ĆWICZENIE I
Enzymy
ZAGADNIENIA DO PRZYGOTOWANIA
1. Budowa enzymów.
2. Zależność szybkości reakcji enzymatycznej od środowiska (temperatura, pH, stężenie enzymu, stężenie substratów, obecność aktywatorów i inhibitorów). Graficzne przedstawienie równania Michaelisa-Menten.
3. Podział enzymów na klasy i ogólne reakcje dla każdej klasy enzymów.
4. Metody wykrywania aktywności enzymów w materiale biologicznym.
WPROWADZENIE
Budowa enzymów
Przemiany biochemiczne przebiegające w organizmie są bardzo skomplikowane i napotykają na ogromne opory fizykochemiczne. Przebieg reakcji umożliwiają bardzo specyficzne biokatalizatory-enzymy, które łącząc się z substratami zmniejszają przeszkody fizykochemiczne uniemożliwiające swobodny przebieg procesu. Obniżenie oporów fizykochemicznych określamy jako obniżenie energii aktywacji. Enzymy zmniejszają barierę energetyczną dla danej reakcji, ale nie wywierają wpływu na zmianę energii swobodnej układu, która jest funkcją stanu początkowego i końcowego reagujących związków, a więc nie zmieniają ilości energii która jest pochłaniana w reakcjach endoenergetycznych lub wydzielana w reakcjach egzoenergetycznych. Enzymy zwiększają jedynie szybkość reakcji i przyśpieszają osiągnięcie stanu równowagi chemicznej, do którego dąży dana reakcja.
Enzymy są zbudowane z białka. W części białkowej enzymu znajduje się zagłębienie - centrum aktywne. Do centrum aktywnego przyłączają się cząsteczki - substraty, które mają ulec przemianie. Powstaje przejściowy kompleks enzym-substrat (ES), następnie zachodzi odpowiednia reakcja i od cząsteczki enzymu odłącza się produkt (P).
E + S ⇔ ES → E + P
Cechą charakterystyczną enzymów jest ich specyficzność, polegająca na katalizowaniu reakcji określonego typu. Enzymy o dużej specyficzności katalizują reakcje tylko dla ściśle określonych substratów. Enzymy o małej specyficzności mogą katalizować ten sam typ reakcji dla wielu różnych substratów.
Często w centrum aktywnym znajdują się dodatkowe cząsteczki, które są niezbędnymi kofaktorami biorącymi czynny udział w katalizowanej reakcji. Część białkowa enzymu nazwana została apoenzymem. Cząsteczki niebiałkowe luźno związane z częścią białkową enzymu w centrum aktywnym i mogące się od niego odłączyć to koenzymy, cząsteczki trwale połączone z apoenzymem to grupy prostetyczne. Bardzo często w skład budowy koenzymów i grup prostetycznych wchodzą witaminy.
Apoenzym + koenzym = holoenzym ( enzym funkcjonalnie czynny)
W reakcjach katalizowanych przez enzymy często uczestniczą dodatkowo jony wapnia, magnezu, cynku, niklu, żelaza, miedzi, manganu, chlorkowe, selenu.. Kofaktorami uczestniczącymi w reakcji mogą być także nukleotydy, np. ATP, CTP, GTP.
Budowa koenzymów i grup prostetycznych jest różnorodna, bardzo wiele z nich zawiera w swoim składzie witaminę z grupy B. Są to: 1) tiamina (witamina B1), 2) ryboflawina (witamina B2), 3) niacyna (kwas nikotynowy, amid kwasu nikotynowego, witamina B3 nazwana także witaminą PP), 4) kwas pantotenowy (witamina B5), 5) witamina B6 (pirydoksyna, pirydoksal, pirydoksamina), 6) biotyna - witamina H, 7) witamina B12 (kobalamina) oraz 8) kwas foliowy (kwas pteroiloglutaminowy). Ponieważ witaminy tej grupy są rozpuszczalne w wodzie, nadmiar ich jest wydalany z moczem. Z tych też przyczyn ich magazynowanie w ustroju jest ograniczone ( z wyjątkiem kobalaminy), co sprawia, że muszą być dostarczane regularnie.
Niedobory witamin powodują zahamowanie wielu procesów katalizowanych przez enzymy, które manifestują się szeregiem objawów chorobowych. Wśród objawów spowodowanych niedoborem wyżej wymienionych witamin są: a) choroba beri-beri (niedobór tiaminy), b) zajady, zapalenie języka, łojotok, światłowstręt (niedobór ryboflawiny), c) pelagra (niedobór niacyny), d) zapalenie nerwów obwodowych (niedobór pirydoksyny), e) niedokrwistość megaloplastyczna, acyduria metylomalonowa i niedokrwistość złośliwa (niedobór kobalaminy), f) niedobór kwasu foliowego powoduje niedokrwistość megaloblastyczną, a w trakcie życia płodowego witamina ta jest niezbędna do prawidłowego rozwoju centralnego układu nerwowego. Niedobory witaminy B12 i kwasu foliowego są czynnikami, które zwiększają zagrożenie miażdżycą.
Występuje także nieliczna grupa kwasów rybonukleinowych (RNA) o właściwościach katalitycznych, są to rybozymy.
Zależność szybkości reakcji enzymatycznej od środowiska
Enzymy są cząsteczkami bardzo wrażliwymi na wpływ środowiska, w którym działają.
Na szybkość reakcji enzymatycznej wpływają:
- pH środowiska,
- temperatura,
- stężenie substratów,
- stężenie enzymu,
- obecność aktywatorów,
- obecność inhibitorów.
Każdy enzym działa najlepiej w środowisku o odpowiednim dla niego odczynie roztworu -pH. Większość enzymów wykazuje największą aktywność w tzw. odczynie bliskim obojętnemu, zbliżonym do pH 7, charakterystycznym dla wody.
Szybkość reakcji katalizowanej przez enzymy zależy od temperatury. Aktywność enzymów rośnie wraz ze wzrostem temperatury, jednak do pewnych granic. Większość enzymów traci zdolność katalizy w temperaturze 600 C, zachodzi wtedy denaturacja (nieodwracalne uszkodzenie) części białkowej enzymu. Wzrost szybkości reakcji biochemicznych można łatwo zaobserwować jako przyśpieszenie tętna u ludzi z podwyższoną temperaturą ciała.
Większość enzymów to enzymy o stałej kinetyce reakcji zwanej hiperboliczną. Szybkość ich działania zależy głównie od stężenia substratów w danych warunkach optymalnych dla działania danego enzymu. Jeśli ilość enzymu w kolejnych próbkach jest stała to szybkość katalizowanej reakcji jest uzależniona od stężenia substratów. Zależność szybkości reakcji od stężenia substratów przedstawiono na rys.1.
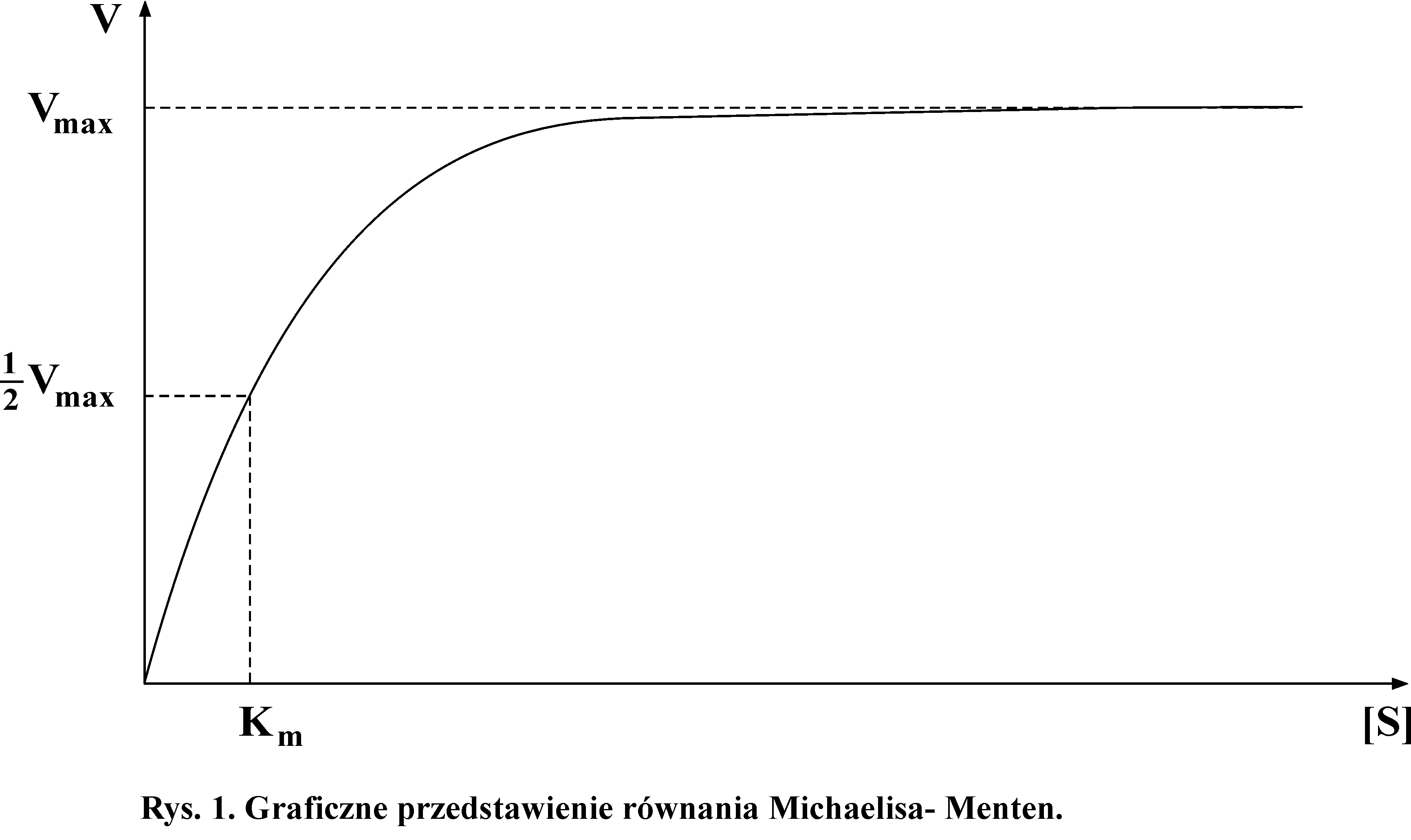
Ponieważ enzym łączy się z substratem tworząc kompleks enzym-substrat, to przy odpowiednio wysokim stężeniu substratu następuje wysycenie substratem enzymu. Dalszy wzrost stężenia substratu już nie powoduje wzrostu szybkości reakcji. Przy maksymalnym wysyceniu enzymu substratem reakcja osiąga szybkość maksymalną (Vmax). Zależność szybkości reakcji od stężenia substratu przedstawia się jako hiperbola. Hiperboliczny przebieg reakcji opisuje równanie wyprowadzone przez Michaelisa -Menten.
Analiza matematyczna powyższego wykresu pozwala na określenie wartości stałej Michaelisa. Stała Michaelisa jest wartością stałą dla danego enzymu i określa jego powinowactwo do substratu, a więc szybkość powstawania kompleksu enzym-substrat.. Mała wartość stałej Michaelisa wskazuje na duże powinowactwo do substratu i dużą szybkość reakcji, duża wartość stałej świadczy o małym powinowactwie do substratu i mniejszej szybkości reakcji. Zmiany wielkości stałej Michaelisa dla danego enzymu wskazują na zmiany struktury białka, a więc na zmiany w obrębie odpowiedniego genu. Zmiana wartości tej stałej może także wskazywać na obecność inhibitorów, związków, które zakłócają przebieg reakcji.
Wartość stałej Michaelisa można stosunkowo łatwo oznaczyć eksperymentalnie, badając zależność szybkości reakcji od stężenia substratu w teście standardowym (rys.1). Stałą Michaelisa można przedstawić w jednostkach stężenia substratu (mol/dm3) wtedy, kiedy reakcja osiąga połowę szybkości maksymalnej.
Niektóre enzymy wymagają do swojego działania aktywatorów, które stabilizują ich właściwą strukturę, są to np. jony magnezu, wapnia, cynku, manganu i inne.
Szybkość przemiany biochemicznej zależy także od ilości cząsteczek enzymów w komórce. Ilość enzymu zależy od aktywności genów kodujących dane białka. Ekspresja poszczególnych genów podlega bardzo skomplikowanym i precyzyjnym mechanizmom regulacyjnym. Stężenie enzymów najważniejszych przemian np. glikolizy, cyklu kwasów trikarboksylowych (Krebsa) jest względnie stałe, charakterystyczne dla danej tkanki. Ilość enzymów biorących udział w procesach detoksykacji ustroju, enzymów cyklu mocznikowego może wzrastać wielokrotnie.
Prawidłowe działanie enzymów może być zmienione za pomocą różnych związków chemicznych. Substancje, które hamują reakcje enzymatyczne można podzielić na inhibitory kompetycyjne i niekompetycyjne.
Mechanizm działania inhibitorów kompetycyjnych polega na współzawodnictwie między substratem a inhibitorem o centrum aktywne enzymu i jest odwracalne. Inhibitor kompetycyjny jest bardzo podobny strukturalnie do właściwego substratu, łączy się z enzymem tworząc nieaktywny kompleks enzym-inhibitor. Stopień zahamowania reakcji zależy od stosunku stężeń inhibitora i substratu. Zwiększenie stężenia substratu może cofnąć tworzenie kompleksu enzym-inhibitor i znieść działanie inhibitora. Mechanizm hamowania kompetycyjnego wykorzystany został do hamowania kluczowego enzymu w syntezie cholesterolu, co pozwala na skuteczne obniżenie tego związku we krwi. Niektóre leki stosowane w terapii nowotworów również działają na tej zasadzie.
Hamowanie niekompetycyjne polega na przyłączaniu się inhibitora w sposób odwracalny do części białkowej poza centrum aktywnym. Zwiększenie stężenia substratu nie cofa działania inhibitora.
Jeśli inhibitor wiąże się w sposób trwały z białkiem, uszkadza nieodwracalnie strukturę enzymu i zmniejsza szybkość katalizowanej reakcji. Często tego typu inhibitory łączą się z aminokwasami występującymi w centrum aktywnym i całkowicie inaktywują enzym. Na przykład diizopropylofluorofosforan (DIPF), składnik gazów bojowych działających na układ nerwowy, reaguje z resztą aminokwasu seryny w centrum aktywnym enzymu - esterazy acetylocholiny . Następuje nieodwracalnie zahamowanie aktywności enzymu, co uniemożliwia przekazywanie impulsów nerwowych.
Regulacja aktywności enzymów
Szybkość przemian biochemicznych jest regulowana przez enzymy. Aktywność większości enzymów zależy od dostępności substratów, odpowiedniego pH środowiska, ewentualnych aktywatorów i ich ilości w komórce.
W organizmie występują szczególne enzymy, których szybkość katalizy zmienia się w zależności od stanu fizjologicznego organizmu. Charakteryzują się zmienną kinetyką działania. Enzymy takie nazywamy enzymami regulatorowymi. Jeśli dany proces biochemiczny składa się z kilku etapów, to zazwyczaj pierwszy etap przemiany jest katalizowany przez enzym regulatorowy, decydujący o tempie całego procesu.
Typowymi enzymami regulatorowymi są enzymy allosteryczne. Zbudowane są one z kilku podjednostek białkowych. Enzymy te posiadają podjednostki katalityczne z centrami aktywnymi, odpowiedzialnymi za katalizę i podjednostki regulatorowe z centrami allosterycznymi, które są odpowiedzialne za odbiór informacji ze strony komórki. Do centrów allosterycznych przyłączają się ligandy allosteryczne. Ligandy allosteryczne to związki chemiczne, które pojawiają się w danym stanie fizjologicznym komórki i są substancjami sygnałowymi. Przyłączenie ligandu allosterycznego do centrum allosterycznego powoduje zmianę kształtu części białkowej enzymu. Zmiana struktury podjednostek katalitycznych wywołuje zmianę szybkości reakcji, adekwatną do informacji docierającej ze strony komórki.
Efektory (ligandy) allosteryczne mogą zwiększać lub zmniejszać szybkość katalizowanej reakcji. Enzymy allosteryczne reagują również na stężenie substratów. Przy niskich stężeniach substratów aktywność tych enzymów jest bardzo mała. Enzymy allosteryczne charakteryzują się zmienną kinetyką dostosowując szybkość reakcji do stanu fizjologicznego panującego we wnętrzu komórki.
Aktywność enzymów może także zależeć od zmian struktury części białkowej enzymu. Wiele enzymów zmienia szybkość reakcji pod wpływem fosforylacji polegającej na przyłączeniu do części białkowej fosforanu pochodzącego z ATP. Odpowiednie enzymy- kinazy białkowe przenoszą jedną resztę fosforanową z ATP na resztę aminokwasową seryny, rzadziej treoniny i tyrozyny. Powstaje ufosforylowana postać enzymu, w zależności od typu enzymu enzym staje się bardziej lub mniej aktywny. Procesy fosforylacji i defosforylacji enzymów są najczęściej wywołane działaniem hormonów. Od fosforylacji i defosforylacji enzymów zależy na przykład synteza i rozpad glikogenu, wielocukru znajdującego się w mięśniach i wątrobie, będącego rezerwuarem glukozy.
Innym sposobem regulacji aktywności enzymów jest ograniczona proteoliza. Polega ona na odłączeniu od białka fragmentu peptydu, który blokuje centrum aktywne. Po odłączeniu peptydu enzym staje się aktywny. Enzymy trawienne są wydzielane w formie proenzymów, (zymogenów) i ulegają aktywacji na skutek ograniczonej proteolizy w świetle przewodu pokarmowego (np. pepsynogen w pepsynę w świetle żołądka po wpływem kwasu solnego).
Klasy enzymów
Typ reakcji katalizowanej przez enzym stał się podstawą klasyfikacji enzymów. Zgodnie z tą zasadą Międzynarodowa Unia Biochemiczna dokonała podziału enzymów na sześć głównych klas jako podstawę klasyfikacji przyjęto typ katalizowanej reakcji przez dany enzym.. Każdy enzym uzyskał ściśle określający go numer klasyfikacyjny (np. glutaminaza EC 3.5.1.2).
Pierwszą klasą enzymów są oksydoreduktazy. Enzymy należące do tej klasy katalizują reakcje utleniania i redukcji, a więc reakcje związane ze zmianą stopni utlenienia cząsteczek.
A- + B ↔ A + B -
Do tej klasy należą takie grupy enzymów między innymi: dehydrogenazy odrywające atomy wodoru od substratów, reduktazy przyłączające do cząsteczek atomy wodoru, peroksydazy i katalazy rozkładające nadtlenki, oksydazy przenoszące elektrony na tlen. Większość oksydoreduktaz wymaga do swojego działania koenzymów takich jak: a) dinukleotyd nikotynamidoadeninowy - NAD+ oraz fosforan dinukleotydu nikotynamidoadeninowego - NADP+, które są aktywnymi postaciami niacyny (witaminy B3 = PP) lub dinukleotyd flawinoadeninowy - FAD (aktywne postacie ryboflawiny witaminy B2).
Drugą klasą są transferazy, enzymy katalizujące reakcje, w których dochodzi do przeniesienia fragmentu czasteczki z jednego substratu na drugi. Niektóre transferazy wymagają do swojego działania koenzymów (np. fosforanu pirydoksalu - witaminy B6, pirofosforanu tiaminy - witaminyB1). Do tej klasy należą takie grupy enzymów jak: aminotransferazy przenoszące grupy aminowe, kinazy (fosfotransferazy, tworzące estry fosforanowe przy użyciu ATP).
Ogólna reakcja katalizowana przez transferazy:
A + B -X → A-X + B.
Hydrolazy (trzecia klasa enzymów) katalizują reakcje hydrolizy, nie wymagają do swojego działania koenzymów, natomiast często współdziałają z jonami wapnia bądź magnezu. Do tej klasy należą np.: glikozydazy rozkładające wiązania glikozydowe wielocukrów, esterazy rozkładające wiązania estrowe, peptydazy rozkładające wiązania peptydowe występujące w białkach. Ogólna reakcja katalizowana przez hydrolazy:
A-B + H2O → A-OH + BH.
Liazy (czwarta klasa) katalizują reakcje rozpadu wiązań lub ich syntezy, która przebiega bez dostarczenia energii z rozpadu ATP. Należą tu takie grupy enzymów jak: syntazy, dekarboksylazy. Ogólna reakcja katalizowana przez liazy:
A-B ↔ A + B.
Izomerazy (piąta klasa) katalizują reakcje wewnętrznej przebudowy cząsteczek - izomeryzacji
A-B-C ↔ A-C-B.
Ligazy (szósta klasa) przeprowadzają reakcje syntezy wiązań, które wymagają energii dostarczanej najczęściej z rozkładu wiązań wysokoenergetycznych ATP. Należą do tej klasy syntetazy i karboksylazy.
Ogólna reakcja katalizowana przez ligazy: A + B + ATP → A-B + ADP + Pi lub
A + B + ATP → A-B + AMP + PPi
Oznaczanie aktywności enzymów odrywa ogromną rolę w badaniach podstawowych, w poszukiwaniu nowych leków zwalniających bądź przyśpieszających dany proces biochemiczny, w analizach toksylogicznych. Oznaczanie aktywności enzymów pomaga w diagnozowaniu bardzo wielu chorób.
Oznaczanie aktywności enzymów w materiale biologicznym polega na pomiarze szybkości reakcji katalizowanej przez enzymy, tzn. na określeniu zmiany stężenia (ilości) substratu lub produktu w jednostce czasu (przy użyciu enzymu pochodzącego z określonej masy materiału biologicznego np. z 1 g tkanki lub 1 cm3 osocza).
Jednostka enzymatyczna (U) jest to ilość enzymu, która katalizuje przekształcenie 1 mikromola substratu w ciągu 1 minuty w odpowiednim dla danego enzymu pH roztworu, w temperaturze 300 C.
W oznaczeniach diagnostycznych często stosuje się jednostki umowne, w których wyskalowano aktywność danego enzymu.
Często wykonywanymi badaniami o znaczeniu diagnostycznym jest oznaczanie w osoczu na przykład poziomu aktywności w osoczu aminotransferazy alaninowej, aminotransferazy asparaginianowej, kinazy kreatynowej, dehydrogenazy mleczanowej, których aktywność rośnie przy schorzeniach serca, mięśni czy wątroby. Natomiast obniżenie aktywności acylotransferazy lecytyna - cholesterol (LCAT) wiąże się z rozwojem procesów miażdżycowych.
Celem ćwiczenia jest oznaczanie aktywności wybranych enzymów z klasy oksydoreduktaz i hydrolaz, w optymalnych dla nich warunkach środowiska.
CZĘŚĆ DOŚWIADCZALNA
Przykłady działania enzymów z różnych klas
Oksydoreduktazy - pierwsza klasa enzymów
1. Wykrywanie aktywności katalazy
Katalaza katalizuje reakcję rozkładu toksycznego nadtlenku wodoru - H2O2:
H2O2 + H2O2 → 2H2O + O2
Katalaza jest hemoproteiną o bardzo dużej aktywności katalitycznej. Występuje we wszystkich tkankach u zwierząt, głównie w krwinkach czerwonych, wątrobie i nerce. Katalaza jest jednym z enzymów chroniących komórki przed toksycznym działaniem reaktywnych form tlenu, które niszczą lipidy tworzące błony komórkowe, białka a nawet kwasy nukleinowe.
Zasada metody
W obecności katalazy następuje rozpad H2O2 do wody i tlenu; wydzielanie się pęcherzyków gazu świadczy o obecności enzymu.
Materiał i odczynniki
1. 3% roztwór H2O2
2. Krew
3. Krew zagotowana
Wykonanie doświadczenia
Do pierwszej probówki odmierzyć 1 cm3 wody i dodać dwie krople krwi.
Do drugiej probówki odmierzyć 1 cm3 wody i dodać dwie krople krwi zagotowanej.
Do obu probówek dodawać kroplami roztwór 3% roztwór wody utlenionej.
O obecności katalazy świadczyć będzie tworzenie się piany.
2. Wykrywanie aktywności peroksydazy
Reakcja katalizowana przez peroksydazę:
Substrat-H2 zred. + H2O2 → Substratutl. + 2H2O
Peroksydaza jest enzymem występującym powszechnie w roślinach, szczególnie dużo tego enzymu zawiera chrzan. Katalizuje reakcję utleniania substratów w obecności nadtlenku wodoru.
U zwierząt w komórkach różnych tkanek występuje peroksydaza glutationowa. Peroksydaza glutationowa jest bardzo ważnym enzymem usuwającym nadtlenki kwasów tłuszczowych, nadtlenek wodoru, chroni grupy -SH w białkach przed niepożądanym utlenieniem. Jest najważniejszym enzymem chroniącym strukturę błon komórkowych, które na skutek rozpadu kwasów tłuszczowych mogłyby ulegać zniszczeniu. W reakcji katalizowanej przez peroksydazę bierze udział glutation (G-SH), który zbudowany jest z trzech aminokwasów i jest dawcą atomów wodoru niezbędnych do przebiegu reakcji.
2 G-SH + H2O2 Ⴎ GS-SG + 2 H2O
W organizmie tworzy się także niezwykle toksyczna, reaktywna forma tlenu-anionorodnik ponadtlenkowy, który jest usuwany przez dysmutazę ponadtlekową . W usuwaniu toksycznych form tlenu biorą udział także związki drobnocząsteczkowe jak na przykład witamina C, E, A, kwas moczowy. Atak tlenu na struktury komórkowe jest jednym z najważniejszych czynników wywołujących starzenie. Długotrwałe podawanie tlenu pacjentowi potęguje procesy degradacyjne i może być niebezpieczne.
Zasada metody
W obecności peroksydazy benzydyna ulega utlenieniu przez nadtlenek wodoru do difenochinonodiiminy, która kondensując z cząsteczką zredukowanej benzydyny tworzy błękit benzydynowy .
Benzydyna H2 (bezbarwna) + H2O2 Ⴎ benzydyna ( niebieska) + 2 H2O
Materiał i odczynniki
1. 1% roztwór benzydyny w lodowatym kwasie octowym
2. 3% roztwór H2O2
4. Ziemniak surowy
5. Ziemniak gotowany
Wykonanie doświadczenia
Przygotować odczynnik benzydynowy: zmieszać równe objętości roztworu benzydyny w lodowatym kwasie octowym i roztworu H2O2.
Na plasterek surowego ziemniaka nanieść kilka kropli odczynnika benzydynowego.
Tę samą czynność wykonać z plasterkiem ziemniaka gotowanego
Porównać obie próby.
3. Wykrywanie aktywności dehydrogenazy bursztynianowej (SDH - ang. succinate dehydrogenase)
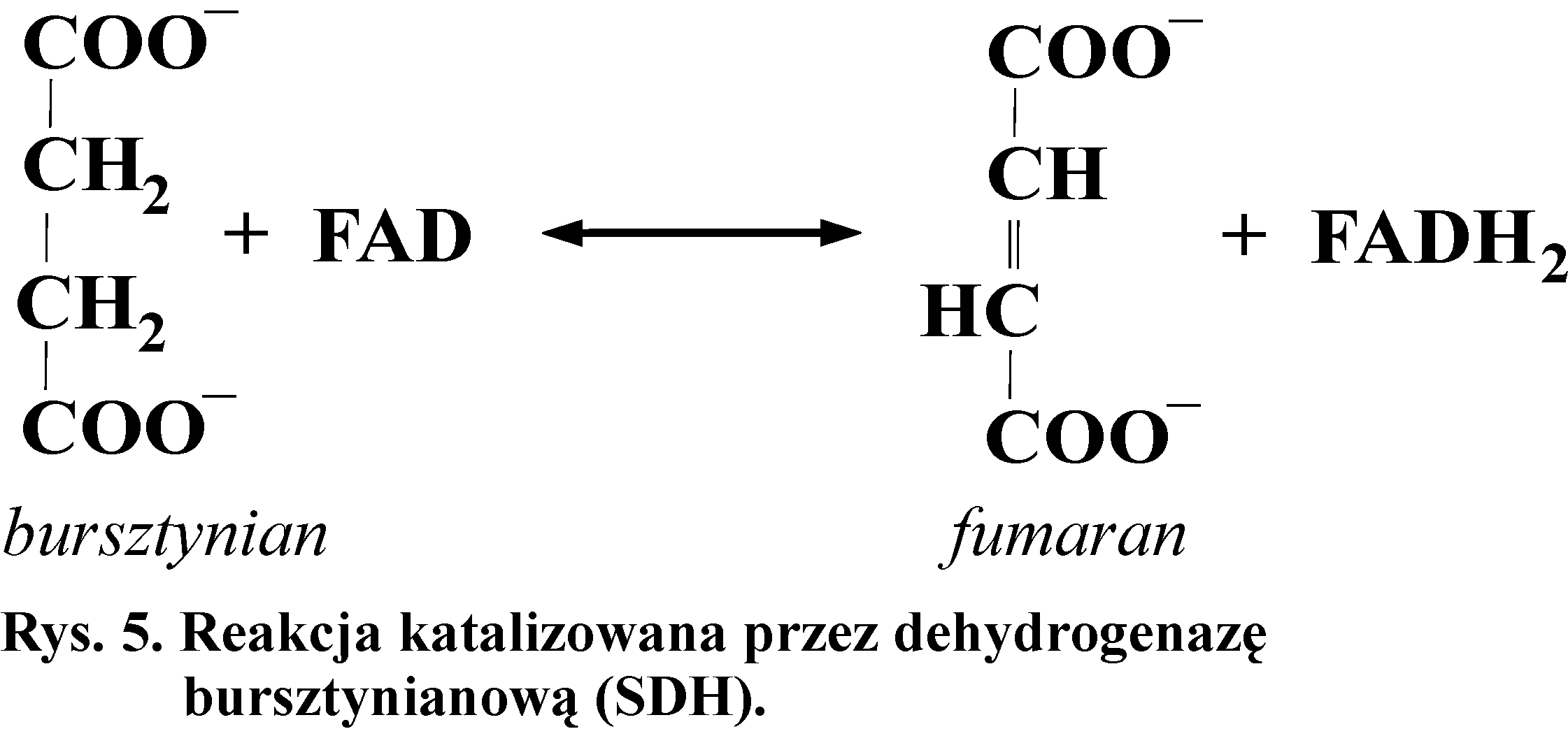
Dehydrogenaza bursztynianowa (SDH) jest mitochondrialną żelazoflawoproteiną. Jest enzymem cyklu Krebsa i stanowi odgałęzienie łańcucha oddechowego. Katalizuje ona odwracalną reakcję utleniania bursztynianu z wytworzeniem fumaranu, grupą prostetyczną enzymu jest FAD (Rys. 5). Inhibitorem kompetycyjnym tego enzymu jest m.in. malonian. Inhibitor kompetycyjny to związek, którego budowa przestrzenna jest podobna do substratu. Hamuje on reakcję enzymatyczną, gdyż przyłącza się, zamiast substratu, do centrum aktywnego enzymu.
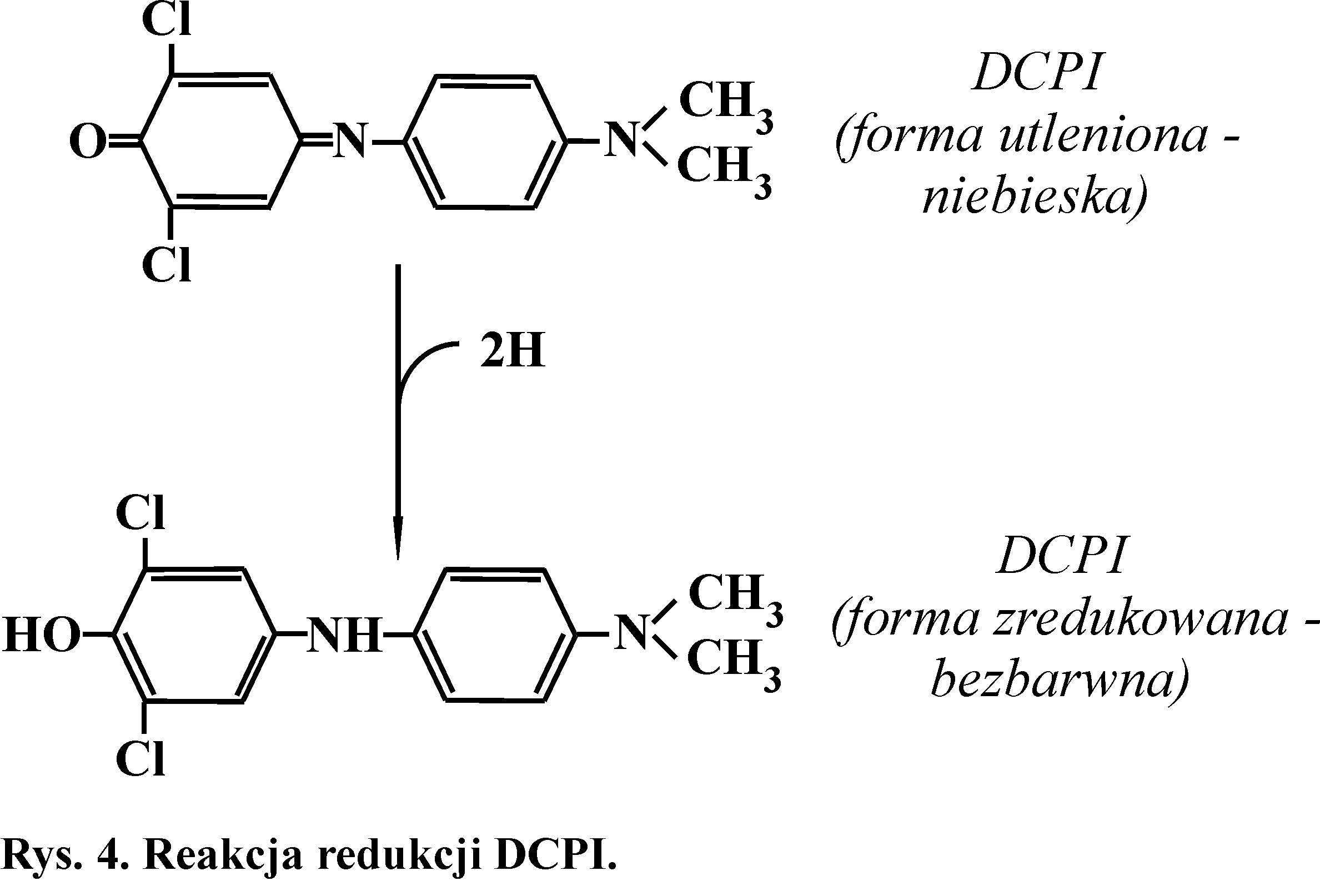
Zasada metody
DCPI jest redukowany przez FADH2 (lub inne zredukowane pośredniki łańcucha oddechowego) (Rys. 4). Sam bursztynian nie redukuje DCPI.
Materiał i odczynniki
1. 0,01 mol/dm3 bufor Tris-HCl pH 7,4
2. 0,01% roztwór DCPI
3. 1 mol/dm3 roztwór bursztynianu sodu
4. 0,5 mol/dm3 roztwór malonianu sodu
5. 5% homogenat wątroby
Wykonanie doświadczenia
Do dwóch probówek dodać odczynniki według tabeli 2.
Tabela 2.
|
1 |
2 |
3 |
|
Ilości dodawanych odczynników w cm3 |
||
0,01 mol/dm3 bufor Tris-HCl pH 7,4 |
1 |
1 |
1 |
0,01% DCPI |
0,6 |
0,6 |
0,6 |
1 mol/dm3 bursztynian |
0,2 |
0,2 |
- |
0,5 mol/dm3 malonian |
- |
0,5 |
- |
zadanie |
- |
- |
0,2 |
5% homogenat wątroby |
0,5 |
0,5 |
0,5 |
H2O |
0,5 |
- |
0,5 |
Zawartość probówek wymieszać, probówki wstawić do łaźni wodnej o temperaturze 370 C i obserwować zmianę zabarwienia. Odbarwienie roztworu świadczy o obecności SDH.
Zwrócić uwagę na hamowanie aktywności enzymu przez malonian.
4. Wykrywanie aktywności oksydazy difenolowej
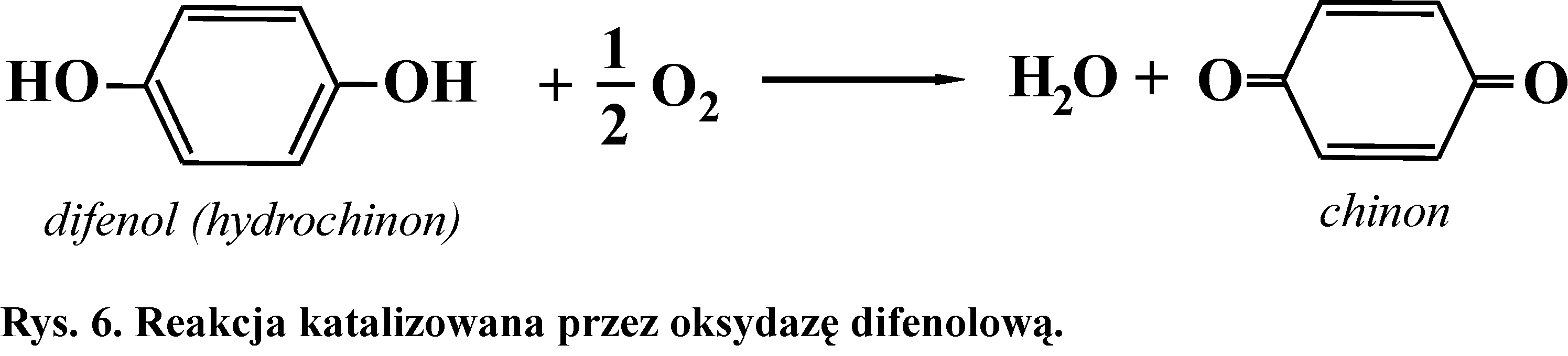
Oksydaza fenolowa (enzym roślinny) jest miedzioproteiną. Katalizuje utlenianie difenoli do chinonów, które są związkami barwnymi. Akceptorem elektronów jest tlen cząsteczkowy (Rys. 6).
Zasada metody
Pod wpływem oksydazy difenolowej znajdującej się w ziemniaku difenole zawarte w żywicy gwajakowej utleniają się do produktów, które mają niebieskie zabarwienie.
Materiał i odczynniki
1. 6% roztwór żywicy gwajakowej w 96% etanolu
2. Ziemniak surowy
3. Ziemniak gotowany
Wykonanie doświadczenia
Kawałek ziemniaka świeżego i ugotowanego zwilżyć kilkoma kroplami nalewki gwajakowej.
Obserwować zmiany.
Hydrolazy - trzecia klasa enzymów
5. Wykrywanie aktywności inwertazy (hydrolazy sacharozy - sacharazy,
β-fruktofuranozydazy)
Inwertaza katalizuje hydrolityczny rozkład sacharozy (dwucukru o właściwościach nieredukujących) (Rys. 8). Produktami reakcji są glukoza (cukier o właściwościach redukujących) i fruktoza.
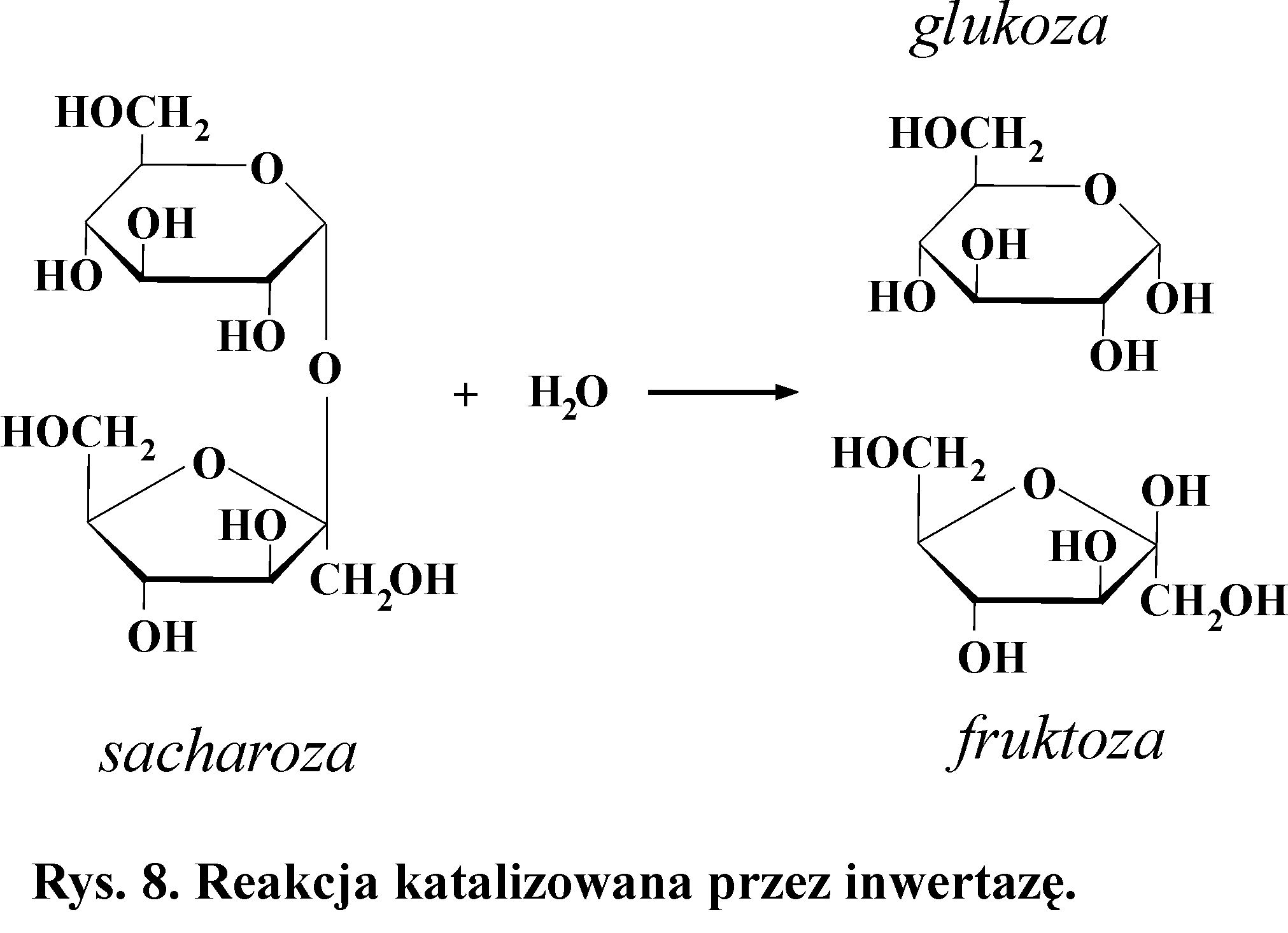
Zasada metody
Glukoza w środowisku zasadowym redukuje Cu2+ do Cu+. Powstaje wtedy trudno rozpuszczalny, czerwony osad Cu2O. (W środowisku zasadowym fruktoza również może dawać pozytywną próbę, gdyż ulega przekształceniu w glukozę).
Materiał i odczynniki
1. 0,2 mol/dm3 roztwór sacharozy
2. 1 mol/dm3 roztwór NaOH
3. Odczynnik Fehlinga I (11% roztwór CuSO4)
4. Odczynnik Fehlinga II (5% NaOH, 1,65% winian sodowo-potasowy)
5. Roztwór inwertazy (w buforze octanowym pH 4,7)
6. Zagotowany roztwór inwertazy (w buforze octanowym pH 4,7)
Wykonanie doświadczenia
Przygotować trzy ponumerowane probówki.
Do pierwszej i drugiej probówki odmierzyć po 2 cm3 0,2 mol/ dm3 roztworu sacharozy, a do trzeciej 2 cm3 zadania.
Do pierwszej i trzeciej dodać 1 cm3 roztworu enzymu, a do drugiej 1 cm3 enzymu zagotowanego.
Wstawić na 15 minut do łaźni wodnej o temperaturze 370 C.
Po inkubacji do wszystkich probówek dodać po 5 kropli 1 mol/dm3 roztworu NaOH, następnie po 1 cm3 odczynnika Fehlinga II, wymieszać i dodać po 1 cm3 odczynnika Fehlinga I (zawierającego Cu2+).
Zagotować.
Pojawienie się czerwonego zabarwienia roztworu (lub czerwonego osadu) świadczy o aktywności enzymu.
Zaobserwować, czy w zadaniu roztwór staje się czerwony (świadczy o obecności sacharozy).
6.Wykrywanie aktywności ureazy
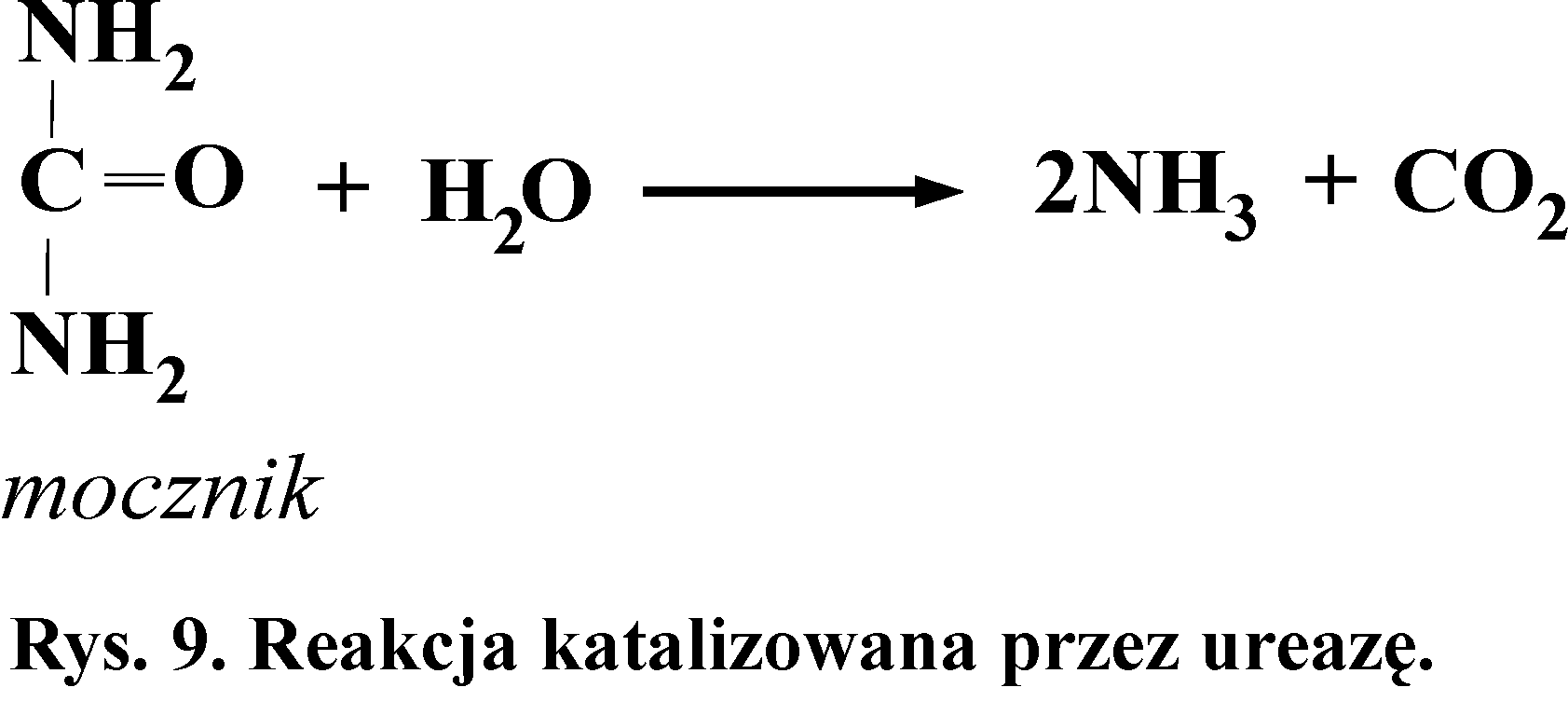
Ureaza (aminohydrolaza mocznika) katalizuje reakcję hydrolitycznego rozpadu mocznika (Rys. 9). Enzym ten nie występuje w tkankach ssaków, znajduje się w nasionach niektórych roślin, np. w soi. Występuje też w bakteriach. Stąd obecność tego enzymu w płytce nazębnej i ślinie.
Zasada metody
Utworzony podczas rozkładu mocznika amoniak reaguje z wodą, a powstające w tej reakcji jony OH- powodują alkalizację środowiska:
NH3 + H2O ↔ NH4+ + OH-
Materiał i odczynniki
1. 2% roztwór mocznika
2. 1% roztwór fenoloftaleiny w 96% etanolu
3. r-r ureazy z ziarna soi w buforze Tris/HCl pH 7,4
Wykonanie doświadczenia
Przygotować dwie probówki.
Do pierwszej dodać 1 cm3 2% roztworu mocznika, a do drugiej 1 cm3 zadania.
Do obu odmierzyć 0,5 cm3 roztworu ureazy.
Następnie do obu dodać jedną kroplę roztworu fenoloftaleiny..
Dla przyspieszenia reakcji probówki umieścić w łaźni wodnej o temperaturze 370 C.
Pojawienie się malinowego zabarwienia roztworu świadczy o aktywności enzymu. (Fenoloftaleina w środowisku zasadowym barwi się na malinowo, a w środowisku kwaśnym i obojętnym jest bezbarwna).
7. Zaobserwować, czy w zadaniu bezbarwny początkowo roztwór staje się malinowy.
7. Zadanie: polega na określeniu substratu dla odpowiedniego enzymu. Student otrzymuje roztwór zawierający jeden z niżej podanych substratów:
1) Bursztynian
2) Sacharoza
3) Mocznik.
ĆWICZENIE II
GLIKOGEN
ZAGADNIENIA DO PRZYGOTOWANIA
1. Struktura glikogenu.
2. Synteza i rozpad glikogenu (reakcje wzorami i enzymy). Hormony
regulujące te procesy.
3. Rola glikogenu wątrobowego i mięśniowego.
4. Metody izolowania i oznaczania procentowej zawartości glikogenu w
wątrobie.
Tematy na samokształcenie do zaliczenia podczas ćwiczenia 2:
II. 3.) ; III. 1. 2. 3. 4. 5.
WPROWADZENIE
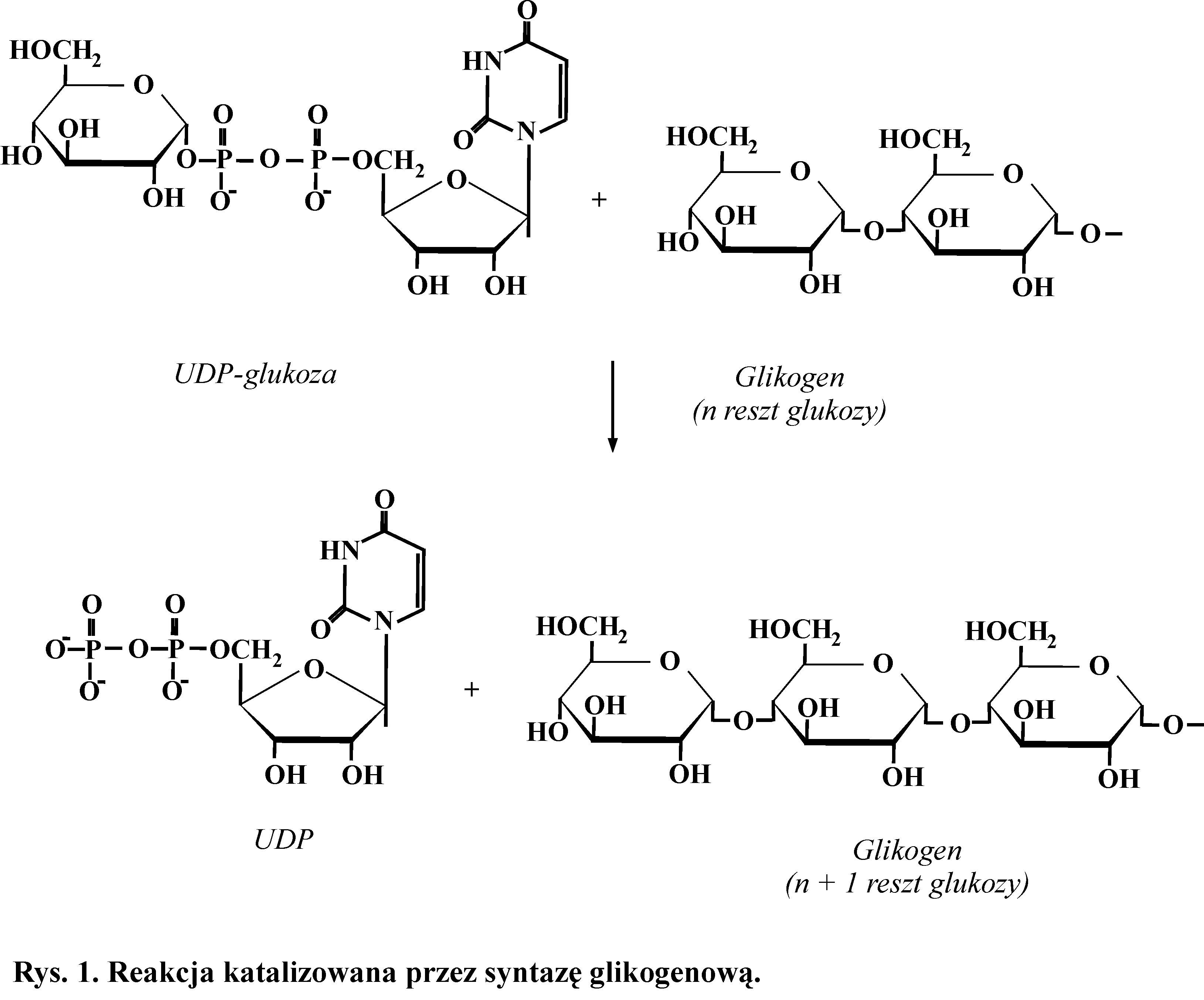
Glikogen jest zapasowym materiałem energetycznym zwierząt i człowieka. Jest to rozgałęziony homoglikan, zbudowany z jednostek α,D-glukopiranozowych, które są połączone dwoma typami wiązań glikozydowych: α-1,4 i występującym średnio co dziewięć reszt glukozowych wiązaniem α-1,6.
Glikogen jest magazynowany głównie w wątrobie i mięśniach. Ilość glikogenu w wątrobie dochodzi do 6-8% masy tego narządu, natomiast w mięśniach jego zawartość może wynosić 0,7%. Jednak mięśnie, z powodu dużej masy, zawierają 3-4-krotnie większy zapas glikogenu niż wątroba. Rezerwa glukozy znajdująca się w glikogenie wątrobowym służy do utrzymywania odpowiedniego stężenia tego związku we krwi w przerwach między posiłkami. Prawidłowe stężenie glukozy we krwi waha się niewielkich granicach i wynosi 3,9-6,1 mmola/dm3 (70-110 mg/100 cm3). Zapas glikogenu zgromadzony w wątrobie wystarcza na uzupełnianie stężenia glukozy przez 12-16 godzin.
Utrzymywanie stałego stężenia glukozy we krwi jest niezwykle istotne dla organizmu. Glukoza stanowi podstawowe źródło energii dla większości tkanek. Ciągłe dostarczanie glukozy jest konieczne do sprawnego funkcjonowania układu nerwowego i erytrocytów, dla których glukoza jest jedynym materiałem energetycznym. Neurony nie katabolizują kwasów tłuszczowych i przy normalnej diecie glukoza jest głównym źródłem energii potrzebnej do podtrzymania procesów życiowych tkanki nerwowej. Już poniżej stężenia 70 mg glukozy na 100 cm3 pojawiają się pierwsze objawy hipoglikemii jak np.: senność, drażliwość, bóle głowy i poczucie głodu. Przy bardzo niskim stężeniu glukozy we krwi występuje zaburzenie czynności mózgu, które może doprowadzić do śpiączki a nawet zgonu.
W stanach przedłużonego głodzenia lub przy braku węglowodanów w pożywieniu większość energii niezbędnej dla życia neuronów pochodzi z katabolizmu tzw. ciał ketonowych - acetooctanu i 3-hydroksymaślanu, których synteza nasila się w tych warunkach. Związki te są także metabolizowane przez inne tkanki z wyłączeniem wątroby i krwinek czerwonych. Jednak podczas głodzenia stężenie glukozy nie powinno spadać znacznie poniżej dolnych granic normy. Niezbędna ilość glukozy wytwarzana jest wtedy na drodze glukoneogenezy - syntezy glukozy z substancji niewęglowodanowych.
Glikogen zmagazynowany w mięśniach nie służy utrzymywaniu stałego stężenia glukozy we krwi, a jedynie dostarcza energii w trakcie przedłużonego skurczu mięśnia. Po 12-18 godzinach głodzenia wątroba zostaje niemal zupełnie pozbawiona glikogenu, natomiast zapas glikogenu w mięśniach zmniejsza się po długotrwałym, intensywnym wysiłku.
Syntezę glikogenu katalizują dwa enzymy z klasy transferaz. Syntaza glikogenowa, która tworzy wiązania α-1,4-glikozydowe i enzym rozgałęziający (amylo[α-1→4]→[α-1→6]-transglukozydaza), który syntetyzuje wiązania α-1,6-glikozydowe. Syntaza glikogenowa może jedynie wydłużać już istniejący łańcuch. Do rozpoczęcia reakcji potrzebny jest inicjator, białko - glikogenina. Dodatkowy enzym, glukozylotransferaza, przyłącza kolejno do grupy -OH specyficznej reszty seryny w tym białku kilka reszt glukozy, łącząc je wiązaniami α-1,4- glikozydowymi. Właściwa reakcja katalizowana jest przez syntazę glikogenową. Substratem do syntezy glikogenu jest UDP-glukoza (Rys. 1).
Gdy łańcuch zostanie przedłużony, do co najmniej 11 reszt glukozowych, wówczas enzym rozgałęziający przenosi część łańcucha z jednego rozgałęzienia na inne, tworząc wiązanie α-1,6 glikozydowe i ustanawiając w ten sposób nowy punkt rozgałęzienia. Powstaje cząsteczka bardzo rozgałęziona. Zwiększenie liczby nieredukujących zakończeń łańcuchów glukozowych umożliwia przyśpieszenie zarówno syntezy glikogenu, jak i jego rozkładu na drodze fosforolizy.
Głównym enzymem przeprowadzającym rozpad glikogenu (glikogenolizę) jest fosforylaza glikogenowa, należąca do klasy transferaz. Enzym ten przy udziale ortofosforanu prowadzi fosforolizę glikogenu od nieredukujących zakończeń cząsteczki tworząc glukozo-1-fosforan.
glikogen(n reszt glukozy) + Pi → glikogen(n-1 reszt glukozy) + glukozo-1-fosforan
Fosforylaza glikogenowa rozłącza tylko wiązania α-1,4-glikozydowe, degradując stopniowo łańcuchy glukozowe, aż do miejsc oddalonych o cztery reszty glukozowe od punktu rozgałęzienia, stworzonego za pomocą wiązania α-1,6. Następnie enzym odgałęziający, α-[1→4]→[1→4]-transferaza glukanowa przenosi fragment trzech reszt glukozy na łańcuch główny. W ten sposób odsłania się miejsce odgałęzienia. Rozkład wiązań α-1,6 katalizuje hydrolaza - amylo-α-[1→6]-glukozydaza, która uwalnia wolną glukozę.
Glukozo-1-fosforan, będący produktem działania fosforylazy, zostaje przekształcony w glukozo-6-fosforan za pomocą fosfoglukomutazy.
Dalsze losy glukozo-6-fosforanu zależą od rodzaju tkanki. W wątrobie występuje enzym fosfataza glukozo-6-fosforanowa, przekształcająca glukozo-6-fosforan w glukozę. Tylko wolna glukoza może być przenoszona przez błonę komórkową; dokonuje tego znajdujący się w błonie komórkowej układ transportujący. Rozkład glikogenu wątrobowego powoduje wzrost stężenia glukozy we krwi.
W mięśniach nie występuje fosfataza glukozo-6-fosforanowej i brak tego enzymu uniemożliwia przekształcenie glukozo-6-fosforanu do wolnej glukozy. Uwolniony glukozo-6-fosforan służy jako materiał energetyczny wykorzystywany podczas skurczu mięśnia.
Rozkład i synteza glikogenu zależy głównie od wpływu hormonów. Kontrolę nad tworzeniem i rozkładem glikogenu prowadzą przede wszystkim dwa hormony- glukagon i insulina. Glikogenolizę w mięśniach i wątrobie może także wywołać hormon stresu - adrenalina.
CZĘŚĆ DOŚWIADCZALNA
1. Izolowanie glikogenu z wątroby zwierzęcej.
Zasada metody
Wiązania glikozydowe występujące w glikogenie są oporne na działanie hydrolityczne jonów OHˉ w podwyższonej temperaturze. W wysokiej temperaturze w środowisku zasadowym wiązania peptydowe białek, estrowe lipidów i fosfodiestrowe kwasów rybonukleinowych ulegają hydrolizie. W wyniku ogrzewania tkanki w roztworze KOH otrzymuje się roztwór glikogenu zanieczyszczony innymi wielocukrami, fragmentami zdenaturowanego DNA oraz związkami małocząsteczkowymi, które znajdowały się w tkance lub powstały na skutek hydrolizy. Dodanie alkoholu etylowego powoduje wytrącenie glikogenu, który jest w niewielkim stopniu zanieczyszczony zdenaturowanym DNA i wysokocząsteczkowymi heteroglikanami, które również strącają się w tych warunkach.
Odczynniki
1. 30% roztwór KOH.
2. 96% etanol.
Wykonanie doświadczenia
1. Do 1 g wątroby umieszczonego w probówce wirówkowej dodać 2,5 cm3 30%
roztworu KOH.
2. Probówkę zakryć korkiem z chłodniczką zwrotną i wstawić na 30 minut do
wrzącej łaźni wodnej, wstrząsać, co kilka minut.
3. Po całkowitym rozpuszczeniu tkanki, probówkę wyjąć z łaźni, oziębić i dodać
4,5 cm3 96% etanolu, dobrze wymieszać.
4. Probówkę ponownie zakryć korkiem z chłodniczką zwrotną i wstawić do gorącej
łaźni wodnej (uwaga - nie odparowywać alkoholu, wyjąć natychmiast
po osiągnięciu wrzenia).
5. Po ostudzeniu wytrącony osad glikogenu odwirować (5 min, 3000 obr./min).
6. Płyn znad osadu wylać, osad rozpuścić w 3 cm3 wody (mieszać bagietką).
7. Wytrącić glikogen przez dodanie 6 cm3 96% etanolu (zawartość probówki dobrze
wymieszać bagietką).
8. Wytrącony glikogen odwirować jak poprzednio (5 min, 3000 obr/min)
9. Płyn znad osadu ostrożnie wylać.
10. Osad osuszyć przez postawienie probówki do góry dnem na bibule, a następnie
rozpuścić w 10 cm3 wody destylowanej. Powstały opalizujący roztwór używać do
dalszych oznaczeń.
2. Wyznaczanie krzywej wzorcowej do oznaczania glukozy.
Zasada metody
Glukoza posiada właściwości redukujące i w środowisku zasadowym redukuje odczynnik dinitrosalicylowy, sama zaś utlenia się do kwasu glukonowego. Odczynnik dinitrosalicylowy po redukcji zmienia barwę z żółtej na pomarańczową o maksimum absorbancji 550 nm. Powstałe zabarwienie roztworu jest proporcjonalne do ilości glukozy w próbce.
Odczynniki
1. 0,01 mol/ dm3 roztwór glukozy.
2. 0,05 mol/dm3 bufor fosforanowy pH 6,9.
3. Odczynnik dinitrosalicylowy (1% kwas 3,5-dinitrosalicylosalicylowy, 1,6% NaOH, 30% winian sodowo-potasowy).
Wykonanie doświadczenia
Do 7 probówek skalowanych na 10 cm3 dodać kolejno odczynniki według tabeli 1:
Tabela 1
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Ilości dodawanych odczynników w cm3 |
||||||
H2O |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
0,8 |
2,0 |
0,01 mol/ dm3 roztwór glukozy |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
- |
Bufor fosforanowy |
0,5 |
0,5 |
0,5 |
0,5 |
0,5 |
0,5 |
0,5 |
Odczynnik dinitro-salicylowy |
2,0 |
2,0 |
2,0 |
2,0 |
2,0 |
2,0 |
2,0 |
1. Po dodaniu odczynników zawartość probówek dobrze wymieszać
2. Wstawić na 10 minut do wrzącej łaźni wodnej.
3. Po wyjęciu z łaźni probówki oziębić.
4. Uzupełnić wodą do 10 cm3, wymieszać.
5. Dokonać pomiarów absorbancji przy długości fali 550 nm, wobec próby ślepej
probówka nr 7.
6. Sporządzić wykres zależności absorbancji od ilości μmoli glukozy w próbie.
3. Hydroliza kwaśna glikogenu. Oznaczanie zawartości procentowej glikogenu w wątrobie na podstawie ilości uwolnionej glukozy.
Zasada metody
Glikogen ulega hydrolizie w 1000 C w środowisku silnie kwaśnym, gdyż w tych warunkach wiązania glikozydowe ulegają rozpadowi. Jeśli hydroliza będzie prowadzona przez odpowiednio długi okres czasu to cała ilość glikogenu ulegnie rozkładowi do wolnej glukozy. Na podstawie ilości uwolnionej glukozy można wyliczyć zawartość procentową glikogenu w wątrobie.
Odczynniki
1. 2 mol/dm3 roztwór HCl.
2. 1,2 mol/dm3 roztwór NaOH .
3. Odczynnik dinitrosalicylowy (1% kwas 3,5-dinitrosalicylosalicylowy,
1,6% NaOH, 30% winian sodowo-potasowy).
4. 0,05 mol/dm3 bufor fosforanowy pH 6,9.
Wykonanie doświadczenia
1. Otrzymany w doświadczeniu 1 roztwór glikogenu rozcieńczyć dwukrotnie:
pobrać 5 cm3, dodać 5 cm3 wody i dokładnie wymieszać.
2. Przygotować trzy probówki.
3. Do probówek 1 i 2 odmierzyć po 0,4 cm3 rozcieńczonego dwa razy roztworu
glikogenu, do probówki nr 3 dodać 0,4 cm3 wody (próba ślepa).
4. Do wszystkich probówek dodać po 0,6 cm3 2 mol/dm3 roztworu HCl i zanotować
czas (t0).
5. Zawartość probówki nr 1 i 3 natychmiast zobojętnić przez dodanie 1 cm3
1,2 mol/dm3 roztworu NaOH.
6. Probówkę nr 2 wstawić do wrzącej łaźni wodnej na 40 minut
7. Probówkę nr 2 wyjąć z łaźni po 40 minutach, następnie zobojętnić przez dodanie 1 cm3
1,2 mol/dm3 roztworu NaOH.
8. Do wszystkich probówek dodać po 0,5 cm3 0,05 mol/dm3 buforu fosforanowego
pH 6,9 w celu wyrównania we wszystkich probówkach wartości pH.
9. Następnie do wszystkich probówek dodać 2 cm3 odczynnika dinitrosalicylowego,
zawartość probówek dobrze wymieszać i wstawić do wrzącej łaźni wodnej na
10 minut.
10. Probówki wyjąć, ochłodzić i uzupełnić do 10 cm3 wodą destylowaną, ponownie
wymieszać.
11. Zmierzyć absorbancję przy długości fali 550 nm, wobec próby ślepej (probówka
nr 7 z doświadczenia nr 2).
Z krzywej wzorcowej do oznaczania glukozy (doświadczenie nr 2), odczytać ilość μmoli glukozy.
Obliczyć ilość μg glukozy uwolnionej podczas hydrolizy próbki glikogenu i obliczyć zawartość procentową glikogenu w wątrobie. Należy uwzględnić wszystkie stosowane rozcieńczenia preparatu glikogenu uzyskanego z jednego grama tkanki i ilość oznaczonej glukozy pomnożyć przez 0,9, aby uwzględnić fakt, że ze 162 g glikogenu powstaje 180 g glukozy (162:180).
4. Zadanie ilościowe - oznaczanie glukozy (indywidualne dla każdego studenta
Wykonanie doświadczenia
Do dwóch probówek dodać po 1 cm3 roztworu zadania i po 1 cm3 wody, 0,5 cm3 0,05 mol/dm3 buforu fosforanowego pH 6,9 i 2 cm3 odczynnika dinitrosalicylowego.
Zawartość probówek dokładnie wymieszać i ogrzewać przez 10 minut we wrzącej łaźni wodnej. Po wyjęciu oziębić i uzupełnić wodą do 10 cm3. Zmierzyć absorbancję przy długości fali 550 nm wobec próby ślepej (probówka nr 7 z doświadczenia 2). Z krzywej wzorcowej odczytać ilość μmoli glukozy w 1 cm3 zadania.
ĆWICZENIE III
Mocz prawidłowy i patologiczny
Każdy student zobowiązany jest do przyniesienia na zajęcia około 200 cm3 moczu, który posłuży do wykonywania doświadczeń.
ZAGADNIENIA DO PRZYGOTOWANIA
1. Powstawanie moczu i prawidłowe składniki moczu.
2. Wykrywanie prawidłowych i patologicznych składników moczu
Tematy na samokształcenie do zaliczenia podczas ćwiczenia 3:
V. 1. 2. 3. 4. 5. 6. 7. 8
WPROWADZENIE
Nerka bierze udział w utrzymywaniu stałej objętości i stałego składu płynów ustrojowych (utrzymywanie homeostazy organizmu). Funkcja ta spełniana jest poprzez wytwarzanie moczu. W ten sposób z organizmu usuwany jest nadmiar wody i związki pochodzące z zewnątrz (np. leki i produkty ich degradacji, substancje toksyczne) które zostały wytworzone w organizmie podczas procesów metabolicznych, jak np. kreatynina, mocznik, kwas moczowy.
Mocz tworzy się w nerce, w nefronach. Nefron jest jednostką czynnościową nerki. U człowieka jest ich około 1 mln. W jego skład wchodzi: 1. kłębek nerkowy (zawierający torebkę Bowmana), 2. kanalik bliższy (proksymalny), 3. pętla Henlego zawierająca ramię zstępujące i ramię wstępujące, 4. kanalik dalszy (dystalny), 5. kanalik zbiorczy. W wyniku ultrafiltracji osocza powstaje mocz pierwotny, który podlega resorpcji zwrotnej i wydzielaniu kanalikowemu (sekrecji kanalikowej) i tworzy się mocz ostateczny.
Szybkość ultrafiltracji zależy od ciśnienia krwi (wzrost ciśnienia krwi powoduje wzrost ultrafiltracji), od ciśnienia koloido-osmotycznego związanego z obecnością w osoczu białek, głównie albumin (zmniejszenie tego ciśnienia prowadzi do wzrostu ultrafiltracji) i od ciśnienia panującego w torebce Bowmana (spadek ciśnienia wewnątrz torebki prowadzi do wzrostu ultrafiltracji). Skuteczne ciśnienie filtracyjne (EFP - ang. effective filtration pressure) = ciśnienie krwi w naczyniach włosowatych kłębuszka (55 mm Hg = 7,3 kPa) - ciśnienie koloido-osmotyczne w naczyniach włosowatych kłębuszka (25 mm Hg = 3,3 kPa) - ciśnienie w torebce Bowmana (15 mm Hg = 2 kPa). Szybkość ultrafiltracji w warunkach prawidłowych wynosi 125 cm3 osocza na minutę. Powstaje w ten sposób mocz pierwotny, który od osocza różni się tylko brakiem białek, które z uwagi na dużą masę cząsteczkową nie mogą przechodzić przez błonę komórkową komórek torebki kłębuszka. Przechodzą przez nią związki, których masa cząsteczkowa nie przekracza 60 000. Podczas przepływu moczu pierwotnego przez kanaliki nerkowe zachodzi wchłanianie zwrotne (resorpcja) wody i rozpuszczonych w niej związków organicznych i jonów. W efekcie tych procesów powstaje mocz ostateczny, w ilości 1,5 dm3 na dobę (1 cm3 na minutę).
W kanalikach proksymalnych zachodzi obligatoryjne (obowiązkowe) zwrotne wchłanianie wody, niepodlegające regulacji hormonalnej, związane z aktywnym wchłanianiem jonów Na+. W ten sposób wchłania się 85% wody. Wraz z wodą resorbowane są z moczu pierwotnego do osocza substancje w niej rozpuszczone. Niektóre z nich są wchłaniane aktywnie i potrzebna do tego procesu energia pochodzi z rozpadu ATP. Substancje te są wchłaniane w kanaliku proksymalnym całkowicie lub prawie całkowicie i w moczu w warunkach prawidłowych nie występują lub ich ilość jest bardzo mała. Związki te noszą nazwę wysokoprogowych i należą do nich: glukoza, aminokwasy, kreatyna, Na+, HCO3-. Pojawiają się one w moczu dopiero wtedy, gdy ich stężenie w osoczu przekroczy próg nerkowy, tzn. będzie wyższe od zdolności kanalików nerkowych do ich wchłonięcia aktywnego. Inną grupą są związki niskoprogowe, które są wchłaniane biernie, bez nakładu energii, w małych ilościach i są one zawsze wydalane z moczem. Należą do nich np. mocznik i kwas moczowy. Natomiast związki nazywane bezprogowymi nie ulegają resorpcji, przykładem takiego związku jest kreatynina.
W pętli Henlego odbywa się dalsze zagęszczanie moczu. Na skutek wchłaniania wody w ramieniu zstępującym rośnie osmolarność moczu pierwotnego (do 1200 mosmoli), a w ramieniu wstępującym, które jest nieprzepuszczalne dla wody osmolarność maleje, gdyż zachodzi resorpcja jonów Na+ i Cl-. Tak więc mocz opuszczający pętlę jest izotoniczny w stosunku do osocza (osmolarność = 300 mosmoli), pomimo tego, że wiele substancji ulega w nim zagęszczeniu.
W kanalikach dystalnych i zbiorczych resorpcja zwrotna wody i jonów Na+ podlega regulacji hormonalnej przez wazopresynę i aldosteron. Wazopresyna jest hormonem peptydowym, wydzielanym przez tylny płat przysadki wtedy, gdy wzrasta osmolarność płynów ustrojowych. Nazywana jest hormonem antydiuretycznym (ADH), gdyż po jej wydzieleniu następuje wchłanianie zwrotne wody - fakultatywne, z wtórnym wchłanianiem jonów Na+. Drugim hormonem jest aldosteron, hormon steroidowy, należący do mineralokortykoidów, syntetyzowany w korze nadnerczy, którego wydzielenie powoduje wchłanianie zwrotne Na+, co pociąga za sobą wtórne wchłanianie wody. Oba te hormony biorą udział w regulowaniu gospodarki wodno-elektrolitowej.
Powstający w kanaliku zbiorczym mocz ostateczny jest hipertoniczny w stosunku do osocza. Jego osmolarność jest cztery razy wyższa niż osocza i nie może przekroczyć 1200 mosmoli. Tak więc wydalany mocz w porównaniu do osocza jest czterokrotnie zagęszczony, natomiast poszczególne związki wydalane w moczu są zagęszczane znacznie bardziej, np. kwas moczowy około 15 razy, mocznik około 80 razy, kreatynina 100 razy, jony NH4+ 400 razy.
Jeżeli w moczu wydalane są związki, które w warunkach prawidłowych są resorbowane całkowicie (np. glukoza), to wiąże się to ze zwiększeniem ilości wytwarzanego moczu. To zjawisko nosi nazwę diurezy osmotycznej.
Mocz wydalany w ciągu doby zawiera substancje nieorganiczne, których ilość waha się od 20 do 30 g i są to jony: sodowy, potasowy, chlorkowy, siarczanowy, fosforanowe. Wydalane są również z moczem substancje organiczne, w ilości 30-40 g na dobę. Głównie są to produkty przemiany azotowej i należą do nich: mocznik, kwas moczowy, kreatynina. amoniak (NH4+).
CZĘŚĆ DOŚWIADCZALNA
1. Oznaczanie pH moczu
Odczyn moczu jest najczęściej lekko kwaśny. Jego pH może się wahać w granicach 5,5 - 6,5 i zależy od stosowanej diety. Mocz może mieć odczyn zasadowy szczególnie przy diecie bogatej w jarzyny, owoce i produkty mleczne. Odczyn bardziej kwaśny ma mocz osób stosujących dietę wysokobiałkową.
Materiał i odczynniki
1. Paski wskaźnikowe pH
2. Mocz
Wykonanie doświadczenia
Na pasek wskaźnikowy nanieść kilka kropli moczu i porównać uzyskane zabarwienie ze skalą barw.
Składniki moczu prawidłowego
2. Wykrywanie jonów Cl-
Jon Cl- jest głównym anionem osocza. Wydalany jest w ilości od 6 do 10 g na dobę. Ilość wydalanych chlorków jest ściśle związana z wydalaniem wody. Im więcej wody organizm usuwa, tym więcej wydala chlorków.
Zasada metody
Jony Cl- tworzą z jonami Ag+ biały, serowaty osad AgCl, nierozpuszczalny w HNO3.
Cl- + Ag+ Ⴎ AgClႯ
Materiał i odczynniki
1. Stężony HNO3
2. 5% roztwór AgNO3
3. Mocz
Wykonanie doświadczenia
Do 2 cm3 moczu dodać 0,5 cm3 stężonego kwasu azotowego, a następnie kilka kropli roztworu azotanu srebra.
3. Wykrywanie jonów SO42-
W ciągu doby wydalanych zostaje od 2 do 3 g jonów siarczanowych. Powstają one głównie podczas przemiany (utleniania) cysteiny, aminokwasu zawierającego grupę -SH i w całości są wydalane z moczem, nie ulegają resorpcji zwrotnej w kanalikach nerkowych.
Zasada metody
Jony SO42- tworzą z jonami Ba2+ biały, drobnokrystaliczny osad BaSO4.
SO42- + Ba2+ Ⴎ BaSO4Ⴏ
Materiał i odczynniki
1. 10% roztwór BaCl2
2. Mocz
Wykonanie doświadczenia
Do kilku cm3 moczu dodać 1-2 cm3 roztworu chlorku barowego.
4. Wykrywanie mocznika (reakcja z podbrominem)
Mocznik powstaje w organizmie w tzw. cyklu mocznikowym. Związek ten pochodzi głównie z przemiany azotowej białek.
Podczas deaminacji aminokwasów tworzy się bardzo toksyczny amoniak. Powstaje on również w mniejszej ilości, w przemianach innych związków, np. nukleotydów purynowych i pirymidynowych oraz amin. Proces syntezy mocznika pełni ważną biologiczną rolę w przekształcaniu amoniaku w łatwo rozpuszczalny, nietoksyczny w stężeniach fizjologicznych związek, który następnie jest wydalany z moczem.
Zdrowy, dorosły człowiek, na prawidłowej diecie jest w stanie tzw. równowagi azotowej. Dobowa ilość azotu wydalana z organizmu jest równa ilości azotu przyjmowanego z pożywieniem. Przy spożyciu białka w ilości około 100 gramów na dobę wydalane jest 16 gramów azotu. 80 - 90% całkowitej puli wydalanego azotu stanowi mocznik. Ilość wytworzonego w organizmie dorosłego człowieka mocznika zależy głównie od zawartości białka w pożywieniu i przeciętnie wynosi od 20 do 40 g na dobę. Stężenie tego związku we krwi w warunkach prawidłowych waha się w szerokich granicach, od 20 do 40 mg/100 cm3, a nawet może osiągać 50 mg/100 cm3. Z moczu pierwotnego mocznik resorbowany jest biernie.
Synteza mocznika zachodzi wyłącznie w wątrobie. Amoniak łączy się z CO2 tworząc karbamoilofosforan, reakcja ta wymaga udziału dwóch cząsteczek ATP, jako źródła energii. Karbamoilofosforan następnie reaguje z ornityną, tworząc cytrulinę. Cytrulina przy udziale energii pochodzącej z hydrolizy ATP kondensuje z asparaginianem, który jest dawcą drugiego atomu azotu. Powstały argininobursztynian ulega rozszczepieniu na argininę i fumaran. Arginina hydrolizuje dając mocznik i ornitynę, zamykając w ten sposób jeden obrót cyklu. Dwie pierwsze reakcje zachodzą w mitochondriach komórek wątroby, pozostałe trzy przebiegają w cytoplazmie.
Zasada metody
Pod wpływem podbrominu sodowego następuje utlenienie i rozkład mocznika z wytworzeniam CO2, H2O i azotu (Rys. 1).
Materiał i odczynniki
1. Roztwór podbrominu sodu (10% Br2 w 40% roztworze NaOH)
2. Mocz
Wykonanie doświadczenia
Do 1 cm3 moczu dodać kilka kropel roztworu podbrominu sodowego.
Zawartość probówki wymieszać.
Pojawienie się pęcherzyków gazu świadczy o obecności mocznika.
Gazem tym będzie tylko azot, bo CO2 w środowisku zasadowym przekształci się w węglany i wodorowęglany.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
5. Wykrywanie kwasu moczowego
Kwas moczowy jest końcowym produktem katabolizmu zasad purynowych w organizmie człowieka. Nukleotydy w komórkach ulegają stałym przemianom. Związki te są rozkładane za pomocą enzymów, a także ulegają samorzutnemu rozpadowi na skutek wpływu środowiska.
Nukleotydazy rozkładają hydrolitycznie nukleotydy do nukleozydów. Z nukleozydów fosforylazy nukleozydowe uwalniają wolne zasady i rybozo-1-fosforan. Większość uwolnionych zasad purynowych zostaje ponownie użyta do tworzenia nukleotydów na drodze rezerwowych szlaków metabolicznych.
Niewielka część uwolnionych zasad ulega przekształceniu do kwasu moczowego i jest wydalana z moczem.
Główne szlaki metaboliczne przemiany AMP i GMP prowadzące do utworzenia kwasu moczowego nieco się różnią. Katabolizm AMP rozpoczyna deaminaza AMP:
AMP + H2O → IMP + NH3
IMP i GMP są substratami dla 5' nukleotydazy, która przekształca je w nukleozydy.
IMP + H2O → inozyna + Pi
GMP + H2O → guanozyna Pi
Następnie nukleozydy są rozkładane za pomocą fosforylazy nukleozydów purynowych.
inozyna + Pi → hipoksantyna + rybozo-1-fosforan
guanozyna + Pi → guanina + rybozo-1-fosforan
Guanina pod wpływem hydrolazy - guanazy (deaminazy guaninowej) przekształcana jest do ksantyny.
guanina + H2O → ksantyna + NH3
Hipoksantyna i ksantyna są przekształcane w kwas moczowy przy udziale oksydazy ksantynowej.
hipoksantyna → ksantyna→ kwas moczowy
Mechanizm tej reakcji jest złożony, enzym do swojego działania wymaga udziału O2, FAD, jonów molibdenu i żelaza. Jeden atom tlenu cząsteczkowego, utleniacza w obu reakcjach, zostaje dołączony do pierścienia puryny, drugi zaś zostaje zredukowany do H2O2. Nadtlenek wodoru następnie jest rozkładany przez katalazę do H2O i O2.
Przyłączenie tlenu do pierścienia puryny powoduje zwiększenie polarności, a zatem zwiększa rozpuszczalność związku w wodzie. Jednak kwas moczowy jest nadal związkiem trudno rozpuszczalnym. Lepiej rozpuszczalne są jego sole - moczany, które tworzą się w pH obojętnym i zasadowym. Kryształy moczanów wytrącają się w roztworach przekraczających stężenie 7 miligramów tego związku na 100 cm3 wody. Dlatego też znaczna część moczanów w osoczu zaadsorbowana jest na albuminach, co przeciwdziała ich wytrącaniu.
Prawidłowe stężenie kwasu moczowego (głównie w postaci moczanów) we krwi dorosłych kobiet wynosi od 2 do 6 mg/100 cm3, u mężczyzn od 3 do 7,5 mg/100 cm3. Stężenie kwasu moczowego w osoczu zależy także od diety, niższe stężenia tego związku występują u ludzi stosujących dietę wegetariańską.
W ciągu doby organizm zdrowego, dorosłego człowieka wytwarza przeciętnie od 400 do 800 miligramów kwasu moczowego, który następnie jest wydalany z moczem. Jest on częściowo wchłaniany aktywnie w kanalikach proksymalnych. Na dobę wydalany jest w ilości 500-800 mg w postaci wolnej lub jako moczany sodu lub potasu.
Nadmierne jego wytwarzanie powoduje podwyższenie jego stężenia we krwi (hiperurykemia) i jest przyczyną zmian chorobowych wywołanych przez wytrącanie się kryształów moczanu sodu, szczególnie w stawach i nerkach. Choroba ta jest znana jako dna moczanowa. Większość ssaków przekształca dalej kwas moczowy do łatwo rozpuszczalnej substancji - alantoiny. Brak możliwości przekształcania kwasu moczowego do alantoiny jest jednak korzystnym czynnikiem selekcyjnym. Moczany bardzo łatwo się utleniają i są bardzo wydajną substancją usuwającą wysoce reaktywne, szkodliwe formy tlenu, np.: rodniki hydroksylowe, aniony ponadtlenkowe. Obecność moczanów w osoczu może wydatnie przyczyniać się do wydłużenia życia ludzkiego i do zmniejszenia zapadalności na choroby nowotworowe. Moczany jak również bilirubina są przykładami reguły, że niektóre produkty końcowe pochodzące ze szlaków metabolicznych odgrywają ważną rolę jako czynniki ochronne.
Zasada metody
Pod wpływem kwasu moczowego kwas fosforowolframowy w środowisku zasadowym (Na2CO3) ulega redukcji do tlenków wolframu o niższym stopniu utlenienia, mających niebieskie zabarwienie. Za reakcję dodatnią uważa się również pojawienię się intensywnego, zielonego zabarwienia, powstającego na skutek nakładania się na barwę niebieską, żółtej barwy pochodzącej od innych składników moczu.
Materiał i odczynniki
1. Odczynnik fosforowolframowy (4% Na2WO4x 2H2O w 3% roztworze H3PO4)
2. 10% roztwór Na2CO3
3. Mocz
Wykonanie doświadczenia
Do 1 cm3 moczu dodać kilka kropli odczynnika fosforowolframowego i 1 cm3 10% roztworu Na2CO3.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
6. Wykrywanie urobilinogenu - reakcja Ehrlicha
Urobilinogen powstaje w przewodzie pokarmowym z bilirubiny wątrobowej w wyniku działania enzymów bakterii jelitowych, skąd częściowo zostaje wchłonięty do krwioobiegu i utleniony w wątrobie do bilirubiny, która po sprzęgnięciu wydalana jest ponownie do przewodu pokarmowego. Niewielka ilość urobilinogenu jest wydalana z moczem, jest to fizjologiczna urobilinogenuria. Podczas niewydolności wątroby maleje jej zdolność do utleniania urobilinogenu i zwiększa się wtedy jego ilość wydalana z moczem. Przyczyną wydalania zwiększonej ilości urobilinogenu może być również wzmożony rozpad krwinek czerwonych np. przy żółtaczce hemolitycznej. Natomiast przy zaczopowaniu przewodów żółciowych ilość wydalanego z moczem urobilinogenu znacznie się obniża.
Zasada metody
Urobilinogen z odczynnikiem Ehrlicha, w obecności HCl, tworzy czerwony kompleks.
Materiał i odczynniki
1. Odczynnik Ehrlicha (2% roztwór aldehydu p-N,N-dimetyloaminobenzoesowego w 1mol/dm3 roztworze HCl)
2. Mocz
Wykonanie doświadczenia
Do 4 cm3 moczu dodać 0,5 cm3 odczynnika Ehrlicha. Zawartość probówki ogrzać. Przy zwiększonej ilości urobilinogenu mocz zabarwia się na czerwono już na zimno.
7. Wykrywanie kreatyniny metodą Jaffego
Kreatynina jest produktem degradacji kreatyny. Kreatyna występuje głównie w mięśniach i w postaci fosforanu jest magazynem energii. Cykl mocznikowy jest także punktem wyjścia do syntezy tego ważnego metabolitu. Arginina, metabolit cyklu mocznikowego, ulega w komórkach nerki kondensacji z glicyną. Powstaje guanidynooctan, który jest metylowany przez S-adenozylometioninę
(SAM) do kreatyny. Kreatyna jest następnie fosforylowana za pomocą ATP i przechodzi w fosforan kreatyny, stanowiący zapas łatwo uruchamianej energii w mięśniu. Ostatni etap przemiany odbywa się bez udziału enzymów. W ciągu doby około 1,6% fosfokreatyny przekształca się w bezwodnik - kreatyninę (Rys. 2).
Kreatynina jest wydalana z moczem, przeciętnie od 1 do 1,8 gramów na dobę. Kreatynina nie ulega wchłanianiu zwrotnemu w kanalikach nerkowych. Na dobę wydala się jej od 1 g do 1,8 g, w zależności od wielkości masy mięśniowej.
Prawidłowe stężenie kreatyniny w surowicy wynosi u dorosłych kobiet od 0,5 do 1 mg/100 cm3, a u mężczyzn 0,6 - 1,2 mg/100 cm3. Znacznie niższe stężenie kreatyniny występuje u małych dzieci do trzech lat (0,2-0,4 mg/100 cm3). W miarę dorastania stężenie to stopniowo wzrasta, aby osiągnąć w wieku 17 lat wartości charakterystyczne dla ludzi dorosłych.
Podwyższone stężenie kreatyniny we krwi może wystąpić w chorobach nerek, mięśni i zatruciach witaminą D.
Zasada metody
Kreatynina w środowisku zasadowym tworzy z kwasem pikrynowym czerwony kompleks.
Materiał i odczynniki
1. 1,5% roztwór kwasu pikrynowego
2. 2 mol/dm3 roztwór NaOH
3. Mocz
Wykonanie doświadczenia
Do 1 cm3 moczu rozcieńczonego dwa razy wodą destylowaną, dodać 1 cm3 nasyconego roztworu kwasu pikrynowego, a następnie zalkalizować kilkoma kroplami 2 mol/dm3 roztworu NaOH.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
8. Wykrywanie kreatyniny metodą Weyla
Materiał i odczynniki
1. 10% roztwór nitroprusydku sodu
2. 2 mol/dm3 roztwór NaOH
3. 30% kwas octowy
4. Mocz.
Wykonanie doświadczenia
Do 2 cm3 moczu dodać 3-4 krople roztworu nitroprusydku sodu oraz 1 cm3 2 mol/dm3 roztworu NaOH. Pojawia się czerwone zabarwienie, które po pewnym czasie blednie, a znika natychmiast po zakwaszeniu 30% kwasem octowym.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
Składniki moczu patologicznego
W stanach patologicznych z moczem wydalane są substancje, które w warunkach prawidłowych praktycznie nie pojawiają się w moczu lub są wydalane w bardzo małych ilościach i taki mocz określamy jako patologiczny. Patologicznymi składnikami moczu są: białko, ciała ketonowe, glukoza, bilirubina, kwasy żółciowe, hemoglobina, zwiększona ilość urobilinogenu. Przyczyny wydalania substancji patologicznych mogą być różne; związane nie tylko z zaburzeniami czynności nerek, ale także ze schorzeniami (zaburzeniami metabolizmu) innych narządów.
9. Wykrywanie ciał ketonowych (acetonu) - reakcja Legala
Ciała ketonowe (Rys. 2) są prawidłowymi produktami metabolizmu, syntetyzowane są w wątrobie z acetyloCoA. Należą do nich acetooctan i ၢ-hydroksymaślan. W warunkach prawidłowych ich stężenie we krwi jest bardzo niewielkie i wynosi od 0,2 do 0,8 mg/100 cm3, gdyż poza wątrobą i erytrocytami są one wykorzystywane jako materiał energetyczny i rozkładane do CO2 i H2O. U zdrowego człowieka z moczem wydalane są bardzo małe ilości tych związków i jest ich nie więcej niż 20 mg na dobę. Jest to tak mała ilość, że nie można jej wykryć stosowanymi metodami.
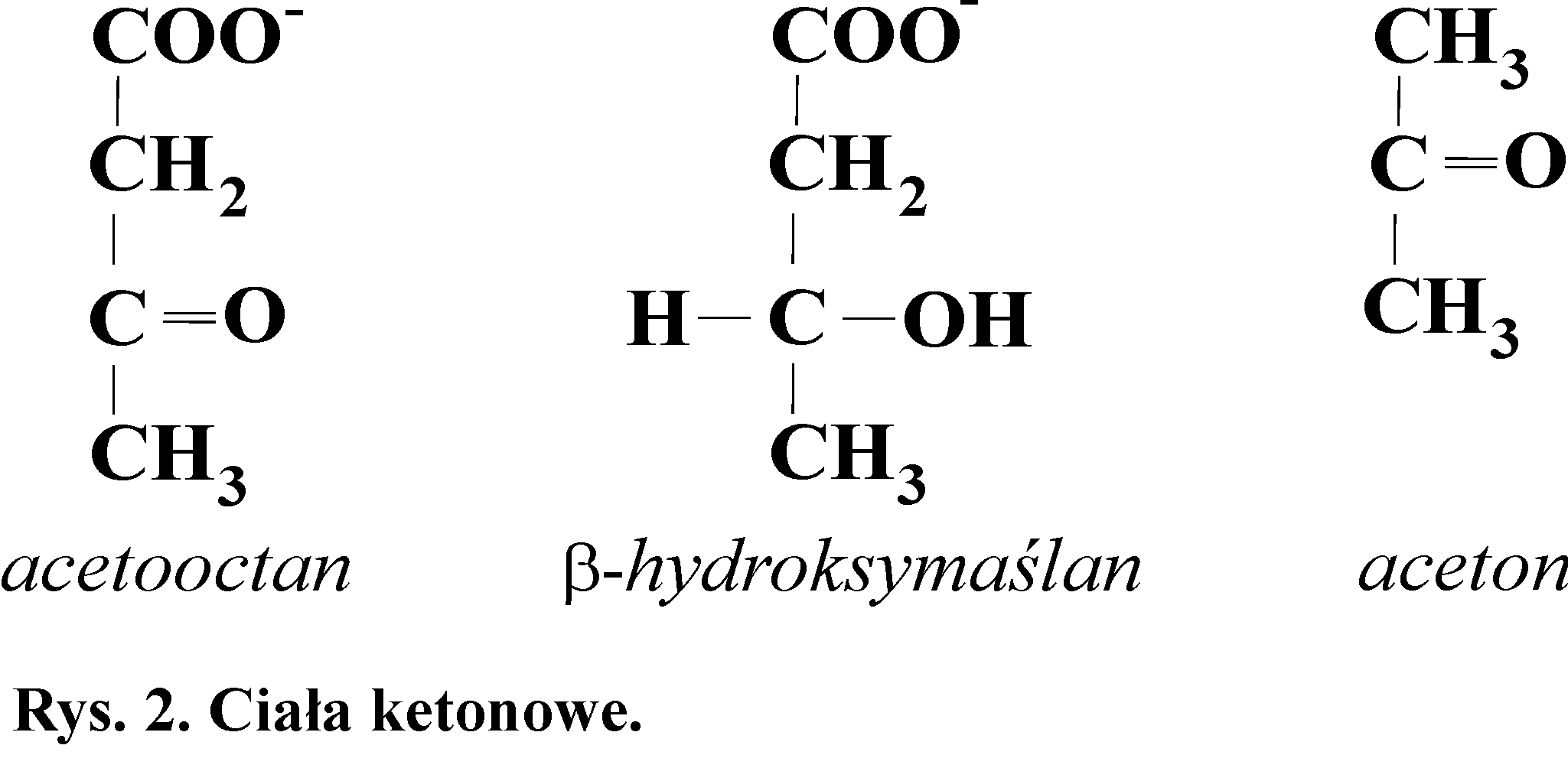
W pewnych warunkach np. w głodzie czy cukrzycy może dochodzić do wzmożonej syntezy tych związków w wątrobie. Ilość wytworzonych wtedy ciał ketonowych przekracza zdolność całkowitego wykorzystywania tych związków przez organizm. Wzrasta stężenie acetooctanu i w wyniku jego spontanicznej dekarboksylacji powstaje aceton. Jest on wydalany z moczem razem z acetooctanem i ၢ-hydroksymaślanem (ketonuria). Jeżeli ilość wytworzonych ciał ketonowych jest tak duża, że nerka nie może ich całkowicie wydalić z moczem, to dochodzi do ketonemii - zwiększenia ich stężenia we krwi, co prowadzi do zaburzenia równowagi kwasowo-zasadowej (kwasicy metabolicznej).
Materiał i odczynniki
1. 30% roztwór NaOH
2. 10% roztwór nitroprusydku sodu
3. 30% CH3COOH
4. Mocz prawidłowy
5. Mocz patologiczny zawierający aceton
Wykonanie doświadczenia
Reakcję wykonać z moczem prawidłowym i patologicznym
3 cm3 badanego moczu zalkalizować 5 kroplami 30% roztworu NaOH.
Dodać 3-5 kropli roztworu nitroprusydku sodu ( powstaje czerwone zabarwienie).
Dodać 10-15 kropli 30% kwasu octowego (zabarwienie nie znika, a nawet pogłębia się).
Zaobserwować różnice w reakcji z moczem patologicznym
Porównać z ćwiczeniem na wykrywanie kreatyniny.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
10. Wykrywanie glukozy (reakcja Benedicta)
Glukoza resorbowana jest w kanaliku proksymalnym aktywnie, przy udziale energii z rozpadu ATP i w warunkach prawidłowych nie jest wydalana z moczem. Jest ona substancją wysokoprogową i dopóki jej stężenie we krwi nie przekroczy 180 mg/100 cm3 nerka jest w stanie zresorbować z moczu pierwotnego całą jej ilość. Glukozuria to wydalanie glukozy z moczem, występuje u ludzi chorych na cukrzycę, u których stężenie glukozy przekroczyło próg nerkowy. Chory może wydalać z moczem nawet kilkadziesiąt gramów glukozy na dobę.
Glukoza może pojawić się w moczu również wtedy, kiedy jej stężenie we krwi jest prawidłowe, ale dochodzi do zaburzenia jej wchłaniania zwrotnego w kanalikach proksymalnych, na skutek dysfunkcji nerek.
Zasada metody
Glukoza redukuje Cu2+ do Cu+ i powstaje nierozpuszczalny, czerwony osad Cu2O.
Materiał i odczynniki
1. Odczynnik Benedicta (17,3% cytrynian trisodowy, 9% Na2CO3, 1,1% CuSO4)
2. Mocz prawidłowy
3. Mocz patologiczny zawierający glukozę
Wykonanie doświadczenia
Do 2 cm3 odczynnika Benedicta (zawiera Cu2+) dodać kilka kropli badanego moczu. Wstawić na kilka minut do wrzącej łaźni wodnej. Próbę wykonać z moczem prawidłowym i patologicznym.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
11. Wykrywanie białka
Mocz zdrowego człowieka nie zawiera białka. Podczas ultrafiltracji osocza przez błonę torebki kłębuszka przechodzą tylko związki drobnocząsteczkowe. Dla białek jest ona nieprzepuszczalna. Niekiedy może przechodzić do moczu pierwotnego i pojawiać się w moczu albumina, której masa cząsteczkowa jest niższa niż globulin. Zjawisko to nosi nazwę białkomoczu fizjologicznego (proteinurii fizjologicznej) i jest związane np. z dużym wysiłkiem fizycznym, gwałtownym oziębieniem ciała.
Białko pojawia się w moczu w stanach patologicznych, np. kiedy zwiększa się przepuszczalność kłębków nerkowych w wyniku ich uszkodzenia. Ilość wydalanego wtedy z moczem białka może dochodzić nawet do kilkudziesięciu gramów na dobę.
Zasada metody
W środowisku kwaśnym pod wpływem kwasu sulfosalicylowego białko ulega denaturacji i wytrąceniu.
Materiał i odczynniki
1. 30% roztwór CH3COOH
2. 20% roztwór kwasu sulfosalicylowego
3. Mocz prawidłowy
4. Mocz patologiczny zawierający białko
Wykonanie doświadczenia
Doświadczenie wykonać z moczem prawidłowym i patologicznym.
Do 4 cm3 moczu dodać 1-2 krople 30% roztworu kwasu octowego.
W razie wystąpienia zmętnienia roztwór przesączyć.
Do klarownego moczu lub przesączu dodać 3-5 kropli 20% roztworu kwasu sulfosalicylowego.
Próba jest dodatnia jeżeli roztwór ulega zmętnieniu (może powstać opalescencja) lub wytrąca się osad zdenaturowanego białka.
Wynik doświadczenia zależy od ilości białka obecnego w moczu.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
12. Wykrywanie śladów krwi
Krew spełnia w organizmie wiele funkcji, umożliwia zachowanie homeostazy, uczestniczy w utrzymaniu równowagi kwasowo-zasadowej (utrzymywanie stałego pH w granicach 7,38-7,42) i w regulacji gospodarki wodno elektrolitowej. Krew transportuje do tkanek wiele różnych substancji oraz umożliwia usunięcie z komórek metabolitów. W składzie krwi można wyróżnić elementy morfotyczne i osocze. Elementami morfotycznymi są erytrocyty, leukocyty i trombocyty (płytki krwi). Głównym składnikiem erytrocytów jest hemoglobina, która odgrywa zasadniczą rolę w transporcie tlenu a także znaczącą w transporcie dwutlenku węgla. Hemoglobina jest hemoproteiną zbudowaną z czterech łańcuchów polipeptydowych, a każdy łańcuch połączony jest z cząsteczką hemu zawierającego dwuwartościowy kation żelaza (Fe2+).
W warunkach prawidłowych hemoglobina, która zostaje uwolniona z erytrocytów do osocza jest wiązana z ၡ2 globuliną osocza - haptoglobiną i wytworzony kompleks nie przenika przez kłębuszki nerkowe. Haptoglobina może związać od 400 mg do 1,8 g hemoglobiny. Jeżeli dochodzi do uwolnienia z krwinek czerwonych większej ilości hemoglobiny (np. przy rozległych poparzeniach, niektórych zatruciach, wstrząsie potransfuzyjnym) to gwałtownie wzrasta stężenie hemoglobiny wolnej w osoczu (hemoglobinemia). Hemoglobina ta ulega filtracji w kłębuszkach nerkowych i resorpcji zwrotnej. Po przekroczeniu we krwi stężenia 70 mg/100 cm3 nadmiar jej nie jest wtórnie wchłaniany w kanalikach nerkowych i dlatego pojawia się w moczu.
Do moczu mogą przedostawać się krwinki czerwone z uszkodzonych odcinków układu moczowego, zaburzenie to nosi nazwę hematurii.
Zasada metody
Metoda oparta jest na właściwościach pseudoperoksydazowych hemoglobiny, która podobnie jak enzym peroksydaza, utlenia benzydynę w obecności nadtlenku wodoru. Katalityczne działanie wykazuje jon Fe2+ znajdujący się w niebiałkowej części hemoglobiny - hemie. W peroksydazie o reakcji benzydynowej decyduje centrum aktywne, znajdujące się w apoenzymie. Dlatego po zdenaturowaniu obu białek (peroksydazy i hemoglobiny) tylko w przypadku hemoglobiny próba benzydynowa będzie dodatnia.
Materiał i odczynniki
1. 10% roztwór benzydyny w lodowatym CH3COOH
2. 3% roztwór H2O2
3. Mocz prawidłowy
4. Mocz patologiczny zawierający krew
Wykonanie doświadczenia
Do kilku kropli odczynnika benzydynowego, przygotowanego przez zmieszanie równych objętości roztworu benzydyny i roztworu H2O2 dodać kilka kropli moczu. Doświadczenie wykonać z moczem: a) prawidłowym, b) patologicznym, c) patologicznym zagotowanym.
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
13. Wykrywanie bilirubiny - odczyn Gmelina
Erytrocyty żyją około 120 dni. Rozpad erytrocytów zachodzi w komórkach układu siateczkowo-śródbłonkowego, głównie wątroby, śledziony i szpiku kostnego. Tam też następuje przemiana hemu w bilirubinę. W pierwszym etapie tego procesu bierze udział zlokalizowana w mikrosomach oksygenaza hemowa, związana z mikrosomalnym łańcuchem transportu elektronów. Przy udziale O2 i NADPH+H+ zachodzi rozerwanie pierścienia hemu. Powstaje werdoglobina, która następnie rozpada się na globinę (część białkową), biliwerdynę powstałą z hemu i jon żelaza. Biliwerdyna ma niebieskozielone zabarwienie. Następnie biliwerdyna redukuje się pod wpływem reduktazy biliwerdyny i powstaje pomarańczowożółta bilirubina, która dyfunduje do krwi.
Bilirubina jest związkiem trudnorozpuszczalnym i transportowana jest do wątroby w połączeniu z albuminami. Jedna cząsteczka albuminy łączy się z dwiema cząsteczkami bilirubiny.
W warunkach prawidłowych we krwi występuje tylko bilirubina wolna (przedwątrobowa, pośrednia), która jest połączona z albuminą i ten kompleks nie ulega przesączaniu kłębkowemu. Bilirubina wolna nie pojawia się w moczu nawet wtedy, kiedy jej stężenie w osoczu wzrośnie powyżej 1 g/100 cm3. Bilirubina wolna w wątrobie ulega przekształceniu głównie w diglukuronidy i siarczany. W tej postaci jest ona wydalana do przewodu pokarmowego. Ta bilirubina nosi nazwę związanej (wątrobowej) i w warunkach prawidłowych we krwi nie występuje. Pojawia się ona we krwi przy zaczopowaniu przewodów żółciowych lub uszkodzeniu komórek wątroby. Ulega ona, tak jak inne związki drobnocząsteczkowe, przesączeniu kłębkowemu i resorpcji zwrotnej. W moczu może pojawiać się dopiero wtedy, kiedy jej stężenie w osoczu będzie wyższe niż 1,6 mg/100 cm3.
Zasada metody
Bilirubina utlenia się pod wpływem HNO3 do biliwerdyny, która ma barwę zieloną.
Materiał i odczynniki
1. Stężony HNO3
2. Mocz prawidłowy
3. Mocz patologiczny zawierający bilirubinę
Wykonanie doświadczenia
Doświadczenie wykonać z moczem prawidłowym i patologicznym.
Do probówki nalać 1 cm3 stężonego HNO3.
Następnie po ściankach probówki ostrożnie nawarstwić badany roztwór.
W obecności barwników żółciowych po kilku minutach na granicy płynów pojawia się tęcza barw (z dominującą barwą zieloną).
Równolegle wykonać próbę z indywidualnym zadaniem na zaliczenie.
14. Wykrywanie kwasów żółciowych - reakcja Haya
Kwasy żółciowe są składnikiem żółci i w warunkach prawidłowych z żółcią są wydalane do przewodu pokarmowego. Powstają one w wątrobie z cholesterolu. Są naturalnymi detergentami, umożliwiają trawienie tłuszczów w przewodzie pokarmowym przez ich emulgację. Przy zablokowaniu drogi odpływu żółci (np. w żółtaczce mechanicznej) wzrasta ich stężenie we krwi i są one częściowo wydalane z moczem.
Zasada metody
Polega na wykazaniu obniżenia napięcia powierzchniowego przez kwasy żółciowe.
Maetriał i odczynniki
1. Siarka sublimowana
2. Mocz prawidłowy
3. Mocz patologiczny z żółcią
Wykonanie doświadczenia
Przygotować trzy probówki.
Do pierwszej nalać 2 cm3 prawidłowego moczu, do drugiej taką samą ilość moczu patologicznego, a do trzeciej 2 cm3 wody destylowanej.
Na powierzchnię każdego płynu wrzucić odrobinę siarki sublimowanej.
Studenci otrzymują jako zadanie „sztuczny mocz”, który jest mieszaniną różnych składników występujących w moczu prawidłowym i patologicznym. W zadaniu są obecne dwa spośród następujących związków:
1) mocznik,
2) kreatynina,
3) aceton,
4) glukoza,
5) krew,
6) bilirubina,
7) kwas moczowy,
8) białko.
4
v
v
Wyszukiwarka
Podobne podstrony:
997
997
997
997
997
997
997
marche 997 p
997 0017
997 0005
BWV 997 Lute Suite Sarabande
BWV 997 Suite Lute No 2
997 0016
997 0006
Lute Suite No 2 BWV 997 3 Sarabande
akumulator do porsche 911 997 36 carrera 36 carrera 4 36 s 38
akumulator do porsche 911 targa 997 36
997 0009
więcej podobnych podstron