
Arkadiusz Ptak
Instytut Fizyki
Politechnika Poznańska
Podstawy nanotechnologii
Modelowanie i symulacje
molekularne

Modelowanie i symulacje molekularne
1.
Wprowadzenie
2.
Metody mechaniki molekularnej (klasycznej)
3.
Metody mechaniki kwantowej
•
metody ab-initio
•
metody półempiryczne
•
Metody DFT
PLAN

1. Wprowadzenie
a.
Czym s
ą
modelowanie molekularne i symulacje
b.
Zalety i ograniczenia
c.
Schemat metodologiczny
d.
Przybli
ż
enia, rodzaje oblicze
ń
e.
Obliczeniowe wyzwania
WPROWADZENIE
Modelowanie i symulacje molekularne
WPROWADZENIE
Modelowanie i symulacje molekularne

Modelowanie molekularne (MM)
- proces tworzenia modeli cz
ą
steczek, nanostruktur lub ich układów.
Modelowanie
- proces tworzenia modeli lub ich stosowanie.
Model
- system zało
ż
e
ń
, poj
ęć
i zale
ż
no
ś
ci mi
ę
dzy nimi pozwalaj
ą
cy opisa
ć
w
przybli
ż
ony sposób jaki
ś
aspekt rzeczywisto
ś
ci.
WPROWADZENIE
WPROWADZENIE
Czym jest modelowanie?

Symulacja
- przybli
ż
one odtwarzanie zjawiska lub zachowania danego obiektu za
pomoc
ą
jego modelu.
Symulacja molekularna jest rodzajem eksperymentu komputerowego,
którego przedmiotem s
ą
teoretyczne modele struktury i własno
ś
ci
cz
ą
steczek lub układów ponadcz
ą
steczkowych.
Symulacja jest implementacj
ą
modelu w czasie; pokazuje zachowanie
modelu.
WPROWADZENIE
WPROWADZENIE
Czym s
ą
symulacje molekularne?

Metody MiSM wykorzystuje si
ę
w:
- fizyce molekularnej (cz
ą
steczki)
- chemii obliczeniowej (cz
ą
steczki)
- biochemii i biologii obliczeniowej (biocz
ą
steczki)
- nanotechnologii (nanostruktury)
Czym s
ą
modelowanie i symulacje molekularne?
WPROWADZENIE
Czym s
ą
modelowanie i symulacje molekularne?
WPROWADZENIE

Ograniczenia:
• weryfikacja (przez falsyfikacj
ę
) – porównanie z danymi eksperymentalnymi;
je
ś
li brak, to sposoby po
ś
rednie:
- dane dla podobnych układów molekularnych (nanostruktur)
- stosowanie wielu metod MiSM
- poleganie na u
ż
yteczno
ś
ci wykorzystywanych w modelach teorii
(znajomo
ść
zało
ż
e
ń
i ogranicze
ń
)
• ograniczenia obliczeniowe, w tym sprz
ę
towe
Zalety i wykorzystanie:
• dodatek do istniej
ą
cych lub uzyskiwanych danych eksperymentalnych
• zdolno
ść
przewidywania wła
ś
ciwo
ś
ci nanoostruktur przed wykonaniem
eksperymentów (cz
ę
sto kosztownych)
• zdolno
ść
przewidywania wła
ś
ciwo
ś
ci nanoostruktur jeszcze nie wytworzonych
• główne
ź
ródło wiedzy w przypadku braku jakichkolwiek danych eksperymentalnych
(np. gdy konieczno
ść
wytworzenia ekstremalnych warunków eksperymentu: bardzo
wysoka lub niska temperatura, UHV, niewa
ż
ko
ść
itp..)
Zalety i ograniczenia
WPROWADZENIE
Zalety i ograniczenia MiSM
WPROWADZENIE
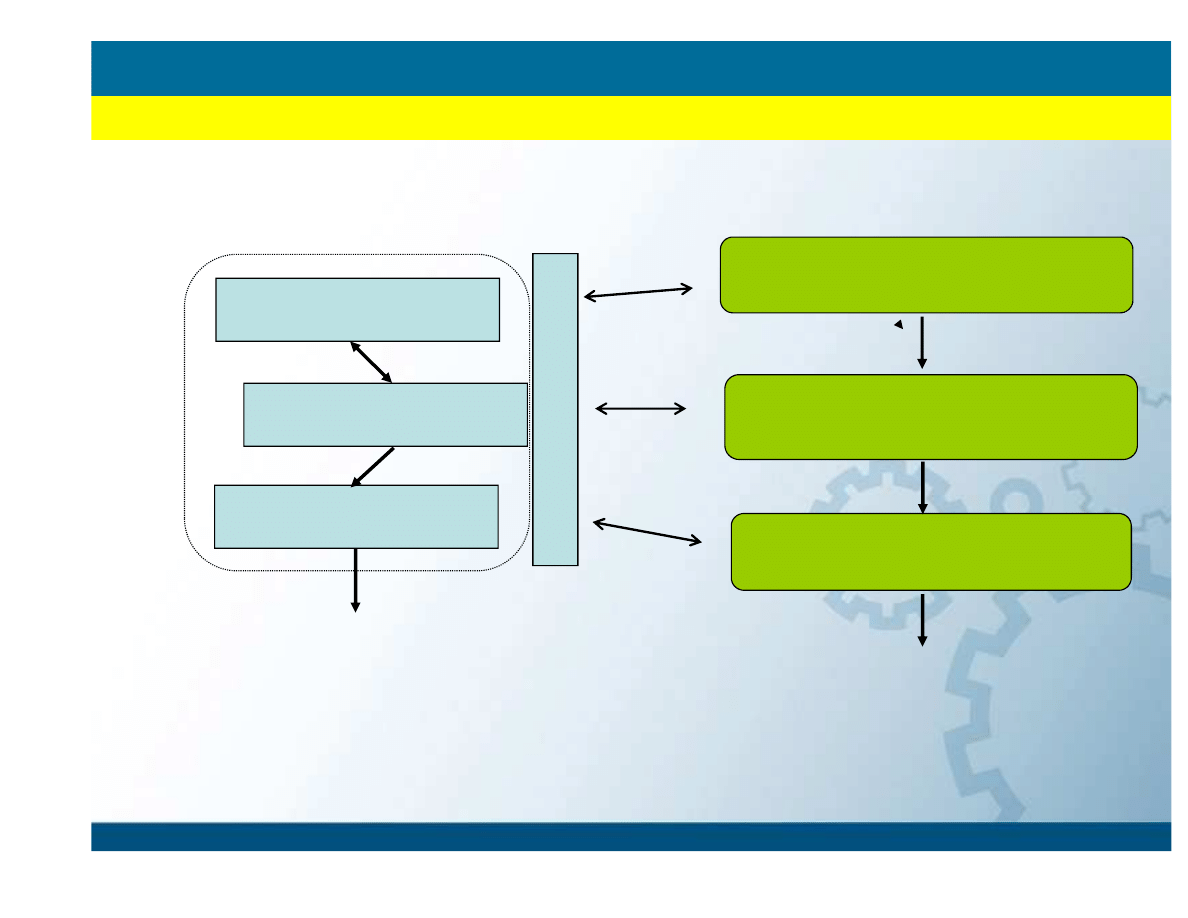
Schemat blokowy:
Visualization Tool
Compute Engine
Analysis Tool
U
se
r
in
te
rf
a
ce
Budowa wst
ę
pnego modelu
molekuły
Wybór silnika obliczeniowego
(rodzaju modelu teoretycznego)
Analiza obliczonych wła
ś
ciwo
ś
ci
Projekt MiSM
WPROWADZENIE

Dla wszystkich metod:
Przestrze
ń
homogeniczna (jednorodna) i izotropowa
Dla metod kwantowych:
Przybli
ż
enie Borna-Oppenheimera:
ruch j
ą
der atomowych znacznie wolniejszy ni
ż
elektronów
Szczegółowe zało
ż
enia dla konkretnych metod MiSM
(np. wprowadzenie dodatkowych pól fizycznych)
Zało
ż
enia ogólne
WPROWADZENIE
Zało
ż
enia ogólne
WPROWADZENIE
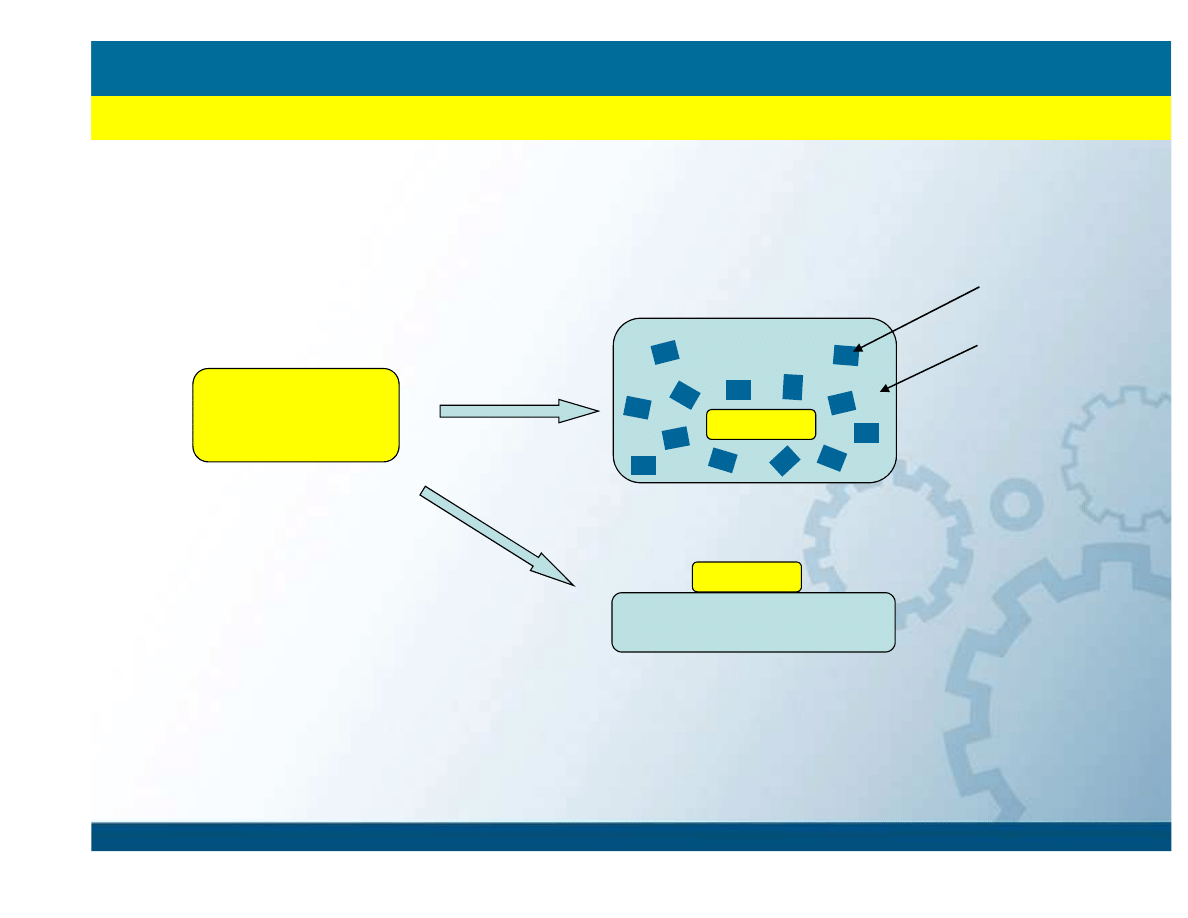
Molecule
in vacuo
Liquid
Metodologia
WPROWADZENIE
Solid
Metodologia
WPROWADZENIE
Otoczenie
explicite
continuous
phase
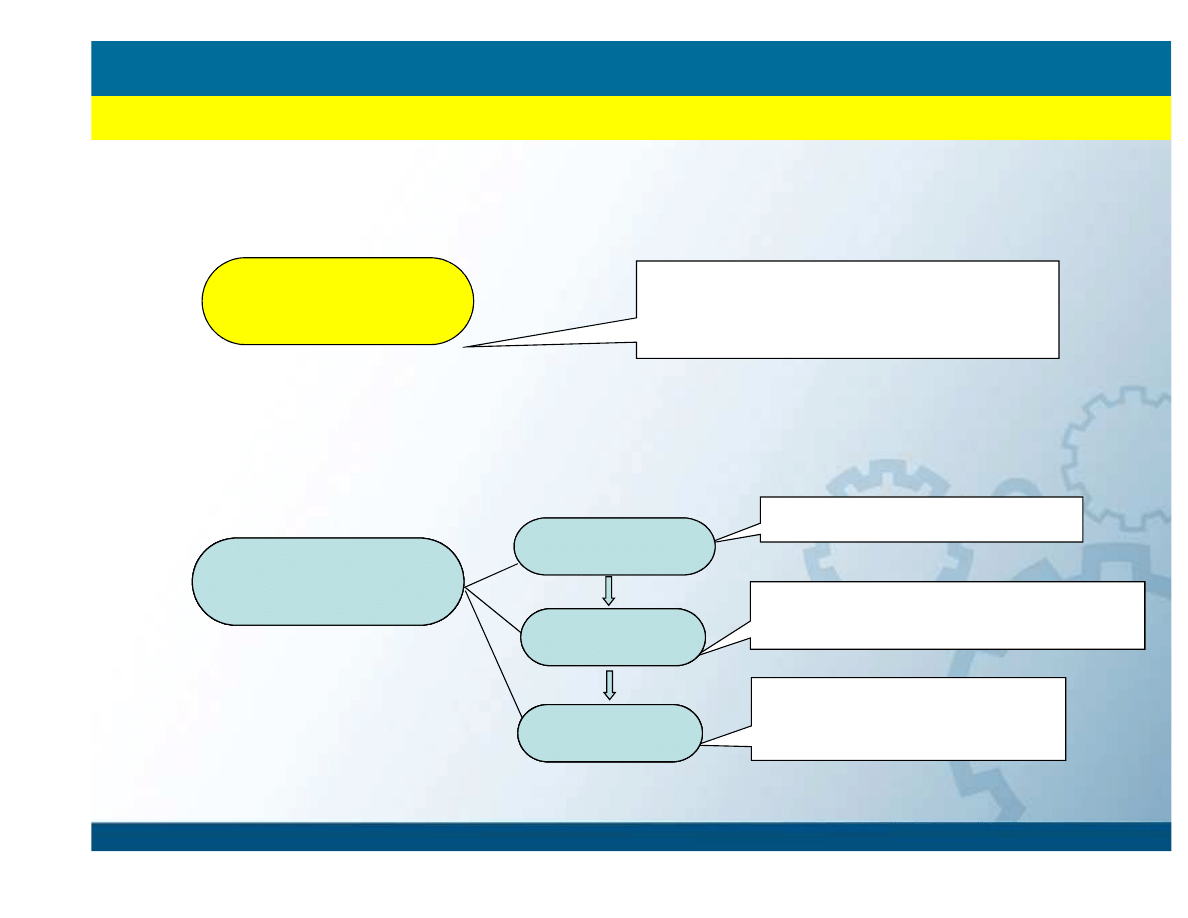
Classical physics
Quantum physics
Potential functions; Only effects related to
the arrangement of atomic nuclei;
not optical spectra for instance.
Ab initio
DFT
Semi-empirical
Self-consistent field (SCF)
or
Hartree-Fock calculations
Density Functional Theory
- based on some empirical knowledge
Based on empirical knowledge
Quantum physics
Ab initio
DFT
Quantum physics
Ab initio
Semi-empirical
DFT
Quantum physics
Ab initio
Metodologia
WPROWADZENIE
Teoria
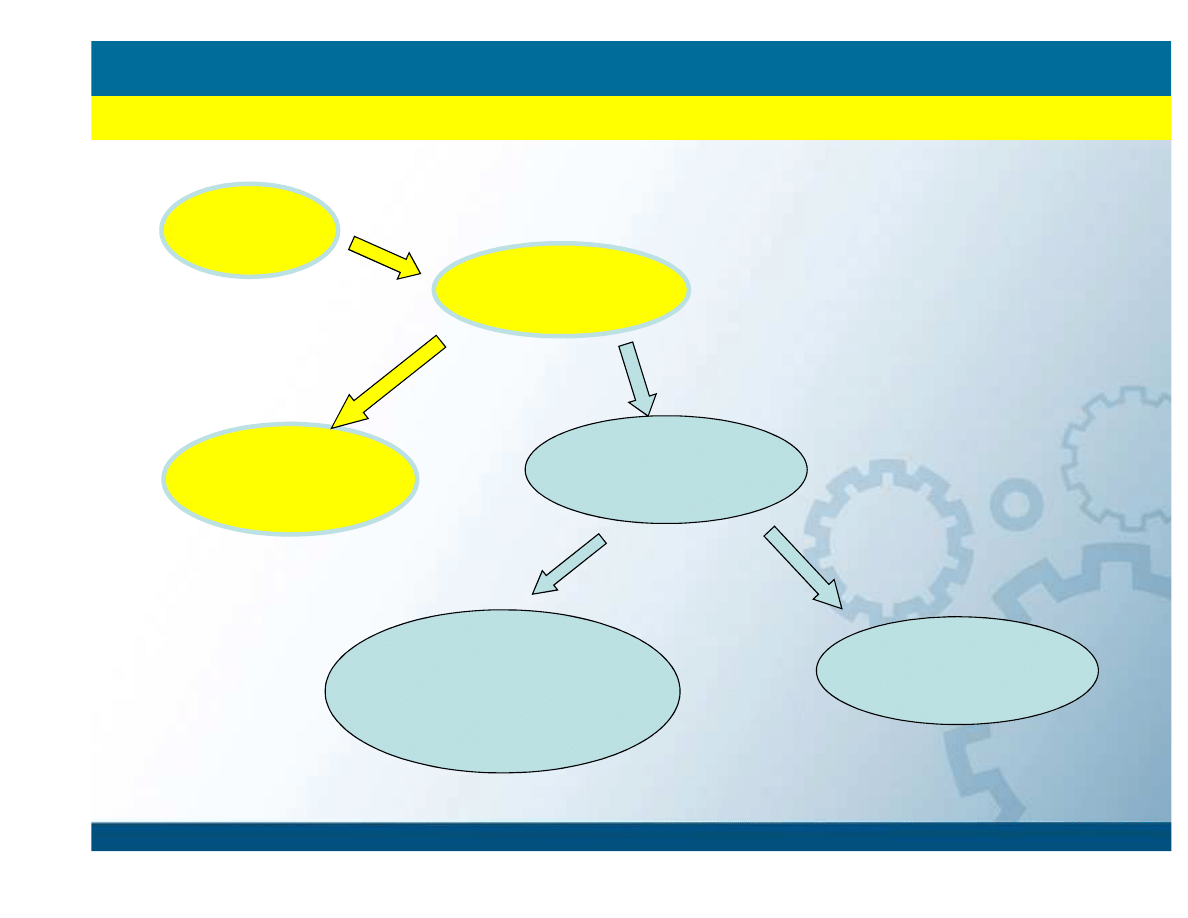
Geometry
optimization
Molecular
Dynamics
Calculation of orbitals
Simulation of spectra
Metodologia
WPROWADZENIE
Density of charge
Density of spin
Electrostatic potentials
Rodzaje oblicze
ń
Single Point
Metodologia
WPROWADZENIE
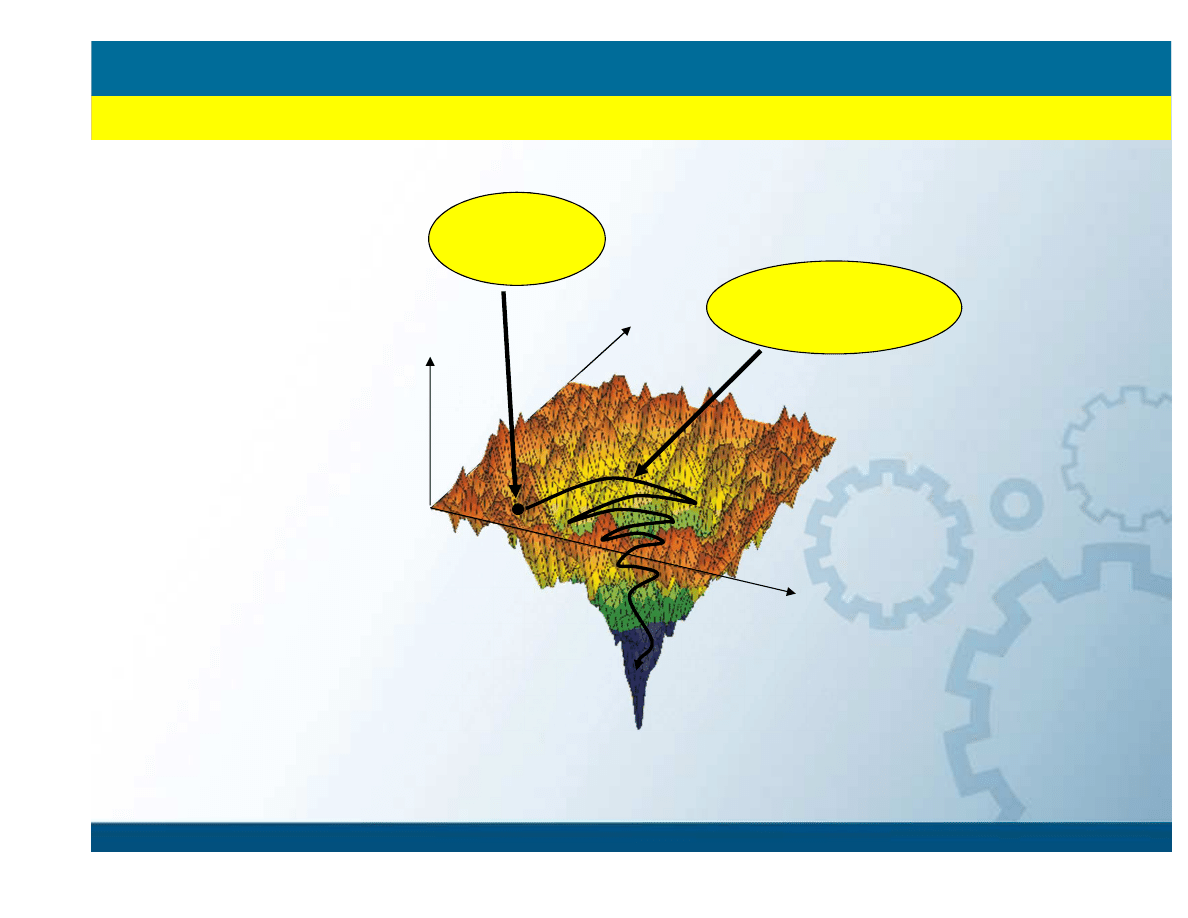
Energy landscape
x
y
E
Single Point
Geometry
optimization

Architektura komputerowa:
• wieloprocesorowe superkomputery (Cray, IBM)
• stacje robocze (Silicon Graphics, Sun, HP, IBM)
• klastery komputerowe (poł
ą
czone w sieci, grid)
• pojedyncze PC
Dwa aspekty:
• obliczenia
• wizualizacja
Hardware
WPROWADZENIE
Hardware
WPROWADZENIE
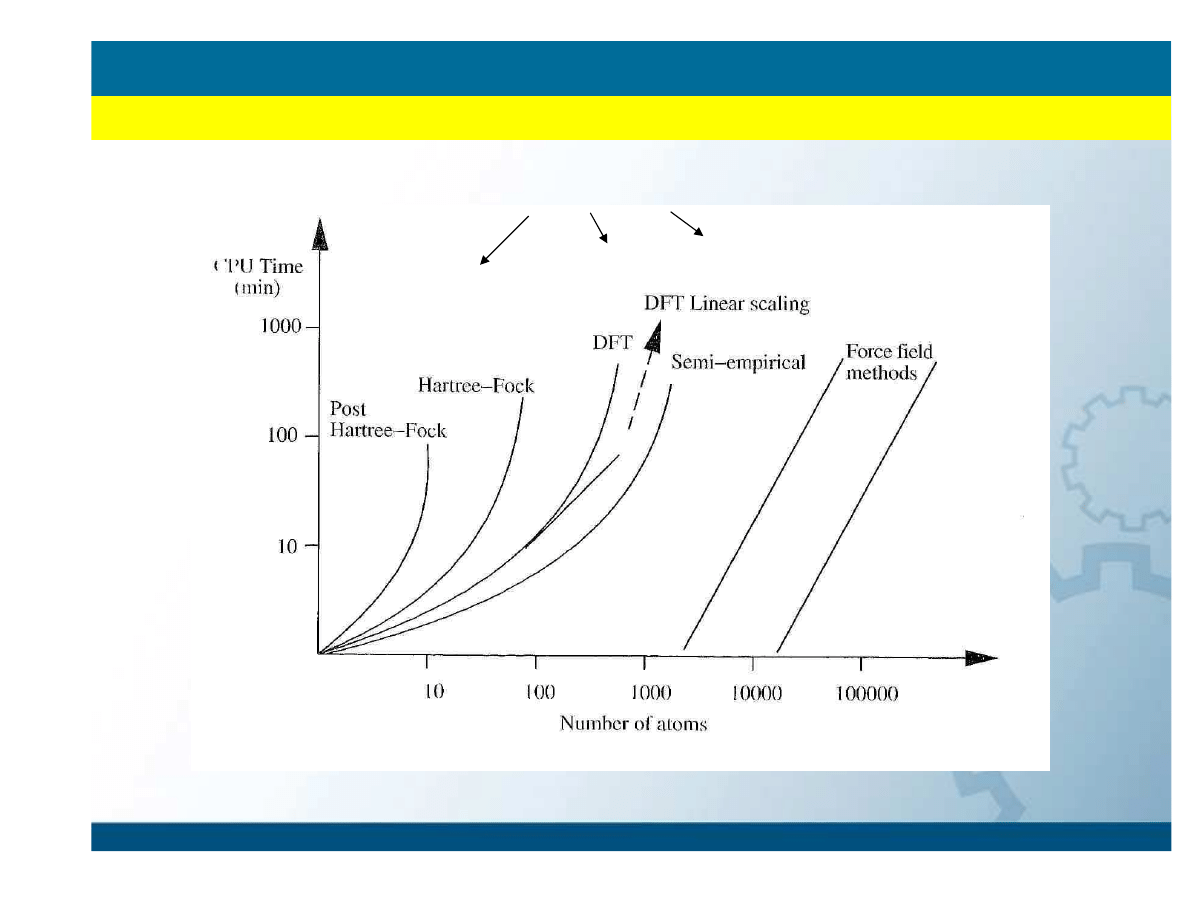
Metody klasyczne
Metody
półempiryczne
Ab initio
Ograniczenia sprz
ę
towe
WPROWADZENIE
DFT
Metody kwantowe
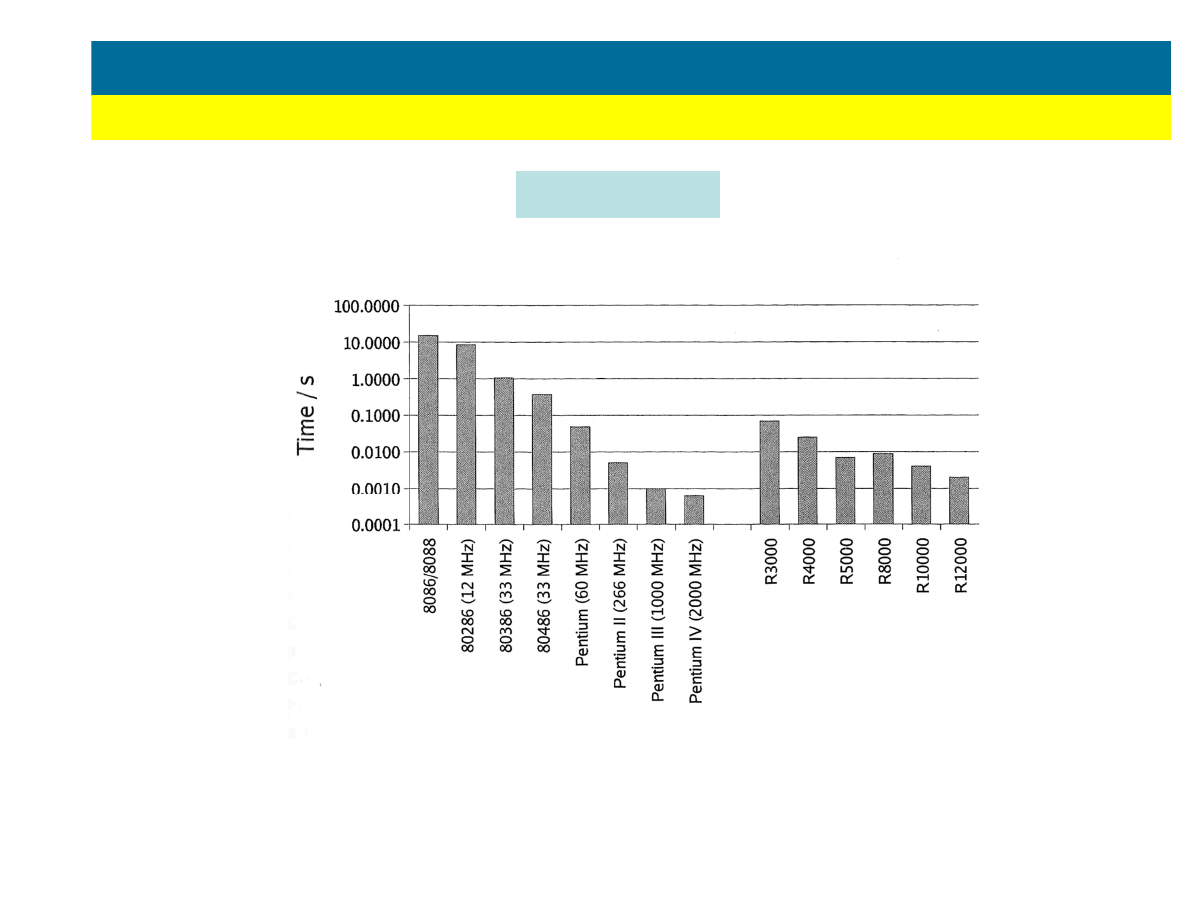
Post
ę
p
Intel
MIPS
WPROWADZENIE
Ograniczenia sprz
ę
towe

Przykłady:
• Insight II
• Gaussian
• NWChem
• SPARTAN
• HyperChem
• CACHe
• Materials Studio
• MAPS
• MaterialsExplorer
• NAMD
• Arguslab
Oprogramowanie
WPROWADZENIE

METODY
MECHANIKI KLASYCZNEJ

METODY MECHANIKI KLASYCZNEJ
1.
Mechanika molekularna
a.
Okre
ś
lenie modelu
b.
Potencjały oddziaływania
•
Potencjały wi
ą
za
ń
atomowych
•
Potencjały k
ą
tów wi
ą
za
ń
•
Potencjały torsyjne
•
Potencjały odziaływa
ń
van der Waalsa
•
Potencjał wi
ą
za
ń
wodorowych
•
Potencjał oddziaływa
ń
elektrostatycznych
c.
Optymalizacja geometrii (energii)
2.
Podstawy u
ż
ytkowania programów MM&S
a.
Budowanie cz
ą
steczek
b.
Optymalizacja struktury
Plan
METODY MECHANIKI KLASYCZNEJ
Plan
Model i Energia
• Cz
ą
steczka jest zespołem oddziałuj
ą
cych na siebie atomów
• Oddziaływania mo
ż
na opisa
ć
za pomoc
ą
prostych funkcji analitycznych
el
bonds
H
vdW
torsion
angle
stretch
tot
E
E
E
E
E
E
E
+
+
+
+
+
=
−
METODY MECHANIKI KLASYCZNEJ
Energia całkowita:
Model i Energia
METODY MECHANIKI KLASYCZNEJ
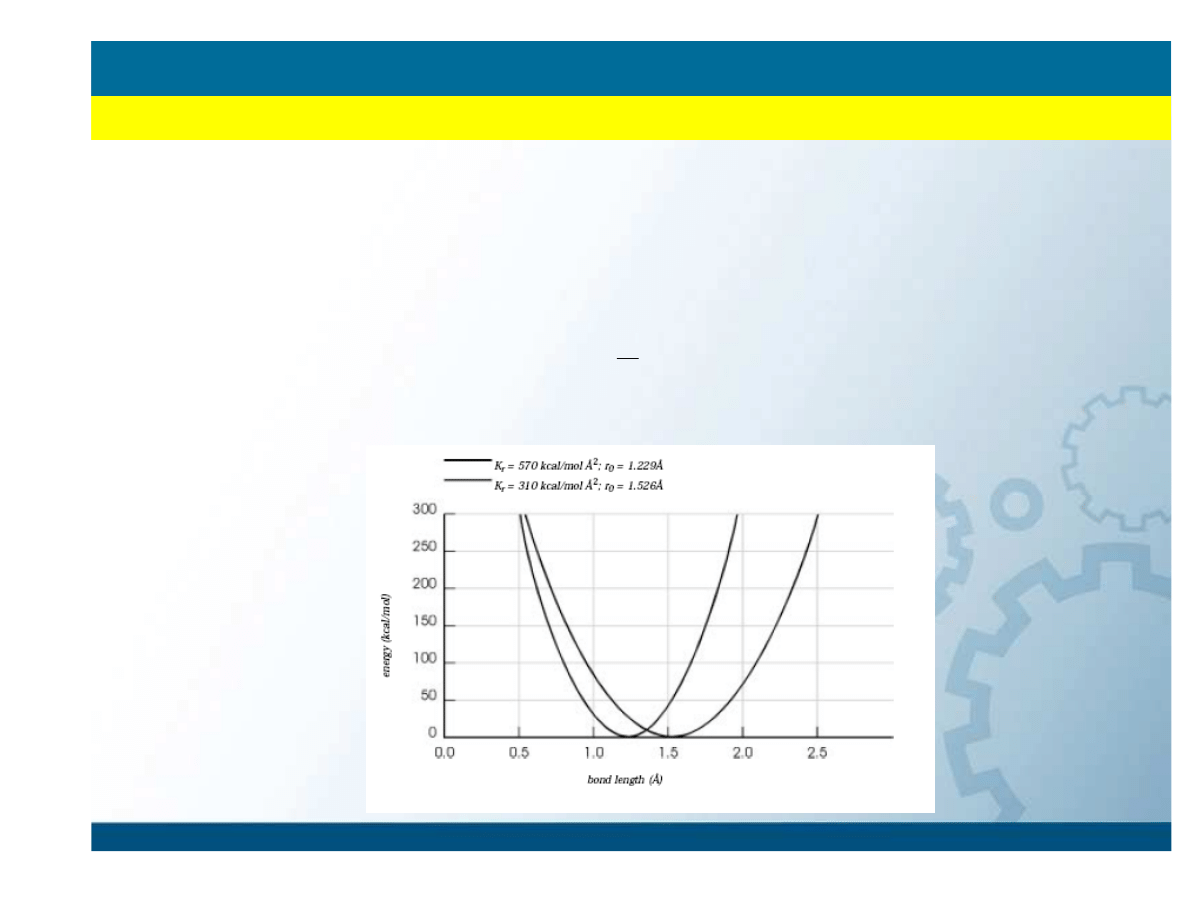
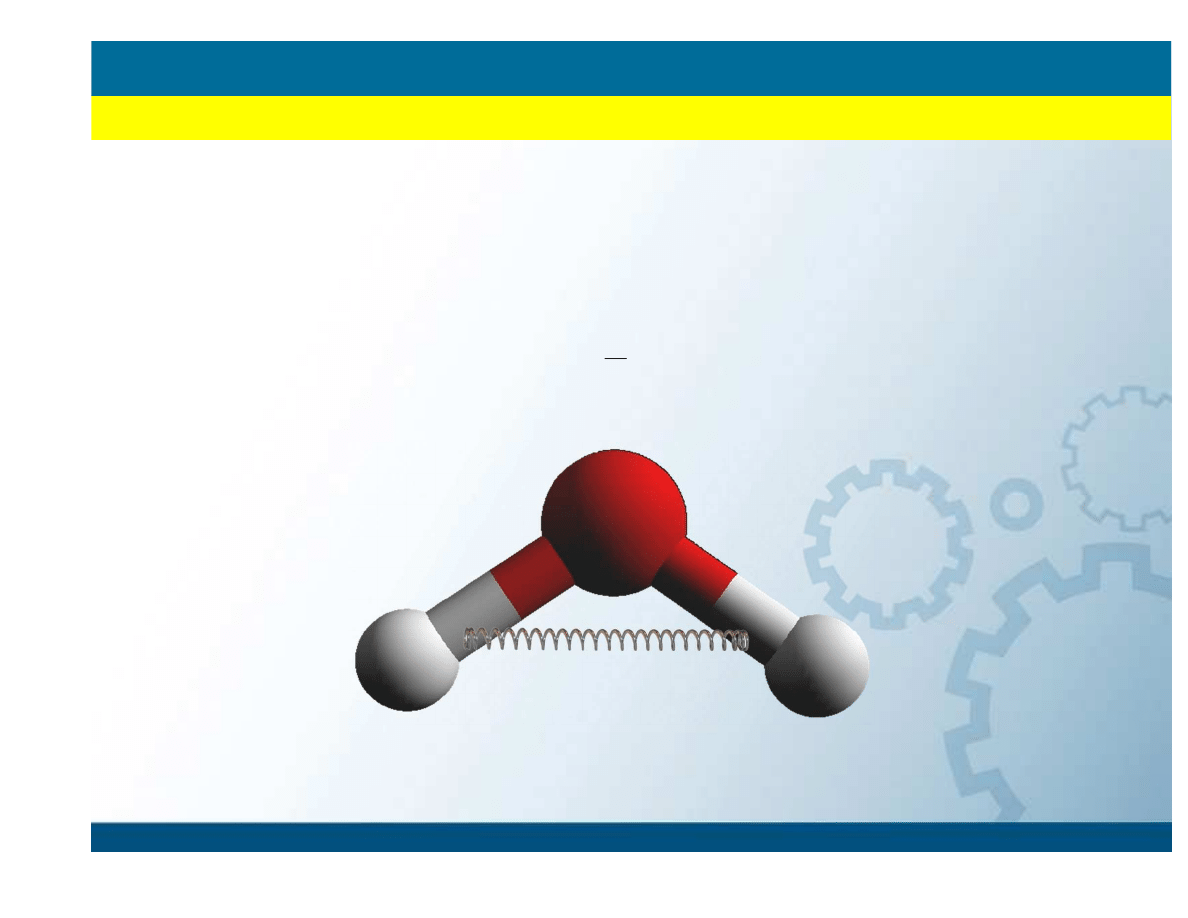
Bond stretching
(rozci
ą
ganie wi
ą
za
ń
)
∑
−
=
bonds
r
stretch
r
r
k
E
2
0
)
(
2
1
C-O
C-C
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
Bending
(zginanie)
∑
−
=
angles
bend
k
E
2
0
)
(
2
1
θ
θ
θ
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
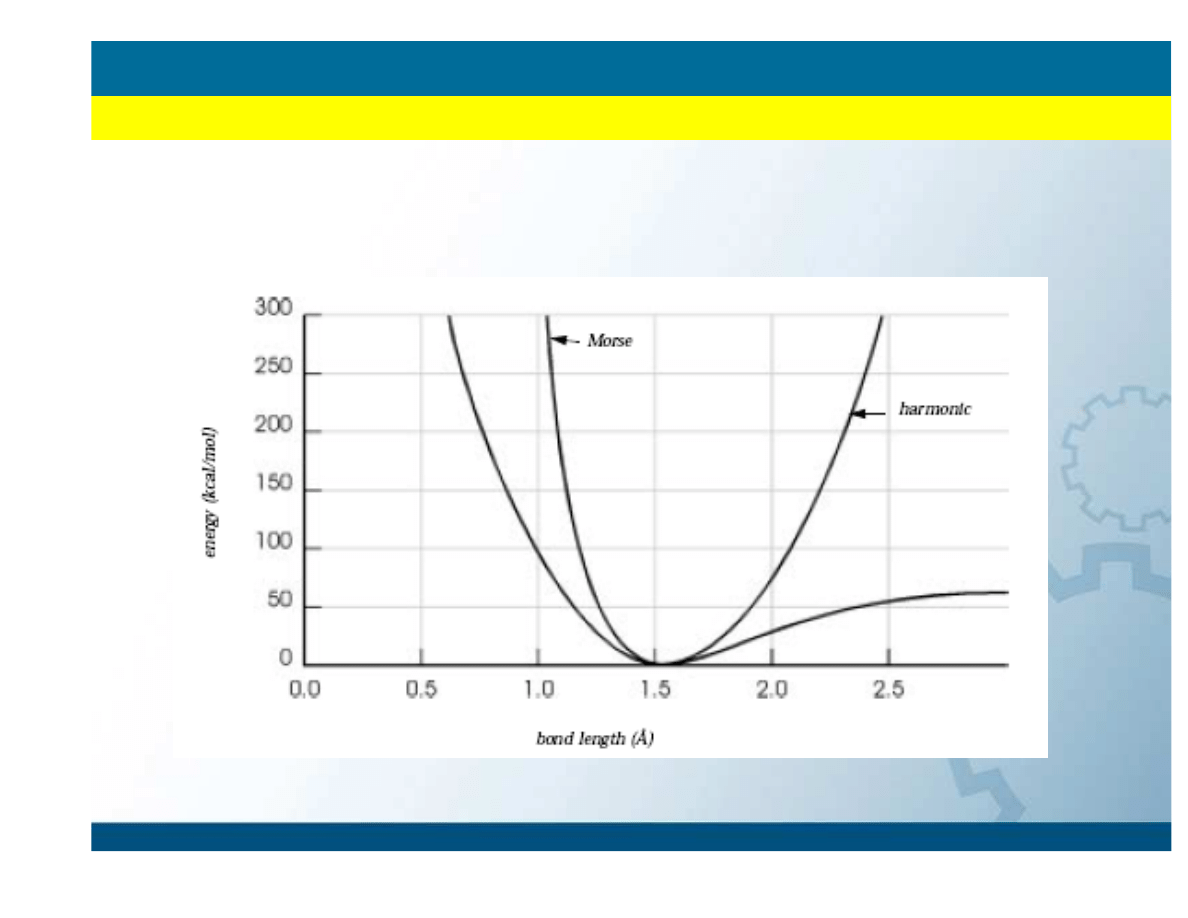
Dlaczego potencjał harmoniczny a nie Morse’a?
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
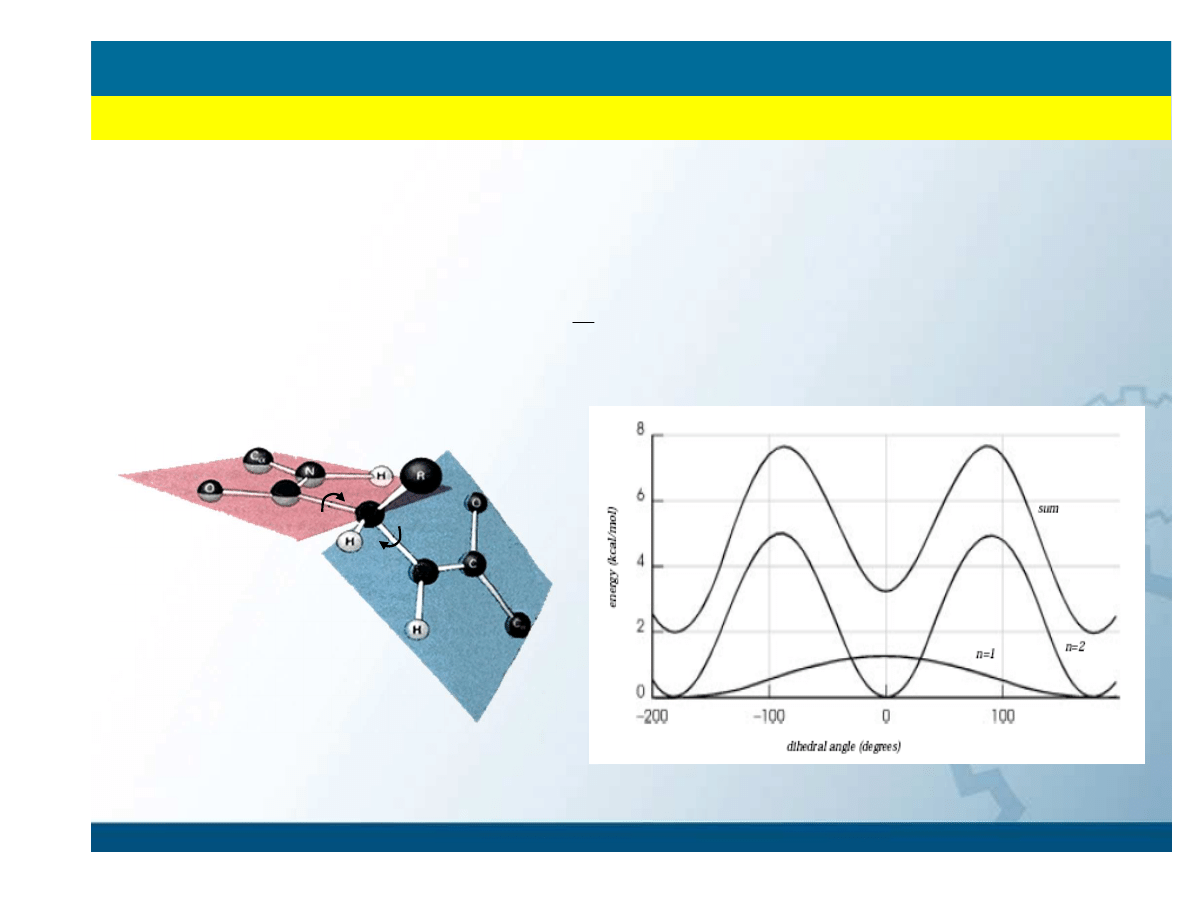
Torsions
(obroty torsyjne)
[
]
∑
Φ
−
Φ
+
=
dihedrals
n
dihedrals
n
V
E
2
0
)
cos(
1
2
1
HN-C=O
cis
trans
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
C
α
αα
α
C
N
Φ
Φ
Φ
Φ
Ψ
Ψ
Ψ
Ψ
Pola siłowe
METODY MECHANIKI KLASYCZNEJ

Van der Waals interactions
(oddziaływania vdW)
∑
<
−
=
j
i
ij
ij
ij
ij
vdW
R
B
R
A
E
6
12
Hydrogen bonding
(wi
ą
zania wodorowe)
∑
−
−
−
=
bonds
H
ij
ij
ij
ij
bonds
H
R
D
R
C
E
10
12
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
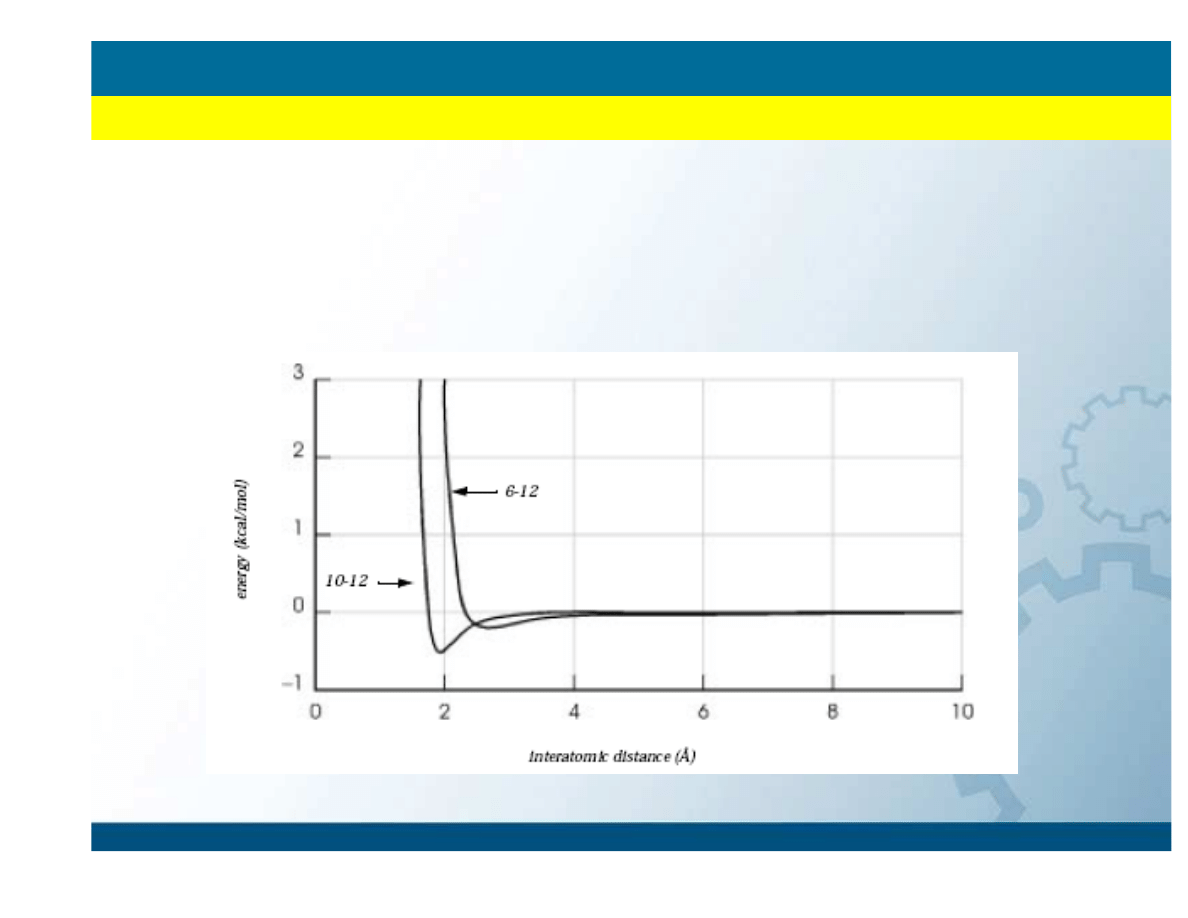
Porównanie potencjałów oddziaływa
ń
Van der Waalsa i
wi
ą
za
ń
wodorowych
Pola siłowe
METODY MECHANIKI KLASYCZNEJ
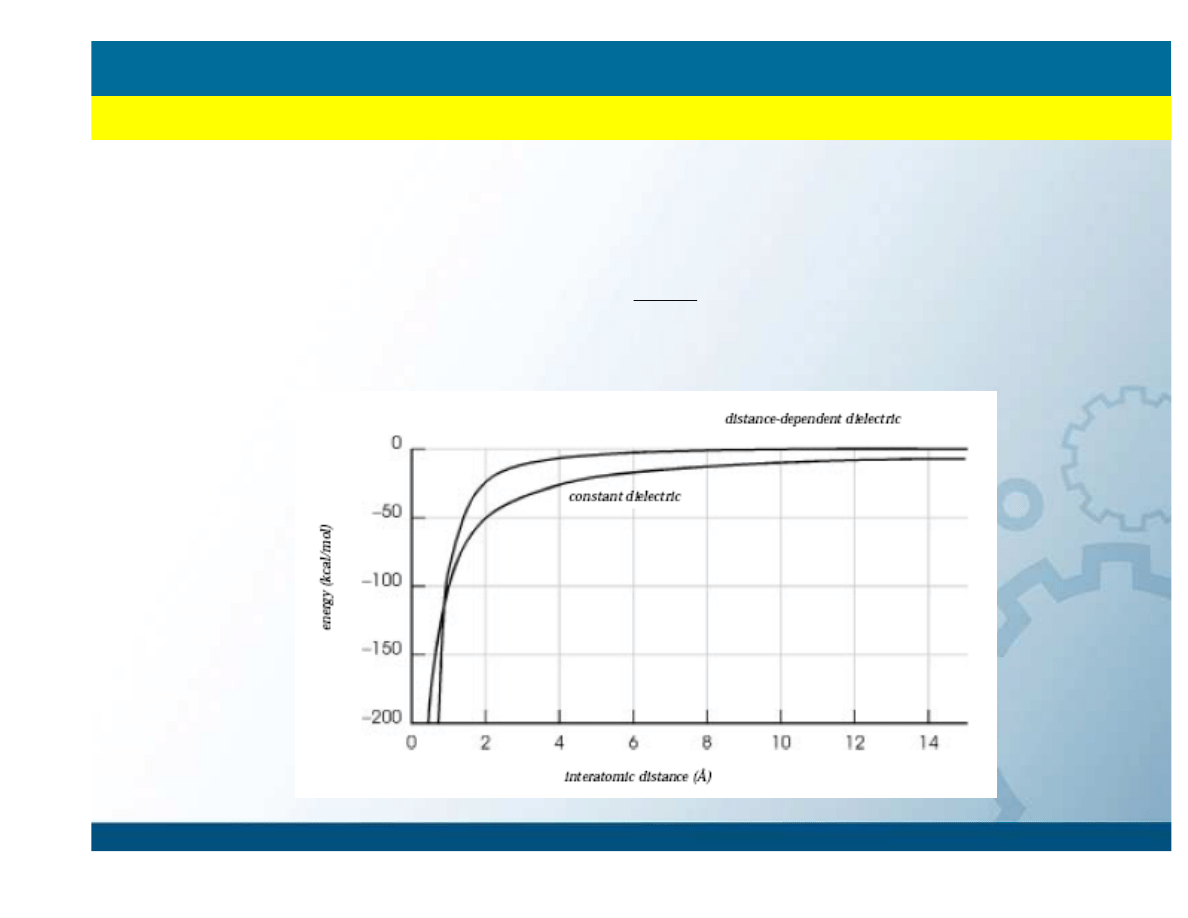
Electrostatic interactions
∑
<
=
j
i
ij
j
i
el
R
q
q
E
ε
)
(
ij
R
ε
ε
=
Pola siłowe
METODY MECHANIKI KLASYCZNEJ

Systematyczne przesuwanie wszystkich atomów cz
ą
steczki
(nanostruktury) dopóki siła wypadkowa działaj
ą
ca na ka
ż
dy atom
nie wynosi zero.
Optymalizacja geometrii
METODY MECHANIKI KLASYCZNEJ
Wyszukiwarka
Podobne podstrony:
MiSM Mech Kwant dla ETI [tryb zgodności]
Cukrzyca WL schematy dla studentów [tryb zgodności]
kora dla stud [tryb zgodnosci]
cwiczenia wiek i tempo wzrostu wyslac dla v roku ochrony tryb zgodnosci
dla stud odruchy i zmysly [tryb zgodnosci]
dla stud odruchy i zmysły [tryb zgodności]
2012 KU W5 tryb dzienny moodle tryb zgodnosci
(W7a Stale do kszta t na zimno cz I [tryb zgodno ci])
2 Sieci komputerowe 09 03 2013 [tryb zgodności]
Microsoft PowerPoint IP5 klasyfikacje tryb zgodnosci
Microsoft PowerPoint IP tryb zgodnosci
PA2 opis matematyczny [tryb zgodności]
ATMOSFERA [tryb zgodnosci]a id Nieznany
(Rachunkowosc podatkowa wyklad 4 5 [tryb zgodności])
Microsoft PowerPoint IP5 bazydanych tryb zgodnosci
więcej podobnych podstron