|
UNIWERSYTET ZIELONOGÓRSKI Wydział Inżynierii Lądowej i Środowiska |
|
|
CHEMIA FIZYCZNA LABORATORIUM
|
|
II ROK INŻYNIERII ŚRODOWISKA STUDIA DZIENNE GR 27B
|
Temat 4 |
|
Współczynnik podziału (Nernsta) kwasu benzoesowego pomiędzy fazą wodną a fazą organiczną.
OPRACOWALI: Kamil Turczyniak Ewa Kołoszyc Piotr Waśkowicz Jakub Woźniak
|
||

I. CZĘŚĆ TEORYTYCZNA
Przedmiotem prawa podziału Nernsta jest zagadnienie składu dwóch współistniejących faz ciekłych w układzie trójskładnikowym.
Niech dwie ciecze, 1 i 2 wzajemnie nierozpuszczalne tworzą po zmieszaniu warstwy ciekłe, będące praktycznie czystymi cieczami 1 i 2. Jeżeli do układu tego dodamy pewną ilość (A1) trzeciego składnika, ciekłego lub stałego, rozpuszczalnego w obu cieczach, pojawi się on w obu warstwach tworząc roztwory A1' i A1”. Stężenia składnika 3 w tych roztworach będzie określone konodą przechodzącą przez punkt A1. Dla inne ilości dodanego składnika 3 (A2, A3,…) składy otrzymanych roztworów będą inne (A2', A2”, A3', A3”,…).
Zakładamy, że roztwory składnika 3 w cieczach 1 i 2 można traktować jako roztwory idealne, rozcieńczone. Warunkiem równowagi między roztworami A' i A” jest równość potencjały chemicznego składnika 3 w obu fazach:
![]()
.
Po odpowiednich przekształceniach otrzymujemy:
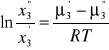
Wielkość po prawej stronie równania jest funkcją temperatury (i w niewielkim stopniu funkcją ciśnienia), nie zależy od stężenia składnika 3. Oznaczając:
![]()
gdzie:
K(T) - współczynnik podziału.
Dla roztworów rzeczywistych prawo podziału Nernsta przyjmuje postać:
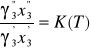
gdzie:
![]()
i ![]()
- współczynniki aktywności składnika 3 w obu fazach.
Prawo to obowiązuje również w przypadku, gdy ciecze 1 i 2 wykazują częściową mieszalność, ponieważ warunek równości potencjału chemicznego składnika rozpuszczonego pozostaje niezmieniony.
Ekstrakcja.
Ekstrakcją nazywamy operację polegającą na częściowym lub całkowitym rozdzieleniu mieszaniny ciekłej lub stałej za pomocą rozpuszczalnika, w którym składniki mieszaniny wykazują różną rozpuszczalność. W przypadku rozdzielania tą metodą mieszaniny stałej proces nazywamy ługowaniem, natomiast w przypadku rozdzielania mieszanin ciekłych - ekstrakcją właściwą lub po prostu ekstrakcją.
Ekstrakcja polega na kontaktowaniu surówki ekstrakcyjnej (roztwór surowy) z odpowiednio dobranym rozpuszczalnikiem wtórnym (C) - ekstrahentem, wykazującym selektywną rozpuszczalność w stosunku do składników surówki oraz ograniczoną rozpuszczalność w roztworze surowym. Po dodaniu rozpuszczalnika wtórnego tworzy się układ dwufazowy (ciecz-ciecz lub ciecz-ciało stałe). Składnik ekstrahowany przechodzi w znacznej części z surówki do rozpuszczalnika wtórnego, dając ekstrakt. Pozostałość poekstrakcyjna jest nazywana rafinatem.
Ekstrakcja w układzie ciecz-ciecz.
W przypadku, gdy rozpuszczalnik wtórny (C) wykazuje zupełną nierozpuszczalność w rozpuszczalniku pierwotnym (A) oraz gdy nie występują zjawiska asocjacji lub dysocjacji składnika ekstrahowanego (B) można stosować prawo podziału Nernsta. Według tego prawa w danej temperaturze stosunek zawartości składnika ekstrahowanego w ekstrakcie i rafinacie jest stały:
![]()
gdzie:
K - współczynnik podziału,
Y - zawartość składnika ekstrahowanego w ekstrakcie,
X - zawartość tego składnika w rafinacie.
Prawo Nernsta jest przypadkiem granicznym, gdyż zwykle zachodzi asocjacja lub dysocjacja i zależność Y = f (X) nie ma charakteru liniowego. Przy założeniu całkowitej niemieszalności rozpuszczalników: pierwotnego (A) i wtórnego (C) proces równowagi ekstrakcyjnej w warunkach izotermicznych można rozpatrywać w układzie współrzędnych prostokątnych.
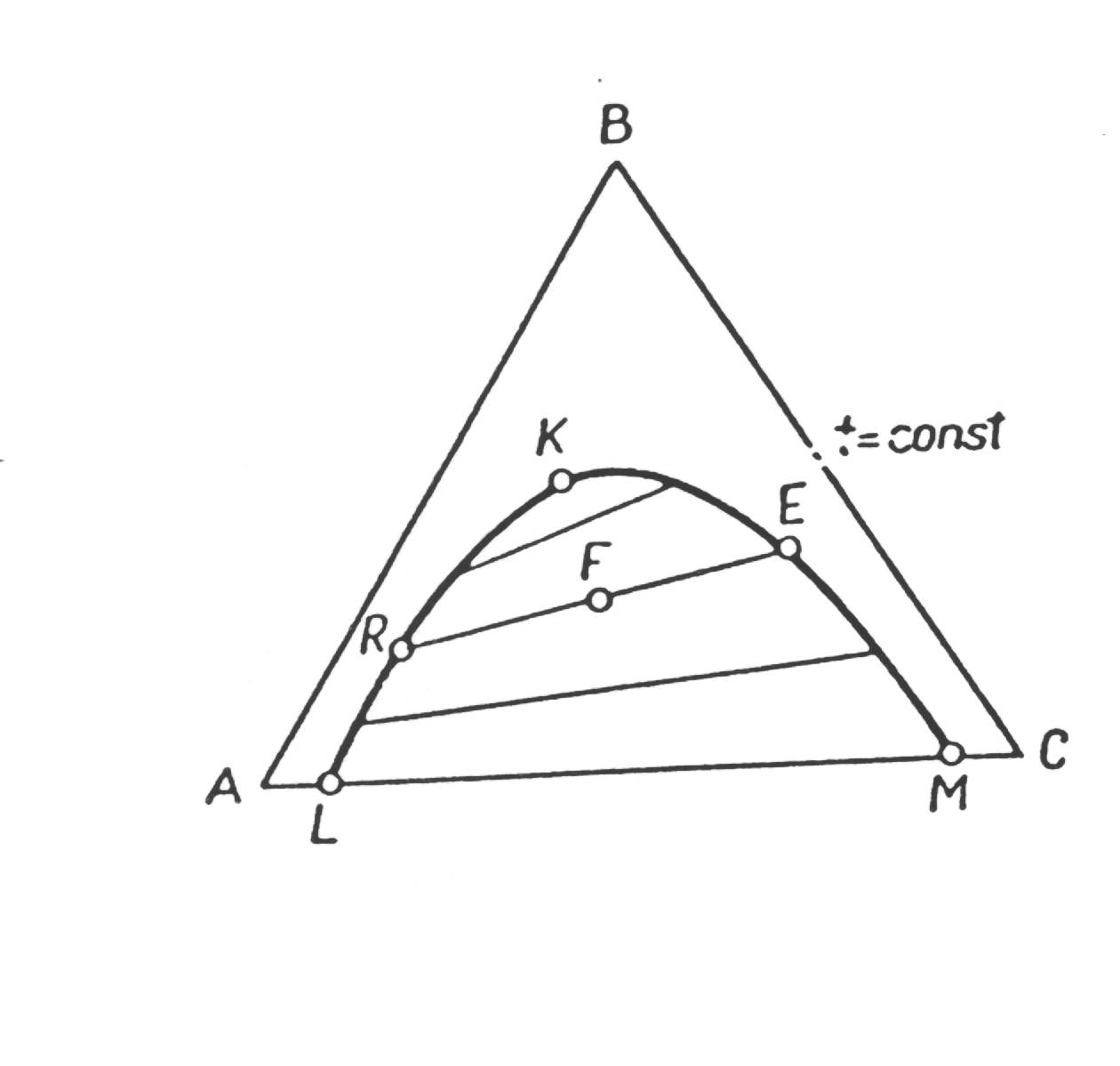
Jeżeli natomiast rozpuszczalnik pierwotny (A) i rozpuszczalnik wtórny (C) rozpuszczają się częściowo w sobie, zachodzi konieczność stosowania trójkąta Gibbsa do przedstawiania równowagi ekstrakcyjnej.
Równowaga ekstrakcyjna w trójkącie Gibbsa.
Krzywa LKM podaje składy obu faz ciekłych, wzajemnie względem siebie nasyconych (dane równowagi ciecz-ciecz). Cięciwy łączą składy obu faz (E - ekstrakt, R - rafinat) odpowiadające stanowi równowagi. Długość tych cięciw, czyli różnica składów tych faz maleje do zera w punkcie krytycznym (K). Jeżeli dana jest mieszanina o składzie (F), wówczas podzieli się ona na rafinat (R) i ekstrakt (E).Na zewnątrz krzywej granicznej mamy roztwory jednofazowe. Poza układem z jedną parą cieczy o ograniczonej mieszalności spotykamy również z dwiema i trzema parami wzajemnie niemieszalnych.
Cechy dobrego ekstrahenta.
Przemysłowe prowadzenie procesu ekstrakcji określa pewne wymagania stawiane dobremu ekstrahentowi. Najważniejszym i zarazem koniecznym warunkiem jest jego ograniczona mieszalność z rozpuszczalnikiem pierwotnym, powinien ekstrahować głównie pożądany składnik (selektywność), współczynnik podziału w danym układzie powinien mieć jak największą wartość, powinien różnić się znacząco gęstością od rozpuszczalnika pierwotnego, istotna jest również łatwość jego regeneracji, pomiędzy rozpuszczalnikiem pierwotnym i wtórnym powinno być optymalne napięcie międzyfazowe tak, aby ciecze łatwo nie emulgowały, ale również zbyt szybko nie zachodziła koalescencja (dłuższy czas kontaktu międzyfazowego ułatwia dyfuzję). Rozpuszczalnik wtórny nie powinien działać niszcząco na aparaturę, ani reagować ze składnikami surówki, powinien być on tani, najlepiej nietoksyczny, ekologicznie bezpieczny. Przy tak licznych wymaganiach należy wybierać optymalne rozwiązania.
Sposoby prowadzenia ekstrakcji.
Ekstrakcja może być prowadzona w sposób okresowy lub ciągły. W metodzie ciągłej stosuje się zasadę przeciwprądu strumieni surówki i ekstrahenta. Aparaty do ekstrakcji nazywamy ekstraktorami. Składają się one z mieszalników, w których zachodzi mieszanie surówki z ekstrahentem oraz rozdzielaczy (odstojników), w których następuje rozwarstwienie. W ekstrakcji wielostopniowej mieszalniki i odstojniki są ustawione na przemian, szeregowo. W ekstrakcji ciągłej wykorzystujemy jedno urządzenie - kolumnę ekstrakcyjną.
Ekstrakcja jednostopniowa.
Prosty sposób jednostopniowy polega na zmieszaniu surówki z ekstrahentem. Po osiągnięciu stanu równowagi fizykochemicznej następuje rozwarstwienie mieszaniny na ekstrakt i rafinat. W przypadku założenia całkowitej niemieszalności dwóch rozpuszczalników, bilans materiałowy składnika ekstrahowanego można zapisać jako:
Sxo = Sx + CY
Gdzie:
S - ilość rozpuszczalnika pierwotnego w surówce,
xo - zawartość składnika ekstrahowanego,
C - ilość rozpuszczalnika wtórnego,
Y - zawartość składnika ekstrahowanego w ekstrakcie,
x - zawartość składnika ekstrahowanego w rafinacie.
Rozwiązując powyższe równanie względem Y otrzymamy:
![]()
.
Zależność tą ilustruje poniższy rysunek.
Graficzny bilans ekstrakcji jednostopniowej.
Linia AB wychodząca z punktu o współrzędnych [x 0, y = 0] przecina krzywą równowagi w punkcie B, wyznaczając przez to skład ekstraktu y i rafinatu x.
Ekstrakcja przeciwprądowa ciągła.
Schemat ekstrakcji przeciwprądowej przedstawia poniższy rysunek.
Schemat ekstrakcji przeciwprądowej:
E - ekstrakt, R - rafinat, S - surówka, C - rozpuszczalnik wtórny.
W przypadku zupełnej niemieszalności rozpuszczalników pierwotnego i wtórnego bilans cząstkowy składnika ekstrahowanego można zapisać dla każdego z n kolejnych stopni:
Sxo + CYn+1 = Sxn + CY1
II. CZĘŚĆ DOŚWIADCZALNA
Aparatura : 4 rozdzielacze (100 cm3), 4 naczyńka wagowe, zestaw do miareczkowania, pipety (5 cm3), 2 erlenmeyerki (100 cm3), statyw do rozdzielaczy, cylinder miarowy ( 50 cm3 ), łaźnia wodna.
Odczynniki: ksylen (cz. d. a .), woda destylowana, 0,01 molowy roztwór NaOH do miareczkowania, wskaźnik
WYKONANIE ĆWICZENIA:
Na wadze analitycznej odważyć w naczyńkach wagowych 4 probki kwasu benzoesowego, od 0,03 do 0,3 g. Wsypać je do 4 rozdzielaczy i do każdego wlać ( cylinder 50 cm3 ) 25 cm3 ksylenu. Po rozpuszczeniu się kwasu benzoesowego, do każdego rozdzielacza wlać 35 cm3 wody. Wstrząsnąć przez ok. 5 min. Po sklarowaniu się faz rozdzielić fazę wodną od fazy ksylenowej i oznaczyć, w każdej z faz, całkowite stężenie kwasu benzoesowego.
W tym celu z każdej fazy ksylenowej należy pobrać dwukrotnie po 2 cm3 roztworu, dodać 20 cm3 wody, odparować na łaźni wodnej ksylen i miareczkować na gorąco 0,01 molowym NaOH wobec fenoloftaleiny.
Z faz wodnych pobrać dwukrotnie po 5 cm3, dodać 20 cm3 wody i po ogrzaniu na łaźni wodnej miareczkować jak wyżej.
TABELA POMIARÓW I OBLICZEŃ:
Lp. |
Naważki kwasu [g] |
c(1) [mol/dm3] |
c(2) [mol/dm3] |
c(1) (1 - α) |
c(2)/c(1) |
c(2)/c(1) (1-α) |
1. |
0,08 |
0,0052 |
0,0245 |
0,0046 |
4,7 |
5,33 |
2. |
0,15 |
0,0076 |
0,0355 |
0,0069 |
4,7 |
5,14 |
3. |
0,21 |
0,0092 |
0,0640 |
0,0084 |
6,96 |
7,62 |
4. |
0,30 |
0,0108 |
0,0840 |
0,0099 |
7,78 |
8,48 |
III. OBLICZENIA
•Stężenie kwasu benzoesowego dla fazy wodnej:
![]()
cz = 0,01 [mol/dm3]
Vz = 5 [cm3]
c(1) = (0,01⋅ 2,6)/5 = 0,0052 [mol/dm3]
c(1) = (0,01⋅ 3,8)/5 = 0,0076 [mol/dm3]
c(1) = (0,01⋅ 4,6)/5 = 0,0092 [mol/dm3]
c(1) = (0,01⋅5,4)/5 = 0,0108 [mol/dm3]
•Stężenie dla fazy ksylenowej:
![]()
cz= 0,01 [mol/dm3]
Vz = 2 [cm3]
c(2) = (0,01⋅4,9)/2= 0,0245 [mol/dm3]
c(2) = (0,01⋅ 7,1)/2= 0,0355 [mol/dm3]
c(2) = (0,01⋅ 12,8)/2= 0,064 [mol/dm3]
c(2) = (0,01⋅ 16,8)/2= 0,084 [mol/dm3]
•Współczynnik α:
![]()
Kt = 6,46 ⋅10-5
α1= 0,11
α2 = 0,092
α3 = 0,084
α4 = 0,077
•Wartość c(1)(1-α):
c(1)(1-α1) = 0,0052 · (1 - 0,11) = 0,0046
c(1)(1-α2) = 0,0076 ⋅(1-0,092) = 0,0069
c(1)(1-α3) = 0,0092 · (1- 0,084) = 0,0084
c(1)(1-α4) = 0,0108 · (1- 0,077) = 0,0099
•Wartość c(2)/c(1) :
c(2)/c(1) =0,0245 /0,0052 = 4,7
c(2)/c(1) = 0,0355/ 0,0076 = 4,7
c(2)/c(1) = 0,0640/ 0,0092 = 6,96
c(2)/c(1) = 0,0840/ 0,0108 = 7,78
•Wartość c(2)/c(1) (1-α)
c(2)/c(1) (1-α) = 0,0245/ 0,0046 = 5,33
c(2)/c(1) (1-α) = 0,0355/0,0069 = 5,14
c(2)/c(1) (1-α) = 0,0640/0,0084 = 7,62
c(2)/c(1) (1-α) = 0,0840/0,0099 = 8,48
•Wyznaczenie wartości Kc:
Współczynnik podziału kwasu Kc kwasu benzoesowego pomiędzy wodą i ksylenem wyznaczamy graficznie z wykresu
- wartość odczytana z wykresu to 1,2
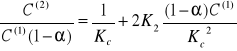
Znając stopień dysocjacji α można z wykresu zależności 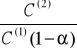
tersus C(1) otrzymać zarówno wartość stałej równowagi reakcji dimeryzacji (K2) jak i współczynnik podziału (Kc)
IV. WYKRES
V. WNIOSKI
Znajomość współczynników podziału substancji pomiędzy dwie niemieszające się fazy ciekłe ma ogromne znaczenie w metodzie rozdziału substancji chemicznych, zwaną metodą ekstrakcyjną. W badanym przypadku rozdział kwasu benzoesowego pomiędzy fazę wodną i ksylemowi, współczynnik podziału jest stosunkowo bliski jedności. Efektywne usunięcie kwasu benzoesowego w tym przypadku z roztworu wodnego do fazy ksylenowej wymagałoby kilkakrotnej ekstrakcji za pomocą kolejnych porcji czystego ksylenu. W wielu jednakże ważnych procesach rozdziału współczynniki podziału są znacznie mniejsze od jedności i nawet jednokrotna ekstrakcja prowadzi do bardzo efektywnego przejścia składnika z jednej fazy do drugiej.
Odchylenia w obliczeniach mogą wynikać z pomyłek w wykonywanym ćwiczeniu.
VI. LITERATURA.
Sobczyk l.,Kisza A.,GatnerK.,Koll A., Eksperymentalna chemia fizyczna, PWN Warszawa 2004,
8
Wyszukiwarka
Podobne podstrony:
553
Oznakowanie IALA Nr 553
ploch 553
FESTOOL 553 pl olejowanie woskowanie
Zeszyty naukowe nr 553 Akademia Ekonomiczna w Krakowie
kpk, ART 553 KPK, III KK 177/05 - wyrok z dnia 7 listopada 2005 r
Juki DDL 552 553 and 555
553
553
553
553
553
552 553
kodeks karny [Dz.U.97.88.553], Licencja Pracownika Ochrony
553
bwv 553 c
553
553
więcej podobnych podstron