ZWIĄZKI KOORDYNACYJNE (KOMPLEKSOWE)
Pojęcia podstawowe i definicje
Wiązanie koordynacyjne jest szczególnym przypadkiem wiązania atomowego (kowalencyjnego). Pojedyncze wiązanie koordynacyjne jest wiązaniem σ, ale mechanizm jego powstawania między atomami A
i B w cząsteczce lub jonie jest nieco inny niż zwykłego wiązania atomowego. Wiązanie to powstaje wtedy, gdy jeden z tych atomów jest dawcą, czyli donorem wiążącej pary elektronów, natomiast drugi atom mający pusty, łatwo dostępny orbital staje się jej akceptorem:
A: + B = A→B,
gdzie strzałka symbolizuje parę elektronową (:) pochodzącą od donora A i wiążącą go z akceptorem B. Powstały produkt A→B jest określany mianem związku koordynacyjnego (kompleksowego).
Rozpatrzmy jako przykład amoniak, NH3, którego cząsteczki mają kształt piramidy trygonalnej.
Atom azotu ma pięć elektronów walencyjnych, które obsadzają cztery orbitale hybrydyzowane typu sp3. Trzy elektrony walencyjne atomu azotu tworzą trzy zwykłe wiązania atomowe σ z atomami wodoru.
Wolna para elektronów walencyjnych atomu azotu może być zaangażowana w utworzenie czwartego wiązania σ z takim atomem lub jonem, który ma pusty orbital, np. jonem H+, który ma nie obsadzony orbital 1s. Istotnie, w wodnych roztworach zachodzi reakcja amoniaku z jonami wodorowymi:
NH3 + H+ = NH4+
Jon amonowy, NH4+, ma strukturę tetraedryczną
z czterema równocennymi wiązaniami N-H typu σ
o jednakowej długości.
{kind=link}
O atomie azotu w jonie amonowym mówimy, że jest atomem centralnym i ma liczbę koordynacji 4, ponieważ jest otoczonym przez cztery atomu wodoru.
Cząsteczka wody może być donorem jednej
z wolnych par elektronów zlokalizowanych na atomie tlenu, a jej akceptorem może być jon metalu mający dostępne orbitale zhybrydyzowane, np. Co2+
o hybrydyzacji oktaedrycznej sp3d2. W wodnych roztworach soli Co(II) zachodzi reakcja:
![]()
Produkt tej reakcji jest oktaedrycznym kationem heksaakwakobaltu(II), w którym Co2+ ze względu
na liczbę związanych, czyli skoordynowanych cząsteczek wody za pośrednictwem sześciu wiązań σ, ma liczbę koordynacji równą 6.
Wiązania koordynacyjne powstają bardzo często
w reakcjach jonów metali z donorami par elektronowych, które mogą być zarówno elektroobojętnymi cząsteczkami różnych związków nieorganicznych, np. wody, amoniaku, tlenku węgla CO, lub anionami, np. anionami F-, Cl-. Donorami par elektronowych mogą być również cząsteczki wielu związków organicznych, np. amin, lub aniony rożnych kwasów organicznych. W powstałych związkach mogą występować nie tylko wiązania koordynacyjne σ. Wiązania koordynacyjne σ i π są obecne w związkach metali z tlenkiem węgla, tzw. karbonylkach, np. Cr(CO)6 i Ni(CO)4 . Wiązania π zapewniają dużą trwałość związków niektórych metali z benzenem, np. Cr(C6H6)2.
Związki z wiązaniami koordynacyjnymi σ i π noszą nazwę związków koordynacyjnych lub kompleksowych.
Jedną z obowiązujących w chemii koncepcji kwasów i zasad jest teoria Lewisa omawiana na pierwszym wykładzie. Zgodnie z tą teorią kwasem jest każdy akceptor wiążących par elektronowych, natomiast zasadą jest każdy donor tych par.
Każda reakcja tworzenia związku koordynacyjnego (kompleksowego) jest zatem reakcją zobojętniania kwasu Lewisa przez zasadę Lewisa, a produktami takich reakcji mogą być:
cząsteczki kompleksowe, np.: Fe(CO)5 - pentakarbonylożelazo(0), [Co(NH3)3Cl3]- triaminatrichlorokobalt(III);
kationy kompleksowe,
np.: [Cu(NH3)4]2+ - tetraaminamiedź(II), [Ni(H2O)6]2+ - heksaakwanikiel(II);
aniony kompleksowe,
np.: [FeF6]3- - heksafluorożelazian(III),
[Ni(CN)4]2- - tetracyjanoniklan(II).
Związek kompleksowy złożony z jonów może mieć: kompleksowy kation i prosty anion, np. [Co(NH3)6]Cl3, kompleksowy anion i prosty kation, K[Au(CN)2], kompleksowy kation i kompleksowy anion, np. [Cr(NH3)6][CoCl6]. Wzory sumaryczne kompleksowych cząsteczek lub jonów zapisuje się zazwyczaj w nawiasie kwadratowym.
W chemii związków koordynacyjnych stosuje się powszechnie następujące terminy i określenia:
Donory par elektronowych, czyli zasady Lewisa, są nazywane ligandami. Ligandy, które są donorami więcej niż jednej pary elektronowej, to ligandy wielomiejscowe lub wielopodstawne.
Związki koordynacyjne, to inaczej związki kompleksowe lub kompleksy, a reakcje ich otrzymywania są określane jako reakcje kompleksowania.
Atom lub jon, który jako kwas Lewisa jest akceptorem wiążących par elektronowych, określa się jako atom lub jon centralny
w powstałym kompleksie.Kompleks z jednym tylko atomem lub jonem centralnym nosi nazwę kompleksu jednordzeniowego. Wszystkie wymienione wyżej przykłady są kompleksami jednordzeniowymi.
Kompleksy z więcej niż jednym atomem lub jonem centralnym są kompleksami wielordzeniowymi.
Niektóre ligandy wielomiejscowe, np. aniony kwasów karboksylowych, mogą pełnić funkcje mostków łączących ze sobą atomy lub jony centralne w kompleksach wielordzeniowych.
Octany miedzi(II) i chromu(II), [M2(CH3COO)4(H2O)2], są kompleksami dwurdzeniowym o strukturze cząsteczkowej typu „koła wodnego” (rys. 1a), z czterema mostkami octanowymi, łączącymi obydwa centralne jony M2+ za pośrednictwem par elektronowych dwóch równocennych atomów tlenu każdej grupy karboksylowej. Octany V(III), Cr(III), Mn(III) i Fe(III) zawierają trójrdzeniową grupą M3O (rys. 1b).
Rys. 1. Struktury cząsteczkowe kompleksów octanowych metali
a) dwurdzeniowe octany Cu(II) i Cr(II) o strukturze „koła wodnego”, gdzie L oznacza cząsteczki wody; b) trójrdzeniowe kompleksy octanowe V(III), Cr(III), Fe(III) i Mn(III), [M3O(CH3CO2)6L3]+, gdzie L to cząsteczka wody, zawierające płaskie ugrupowanie oksokationowe M3O.
Funkcję mostków w kompleksach wielordzeniowych mogą spełniać nawet najprostsze ligandy, takie jak tlenek węgla w karbonylowych kompleksach metali lub jony OH- w hydroksokompleksach metali.
Jeden z karbonylków żelaza, mianowicie dodekakarbonylotriżelazo(0), Fe3(CO)12, jest kompleksem trójrdzeniowym, w którym cząsteczki CO pełnią zarówno funkcję ligandów jednomiejscowych, jak i funkcję mostków, łączących trzy centralne atomy żelaza (rys. 2).
Rys. 2. Trójrdzeniowy karbonylek żelaza(0)
Wielomiejscowe ligandy organiczne
z odpowiednią konfiguracją heteroatomów tlenu i azotu jako donorów dwu lub więcej par elektronowych są zdolne do wiązania się
z atomem lub jonem centralnym tworząc kompleksy z co najmniej jedną pięcio- lub sześcioczłonową strukturą pierścieniową
o znacznej trwałości. Ligandy te noszą nazwę ligandów kleszczowych lub chelatowych,
a powstałe kompleksy określa się jako kompleksy chelatowe lub chelaty.
Dwumiejscowym ligandem chelatowym jest między innymi etylenodiamina, H2N-CH2-CH2-NH2 (en), która dzięki giętkiej strukturze cząsteczki może
z jonem centralnym w kompleksie tworzyć dwa wiązania koordynacyjne za pośrednictwem par elektronowych zlokalizowanych na obydwu terminalnych atomach azotu.
Mn+
H2N NH2
\ /
CH2 --- CH2
Ligandem dwumiejscowym jest również dimetylo-
glioksym (dmg):
Anion kwasu etylenodiaminatetraoctowego (EDTA)
jest ligandem sześciomiejscowym:
Każdy atom lub jon centralny w kompleksie ma ściśle określoną hybrydyzację orbitali atomowych, która wyznacza strukturę geometryczną kompleksu i liczbę utworzonych wiązań koordynacyjnych z ligandami. Najważniejsze typy hybrydyzacji są przedstawione na rys. 2-5 i w tabeli 1.
Rys. 2. Hybrydyzacja digonalna (liniowa) sp
Rys.3. Hybrydyzacja trygonalna sp2
Rys. 4. Hybrydyzacja tetraedryczna sp3
Rys. 5. Hybrydyzacja: a) dsp2 - tetragonalna (płaska, kwadratowa); b) d2sp3 - oktaedryczna; c) na dole rysunku: dz2sp3 i dx2-z2sp3 - piramida tetragonalna
Tabela 1. Struktura cząsteczkowa kompleksu w zależności od typu hybrydyzacji orbitali atomu centralnego
Najczęściej kompleksy mają strukturę oktaedryczną z liczbą koordynacji 6 lub strukturę tetraedryczną
z liczbą koordynacji 4. Płaskiej strukturze tetragonalnej również odpowiada liczba koordynacji 4. Inne struktury geometryczne i inne liczby koordynacji atomu lub jonu centralnego spotyka się rzadziej. Liczbie koordynacji 6 odpowiadają często odkształcone struktury okteadryczne, np. rozciągnięcie osiowe oktaedru przekształca go
w bipimiramidę tetragonalną.
Nomenklatura
Każdy związek koordynacyjny powinien być nazywany zgodnie z obowiązującą nomenklaturą (patrz: A. Bielański, Podstawy chemii nieorganicznej).
Jako nienaruszalną zasadę przyjmuje się podawanie nazwy kompleksu wraz
z umieszczonym w nawiasie okrągłym formalnym stopniem utlenienia atomu centralnego. Zasadę tę ilustrują podane
w niniejszym wykładzie nazwy niektórych kompleksów, np. tetracyjanoniklan(II)
i dodekakarbonylotriżelazo(0), co oznacza, że atomy centralne są na stopniach utlenienia równych odpowiednio 2+ i 0.Ligandy, za wyjątkiem wody i amoniaku,
w nazwach kompleksów mają końcówkę -o.
Wodę i amoniak jako ligandy skoordynowane
w kompleksach określa się jako akwa i amina.
Tabela 2. Nazwy ważniejszych ligandów nieorganicznych
Ligand |
Nazwa |
Ligand |
Nazwa |
H2O NH3 CO NO O2- O22- OH- S2- F- Cl- Br- I- NH2- |
akwa amina karbonylo nitrozylo okso perokso hydrokso tio fluoro chloro bromo jodo amido |
CN- SCN- CNS- H-
CO32- C2O42- NO3- ONO- NO2- SO42- S2O32- |
cyjano tiocyjaniano izotiocyjaniano hydrydo lub hydro węglano szczawiano azotano nitrito-O nitrito-N siarczano tiosiarczano |
Ligandy wymienia się w porządku
alfabetycznych. W celu określenia liczby
poszczególnych ligandów w kompleksie
stosuje się przedrostki greckie: mono-, di-, tri-,
tetra-, penta-, heksa-, hepta-, okta-, nona-, deka-,
dodeka-. Przedrostek mono- zazwyczaj się pomija.
Nazwy bardziej skomplikowanych ligandów
ujmuje się w nawias i poprzedza przedrostkami
bis-, tris-, tetrakis. Gdy ligand spełnia rolę mostka
w kompleksach wielordzeniowych, to jego nazwa
jest poprzedzona grecką literą μ.
- Nazwy cząsteczek i kationów kompleksowych
zawierają nazwy atomów lub jonów centralnych. Ujemnie naładowane kompleksy są anionami, dlatego ich nazwy mają końcówkę -an.
Przykłady:
[Cr(NH3)3Cl3] - triaminatrichlorochrom(III)
Co(C6H6)2 - dibenzokobalt(0)
[Ag(NH3)2]+ - kation diaminasrebra(I)
[Co(H2O)4Cl2]+ - kation tetraakwadichlorokobaltu(III)
[Co(NH3)4-μ-NH2-μ-OH-Co(NH3)4]4+
kation μ-amido-μ-hydrokso-bis{tetraaminakobaltu(III)}
[Fe(CN)6]4- - anion heksacyjażelazianowy(II)
[Fe(CN)6]3- - anion heksacyjanożelazianowy(III)
PtCl42- - anion tetrachloroplatynianowy(II)
PtCl62- - anion heksachloroplatynianowy(IV)
Teoria pola krystalicznego
Wyjątkową zdolność do tworzenia różnych związków koordynacyjnych wykazują metale przejściowe, tzn. należące do rodzin bloku d.
W wielu przypadkach są to połączenia barwne
i wykazujące zróżnicowane właściwości magnetyczne. Barwa i właściwości magnetyczne kompleksu zależą od stopnia utlenienia i struktury elektronowej jonu metalu przejściowego oraz rodzaju ligandów, które wokół jonu centralnego wytwarzają pole elektrostatyczne o określonej symetrii, zwane polem ligandów. Pole ligandów powoduje częściowe zniesienie pięciokrotnej degeneracji orbitali d jonu metalu przejściowego przez rozszczepienie ich na dwie grupy, odpowiednio o energii wyższej i niższej względem energii nierozszczepionych orbitali d w jonie swobodnym.
Jon znajdujący się w polach ligandów o symetrii oktaedrycznej (Oh) i tetraedrycznej (Td) przedstawiono na rys. 6, na którym widać w jaki sposób pole oktaedryczne oddziaływuje na orbitale dz2 i dx2-y2. Rozszczepienie orbitali d jonu metalu przejściowego w tych polach ligandów przedstawiono na rys. 7 i 8.
Rys. 6. Jon metalu przejściowego w polach ligandów o symetrii oktaedrycznej i tetraedrycznej
Rys. 7. Rozszczepienie orbitali d w polu o symetrii oktaedrycznej
Rys. 8. Rozszczepienie orbitali d w polu o symetrii tetraedrycznej
Odstęp energetyczny między rozszczepionymi orbitalami d, oznaczony symbolem Δ, nosi nazwę parametru rozszczepienia. Parametr Δ wyznacza się doświadczalnie z pomiarów spektroskopowych
i tradycyjnie podaje w cm-1, gdzie 1 cm-1 = 11,931 J/mol. Jeśli rozważa się te same ligandy, to parametr rozszczepienia w polu tetraedrycznym, ΔTd, jest mniejszy od parametru rozszczepienia w polu oktaedrycznym, ΔOh, a w przybliżeniu zachodzi między nimi następująca zależność:
ΔTd = 4/9 ΔOh
Parametr rozszczepienia zależy od rodzaju ligandów, które uszeregowane na podstawie wzrostu jego wartości tworzą tzw. szereg spektrochemiczny:
I- < Br- < Cl- < F- < OH- < H2O < NH3 < NO2- < CN-
Przy małych wartościach Δ elektronowe widma kompleksów wykazują pasma absorpcyjne
w zakresie długofalowym promieniowania widzialnego i w bliskiej podczerwieni. Przy dużych wartościach Δ pasma absorpcyjne w elektronowych widmach kompleksów są położone w zakresie krótkofalowym promieniowania widzialnego
i w ultrafiolecie.
W przypadku oktaedrycznych kompleksów metali przejściowych należy mieć na uwadze strukturę elektronową dq jonu centralnego i wielkość parametru rozszczepienia, ponieważ dla q = 4, 5, 6
i 7, decydują one o właściwościach magnetycznych kompleksu. W kompleksach oktaedrycznych q elektronów obsadza rozszczepione orbitale d zgodnie z regułą Hunda, ale gdy parametr ΔOh jest większy od energii sparowania elektronów Π, to od tej reguły są odstępstwa dla q = 4, 5, 6 i 7.
Rozważmy jako przykład kobalt, którego atomy w stanie podstawowym mają następującą strukturę elektronową powłok walencyjnych: 3d74s2. Kation Co2+ ma strukturę 3d7 , a wówczas, gdy jest jonem centralnym w kompleksie oktaedrycznym, to jego struktura elektronowa zależy od parametru Δ:
ΔOh < Π, 7 elektronów obsadza orbitale t2g i eg zgodnie z regułą Hunda, co ostatecznie daje strukturę (t2g)5(eg)2 z trzema elektronami
o niesparowanych spinach, zapewniającą silny paramagnetyzm kompleksu.ΔOh > Π, reguła Hunda nie obowiązuje, co skutkuje strukturą elektronową (t2g)6(eg)1, zapewniająca słabszy niż poprzednio paramagnetyzm kompleksu, ponieważ obecnie tylko jeden elektron ma niesparowany spin.
W przeciwieństwie do kompleksów Co(II), kompleksy oktaedryczne Co(III), jonu o strukturze d6 - elektronowej, są zawsze diamagnetyczne, ponieważ dla każdego rodzaju ligandów ΔOh > Π
i sześć elektronów d musi całkowicie obsadzić orbital t2g.
W kompleksach tetraedrycznych jest spełniana bez wyjątku relacja ΔTd < Π, dlatego q elektronów d jonu metalu przejściowego obsadza rozszczepione orbitale zgodnie z regułą Hunda, wskutek czego kompleksy te są zawsze paramagnetyczne.
Pola ligandów o niższej symetrii w większym stopniu znoszą degenerację orbitali d niż pola
o symetrii tetraedrycznej lub oktaedrycznej. Pole
o płaskiej symetrii tetragonalnej powoduje rozszczepienie orbitali d na cztery grupy, przy czym orbital dz2 uzyskuje energię znacznie wyższą od pozostałych. Z tego powodu płaskie, tetragonalne kompleksy Ni(II), jonu d8 - elektronowego, są zawsze diamagnetyczne, ponieważ 8 elektronów obsadza wyłącznie orbitale o energii niższej niż energia orbitalu dz2.
Izomeria związków kompleksowych
Fascynującym zagadnieniem w chemii koordynacyjnej jest izomeria. W szczególności dotyczy to związków, w których atom lub jon centralny koordynuje kilka różnych ligandów, ponieważ często mamy do czynienia z kilkoma izomerami różniącymi się między sobą właściwościami fizycznymi i chemicznymi.
Izomeria geometryczna
Ten typ izomerii jest związany z położeniem różnych ligandów względem atomu lub jonu centralnego w płaskich tetragonalnych kompleksach typu [MA2B2], [MA2BC] i [MABCD] oraz
w kompleksach oktaedrycznych typu [MA4B2], [MA4BC], [M(A-A)2B2] i [M(A-A)2BC], gdzie A-A oznacza ligand dwumiejscowy. Jeśli dwa różne ligandy są obok siebie, to mają położenią cis, gdy naprzeciw -
w położeniu trans. Wybrane przykłady pokazano na rys. 9.
Rys. 9. Izomery geometryczne cis i trans płaskich, tetragonalnych kompleksów MA2B2
Dwa izomery geometryczne cis i trans posiada diaminadichloroplatyna(II) - [Pt(NH3)2Cl2]. Izomer cis, zwany cis-platyną, oraz jego analogi znajdują zastosowanie jako leki przeciwnowotworowe.
Dwa izomery geometryczne cis i trans ma oktaedryczny kompleks [Co(NH3)4Cl2]+ (rys.10)
Rys. 10. Izomery cis i trans kompleksu [Co(NH3)4Cl2]+
Izomeria koordynacyjna
Izomeria ta występuje w związkach mających zarówno kation, jak i anion kompleksowy różniące się koordynacją ligandów z jonami centralnymi. Jony centralne w kationie i anionie mogą być jonami tego samego metalu na jednakowych lub różnych stopniach utlenienia lub też jonami dwóch różnych metali:
[PtII(NH3)4][PtIVCl6] i [PtIV(NH3)4Cl2][PtIICl4]
[Cr(NH3)6][Cr(NCS)6] i [Cr(NH3)4(NCS)2][Cr(NH3)2(NCS)4]
[Cr(NH3)6][Co(NCS)6] i [Co(NH3)6][Cr(NCS)6]
Izomeria jonowa i hydratacyjna
W pierwszym przypadku (a) izomery dysocjują
w roztworze na dwa różne jony; a w drugim (b), gdy woda koordynuje do jonu centralnego, liczba powstałych jonów w roztworze jest różna, np.:
a) [Co(NH3)5Br]SO4 i [Co(NH3)5SO4]Br
b)[Cr(H2O)6]Cl3,[Cr(H2O)5Cl]Cl2∙H2O, [Cr(H2O)4Cl2]Cl∙2H2O
Izomeria wiązania
Niektóre ligandy jednomiejscowe mają dwa różne heteroatomy, np. N i O w grupie nitrowej NO2
z donorowym atomem azotu, M-NO2, i w anionie NO2- z donorowym atomem tlenu, M-ONO. Inny przykład to aniony NCS-, które są zdolne do koordynacji przez atom azotu lub siarki.
Izomeria polimeryzacyjna
Znane są przypadki występowania kompleksów,
które formalnie mają taki sam skład jakościowy
i ilościowy, ale różnią się masami cząsteczkowymi, np.: [Pt(NH3)2Cl2] i [Pt(NH3)4][PtCl4]
Izomeria optyczna
Interesującym przykładem izomerii związków kompleksowych jest izomeria optyczna kompleksów oktaedrycznych. Cząsteczki lub jony izomerów optycznych mają się do siebie tak, jak przedmiot
i jego obraz w lustrze. Izomery optyczne są związkami chiralnymi, tzn. wykazują zdolność do skręcania płaszczyzny polaryzacji światła spolaryzowanego, przy czym pierwszy z nich skręca ją o pewien kąt w prawo, natomiast drugi skręca ją w lewo.
Rys. 11. Izomeria optyczna kompleksów oktaedrycznych MX2Y2Z2 oraz ML3, gdzie L jest dwumiejscowym ligandem chelatowym
Równowagi w wodnych roztworach związków kompleksowych
Gdy do roztworu FeCl3 dodamy roztwór KSCN, ![]()
to mieszanina zabarwia się na kolor krwistoczerwony. Przyczyną tego zjawiska jest kompleksowanie kationów Fe3+ przez aniony tiocyjanianowe SCN-:
Fe3+ + SCN- ↔ [Fe(SCN)]2+ (1)
[Fe(SCN)]2+ + SCN- ↔ [Fe(SCN)2]+ (2)
[Fe(SCN)2]+ + SCN- ↔ [Fe(SCN)3] (3)
[Fe(SCN)3 ] + SCN- ↔ [Fe(SCN)4]- (4)
[Fe(SCN)4] - + SCN- ↔ [Fe(SCN)5]2- (5)
[Fe(SCN)5]2- + SCN- ↔ [Fe(SCN)6] 3- (6)
Jak widać, produkt i-tej reakcji jest substratem
w reakcji (i+1), gdzie i = 1, 2, 3, 4, 5. Z tego powodu produkty reakcji (1) - (6) nazywamy kompleksami następczymi. W reakcjach tych ustalają się stany równowagi, dlatego zgodnie z prawem prawa działania możemy napisać:
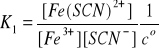
(7)
![]()
(8)
![]()
(9)
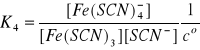
(10)
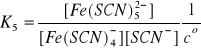
(11)
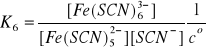
(12)
Stałe równowagi Ki , i = 1, 2, … , 6, są następczymi stałymi trwałości kolejnych kompleksów powstałych
w reakcjach (1) - (6). W wyrażeniach (7) - (12)
w nawiasach występują stężenia w stanie równowagi, mianowicie:
●stężenie wolnych, nieskompleksowanych jonów Fe3+; ●stężenie wolnego liganda czyli jonów SCN-;
●stężenia odpowiednich kompleksów [Fe(SCN)i]3-i oraz
[Fe(SCN)i+1]3-(i+1).
Reakcje tworzenia poszczególnych kompleksów tiocyjanianowych Fe(III) można przedstawić równaniem reakcji sumarycznej:
Fe3++ i SCN- ↔ Fe(SCN)i3-i (13)
ze stałą równowagi βi czyli ogólną (lub kumulatywną) stałą trwałości i-tego kompleksu:
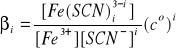
(14)
Między następczymi i ogólnymi stałymi trwałości kompleksów zachodzi następujący związek:
![]()
(15)
Na podstawie wzoru (15) dla sześciu kompleksów
w ukladzie Fe3+ - SCN- mamy:
β1 = K1 , β2 = K1K2 , …, β6 = K1K2K3K4K5K6.
UWAGA: W ogólnym przypadku stosuje się uproszczony zapis pomijając ładunki jonów, a jon metalu i ligand oznaczając odpowiednio symbolami M i L, np.:
M + L ↔ ML; ML + L ↔ ML2 lub M + 2L ↔ ML2, itd.
Definicje
Analityczne stężenie metalu w roztworze
cM = [M] + [ML] + [ML2] + … + [MLn] (16)
[MLi] = βi[M][L]i (17)
cM = [M] + β1[M][L] + β2[M][L]2 + … + βn[M][L]n =
(1 + β1[L] + β2[L]2 + … + βnL]n)[M] = Φ[M] (18)
gdzie [M] jest stężeniem wolnych jonów M, a parametr Φ jest funkcją kompleksowania zależną od stężenia wolnego liganda L.
Ułamek molowy jonów wolnego metalu
![]()
(19)
Ułamek molowy kompleksu MLi
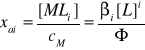
(20)
Suma ułamków molowych jonów wolnego metalu
i poszczególnych kompleksów jest z definicji równa 1.
Σxαi = 1, i = 0, 1, … , n (21)
Udziały jonów wolnego metalu i kompleksów można również przedstawić w procentach molowych.
Analityczne stężenie liganda w roztworze
cL = [L] + [ML] + 2[ML2] + … + n[MLn] (22)
Niektóre ligandy, np. amoniak, aniony słabych kwasów, mogą ulegać w roztworze protonowaniu: L + H+ ↔ LH,
dlatego należy to uwzględnić w równaniu bilansowym.
cL = [LH] + [L] + [ML] + 2[ML2] + … + n[MLn] (23)
Powróćmy do kompleksów w układzie Fe3+ - SCN-. Ogólne stałe trwałości tych kompleksów są następujące:
log β1 |
log β2 |
log β3 |
log β4 |
log β5 |
log β6 |
3,1 |
5,2 |
6,2 |
6,2 |
6,1 |
6,0 |
Na podstawie przedstawionych wzorów można obliczyć w ułamkach lub procentach molowych udziały wolnych, niezwiązanych jonów Fe3+ i wszystkich kompleksów
w zależności od stężenia wolnych, niezwiązanych jonów SCN-. Zależność tę przedstawia rys. 12.
Rys. 12. Udziały kompleksów następczych w układzie Fe3+ - SCN-
1 - [Fe(SCN)]2+, 2 - [Fe(SCN)2]+, 3 - [Fe(SCN)3],
4 - [Fe(SCN)4]-, 5 - [Fe(SCN)5]2-, 6 - [Fe(SCN)6]3-
Kompleksy chelatowe
Kompleksy chelatowe charakteryzuje znaczna trwałość mierzona dużo większymi stałymi trwałości
w porównaniu z kompleksami z ligandami prostymi. Łatwo można się o tym przekonać porównując reakcje akwakompleksów metali z etylenodiaminą (en)
i amoniakiem, np.:
[Co(H2O)6]2+ + 6NH3 ↔ [Co(NH3)6]2+ + 6H2O (24)
[Co(H2O)6]2+ + 3en ↔ [Co(en)3]2+ + 6H2O (25)
Zwraca uwagę fakt, że w reakcji (25) liczba cząsteczek
i jonów po lewej stronie równania jest mniejsza niż po prawej stronie. Jest to tak zwany efekt chelatowy, wynikający ze wzrostu stopnia nieuporządkowania
w układzie.
Innymi słowy, reakcja kompleksowania przez etylenodiaminę skutkuje wzrostem entropii układu,
a w konsekwencji większą trwałością kompleksu [Co(en)3]2+w porównaniu z kompleksem [Co(NH3)6]2+.
Stałe trwałości kompleksów niektórych metali
z amoniakiem i etylenodiaminą zebrano w tabeli 3.
Tabela 3. Stałe trwałości kompleksów metali z etylenodiaminą i amoniakiem
Kation |
Liczba koordynacji
|
log βn |
|
|
|
NH3 |
en |
Ni2+ Co2+ Co3+ Zn2+ Cu2+ |
6 6 6 4 (tetraedr) 4 (kwadrat) |
9 (n=6) 5 (n=6) 34 (n=6) 9 (n=4) 13 (n=4) |
19 (n=3) 14 (n=3) 49 (n=3) 11 (n=2) 20 (n=2) |
Wyszukiwarka
Podobne podstrony:
Związki kompleksowe (kompleksy, związki koordynacyjne
gr2, Związki kompleksowe (inaczej kompleksy, związki koordynacyjne) to związki chemiczne, w których
związki koordynacyjne
Chemia związków koordynacyjnych
07 Otrzymywanie i metody badania związków koordynacyjnychid 6732 ppt
Chemia związków koordynacyjnych
związki koordynacyjne
07 ZWIĄZKI KOMPLEKSOWE (KOORDYNACYJNE)
zwiazki kompleksowe 2
Sprawozdanie 6 związki kompleksowe
Cw2 Zwiazki kompleksowe
otrzymywanie i właściwości związków kompleksowych
7 Związki kompleksowe
Sprawozdanie z ChOiA zwiazki kompleksowe ćw 3
Sprawozdanie NR 5 związki kompleksowe
Zwiazki kompleksowe
więcej podobnych podstron