1
Kataliza
Kataliza - zjawisko które polega na zmianie szybko
ś
ci reakcji przez
obecne substancje, które nie zmieniaj
ą
si
ę
w czasie reakcji ani ilo
ś
ciowo
ani jako
ś
ciowo.
Katalizatory – substancje, które powoduj
ą
zmian
ę
szybko
ś
ci reakcji
chemicznej a nie s
ą
uwzgl
ę
dnione w jej równaniu stechiometrycznym
Ze wzgl
ę
du na wpływ katalizatora na szybko
ść
reakcji chemicznych
katalizatory dzielimy na:
•dodatnie – przyspieszaj
ą
ce szybko
ść
reakcji chemicznych. Ten typ katalizy
ma du
ż
e zastosowanie w przemy
ś
le chemicznym.
•ujemne (inhibitory) spowalniaj
ą
ce szybko
ść
reakcji chemicznych. Ten typ
katalizy ma du
ż
e zastosowanie w przemy
ś
le spo
ż
ywczym, do stabilizacji
produktów spo
ż
ywczych.
Podział katalizatorów ze wzgl
ę
du na stan
skupienia katalizatorów oraz substratów
reakcji :
•heterogeniczne – takie które znajduj
ą
si
ę
w innej frazie ni
ż
substraty
reakcji. Takie katalizatory działaj
ą
ce w układzie wielofazowym cz
ę
sto
nazywamy katalizatorami kontaktowymi, co ma za zadanie obrazowa
ć ż
e ich
katalityczne działanie oparte jest na zetkni
ę
ciu si
ę
reagentów z
powierzchni
ą
katalizatora.
•homogeniczne – takie które znajduj
ą
si
ę
w tej samej fazie co substraty
reakcji – taki rodzaj katalizy mo
ż
e zachodzi
ć
w fazie gazowej lub ciekłej
•Katalizatory mikrowielofazowe – katalizatory heterogeniczne o
rozdrobnieniu koloidalnym (m. innymi do tego typu katalizy zalicza si
ę
kataliza enzymatyczna).
Katalizator wpływa jedynie na szybko
ść
reakcji, nie wpływa
natomiast na stał
ą
równowagi reakcji.
Na czym polega działanie katalizatora?
zmianie energii aktywacji procesu.
Je
ś
li proces z udziałem katalizatora ma mniejsz
ą
energi
ę
aktywacji ni
ż
bez
katalizatora – katalizator przyspiesza szybko
ść
reakcji – jest katalizatorem
dodatnim.
Je
ś
li natomiast energia aktywacji reakcji z udziałem katalizatora jest
wy
ż
sza ni
ż
bez katalizatora – katalizator powoduje zmniejszenie szybko
ś
ci
reakcji – jest on inhibitorem.
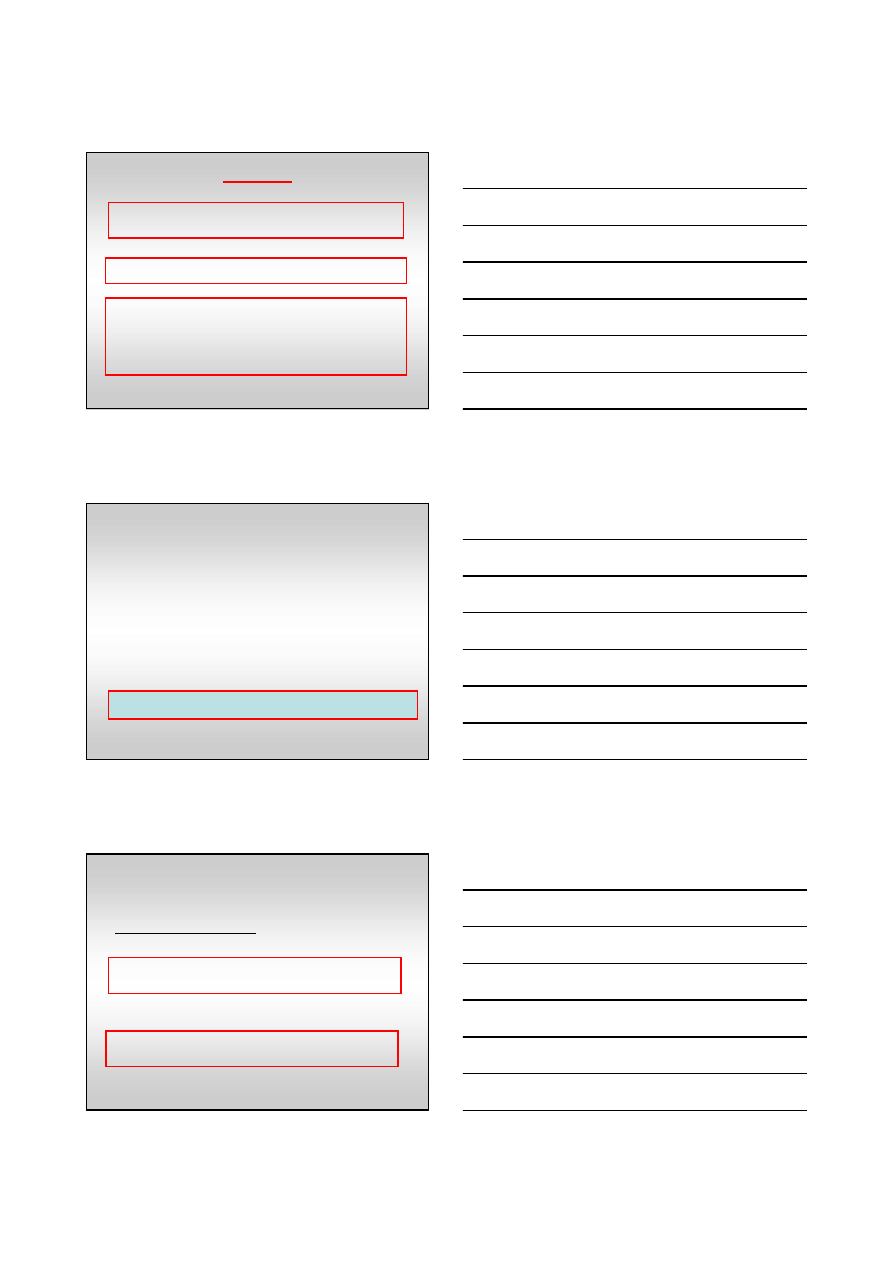
2
Reakcja
Katalizator
Energia aktywacji kJ/mol
brak
335
wolfram
163
osm
197
brak
184
platyna
105
złoto
59
brak
247
platyna
138
złoto
121
Aktywno
ść
katalizatora
Im szybciej katalizator doprowadza dan
ą
reakcj
ę
do stanu równowagi tym
jest aktywniejszy.
Aktywno
ść
katalizatora A
k
okre
ś
la si
ę
jako ró
ż
nic
ę
mi
ę
dzy szybko
ś
ciami
reakcji chemicznej zachodz
ą
cej w obecno
ś
ci katalizatora, v
k
, i bez niego, v
A
k
= v
k
– v
3
Selektywno
ść
katalizatora
zdolno
ść
katalizatora do tworzenia poszczególnych produktów i
definiuje si
ę
jako stosunek ilo
ś
ci (C
Pi
) jednego z kilku mo
ż
liwych
produktów reakcji (Pi) do całkowitej ilo
ś
ci produktów
ΣΣΣΣ
C
Pi
:
∑
=
Pi
Pi
i
C
C
S
Kataliza homogeniczna
katalizator jest w tej samej fazie co substraty
przyk
ł
ady:
•reakcja utleniania SO
2
do SO
3
wobec tlenków azotu,
•reakcja estryfikacji mieszaniny alkoholu etylowego i kwasu octowego
wobec st
ęż
onego kwasu siarkowego
•reakcja hydrolizy sacharozy wobec kwasu solnego.
Etapy analizy homogenicznej
Etap pierwszy polega na tworzeniu przez katalizator z jednym z substratów
reakcji zwi
ą
zku po
ś
redniego,
I .
S + K → SK
Etap drugi to reakcja w której utworzony w pierwszym etapie zwi
ą
zek po
ś
redni
wchodzi w reakcj
ę
z drugim substratem. Podczas tego etapu nast
ę
puje
„odtworzenie” katalizatora
II.
SK + B → SB + K
4
Zasada działania katalizatora homogenicznego
Dodatni – obni
ż
a energi
ę
aktywacji
Zmiany energii aktywacji układu w reakcji chemicznej z
udziałem katalizatora dodatniego.
kataliza kwasowo-zasadowa
. S
ą
to reakcje katalizowanych przez kwasy lub zasady.
Mechanizm takiej reakcji polega na przył
ą
czeniu protonu do cz
ą
steczki
(kataliza kwasowa) lub jego odł
ą
czeniu (kataliza zasadowa).
.Mechanizm takiej reakcji polega na przył
ą
czeniu protonu do cz
ą
steczki
(kataliza kwasowa) lub jego odł
ą
czeniu (kataliza zasadowa).
Reakcje katalizowane przez kwas
Etap I – tworzenie zwi
ą
zku po
ś
redniego przez przy
łą
czenie protonu – jest to
reakcja odwracalna
S + AH
↔
SH
+
+A
Etap II – reakcja zwi
ą
zku po
ś
redniego i powstawanie produktu reakcji.
Jednocze
ś
nie jest odbudowywany katalizuj
ą
cy reakcj
ę
kwas
SH
+
+A
-
+ S`
→
P+ AH
gdzie S, S` - substraty, P – produkt.
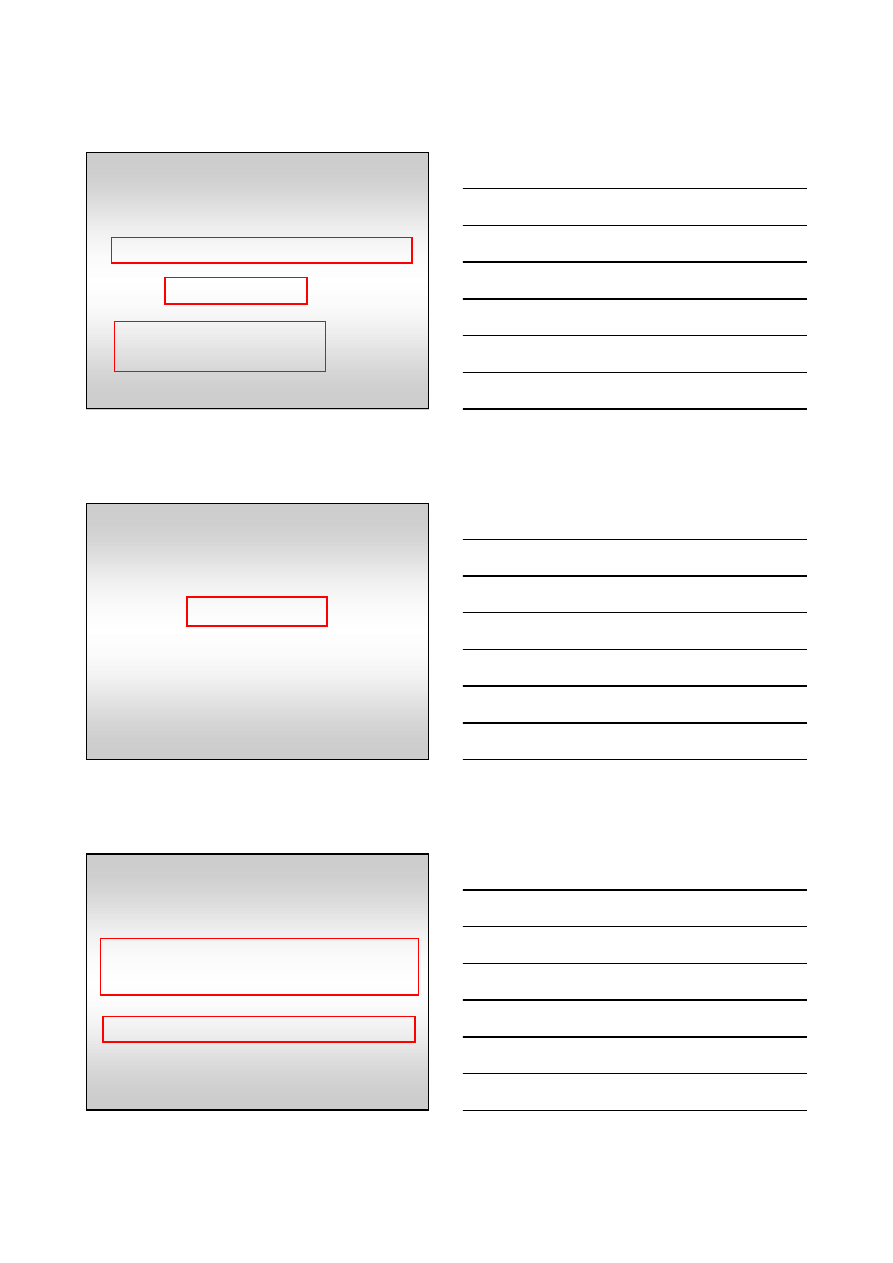
5
Szybko
ść
reakcji katalizy kwasowo zasadowej
Sta
ł
e szybko
ś
ci reakcji katalizy kwasowo- zasadowej zale
żą
liniowo od
st
ęż
e
ń
wszystkich kwasów i zasad w roztworze.
[
]
∑
+
=
i
i
n
BOH
HA
k
k
k
lub
gdzie k –szybko
ść
reakcji katalitycznej
kn – szybko
ść
reakcji przebiegaj
ą
cej bez katalizatora
ki – stała szybko
ś
ci reakcji katalizowanych przez
wszystkie kwasy lub zasady w znajduj
ą
ce si
ę
w
ś
rodowisku
reakcji.
Równanie to jest równaniem ogólnym. Szczegó
ł
owo mo
ż
na je zapisa
ć
w
postaci:
∑
∑
+
+
+
+
=
−
+
j
j
i
HA
OH
O
H
n
B
k
k
k
k
k
i
3
gdzie
- sta
ł
a szybko
ś
ci reakcji katalizowanej przez jon hydroniowy
- sta
ł
a szybko
ś
ci reakcji katalizowanej przez jon wodorotlenowy
-sta
ł
a szybko
ś
ci reakcji katalizowanej przez wszystkie kwasy Brönsteda, z wyj
ą
tkiem jonu H
3
O
+
- sta
ł
a szybko
ś
ci reakcji katalizowanych przez wszystkie zasady Brönsteda z wyj
ą
tkiem jonu OH
-
.
+
O
H
k
3
−
OH
k
HA
k
B
k
typy katalizy kwasowo-zasadowej
specyficzna kataliza kwasowo-zasadowa je
ś
li stałe szybko
ś
ci reakcji
katalizowanych przez jony H3O+ () lub OH- () s
ą
znacznie wi
ę
ksze ni
ż
stałe
szybko
ś
ci reakcji katalizowanych przez inne ni
ż
jony H3O+ i OH- kwasy (kHA)
specyficzna kataliza kwasowa i zasady (kB) Bronsteda, specyficzna kataliza
zasadowa.
ogóln
ą
kataliz
ą
kwasow
ą
lub ogóln
ą
kataliz
ą
zasadow
ą
-je
ś
li na szybko
ść
reakcji ma głównie wpływ obecno
ść
innych kwasów lub zasad Brønsteda
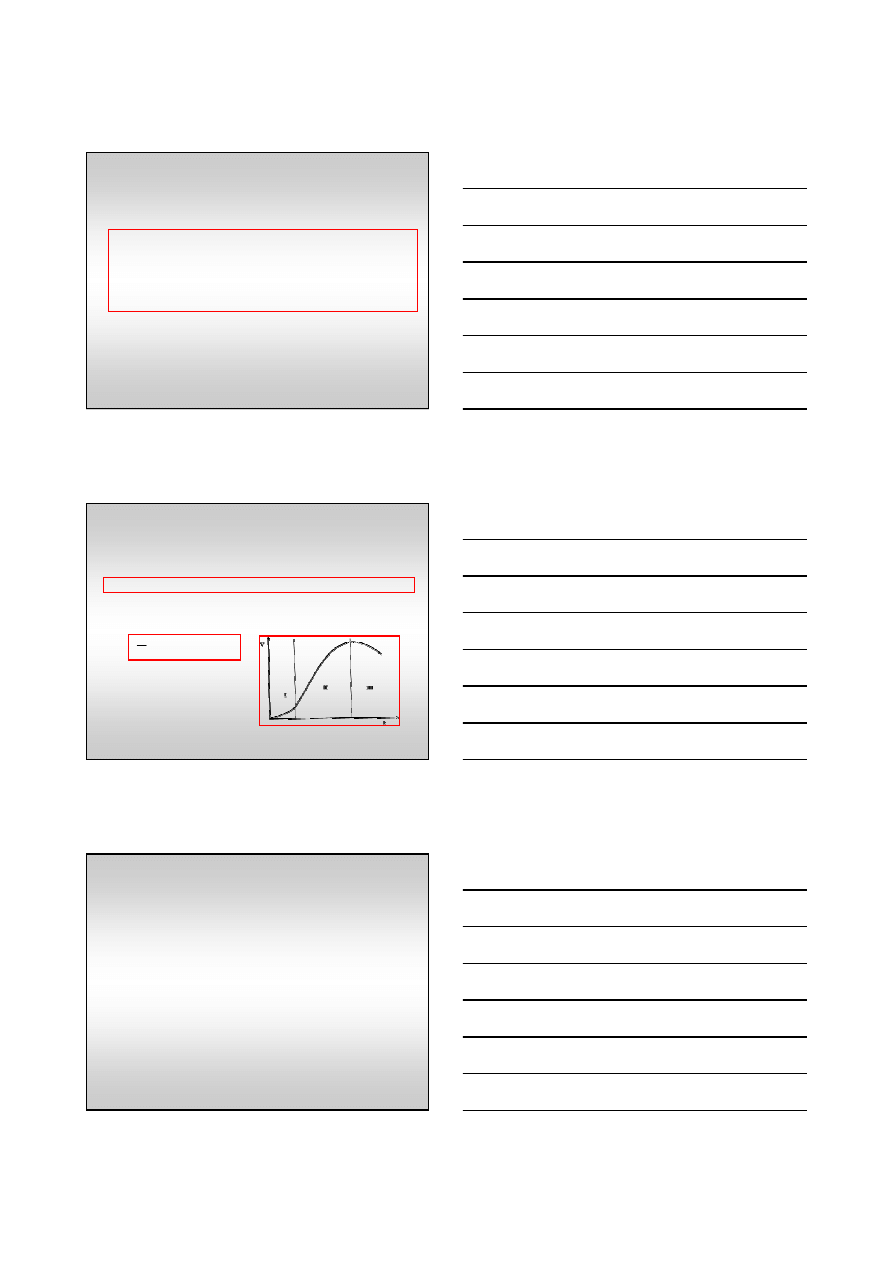
6
Od czego zale
ż
y katalityczne działania kwasu
lub zasady?
od
ł
atwo
ś
ci odczepiania protonu tzn. od sta
ł
ej równowagi dysocjacji kwasu K
a
nie od jego rodzaju.
Brønsted wyprowadzi
ł
zale
ż
no
ść
empiryczn
ą
pomi
ę
dzy sta
łą
katalityczn
ą
kwasu
k’ a jego sta
łą
dysocjacji K
a
:
log k’ = a + b log K
a
,
gdzie a i b sta
ł
e zale
ż
ne od rodzaju reakcji.
Autokataliza
Autokataliza - produkt reakcji jest katalizatorem
Ogólne wyra
ż
enie na szybko
ść
reakcji autokatalitycznej
= [k
1
+ k
2
x
p
] (a – x)
n
dt
dx
gdzie k
1
i k
2
- sta
ł
e szybko
ś
ci reakcji bez
katalizatora i z katalizatorem,
p
–
liczba
cz
ą
steczek
produktu
uczestnicz
ą
cych
w
elementarnej
przemianie katalitycznej, n- rz
ą
d reakcji,
a.
st
ęż
enie pocz
ą
tkowe reagenta i x-
st
ęż
enie produktu.
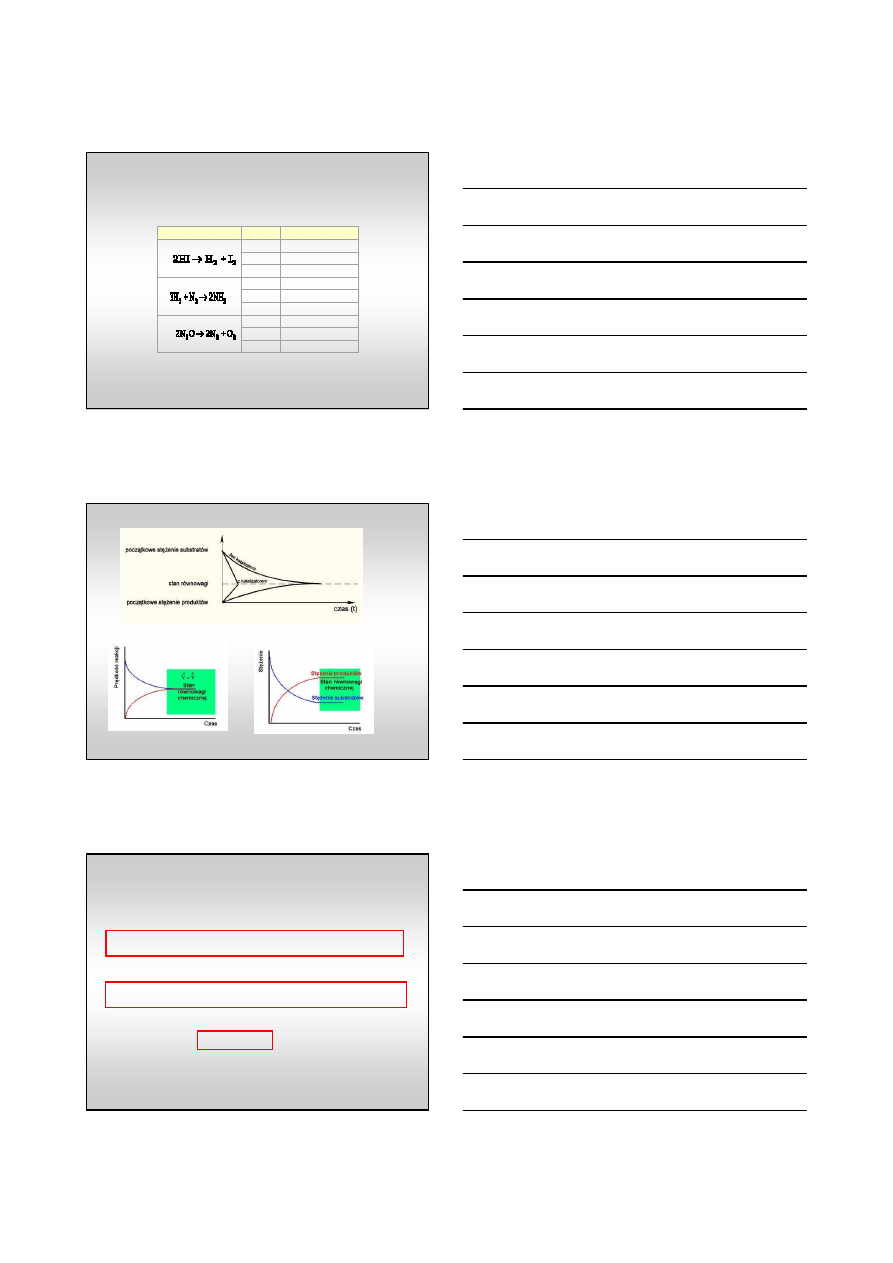
krzywa kinetyczna reakcji autokatalizy
Rodzaje reakcji autokatalizy
autokataliza prosta - reakcja praktycznie nie przebiega bez udziału
katalizatora
autokataliza zło
ż
ona - reakcja zachodzi bez autokatalizatora i z
autokatalizatorem.
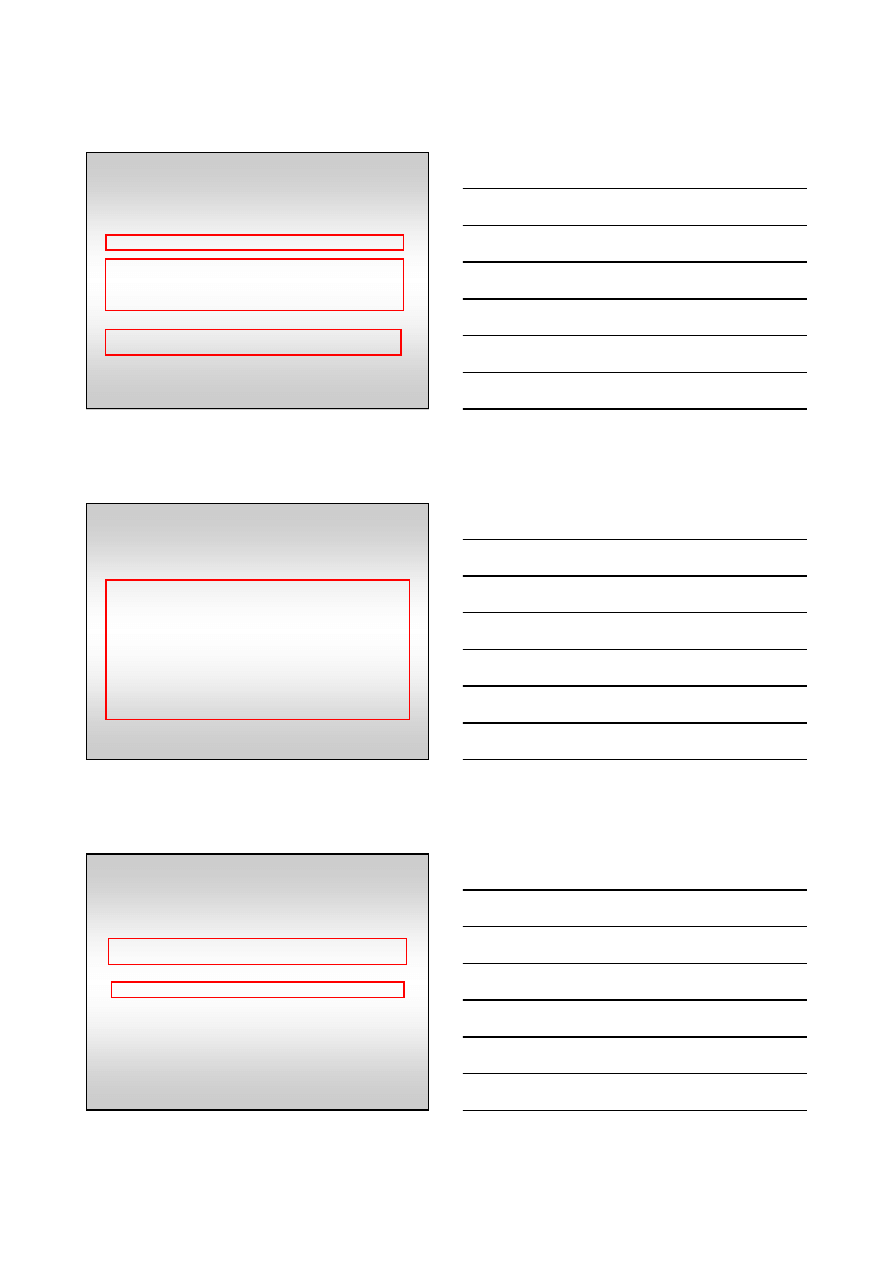
7
Kataliza heterogeniczna
katalizator stanowi odr
ę
bn
ą
faz
ę
.
-
zdecydowana wi
ę
kszo
ść
reakcji przebiega w obecno
ś
ci katalizatorów
sta
ł
ych nazywanych kontaktami
-
czasami katalizator mo
ż
e stanowi
ć
faz
ę
ciek
łą
b
ą
d
ź
katalizowa
ć
mog
ą
ś
cianki naczynia reakcyjnego.
Szybko
ść
reakcji heterogenicznej zale
ż
y od wielko
ś
ci powierzchni na
której proces przebiega,
Przykłady katalizy heterogenicznej
Wi
ę
kszo
ść
procesów przemys
ł
u chemicznego przebiega w obecno
ś
ci
katalizatorów sta
ł
ych.
konwersja tlenku w
ę
gla z para wodn
ą
w obecno
ś
ci katalizatora Fe
2
O
3
aktywowanego Cr
2
O
3
.
W metodzie kontaktowej otrzymywania kwasu siarkowego podczas
utleniania SO
2
do SO
3
stosuje si
ę
jako kontakt V
2
O
5
aktywowany K
2
O.
spalanie amoniaku wobec platyny jako kontaktu:
4NH
3
+ 5 O
2
= 4 NO + 6 H
2
O
reakcja bez zastosowania katalizatora
4NH
3
+ 3 O
2
= 2 N
2
+ 6 H
2
O
Aktywno
ść
kontaktów zwi
ą
zana jest z adsorpcj
ą
cz
ą
steczek na aktywnych
miejscach powierzchni kontaktu czyli centrach aktywnych
Promotory – substancje, które zwi
ę
kszaj
ą
aktywno
ść
katalizatora
8
Etapy reakcji katalizy heterogenicznej na
katalizatorze stałym dla gazowych substratów
reakcji:
1. dyfuzja
zewn
ę
trzna
–
transport
reagentów
do
powierzchni
katalizatora,
2. dyfuzja wewn
ę
trzna – dyfuzja substratów w porach katalizatora,
3. adsorpcja – chemisorpcja przynajmniej jednego z substratów na
powierzchni katalizatora,
4. reakcja powierzchniowa –
nast
ę
puje w niej przekszta
ł
cenie
zaadsorbowanych substratów i powstanie produktów reakcji
adsorbowanych na powierzchni katalizatora,
5. desorpcja produktów,
6. dyfuzja wewn
ę
trzna produktów – transport od wewn
ę
trznej do
zewn
ę
trznej powierzchni katalizatora,
7. dyfuzja zewn
ę
trzna – z powierzchni katalizatora do fazy gazowej.
Typy katalitycznych reakcji kontaktowych:
oksydakcyjno-redukcyjne – zwi
ą
zanych z przej
ś
ciem elektronu od katalizatora
do substratu np. uwodornianie, utlenianie, odwodornianie. Reakcje takie
przebiegaj
ą
na powierzchni katalizatorów metalicznych np. Fe, Pt, Ni, lub
pó
ł
przewodnikowych np. V
2
O
4
.
kwasowo-zasadowe – zwi
ą
zanych z przej
ś
ciem protonu pomi
ę
dzy kwasem i
zasada wg Brønsteda albo zgodnie z definicja Lewisa tworzy si
ę
lub zrywa
wi
ą
zanie koordynacyjne. Dlatego katalizatorami mog
ą
by
ć
zwi
ą
zki, które mog
ą
uzyskiwa
ć
lub traci
ć
proton lub woln
ą
par
ę
elektronow
ą
np. glinokrzemiany lub
zeolity. Do tej grupy reakcji nale
żą
procesy hydrolizy, izomeryzacji, hydratacji,
dehydratacji, polimeryzacji.
Zasada działania katalizatora heterogenicznego
Droga syntezy amoniaku.
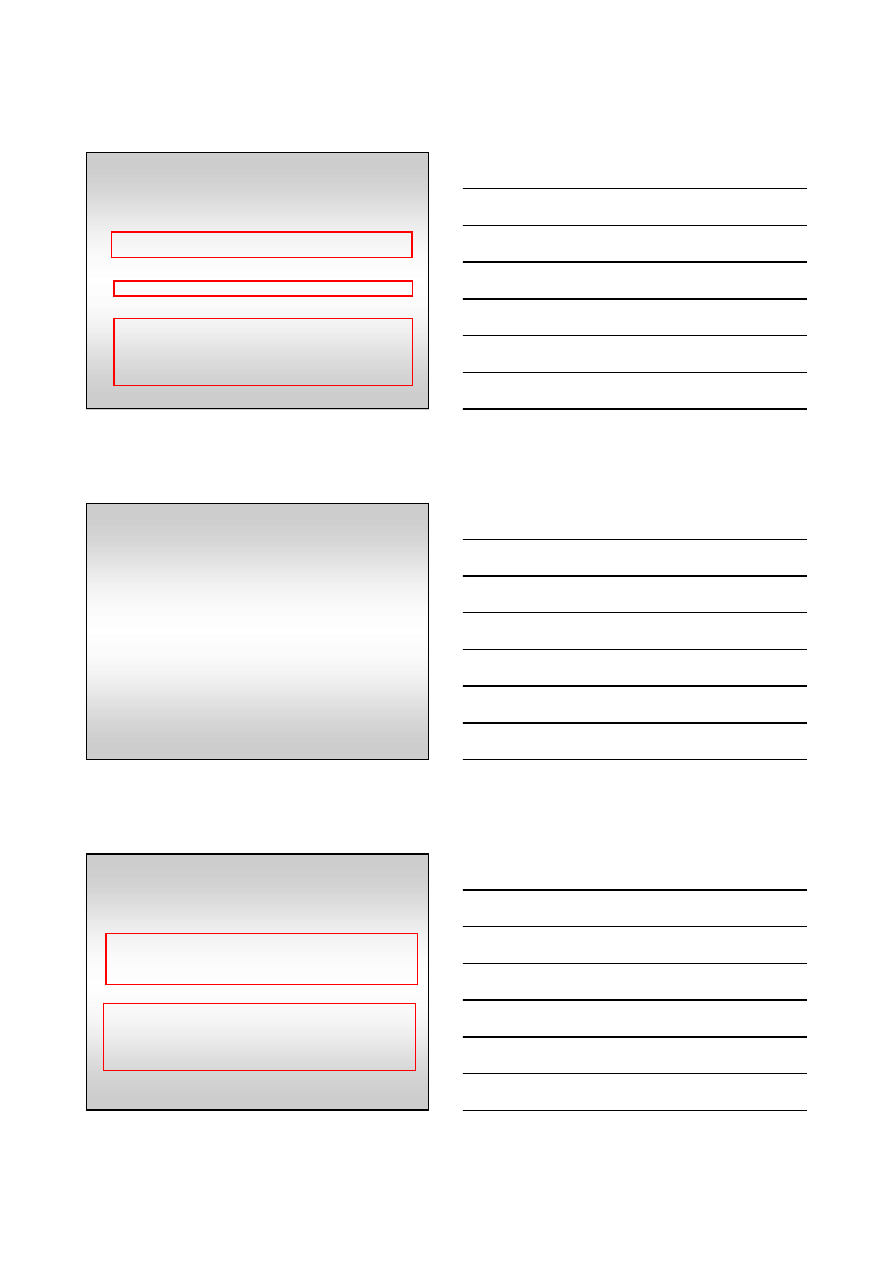
9
Aktywno
ść
katalizatorów mo
ż
na zmienia
ć
przez dodatek obcych atomów lub
cz
ą
steczek zwanych promotorami
Promotory – substancje, które zwi
ę
kszaj
ą
aktywno
ść
katalizatora
promotor chemiczny – zmienia katalityczne w
ł
a
ś
ciwo
ś
ci kontaktu
promotor strukturalny (teksturalny) zmieniaq w
ł
a
ś
ciwo
ś
ci strukturalne
kontaktów, jego
ś
rednic
ę
porów oraz powierzchni
ę
w
ł
a
ś
ciw
ą
katalizator mieszany – katalizator sk
ł
adaj
ą
cy si
ę
a co najmniej dwóch
katalizatorów o porównywalnych ilo
ś
ciach.
Przykłady reakcji katalitycznych z dodatkiem
promotorów
1. synteza NH
3
z pierwiastków . katalizator Fe, dodatki – tlenki: glinu, potasu,
magnezu, wapnia, krzemu
2. utlenianie amoniaku do tlenku azotu II . katalizator Pt, dodatki – KVO
3
osadzony na ziemi okrzemkowe
3. synteza metanolu z H
2
i CO. Katalizator mieszanina tlenku cynku i chromu
Teorie katalizy heterogenicznej
Teoria Faradaya (1833 r.) - teori
ę
zwi
ą
zków po
ś
rednich
proces katalizy polega na adsorpcji reagentów w warstwie powierzchniowej
katalizatora w wyniku czego powstaj
ą
zwi
ą
zki po
ś
rednie a szybko
ść
reakcji
wzrasta.
Teoria Taylora (1925 r) - teoria centrów aktywnych
powierzchnia katalizatora jest chemicznie i fizycznie niejednorodna, (latego
w
ł
a
ś
ciwo
ś
ci ró
ż
nych centrów adsorpcyjnych mog
ą
si
ę
ró
ż
ni
ć
).
tylko niewielka cz
ęść
centrów adsorpcyjnych na powierzchni jest aktywna
katalitycznie.
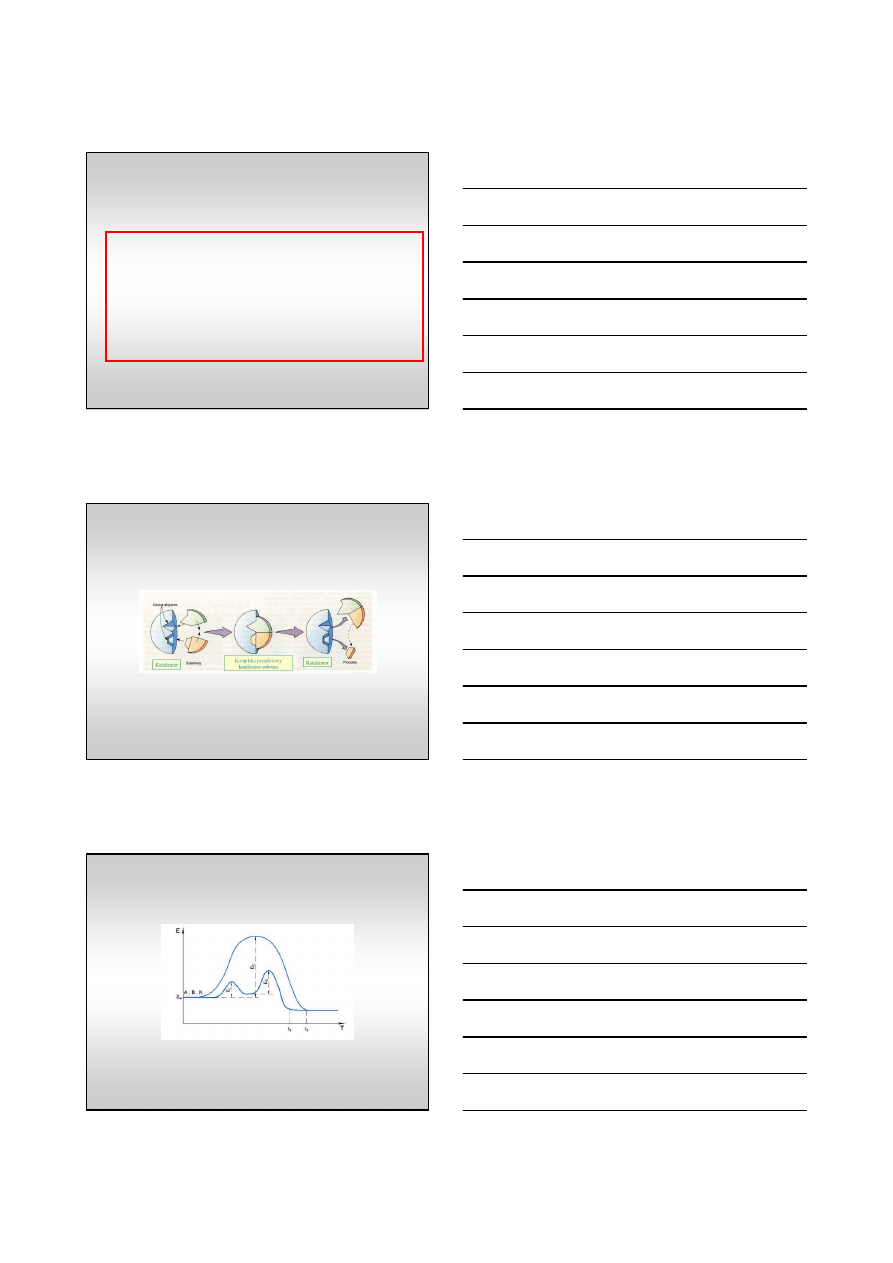
10
Teoria multipletu A.A. Ba
ł
andina (1929)
Teoria multipletu A.A. Ba
ł
adina (1929) - centrum aktywnym na powierzchni
katalizatora jest grupa kilku miejsc aktywnych tzw. multiplet a nie jedno miejsce
adsorpcji o okre
ś
lonej energii chemisorpcji.
-
rozpatruje si
ę
ogólny typ adsorpcji uwzgl
ę
dniaj
ą
cy oddzia
ł
ywanie cz
ą
steczek
reagentów z kilkoma centrami na powierzchni katalizatora, tzw. adsorpcja na
multiplecie. - tworzy si
ę
zwi
ą
zek przej
ś
ciowy w sk
ł
ad którego wchodzi kilka
atomów katalizatora.
adsorpcja na multiplecie wymaga spe
ł
nienia dwóch zasad:
zgodno
ś
ci geometrycznej oraz
zgodno
ś
ci energetycznej.
Działania katalizatora

11
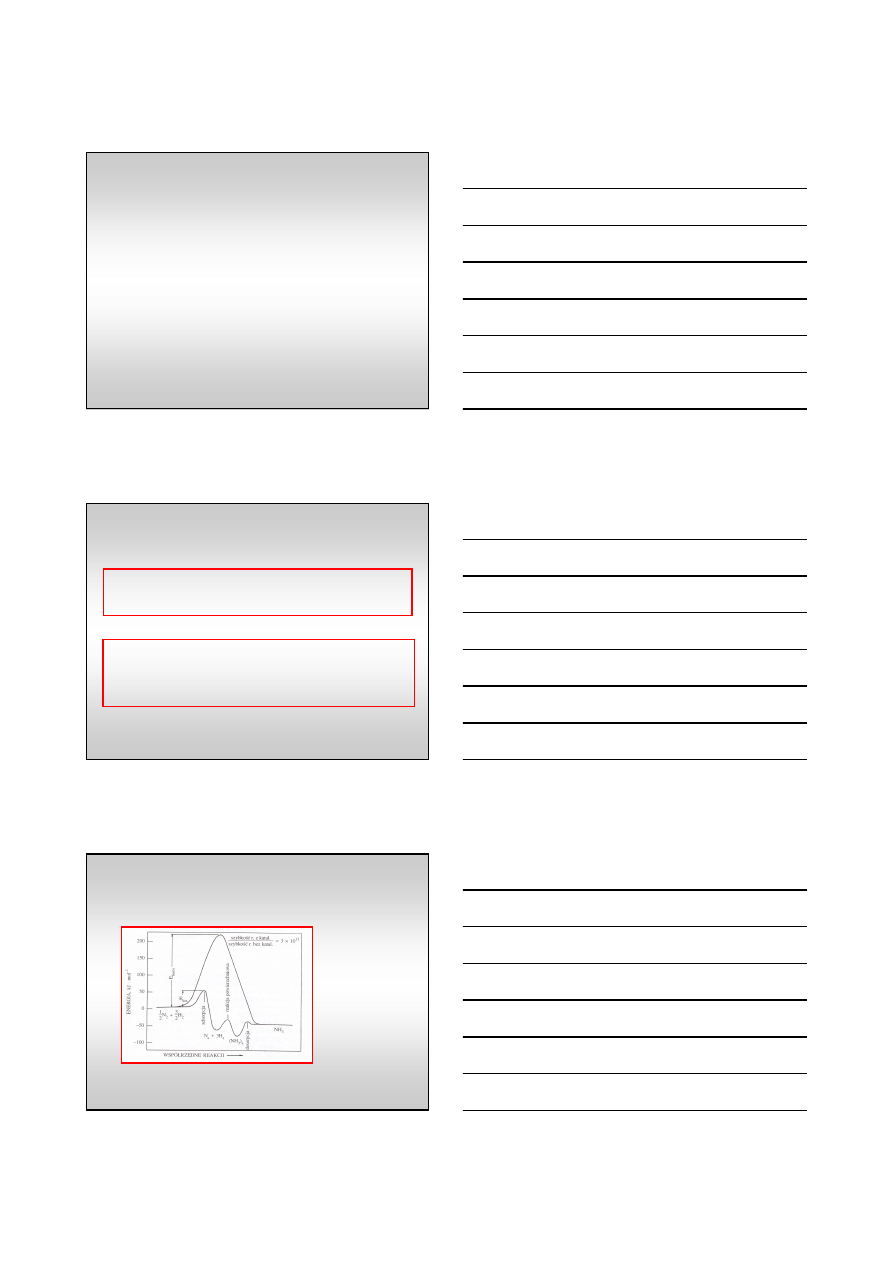
mechanizm katalitycznego uwodorniania
etylenu C
2
H
2
CH
2
=CH
2
+H
2
CH
3
CH
3
etap pierwszy - adsorpcja etylenu
i wodoru na powierzchni
katalizatora
W wyniku oddziaływania z
katalizatorem cz
ą
steczka wodoru
ulega katalitycznej dysocjacji, której
wynikiem jest powstanie dwóch
zaadsorbowanych atomów wodoru
Etap II - w wyniku oddziaływania z
katalizatorem cz
ą
steczka wodoru
ulega katalitycznej dysocjacji, której
wynikiem jest powstanie dwóch
zaadsorbowanych atomów wodoru
Etap III -atomy wodoru s
ą
kolejno
podstawiane do grup
metylenowych
Etap IV - nowo powstała cz
ą
steczka etanu
ulega desorpcji z powierzchni katalizatora
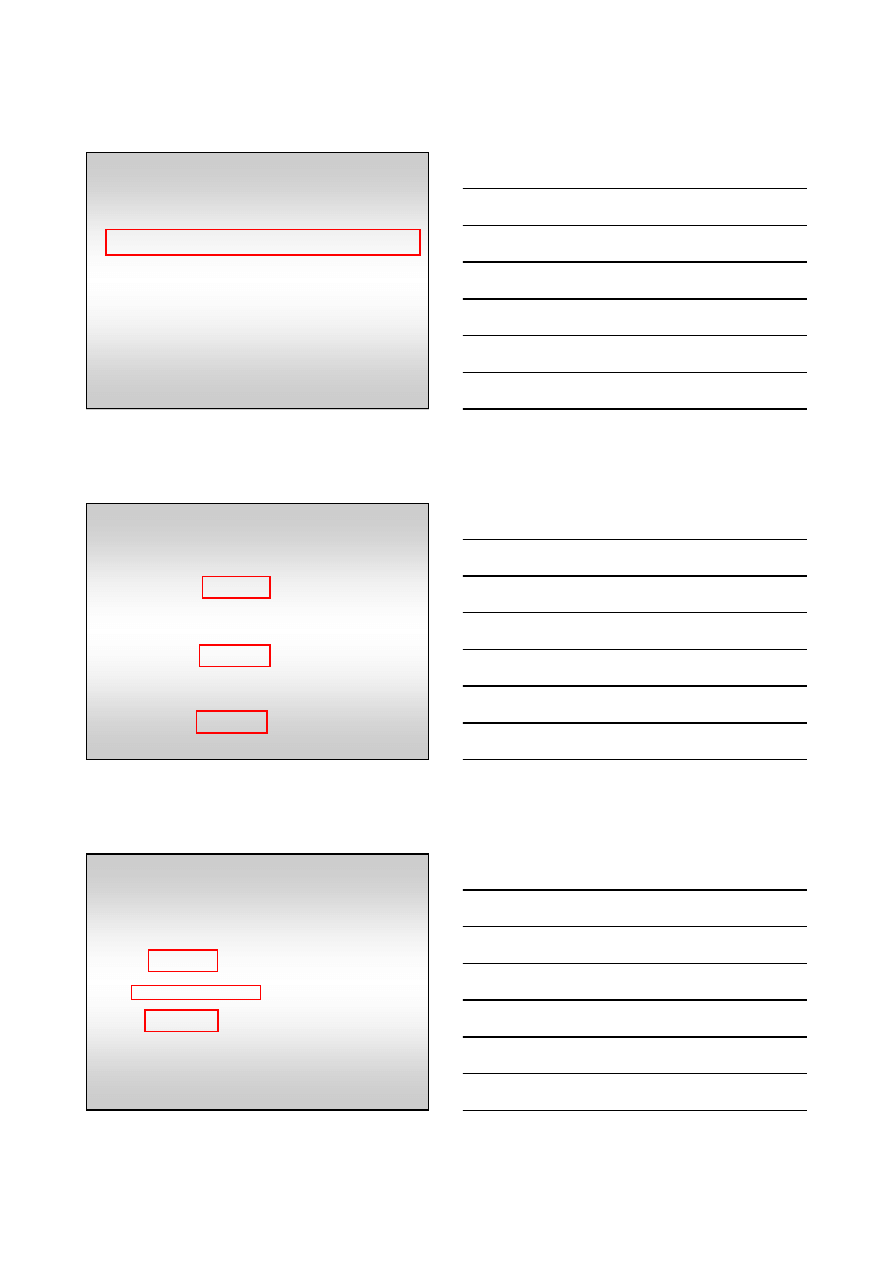
12
Kataliza mikroheterogeniczna
Dotyczy ona uk
ł
adów w których katalizator wyst
ę
puje w stanie rozdrobnienia
koloidalnego. Typowymi takimi katalizatorami s
ą
enzymy.
mechanizm Michaelisa-Menten reakcji
enzymatycznej
P
S
E
→
Etap I – tworzenie kompleksu enzymu z substratem
ES
S
E
→
+
Szybko
ść
tworzenia ES – reakcja dwucz
ą
steczkowa, pierwszego
rz
ę
du wzgl
ę
dem substratu oraz enzymu
]
][
[
S
E
k
v
I
ES
=
Etap II –rozpad kompleksu aktywnego na enzym i substrat
mechanizm Michaelisa-Menten reakcji
enzymatycznej-rozpad kompleksu aktywnego
S
E
ES
+
→
Szybko
ść
rozpadu ES na E iS
]
[
'
ES
k
v
I
RES
=
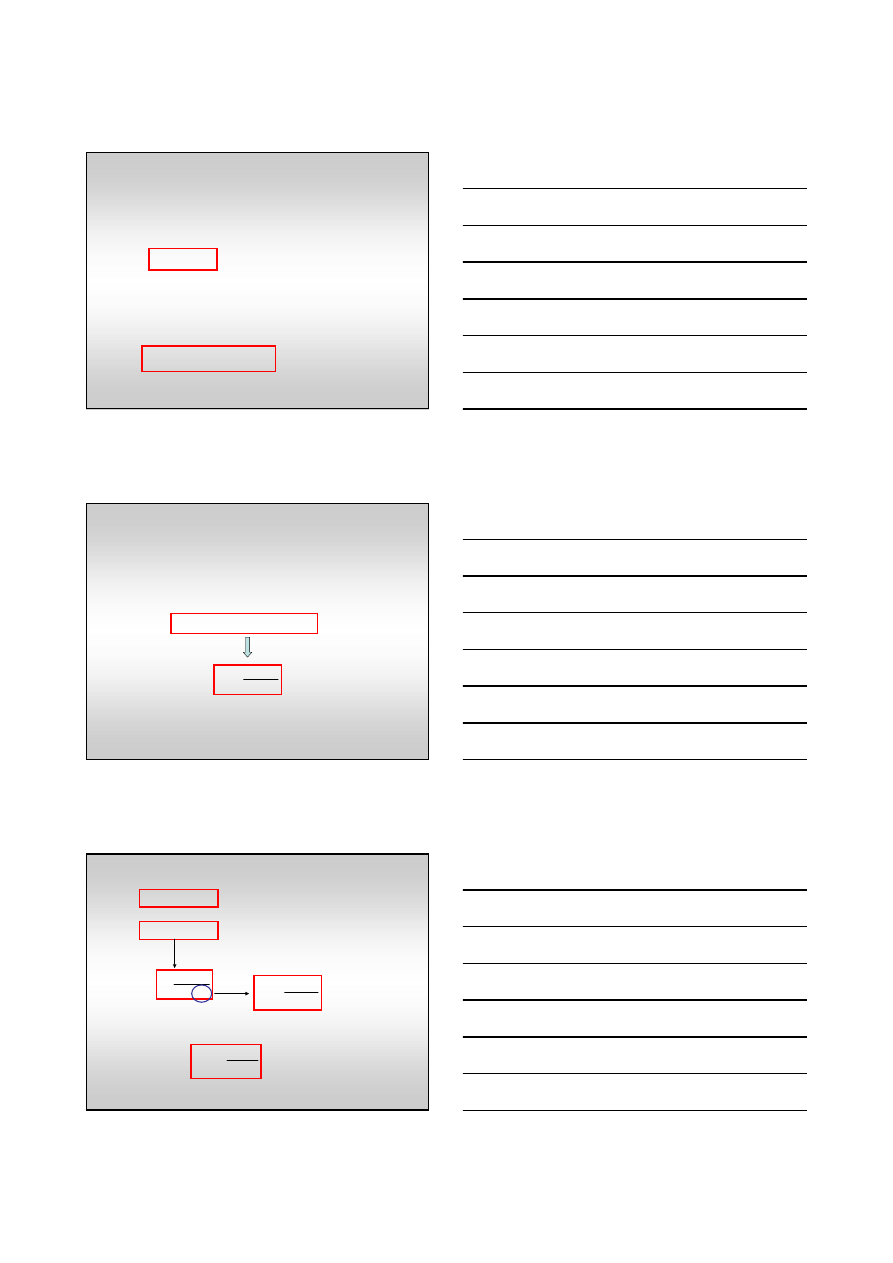
13
Etap III – reakcja tworzenia produktu i odtwarzania si
ę
enzymu
S
P
ES
+
→
]
[
)
(
ES
k
v
v
II
ES
Z
P
=
=
Szybko
ść
tworzenia si
ę
produktu P (v
P
) = szybko
ś
ci zu
ż
ywania kompleksu
ES V
Z(ES)
W stanie stacjonarnym szybko
ść
tworzenia si
ę
ES i zu
ż
ywania si
ę
ES
na skutek procesów zachodz
ą
cych w etapach 2 i 3 s
ą
sobie równe
]
[
]
[
]
][
[
'
ES
k
ES
k
S
E
k
II
I
I
+
=
II
I
I
k
k
S
E
k
ES
+
=
'
]
][
[
]
[
]
[
]
[
]
[
0
ES
E
E
+
=
]
[
0
E
k
P
=
M
II
K
S
S
k
k
+
=
]
[
]
[
I
II
I
M
k
k
k
K
+
=
'
M
K
S
E
ES
]
][
[
]
[
=
Wyszukiwarka
Podobne podstrony:
KINETYKA wykladI id 235063 Nieznany
LOGIKA wyklad 5 id 272234 Nieznany
ciagi liczbowe, wyklad id 11661 Nieznany
AF wyklad1 id 52504 Nieznany (2)
Neurologia wyklady id 317505 Nieznany
ZP wyklad1 id 592604 Nieznany
CHEMIA SA,,DOWA WYKLAD 7 id 11 Nieznany
or wyklad 1 id 339025 Nieznany
II Wyklad id 210139 Nieznany
cwiczenia wyklad 1 id 124781 Nieznany
BP SSEP wyklad6 id 92513 Nieznany (2)
MiBM semestr 3 wyklad 2 id 2985 Nieznany
algebra 2006 wyklad id 57189 Nieznany (2)
olczyk wyklad 9 id 335029 Nieznany
Kinezyterapia Wyklad 2 id 23528 Nieznany
AMB ME 2011 wyklad01 id 58945 Nieznany (2)
AWP wyklad 6 id 74557 Nieznany
PRAWO SPORTOWE Wyklady(1) id 38 Nieznany
więcej podobnych podstron