczynnik podziału KA - dłuższy czas elucji składnika tA
(tj. większe powinowactwo składnika A
do fazy stacjonarnej i cS > cM )
Składniki o tych samych wartościach współczynnika podziału w danym układzie chromatograficznym będą miały takie same czasy elucji i nie zostaną rozdzielone
Jeśli KA = KB to tA = tB
Podstawowym warunkiem rozdziału składników mieszaniny jest:
różnica ich wpółczynników podziału
Rys.2 Kolejne etapy rozdziału dwuskładnikowej mieszaniny oraz sygnał detektora dla poszczególnych etapów
Współczynnik podziału K a tym samym prędkość migracji składnika przez układ chromatograficzny i czas elucji zależy od:
rodzaju i składu fazy ruchomej
rodzaju fazy stacjonarnej
Zmieniając rodzaj i skład fazy ruchomej oraz stacjonarnej można wpływać na współczynnik podziału składników mieszaniny i ich rozdział
Wniosek powyższy jest słuszny przy następujących założeniach:
K = const
a więc w każdej chwili procesu chromatograficznego (elucji) stosunek stężeń
substancji w obu fazach jest taki sam,
brak dyfuzji
w układzie chromatograficznym nie zachodzi wyrównywanie stężeń na
drodze dyfuzji,
tzn. izoterma podziału jest liniowa
cS
A
B
cM
Założenie 1) jest z praktycznego punktu widzenia nie możliwe do spełnienia, ze względu na fakt, że jedna z faz jest ruchoma.
Można jednakże przyjąć, że układ chromatograficzny składa się z szeregu warstw (półek), w których ustala się stan równowagi zgodny ze współczynnikami podziału.
Proces elucji (rozwijania chromatogramu) realizowany jest w kolejnych warstwach (półkach), a wiec jest to szereg kolejno następujących po sobie równowag podziału.
Przykład
KA = 9
KB = 1
KC = 1/9
Przyjmijmy, że faza ruchoma nie przepływa w sposób ciągły, lecz jest dodawana porcjami ∆V. Po dodaniu kolejnych objętości ∆V ustala się stan równowagi zgodny ze współczynnikami podziału:
∆V = 1
faza stacjonarna faza ruchoma
A 90% 10%
1 warstwa B 50% 50%
C 10% 90%
∆V = 2
faza stacjonarna faza ruchoma
A 81% 9%
1 warstwa B 25% 25%
C 1% 9%
A 9% 1%
2 warstwa B 25% 25%
C 9% 81%
Krzywa obrazująca rozkład stężeń każdego składnika w kolejnych (n) warstwach w fazie ruchomej lub stacjonarnej w funkcji czasu (lub objętości fazy ruchomej) ma postać krzywej Gausa:
profil stężenia
faza ruchoma w fazie ruchomej
profil stężenia
w fazie stacjonarnej
Każdy składnik przesuwając się wzdłuż układu chromatograficznego ulega serii podziałów zgodnie z wartością jego K. W konsekwencji czas przebywania składnika w fazie stacjonarnej powoduje jego opóźnienie w stosunku do prędkości przesuwania się fazy ruchomej.
Zatrzymywanie składnika w fazie stacjonarnej (wydłużenie czasu elucji) jest tym większe im większa jest liczba jego kolejnych podziałów.
Składniki mieszaniny będą w różnym stopniu zatrzymywane przez fazę stacjonarną (zgodnie z ich współczynnikami podziału K), przy czym to zróżnicowanie czasu elucji (rozdzielenie) będzie tym większe im większa będzie liczba kolejnych podziałów
c
C B A C B A C B A
n
4. Parametry retencji (cd)
Kolejnym parametrem retencji jest współczynnik retencji (pojemnościowy) K' (lub k)
a - masa adsorbenta
mr - masa składnika
w fazie ruchomej
ms - masa składnika
zaadsorbowanego
przez adsorbent
Prędkość migracji (oraz czas elucji) składnika zależy od współczynnika podziału K oraz objętości fazy stacjonarnej VS (lub masy adsorbenta) w kolumnie.
Uzyskanie skutecznego rozdzielenia zależy od K (rodzaju fazy stacjonarnej, rodzaju fazy ruchomej) oraz od objętości fazy stacjonarnej VS w kolumnie (GC)
4.1 Definicja chromatogramu (Parametry retencji cd.)
Chromatogram to graficznie przedstawiony obraz rozdziału mieszaniny. To zależność sygnału detektora S (mierzącego w sposób ciągły zmianę stężenia (A, n, I, i inn.) składnika w fazie ruchomej od czasu t lub objętości fazy ruchomej VR
S
h
b0.5 =2.35 δ
tR w=4δ
VR
tM
tRA δ = parametr kształtu,
odchylenie standardowe
-(x-m)2 m(współczynnik położenia) = tR
y = h e 2δ2 gdy x = m tj. w punkcie odpowiadającym
czasowi retencji - wartość funkcji
jest największa i określa maksy-
malne stężenie związku
Parametr |
Symbol |
Zależność |
martwy czas retencji |
tM |
|
martwa objętość retencji |
VM |
VM = tMF |
czas retencji skł. A |
tRA |
|
objętość retencji skł. A |
VA |
VA = tRA F |
zredukowany czas retencji |
t'A |
t'A = tA - tM |
zredukowana objętość retencji |
V'A |
V'A= VA - VM |
objętościowe natężenie przepływu |
F |
|
liniowa prędkość przepływu |
u |
u = L/tM |
selektywność |
α |
|
Interpretacja parametrów retencji:
martwy czas retencji tM = L/u to czas retencji składnika nie zatrzymywanego w kolumnie, tj. składnika dla którego k=0. Szybkość elucji takiego składnika jest równa prędkości fazy ruchomej
martwa objętość retencji VM - objętość fazy ruchomej konieczna do wymycia składnika martwego z kolumny (nie zależy od u). Jest to objętość między cząsteczkami fazy stacjonarnej oraz objętość porów fazy stacjonarnej dostępna dla fazy ruchomej.
czas retencji składnika tRA - czas upływający od momentu wprowadzenia do kolumny do pojawienia się maksimum piku na chromatogramie
(zależy od L, u, K)
objętość retencji składnika VA - objętość fazy ruchomej konieczna do wymycia składnika z kolumny (nie zależy od u)
4.2 Zależności parametrów retencji
Przy spełnieniu liniowości izotermy podziału (K = const, tR nie zależy od masy składnika):
→ K = (VR -VM)/VS
K' = (VR - VM)/VM
jeśli K = O (składnik nie zatrzymywany w kolumnie) to tR = tM
tM = L / u
tR = L/u (1 + K')
5. Parametry opisujące układ chromatograficzny
5.1 Selektywność (współczynnik rozdzielania) α
(1) α = K2 / K1 = K'2 / K'1 = t'R2/ t'R1
α charakteryzuje względny rozdział składników mieszaniny w danym układzie chromatograficznym (zdolność układu chromatograficznego dla rozdzielenia składników mieszaniny)
Dla K2 > K1 im większa wartość α tym lepsze rozdzielenie składników mieszaniny
Jeśli K2 = K1 to α = 1 brak rozdzielenia
5.2 Rozdzielczość układu chromatograficznego (równanie
Prunella) Rs
Rozdzielczość pików RS określona jest zależnością:
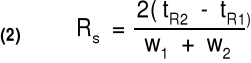
ΔtR - sprawność termodynamiczna - f(K, α)
w1, w2 - sprawność kolumny związana
ΔtR z poszerzaniem pików
t
w2
t
Dla całkowitego rozdzielenia pików chromatograficznych konieczne jest:
RS = 1.5 (piki gausowskie)
RS = 1 (piki o kształcie trójkąta)
RS = 0.8 dla analizy jakościowej
RS = 1.2 dla analizy ilościowej
Powyższa zależność nie podaje korelacji pomiędzy rozdzielczością i warunkami analizy, nie mówi jak poprawić zdolność rozdzielczą. Umożliwia jedynie wyznaczanie rozdzielczości.
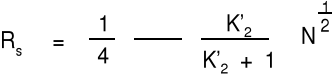
indeks 2 odnosi się do piku bardziej zatrzymywanego
α - selektywność
N - sprawność kolumny (liczba półek teoretycznych)
jeśli α = 1 to Rs = 0
jeśli K'2 = 0 to Rs = 0
jeśli N = 0 ? to Rs = 0
5.3 Interpretacja równania Prunell'a. Zwiększenie rozdzielczości
zwiększenie K' dla K'>10 {K'/(1 + K')} → 1
optymalny zakres K' = {1 - 10} chromatografia kolumnowa
K' = {0.1 - 10} chromatografia cienkowarstwowa
tM/tR
ΔtR
ΔtR
ΔK
ΔK ΔK
0.01 0.1 1 10 100 K'
zwiększenie α
RS = 0.25 (α-1)/ α * Nef 0.5
Nef
104 R = 1
103
1.04 1.08 1.2 1.4 α
W przypadku gdy α jest bardzo małe konieczna jest bardzo duża liczna półek (sprawność kolumny) dla zrealizowania rozdziału.
- zwiększenie N (sprawności kolumny)
5.3 Sprawność kolumny. Liczba półek teoretycznuch N
Ponieważ w miarę przesuwania się pasma wzdłuż kolumny (zwiększania tR) jego szerokość w rośnie, wobec tego wartość względnego poszerzania pasma przyjęto jako miarę sprawności kolumny.
Sprawność kolumny chromatograficznej mierzy się dla każdej substancji liczbą półek teoretycznych N w kolumnie.
Po przebyciu przez próbkę pewnej drogi w kolumnie, rozkład jej stężenia w wycieku ma kształt krzywej Gaussa. Odchylenie standardowe tego piku σ (w jednostkach czasu) powiązane z liczbą półek N zależnością:
σ2 = tR2 /N
Dla piku Gausowskiego jego szerokośc w, na określonej wysokości jest związana z kwadratem odchylenia standardowego σ (wariancją) zależnością:
w2 = a σ2
w której współczynnik proporcjonalności a zależy od wysokości, na jakiej mierzone jest w.
N = a (tR/w)2 w a
wh = 0.5 h 5.54 (wh = 2.36 σ)
wb 16 (wb= 4 σ)
Powyższe metody wyznaczania sprawności powinny być stosowane dla pików gausowskich (pierwszych pików nba chromatogramie).
Wysokość półki (wysokość równoważna półce teoretycznej (HETP) jest zdefiniowana jako
H = L / N
Jak zwiększyć sprawność kolumny?
Zgodnie z teorią półek N = L/H, wobec tego, jak zmniejszyć wysokość półki H?
6. Teoria kinetyczna procesu chromatograficznego
Sprawność kolumny wiąże się z poszerzaniem się pików chromatograficznych. Im dłużej dany składnik przebywa w kolumnie tym szersze będzie jego pasmo na chromatogramie. Stopień poszerzania pików jest parametrem kinetycznym.
6.1 Przyczyny poszerzania pików chromatograficznych
- brak liniowości izotermy podziału (K ≠ const, K zależu od stężenia)
CM
CS
czynniki hamujące ustalanie się równowagi podziału - dyfuzja
składników rozdzielanej mieszaniny w fazie ruchomej oraz fazie
stacjonarnej, opory w przenoszeniu masy. Wpływ tych czynników
na poszerzanie pików opisuje teoria kinetyczna.
6.2 Podstawy teorii kinetycznej
7. Dobór optymalnych warunków rozdziału - mechanizm retencji
Rozdział składników mieszaniny w chromatografii realizowany jest w serii kolejno po sobie nastepujących równowag podziału pomiędzy dwie fazy układu chromatograficznego, zgodnie z wartościami K.
Podczas transportu składników mieszaniny przez układ chromatograficzny występuje dyfuzja (hamująca ustalanie się równowag podziału), której efektem jest poszerzanie się pików chromatograficznych i niekompletny rozdział.
Optymalny rozdział zależy od zarówno od parametrów termodynamicznych (K,K',α) wpływających na retencję oraz kinetycznych (poszerzanie pasmchromatograficznych):
1. α > 1 czynniki
K' = {1 - 10 lub O.1 - 10} termodynamiczne
N, H czynniki kinetyczne
Dwa pierwsze parametry określają retencję składnika, którą można zmieniać (zwiększać) przez:
zmianę rodzaju i składu fazy ruchomej rozpuszczalnika -HPLC
- zmianę typu i rodzaju fazy stacjonarnej - HPLC,GC
zmianę temperatury - GC
proces rozpuszczania jest egzotermiczny
lnK = - Hr /RT + lnc → k ~ 1/T
zatem, podwyższenie temperatury powoduje skrócenie czasu retencji
Rozdział chromatograficzny można uzyskać przez zmianę parametrów wpływających na retencję składników mieszaniny, a więc charakter niekowalencyjnych oddziaływań międzycząsteczkowych w danym układzie chromatograficznym.
- elektrostatyczne oddziaływania w układzie chromatograficznym:
polarne oddziaływania Van der Waals'a (pomiędzy cząsteczkami
mającymi ładunek powierzchniowy,
niepolarne oddziaływania dyspersyjne pomiędzy neutralnymi
czasteczkami (grupami funkcyjnymi)
wiązania wodorowe pomiędzy cząsteczkami składnika oraz
cząsteczkami fazy stacjonarnej (lub ruchomej) należą do silnych
oddziaływań przyczyniające się do powolnego ustalania się
równowagi podziału i ogonowania pików.
Składniki zawierające polarne grupy funkcyjne będą silniej zatrzymywane (oddziaływać) przez polarną fazę stacjonarną. Związki o charakterze niepolarnym łatwiej rozdzielić na niepolarnej fazie stacjonarnej. Wiele związków zawiera zarówno polarne jak i niepolarne fragmenty struktury. Ponadto oddziaływania między cząsteczkami składników mieszaniny oraz fazą stacjonarną zależą od rodzaju rozpuszczalnika (fazy ruchomej), w którym przebywają.
W chromatografii prostej decydują polarne oddziaływania międzycząsteczkowe
W chromatografii z odwróconymi fazami - dyspersyjne (hydrofobowe) oddziaływania międzycząsteczkowe.
W rzeczywistości model oddziaływań międzycząsteczkowych w układzie chromatograficznym jest bardziej złożony
7.1 Oddziaływania międzycząsteczkowe a mechanizm retencji .
RODZAJ fazy ruchomej oraz fazy stacjonarnej poprzez oddziaływania między cząsteczkami obu faz oraz cząsteczkami składników mieszaniny determinują ich zatrzymywanie (retencję) w kolumnie a w konsekwencji ich rozdział. Na przykład te składniki, których oddziaływania z fazą ruchomą są silniejsze niż z fazą stacjonarną będą wymywane szybciej. W uproszczonym modelu oddziaływań w układzie chromatograficznym zarówno rodzaj fazy stacjonarnej jak i rodzaj fazy ruchomej (moc elucyjna) warunkuje skuteczność rozdziału.
Chromatografia podziałowa
cząsteczki składnika
mieszaniny X
cząsteczki fazy cząsteczki fazy
ruchomej M stacjonarnej S
Można przyjąć, że o rozdziale decydują konkurencyjne oddziaływania
składnika X z fazą ruchomą M oraz składnika X z fazą stacjonarną S.
X M >> X S składnik X będzie lepiej rozpuszczał się w fazie ruchomej M (i będzie słabiej zatrzymywany przez fazę stacjonarną S) lub
X S >> X M składnik X będzie się lepiej rozpuszczał w fazie stacjonarnej (i będzie mocniej zatrzymywany przez fazę stacjonarną)
Chromatografia adsorpcyjna
cząsteczki składnika
mieszaniny X
cząsteczki fazy cząsteczki fazy
ruchomej M stacjonarnej S (ciało stałe)
Można przyjąć, że o rozdziale decydują konkurencyjne oddziaływania składnika X z fazą stacjonarną S oraz rozpuszczalnika M z fazą stacjonarną S
X S >> M S składnik X będzie silniej zatrzymywany przez
fazę stacjonarną
M S >> X S składnik X będzie słabiej zatrzymywany przez
fazę stacjonarną
Cząsteczki składnika będą się adsorbowały na centrach aktywnych żelu jeśli ich oddziaływania z adsorbentem są silniejsze niż oddziaływania cząsteczek rozpuszczalnika z adsorbentem.
Oddziaływania międzycząsteczkowe rozpuszczalnika oraz składników mieszaniny z adsorbentem zależą od właściwości chemicznych (rodzaj centrów aktywnych) oraz fizycznych adsorbenta
Wpływ rodzaju fazy ruchomej (rozpuszczalnika) na rozdział
składników mieszanin. Elucja izokratyczna i gradientowa
Rodzaj rozpuszczalnika wpływa na wartość współczynnika podziału oraz pojemnościowego składników mieszaniny i w konsekwencji na ich retencję w układzie chromatograficznym.
Optymalny zakres K' to: {1-10} dla chromatografii kolumnowej lub {0.1-10} dla cienkowarstwowej. Zatem właściwy rozpuszczalnik to taki, dla którego wartości K' składników mieszaniny znajdują się w tym zakresie.
W chromatografii podziałowej rozpuszczalnik wpływa na K poprzez oddziaływania tego rozpuszczalnika z cząsteczkami składników mieszaniny a zatem:
moc elucyjna rozpuszczlnika jest (w przybliżeniu) zależna od oddziaływań miedzycząsteczkowych tego rozpuszczalnika z cząsteczkami składnika (ów) mieszaniny.
W chromatografii adsorpcyjnej rozpuszczalnik wpływa na K' poprzez oddziaływania z adsorbentem, stąd:
moc elucyjna rozpuszczalnika zależy od oddziaływań tego rozpuszczalnika z adsorbentem.
Uszeregowanie rozpuszczalników wg. mocy elucyjnej (polarności, stałej dielektrycznej, parametru rozpuszczalności Hildebranda) to szereg eluotropowy.
Chromatografia prosta
K' {1-10}
0 PM PS 100
PX
1. Jeśli K' >> 10
0 PM PX PS 100
PM
Jeśli K' >> 10 (zbyt długi czas retencji) : składnik ma większe powinowactwo do fazy stacjonarnej (polarność zbliżoną do polarności fazy stacjonarnej), rozpuszczalnik o słabej mocy elucyjnej. Rozdział można poprawić stosując rozpuszczalnik o większej mocy elucyjnej - większej polarności
2. Jeśli K' << 1
0 PM PX PS 100
PM
Jeśli K' << 1 (zbyt krótki czas retencji) składnik ma większe powinowactwo do rozpuszczalnika, (polarność zbliżoną do polarności rozpuszczalnika), rozpuszczalnik o zbyt dużej mocy elucyjnej). Rozdział można poprawić stosując rozpuszczalnik o mniejszej mocy elucyjnej - polarności, w powyższym przykładzie P<0. Taki rozdział można zrealizować stosując chromatografię z odwróconymi fazami.
W odwróconym układzie faz, długie czasy retencji można skrócić przez zastosowanie rozpuszczalnika o większej mocy elucji (mniej polarnego, lub w przypadku eluentu metanol/woda o mniejszej zawartości wody). Natomiast zbyt krótkie czasy retencji można wydłużyć stosując rozpuszczalnik o mniejszej elucji (bardziej polarny lub zawierający więcej wody)
W obu typach chromatografii, jeśli czasy retencji są zbyt krótkie to rozdział można poprawić wydłużając czasy retencji przez zastosowanie rozpuszczalnika o mniejszej sile elucji ( w układzie normalnym jest to rozpuszczalnik jest to eluent mniej polarny a w odwrotnym układzie faz - eluent bardziej polarny, lub większej zawartości wody.
w praktyce chromatograficznej stosuje się fazy stacjonarne o polarności zbliżonej do polarności składników mieszaniny
związki polarne lepiej się rozdzielają na polarnych fazach stacjonarnych i wymagają mało polarnych rozpuszczalników
związki niepolarne rozdzielają się na niepolarnych fazach stacjonarnych z zastosowaniem polarnych rozpuszczalników (chromatografia z odwróconymi fazami)
związki średnio polarne lepiej rozdzielać stosując chromatografię z odwróconymi fazami stosując mocniejsze bardziej polarne rozpuszczalniki.
Jako fazę ruchomą można zastosować mieszaninę rozpuszczalników. Np. heksan/CCl4, dioksan/CH2Cl2, metanol/woda Siła elucyjna mieszaniny rozpuszczalników różniących się polarnościami zmienia się w przybliżeniu liniowo (w skali logarytmicznej) wraz ze zmianą stężenia jednego ze składników.
Elucja izokratyczna - rozdział odbywa się przy stałej sile elucyjnej rozpuszczalnika (pojedynczy rozpuszczalnik lub mieszanina rozpuszczalników o tym samym składzie)
Elucja gradientowa
Polega na ciągłej zmianie mocy elucyjnej rozpuszczalnika podczas rozdziału chromatograficznego mieszaniny.
a) b)
K'<<1 K'>>10
dla poprawy rozdzielczości należy dla poprawy rozdzielczości należy
zastosować rozpuszczalnik o zastosować rozpuszczalnik o
mniejszej mocy elucyjnej większej mocy elucyjnej
(o mniejszym powinowactwie
do składników lub adsorbenta)
c)
K'<<1 K'{1-10} K”>>10
dla poprawienia rozdzielczości należy zastosować elucję gradientową
umożliwia ona wolniejszą elucję składników szybko wymywanych z kolumny i jednocześnie szybszą elucję składników długo wymywanych.
(w przypadku chromatografii gazowej elucję z gradientem temperatury, np. t0=60˚C, Δt =10˚C/min, tk=150˚C.
Wpływ rodzaju stacjonarnej na rozdział
składników mieszanin.
Podstawowe parametry fizykochemiczne adsorbentów:
średnica ziaren (3 -10μm)
rozrzut ziaren (10%)
rozmiary porów (70 - 300L) oraz objętość porów
powierzchnia właściwa (50 - 250 m2/g) (im większa powierzchnia właściwa tym większa retencja)
powierzchnia właściwa po chemicznej modyfikacji zmniejsza się nawet o ok. 50%
gęstość centrów aktywnych (bonding phase density) (1 - 5 na nm2)
określa hydrofobowość adsorbenta, w przypadku chromatografii prostej
centrami aktywnymi mogą być np. grupy -OH, -NH2, -CN
w chromatografii z odwróconymi fazami - C4, C8, C18, fenylowa
Klasyfikacja adsorbentów
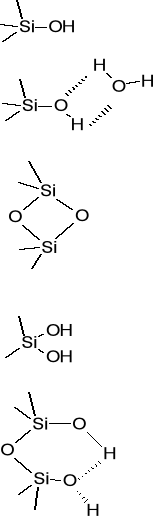
Właściwości adsorpcyjne SiO2*H2O
Silikażel jest najczęściej stosowanym adsorbentem w praktyce chromatograficznej. Higroskopijność i zawartość wody w składzie SiO2*H2O tego adsorbenta decyduje o ilości wolnych, nie związanych mostkami wodorowymi grup silanolowych odpowiedzialnych za jego właściwościach adsorpcyjnych.
Rodzaje centrów aktywnych SiO2:
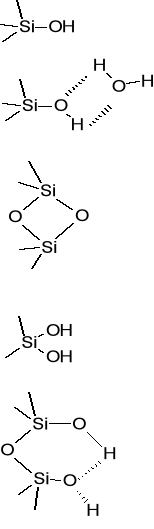
Właściwości adsorpcyjne tego adsorbenta zależą od oddziaływań z wolnymi (nie związanymi) grupami silanolowymi, które wykazują słabo kwasowy charakter.
Maksymalna zawartość grup silanolowych na powierzchni żelu krzemionkowego - 8 - 9 μmol/m2 lub średnio ok. 5 grup -OH na 100 Å
Inne adsorbenty
7.4 Chemiczna modyfikacja silikażeli - chemicznie związane fazy stacjonarne
Żele krzemionkowe poddawane są modyfikacji:
termicznej , powodującej zmianę ilości wolnych grup silanolowych,
chemicznej przez dołączenie organicznych ligandów, nowych centrów aktywnych, zmieniających własności adsorpcyjne silikażeli
Kowalencyjnie związane z fazą stacjonarną (żel krzemionkowy) grupy to najczęściej: C8, C10, C18 , o dł. łancuchów 21 Å. Przyjmuje się, że cząsteczki składników mieszaniny penetrują pomiędzy tymi łańcuchami - a więc proces ten jest traktowany termodynamicznie jak rozpuszczanie.
Po chemicznej modyfikacji
- zmienia się powierzchnia właściwa adsorbenta
np. powierzchnia właściwa żelu krzemionkowego - 350 m2/g
powierzchnia wł. po modyfikacji chemicznej - 170 m2/g.
pozostaje ok. 50% wolnych grup silanolowych (które mogą być dostępne
jeśli grupy R są krótkie, lub niedostępne - jeśli grupy R są długie, jak np.
C10, C18, w oddziaływaniach z rozpuszczalnikiem lub składnikiem.
np. silikażele zawieraja ok. 5 grup -OH na 100 Å,
najczęściej ok. 50% nie przereagowanych grup -OH pozostanie
po chemicznej modyfikacji silikażeli.
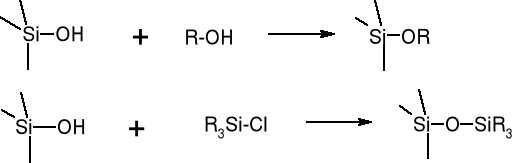
Typy połączeń grupy R z atomami Si silikażeli:
Si - R
Si-O-R
Si-O-Si-R
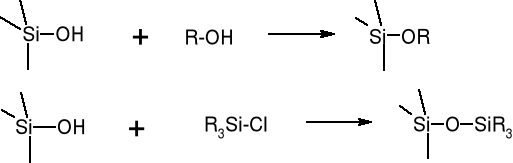
Można kontrolować właściwości tego typu faz poprzez rodzaj, ilość podstawników R na jednostkę powierzchni adsorbenta, długość łańcuchów alkilowych.
Podstawnik R może zawierać różnorodne grupy funkcyjne (C18, C14, C8, C4, C1, fenylowa, -CN, -NH2, i inn.), zatem faza stacjonarna może mieć rózną polarność lub hydrofobowość.
Wpływ różnych czynników na retencję
struktury składników mieszaniny
VR 2
1
3
n
1 - alkilobenzeny
2 - alkilofenony
3 - estry kwasu p-hydroksybenzoesowego
Hypersil - C18 (5µm, 150x4.6mm), eluent: MeCN/woda 60/40, 1 ml/min
Alkilobenzeny wykazują silniejszą retencje niż alkilofenony. Obecność grupy karbonylowej powoduję słabsze oddziaływania z niepolarną (hydrofobową) fazą stacjonarną.
Najmniejsza retencja parabenów wynika z obecności bardziej polarnej(od gr. karbonylowej) estrowej, która słabo oddziałuje z niepolarną fazą stacjonarną
charakteru fazy ruchomej oraz rodzaju oraz ilości centrów aktywnych
jeśli powierzchnia adsorbenta ma dużą gęstość centrów aktywnych R (niepolarnych) wtedy o retencji decydują niepolarne oddziaływania dyspersyjne
(jest to charakterystyczna cecha metod z odwróconymi fazami)
4
3
2
1
Zmiana retencji kwasu dodecylowego (faza stacjonarna: C18, duża gęstość centrów aktywnych - ok. 2 grupy R18 na 100 Å) w zależności od składu fazy ruchomej:
- 100% MeCN tR - 3.18 zmniejszenie
- 99/1 MeCN/H2O tR - 3.20 mocy elucyjnej
- 95/5 MeCN/H2O tR - 3.36 (tj. oddziaływań
- 90/10 MeCN/H2O tR - 3.88 z fazą stacjonarną)
(zwiększenie polarności)
Kwas dodecylowy jest polarny lecz w jego retencji nie biorą udziału polarne (lub jonowe) oddziaływania z niepolarną (C18) fazą stacjonarną; Symetria pików potwierdza występowanie wyłączne oddziaływań dyspersyjnych.
Zmniejszając udział acetonitrylu (zwiększając udział wody) w fazie ruchomej obserwujemy zwiększanie się retencji kwasu dodecylowego
3
2
1
Retencja tego samego składnika (faza stacjonarna: Hypersil-C1) w zależności od składu fazy ruchomej
- 100% MeCN tR - ?
- 99/1 MeCN/H2O tR - 7.30
- 95/5 MeCN/H2O tR - 2.72
Gęstość centrów aktywnych w trójmetylosilanu (Hypersil-C1) wynosi 3 grupy C1 na 100 Å(a więc jest większa niż poprzednio) lecz grupy C1 są 18 razy krótsze. Powierzchnia SiO2 ma 5 - 8 grup OH na 100 Å, lecz tylko połowa została podstawiona (podczas chemicznej modyfikacji) grupami C1, natomiast pozostałe grupy -OH są dostępne dla cząsteczek składnika ze względu na małe rozmiary grup R1.
Z tego względu kwas dodecylowy jest silniej zatrzymywany w przypadku czystego MeCN (1) Jego retencja zmniejsza się ze wzrostem zawartości wody w fazie ruchomej. Gdy zawartość wody nie jest zbyt duża wtedy decydują polarne oddziaływania z adsorbentem (jego wolnymi grupami silanolowymi). Pik 3 (95/5 MeCN/H2O) jest symetryczny co świadczy o tym, że 5% H2O wystarczyło do stłumienia polarnych oddziaływań grup silanolowych adsorbenta z kwasem dodecylowym.
składu fazy ruchomej
faza ruchoma MW BP R.I UV η μ
[ºC] [nm] [cP] [Debye]
acetonitryl 41 82 1.341 195 0.358 3.37
dioxan 88 101 1.421 215 1.26 0.45
etanol 46 78 1.359 205 1.19 1.68
metanol 32 65 1.326 205 0.584 1.66
izopropanol 60 82 1.375 205 2.39 1.68
tetrahydrofuran 72 66 1.404 215 2.20 1.70
woda 18 100 1.33 185 1.00 1.84
Polarność fazy ruchomej nie jest jedynym czynnikiem warunkującym oddziaływania z adsorbenten. Np. moment dipolowy wody jest mniejszy niż acetonitrylu lecz woda nie bierze udziału w hydrofobowych oddziaływaniach z adsorbentem natomiast acetonitryl tak.
Chromatografia gazowa
Obejmuje metody chromatograficzne, w których fazą ruchomą jest gaz.
Fazą stacjonarna może być adsorbent (chromatografia adsorpcyjna)lub faza ciekła na nośniku (chromatografia podziałowa)
Składniki mieszaniny muszą być przeprowadzone w stan gazowy
Cechy chromatografii gazowej są konsekwencją zastosowania gazu jako fazy ruchomej - a więc substancji o znacznie niższej niż ciecz lepkości i większym współczynniku dyfuzji. Czas retencji oraz współczynnik retencji rozdzielanych składników jest zatem funkcją ich prężności pary nad fazą stacjonarną (zależną od temperatury) oraz rozpuszczalności w fazie stacjonarnej. Powinowactwo do fazy ruchomej (obojętnego gazu) odgrywa znaczącą rolę w chromatografii cieczowej
Gaz nośny
Jego rola polega wyłącznie na przenoszeniu składników przez kolumnę natomiast nie bierze on udziału w specyficznych oddziaływaniach ze składnikami mieszniny i nie wpływa na jakość rozdziału.
Rodzaj gazu nośnego (azot, hel, argon, wodór) zależy od stosowanego detektora: dla katarometru najlepszym gazem jest wodór i hel, których przewodnictwo cieplne (7.14, 5.97) najbardziej różni się od przewodnictw cieplnych rozdzielanych substancji zapewniając dużą wykrywalność. W detektorze argonowym, gazem nośnym jest argon.
Wybór gazu powinien uwzględniać wielkość współczynników dyfuzji składników w fazie ruchomej dla obniżenia dyfuzji podłużnej i zwiększenia sprawności kolumny.
Detektory
Obecnie stosuje się prawie wyłącznie detektory różniczkowe, mierzące zmiany właściwości eluatu - gazu nośnego zawierającego poszczególne składniki.
Cechy detektorów:
- czułość (stosunek przyrostu sygnału do przyrostu stężenia lub masy)
wykrywalność (najmniejsza ilość substancji wywołująca sygnał, którego wysokość jest 2x większa od poziomu szumów
zakres liniowości (proporcjonalność sygnału do stężenia substancji)
mała objętość celki pomiarowej
stabilność i odtwarzalność wskazań
stała czasowa (0.01 dla kolumn kapilarnych)
Detektor cieplno-przewodnościowy (katarometr TCD)
Mierzy zmianę przewodnictwa cieplnego przepływającego gazu na skutek pojawienia się w nim składnika mieszaniny.
Zasadniczym elementem są cztery, włączone w obwód mostka Wheatstone'a, czujniki (drut wolframowy, platynowy, termistory z tlenków manganu, kobaltu niklu), których cechą jest znaczna zmiana oporu przy niewielkiej zmianie temperatury czujnika (tmp. detektora musi by ć utrzymywana z dokładnością do dziesiątych stopnia Celsjusza).
Jeśli przez celkę detektora przepływa gaz nośny to czujniki wykazują jednakowy opór elektryczny i układ jest w równowadze. Gdy w gazie nośnym pojawi się składnik mieszaniny o innym przewodnictwie cieplnym niż gaz nośny), wówczas temperatura i w konsekwencji opór elektryczny czujnika zmienia się.
Sygnał tego detektora zależy od prędkości przepływającego gazu, dlatego konieczna jest zachowanie stałości warunków (tmp., prędkości przeplywu)
Wykrywalność zależy od rodzaju gazu nośnego; najkorzystniejszy jest wodór i hel.
Czułość - zależy od temperatury czujników, którą można regulować.
Podstawowe cechy:
mała wykrywalność 10-7 - 10-8 g/cm3 (nie można stosować kolumn kapilarnych)
uniwersalność, można analizować wszystkie substancje, powszechnie stosowany w analizie gazów
-nie niszczy próbki
zakres liniowości - 105
Detektor płomieniowo-jonizacyjny (FID)
Jest najbardziej rozpowszechniony detektorem w chromatografii gazowej.
Mierzy zmiany natężenia prądu jonowego powstającego w celce detektora podczas spalania składników w gazie nośnym.
Podstawowym elementem jest palnik z temperaturą płomienia zależną od natężenia przepływu doprowadzonego do niego tlenu (powietrza) i wodoru oraz gazu nośnego.
W palniku umieszczonym między dwiema elektrodami i zasilaczem spala się badana substancja w gazie nośnym w wyniku czego powstają termojony (karbojony), zbierane na odpowiedniej elektrodzie i rejestrowany jest powstający prąd jonowy. Sygnał detektora jest proporcjonalny do ilości atomów węgla w cząsteczce, przy czym każdy szereg homologiczny ma inny współczynnik proporcjonalności.
Jest to detektor masowy, którego sygnał zależy od szybkości z jaką docierają do niego wykrywane substancje.
Cechy detektora
wysoka wykrywalność (10-12 g/s)
zakres liniowości 107
stosowany dla związków organicznych (największa czułość dla węglowodorów)
mniejsza czułość dla chlorowcopochodnych oraz pochodnych tlenowych, siarkowych, azotowych
nie można wykrywać gazów szlachetnych, tlenu, azotu, tlenku i dwutlenku węgla, amoniaku, tlenków azotu, siarkowodoru, dwutlenku siarki, aldehydu i kwasu mrówkowego, wody i dwusiarczku węgla
niszczy próbkę
Detektor płomieniowo-fotometryczny FPD
Jest odmianą detektora płomieniowo-jonizacyjnego, wykorzystywanego dla związków siarki i fosforu z dużą czułością.
Eluat z kolumny spala się w płomieniu z nadmiarem wodoru, tak że ze związków siarki i fosforu powstają wzbudzone cząsteczki S2 lub tlenek fosfiny (HPO), które emitują charakterystyczne dla siebie promieniowanie (394 nm dla związków siarki lub 526 nm dla związków fosforu) rejestruje się za pomocą fotopowielacza. W niektórych detektorach możliwe jest jednoczesne oznaczanie tych związków.
Detektor termojonowy TID (alkaliczny detektor płomieniowo-jonizacyjny AFID)
Jest również odmianą detektora płomieniowo-jonizacyjnego. Stosowany jest detektorem azotowo-fosforowym umożliwiającym oznacznie śladowych ilości tych związków oraz halogenopochodnych.
Detektor argonowy i helowy (ArD, HeD)
Mierzą natężenie prądu jonowego
Źródłem jonizacji są meta-stabilne atomy argonu Ar*(lub helu He*), które otrzymuje się przez jonizację argonu (helu) promieniowaniem α lub β (izotopy radu, strontu).
Wzbudzone atomy argonu (helu) tracą energie w zderzeniach z cząsteczkami badanych substancji. Jeżeli potencjał jonizacji tych substancji jest niższy od potencjału wzbudzenia argonu (helu) to ulegają one jonizacji:
Ar + e → Ar* + e- (energia metatrwałych at. argonu - 11.6 eV)
Ar* + S → Ar + S+ + e
W detektorze argonowym jonizacji ulegają związki, których potencjał jonizacji jest niższy od energii wzbudzonych atomów argonu (11.6 eV).
W praktyce sprowadza się to do oznaczania wszystkich związków organicznych, natomiast nie mogą być oznaczane gazy trwałe (wodór, tlen, azot).
Mniejsza czułość dla pochodnych azotowych i chlorowcopochodnych (rekombinacja)
W detektorze helowym (wyższy potencjał wzbudzenia atomów helu 19.6 eV) można oznaczać wszystkie gazy trwałe (z wyj, neonu)
Gazem nośnym może być tylko argon (hel)
Detektor fotojonizacyjny (PID)
Substancje ulegają jonizacji pod wpływem promieniowania nadfioletowego. Dobierając źródło emitujące promieniowanie o różnej długości można selektywnie i z dużą wykrywalnością oznaczać określone związki (10-11 g/s).
Detektor wychwytu elektronów (rekombinacyjny) ECD
Źródło jonizacji - cząstki α, β żródła promieniotwórczego
Gaz nośny - azot, argon z metanem
W wyniku jonizacji gazy nośnego, jony dodatnie i elektrony zbierane są przez odpowiednie elektrody i tworzy się prąd tła.
N2 + β → N2+ + 2e
Wprowadzona do detektora substancja (o powinowactwie do elektronowym) wychwytuje elektrony, tworząc jony ujemne, które z kolei zderzają się z jonami dodatnimi (rekombinacja) tworząc cząsteczki obojętne. W wyniku tych przemian następuje obniżenie natężenia prądu jonowego.
S + e → S-
S- + N2+ → S + N2
Umożliwia wykrywanie śladowych ilości substancji ( 10-13 - 10-14 g/s) mających powinowactwo elektronowe, pochodnych tlenowych, siarkowych, azotowych, chlorowcopochodnych. Nie jest przydatny do oznaczania węglowodorów, estrów eterów.
Kolumny i ich wypełnienie
W chromatografii gazowej stosowane są:
kolumny pakowane (l =1-3m, d =2-6mm)
mikropakowane (d = 0.8-1,2 mm, długość kilkunastu metrów)
preparatywne (pakowane, d>6mm, dł. kilka metrów)
kapilarne (d = 0.2 - 0.6 mm długość kilkadziesiąt metrów, mają większą od mikrokapilarnych pojemność sorpcyjną)
mikrokapilarne (d>0.1mm, długość kilkadziesiąt metrów, większą od
kapilarnych rozdzielczość)
Kolumny kapilarne (metalowe lub najczęściej szklane, ze stopionej czystej krzemionki) mogą mieć ścianki pokryte ciekłą fazą stacjonarną lub porowatą warstwę adsorbenta na ściankach .
Fazę stacjonarną wiąże się chemicznie w reakcjach z grupami hydroksylowymi krzemionki, lub przez sieciowanie fazy stacjonarnej z zastosowaniem inicjatorów reakcji rodnikowych, promieniowania UV.
Adsorbenty
Różnią się:
powierzchnią właściwą (kilkadziesiąt do kilkuset m2/g)
rodzajem i ilością centrów aktywnych
aktywnością (aktywacja polega na wygrzewaniu w odpowiedniej temperaturze i usunięciu wody, dwutlenku węgla)
Adsorbenty nieorganiczne
sita cząsteczkowe (naturalne lub syntetyczne zeolity - glinokrzemiany, najczęściej wapnia) o określonej średnicy porów (1.3 μm , 0.5μm)
Stosowane są do rozdzielania gazów (azotu, tlenu, helu, tlenków węgla,
tlenków azotu, gazów szlachetnych
żele krzemionkowe (Spherosil, Corasil, Porasil, Chromosil) o różnych właściwościach w zależności od średnicy cząstek, powierzchni właściwej, średnicy porów i inn.
tlenek glinu, ma mniejsze znaczenie (ze względu na trudności w utrzymaniu aktywności na stałym poziomie)
Powyższe adsorbenty mają właściwości hydrofilowe i nie mogą być stosowane jeśli gaz nośny jest wilgotny lub próbka zawiera wodę
Thermo-trap TA - adsorbent fydrofobowy do analiz śladowych
substancji w wodzie i powietrzu
Adsorbenty organiczne
Obecnie najczęściej stosowany rodzaj adsorbentów w chromatografii gazowej. Są to porowate polimery, kopolimery (np. styrenu i dwuwinylobwnzenu - Porapak P, PS, Q, QS, R, N, T, Chromosorb 101, 102, 103 itd.), różniące się powierzchnią właściwą, porowatością, temperaturą stosowania.
Fazy stacjonarne
Rozdzielanie składników mieszanin na ciekłych fazach stacjonarnych zależy od różnic w rozpuszczalności tych składników. Stąd najważniejszą cechą faz stacjonarnych wpływającą na ich rozdzielczość jest ich polarność. Drugą cecha determinującą przydatność w chromatografii gazowej jest mała prężność pary i duża odporność termiczna.
Fazy stacjonarne sklasyfikowano w oparciu o indeks polarności Rohrschneidera, wg umownej skali, w której skwalan ma polarność = 0 a β, β' -oksy-dwupropionitryl ma polarność = 100.
Inne systemy klasyfikacji uwzględniają ich elektrodonorowe i akceptorowe własności
Węglowodory - Skwalan, Apiezony (węglowodory alkanowe)
Silikony - oleje silikonowe i polimery, dimetylo oraz metylofenylosiloksany są najczęściej stosowanym rodzajem faz stacjonarnych. Różnią się zawartością grup funkcyjnych.
Pierwsze są stosowane do rozdzielania związków niepolarnych drugie - aromatycznych i średniopolarnych. Do tej grupy zalicza się także fazy fluoroalkilosilikonowe (średnia polarność), nitrylosilikony (wysoka polarność)
Poliglikole (glikole polietylenowe - Carbowax PEG, propylenowe - dzięki obecności atomów o charakterze donorów i akceptorów elektronów mogą oddziaływać ze związkami o różnym charakterze.
Estry kwasów karboksylowych (sebacynowego, adypinowego, ftalowego, bursztynowego) - mają zdolność tworzenia wiązań wodorowych, wadą jest reagowanie z alkoholami i aminami w tmp..120 oC
Inne fazy stacjonarne:
optycznie czynne (dipeptydy, diaminy) - do rozdzielania związków
racemicznych
ciekłe kryształy ( o charakterystycznym dla kryształów stałych
uporządkowaniu cząsteczek, wykazują anizotropię różnych właściwości)
Ich oddziaływanie ze składnikami mieszaniny zależy w mniejszym
stopniu od ich polarności a w większym od kształtu cząsteczek (łatwość
rozdzielania izomerów geometrycznych)
W wyborze fazy stacjonarnej należy uwzględnić oprócz jej polarności, także elekrono-donorowe/akceptorowe właściwości, stałą dielektryczną i in.
Czynniki wpływające na rozdzielczość w chromatografii gazowej
RS = f (α, K', N)
- selektywność układu chromatograficznego (fazy
stacjonarnej), współczynnik rozdzielenia
K' - współczynnik retencji (pojemnościowy)
N lub H - sprawność kolumny
Miarą selektywności fazy stacjonarnej jest różnica czasów retencji dwóch związków o różnej budowie lecz o identycznych temperaturach wrzenia. Jeśli ta różnica jest duża to świadczy to o dobrej selektywności fazy stacjonarnej
α oraz K' zależą od:
rodzaju fazy stacjonarnej (polarność, stała dielektryczna,
własności elektrono-donorowe/akceptorowe, oraz
temperatury
VM
VR = VM + KVS = VM + K' VS = VM (1 + K')
VS
tR = tM (1 + K')
proces rozpuszczania jest egzotermiczny
lnK = - Hr /RT + lnc → k ~ 1/T
zatem
podwyższenie temperatury powoduje skrócenie czasu
retencji i pogorszenie rozdzielczości
obniżenie temperatury powoduje wydłużenie czasu retencji i poprawę rozdzielczości
Zbyt niska temperatura powoduje poszerzanie i niesymetryczność pików i wydłuża czas analizy
Wzrost tmp. o 30oC skraca czas analizy o ok. połowę
Dobór odpowiedniej temperatury zależy od temperatury wrzenia składników rozdzielanej mieszaniny oraz termicznej odporności fazy stacjonarnej
W chromatografii podziałowej
temperatura kolumny powinna być zbliżona do temperatury wrzenia składników mieszaniny
W chromatografii adsorpcyjnej
temperatura kolumny może być wyższa od temperatury wrzenia składników mieszaniny
Chromatografia izotermiczna (w stałej temperaturze) oraz z programowaniem temperatury
Gdy temperatury wrzenia składników mieszaniny nie różnią się więcej niż o kilkadziesiąt stopni, wówczas można rozdzielić mieszaninę stosując stałą temperaturę kolumny w czasie analizy
Gdy temperatury wrzenia składników mieszaniny różnią się znacznie (np. 100oC) wówczas należy zastosować programowanie temperatury
K = cS / cM
Jeśli KA > KB to tA > tB
Jeśli KA = KB to tA = tB
faza
stacjonarna
K = Cs/CM
K' = K * VS/VM
K' = cS*VS / cM*VM = cS/cM * VS/VM
K' = NS/NM
VR = VM + K VS
VR = VM + K AS
VM
VR = VM + K' VS = VM (1 + K')
VS
tR = tM (1 + K') stąd
K' = (tR -tM) / tM
ms/a
Ka =
mr/VM
ms/a a ms
K'a = Ka * a/VM = * =
mr/VM VM mr
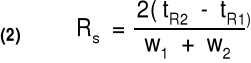
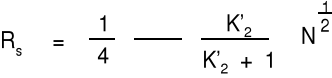
tR = f (K, K', Vs lub As, L, u)
Wyszukiwarka
Podobne podstrony:
Chromatografia #2, Technologia chemiczna, 5 semestr, analiza instrumentalna, notatki
Chromatografia #3, Technologia chemiczna, 5 semestr, analiza instrumentalna, notatki
Chromatografia #1, Technologia chemiczna, 5 semestr, analiza instrumentalna, notatki
Potencjometria zad, Technologia chemiczna, 5 semestr, analiza instrumentalna, notatki
MS, Technologia chemiczna, 5 semestr, analiza instrumentalna, notatki
Chromatografia gazowa przerobka, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozda
chromatografia zestawienie, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
1(1), Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
sprawozdanie1 cw.4, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
Cw9, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
CWGC, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
ćw 5, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
se, Technologia chemiczna, 5 semestr, analiza instrumentalna, zaliczenia
1(2), Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
cw 2(1), Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
saa, Technologia chemiczna, 5 semestr, analiza instrumentalna, zaliczenia
cw 1, Technologia chemiczna, 5 semestr, analiza instrumentalna, sprawozdania
więcej podobnych podstron