ENZYMY
Enzymy są katalizatorami, które zwiększają szybkość reakcji chemicznej, same nie ulegając zmianie. W obecności enzymu szybkość reakcji znacznie wzrasta nawet do 107 razy. Reakcje katalizowane przez enzymy zazwyczaj przebiegają we względnie łagodnych warunkach (temp.<100 st.C, ciśnienie atmosferyczne, obojętne pH). Enzymy są zwykle wysoce specyficzne względem substratów, na które działają i produktów, które tworzą. Aktywność enzymatyczna może być regulowana, zmieniając się w zależności od stężenia substratów, lub innych cząst. Prawie wszystkie enzymy są białkami. Zidentyfikowano też kilka rodzajów cząst. RNA aktywnych katalitycznie.
Zmiany energii zachodzące w przebiegu reakcji biochemicznej:
Dla wszystkich reakcji istnieje bariera energetyczna, którą należy pokonać, aby umożliwić przebieg reakcji. Jest to ilość energii potrzebna do przeprowadzenia cząst. substratu w stan przejściowy, który jest niestabilną formą chemiczną, prowadzącą od substratów do produktów. Stan przejściowy ma największą energię swobodną ze wszystkich związków w drodze reakcji. Energia aktywacji (∆G++) równa się różnicy energii swobodnej między stanem przejściowym, a substratem. Enzym działa w drodze stabilizowania stanu przejściowego reakcji chem. i zmniejsza ∆G++. Enzymy nie zmieniają poziomu energii zarówno substratu, jak i produktu. Enzym zwiększa szybkość przebiegu reakcji, ale nie wpływa na ogólną zmianę energii w tej reakcji. Zmiana energii swobodnej (Gibbsa) (∆G=kJ*mol-1) decyduje czy reakcja będzie energetycznie korzystna, czy niekorzystna. Reakcja energetycznie korzystna - tzn. produkty mają niższy poziom energii niż substraty, a wartość ∆G jest ujemna. ∆G jest pojęciem innym niż ∆G++. Wartość ∆G jest niezależna od drogi reakcji i nie dostarcza żadnej informacji o szybkości reakcji. O szybkości reakcji decyduje energia aktywacji (∆G++). Ujemna wartość energii swobodnej (∆G) wskazuje, że reakcja jest termodynamicznie korzystna we wskazanym kierunku (ma dużą szansę przebiegu spontanicznego), natomiast dodatnia wartość ∆G - reakcja jest niekorzystna termodynamicznie i wymaga nakładu energii, aby zajść we wskazanym kierunku. W układach biochemicznych ten nakład energii uzyskuje się często przez sprzężenie reakcji energetycznie niekorzystnej z reakcją energ. korzystną. Często jest korzystne odwoływanie się do wartości ∆G aktualnej dla standardowego zestawu warunków (tj. jednakowe stężenie [1M] zarówno substratów jak i produktów reakcji, stałe pH ≠ 7,0). W tych warunkach wartość stwierdzona dla ∆G jest nieco odmienna niż w innych warunkach i jest określana jako ∆G0'. Przykładem reakcji energ. korzystnej, o dużej ujemnej wartości ∆G0' i często używanej do napędzania reakcji energ. niekorzystnych, jest hydroliza ATP, tworząca ADP i Pi. [ ATP + H2O ADP + Pi ; ∆G0'= -30,5 kJ*mol-1 ].
Reakcja chemiczna istnieje zazwyczaj w stanie równowagi dynamicznej, w której mimo ustawicznego przekształcania się nowych cząst. substr. i tworzenia nowych cząst. prod., proporcja substratu do produktu pozostaje stała.
Rozważmy następującą sytuację: A B [10-4s-1]; A B [10-6s-1]; gdzie szybkość reakcji w kierunku wprost (od A do B) wynosi 10-4 na sekundę, a szybkość reakcji w kierunku odwrotnym wynosi 10-6 na sek. W równowadze stosunek stężeń substr. i prod stanowi wartość stałą, znaną jako stała równowagi (K). Stałą równowagi dla danej reakcji definiuje się jako: K=[produkty]/[substraty]=[B]/[A]; „[]” - stężenie. Stała równowagi jest określana przez stosunek szybkości reakcji w kierunku wprost (Kf) do szybkości reakcji w kierunku odwrotnym (Kb): K=Kf/KB=10-4/10-6=100. Dla tej reakcji w stanie równowagi istnieje 100x więcej produktu B niż substratu A, niezależnie od tego, czy enzym jest obecny, czy nie. Dzieje się tak dlatego, że enzymy nie zmieniają położenia równowagi reakcji, ale przyspieszają szybkość reakcji w obu kierunkach w tym samym stopniu. Enzymy przyspieszają osiągnięcie stanu równowagi, ale nie przesuwają jego położenia.
Miejsce aktywne enzymu jest regionem, który wiąże substrat i przemienia go w produkt. Zazwyczaj jest to względnie niewielka część całej cząsteczki enzymu i stanowi określoną trójwymiarową przestrzeń, utworzoną przez reszty aminokwasów, które w liniowym łańcuchu polipeptydowym mogą leżeć daleko od siebie. Miejsce aktywne jest często szczeliną lub zagłębieniem w cząst. enzymu, które tworzą środowisko w znacznym stopniu niepolarne, co ułatwia wiązanie substratów. Substr. jest wiązany w miejscu aktywnym przez liczne słabe siły (oddziaływania elektrostatyczne, wiązania wodorowe, siły Van der Waalsa, oddział. hydrofobowe), a w pewnych przypadkach przez odwracalne wiązania kowalencyjne. Po związaniu cząst. substratu i utworzeniu kompleksu enzym - substrat, katalitycznie czynne reszty w obrębie miejsca aktywnego enzymu działają na cząst. substratu tak, aby przekształcić go początkowo w stan przejściowy, a następnie w produkt, który zostaje uwolniony do roztworu. Potem enzym, już wolny, może związać kolejną cząst. substratu i rozpocząć nowy cykl katalityczny.
Zaproponowano dwa modele wyjaśniające jak enzym może związać swój substrat: (1) model zamka i klucza - Fischer 1894 - kształt substratu i aktywnego miejsca enzymu miałby pasować do siebie jak klucz do zamka; oba kształty są sztywne i utrwalone oraz pasują do siebie idealnie po odpowiednim zestawieniu; (2) model indukowanego dopasowania - Koshland 1958 - związanie substr. indukuje zmianę konformacyjną w aktywnym miejscu enzymu; enzym może zniekształcać substrat wymuszając w nim konformację podobną do stanu przejściowego (np. związanie glukozy z heksokinazą indukuje taką konformacyjną zmianę w strukturze enzymu, że jego miejsce aktywne przyjmuje kształt komplementarny do substratu (glukozy) tylko po jego związaniu z enzymem). Różne enzymy wykazują cechy charakterystyczne dla obu modeli, a więc pewną komplementarność i pewne zmiany konformacji.
SPECYFICZNOŚĆ SUBSTRATOWA
Właściwości ułożenia przestrzennego reszt aminokwasów tworzących aktywne miejsca enzymu determinują rodzaj cząsteczki, która może zostać związana i stać się substratem dla tego enzymu. O specyficzności substratowej często decydują zmiany stosunkowo niewielkiej liczby aminokwasów w miejscu aktywnym. Widać to wyraźnie w trzech enzymach trawiennych (trypsynie, chymotrypsynie, elastazie). Te 3 enzymy należą do rodziny enzymów o nazwie proteazy serynowe - „serynowe”, ponieważ mają w miejscu aktywnym resztę seryny, której udział w katalizie jest krytyczny; „proteazy”, ponieważ katalizują hydrolizę wiązań peptydowych w białku. Te 3 enzymy rozszczepiają wiązania peptydowe w białkowych substratach, po karboksylowej stronie pewnych reszt aminokwasów. Trypsyna rozszczepia (tnie) po karboksylowej stronie dodatkowo naładowanych reszt Lys lub Arg. Chymotrypsyna - po karboksylowej stronie dużych reszt aminokw. aromatycznych i hydrofobowych; elastaza - po karboksylowej str. reszt o krótkich, nienaładowanych łańcuchach bocznych. Różnica specyficzności tych enzymów jest narzucana przez charakter grup aminokwasów w miejscach wiązania substratu, komplementarnych względem substratu, na który działają. Tak więc trypsyna ma w swoim miejscu wiązania substratu ujemne naładowaną resztę ASP, która oddziałuje z dodatnimi ładunkami Lys i Arg substratu. Chymotrypsyna ma w miejscu wiązania substr. reszty aminokwasów o krótkich łańcuchach bocznych, tj. Gly i Ser, co umożliwia wejście do tych miejsc dużych łańcuchów bocznych substratu. Elastaza ma względnie duże, nienaładowane boczne łańcuchy aminokwasów Wal, Thr, wystające do miejsca wiązania substr., co pozwala na wejście do tego miejsca wyłącznie krótkich łańcuchów bocznych Gly i Ala substratu.
SPECYFICZNOŚĆ KIERUNKU DZIAŁANIA ENZYMÓW
Swoistość kierunku działania polega na zdolności enzymu do katalizowania tylko jednej z termodynamicznie możliwych reakcji, jakim może podlegać substrat wchodzący z nim w kompleks. Jeżeli dany substrat może przekształcić się w kilku różnych kierunkach, każda z reakcji jest katalizowana przez odrębny enzym zdolny do przekształcania wiązania określonego typu. Np. dowolny aminokwas ulega przemianom w co najmniej trzech kierunkach, każda z reakcji jest katalizowana przez inny enzym: dehydrogenazę, dekarboksylazę, aminotransferazę. Dwa ostatnie współdziałają z tym samym koenzymem - fosforanem pirydoksalu. Co jest dowodem, że swoistość kierunku działania jest funkcją części białkowej enzymu, a w mniejszym stopniu wiąże się z koenzymem.
Zmiany energii zachodzące podczas przebiegu reakcji biochemicznej:
1. Model klucza i zamka
2.Model indukowanego dopasowania
Schemat miejsc wiążących substrat w proteazach serynowych:
Czynniki wpływające na aktywność enzymów:
1.Oznaczanie enzymów. Ilość obecnego białka enzymatycznego można oznaczyć mierząc uzyskany efekt katalityczny, a więc ilość substratu przetworzonego w produkt. Aby oznaczyć enzym (mierząc jego aktywność) musimy znać sumaryczne równanie katalizowanej reakcji i dysponować procedurą analityczną umożliwiającą określenie albo ubytku substratu, albo pojawienie się produktu. Poza tym należy wziąć pod uwagę czy enzym wymaga jakichś kofaktorów oraz znać pH i temp., w której enzym jest optymalnie aktywny. Dla enzymu ssaków optimum takie przypada zazwyczaj na 25-37st.C. Istotne jest również, aby mierzona szybkość reakcji stanowiła miarę badanej aktywności enzymatycznej i nie była ograniczona niedostatecznym dopływem substratu. Dlatego też, zwykle są konieczne duże stęż. Substratu, tak, aby początkowa szybkość reakcji, którą właśnie mierzy się doświadczalnie była proporcjonalna do stęż. enzymu. Zazwyczaj enzym oznacza się przez pomiar szybkości pojawienia się produktu, lub szybkości zużywania substratu. Jeśli substrat, lub produkt absorbuje światło o specyficznej dł. fali, to zmiany stęż. tego związku można mierzyć śledząc zmianę absorbancji przy tej dł. fali, dokonuje się to zazwyczaj za pomocą spektrofotometru. Ponieważ absorbancja jest w pewnym zakresie proporcjonalna do stęż., szybkość zmiany absorbancji jest proporcjonalna do aktywności enzymu wyrażonej w molach zużytego substratu (lub wytworzonego produktu) w jednostce czasu. Cząsteczkami najczęściej używanymi do pomiarów absorbancji, w celu oznaczania enzymu są 2 koenzymy: zredukowany dinukleotyd nikotynoamidoadeninowy (NADH), oraz zredukowany fosforan dinukleotydu nikotynoamidoadeninowego (NADPH), które absorbują w obszarze ultrafioletu (UV) przy 340nm., Jeśli więc w przebiegu reakcji zostanie wytworzony NADH, lub NADPH nastąpi odpowiedni wzrost absorbancji przy 340nm,a jeśli reakcja dotyczy utleniania NADH do NAD+,lub NADPH do NADP+ wystąpi odpowiedni spadek absorbancji, ponieważ te utlenione formy nie absorbują promieniowania przy 340nm,np. pomiaru aktywności dehydrogenazy mleczanowej z mleczanem jako substratem można dokonać śledząc wzrost absorbancji przy 340nm zgodnie z następującym równaniem:
Pomiar połączonych reakcji enzymatycznych
Substraty i produkty wielu reakcji nie absorbują światła o dł. fali dogodnej do zmierzenia. W tym przypadku często pomiar enzymu katalizującego tę reakcję jest możliwy dzięki związaniu (lub sprzężeniu jej) z drugą reakcją enzymatyczną, w której zachodzi charakterystyczna zmiana absorbancji, np. działanie enzymu oksydazy glukozowej często stosowanej do pomiaru stężenia glukozy we krwi u pacjentów cierpiących na cukrzycę, nie powoduje żadnej zmiany absorbancji, podczas przemiany substratu w produkt. Jednakże wytworzenie w tej reakcji nadtlenek wodoru można poddać działaniu kolejnego enzymu- peroksydazy przemieniającej jednocześnie związek bezbarwny w barwny, którego absorbancję można łatwo zmierzyć
Jeśli aktywność oksydazy glukozowej ma być mierzona dokładnie, to peroksydaza oraz jego dalsze substraty lub koenzymy muszą być podane w nadmiarze, tak, aby ten drugi enzym nie stanowił ograniczającego etapu w tym złączonym pomiarze. Zachowanie tego warunku spowoduje, że szybkość powstawania barwnego chromogenu, będzie proporcjonalna do szybkości powstawania H2O2,która to szybkość jest z kolei proporcjonalna do aktywności oksydazy glukozowej.
Szybkość działania enzymów.
Tempo przebiegu reakcji katalizowanej przez enzym jest określane jako szybkość reakcji. Szybkość działania enzymu podaje się zazwyczaj jako wartości w czasie zerowym, bowiem szybkość jest największa wtedy, gdy nie ma jeszcze produktu. Dzieje się tak dlatego, że w nieobecności produktu, nie może występować efekt hamowania przez sprzężenie zwrotne własnym produktem- co jest częstym zjawiskiem w enzymach, ani też reakcja nie może przebiegać w kierunku odwrotnym zasilanym przez produkt. Typowy wykres powstawania produktu jako funkcji czasu dla reakcji katalizowanej enzymatycznie, wykazuje początkowy okres szybkiego narastania produktu, co odpowiada prostoliniowemu odcinkowi wykresu. Po okresie tym następuje stopniowe zwolnienie aktywności enzymu w miarę zużywania substratu i/lub osłabiona aktywność enzymu. Wartość V0 uzyskuje się kreśląc linię prostą styczną do początkowego odcinka krzywej zaczynając od czasu zerowego. Nachylenie tej prostej ma właśnie wartość V0.
Zależność między powstawaniem produktu a czasem trwania reakcji katalizowanej przez enzym (szybkość działania enzymu).
Jednostki enzymatyczne
Aktywność enzymatyczną można wyrazić na wiele sposobów, najpowszechniejsze jest podawanie początkowej szybkości (V0) katalizowanej reakcji. Istnieją też 2 standardowe jednostki aktywności enzymatycznej. Jednostką enzymatyczną jest ilość enzymu, która katalizuje przekształcenie 1µmola substratu, w ciągu 1 min.,w 25stC,w warunkach optymalnych dla danego enzymu. Katal jest przyjętą w układzie SI jednostką aktywności enzymu zdefiniowaną jako aktywność katalityczna, która zwiększa szybkość reakcji, w specyficznym układzie o 1mol/s. Różne jednostki aktywności można wzajemnie przeliczać używając równoważności 1µmol * min-1= 1U= 16,67nkat - nanokatali. Nazwa aktywność (lub aktywność całkowita) odnosi się do całkowitej liczby jednostek enzymatycznych w próbie, natomiast aktywność specyficzna jest liczbą jednostek enzymatycznych na 1 mg białka. Aktywność specyficzna jest miarą czystości enzymu. Wzrasta ona podczas oczyszczania enzymu, a staje się maksymalna i stała po całkowitym oczyszczeniu enzymu.
Stężenie substratów
Normalny układ zależności szybkości działania enzymu od stężenia substratu [S] polega na tym, że przy małych stężeniach substratu podwojenie [S] powoduje podwojenie początkowej szybkości reakcji[vo]
Jednakże przy większych stężeniach substratu enzym ulega wysyceniu i dalszy wzrost [S] powoduje tylko małą zmianę [vo]. Dzieje się tak, ponieważ przy wysycających stężeniach substratu praktycznie wszystkie cząsteczki enzymu zawierają związany substrat. Sumaryczna szybkość działania enzymu jest teraz zależna od szybkości, z jaką produkt może oddysocjować i dalsze dodanie substratu nie będzie już miało na to wpływu. Kształt wykresu przedstawiającego wartość [vo] jako funkcję[S] jest nazywany krzywą hiperboliczną.
Zależność między stężeniem substratu [S] a początkową szybkością reakcji [V0]
Stężenie enzymu
Kiedy stężenie substratu jest wysycające (wszystkie cząsteczki enzymu mają związany substrat), podwojenie stężenia powoduje podwojenie [vo]. Zależność ta ujawnia się jako linia prosta na wykresie przedstawiającym [vo] jakom funkcją stężenie enzymu.
Temperatura
Wpływa na szybkość reakcji katalizowanych enzymatycznie
1) wzrost temperatury zwiększa energię chemiczną cząsteczek substratu. To powoduje zwiększenie proporcji cząsteczek substratu mających dostateczną ilość energii, aby przekroczyć G (energię aktywacji, a przez to zwiększyć szybkość reakcji.Równocześnie jednak w wyższych temp. Ujawnia się drugi efekt
2) wzrastająca energia termodynamiczna cząsteczek, które tworzą strukturę samego białka enzymatycznego zwiększa szansę zerwania licznych, słabych wiązań niekowalencyjnych
(wiązania wodorowe, siły Van der Wellsa)utrzymujących 3D strukturę enzymu. Końcowym efekcie będzie to prowadzić do denaturacji (rozfałdowania) enzymu, a nawet małe zmiany 3D kształtu enzymu mogą zmieniać strukturę miejsca aktywnego i doprowadzić do spadku aktywności katalitycznej. Ostateczny wpływ podwyższonej temperatury na szybkość reakcji enzymatycznej jest wynikiem równowagi między tymi dwoma przeciwnymi działaniami. Dlatego też wykres zależności [vo] od temperatury będzie miał kształt krzywej o wyraźnie zaznaczonym optimum termicznym.(rys.4a). Dla wielu enzymów ssaków przypada ono na ok.37OC, ale istnieją organizmy, których optimum termiczne jest wyższe lub niższe np. polimeraza, której używa się do łańcuchowej reakcji polimeryzacji, występuje u bakterii żyjących w gorących źródłach, źródłach, więc przystosowanych do optymalnej pracy w wysokich temperaturach.
8. Wartość pH
Każdy enzym ma optymalne pH działania, w którym szybkość katalizowanej przez niego reakcji jest maksymalna. Małe odchylenie pH od wartości optymalnej powoduje spadek aktywności wywieranej zmianami jonizacji grup w aktywnym miejscu enzymu. Większe odchylenie pH prowadzi do denaturacji białka enzymu. W wyniku zakłócenia licznych słabych oddziaływań niekowalencyjnych utrzymujących strukturę 3D białka. Wykres Vo jako funkcję pH daje zazwyczaj krzywą w kształcie dzwonu. (Rys4B). Wiele enzymów wykazuje optimum aktywności w pobliżu pH=6,8, ale ogólnie istnieje duże zróżnicowanie optimum pH enzymów wywołane różnicami środowiska, w którym enzymom przyszło działać, np. enzym trawienny pepsyna jest przystosowany do działania w kwaśnym pH żołądka (~ 2,0)
Rysunek 4 wpływ temperatury (a) oraz pH (b) na aktywność enzymu.
Izoenzymy (izozymy)
Są różnymi formami enzymów, które katalizują tę samą reakcję, ale wykazują odmienne właściwości fizyczne i kinetyczne takie jak: pJ, optimum pH, powinowactwo do substratu lub wrażliwość na inhibitory. Różne formy izoenzymu danego enzymu pochodzą zazwyczaj z różnych genów i często występują w różnych tkankach ciała. Przykładem enzymu, który ma różne formy izozymowe jest dehydrogenaza mleczanowa (LDH) katalizująca odwracalnie przekształcenie pirogronianu w mleczan w obecności NADH jako koenzymu. LDZ. Jest tetrametrem dwóch różnych typów podjednostek określanych jako H oraz M, które wykazują małe różnice w sekwencji aminokwasów. Podjednostki mogą się łączyć ze sobą tworząc 5 izoenzymów o składzie: H4, H3M, H2M2, HM3, M4. Te 5 izoenzymów można rozdzielić elektroforetycznie. Podjednostka M występuje w mięśniach szkieletowych i wątrobie, w sercu. Izoenzymy H4 oraz H3M występują głównie w sercu i erytrocytach, H2M2 jest obecny głównie w mózgu i nerkach, natomiast HM3 oraz M4 występują przede wszystkim w wątrobie i mięśniach szkieletowych. Tak więc wzór izoenzymów jest charakterystyczny dla każdej tkanki, co ma ogromne znaczenie diagnostyczne w medycynie. Zawał serca mięśniowego, żółtaczka zakaźna i choroby mięśni, powodują w uszkodzonej tkance śmierć komórek, których zawartość przedostaje się do krwi. Ponieważ LDH jest rozpuszczalnym białkiem cytozolowym, łatwo zostaje w tych warunkach uwalniane do krwi, która w normalnych warunkach zawiera mało LDZ. Dlatego też wzór izoenzymów we krwi wskazuje, z której tkanki izozymy pochodzą i można go użyć do postawienia diagnozy np. zawału mięśnia sercowego oraz monitorowania przebiegu leczenia.
Flawinomononukleotyd (FMN) i dinukleotyd flawinoadeninowy (FAD)
Kinetyka i inhibicja enzymów
Model Michaelisa- Menten opiera się na następującej koncepcji katalizy enzymatycznej
E+S k1,k2ESk3E+P
Enzym (E) wiąże się ze swym substratem(S) tworząc kompleks (ES) enzym-substrat. Kompleks ES może dysocjować z powrotem do E+S lub przekształcać się w sam E i produkt (P) Stałe szybkości k1, k2, k3 opisują szybkości przebiegu każdego etapu procesu katalitycznego. Przyjmuje się, iż reakcja odwrotna, w której enzym i produkt (E+P) byłyby przekształcane w kompleks ES, przebiega z tak małą szybkością, że reakcję można pominąć. Na podstawie obserwacji właściwości wielu enzymów wiadomo było, że przy małych stężeniach substratu początkowa szybkość reakcji jest wprost proporcjonalna do [S]. Natomiast przy dużych stężeniach substratu szybkość zmierza do wartości maksymalnej tzn., że szybkość staje się niezależna od [S] (rys 1a). Szybkość maksymalną oznacza się jako Vmax. W celu opisania tych obserwacji Michaelis i Menten wprowadzili równanie nazwane od ich nazwisk. Vo= Max *[S]/ Km + [S]. Równanie to opisuje krzywą hiperboliczną typu pokazanego dla danych doświadczalnych na rys 1a. Wprowadzając to równanie M i M zdefiniowali nowa stałą Km zwaną stała Michaelisa (jednostki molarne M). Km= k2 +k3/k1
Km jest miarą stabilności kompleksu ES stanowiąc iloraz sumy szybkości rozkładu ES i szybkości jego powstawania. Dla wielu enzymów k2 jest znacznie większe niż k3. W tych warunkach wartość Km staje się miarą powinowactwa enzymu do substratu, ponieważ wartość ta zależy od względnych wartości k1 o k2 dla odpowiedniego tworzenia i dysocjacji kompleksu ES. Duża wartość Km wskazuje na słabe wiązanie substratu (k2 przeważa nad k1). Wartość Km można wyznaczyć doświadczalnie na podstawie tego, że wartość ta równa się stężeniu substratu, przy którym szybkość reakcji jest równa połowie Max.
Ponieważ Max jest osiągana przy nieskończenie dużym stężeniu substratu, nie można wyznaczyć wartości Max (a więc również Km). Z wykresu hiperbolicznego przedstawionego na Rys.1e. Jednakże wartość Vmax oraz Km można wyznaczyć doświadczalnie mierząc Vo
przy różnych stężeniach. Następnie wykorzystuje się wykres kinetyki enzymu w układzie współrzędnych, które SA odwrotnościami V i S, czyli wykres alfa-beta podstawiając 1 do Vo jako funkcję 1 do [S] (1/Vo=1/[S]) (rys 1b). Wykres taki pochodzi z przekształcenia równania M-M do postaci 1/Vo =1/Vmax + Km/Vmax * 1/[S], dając linię prostą, której przecięcie z osią Y równa się 1/Vmax Max przecięcia z osią X= -1/Km. Nachylenie linii ma wartość Km/Max (rys.1b). Wykres alfa-beta stanowi również użyteczną metodę określania typu wiązania się inhibitora z enzymem (patrz niżej). W prawdzie dla wielu enzymów model M-M stanowi b. dobry model danych doświadczalnych, ale istnieją enzymy, które nie podlegają kinetyce M-M. Enzymy te (np. ATCaza) są nazywane enzymami allosterycznymi.
INHIBICJA ENZYMÓW
Istnieje wiele typów cząsteczek, które są zdolne do zakłócania aktywności danego enzymu. Każda cząsteczka działająca bezpośrednio na enzym w kierunku zmniejszenia jego aktywności katalitycznej to inhibitor. Pewne inhibitory enzymów są normalnymi metabolitami komórkowymi, które hamują dany enzym w ramach naturalnej metabolicznej kontroli odpowiedniego szlaku. Inne inhibitory mogą być substancjami obcymi dla organizmu takimi jak toksyny i leki i w tym przypadku hamowanie enzymu może mieć działanie terapeutyczne, ale również letalne. Rozróżnia się 2 główne typy inhibicji enzymów: nieodwracalna i odwracalna, przy czym inhibicja odwracalna dzieli się na kompetycyjną i niekompetycyjną. Inhibicję odwracalną można przezwyciężyć usuwając inhibitor z enzymu np. w drodze dializy, ale jest to z definicji niemożliwe w inhibicji nieodwracalnej.
INHIBICJA NIEODWRACALNA
Te inhibitory, które wiążą się z enzymem nieodwracalnie często tworzą kowalencyjne wiązania z resztami aminokwasów znajdujących się w miejscu aktywnym lub jego pobliżu i inaktywują enzym na stałe. W wiązaniu tym biorą udział reszty Ser i Cys mające odpowiednio reaktywne grupy OH i SH np. związek diizopropylofluorofosforan, składnik gazów bojowych działających na ukla nerwowy, reaguje z reszta Ser w miejscu aktywnym enzymu uniemożliwiając przekazywanie impulsów nerwowych. Amid kwasu jodooctowego modyfikuje reszty Cys i może być stosowany jako czynnik diagnostyczny w określeniu czy aktywność enzymatyczna wymaga 1 tylko czy tez więcej reszt Cys. Antybiotyk penicylina nieodwracalnie hamuje enzym transpeptydazę peptydoglikanu, która tworzy poprzeczne wiązania w ścianie kom bakteryjnych, przy czym hamowanie to polega na łączeniu się penicyliny z reszta Ser w miejscu aktywnym enzymu.
ODWRACALNA INHIBICJA KOMPETYCYJNA
Inhibitor kompetycyjny jest zazwyczaj strukturalnie podobny do normalnego substratu danego enzymu. Dzięki temu współzawodniczy z cząsteczkami substratu o wiązanie się z miejscem aktywnym. Enzym może wiązać albo cząsteczkę substratu albo cząsteczkę inhibitora, ale nie oba jednocześnie. Inhibitor ten wiąże się z miejscem aktywnym odwracalnie. Przy dużych stężeniach substratu działanie inh. komp. Zostaje przezwyciężone ponieważ duże stężenie substratu będzie z powodzeniem współzawodniczyć z cząsteczka inhibitora o wiązanie się w miejscu aktywnym. Nie nastąpi wiec żadna zmiana wartości Vmax enzymu, ale w obecności inh. komp. pozornie zmniejsza się powinowactwo enzymu do jego substratu i dlatego wartość Km wzrasta.
Dobrego przykładu hamowanie kompetycyjnego dostarcza dehydrogenaza bursztynianowa. Enzym ten używa bursztynianu jako substratu i jest hamowany kompetycyjnie przez malonian, który różni się od bursztynianu posiadaniem tylko 1 a nie 2 grup metylenowych. Wiele leków działa przez naśladowanie struktury substratu specyficznego dla enzymu docelowego i stad działaj one jako inh. komp. tego enzymu. Kompetycyjny charakter inhibicji można rozpoznać z użyciem wykresu LB. Mierzy się wtedy V0 przy rożnych stężeniach substratu w obecności stałego stężenia inhibitora. Inhibicja komp. zwiększa nachylenie linii na wykresie LB i zmienia jej przecięcie z osią X (bo Km wzrasta) ale nie zmienia pozycji przecięcia tej linii z osią Y bo wartość Vmax pozostaje stała.
Inhibicja dehydrogenazy bursztynianowej przez malonian:
Bursztynian + FAD -deh bursztynianowa--> fumaran + FADH2
Malonian -||deh. bursztynianowa--> r nie zach.
ODWRACALNA INHIBICJA NIEKOMPETYCYJNA
Inhibitor niekompetycyjny wiąże się odwracalnie w innym miejscu enzymu niż jego miejsce aktywne i powoduje zmianę przestrzennego kształtu enzymu ,co prowadzi do zmniejszenia aktywności katalitycznej. Ponieważ inhibitor wiąże się w innym miejscu niż substrat enzymu może wiążąc albo inhibitor albo substrat lub oba jednocześnie. Efektu działania tego inhibitora nie można przezwyciężyć przez zwiększenie stężenia substratu i dlatego zmniejsza się wartość Vmax. W tej inhibicji powinowactwo enzymu do substratu pozostaje niezmienione a wiec wartość Km nie zmienia się. Przykładem takiej inhibicji jest działanie pepsatyny na enzym reninę.
Pepstayna - większość proteaz aspartylowych, karboksylowych, kwasowych hamowana jest przez bardzo małe stężenia izolowanego z mikroorg peptydu nazwanego pepsatyną.
Niekompetycyjny charter inhibicji można latwo rozpoznać na wykresie LB, bo następuje wtedy wzrost nachylenia linii wyznaczonej doświadczalnie i zmienia się miejsce jej przecięcia z osią Y (bo wartość Vmax maleje) ale miejsce jej przecięcia z osią X nie zmienia się (bo Km jest stałą).
KONTROLA AKTYWNOŚCI ENZYMATYCZNEJ
Regulacja przez sprzężenie zwrotne.
W systemach biologii szybkości wielu enzymów mogą być zmieniane przez obecność innych cząsteczek tj. aktywatory i inhibitory (określanych jako efektory).
Istotę kontroli szlaków metabolicznych jest to, że enzym działający przy początkowym szlaku jest hamowany przez końcowy produkt tego szlaku metabolicznego.
Proces ten nazywamy hamowaniem w drodze sprzężenia zwrotnego. Ma często miejsce na decydującym etapie reakcji(przemiana A w B)
Etap decydujący jest pierwszym, na którym powstaje intermedia występujący tylko w danym szlaku i dlatego etap ten wymusza dalsze przemiany tego związku wzdłuż danego szlaku.
Kontrola enzymu, którą przeprowadza decydujący etap szlaku metabolicznego umożliwia zaoszczędzenie w organizmie energii metabolicznej i zapobiega nagromadzaniu się jej w dalszym przebiegu szlaku dużych ilości zbędnych intermediatów metabolicznych.
Ponieważ wiele szlaków metabolicznych jest rozgałęzionych hamowanie przez sprzężenie zwrotne musi umożliwiać dalszy przebieg syntezy 1 produktu rozgałęziającego się szlaku, nawet wtedy gdy 2 produkt jest obecny w nadmiarze.
W tej sytuacji musi działać proces inhibicji przez sekwencyjne sprzężenie zwrotne, w którym końcowy produkt jednego rozgałęzienia szlaku będzie hamował 1 enzym działający poza miejscem rozgałęzienia.(przemian C w D lub C w E ) Gdy ten intermedia miejsca rozgałęzienia nagromadzi się hamuje on z kolei pierwszy decydujący etap całego szlaku(przemian A w B).
Ponieważ produkt końcowy szlak metabolicznego, obejmującego wiele reakcji enzymatycznych ma małą szanse strukturalnego podobieństwa do związku wyjściowego, produkt końcowy będzie wiązał się z enzymem w punkcie kontrolnym, którym jest miejsce inne niż miejsce aktywne.Takie enzymy są zawsze enzymami allosterycznymi.
A->B->C->D->Z
a) E1 hamowany przez produkt Z (przez sprzężenie zwrotne)
b) sekwencyjne hamowanie przez sprzężenie zwrotne
ENZYMY ALLOSTERYCZNE
Wykres wartości V0 jako funkcji (S) dla enzymu allosterycznego daje krzywą sigmoidalna a nie krzywą hiperboliczną wynikającą z równania Michelisa-Michtona.
W środkowym zakresie stężeń substratu krzywa ta ma stromy odcinek, odzwierciedlający gwałtowny wzrost szybkości enzymu zachodzący w wąskim zakresie stężeń substratu.
To właśnie nadaje enzymom allosterycznym szczególną wrażliwość na małe zmiany stężenia w zakresie fizjologicznym
W enzymach allosterycznych związanie cząsteczki substratu do jednego miejsca aktywnego wpływa na wiązanie cząsteczki substratu do innych miejsc aktywnych enzymu
Mówi się że różne miejsca aktywne zachowują się kooperatywnie w zakresie wiązania cząsteczek substratu i oddziaływania na nie( np. wiązanie O2 do 4 podjednostek hemoglobiny)
Enzymy allosteryczne są w związku z tym często białkami złożonymi z wielu podjednostek, z których każda ma 1 lub więcej miejsc aktywnych.
Związanie substratu w jednym miejscy aktywnym, indukuje w białku zmianę konformacyjną, które jest przenoszone do innych miejsc aktywnych zmieniając ich powinowactwo do cząsteczek substratu.
Poza tym enzymy allostertczne mogą być kontrolowane przez cząsteczki efektorów9aktywatory i inhibitory), które wiążą się z enzymem w miejscu odmiennym od miejsca aktywnego, na tej samej podjednostce lub innej, powodując przez to zmianę konformacji miejsca aktywnego-co zmienia szybkość działania enzymu9n wiązanie CO2, H+, oraz 2,3-bisfosfoglicerynianu do hemoglobiny)
Aktywator allosteryczny zwiększa aktywność enzymu, natomiast inhibitor allosteryczny zmniejsza ją.
TRANSKARBAMOILAZA ASPARAGINIANOWA- karbamoilotransferaza asparaginianowa-Kluczowy enzym w biosyntezie pirymidyn.
Dostarcza dobrego przykładu regulacji allosterycznej. ATC-aza katalizuje tworzenia N-karbamoiloasparaginianu z asparaginianu i karbamoilofosforanu, co jest decydującym etapem w biosyntezie pirymidyn.
Związanie 2 substratów asparaginianu i karbamoilofosforanu jest seperatywne na co wskazuje sigmoidala krzywa V0 jako funkcja stężenia substratu.
ATC-aza jest złożona z 6 podjednostek katalitycznych i 6 podjednostek regulatorowych
Enzym podlega inhibicji na drodze sprzężenia zwrotnego przez końcowy produkt szlaku cystydynotrifosforanu(CTP), który działa jako inhibitor allosteryczny.
Jego cząsteczka wiąże się z podjednostkami regulatorowymi i powoduje zmniejszenie katalitycznej aktywności ATC-azy, zmniejszając powinowactwo podjednostek katalitycznych do cząsteczek substratu.
Odwrotnie ATP, jeden z intermediatów pojawiających się wcześniej na szlaku działa jako aktywator allosteryczny zwiększając powinowactwo ATC-azy do jej substratów, powodując wzrost jej aktywności
ATP współzawodniczy z CTP o to samo miejsce wiążące na podjednostce regulatorowej. Wysoki poziom ATP stanowi dla komórek sygnał ze jest dstepna energia replikacji DNA równocześnie ATC-aza zostaje uaktywniona co prowadzi do syntezy nukleotydów pirymidynowych niezbędnych do tej replikacji.
Gdy pirymidyny są obecne w dużej ilości wysoki poziom CTP hamuje ACT-azę zapobiegając przez to zbędnej syntezie N-karmamoiloasparagininu i dalszych intermediatów szlaku.
Powstawanie N-karmamoiloasparagininu
Wykres poczatkowej szybkosci reakcji(Vo) allosterycznego enzymu ATCazy jako funkcji stezenia substratu
ODWRACALNA MODYFIKACJA KOWALENCYJNA
Polega na tworzeniu i rozcinaniu wiązań kowalencyjnych między grupami niebiałkowymi a cząsteczką enzymu.
Chociaż wiele niebiałkowych grup może być odwracalnie dołanczanych do enzymu, wpływając na ich aktywność najczęstszą modyfikacją jest dodawanie i usuwanie grupy fosforanowej(fosforylacja i defosforylacja). Fosforylacja katalizowana jest często przez kinazy białkowe, używając często ATP jako donora grupy fosforanowej. Natomiast defosforylacja jest katalizowana przez fosfatazy białkowe. Dodawanie i usuwanie gr. Fosforanowej powoduje zmiany w trzeciorzędowej strukturze enzymu co zmienia jego aktywność katalityczną.
Jedna klasa kinaz przenosi gr. Fosforanową specyficznie na grupy hydroksylowe reszt seryny lub treoniny docelowego enzymu. Kinazy białkowe serynowo-treoninowe, których przykładem jest kinaza białkowa zależą od cAMP. Natomiast 2klasa przenosi gr fosforanową na grupy hydroksylowe reszty tyrozynowej(kinazy tyrozynowe)
Fosfatazy białkowe katalizują hydrolizują katalityczne usuwanie grupy fosforanowej z białek, regenerując niezmodyfikowane gr. Hydroksylowe aminokwasów i uwalniają Pi. Ufosforylowana forma enzymu może być albo mniej albo bardziej aktywna w porównaniu z forma zdefosforylowaną..Tak więc cykl fosforylacji/defosforylacji może być użyty jako szybki odwracalny przełącznik dla szlaku metabolicznego, w zależności od potrzeb komórki.Np. fosforylaza glikogenowa enzym działający podczas rozkładu glikogenu jest aktywna w formie ufosforylowanej a synteza glikogenowa jest najbardziej aktywna w formie nieufosforylowanej.
Do innych typów odwracalnej modyfikacji kowalencyjnej używanych do regulacji aktywności pewnych enzymów należą:1.adenylacja przenoszenia grupy adenylowej z ADP
2.ADP-rybozylacja(przeniesienie jednostki adenozynodifosforybozylowej z NAD+)
Odwracalna fosforylacja i defosforylacja enzymu
AKTYWACJA PROTEOLITYCZNA
Enzymy są syntetyzowane jako większe nieaktywne formy prekursorowe o nazwie proenzymy lub zymogeny . Aktywacja zymogenu polega na nieodwracalnej hydrolizie jednego lub więcej wiązań peptydowych.
PROTEAZY TRZUSTKOWE
Enzymy trawienne trypsyna, chymotrypsyna i elastaza powstają w trzustce jako zymogeny. Potem są transportowane do jelita cienkiego, gdzie ich formy zymogenowe zostają zaktywowane przez rozszczepienie specyficznych wiązań peptydowych.trypsyna jest syntetyzowana jako zymogen trypsynogen i w jelicie jest rozcinana( i w ten sposób jest aktywowana) przez enzym enteropeptydazę, wytwarzany tylko w jelicie. Po zaktywowaniu trypsyna może rozcinać i aktywować dalsze cząsteczki trypsynogenu jak i chymotrypsynogen i proelastazę (RYS5)
Centralna rola trypsyny w aktywowaniu zymogenów trzustkowych
Chymotrypsyna jest syntetyzowana jako zymogen, chymotrypsynogen, pojedynczy łańcuch polipeptydowy o 245 resztach aminokwasowych (rys 6)po znalezieniu się w jelicie chymotrypsynogen jest rozszczepiany najpierw na trypsynę, po karboksylowej stronie reszty Arg 15 tworząc πchymotrypsynę, która jest w pełni aktywna.
Następnie z polipeptydowego łańcucha πchymotrypsyny są usuwane dwa peptydy, przez co powstaje stabilna forma σ-chymotrypsyna. Ulega ona zmianom konformacyjnym przechodząc w dojrzała, aktywna αchymotrypsynę. 3 fragmenty wyjściowego, pojedynczego łańcucha polipeptydowego są w αchymotrypsynie utrzymywane razem przez działanie kowalencyjnych wiązań dwusiarczkowych oraz interakcji niekowalencyjnej
Aktywacja chymotrypsynogenu przez rozszczepienie proteolityczne.
Trzustka syntetyzuje też nowe białko - inhibitor trypsyny. to białko wiąże się bardzo silnie z miejscem aktywnym trypsyny chroniąc trzustkę przed zniszczeniem przez przedwcześnie zaktywowane cząsteczki trypsyny. jeśli ten ochronny mechanizm zawiedzie np. z powodu niedrożności przewodu trzustkowego zymogen może zostać zaktywowany i strawić trzustkę w procesie zwanym ostrym zapaleniem trzustki.
KASKADA KRZEPNIECIA KRWI
Cały ten proces przebiega w postaci serii aktywizacji zymogenowych. aktywacja zymogenów powoduje wzmocnienie początkowego sygnału, gdyż pojedynczy zaktywowany enzym może działać z wieloma cząsteczkami substratu wywołując dalsza aktywacje. ponieważ rozszczepienie proteolityczne nie wymaga ATP rozszczepienie to jest mechanizmem właściwym dla aktywacji białek poza obrębem komórek. Enzym raz zaktywowany pozostaje aktywny.
REGULACJA ENZYMATYCZNEJ SYNTEZY I ROZKLADU
Ilość poszczególnego enzymu w komórce lub tkance zmienia się zależnie od szybkości jego syntezy i degradacji. czynniki wpływające na szybkość syntezy obejmują indukcje lub represje genu kodującego enzym, a także szybkość degradacji mRNA wytwarzanego przez ten gen. Wiele kluczowych enzymów działających w konkretnych punktach szlaków metabolicznych ma mRNA o szczególnie krótkim okresie życia, dzięki czemu szybkość syntezy enzymu jest łatwo kontrolowana przez czynniki wpływające na szybkość transkrypcji genu. Szybkość degradacji enzymu znajduje swój wyraz w jego półokresie życia- w czasie potrzebnym do degradacji polowy1/2 ilości białka. gdy krotki półokres życia to enzymy nazywane są labilnymi.
KLASYFIKACJA ENZYMOW
Nazwy utworzone przez dodanie końcówki -aza do nazwy substratu. Np. ureaza jest enzymem katalizującym hydrolizę mocznika, a fruktazo-1,6-bisfosfataza hydrolizuje fruktozo-1,6-bisfosforan.Pewne enzymy jak trypsyna i chymotrypsyna maja nazwy nieodnoszące się do ich substratów (nazwy zwyczajowe). Są też enzymy, które maja po kilka różnych nazw. Nazewnictwo enzymów uzgodniono międzynarodowo-EC. System ten dzieli enzymy na 6 klas(tabela1)Następnie każdy enzym zostaje zidentyfikowany przez czteroliczbowy numer, np. trypsyna ma nr EC3.4.21.4. przy czym liczba 3 oznacza ze jest hydrolaza, liczba 4 ze hydrolizuje wiązanie peptydowe, 21 ze enzym jest proteza serynowa, mająca w miejscu aktywnym krytyczna resztę seryny, a kolejna 4 ze był to czwarty enzym historycznie przypisany do tej klasy
Międzynarodowa klasyfikacja enzymów
klasa |
nazwa |
Typ kat. reakcji |
przykłady |
1 |
OOksyreduktazy |
Przenoszenie elektronów |
Dehydrogenaza alkoholowa |
2 |
Transferazy |
Przenoszenie grup funkc. |
Heksokinaza |
3 |
Hydrolazy |
R. hydrolizy |
Trypsyna |
4 |
Liazy |
Rozszczepienie wiązań C-C,C-O,C-N |
Dekarboksylaza pirogronianowa |
5 |
Izomerazy |
Przenoszenie grup w obrębie cząsteczki |
Izomeraza maleinianowa |
6 |
Ligazy (syntetazy) |
Tworzenie wiąz, sprzężone z hydrolizą ATP |
Karboksylaza pirogronianowa |
Oksydoreduktazy
Do tej klasy należą enzymy katalizujące reakcję oksydoredykcyjne a więc przemiany związane z przeniesieniem e i tlenu. W przenoszeniu tych składników towarzyszą zwykle charakterystyczne koenzymy. Klasa ta obejmuje enzymy o potocznych nazwach dehydrogenazy, reduktazy, oksydazy, oksygenazy, hydrolazy i peroksydazy. Dehydrogenazy przenoszą p+ i e- na tlen lub częściej z substratu na koenzymy lub odwrotnie. Reduktazy katalizują przenoszenie p+ i e- lub samych e- z przenośników (np. łańcucha oddechowego) na dalsze układy oksydoredykcyjne. Oksydazy aktywują tlen cząsteczkowy przez przenoszenie nań e- w skutek tego może się on łączyć z p+ wydzielonymi uprzednio do roztworu tworząc cząsteczkę wody lub rzadziej H2O2 (oksydazy flawinowe). Oksygenazy i hydrolazy katalizują przyłączenie tlenu do związku org. z przeniesieniem p+ i e- i z udziałem koenzymu, a peroksydazy działają utleniająco na zw. organiczne z udziałem H2O2. jak już wspomniano pełna nazwa klasy oksydoreduktaz powinna zawierać nazwy dawcy i akceptora (przenoszonych p+i e-), oraz nazwę klasy. Dehydrogenaza alkoholowa EC 1.1.1.1. powinna mieć nazwę oksydoreduktaza alkohol:NAD+
Transferazy
Enzymy te katalizują reakcję przeniesienia grup pomiędzy poszczególnymi związkami i to zwykle z udziałem specyficznych koenzymów. Typowymi przedstawicielami tej klasy są: aminotransferazy, fosfotransferazy- kinazy, acylotransferazy i glikozylotrnsferazy katalizując odpowiednio przeniesienie grup aminowej, fosforanowej z udziałem ATP, arylowej lub glikozylowej. Duże znaczenie mają enzymy przenoszące grupy jednowęglowe, z których najważniejsze są metylotransferazy (CH3-) oraz formylotransferazy (HCO) Przeniesienie wymienionych grup odbywa się z reguły za pośrednictwem typowych koenzymów. Nazwa enzymów tej klasy powinna zawierać określenie grupy przenoszonej, dawcy oraz akceptora np. prawidłową nazwą heksokinazy jest 6-fosfotransferaza: ATP:D-heksoza.
Hydrolazy
Enzymy te katalizują reakcję hydrolizy, czyli rozkłady wiązań z udziałem cząsteczki wody. Z ważniejszych enzymów należy wymienić esterazy rozkładające wiązanie sterowe, enzymy rozkładające wiązania peptydowe (peptydazy), amidowe (amidazy) i inne. Hydrolazy nie wymagają zwykle współdziałania koenzymów, co jest wyjątkiem w stosunku do innych klas. Nazwa enzymu obejmuje w tym przypadku określenie klasy i substratu np. hydrolaza aminoacetylokoenzymu-A. jak wspomniano skrócone nazwy enzymów hydrolitycznych można tworzyć przez dodanie do nazwy substratu -aza np. arginaza, czy ureaza, tzn. enzymy rozkładające hydrolitycznie argininę i mocznik.
Liazy
Klasa ta obejmuje enzymy, które odwracalnie lub nieodwracalnie katalizują odłącznie grup od substratu bez udziału H2O. należą tu enzymy katalizujące wiązania C-C,C-O,C-N np. kolejno dekarboksylaza aminokwasów lub oksokwasów C-O, czyli przyłączające lub odrywające cząst.H20 np.hydro-liaza jabłczanu, enzymy katalizujące rozkład wiązań C-N np. deaminazy (amoniakoliaza asparaginianowa) oraz rozkładające niektóre inne wiązania np.C-S prawidłowa nazwa enzymów klasy liaz zawiera określenie substratu i rodzaju odłączonej grupy oraz nazwę klasy, np. hydroliza L-jabłczanu.
Izomerazy
Do tej klasy należą enzymy katalizujące rekcję izomeryzacji, jak: racemizacja, epimeryzacja, izomeryzacja cis-trans oraz wewnątrzcząsteczkowe przemiany oksydoredukcyjne i przeniesienie grup. Pełna nazwa obejmuje określenie substratu i rodzaju izomeryzacji np. izomeraza maleinianowa cis-trans.
Ligazy
Zwane inaczej syntetazami. Enzymy tej klasy katalizują wytwarzanie wiązań między dwiema cząst., co jest powiązane z rozpadem bogatego w energię związku makroergicznego, a więc z udziałem ATO, do tej klasy należą więc enzymy aktywujące powstawanie wiązania C-O (np. syntetazy aminoacylo-tRNA) wiązania C-S (np. ligaza, kwas:CO-A-SH,ATP) wiązania C-N (np. ligazy:kwas:aminokwas,ADP)i wiązania C-C (karboksylazy). Pełna nawa enzymu obok określenia klasy powinna zawierać nazwy obu łączących się substratów oraz związku tworzącego się w wyniku rozkładu związku makroergicznego np. ligaza-S-CoA:CO2,ADP.
Koenzymy i grupy prostetyczne
Wiele enzymów wymaga do swego specyficznego działania obecności małych jednostek niebiałkowych o nazwie kofaktory. Kofaktorami może być jeden lub więcej jonów nieorganicznych np. Zn lub Fe, albo złożona cząsteczka organiczna o nazwie koenzym. Taki metal luk koenzym, który jest kowalencyjnie związany z enzymem nazywamy grupą prostetyczną. Całość katalitycznie aktywnego enzymu wraz z jego koenzymem lub jonem metalu określa się jako holoenzym. Sama białkowa część enzymu bez jego kofaktora jest nazywana apoenzymem. Pewne koenzymy tj. NAD+ są wiązane i uwalniane przez enzym czasie jego cyklu katalitycznego, dlatego działają one właściwie jako kosubstraty. Wiele koenzymów jest pochodnymi prekursorów- witamin (tabela 1), które są często istotnym składnikiem pożywienia, a których niedostateczny dopływ powoduje choroby niedoboru
Niektóre powszechnie wyst. Koenzymy, ich witaminowe prekursory i choroby spowodowane ich niedoborem.
Koenzymy |
Prekursor |
Choroby z niedoborem |
Koenzym A FAD, FADH
NAD+,NADP+ Pirofosforan tiaminy Tetrahydrofolian Deoksyadenozyno- kobalamina ?Kosubstr do hydroks Pro w kolagenie Fosforan pirydoksalu |
Kw.Pantotenowy Ryboflawina (witB2) Niacyna Tiamina (witB1) Kw.foliowy Kobalamina (witB12) Kw. Askorbinowy (witC) Pirydoksyna (witB6) |
Zapalenie skory Niedobór wzrostu Pelagra Bri-beri Niedokrwistość Niedokrwistość złośliwa Szkorbut
Zapalenie skory |
KOFAKTORY ENZYMOW-WITAMINY I ELEMENTY SLADOWE (MIKROELEMENTY)
Nazwa kofaktor lub koenzym określa się część enzymu niebędącą łańcuchem polipeptydowym, może być ona silnie związaną z białkiem enzymatycznym przez cały czas istnienia enzymu( grupa prostetyczna) lub być wiązana przez koenzym tylko w czasie procesu katalitycznego. Pełni funkcje drugiego substratu reakcji. Często kofaktorami są jony metali np. z karbopeptydaza związane są jony Zn2+. Takie jony zaliczane są często do mikroelementów stanowiących ważny składnik pokarmowy. Wiele innych kofaktorów to specyficzne zw. organiczne, często pochodne witamin np. fosforan pirydoksalu jest pochodna wit.B6. Ponieważ pewne witaminy są prekursorami kofaktorów, których organizm sam nie jest w stanie syntetyzować, stanowią one niezbędny składnik pokarmowy. Kofaktory jako składniki enzymu nie zużywają się w czasie reakcji enzymatycznych. Mogą wiec być wykorzystywane wielokrotnie, dlatego nie sa wymagane w dużych ilościach. Tiamina (wit.B1) jest składnikiem pokarmowym niezbędnym kręgowcom. Ponieważ przeciętna codzienna dieta człowieka zawiera tiaminę zaledwie w ilościach zbliżonych do zalecanego min., niektóre produkty spożywcze są sztucznie wzbogacane w ta witaminę. Reakcje wykorzystujące tiaminę w jednych tk. są bardziej kluczowe niż w innych. Także intensywności jego wykorzystania jest różna w różnych kom. Objawy awitaminozy B1(beri-beri) zaleza od cech osobniczych i mogą ograniczac się do niewielkich zaburzen ze strony ukl. nerwowego, albo obejmowac powazniejsze zmiany tj. paraliz i zanik miesni lub w stanach znacznych niedoborow tiaminy uszkodzenia ukl. krazenia. Objawy beri-beri wyst. zazwyczaj na skutek ograniczenia diety do luskanego i polerowanego ryzu lub ziaren pszenicy pozbawionych lusek, ponieważ najbogatszym źródłem tiaminy jest zew. warstwa ziarniakow. Niedobor tiaminy może być również spowodowany nadużywaniem alkoholu. Może tez występować u ludzi odżywianych roztw. glukozy nieuzupelnianych witaminami. Tiamina zaw. Podstawowe grupy tiazolowi, które uczestnicza m.in. w reakcji dekarboksylacji kw. Pirogronowego katalizowanej przez dekarboksylaze pirogronianowa. Tiamina wystepuje w kom gl. jako aktywny koenzym pirofosforan tiaminy (TTP). Innymi kluczowymi koenzymami sa FMM,FAD,NAD+,CoA. FMM i FAD sa koenzymami silnie związanymi z bialkiem enzymatycznym, czyli gr. Prostetycznymi. Uczestnicza w reakcjach oksydoredukcyjnych, których redukcji lubutlenianiu substratu towarzyszy utlenianie lub redukcja czesci flawinowej koenzymu. Enzymy zaw. jako gr. prostetyczne FMM lub FAD naleza do flawoproteid. W postaci utlenianej maja intensywny zolty kolor, czerwony albo zielony. Podczas gdy forma zredukowana jako bezbarwny wazny przykład flawoproteidy jest dehydrogenaza. Naleza do nich również enzymy uczestniczace w utlenianiu kw. pirogronowego, kw. tlenowych i aminokwasow. Kw. nikotynowy ( niacyna) jest prekursorem NAD+). Koenzym pelniacy centralna funkcje w metabolizmie kom. Kw. nikotynowy może być wytwarzany z tryptofanu, brak obu tych składników w pożywieniu prowadzi do schorzenia zwanego pelagra. Objawy tej choroby wyst. u osob , których dieta sklada się gł. z kukurydzy szczególnie ubogiej w tryptofan. Kw. nikotynowy produkowany w dużych ilościach jest toksyczny, natomiast amid kw. nikotynowego jest skl. 2 koenzymow NAD+ i NADP+. Oba uczestnicza w rekacjach oksydoredukcyjnych, podczas których przyłączają lub odłączają 2elektrony i tylko 1 proton. Drugi proton pozostaje w roztworze. Dlatego w wyniku redukcji NAD+ powstaje NADH+H+. Kw. pantotenowy jest jednym ze skl. koenzymu A. Nie sa znane przypadki niedoboru tego koenzymu. Jest jednym z kofaktorow w metabolizmie kom. Uczestniczacym w licznych procesach enzymatycznych lezacych na skrzyżowaniu szlakow anabolicznych i katabolicznych. Jest nosnikiem gr. acylowych, które lacza się z koenzymem A makroergicznym wiazaniem tioestrowym. Wiazanie to jest utworzone miedzy gr. karboksylową kwasu a gr. tiulową koenzymu.
PIERWIASTKI SLADOWE
Liczne pierwiastki śladowe są niezbędnymi skł. pokarmowymi dla org., ponieważ pełnią rolę swoistych aktywatorów poszczególnych enzymów. Są one najczęściej czynnikami ukł. Aktywującego tzn. białko enzymatyczne jest niezdolne do aktywacji substratu i jeżeli brakuje odpowiedniego jonu metalu. Usuniecie przez dializę powoduje odwracalna utratę aktywności, a ponowne dodanie przywraca aktywność. Dla niektórych enzymów metale są niezbędne do wytwarzania wiązań miedzy enzymem a substratem lub enzymem, koenzymem a substratem, w których enzymach metal stanowi istotna część skł. centrum katalitycznego enzymu, który bez metalu jest nieczynny. Jest to typowa funkcji metalu jako koenzymu np. w enzymach przenoszących elektrony. Enzymy są najczęściej aktywowane przez następujące jony: Mg2+(fosfatazy, fosforylany, fosfokinazy, syntetazy) Zn2+( anhydraza weglanowa,dehydrogenaza mleczanowa i alkoholowa oraz proteazy) Mn2+ (peptydazy,arginazy)Ca2+(lipaza)Cu2+(oksydaza) oraz niekiedy Fe2+,Fe3+,Co2+,Ni2+,Na+,K+. Kationy metali ciezkich maja na ogol dzialanie hamujące, aniony maja maly wpływ na aktywność enzymow. Wyjątkiem jest amylaza aktywowana przez chlorki.
Wiele enzymow jest wrażliwych na male stężenia soli metali ciezkich, mniejsze niż 10-3mola co tłumaczy się katalitycznym wpływem matali na utlenianie np. gr. skl. tlenem atmosferycznym i jest czesta przyczyna utraty aktywności enzymow w procesie oczyszczania ( wersenian EDTA oraz 8-hydroksychiralina). Dzialanie tych inhibitorow cechuje odwracalność, daje się ona znieść przez nadmiar srodkow tiulowych tj. cysteina czy dimerkaptoetanol. Inhibitorami sa również zw. tworzące kompleksy z metalami będącymi czescia centrum katalitycznego lub biorącymi udzial w procesie katalitycznym. Naleza tu np. cjanki, azydki, H2S, CO2 , które upośledzają oddychanie kom. hamując większość oksydaz z Fe lub Ca w grupie czynnej.
PRZYKLADY ZASTOSOWAN INHIBITORÓW ENZYMOW
Leczenie ofiar enzymów - przykład ten dotyczy zastosowania inhibitorów kompetencyjnych. Glikol etylenowy jest stosowany jako składnik płynów chłodniczych w samochodach obniżając temp. Zamrażania. Sam nie jest trujący, ale w organizmie ulega utlenieniu do silnie toksycznego kw. szczawiowego. Reakcja ta może być hamowana przez alkohol etylowy, który jest w tym przypadku inhibitorem kompetencyjnym enzymu. Etanol podany dożylnie w b. wysokich subtoksycznych dawkach współzawodniczy z glikolem etylenowym, który zostaje wydalony w postaci niezmienionej(rys.) Podobna terapie stosuje się w przypadku zatrucia alkoholem metylenowym (metanolem).
W gornej czesci rysunku przedstawiono szlak metaboliczny, w którym glikol etylenowy jest przekształcony w toksyczny kw. szczawiowy. Proces ten można zahamowac, podając etanol, który jest inhibitorem współzawodniczącym pierwszego enzymu tego szlaku.
HAMOWANIE SYNTEZY TTP(timidynotrifosforanu)W CHEMIOTERAPII NOWOTWOROW
Kom. nowotworowe rosną i namnażają się(proliferują) zwykle szybciej niż większość kom org. Dlatego tez jedna ze strategii terapi antynowotworowej polega na selektywnym spowalnianiu podziałów szybko dzielących się komórek. Podczas podziałów musi dojść do replikacji DNA niezachodzącej w kom. Niedzielących się. Jedna z substancji koniecznych jedynie podczas syntezy DNA jest tymidynotrifosforan (TTP). Dwa leki stosowane w chemioterapii nowotworow hamuja synteze tego zw. Jeden ze stosowanych leków 5-fluorouracyl jest prekursorem fluorodeoksyurydylanu, który hamuje aktywność syntazy tymidynalowej, ponieważ wiąże się z enzymem podobnie jak tymidylan lecz altom fluoru występujący w jego czast. Uniemożliwia przyłączenie grupy metylowej. Inny lek ametopteryna (metotreksa) stosowany w terapii niektórych bialaczek dziala w sposób pośredni. Jest on inhibitorem współzawodniczącym reduktazy dihydrofolianowej. Inhibicja tego enzymu uniemozliwia odtworzenie tetrahydrofolianu z dihydrofolianu i tym samym hamuje reakcje katalizowana przez sytaze tymidylanowa. Oczywiście oba te leki wpływają tez na zdrowie kom., ale ostrożnie dozowane mogą Stanowic użyteczne narzedzie chemioterapeutyczne do czasu wynalezienia bardziej skutecznej i mniej szkodliwych srodkow farmaceutycznych. Fluorouracyl jest obecnie testowany klinicznie na dużej grupie osob chorych na raka jelita grubego.
ENZYMY PASZOWE
Wiadomo, ze polisacharydy pochodzenia nieskrobiowego zawarte w paszach roślinnych ( celuloza), hemicelulozy, arabino-ksylany, B-glukany oraz pektyny i lignina, składniki wlokna pokarmowego sa odporne na dzialanie enzymow trawiennych wytwarzanych przez org. zwierzat. Dot. to gł. zwierzat monogastrycznych, gdyz bogata mikroflora zwłaszcza zwierząt przeżuwających wytwarza większość enzymów zdolnych rozkładać cukry włókna pokarmowego. Jedynie bakterie i grzyby niedoskonale maja zdolność syntezy enzymów rozkl. polisacharydy pochodzenia nieskrobiowego. W wielu doświadczenia przeprowadzonych na drobiu i świniach wykazano pozytywny wpływ dodawania brakujących enzymów do mieszanek paszowych na wskaźniki produkcyjne. Enzymy te pochodzenia mikrobiologicznego należą do klasy hydrolaz. W degradacji włókna paszowego uczesnicza takie enzymy jak: celulolaza ( degradujaca celuloze) B-1,3-glukanaza (hydrolizująca B-glukan) hemicelulozy (mieszanina enzymów rozkładających hydrolitycznie różne formy hemiceluloz). Ponadto w praktyce żywieniowej stosuje się także inne hydrolazy rozkładające skrobie (α-amylaza, glukoamylaza), białka ( proteazy) i lipidy (lipazy). Preparaty enzymatyczne stosowane do pasz składają się 1,2lub 3 enzymów np. amylazy, proteazy, B-glukanazy. Produkcja preparatów enzymatycznych rozkładających te związki zajmuje się wiele firm biotechnologicznych. Stosunkowo tanio enzymy można otrzymac na drodze mikrobiologicznej ( wykorzystując bakterie i grzyby) Często wykorzystuje się szczepy bakteryjne z rodzaju Bacillus czy tez Aspergillus oryzae, czy tez inne. Powstaly produkt(związany z działalnością mikrofermentacyjna) jest zwykle mieszanina enzymow i w takiej postaci może być dodawany do pasz.
KLASYFIKACJA KOENZMÓW
Koenzymy dzieli się na kilka grup przyjmując za podstawę typy reakcji , w których biorą udział :
koenzymy przynoszace p-i e- (wspólpracujące z oksydoreduktazami )
koenzymy przynoszace grupy (współpracuące z transferazami)
koenzymy liaz, izomeraz i ligaz
Enzymy klasy hydrolaz nie wymagają udziału koenzymów a jedynym w wielu przypadkach aktywacji kationami metalu lub anionami nieorg.
Koenzymy oksydoreduktaz nukleotydy nikotynaamidowe oraz wit PP.
Do nukleotydów tych należą NAD+ i NADPH. Witamina PP czyli kwas nikotynowy (pirydyno-3-karboksylowy) wzglednie jego amid, gdy oba zw. jednakowo czynne biologicznie , zwane ogólnie niacyną, zapobiegają chorobie skory zwanej pelagrą. Tryptofan rownierz skutecznie zapobiega tej chorobie gdyż może ona być łatwo przekształcony w kw. nikotynowy, a nastepnie w jego amid.
Amid kwasu nikotynowego jest częscią składową i gr. aktywną 2 koenzymów współdziałających z dehydrogenazami a mianowicie z NAD+, NADP+ sa zbudowane z amidu kw. nikotynowego który jest przyłączany wiązaniem N-glikozydowym C1 rybozy, ta z kolei atomem C5 łączy się wiazaniem estrowym kolejno z dwiema resztami fosforowymi. Do II reszty fosforanowej jest dołączone również estrowo następna cząst. rybozy przez atom C5, która z kolei wiazaniem N-glikozydowym łączy się z zasadą purynową- adeniną. NADP+ rożni się od NAD+ zawartością III reszty fosforanowej przyłączonej estrowo do C2 rybozy wiązniem z adaniną. Ponieważ azot pierścienia pirydynowego występuje w postaci amoniowej (IV rzedowej) ma on ładunek dodatni nadając go całej cząsteczce, dlatego skróty NADP+, NAD+ przyjęto pisać z ładunkiem dodatnim. Podstawową funkcję NADP+ i NAD+ jest wspołdziałanie a dehydrogenazami przy odwodorowaniu substratu np.: dehydrogenaza alkoholowa katalizuje odwaracalną reakcję przeniesienia 2 protonów i 2e- z etanolu na NAD+ , a tym samym przemianę jego do alde. octowego. Jeden z H+ oraz 2e- przyłączają się do NAD+, a drugi H+ pozostaje w roztworze. W cząsteczce NAD+ przemianie ulega tylko pierścień pirydynowy amidu kwasu nikotynowego
NAD NADP
Typowym przykładem dehydrogenazy o zmiennym kierunku działania jest dehydrogenaza glutaminianowa katalizująca przemianę 2-oksoglutaranu do glutaminianu lub odwrotnie z udziałem NAD+ lub NADP +. Reakcja w prawo nazywana jest deaminacją, a odwrotnie aminacja redukcyjną
NUKLEOTYDY FLAWINOWE - FMN, FAD, WIT. B2 (RYBOFLAWINA)
Wit. B2 - 7,8-dimetylo-10-rybitolo-izoalloksazyna. Jest składnikiem 2 koenzymów (FMN,FAD) współdziałających z oksydoreduktazami przenoszącymi e- i H+ ze zredukowanego NAD+ (reduktazy) lub z substratu (dehydrogenazy).
FMN - mononukleotyd flawinowy, FAD - dinukleotyd flawinoadeninowy
PROCES OKSYDOREDUKCJI FLAWINY
Grupą czynna przy przenoszeniu H+ i e- jest ukł. izoalloksazyny, który odwracalnie może je przyłączać do at. N w pozycjach 1 i 5 zgodnie z reakcją:

KWAS LIPONOWY
Jest koenzymem enzymów oksydoredukcyjnych biorących udział w przenoszeniu H2, a także uczestniczy w przenoszeniu rodników arylowych (R-CO-). Pod względem chemicznym jest to disulfidowa pochodna kwasu oktanowego (kw. 6,8-ditiooktanowy).
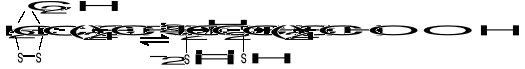
W formie skróconej:
Podczas transportu rodników arylowych (R-CO-) np. acetylu (CH3-CO-) powstaje kwas acetyloliponowy o wzorze:
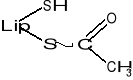
KOENZYMY TRANSFERAZ
Transferazy są to enzymy katalizujące przeniesienie grup atomów z 1 substratu na 2.
KOENZYM A (Co-A) I KWAS PANTOTENOWY
Kw. pantotenowy został wykryty jako 1 ze składników kompleksu witamin B.
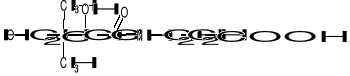
Zbudowany jest z kwasu 2,4-dihydroksy-3,3-dimetylomasłowego i β-alaniny, które są związane ze sobą za pomocą wiązania peptydowego. Kwas pantotenowy wchodzi w skład Co-A, który ma następującą budowę:
BUDOWA Co-A
Kw pantotenowy:
CH3 H O O
-O-CH2 -C- CH-C-N-CH2-CH2-C-
CH3 H
Zbudowany z cząsteczki adenozynomonofosforanu zestryfikowanej dodatkową resztą fosforanową przy C3 rybozy połączonej bezpośrednio z fosforanem kwasu pantotenowego, a ten z kolei z udziałem wiązania peptydowego przyłącza cysteaminę (produkt dekarboksylacji cysteiny). Grupą czynną Co-A, która związuje się z resztami arylowymi jest należąca do cysteaminy grupa -SH.
Funkcja biologiczna Co-A polega na aktywowaniu i przenoszeniu reszt kwasu octowego (CH3-CO- acetyl) oraz reszt innych kwasów (R-CO- acyl). Atom H w grupie -SH może być podstawiony dowolnym rodnikiem arylowym, najczęściej resztą kwasu octowego (acetylem). Utworzony wówczas związek nosi nazwę acylo-S-CoA. Ponieważ Acetylo-CoA zawiera w cząsteczce wiązanie makroergiczne ≡CNS- dla jego utworzenia jest konieczne dostarczenie energii. Pochodzi ona ze sprzężenia z niektórymi reakcjami egzoergicznymi, np. z oksydacyjną dekarboksylacją pirogronianu lub β-utleniania kwasów tłuszczowych.
Wytworzony we wspomnianych reakcjach aktywny octan jest zużywany w komórce do produkcji energii oraz jako związek wyjściowy do biosyntezy dużej liczby związków węgla.
W pierwszym przypadku reszta dwuwęglowa (acetyl) może być przeniesiona z C0-A na kwas szczawiooctowy, dając kwas cytrynowy (cykl Krebsa):
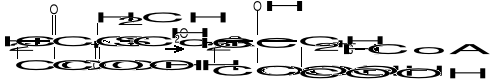
W przemianie tej grupa metylowa (-CH3) aktywnego octanu reaguje z grupą ketonową kwasu szczawiooctowego, natomiast cz. H2O powoduje hydrolizę wiązania tioestrowego :
(-CO~S-CoA + H2O = -COOH + HSCoA).
W poszczególnych reakcjach cyklu Krebsa wbudowana w cytrynian reszta acetylowa zostaje rozłożona do CO2 i H2 związanych z NAD i FAD. Dopiero w łańcuchu oddechowym uwalnia się w postaci ATP energia pochodząca z utleniania wodorów z NADH2. jak wspomniano wcześniej acetylo-CoA dostarcza reszt kwasu octowego (acetylowych) do syntezy różnych zw. org. i jest on m.in. prekursorem kwasu mewalonowego (na drodze kondensacji 3 cząst. Acetylo-CoA z którego powstaje cholesterol i cała grupa hormonów sterydowych). Z kwasu mewalonowego tworzą się również karotenoidy oraz niektóre regulatory wzrostu (GA i ABA).
FOSFORANY NUKLEOZYDÓW
Koenzymy, podstawową ich funkcją jest przenoszenie reszt fosforanowych. Najważniejszym z nich jest adenozynotrifosforan (ATP). Enzymy, w których ATP pełni funkcję koenzymu należą do klasy transferaz (kinazy). ATP ma dwa wiązania bogate w energię. Wyróżniamy 4 typy reakcji, w których ATP przekazuje na substrat 1 z odłączonych grup:
Typy reakcji przenoszenia grup z udziałem ATP
[1] przeniesienie reszty monofosforanu
[2] przeniesienie reszty difosforanu
[3] przeniesienie reszty adenozynomonofosforanu
[4] przeniesienie reszty adenozylowej
- PRZENIESIENIE RESZTY FOSFOROWEJ (ortofosforanowej) H2PO41- z wydzieleniem ADP. Najczęściej spotykana. Katalizują ją fosfotransferazy (kinazy), które przenoszą resztę fosforową na grupy alkoholowe, kwasowe lub aminowe wielu substratów.
np. odwracalne przeniesienie reszty fosforanowej ATP na kreatynę
nieodwracalna przemiana glukozy w 6-fosforan glukozy
- PRZENIESIENIE RESZTY DIFOSFOROWEJ (pirofosforowej) H3P2O7 z wydzieleniem AMP. Spotykane rzadziej w metabolizmie.
np. przeniesienie reszty difosforanowej i ATP na 5-fosforan rybozy, tworzy się 5-fosforybozylo-1-difosforan (PRPP). Reakcja katalizowana jest przez pirofosfokinazę 5-fosforybozową
- PRZENIESIENIE RESZTY ADENOZYNOMONOFOSFORANU - AMP z wydzieleniem reszty difosforowej.
np. przeniesienie AMP na kwasy tłuszczowe lub aminokwasy
Powstaje aminoacylo- AMP. AMP wymienia się na odpowiedni tRNA, który przenosi resztę aminoacylową na matrycowe RNA. Przy aktywacji kwasów tłuszczowych acylo-AMP, nukleotyd wymienia się z Co-A dając aktywny acyl (acylokoenzym A).
- PRZENIESIENIE RESZTY ADENOZYLOWEJ z wydzieleniem reszty difosforowej
np. aktywacja metioniny do reaktywnego związku sulfonowego - adenozylometioniny (dawca grup metylowych)
WITAMINA B6 - FOSFORAN PIRYDOKSALU
Strukturę witaminy stanowi pierścień pirydynowy, podstawiony w pozycji 2 grupą metylową, w pozycji 3 grupą hydroksylową, w pozycji 5 hydroksymetylową i w pozycji 4 grupą aldehydową. Fosforan pirydoksalu powstaje w wyniku działania ATP na pirydoksal.
Koenzym ten katalizuje transaminację i dekarboksylację aminokwasów. Zaliczany jest do grupy koenzymów transferaz.
IZOLOWANIE ENZYMÓW
Stosuje się metody pozwalające na utrzymanie ich w stanie jednorodnym, bez utraty aktywności katalitycznej. Enzymy:
nietrwałe w pH poniżej 5 i powyżej 9
ulegają denaturacji cieplnej i powierzchniowej
inaktywują się w obecności soli metali ciężkich
Preparatykę przeprowadza się w buforach w pH ok. 7, niskiej temperaturze, stosując wodę podwójnie destylowaną i środki kompleksujące metale ciężkie, w razie potrzeby związki tiolowe. Rozpuszczalne enzymy (z cytoplazmy) można ekstrahować wodą lub rozcieńczonymi roztworami soli o wartościach pH oddalonych od punktu izoelektrycznego, ekstrakcję przeprowadza się po rozdrobnieniu tkanek w homogenizatorze. Aby oddzielić enzymy mocno wbudowane w ziarnistości komórkowe trzeba stosować dodatkowo: zamrażanie, odtajanie, ekstrakcję butanolem, wprowadzenie środków zmniejszających napięcie powierzchniowe itp. Do oczyszczania enzymów najczęściej stosuje się frakcjonowanie zwłaszcza siarczanem amonowym i rozpuszczalnikami organicznymi (głownie acetonem, etanolem w niskiej temperaturze lub eterem). Dodatkowo stosuje się chromatografię, elektroforezę, ultrawirowanie. Duże możliwości otrzymywania jednorodnych preparatów enzymatycznych stworzył rozwój chromatografii powinowactwa. Ostatnim etapem oczyszczania może być krystalizacja. Enzymy typu albumin łatwo krystalizują przez dosycanie roztworu siarczanem amonu w odpowiedniej temperaturze i pH, a enzymy typu globulin można wykrystalizować przez dializę wobec wody. Jednym z kryteriów jednorodności enzymów jest osiągnięcie wyniku oczyszczania maksymalnej liczby jednostek aktywności przypadających na 1 mg azotu białkowego, niezwiększającej się przez rekrystalizację, czy inne sposoby oczyszczania. Handlowe preparaty zawierają zwykle domieszki innych enzymów. Dodatkową trudnością jest występowanie tego samego enzymu w kilku odmianach (izoenzymy).
CHROMATOGRAFIA POWINOWACTWA
Chromatografia powinowactwa ze względu na swe unikatowe właściwości pozwala znacznie uprościć procedurę izolowania danej substancji, jednocześnie zachowując jej strukturę biologiczną. Typ chromatografii adsorpcyjnej, w której wykorzystuje się powinowactwo dwóch substancji. Jedna z nich chemicznie związana z nierozpuszczalnym nośnikiem (zwana ligandem) specyficznie absorbuje drugą substancję izolując ją z mieszaniny innych związków. Oddziaływanie substancji rozpuszczonych i nierozpuszczalnych w fazie unieruchomienia z unieruchomionym ligandem może mieć różny charakter jak między: 1) hormonem a receptorem 2) enzymem a substaratem 3) enzymem a inhibitorem 4) przeciwciałem a antygenem 5) komplementarnymi odcinkami kwasów nukleinowych 6) kwasami nukleinowymi a białkami.
Schemat procesu chromatografii powinowactwa:
immobilizacja ligandu
specyf wiąz substr do unieruchomionego ligandu i odmycie niespecyf zaadsorbowanych cząsek białkowych
elucja specyf związanego substr.
ROLA ONACZANIA ENZYMÓW W DIAGNOSTYCE KLINICZNEJ
W stanach patologicznych zmiany aktywności enzymów we krwi są często odbiciem zmian zachodzących w narządach i dlatego badania enzymatyczne mogą dostarczyć lekarzom wielu cennych info, które ułatwią im rozpoznanie schorzenia, wyciągnięcie wniosków prognostycznych i kontrolę leczenia. Nowoczesna diagnostyka enzymatyczna opiera się na założeniu, że uszkodzenie narządu pociąga za sobą uszkodzenie struktur komórkowych lub zmianę przepuszczalności błon komórkowych. Przez uszkodzone błony następuje ucieczka enzymów i zwiększa się ich ilość w cieczach ustrojowych, czy wydalinach (krew, płyn mózgowo-rdzeniowy, mocz, ciecze wysienkowe, sok żołądkowy i dwunastniczy). Pozwala to wyciągnąć pewne wnioski o lokalizacji, rozmiarach i intensywności uszkodzenia. W niektórych wypadkach przeciwnie, zmniejszenie aktywności pewnych enzymów (sekrecyjnych) surowicy świadczy o uszkodzeniu i niewydolności czynnościowej danego narządu. Należy jednak pamiętać, że na aktywność danego enzymu surowicy mogą mieć ponadto wpływ zmiany ilościowe ich aktywatorów lub inhibitorów oraz intensywność produkcji enzymów przez organizm.
ENZYMY SUROWICY KRWI
ENZYMY SEKRECYJNE
Są normalnie wydzielane do krwi, gdzie spełniają ważną rolę fizjologiczną. Należą do nich: czynnik krzepnięcia krwi oraz fibrynolizy (esteraza cholinowa, ceruloplazmina, lipaza lipoproteinowa). Ilość tych enzymów zależy od procesu ich syntezy w rybosomach wątroby. Stąd po uszkodzeniu komórki wątroby zachodzi spadek aktywności tych enzymów w surowicy, a nie wzrost jak pozostałych.
ENZYMY WSKAŹNIKOWE
Pojawiają się w dużych ilościach po uszkodzeniu narządów, wskazując niekiedy na specyficzność. Aktualna zawartość tych enzymów w surowicy jest wypadkową wielu czynników: stężenie danego enzymu w uszkodzonym narządzie, liczba uszkodzonych komórek i stopień ich uszkodzenia oraz rozmieszczenie enzymów w różnych strukturach komórki. W wyniku działania czynników cytotoksycznych jakiegokolwiek pochodzenia (bakteryjnego, chemicznego, wirusowego, fizycznego iitp) dochodzi do większego, czy mniejszego uszkodzenia struktur. Po wybiórczym uszkodzeniu błony komórkowej uciekają enzymy luźno występujące w cytoplazmie, ponieważ enzymy mocno związane z siateczką endoplazmatyczna, mitochondriami, rybosomami, czy lizosomami pojawiają się w krwiobiegu jedynie po bardzo poważnym uszkodzeniu komórki lub po ich śmierci biologicznej. Można na tej podstawie wysnuć wnioski o stopniu uszkodzenia komórek.
ENZYMY EKSKRECYJNE
Wzrost ich aktywności w surowicy krwi obserwuje się wskutek przeszkody w normalnym odpływie różnych wydzielin ustrojowych tj. żółć, sok trzustkowy, ciecz sterczowa czy ślina. W wyniku istnienia przeszkody w drogach odprowadzających wydzielinę dochodzi do jej zastoju, co doprowadza do przejścia enzymów do krwiobiegu. Należą tu enzymy soku trzustkowego (amylaza, lipaza, RNAza, DNAza), żółci (fosfataza zasadowa, γ-glutamylotranspeptydaza, aminopeptydaza leucynowa) oraz fosfataza kwasowa cieczy sterczowej. Stosunkowo najlepiej rozwinęła się diagnostyka enzymologiczna schorzeń mięśnia sercowego, wątroby, trzustki, czy żołądka.
ENZYMY RESTRYKCYJNE
Dzięki rekombinacyjnej technologii DNA izolacja, badanie i dowolne zmiany genów stały się powszechne. Duży postęp dokonał się po opracowaniu metod - sposób rozcinania DNA w specyf miejscach za pomocą restrykcyjnych endonukleaz (e restrykcyjnych), procedury umożliwiające wykrycie z dużą dokł specyf sekwencji DNA czy RNA (hybrydyzacja kw nukleinowych), uzyskiwanie specyf sekw DNA w dużej il i czystości (klonowanie DNA) i szybkie sekwencjonowanie DNA (okr kolejności w genach i fr DNA). Rewolucją jest ostatnio łańcuchowa reakcja polimerazy (PCR). Są to zagadnienia biol mol.
TRAWIENIE E RESTRYKCYJNYMI
E restr rozpoznają w dwuniciowym DNA specyf sekw nukleotydowe (miejsca restrykcyjne) o dł 4, 5, 6 nukleotydów i rozcinają obydwie nici DNA w ściśle okr miejscach, znajdującej się w obrębie rozpoznanej sekw. Istnieją 3 sposoby rozcinania DNA:
1. Nierównomierne „na ukos” pozostawiające niesparowany koniec 5'
2. Nierównomierne z wolnym końcem 3'
3. Rozcięcie obu nici w tym samym miejscu pozostawiając „tępe końce”.
Końce przy rozcinaniu „na ukos” są nazywane końcami kohezyjnymi („lepkimi”), bo umożliwiają dwóm dowolnym fr DNA, uzyskanym z użyciem tego samego e restr , utw komplementarnych par zasad. Końce takie moga być następnie połączone kowalencyjnie przez ligazę DNA - jest to proces ligacji. Nową cz DNA nazywamy cz zrekombinowanego DNA (cz zrekombinowaną). Cz o tępych końcach także można łączyć za pomocą ligazy, ale r ta przebiega trudniej niż w przypadku końców kohezyjnych.
Trzy sposoby rozcinania:
1. BamHI
G↓GATCC → G3' 5'GATCC
CCTAG↑G CCTAG5' 3'G
2. KpnI
GGTAC↓C → GGTAC3' 5'C
C↑CATGG C5' 3'CATGG
3. PvuII
CAG↓CTG → CAG3' 5'CTG
GTC↑GAC GTC5' 3'GAC
NAZEWNICTWO
E restr izoluje się z bakterii, gdzie ich rola polega na ochronie komórki gospodarza przed infekcją wirusową. Dotychczas wyizolowano >100 e restr. Ich 3-literowe nazwy pochodzą od nazw gat bakterii, z których zostały wyizolowane. 1 litera to pierwsza lit nazwy rodzaju, a nast dwie - nazwy gatunku. Ponieważ bakteria może wytw >1 e, są one oznaczone lit rzymskimi. Np EcoRI to pierwszy e wyizolowany od E. coli (R-szczep szorstki).
Popularne e restr i wytwarzające bakterie:
Bam III G|GATCC Bacillus amyloliquefaciens
CCTAG|G
Bgf II A|GATCT Bacillus globigii
TCTAG|A
EcoRI G|AATTC Escherichia coli RY13
CTTAA|G
Hind III A|AGCTT Haemophilus influenzae Rd
TTCGA|A
Hha I GCG|C Haemophilushaemolyticus
CGC|G
HpaI GTT|AAC Haemophilus parainfluenzae
CAA|TTG
Fr DNA nazywane fr restrykcyjnymi powst po rozcięciu cz DNA e restr można rozdzielic za pomocą elektroforezy żelowej. El-for w żelu poliakryloamidowym umożliwia rozdział małych fr o dł <500pz, do większych fr potrzebny jest żel agarozowy (ma większe pory). Produkty trawienia DNA rozdzielają sie na serię prążków będących fr DNA. Ponieważ małe el migrują dalej niż większe, można oszacować ich wielkość na podstawie drogi migracji w porównaniu ze standardową mieszaniną fr o znanej dł. Po el-for fr DNA można uwidocznić bromkiem etydyny, który wiąże się z DNA silnie fluoryzując dając jasnopomarańczowe światło. Alternatywnie, jeśli DNA był znakowany izotopem np. 32P, fr restr mozna zlokalizować w żelu nakładając na niego film rentgenowski; radioakt zaw w pasmach DNA powoduje rozkład związków srebra w emulsji filmu, prowadząc do powstania ciemnych plam w miejscach odp radioakt prążkom
(autoradiografia).
MAPY RESTRYKCYJNE
2-niciowy DNA może być rozcięty przez wiele e restr rozpoznających różne sekw. Rozdzielając te fr i określając ich długość elektroforetycznie, można wnioskować, w jakich miejscach łańcuch był rozcięty. Na tej podstawie można sporządzić mapę restrykcyjną, pokazującą lakalizację miejsc rozcięcia. Mapy ułatwiaja porównanie dwóch cz DNA (np. podczas badania ewolucyjnego pokrewieństwa między dwoma gat) bez konieczności określania każdej sekw nukleotydowej. Są też ważne podczas eksperymentów prowadzonych technikami rekombinacji DNA (planowanie miejsc rozcięcia i śledzenie eksperymentu).
![]()
E BX E SCX B
0 5 10 15 20
Typowa mapa restr cz DNA. Miejsca cięć ozn literami.
POLIMORFIZM DŁUGOŚCI FR CZ RESTR
Analiza genomowego DNA człowieka ujawniła, że DNA poszczeg osób wykazuje w sekw wiele różnic nie wywierających widocznych efektów (różnice w obrębie intronów lub między genami). Niektóre b powszechnie wyst u poszczeg osób w populacji - te międzyosobnicze różnice nazwano polimorfizmem. Niektóre jego rodzaje wpływają na dł fr restr, np. gdy wskutek zmiany nukleotydu w miejscu rozpoznawanym przez restryktazę nastąpi eliminacja określonego miejsca rozcinania DNA. Zamiast 2 odc powst 1 dłuższy fr. Alternatywnie polimorfizm może wynikać z insercji lub delecji pewnych sekw zaw między dwoma miejscami rozcinania, w wyniku czego zwiększa/zmniejsza się wielkość powst fr. Polimorfizm wpływający na wielkość fr restr jest określany jako RFLP - pilomorfizm dł fr restrykcyjnych. Można go wykryć za pomocą hybrydyzacji metodą Southerna, pod warunkiem dysponowania sondą molek w post odc DNA o sekw komplementarnej do fr restr zaw miejsca decydujące o polimorfizmie.
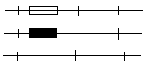
Hpa I Hpa I* Hpa I
Prawidłowy gen
β-globiny
Gen sierpowatej
Sonda DNA
7,6 kpz 6,4 kpz
Analiza genet schorzenia za pomocą RFLP. Nieprawidłowość genu β-globiny jest sprzężona ze zmianą sekw powodującą utratę m-sca restr Hpa I*. Obecność lub brak tego m-sca można wykryć metodami Soultherna stosując sondę komplementarną do fr wielkości 7,6 kpz.
Prawidłowy Sierpowaty
------ 14
7,6 -------
Prawidłowy DNA z 3 m-scami Hpa I daje fr dł 7,6 kpz wykrywalny sondą, a DNA z mutacją sierpowatości daje fr dł 14 kpz wskutek zaniku miejsca Hpa I.
Duże znaczenie ma możliwość wykorzystania go jako markera okr genet schorzeń człowieka. Gdy polimorfizm dot odc DNA zn się w pobliżu kluczowego genu (zmiany=schorzenia). Obydwie zmiany, tj. powodująca polimorfizm i wywołująca schorzenie, leżą blisko siebie na tym samym chromosomie, to wykazują tendencję do współdziedziczenia. Identyfik takich ścisłych sprzężeń RFLP jest korzystna, bo można ukierunkować eksperymenty zmierzające do klonowania rejonów DNA, zn się w pobliżu RFLP z nadzieją identyfikacji samego genu, który następnie można by zsekwencjonowac i dokładnie zbadać. Po drugie, nawet gdy gen taki nie występuje, RFLP funkcjonuje jako przesiewowy marker, umożliwiający wykrycie schorzenia. U osób, u których występuje dany RFLP, istnieje duże prawdopodobieństwo powst sprzężonej z nim wady genu. Inne RFLP, wyst w b dużej odl od genu lub zlokalizowane na innych chromosomach nie będą z nim sprzężone, więc wyszukiwanie i skorelowanie RFLP z okr chorobą wymaga dużo pracy. Należy przeprowadzić testy na próbkach pobranych od poszczególnych członków dużej liczby rodzin, w których schorzenie to występowało. W pobranych próbach należy zbadać cały zestaw RFLP, aż wykryje się taki, który jest dziedziczony razem z uszkodzonym genem, wywołującym schorzenie.
2
Wyszukiwarka
Podobne podstrony:
ściąga na egz, studia, ściągi
Sciaga na mozaka, aaa, studia 22.10.2014, Materiały od Piotra cukrownika, materialy Kamil, płytkas V
Ściąga na EGZ. 97-2003 do wysłania, studia rolnictwo, semestr 5
Technologia remediacji druga ściąga na 2 koło całość, Studia, Ochrona środowiska
SiAM sciaga na egz
ŚCIĄGA NA EGZ
ściąga na egz
sciaga na egz
ściąga na egz
sciaga na egz
SiAM sciaga na egz (1)
PEDEUTOLOGIA śćiąga na egz
ściaga na egz
sciaga na lab.ps, STUDIA, SEMESTR II, Materiały Metalowe, mm
ściąga na III koło z anatomiill, biologia
Sciaga do wydruku, Księgozbiór, Studia, Biologia i Ekologia
ściąga na egz z funamentów
więcej podobnych podstron