CHOROBY
CHROMOSOMOWE
CZŁOWIEKA II

WSKAZANIA DO BADANIA
KARIOTYPU:
kliniczne podejrzenie określonej aberracji
chromosomów (np.zespół Downa).
fenotyp dziecka określający bliżej nieokreśloną
aberrację chromosomową (np.dysmorfia twarzy,
dysplastyczne dłonie i/lub małżowiny uszne,
mnogie wady rozwojowe, opóźnienie rozwoju
psychomotorycznego lub upośledzenie umysłowe).
upośledzenie umysłowe.
fenotyp charakterystyczny dla zespołu
mikrodelecji (np. retinoblastoma, guz Wilmsa
zespół WolfaHirschhorna, zespół PraderaWilliego
itp.)

Niepowodzenia rozrodu: brak ciąży, poronienia
samoistne, porody martwe, zgony dzieci z wadami
(badanie kariotypu dotyczy obojga małżonków).
Znaczny niedobór wzrostu u fenotypowych kobiet
(możliwość występowania zespołu Turnera).
Rodzice (i ew. inni członkowie rodziny) dzieci z
wykrytymi aberracjami struktury chromosomów (w
celu wykrycia ew. nosicielstwa translokacji
zrównoważonej wśród zdrowych członków
rodziny).
Zespoły z łamliwością chromosomów (np. zespół
Wernera).
Zaburzenia determinacji i różnicowania płci
(badanie kariotypu pozwala określić płeć
chromosomową i rozpoznać ewentualne aberracje
chromosomów płciowych).

UWAGA !
Generalnie nie ma wskazań do badania kariotypu w
przypadku chorób uwarunkowanych jednogenowo.
Wyjątek zespoły z łamliwością chromosomów
t.j.zespół Wernera,zespół Blooma, które choć
wykazują etiologię jednogenową, to obserwuje się w
ich przebiegu łamliwość chromosomów. W
niektórych przypadkach, przy podejrzeniu, że zespół
wad jest uwarunkowany jednogenowo, badanie
kariotypu przeprowadza się jako element
rozpoznania różnicowego.
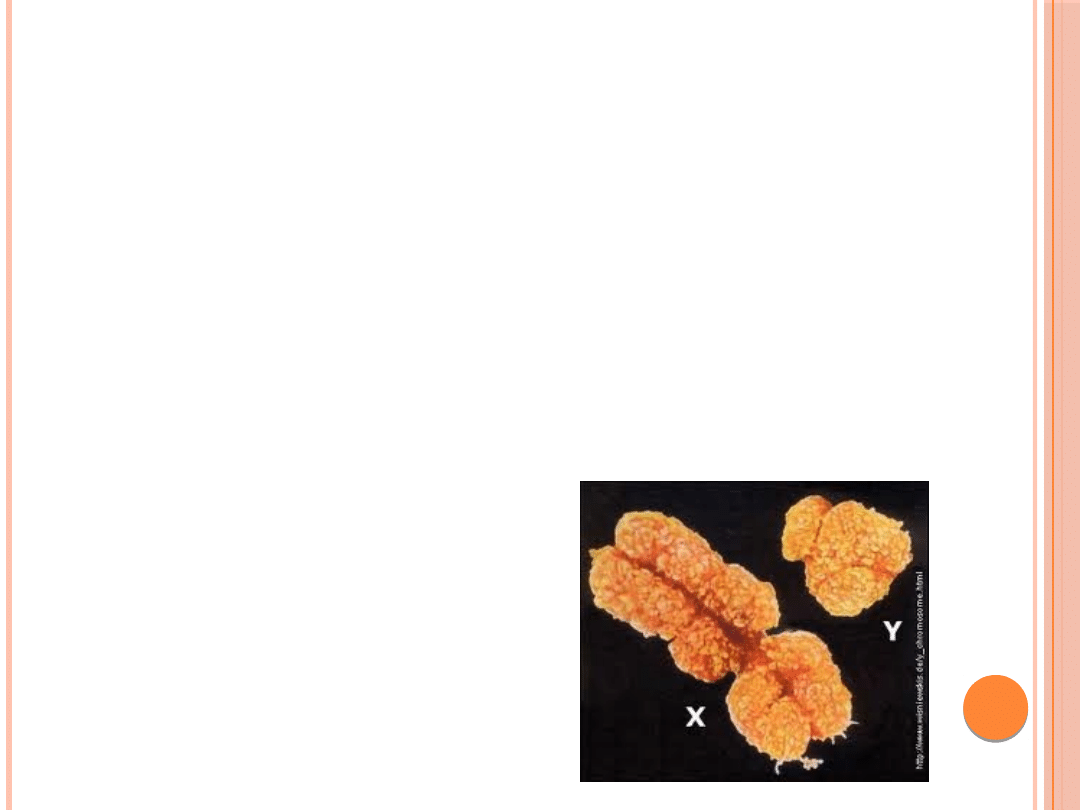
Chromatyna płciowa:
Odnosi się ona do odpowiednio wybarwionych
struktur chromatyny widocznych w jądrze
interfazowym, odpowiadającym chromosomom
X i Y.
Można ją wykryć niemal we wszystkich
komórkach.

Chromatyna X :
Inaczej ciałko Barra,znajduje się w komórkach niemal
wszystkich tkanek.
U człowieka bada się w rozmazach nabłonka jamy
ustnej lub w komórkach płynu owodniowego.
W granulocytach obojętnochłonnych po odpowiednim
wybarwieniu , chromatyna uwidacznia się poza odręb
jądra,ale pozostających z nim włączności grudek
chromatyny
tzw.pałeczek Dobosza.
Część chromatyny jądrowej uległa inaktywacji w
pierwszych dniach rozwoju zygoty. Ma to na celu
wyrównanie informacji genetycznej zawartych w dwóch
chromosomach X u kobiety w stosunku do jednego
chromosomu X u mężczyzny.
Inaktywacji nie podlega grupa genów nabłonka jamy
ustnej kobiety(2060% badanych jąder komórkowych)

W przypadku patologicznych zmian stwierdza się zmiany w
liczbie i morfologii ciałek Barra (np. w zespole Turnera w
którym stwierdzono izochromosom ramion długich chromosomu
X,chromatyna płciowa X jest większa i zawiera więcej DNA,niż
chromatyna utworzona przez prawidłowy chromosom).
Badanie chromatyny X jest :
najprostszą,pośrednią formą wykrywania aberracji
liczbowych chromosomu płciowego X, wykorzystywana
jest także w genetce klinicznej do określenia płci oraz
zaburzeń rozwoju cielesnopłciowego teraz zastąpiona
przez inne metody (analiza kariotypu i technik hybrydyzacji in
situ FISH)
wykorzystuję się ją też w medycynie sądowej (trudność
wykonania pełnej analizy chromosomalnej).
jako pomoc w badaniach przesiewiowych wykrywania
chromosomu X w komórkach interfazowych tkanek
prawidłowych i nowotworowych(niski odsetek ciałek
Barra ma znaczenie prognostyczne i świadczy o
złośliwości guza)

nieczynne chromosomy X charakteryzują się
opóźnioną replikacją DNA,która zachodzi w
późnej fazie S.
na podstawie kryterium cytologicznego
stwierdzamy inaktywację chromosomu X
zaobserwoanie w stadium metafazy innego wzoru
prążków tzw.prążków replikacyjnych w
zinaktywowanym chromosomie i jego aktywnym
homologu.
na podstawie kryterium molekularnegoróżnice w
metylacji genów na chromosomie X(metoda
PCR,fluorescencji hybrydyzacji in situ)

Chromatyna Y:
Pojawia się ona w interfazowym jądrze komórkowym
wybarwionym barwnikami fluorescencyjnymi .
stanowi ją dystalna,heterochromatynowa część
długich ramion chromosomu Y q12
stwierdza się go u 3050 % badanych komórek
męskich
badanie chromatyny Y jest jedną z metod służacych
do określania płci i diagnostyki aberracji liczbowych
chromosomów płciowych
w jądrach granulocytów obojętnochłonnych tworzy
się pałeczka dobosza Y, którą odróżnia się od
pałeczki dobosza X
zdolnością do fluorescencji.

GENETYCZNA DETERMINACJA
PŁCI :
Zależna jest od genów zlokalizowanych na
chromosomach X i Y oraz chromosomach
somatycznych.
Geny te odpowiedzialne są za syntezę
białek struktualnych i regulatorowych niezbędnych
do prawidłowego różnicowania się pierwotnej
gonady w
kierunku jądra lub jajnika oraz
rozwoju wewętrznych i zewnętrznych narządów
płciowych.
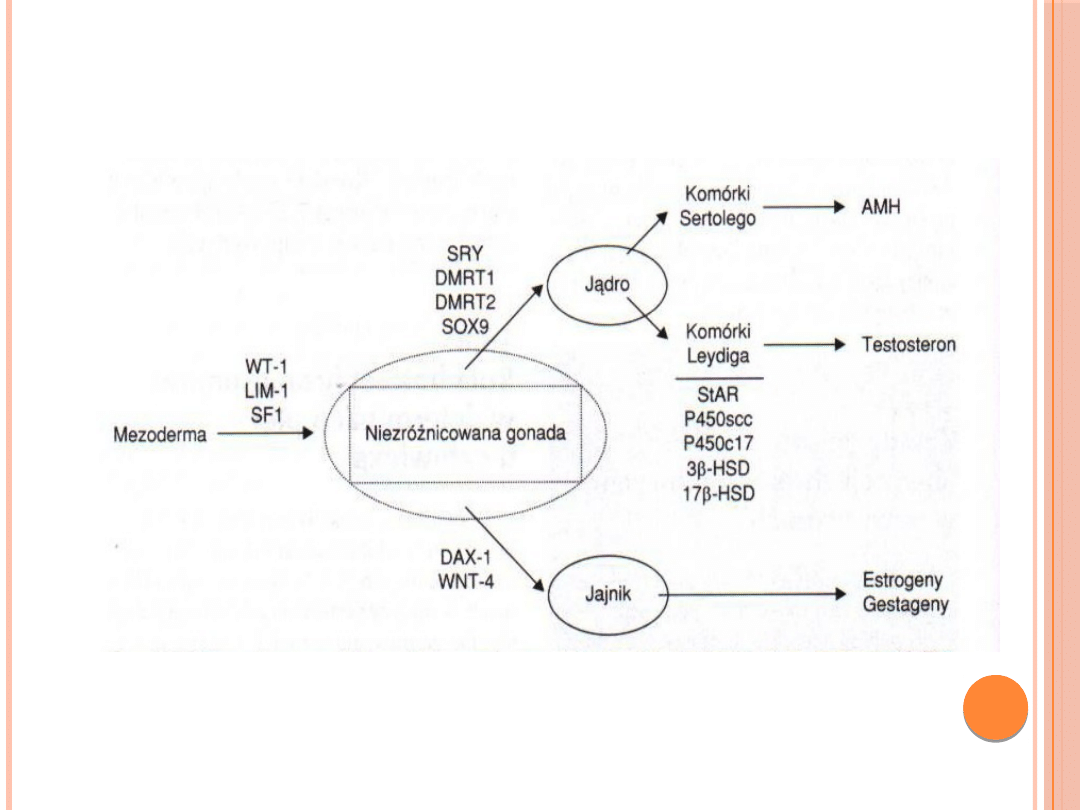
GENY BIORĄCE UDZIAŁ W
RÓZNICOWANIU GONAD:
W rozwój układu płciowego jest zaangażowane około 150 genów !

PAR – regiony peseudoautosomalne to niewielkie
rejony wykazujące homologię sekwencji DNA w
odrębie krótkich ramion chromosomów X i Y.
U mężczyzn podczas mejozy w procesie
spermatogenezy dochodzi do koniungacji obu
chromosomów za pomocą regionów PAR i tworzy
się struktura zwana pęcherzykiem płciowym
następnie zachodzi rekombinacja crossingover
między regionami PAR.
PRAWIDŁOWY
ROZWÓJ GONADY:
1) żeńskiej 46 XX
2) męskiej 46 XY

Gen SRY
Znajduje się w regionie p11.3
Nie zawiera intronów i koduje białko SRY o
wielkości 204 aminokwasów i masie 23,9 kD.
Należy do czynników transkrypcyjnych (HMG
box) i wykorzystuje pokrewieństwo z kilkoma
czynnikami transkrypcyjnymi np.LEF1.
W części środkowej jest łańcuch 80 aminokwasów
homologiczny z odpowiedzialnym za wiązanie
DNA motywem aminokwasów występujących w
grupie białek transkrypcyjnych HMG.

Rola genu SRY w różnicowaniu pierwotnej
gonady:
na drodze ekspresji szeregu genów są syntezowane
produkty w odpowiednim stężeniu i sekwencji
czasowej na szlaku różnicowania komórkowego.
miejscem transkrypcji genu SRY są komórki
Sertolego.
rola produktu białkowego genu SRY sprowadza się
do funkcji regulacyjnych wobec genów biorących
udział w różnicowaniu i rozwoju wewnętrznych
narządów płciowych męskich.
białko SRY może być aktywatorem transkrypcji
genu kodującego wytwarzanie czynnika AMH.
białko SRY jest negatywnym regulatorem
transkrypcji genu aromatazy P450.

Rozwój układu płciowego:
1)
Powstają zawiązki niezróżnicowanych gonad i
zewnętrznych narządów płciowych, które się
między sobą nie różnią morfologicznie.
2)
Następuje proces różnicowania morfologicznego
przebiega odzielnie u płci męskiej i żeńskiej.
Proces różnicowania odbywa się w 9 tyg. życia
płodowego !

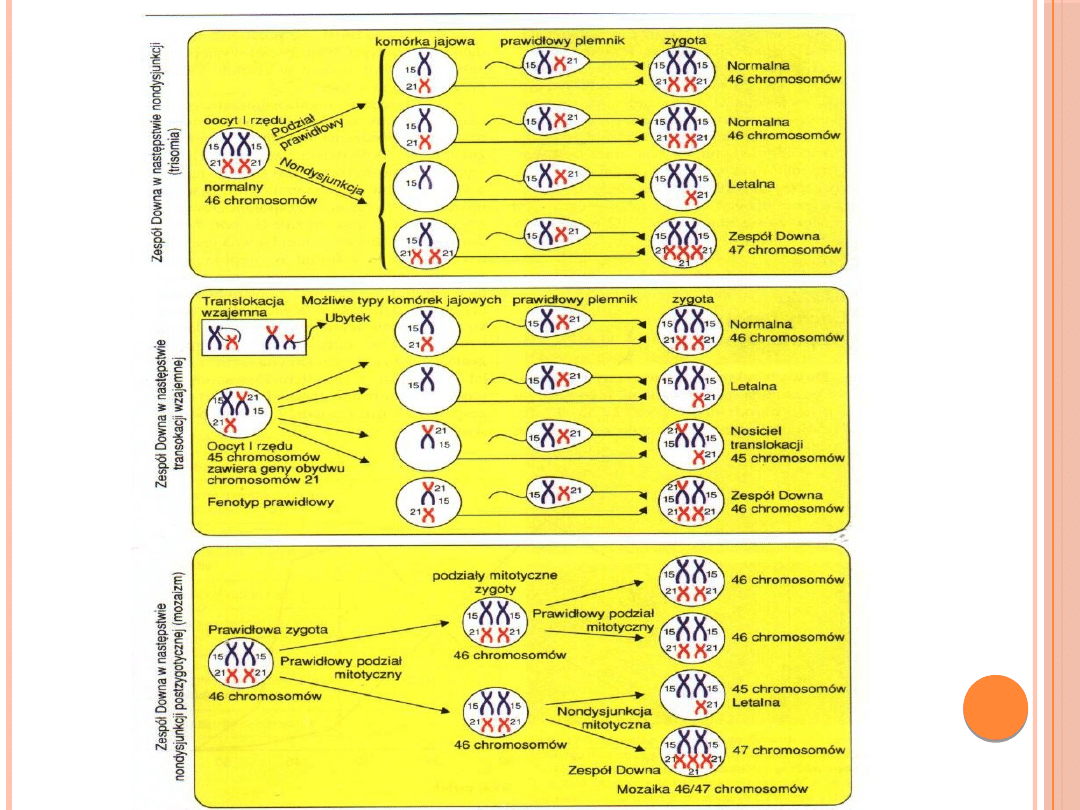
Aberracje liczbowe autosomów:
Zespół Downa
Zespół Pataua
Zespół Edwardsa
Trisomia chromosomu 8

Zespół Downa
Częstość urodzeń 1:700
Przyczyną dodatkowy chromosom pary 21
Do wystąpienia tej choroby przyczyną może być każda
z trzech wymienionych aberracji chromosomowych:
1)
Trisomia 21 pary 47,XX,+21 lub 47,XY,+21
2)
Translokacja niezrównoważona 46,XX,der(21;21)
(q10;q10),+21
3)
Kariotyp mozaikowy 46,XX/47,XX,+21
PRAWDOPODOBIEŃSTWO URODZENIA DZIECKA Z
ZESPOŁEM DOWNA WZRASTA Z WIEKIEM
MATKI!!!

CECHY FENOTYPOWE ZESPOŁU
DOWNA:
skośne ustawienie szpar powiek
bruzda poprzeczna na dłoni tzw.bruzda małpia
nisko usadzone i zniekształcone małżowiny uszne
obniżone napięcie mięśniowe
opuszczone kąciki ust
szeroka przestrzeń między I a II palcem stopy
duży pobrużdżony, wystający język
krótkie szerokie dłonie
zmarszczka nakątna
krótkogłowie
zapadnięty grzbiet nosa

upośledzenie umysłowe
niski wzrost ok.150 cm
przedwczesne starzenie się
większa zapadalność na chorobę Alzheimera i
ostrą białaczkę szpikową
padaczka ,zaćma, niedoczynność
tarczycy
mężczyźni bezpłodni
średnia długość życia 3540 lat

Zespół Pataua
Częstość występowania 1:800010000
Przyczyną dodatkowy chromosom
13 pary
u 20 % stwierdzono translokację
niezrównoważoną w odrębie chromosomu 13
kariotypy mozaikowe z linia disomiczną
chromosomu 13 i trisomiczną 46,XX/47,XX,+13
duży odsetek ciąż których zarodek posiada
trisomię 13 ulega poronieniu
u noworodka z zespołem Pataua występują wady
rozwojowe i cechy dysmorficzne

CECHY DYSMORFICZNE DLA
ZESPOŁU PATAUA :
mikrocefalia
ubytki skóry na głowie
wystające czoło
rozszczep wargi i podniebienia
wady gałek ocznych (częściowy ubytek siatkówki
i tęczówki oka)
hipoteloryzm
anomalie palców (polidaktylia i syndaktylia)
nisko osadzone uszy

Wady wrodzone narządów
wewnętrznych:
NEREKnerki torbielowate i wodonercze
SERCEubytki w przegrodzie serca
MACICYmacica dwurożna
anomalie w budowie anatomicznej mózgu
(złączenie płatów czołowych mózgowia,brak
opuszek węchowych,niedorozwój mózgu)
hipotonia mięśniowa
głuchota
wodogłowie
CZĘSTOŚĆ WYSTĄPIENIA ZESPOŁU PATAUA
ROŚNIE Z WIEKIEM MATKI, OKOŁO 70%
UMIERA W I PÓŁROCZU ŻYCIA !
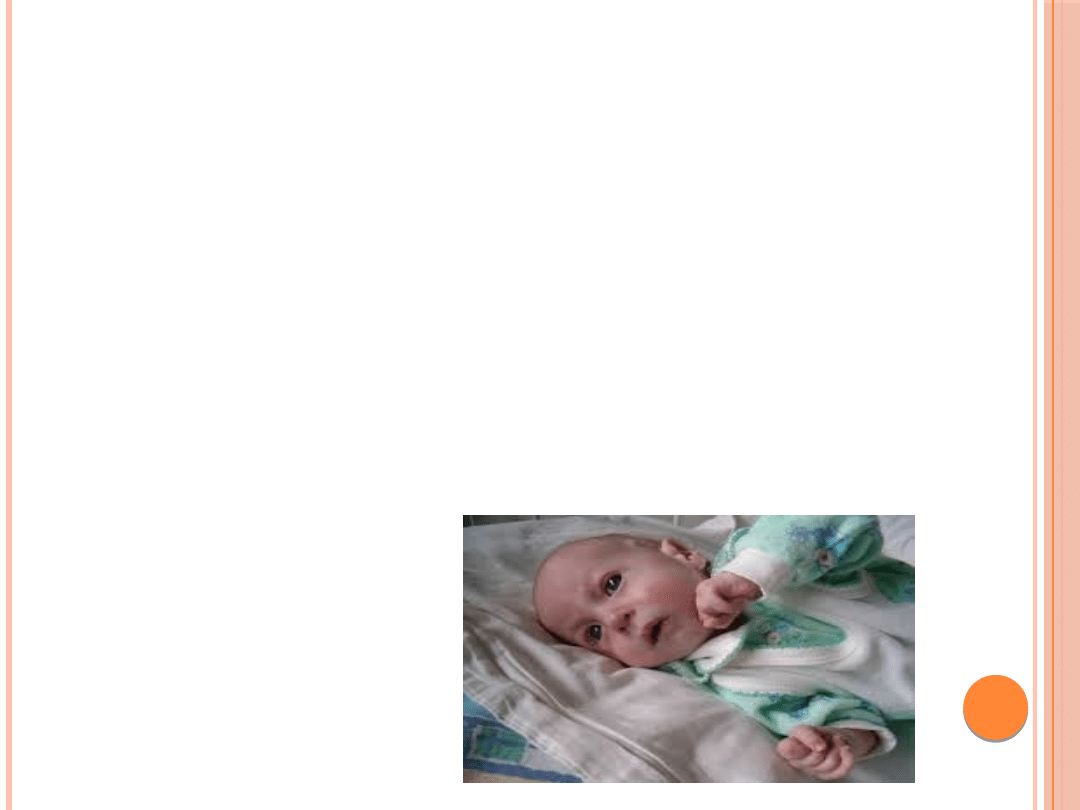
ZESPÓŁ EDWARDSA :
częstość występowania 1:5000 urodzeń
uwarunkowany trisomia 18 chromosomu
duże znaczenie ma wiek matki w okresie poczęcia
kariotyp mozaikowy występuje w niewielkim
stopniu
duży odsetek ciąży ulega samoistnemu
poronieniu

Cechy dysmorficzne :
niska waga urodzeniowa
małogłowie
wystająca potylica
mała bródka
nisko osadzone i zniekształcone małżowiny uszne
hiperteloryzm
zmarszczka nakątna
krótka szyja z widocznym fałdem skórnym
zaciśnięte w pięści dłonie i nakładanie się na siebie
palców ręki
stopa cepowata z charakterystycznymi zrostami
palców,wystającą kością piętową,krótkim paluchem
niedorozwój płytki paznokciowej

Wady wrodzone płodu :
SERCAubytki w przegrodzie
NEREKnerka podkowiasta
PRZEWODU POKARMOWEGO
zaburzenia rozwoju psychoruchowego
niezstąpienie jąder u chłopców i niedorozwój
narządów płciowych zewnętrznych u dziewcząt
TYLKO 10 % PRZEŻYWA 1 ROK ŻYCIA !
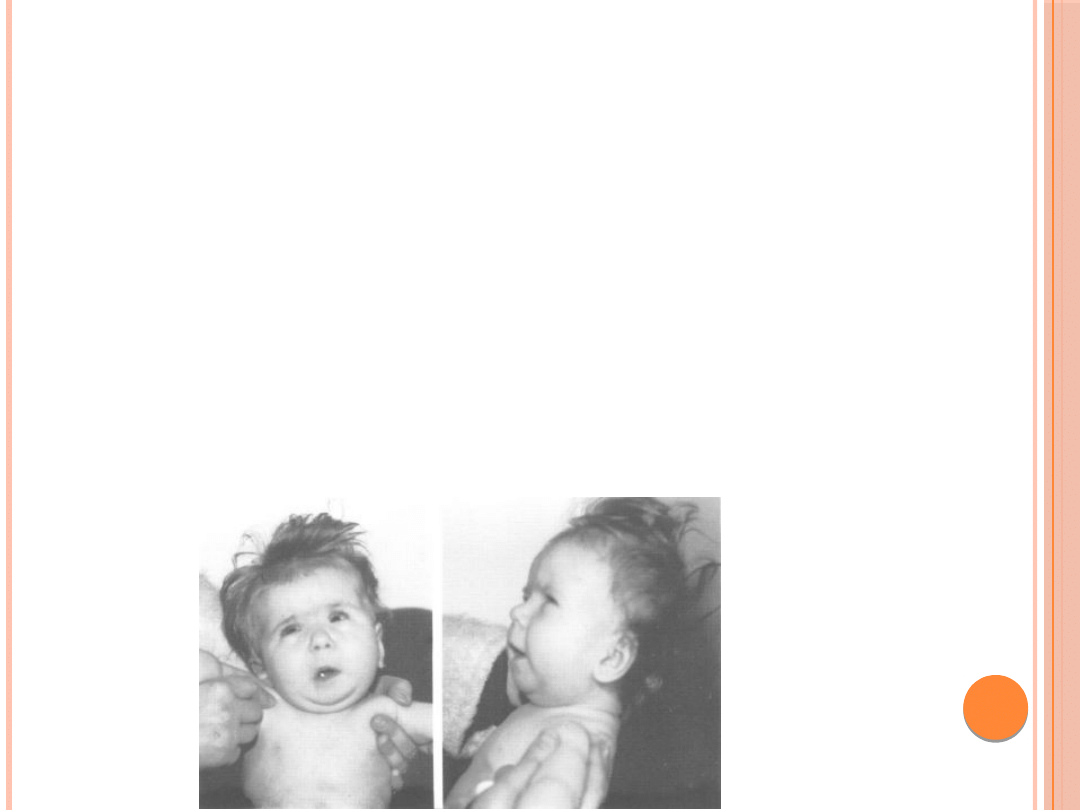
Trisomia 8 chromosomu :
47,XX,+8 lub 47,XY,+8
przypadki z kariotypem mozaikowatym
46,XX/47,XX,+8 lub 46,XY/47,XY,+8
cecha charakterystyczna to bruzda w odrębie
skóry dłoni i stóp
upośledzenie umysłowe niewielkiego stopnia

Poliploidia
u człowieka rzadko dotyczą spontanicznie poronionych
płodów i martwych urodzeń
występowanie triploidii lub tetraploidii stwierdza się u około
15% wszystkich spontanicznie poronionych płodów(triploidia
20 %, tetraploidia 6 % samoistnie poronionych zarodków).
mają one zwielokrotniony cały zestaw chromosównp.3n, 4n
wyróżniamy autopoliploidie oraz allopoliploidie (nie
występują u człowieka)
triploidia są wynikiem nieprawidłowego podziału
mejotycznego gamet lub zapłodnienia komórki jajowej przez
dwa plemniki –dispermia
tetraploidia gdy brak jest pierwszego podziału zygoty lub
zapłodnione przez 3 plemniki(polispermia –brak dowodów)
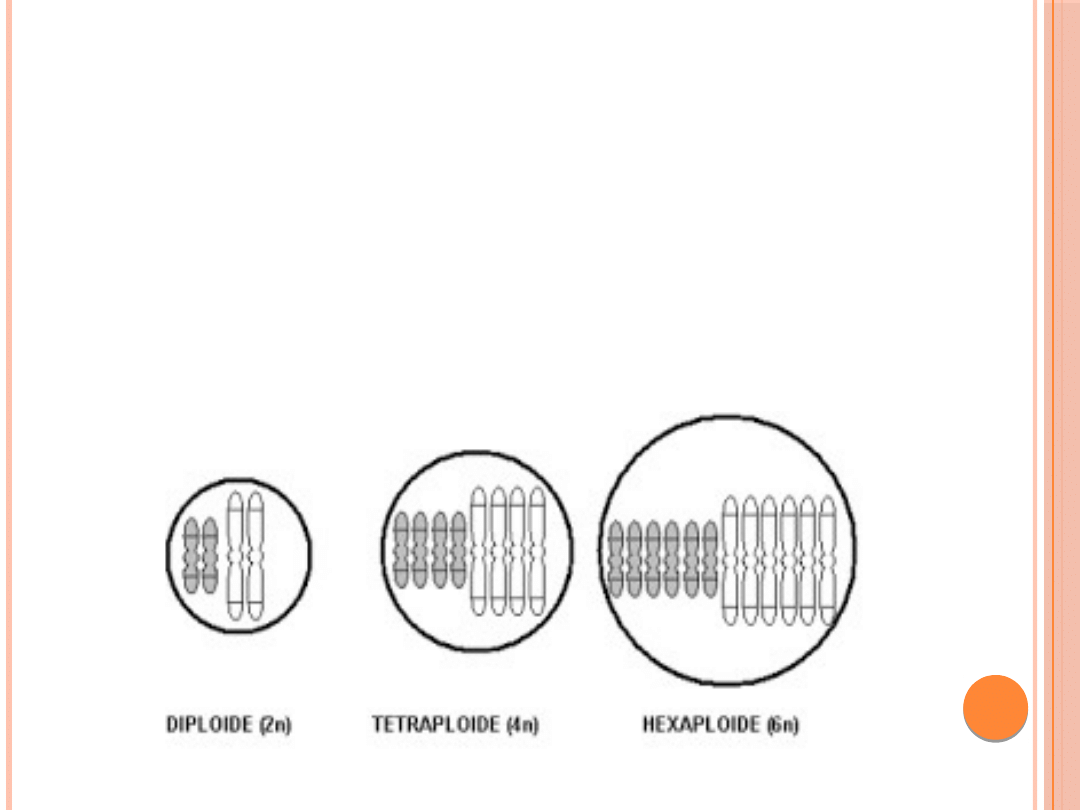
KOMÓRKI TETRAPLOIDALNE SĄ SPOTYKANE
W REGENERUJĄCEJ SIĘ WĄTROBIE A
POLIPLODIA W MEGAKARIOCYTACH (JĄDRA
SĄ POLIPLOIDALNE I LICZBA CHROMOSOMÓW
RÓWNA SIĘ 8 LUB 16KROTNEJ LICZBIE
HAPLOIDALNEJ

DZIĘKUJĘ ZA UWAGĘ
PAULINA RABIEJ, GR.45, LEKARSKI III
Document Outline
- Slide 1
- Wskazania do badania kariotypu:
- Slide 3
- Slide 4
- Chromatyna płciowa:
- Chromatyna X :
- Slide 7
- Slide 8
- Chromatyna Y:
- GENETYCZNA DETERMINACJA PŁCI :
- Geny biorące udział w róznicowaniu gonad:
- Slide 12
- Gen SRY
- Rola genu SRY w różnicowaniu pierwotnej gonady:
- Rozwój układu płciowego:
- Aberracje liczbowe autosomów:
- Zespół Downa
- Slide 18
- CECHY FENOTYPOWE ZESPOŁU DOWNA:
- Slide 20
- Zespół Pataua
- Cechy dysmorficzne dla zespołu pataua :
- Wady wrodzone narządów wewnętrznych:
- Zespół Edwardsa :
- Cechy dysmorficzne :
- Wady wrodzone płodu :
- Trisomia 8 chromosomu :
- Poliploidia
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
2 Ch chromosomowe cz 1 Stec
2 Ch chromosomowe cz 1 Puzio
5 Ch jednogenowe cz 2 Ch wieloczynnikowe Skrzypek
4 Ch jednogenowe cz 1 Ładosz
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Genetyka
Ch zw krŕg cz 2 do wydruku
ch stawow o podl imm cz II
ch stawow o podl imm psow i kotow cz. II
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Lekarski I rok ŚUM, biologia
Geometria w praktyce, cz 2 ?ch czterospadowy i kopertowy
Ch zw krŕg cz 1 do wydruku
więcej podobnych podstron