
TTTTTTTT
AAAAA
TTTTTTT
C
TTTT
AAA
#6 Metody wiarygodnościowe
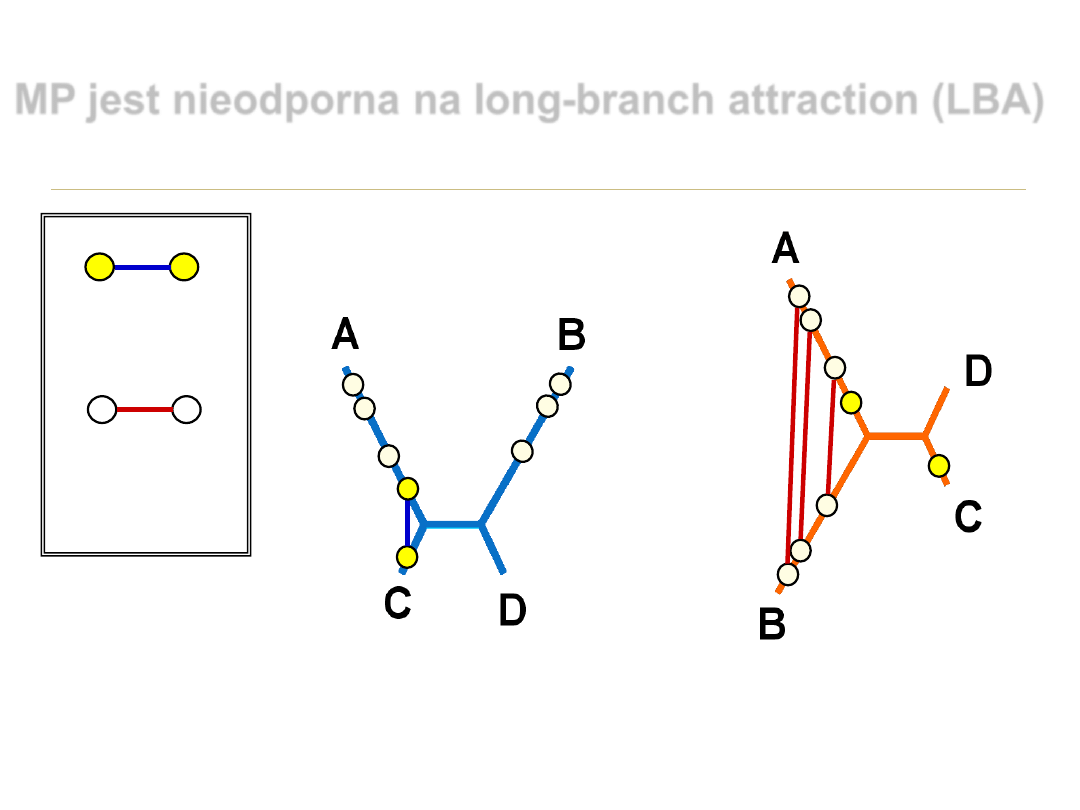
błędne drzewo z LBA
prawdziwe drzewo
MP jest nieodporna na long-branch attraction (LBA)
synapomorfia
homoplazja
– pozorna
synapomorfia
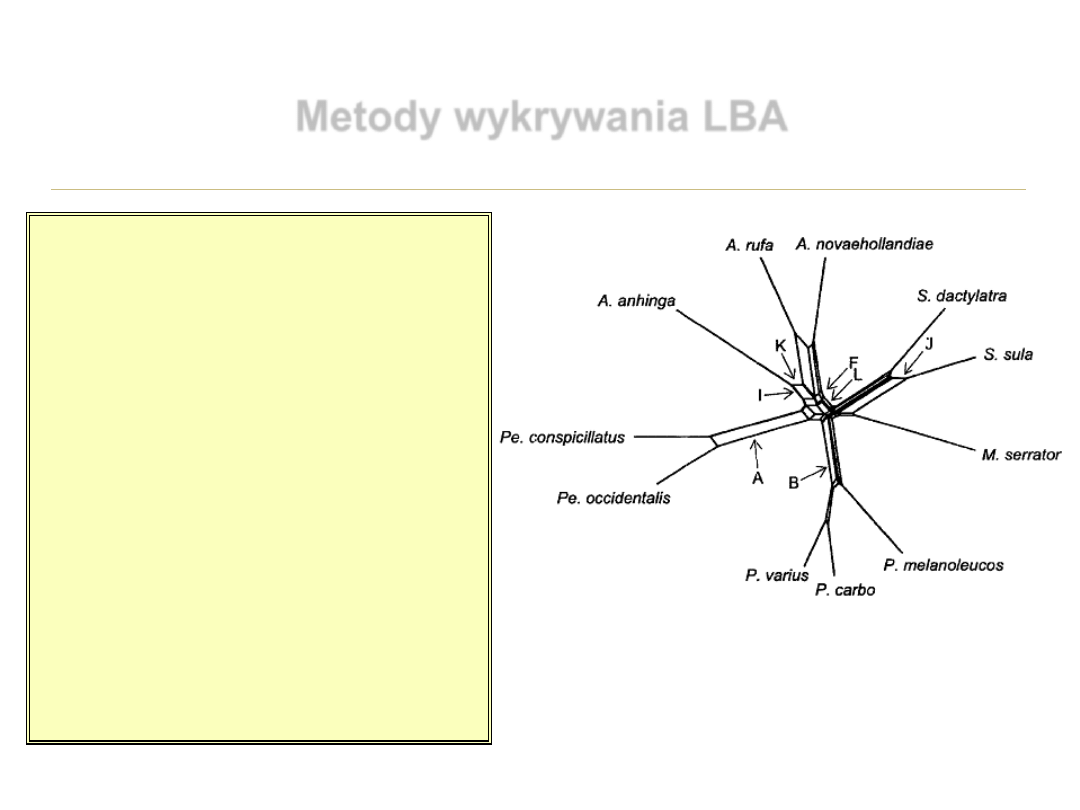
Metody wykrywania LBA
Neighbor-nests generowane
przez SplitTree4
1.
Osobne analizy partycji
2.
Usuwanie potencjalnych
długich gałęzi
3.
Dobór grup zewnętrznych
4.
Symulacje parametryczne
(bootstrap)
5.
Split decomposition +
analiza spektralna
6.
Niezgodność wyników z
różnych metod
7.
Porównanie z filogenią
morfologiczną

Jak uniknąć wpadnięcia w „strefę
Felsensteina”
1.
Wyłączanie pewnych fragmentów
sekwencji np. 3-cia pozycja kodonu,
fragmenty hiperzmienne
2. Dodawanie nowych taksonów (?)
3.
Dodawanie nowych źródeł danych i
analiza total evidence
4.
Zastosowanie metod biorących pod
uwagę długość gałęzi
1.
Wyłączanie pewnych fragmentów
sekwencji np. 3-cia pozycja kodonu,
fragmenty hiperzmienne
2. Dodawanie nowych taksonów
3.
Dodawanie nowych źródeł danych i
analiza total evidence
4.
Zastosowanie metod biorących pod
uwagę długość gałęzi
Metody wiarygodnościowe
są najbardziej odporne na ten problem,
jeśli zostanie zastosowany odpowiedni
model substytucji

Modele ewolucji sekwencji
•
Modele mogą dotyczyć różnych aspektów ewolucji
sekwencji:
–
Różnorodnego stosunku transwersji do tranzycji.
– Odmiennej frekwencji nukleotydów.
–
Różnorodnego tempa ewolucji w poszczególnych
miejscach sekwencji.
–
Różnorodnego tempa ewolucji (=substytucji) w
ramach linii (poszczególnych taksonów) czy całych
partii drzewa.

Modele ewolucji sekwencji
• Bogactwo parametrów modeli ma pozytywne i
negatywne strony:
im więcej parametrów do testowania tym lepiej można
dopasować model do konkretnych danych.
im więcej parametrów do testowania tym wyższa
wariancja oszacowania.
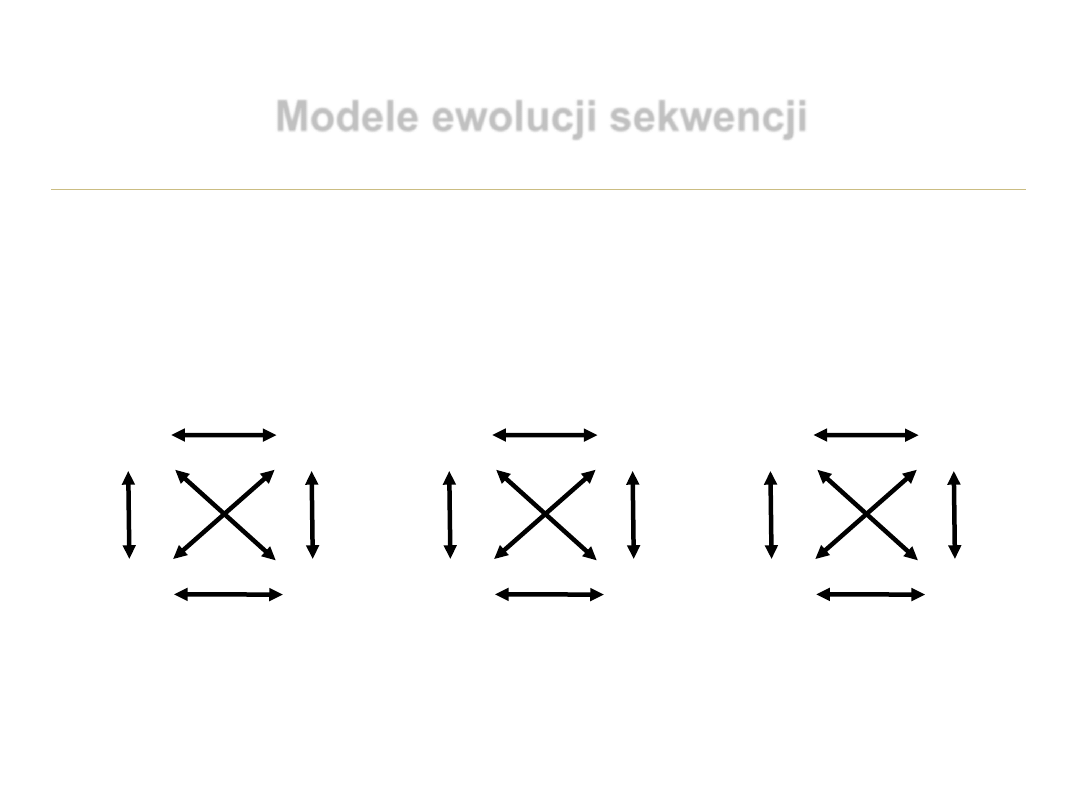
Modele ewolucji sekwencji
T
C
A
G
a
a
a
a
a
a
Jukes-Cantor
T
C
A
G
a
f
e
b
c
d
Generalny
T
C
A
G
2α
α
α
Kimura 2 parametrowy
2α
2α
2α

Metody wiarygodnościowe
1. Maximum Likelihood (ML, metoda
maksymalnej wiarygodności)
2. Bayesian Phylogenetic Inference +
Markov Chain Monte Carlo (BPI+ MCMC,
wnioskowanie Bayesowkie z łańcuchem
Markowa Monte Carlo)

Metody wiarygodnościowe
Zalety metod:
–
Dobrze pracują z danymi zawierającymi zarówno odlegle
spokrewnione sekwencje jak i bliskie sobie gatunki – najbardziej
uniwersalne
–
Wykorzystują wszystkie dane – zmienne i niezmienne, informatywne
parsymonicznie i nieinformatywne
–
Dobrze sprawują się przy niejednorodnym tempie substytucji (biorą
pod uwagę długość gałęzi) i w związku z tym są najbardziej odporne
na LBA spośród wszystkich metod filogenetycznych
–
Mogą stosować szeroką gamę modeli ewolucji sekwencji (DNA i
aminokwasy), a nawet modele ewolucji cech morfologicznych
Wady metod:
–
Czułe na źle dobrany model ewolucji cech
–
Bardzo powolne i wymagają dużej mocy obliczeniowej komputera

Metoda maksymalnej wiarygodności
(Maximum Likelihood, ML)
•
Metoda do szacowania parametrów w statystyce: najlepszą
estymacją parametru jest wartość, która najbardziej
prawdopodobnie wygeneruje obserwowane dane
•
Zaaplikowana do filogenetyki molekularnej przez Joe Felsensteina
(1981)
•
ML
zakłada określony, niekiedy złożony model ewolucji sekwencji.
•
Celem analizy ML
jest odpowiedź na pytanie:
Jakie jest prawdopodobieństwo P powstania obserwowanych
danych D (w tym wypadku alignmentu wielu sekwencji) dla danej
topologii drzewa filogenetycznego T
przy określonym modelu
ewolucji ME?

Jak działa ML?
1.
Warunkiem wstępnym analizy jest otrzymanie
odpowiedniego modelu ewolucji sekwencji ME (np.
Modeltest).
2.
Dla każdej pozycji j w alignmencie generowane są
wszystkie możliwe topologie drzewa dla danej liczby
taksonów (sekwencji).
3.
Opierając się na modelu ewolucji ME obliczamy
prawdopodobieństwo układu nukleotydów L(j) dla
każdego z tych drzew w pozycji j i je sumujemy.
4.
Obliczamy prawdopodobieństwo całkowite powstania
obserwowanego alignmentu dla topologii L poprzez
iloczyn wszystkich prawdopodobieństw z
poszczególnych pozycji.

Przykładowa analiza MJ dla przypadku 4 taksonów
1 j N
---------------------------------------
Takson 1:
A... C G C G C T G G G ... C
Takson 2:
A... C G C G C T G G G ... C
Takson 3:
A... C G C A A T G A A ... C
Takson 4:
A... C A C A G G G A A ... C
Wybieramy dowolną cechę j = pozycję w alignmencie

1 2 3 4
C C A G
j
----
1:
C
2:
C
3:
A
4:
G
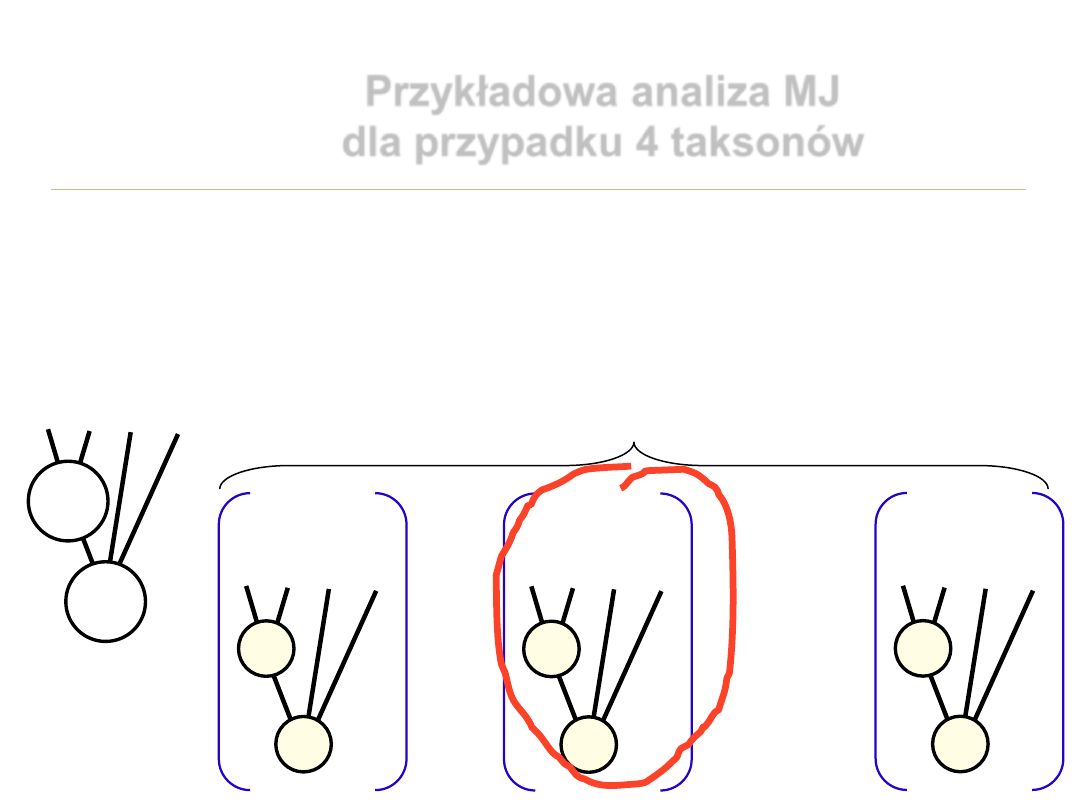
Przykładowa analiza MJ
dla przypadku 4 taksonów
Wyszukujemy wszystkie ukorzenione
topologie dla 4 taksonów i dodatko-
wo oznaczamy liście stanem cechy j
1 2 3 4
C C A G
1 3 2 4
C A C G
2 3 1 4
C A C G
1 4 2 3
C G C A
2 4 1 3
C G C A
3 4 1 2
A G C C
1 2 3 4
C C A G
1 2 4 3
C C G A
3 4 1 2
A G C C
2 3 4 1
C A G C
1 2 3 4
C C A G
1 4 3 2
C G A C
1 3 2 4
C A C G
1 2 3 4
C C A G
1 3 2 4
C A C G
1 4 2 3
C G C A
2 1 3 4
C C A G
2 3 1 4
C A C G
2 4 1 3
C G C A
3 1 2 4
A C C G
3 2 1 4
A C C G
3 4 1 2
A G C C
4 1 2 3
G C C A
4 2 1 3
G C C A
4 3 2 1
G A C C
1 węzeł – 1
2 węzły – 10
3 węzły – 15
-----------
26
j
----
1:
C
2:
C
3:
A
4:
G
Przykładowa analiza MJ
dla przypadku 4 taksonów
Dla każdego z tych drzew obliczamy prawdopodobieństwo L
(j)
układu
nukleotydów w pozycji j alignmentu
jako sumę prawdopodobieństw
wszystkich możliwych pośrednich stanów cechy prowadzących do tej
topologii przy założonym modelu ewolucji sekwencji ME.
1 2 3 4
C C A G
L(j) = P
(1)
+ P
(2)
+ ... + P
(16)
1 2 3 4
C C A G
A
A
1 2 3 4
C C A G
A
C
1 2 3 4
C C A G
T
G
Liczba aranżacji k= 4 dla danej
topologii o n
węzłach
n
AC
GT
AC
GT
j
----
1:
C
2:
C
3:
A
4:
G
Przykładowa analiza MJ
dla przypadku 4 taksonów
P
(i)
= P
A-C(5)
x P
C-C(1)
x P
C-C(2)
x P
A-A(3)
x P
A-G(4)
1 2 3 4
C C A G
A
C
5
Obliczanie prawdopodobieństwa końcowego dla danej
topologii drzewa
Przykładowa analiza MJ dla przypadku 4 taksonów
Ponieważ L jest bardzo małą wartością przedstawia się je w
postaci logarytmu naturalnego:
Preferowane jest drzewo o największej wartości
prawdopodobieństwa L (Maximum Likelihood)
albo najmniejszej –lnL.
L=L
(1)
× L
(2)
× .... × L
(N)
=
∏
=
N
j
L
(j)
1
∑
=
N
j 1
lnL
(j)
lnL=lnL
(1)
+ lnL
(2)
+ ... + lnL
(N)
=
∑
=
N
j 1
lnL
(j)
lnL=lnL
(1)
+ lnL
(2)
+ ... + lnL
(N)
=

Wnioskowanie bayesowskie (BPI, Bayesian
inference)
• Jedna z najstarszych metod statystycznych – 1790,
Thomas Bayes, zastosowana do filogenetyki od 1968.
•
Metoda zbliżona koncepcyjnie do ML
• Celem analizy BI
jest odpowiedź na pytanie:
Jakie jest prawdopodobieństwo P, że dana topologia
drzewa T
przy określonym modelu ewolucji jest
prawdziwa dla obserwowanych danych D (w tym
wypadku alignmentu wielu sekwencji)?
ML: P(D/T)
BPI: P (T/D)
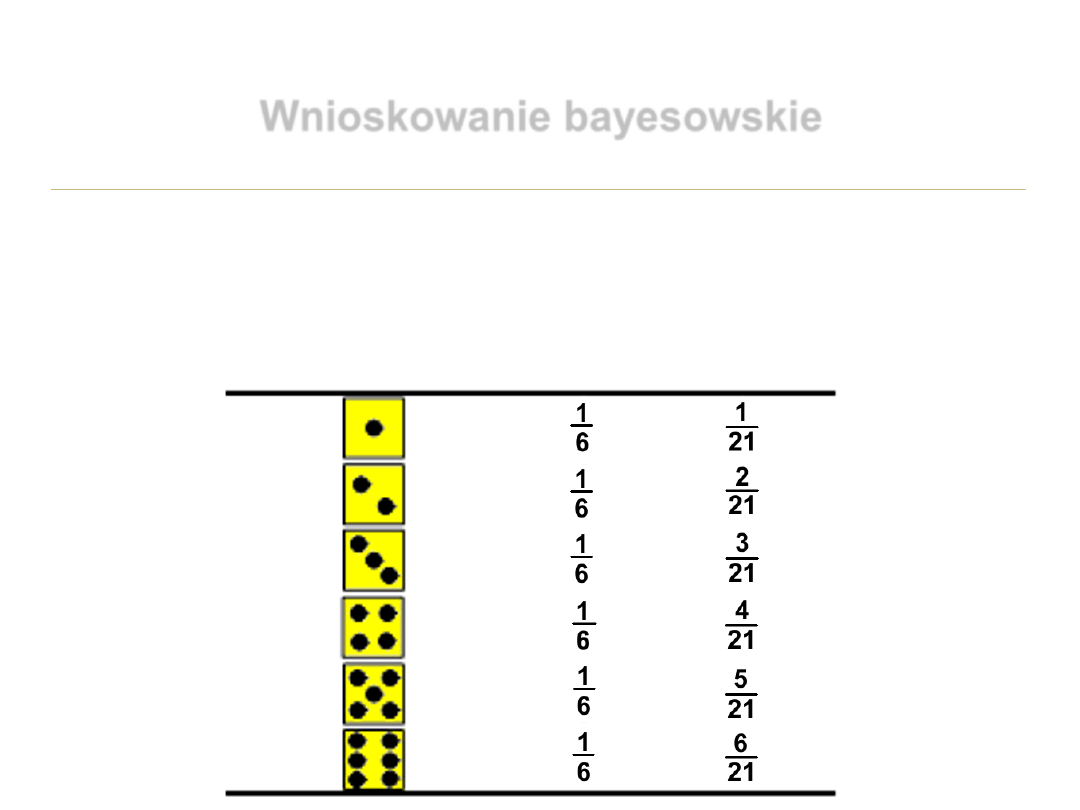
Wnioskowanie bayesowskie
100 kostek, 90 prawdziwych, 10 fałszywych
Prawdopodobieństwo P wyrzucenia oczek wynosi:
prawdziwa fałszywa

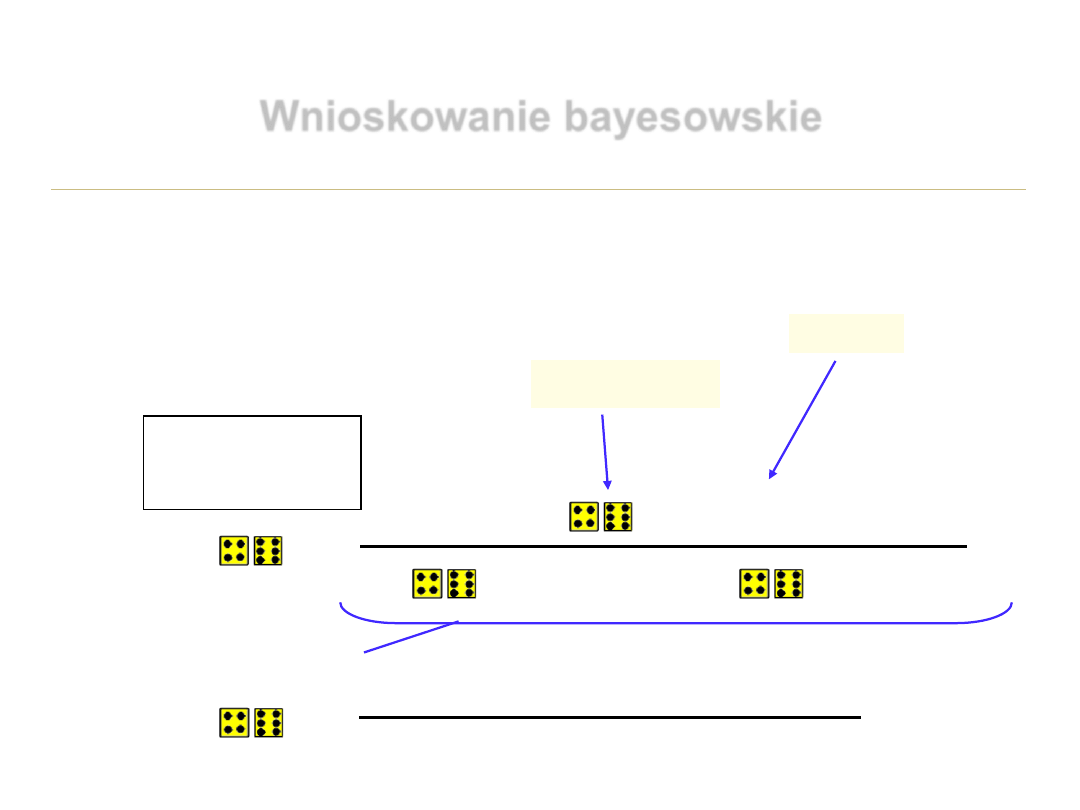
Wnioskowanie bayesowskie
Wyjęto jedną kostkę i dwa razy rzucono – za
pierwszym rzutem 4 oczka, za drugim 6.
Jakie jest prawdopodobieństwo, że kostka jest
fałszywa?
P [ | prawdziwa] = 1/6 x 1/6 = 1/36
P [ | fałszywa] = 4/21 x 6/21 = 24/441
P [fałszywa] / P [prawdziwa] = 1,93
Wnioskowanie bayesowskie
Wnioskowanie bayesowskie opiera się na prawdopodobień-
stwie a posteriori
(końcowym), które obrazuje jak zmieniła
się nasza opinia, czyli prawdopodobieństwo a priori
(początkowe) pod wpływem obserwacji.
prawdziwa +
fałszywa -
P [-| ] =
P [ |-] x P [-]
P [ |-] x P [-]x P [ |+ ] x P [+]
P [-| ] =
24/441 x 1/10
24/441 x 1/10 + 1/36 x 9/10
= 0,179
stała normalizująca
a priori
obserwacji
.

Zastosowanie formuły Bayesa
do rekonstrukcji filogenii
Bayesowskie oszacowanie filogenii opiera się na określeniu
wartości prawdopodobieństwa a posteriori drzewa.
s – liczba gatunków
B(s) –
liczba wszystkich możliwych drzew t
1
, t
2
, ... t
B(s)
dla s
gatunków
D – obserwowane dane tzn. alignment sekwencji lub macierz
morfologiczna
P (D|t
j
) x P (t
j
)
P (D|t
i
) x P (t
i
)
B(s)
j=1
∑
P (t
i
|D) =
Łańcuch Markowa Monte Carlo (MCMC)
Obliczenie prawdopodobieństwa a posteriori
obejmuje sumę wszystkich możliwych drzew i
wszystkich kombinacji długości gałęzi wraz z
modelem ewolucyjnym dla każdego z drzew, co
jest oprócz kilku bardzo prostych przypadków
niemożliwe do uzyskania drogą analityczną.
Zatem prawdopodobieństwo końcowe drzew z
konieczności musi być szacowane.
Konstrukcja MCMC
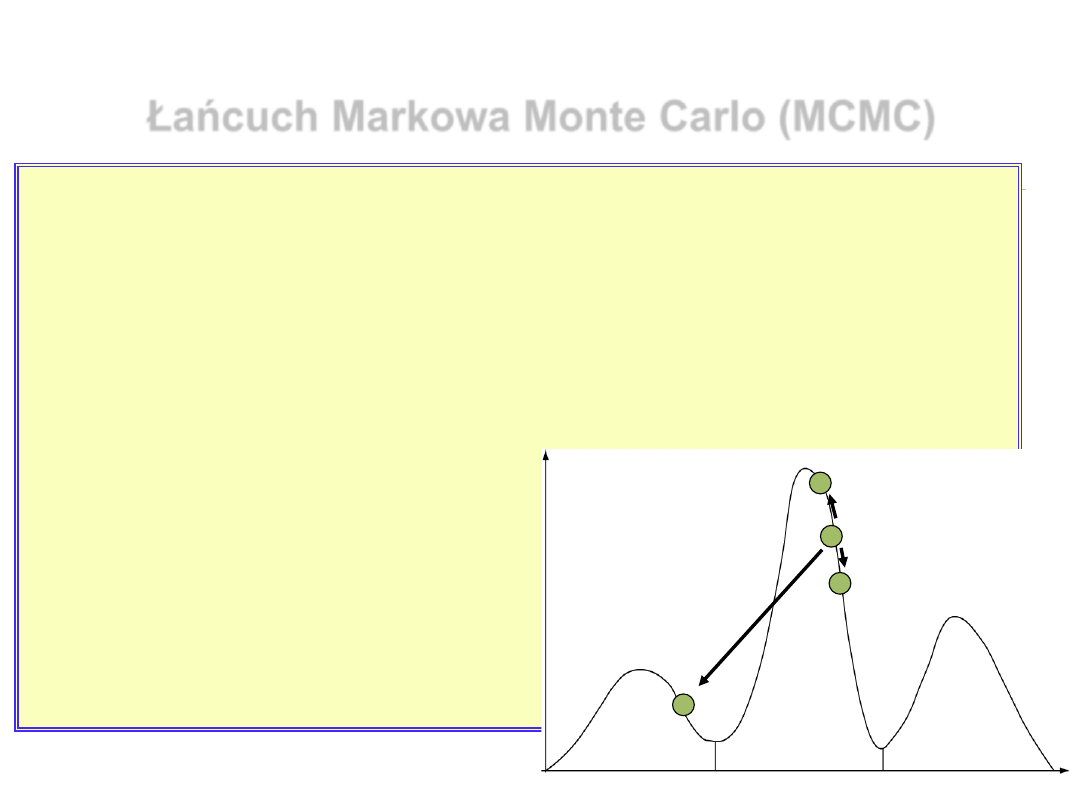
1. Zacznij w dowolnym miejscu przestrzeni parametrów = wybierz dowo lne
drzewo T ze zbi
oru wszystkich możliwych drzew dla danej liczby taksonów
2. Wybierz losowo drzewo T’
, które jest sąsiadem drzewa T z pkt . 1
3.
Przelicz stosunek prawdopodobieństw dla zaproponowanych drzew pr zy
danym modelu ewolucji cech R=P(T’)/P(T)
4.
Jeżeli R>=1 zaakceptuj nowe
drzewo ( T=T’
) i przejdź do pkt . 2
5.
Jeżeli R<1 wybierz losowo liczbę
z przedziału (0,1)
•
jeśli liczba ta jest mniejsza
od R to zaakceptuj nowe drzewo
•
jeśli nie, to obecne drzewo
pozostaw jako T
6.
Wróć do pkt . 2
Łańcuch Markowa Monte Carlo (MCMC)
zbiór wszystkich topologii
zawsze akceptuj
R>1
niekiedy akceptuj
R~1
pr
aw
dopodob
ieńs
tw
o
a
pos
ter
ior
i
nigdy nie
akceptuj
R<<1
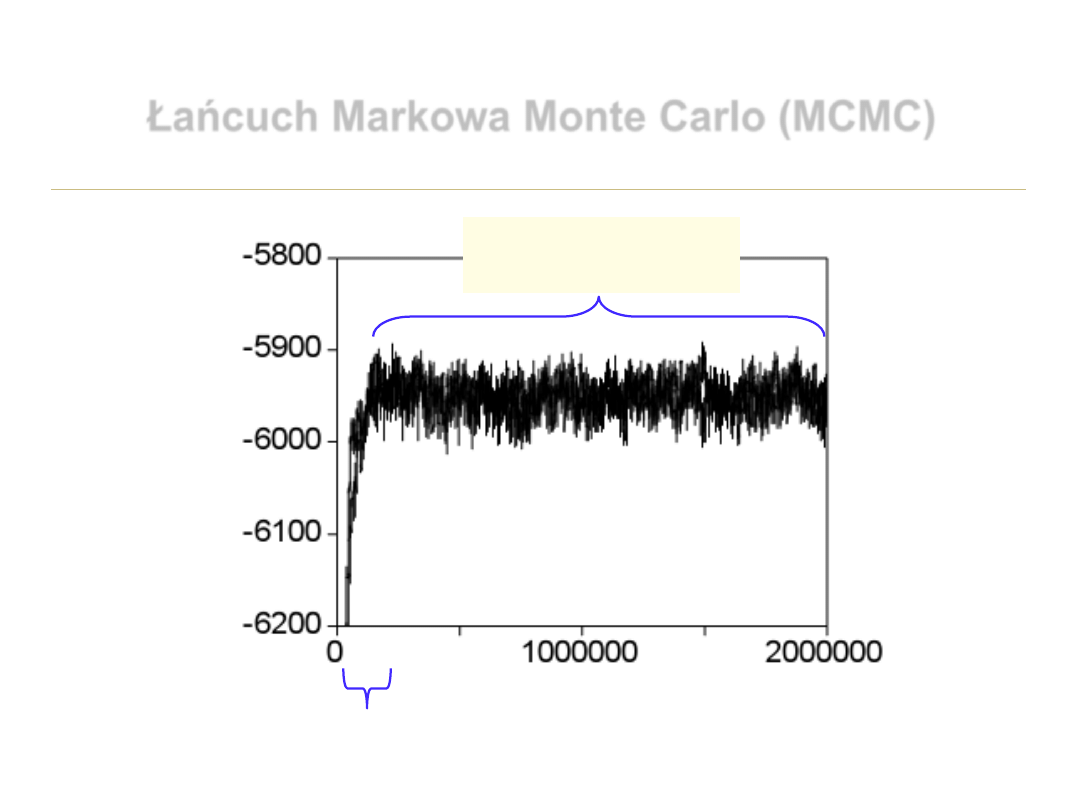
Łańcuch Markowa Monte Carlo (MCMC)
okres początkowy
(burn-in period)
okres zbieżności
(convergence period)
generacje
LnP
a
pos
ter
ior
i

Powiązany MCMC Metropolisa (Metropolis
Coupled MCMC, MCMCMC, (MC)
3
Idea algorytmu polega równoległym uruchomieniu n
łańcuchów Markowa, z czego jeden łańcuch operuje na
badanym rozkładzie prawdopodobieństwa P(t
i
|DX) jako
dystrybucji referencyjnej (tzw. zimna dystrybucja,
cold
distribution
), zaś pozostałe łańcuchy operują na
zmienionej dystrybucji, „podgrzanej” (
heated distribution
)
otrzymanej poprzez podniesienie rozkładu referencyjnego
do potęgi
β
i
, której wartość zawiera się w przedziale od 0
do 1.
T – czynnik „temperatury”
β
i
= 1/1+(i-1)T
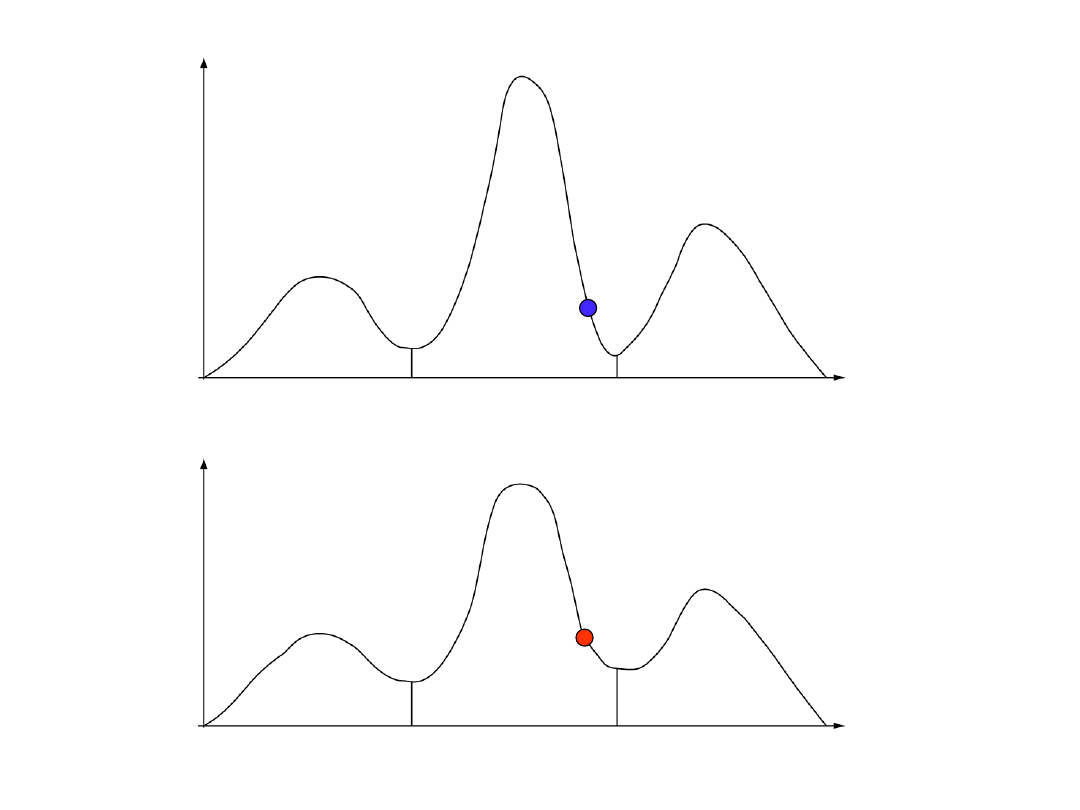
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
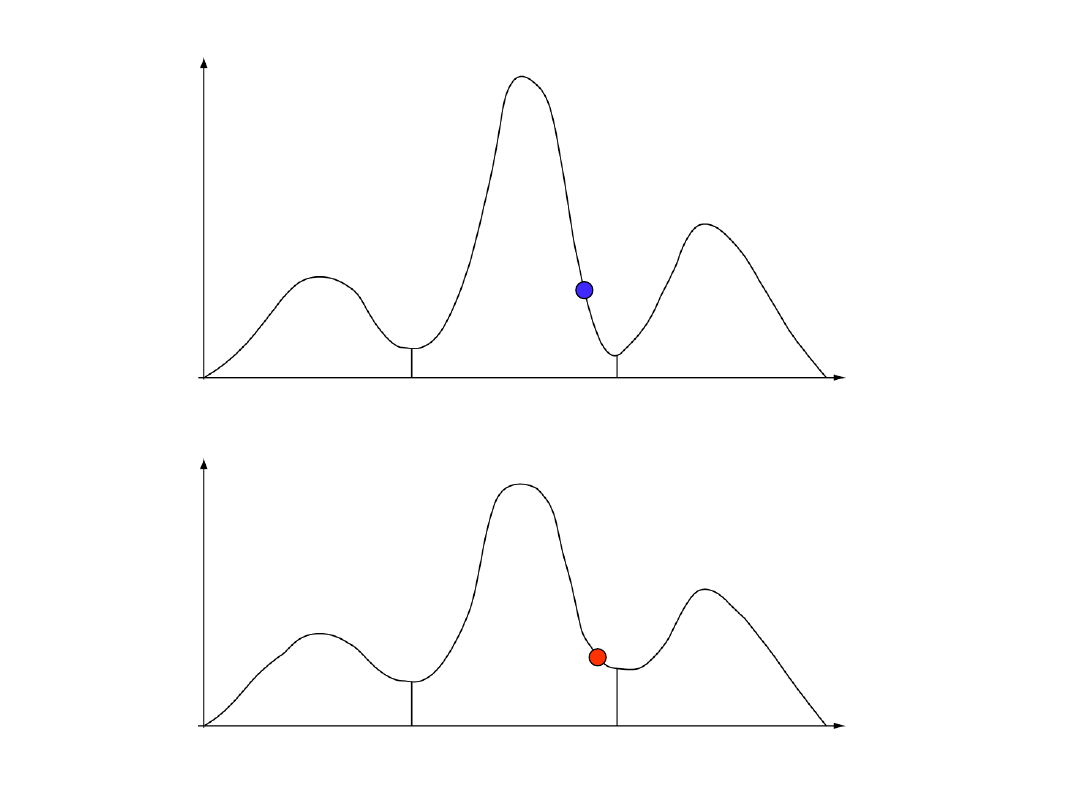
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
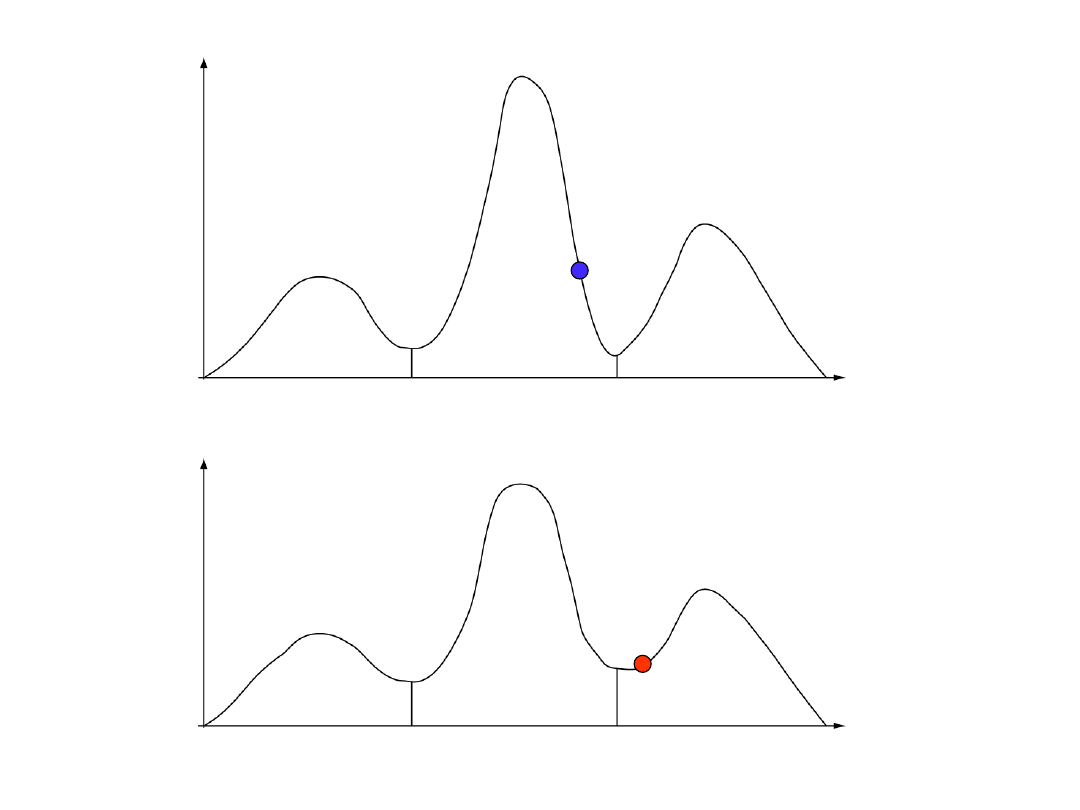
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
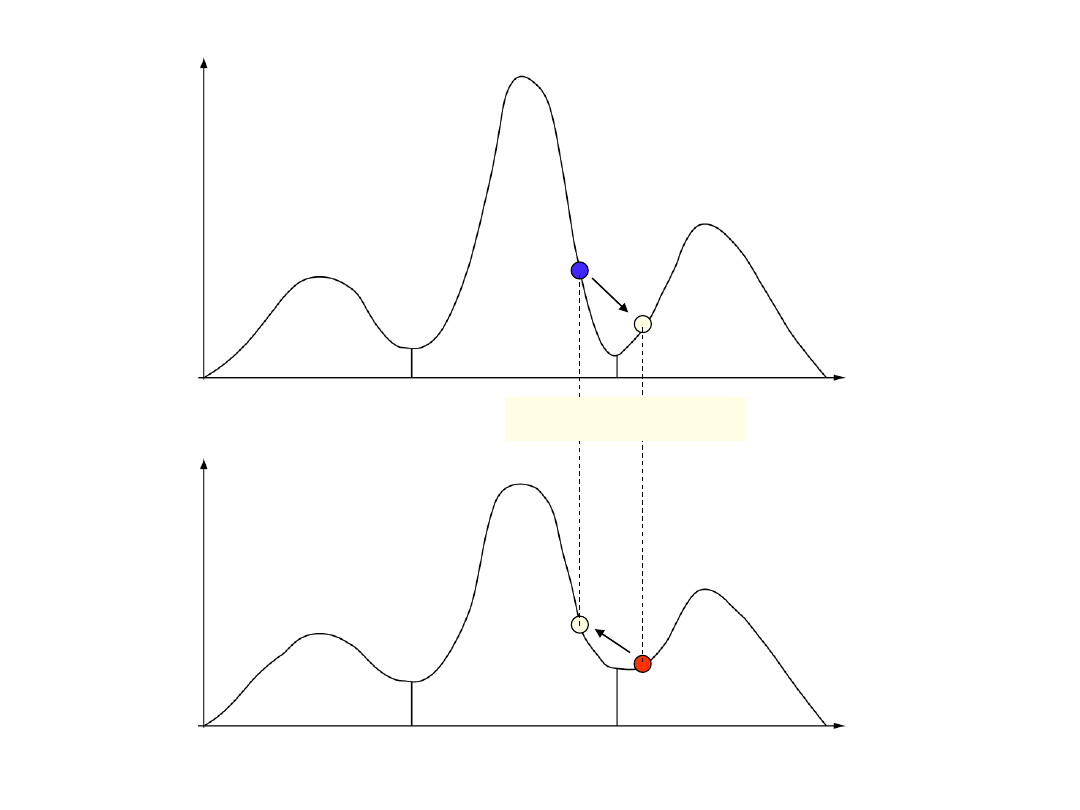
nieudana wymiana
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
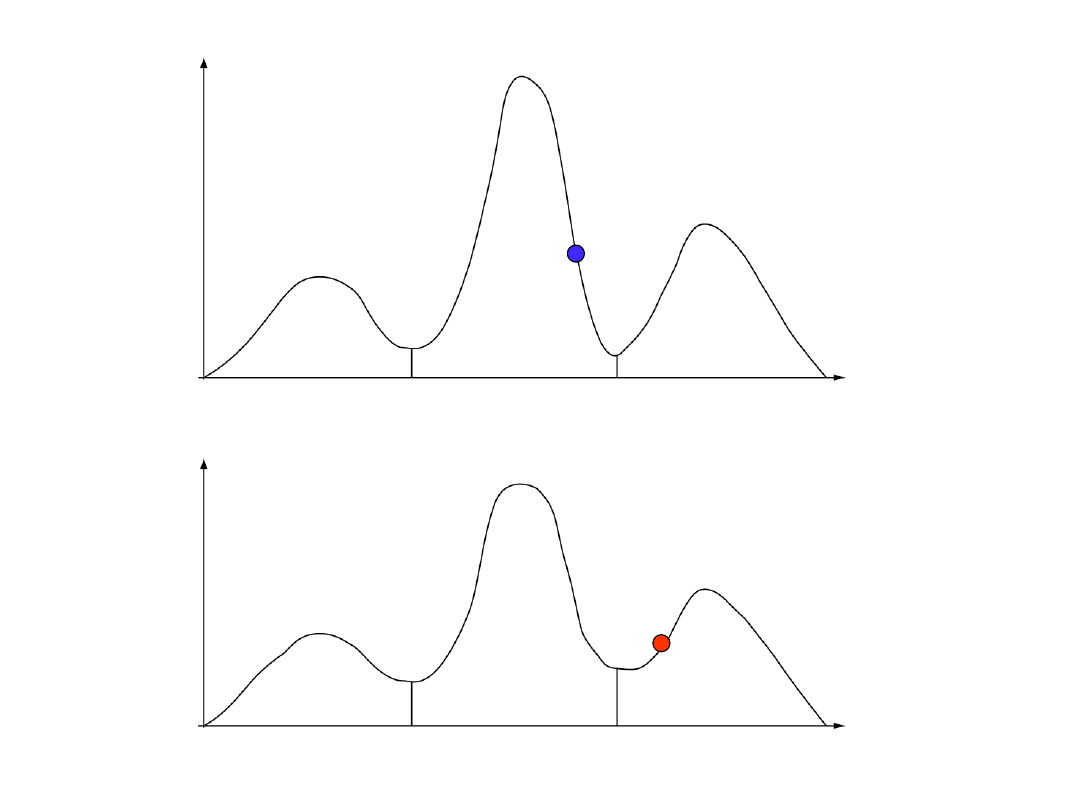
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
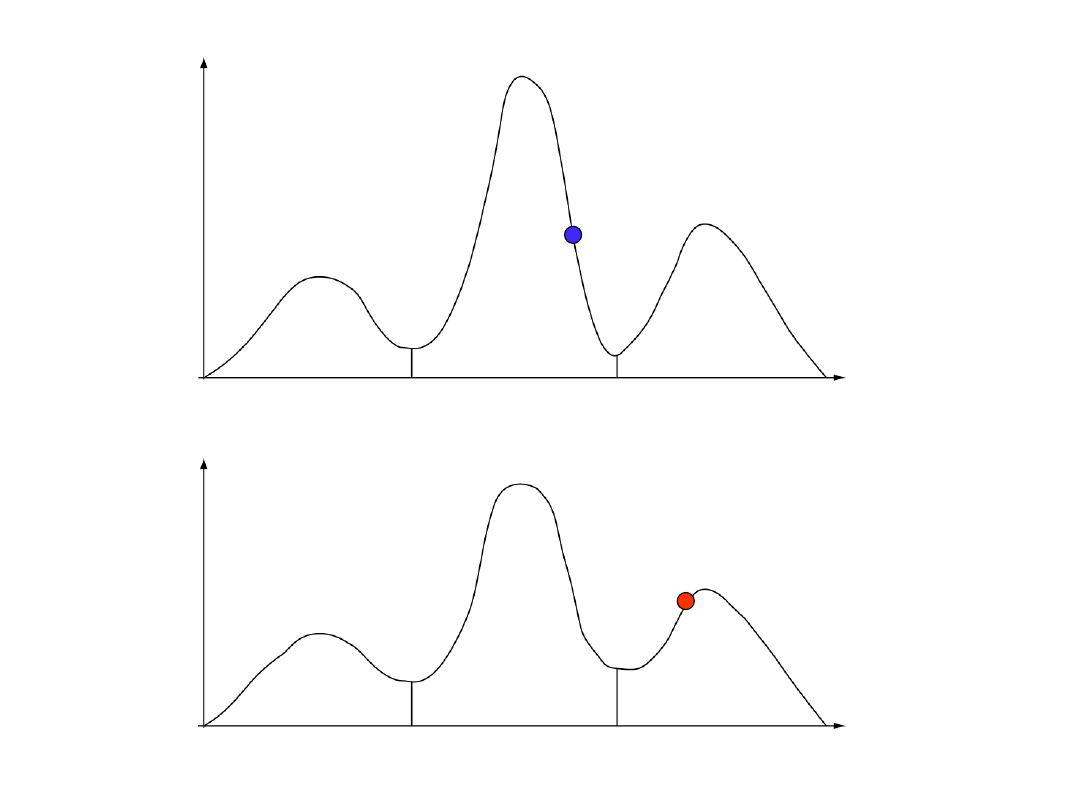
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
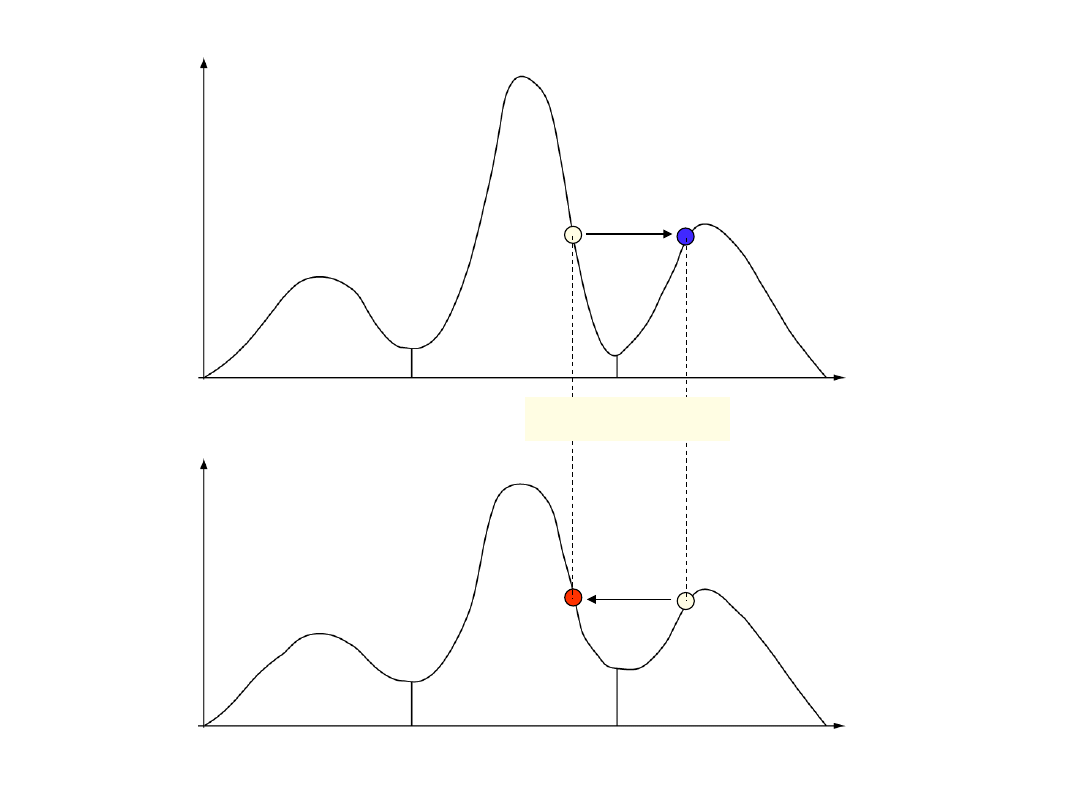
łańcuch zimny
łańcuch gorący
udana wymiana
wg. prezentacji Freda Ronquista
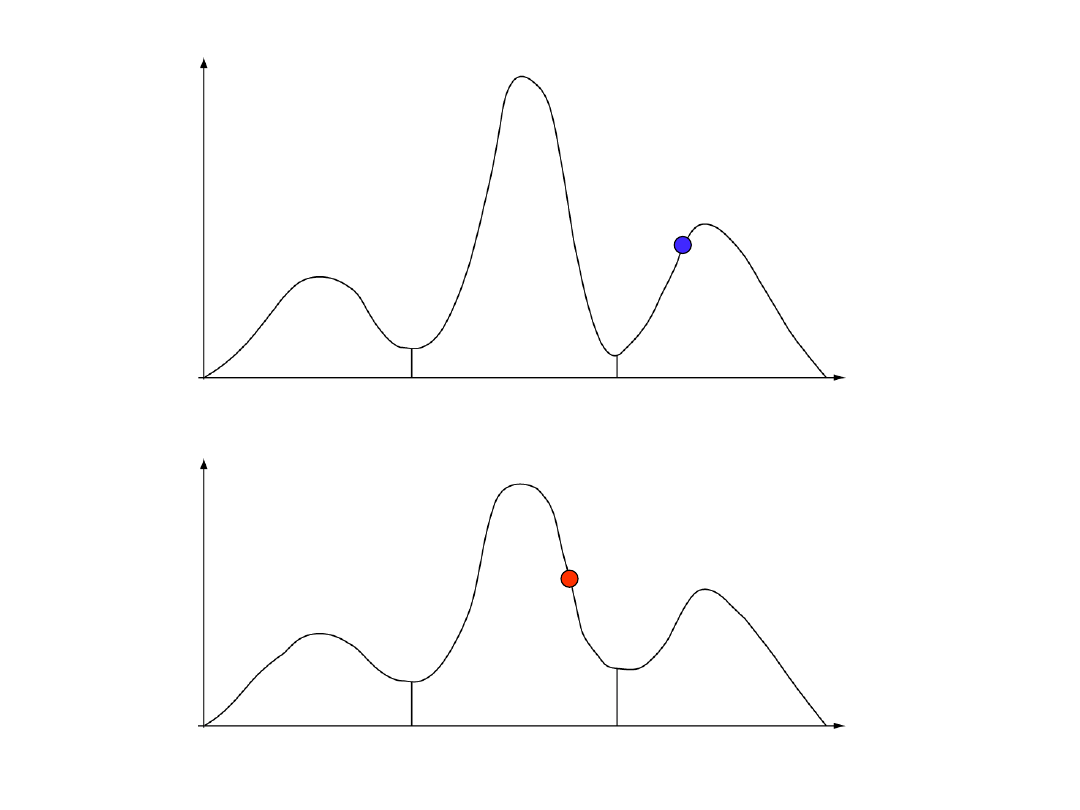
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
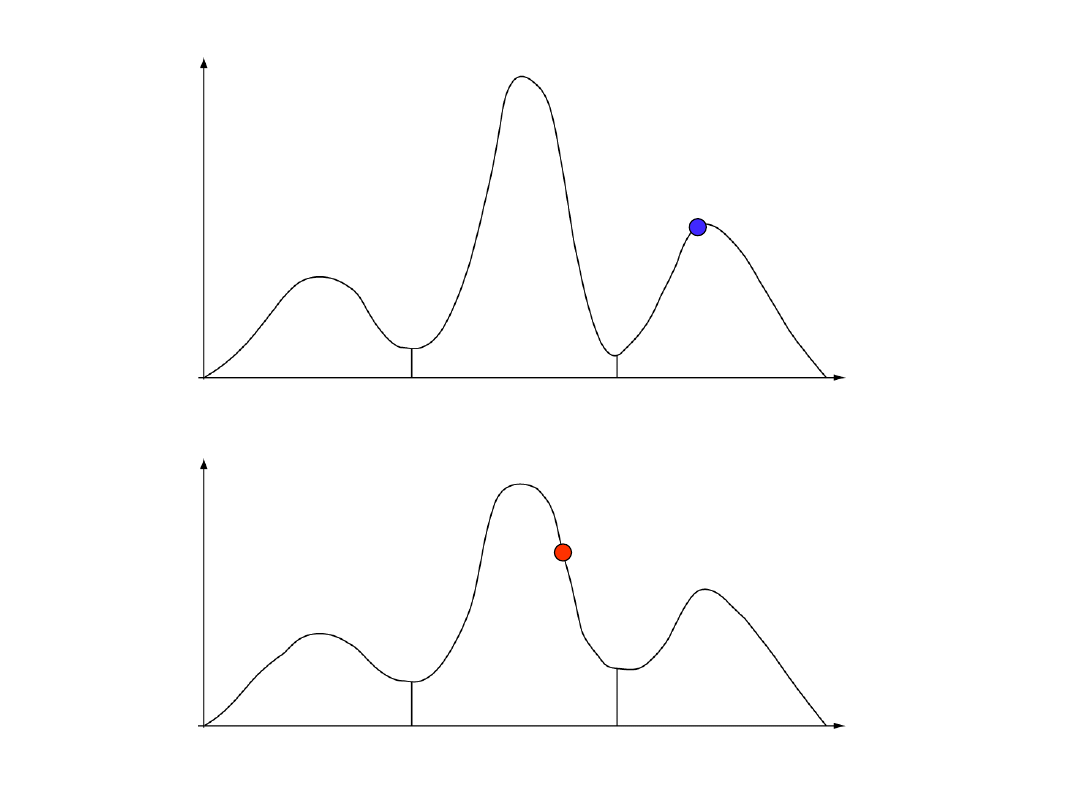
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
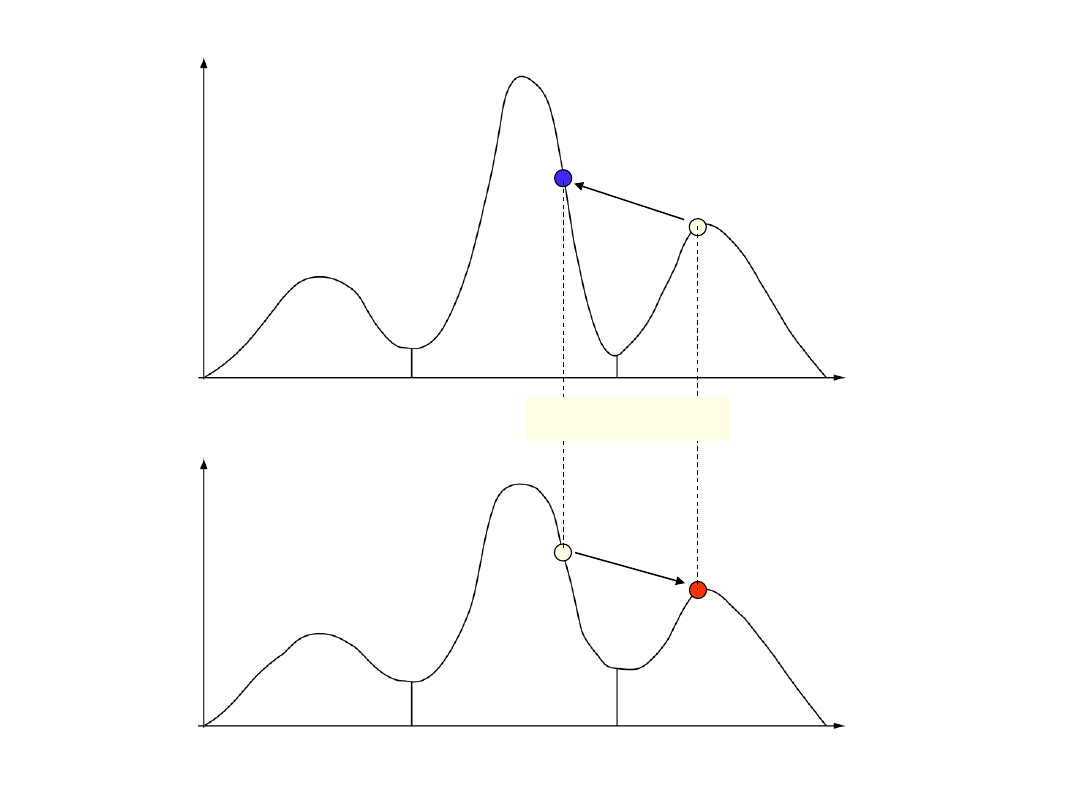
łańcuch zimny
łańcuch gorący
udana wymiana
wg. prezentacji Freda Ronquista
łańcuch zimny
łańcuch gorący
wg. prezentacji Freda Ronquista
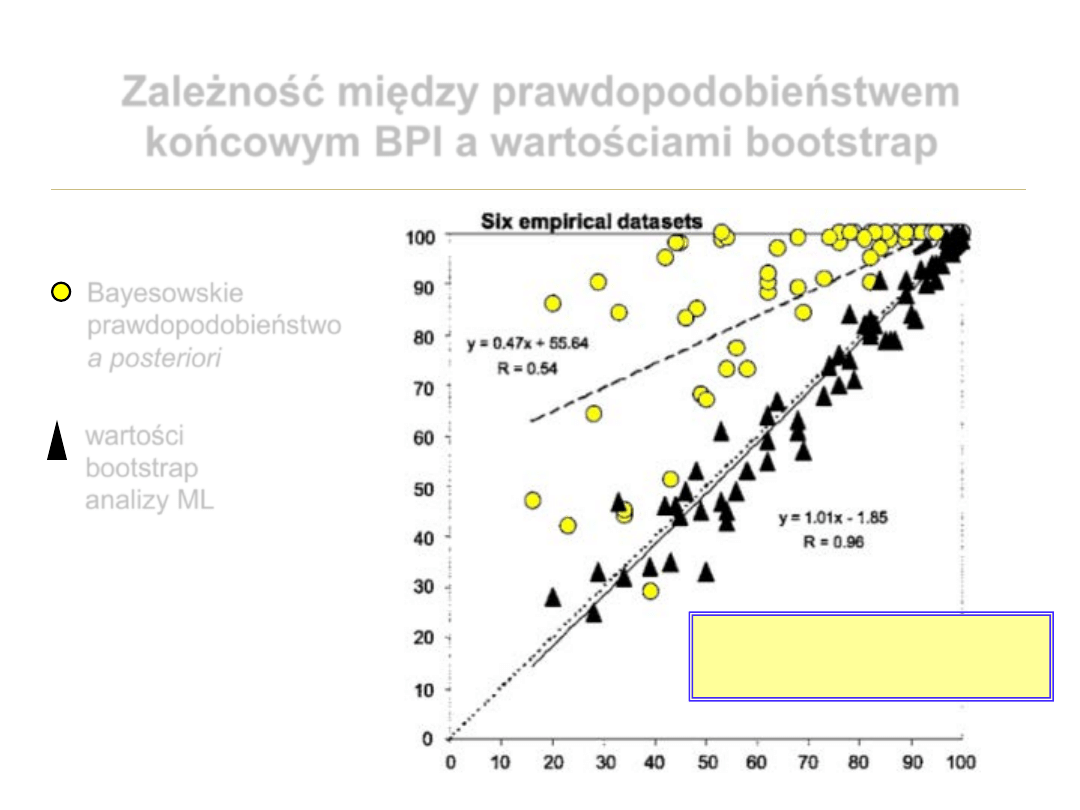
Zależność między prawdopodobieństwem
końcowym BPI a wartościami bootstrap
Bayesowskie
prawdopodobieństwo
a posteriori
wartości
bootstrap
analizy ML
wg. Douady et al. (2003)
95% BPI
≈ 70% boot.
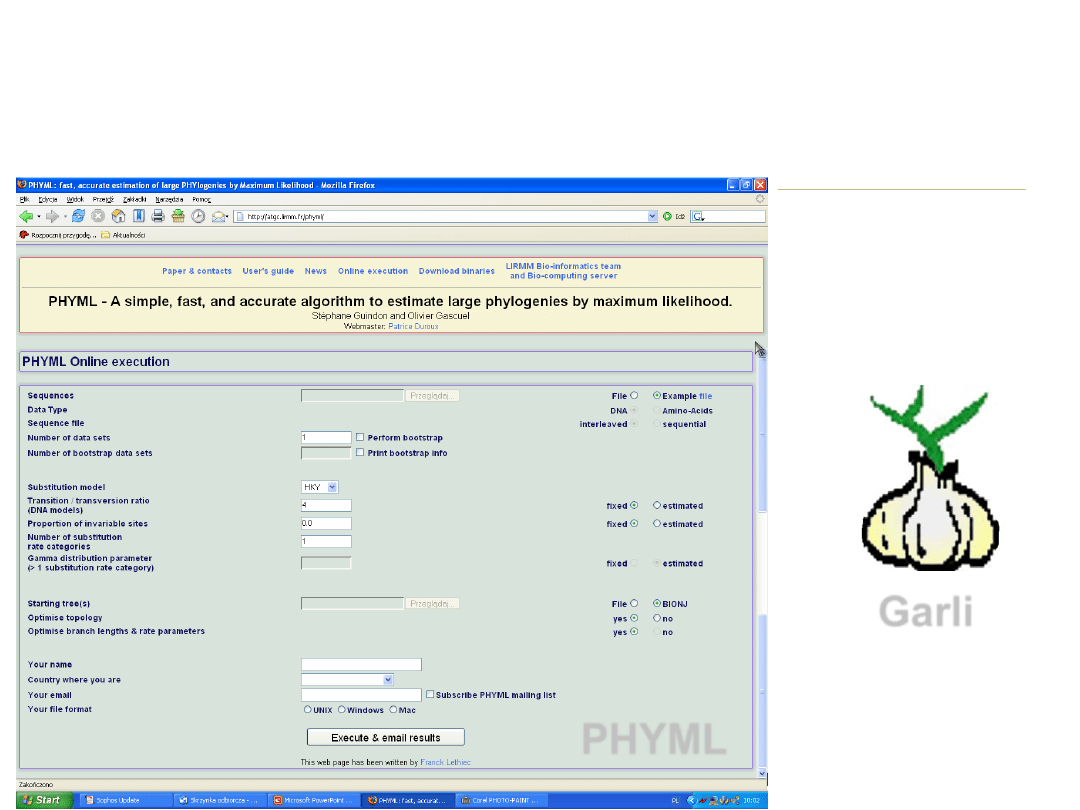
• Maximum Likelihood
PHYML
Garli
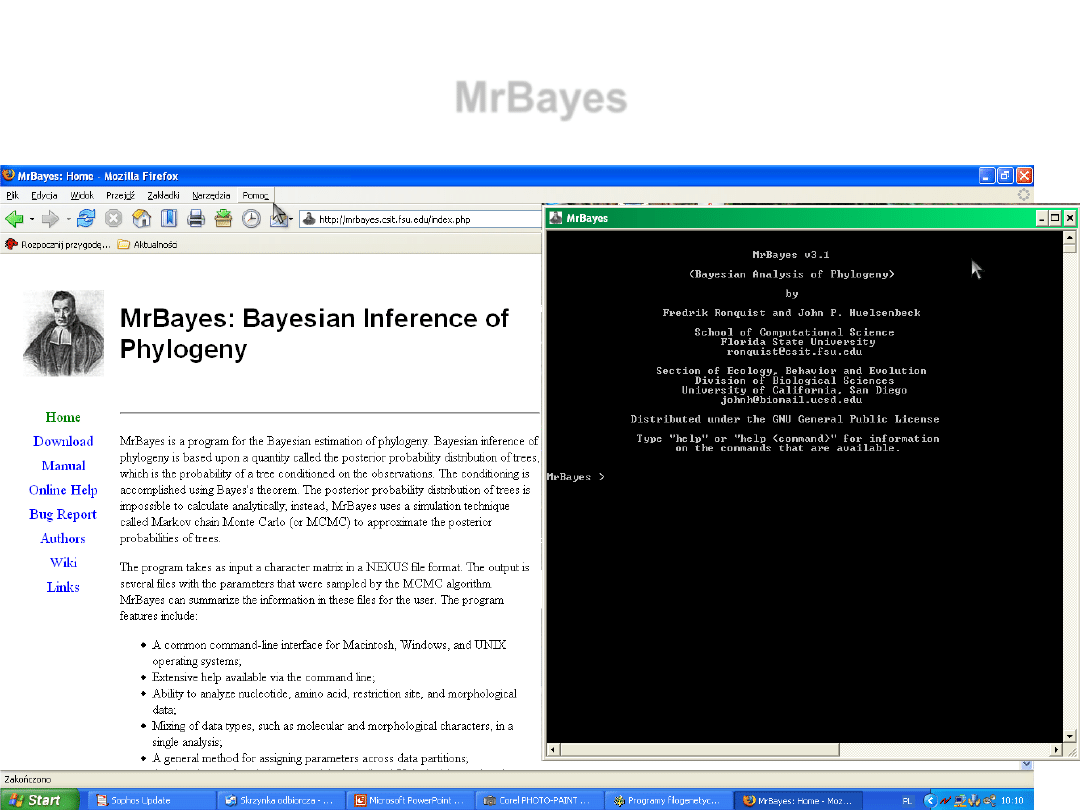
MrBayes
• Wnioskowanie
Bayesowskie
MrBayes
Kalkulacja modeli substytucji
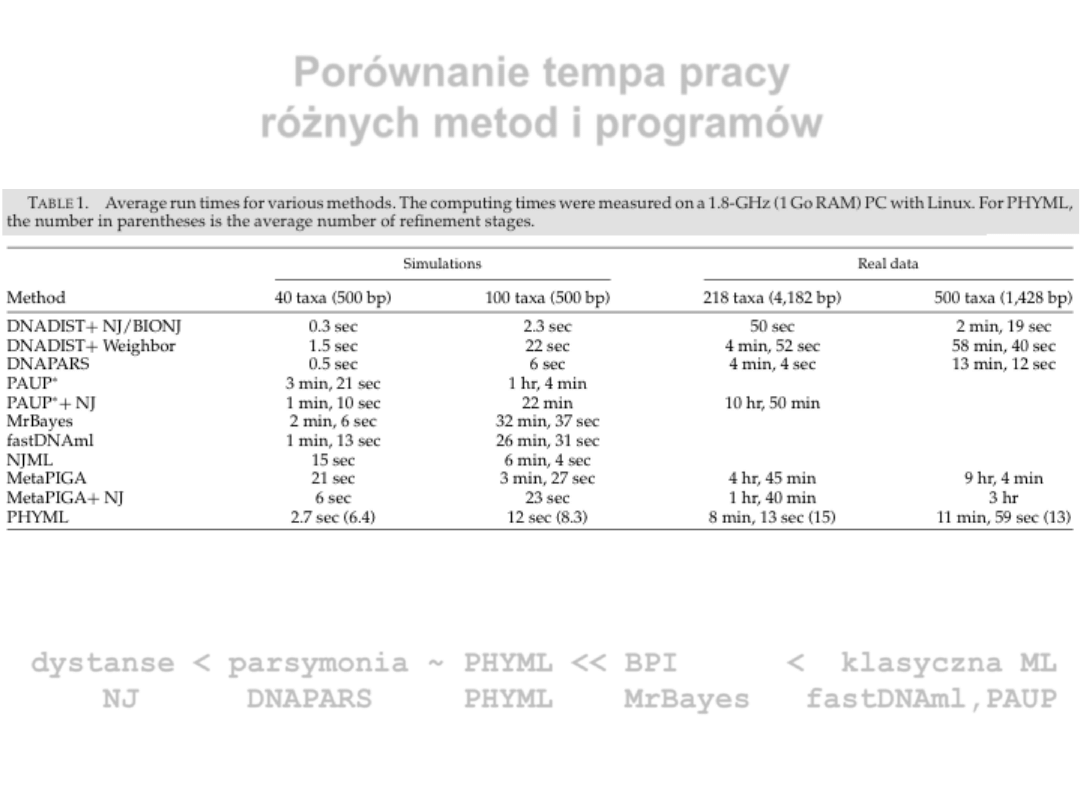
Porównanie tempa pracy
różnych metod i programów
dystanse < parsymonia ~ PHYML << BPI < klasyczna ML
NJ DNAPARS PHYML MrBayes fastDNAml,PAUP
Document Outline
- Slajd numer 1
- Slajd numer 2
- Metody wykrywania LBA
- Jak uniknąć wpadnięcia w „strefę Felsensteina”
- Modele ewolucji sekwencji
- Modele ewolucji sekwencji
- Modele ewolucji sekwencji
- Metody wiarygodnościowe
- Metody wiarygodnościowe
- Metoda maksymalnej wiarygodności (Maximum Likelihood, ML)
- Jak działa ML?
- Przykładowa analiza MJ dla przypadku 4 taksonów
- Przykładowa analiza MJ dla przypadku 4 taksonów
- Przykładowa analiza MJ dla przypadku 4 taksonów
- Przykładowa analiza MJ dla przypadku 4 taksonów
- Obliczanie prawdopodobieństwa końcowego dla danej topologii drzewa
- Wnioskowanie bayesowskie (BPI, Bayesian inference)
- Wnioskowanie bayesowskie
- Wnioskowanie bayesowskie
- Wnioskowanie bayesowskie
- Zastosowanie formuły Bayesa do rekonstrukcji filogenii
- Łańcuch Markowa Monte Carlo (MCMC)
- Łańcuch Markowa Monte Carlo (MCMC)
- Łańcuch Markowa Monte Carlo (MCMC)
- Powiązany MCMC Metropolisa (Metropolis Coupled MCMC, MCMCMC, (MC)
- Slajd numer 26
- Slajd numer 27
- Slajd numer 28
- Slajd numer 29
- Slajd numer 30
- Slajd numer 31
- Slajd numer 32
- Slajd numer 33
- Slajd numer 34
- Slajd numer 35
- Slajd numer 36
- Zależność między prawdopodobieństwem końcowym BPI a wartościami bootstrap
- PHYML
- MrBayes
- MrBayes
- Slajd numer 41
- Kalkulacja modeli substytucji
- Porównanie tempa pracy różnych metod i programów
Wyszukiwarka
Podobne podstrony:
06 metody diagnostyczne(1)
06 Metody wyznaczania pol powierzchni
06 Metody separacji enancjomerów
06 metody dodatkowe
06 Metody zabezpieczania przed korozją, BHP, bhp, 05 03 2011
06 METODY NACIAGU ŚRUB I II Anex G 1591
06 Metody separacji enancjomerów
2008 06 Java Microedition – metody integracji aplikacji [Inzynieria Oprogramowania]
Cichy B Metody numeryczne, mn 06
metodyka pracy opeikunczo wychowaczej, Metodyka pracy opiekuńczo - wychowawczej 06.12.2008
Metody numeryczne PDF, MN mnk1 06
06 EW ZEW Metody i narzędzia w ewaluacji
Projekt numeryczny, IŚ Tokarzewski 27.06.2016, III semestr, Informatyka (Matlab), Projekty, Matlab -
2008 Metody obliczeniowe 06 D 2008 10 22 20 13 23
Rosja nie jest wiarygodna w tym śledztwie(rozmowa z Mariuszem Pilisem), Katyń - Smoleńsk 2010, Katyń
Metodyka WF studia I stopnia wyklad 06
więcej podobnych podstron