
CHOROBY
CHROMOSOMOWE
CZŁOWIEKA I
Szymon Stec, gr. 47

Miejsca łamliwe chromosomów
•
Miejsca kruche chromosomów to obszary o zwiększonej
częstości pęknięć lub złamań w obrębie chromatyd, które
mogą występować samoistnie i/lub powstawać w
określonych warunkach hodowlanych, pod wpływem
substancji chemicznych.
•
Miejsca kruche podlegają dziedziczeniu mendlowskiemu i mają
charakterystyczne lokalizacje chromosomowe.
Są
konserwatywne ewolucyjnie.
Ze względu na częstość
występowania w populacji i wrażliwość na związki chemiczne
dzieli się je na:
•
1
. rzadko występujące miejsca kruche:
•
a
. wrażliwe na działanie folianów
•
b
. niewrażliwe na działanie folianów
•
2
. często występujące miejsca kruche:
•
a. indukowane przez
afidokolinę,
•
b. indukowane przez
bromodezoksyurydynę (BrdU)
•
c. indukowane przez 5-
azacytydynę.

Miejsca łamliwe chromosomów
•
Rzadko występujące miejsca kruche obserwowane są w
mniej niż 5% populacji.
•
Często występujące miejsca kruche uznawane są za
swoistą, niepatogenną cechę chromosomów i uważa się,
że mogą być obecne u wszystkich członków populacji,
przy czym poziom ich ekspresji jest różny u
poszczególnych osób. U niektórych osób miejsce kruche
może być widoczne w preparacie cytogenetycznym nawet
w 30% komórek

Miejsca łamliwe chromosomów
•
Do dokładnej lokalizacji miejsc kruchych stosowany jest
zapis według ISCN (ang. International System for Human
Cytogenetic Nomenclature)
z użyciem skrótu „fra” (ang.
fragile
– kruchy) oraz określeniem chromosomu i miejsca
złamania: regionu, prążka i podprążka w tym
chromosomie, np. fra(X)(q27.3) lub fra(16)(q22.1)
•
Z wyjątkiem miejsc spontanicznie ulegających ekspresji,
do których należą FRA16B i FRA17A, specyficzna
ekspresja wszystkich opisanych do tej pory miejsc
kruchych następuje wskutek indukcji podczas hodowli
tkankowej.

Miejsca łamliwe chromosomów
•
W badaniach in vitro wykazano, że pojawiające się po indukcji
miejsca kruche częściej niż inne obszary chromosomów mogą
być zaangażowane w delecje i translokacje, wymianę
chromatyd siostrzanych (SCE
– Sister Chromatid Exchange),
amplifikacje genów oraz integrację plazmidów.
•
Charakterystyka
molekularna miejsc kruchych umożliwiła
badania patogenności tych miejsc in vivo i określenie ich
znaczenia dla powstawania aberracji chromosomowych,
istotnych dla rozwoju wad wrodzonych lub zmian
nowotworowych.
Wykazano, że wiele miejsc kruchych
zlokalizowanych jest w punktach złamań, zaangażowanych
w te aberracje
. Opisano także zależność pomiędzy
obecnością często występujących miejsc kruchych i miejscami
złamań charakterystycznymi dla nowotworów oraz rzadko
występujących miejsc kruchych i niepełnosprawnością
intelektualną.

Struktura molekularna miejsc kruchych
•
Rzadkie miejsca kruche:
Molekularne
podłoże powstawania rzadko występujących miejsc
kruchych jest
ściśle związane z ich sekwencją nukleotydową. W
przypadku miejsc
wrażliwych na foliany są to powtarzające się
sekwencje
trójnukleotydowe CGG a w przypadku miejsc
niewrażliwych na foliany - powtórzenia minisatelitarne -
motywy powtarzalne bogate w pary AT (VNTR - variable number
of tandem repeats).
Oba typy sekwencji
charakteryzują się zdolnością do tworzenia
specyficznych
drugorzędowych struktur, takich jak struktura
„spinki do włosów” czy struktura tetrahelikalna. Cechuje je
wysoka
elastyczność, która wpływa na dynamikę replikacji i
oddziaływania z białkami histonowymi, a co za tym idzie na
dekondensację materiału genetycznego, która może być
widoczna w preparacie cytogenetycznym jako miejsce kruche.

•
Częste miejsca kruche:
Często występujące miejsca kruche zbudowane są z sekwencji
bogatych w AT, ale w
przeciwieństwie do miejsc rzadko
występujących, nie zawierają motywów powtarzalnych i nie
wykazują tendencji do ekspansji. Ze względu na układ par AT w
postaci wysp
zwiększających elastyczność sekwencji, ich DNA
może
także
formować
charakterystyczne
struktury
drugorzędowe, co prawdopodobnie zaburza proces replikacji
oraz
wysokorzędową organizację chromatyny.

Konsekwencje kliniczne występowania
miejsc kruchych:
•
Zespół łamliwego chromosomu X:
•
Powtórzenia CGG w obszarze FRAXA zlokalizowane są w regionie 5’UTR
genu FMR1 (fragile X mental retardation 1).
•
U
zdrowych ludzi liczba powtórzeń trójnukleotydowych w tym miejscu waha się
od 6 do 54. Triplety CGG przerywne
są często tripletami AGG, których liczba
ma istotny wpływ na stabilność tego obszaru. Występowanie od 55 do 200
powtórzeń CGG określa się jako premutację. Występowanie powyżej 200
powtórzeń, czyli pełna mutacja, warunkuje hipermetylację promotora FMR1 i
zahamowanie ekspresji tego genu,
czyli brak produktu białkowego - FMRP
(fragile X mental retardation 1 protein).
•
FMRP
jest białkiem wiążącym RNA (RNA-binding protein), wchodzącym w
interakcje z licznymi cząsteczkami mRNA oraz różnymi białkami, z którymi
tworzy kompleksy oddziałujące z polirybosomami. FMRP ulega wysokiej
ekspresji w neuronach, gdzie uczestniczy w syntezie białek oraz w
gonadach.
•
Wykazano, że FMR2 ulega silnej ekspresji w mózgu a jego produkt białkowy
może działać jako aktywator transkrypcji. Obecność więcej niż 200 powtórzeń
CGG powoduje
hipermetylację promotora FMR2 i wyciszenie genu.

Konsekwencje kliniczne występowania
miejsc kruchych
•
Choroba jest dziedziczona w sposób recesywny w sprzężeniu z
chromosomem X. Ekspansja premutacji
do pełnej mutacji odbywa się w mejozie,
wyłącznie w żeńskich komórkach rozrodczych. Niepełnosprawność
intelektualną obserwuje się głównie u mężczyzn oraz u niewielkiej liczby kobiet –
nosicielek pełnej mutacji. W pełnoobjawowym zespole, klinicznie najczęściej
stwierdzana jest niepełnosprawność intelektualna w stopniu umiarkowanym lub
głębokim), zachowania o typie autystycznym, nadaktywność psychoruchowa,
zaburzenia uwagi, opóźnienie i zaburzenie rozwoju mowy oraz cechy
dysmorficzne w postaci
wydłużonej twarzy, dużych uszu i powiększenia jąder
u
mężczyzn. U mężczyzn, u których stwierdza się premutację, obserwuje się
niecharakterystyczne objawy w postaci zespołów lękowych, a w późniejszym
okresie życia, u prawie 30% nosicieli, objawy ataksji pochodzenia móżdżkowego.
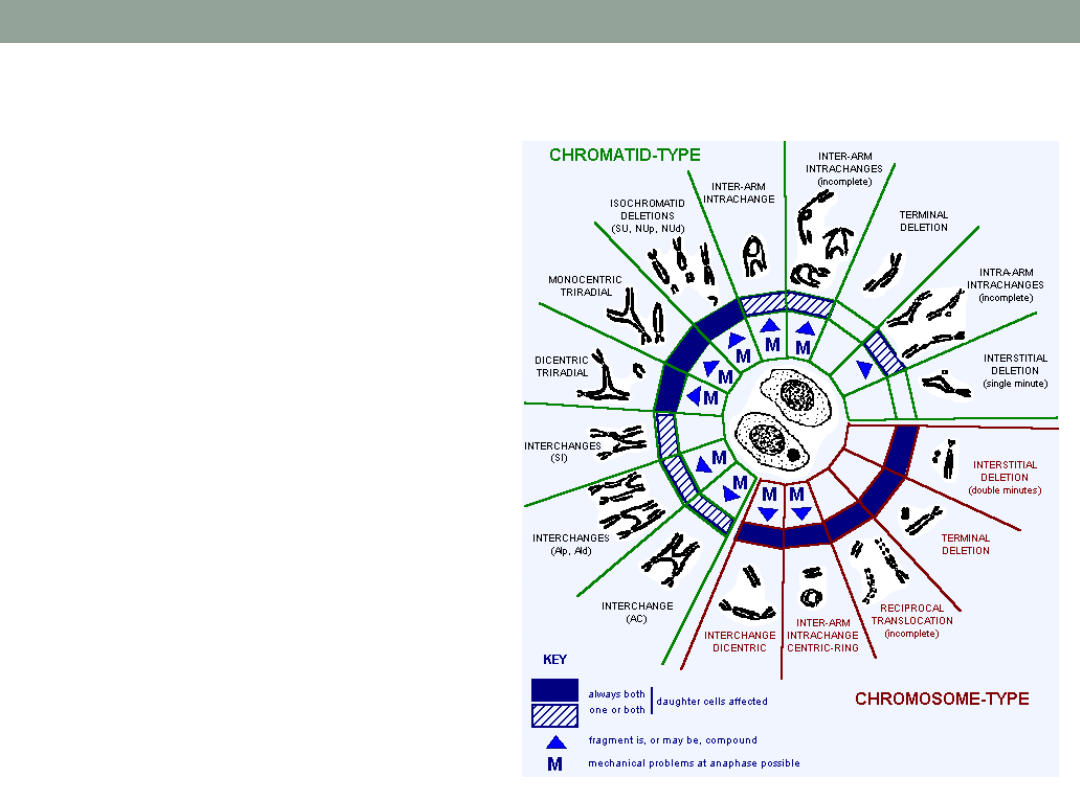
Aberracje chromatydowe
•
Jeśli
założymy,
że
wszystkie
zmiany
w
chromosomach
są
wynikiem
"złamań"
i
ponownych
połączeń
nici
chromosomowych,
możemy wyodrębnić dwa
typy aberracji:
•
Typ chromosomowy , gdzie
zmiany dotyczą
dwóch chromatyd siostrzany
ch w dowolnym locus
•
Typ chromatydowy , gdzie
zmiany dotyczą tylko
jednej chromatydy siostrzan
ej w dowolnym locus

Mozaicyzm
•
Jest to
obecność u jednej osoby dwóch lub większej liczby linii komórkowych wywodzących
się z pojedynczej zygoty, które różnią się kariotypem (składem ilościowym bądź strukturalnym
chromosomów). Spowodowana jest błędem „postzygotycznym” i może się pojawić w każdym
okresie życia - u zarodka, płodu lub po urodzeniu.
•
Okres
, w którym mozaikowatość się pojawia, determinuje proporcje, w jakich występują te dwie linie
komórkowe (lub większa ich liczba), co ma bezpośredni wpływ na ciężkość zmian fenotypu
spowodowanych obecnością nieprawidłowej linii komórkowej.
•
Funkcjonalna mozaikowatość występuje u kobiet, a polega na inaktywacji jednego chromosomu
z pary X w każdej komórce kobiety. Proces inaktywacji zachodzi w trofoblaście w 12 dniu po
zapłodnieniu, a w zarodku po 16 dniach. Do inaktywacji dochodzi tylko w komórkach somatycznych,
ponieważ rozrodcze potrzebują do swojej aktywności dwóch chromosomów X. Jest kwestią
przypadku (losu), czy w danej komórce ulega inaktywacji chromosom ojcowski, czy matczyny, ale
ten wybór jest stały dla wszystkich potomnych komórek. Nosicielki mutacji recesywnych,
sprzężonych z chromosomem X, są zazwyczaj klinicznie prawidłowe, ponieważ tylko pewna część
komórek ma zmutowany gen na aktywnym chromosomie X. Niekiedy zdarza się, że dochodzi do
inaktywacji w większości komórek chromosomu X z prawidłowym genem; wówczas objawy choroby
obserwuje się u nosicielek zmutowanych genów sprzężonych z chromosomem X.
•
Częstym zjawiskiem jest mozaikowatość chromosomowa, powstająca w wyniku
postzygotycznego
błędu podczas mitotycznych podziałów zygoty. Można ją wykryć w kariotypie
uzyskanym sposobem rutynowym bądź potwierdzić w razie wątpliwości za pomocą kariotypu
uzyskanego z hodowli fibroblastów. Obserwuje się ją np. w zespołach Downa, Turnera - wówczas
fenotyp chorego jest zmieniony mniej niż w aneuploidii całkowitej. Często dotyczy aberracji
chromosomowych, które jeżeli wystąpią w każdej komórce organizmu, są zazwyczaj letalne.
Istnieją schorzenia wywołane mozaikowatością chromosomową możliwą do wykazania jedynie w
kariotypie
uzyskanym z hodowli fibroblastów bądź innych tkanek.

Badanie cytogenetyczne
•
Cytogenetyka człowieka jest działem zajmującym się
badaniem chromosomów człowieka. Badania
cytogenetyczne dotyczą liczby, budowy oraz aberracji w
strukturze chromosomów.
•
Badane są zwykle chromosomy metafazowe lub
prometafazowe. Jest to najbardziej skondensowana
forma chromatyny.
•
Chromosomy metafazowe składają się z dwu
siostrzanych chromatyd połączonych centromerem (cen).
Każdy prawidłowy chromosom ma jeden centromer
położony charakterystycznie dla danej grupy.

Badanie cytogenetyczne
•
Oznaczanie kariotypu
– określenie liczby i struktury
chromosomów w komórkach badanej osoby. Analizowane
są chromosomy z dzielących się mitotycznie kom.
somatycznych.
•
Rutynowo wykorzystywane są komórki: limfocyty krwi
obwodowej, rzadziej fibroblasty
skóry lub komórki innych
tkanek.
•
Prenatalnej oceny kariotypu
dokonuje się na podstawie
badania chromosomów amniocytów z płynu
owodniowego lub
komórek trofoblastu. W pewnych
określonych warunkach analizowany jest on na podstawie
komórek krwi pępowinowej.

Badanie cytogenetyczne
Postępowanie laboratoryjne jest w zasadzie niezależne od
rodzaju badanych komórek i przebiega wg schematu:
1.
N
amnażanie komórek z pobranego materiału in vitro w celu
uzyskania komórek mitotycznych. Czas hodowli zależy od
rodzaju komórek. w tkankach o dużej aktywności mitotycznej
in vivo (np. szpik kostny)
hodowla może być zastąpiona
techniką bezpośrednią uzyskiwania chromosomów lub
24h inkubacją.
2.
Nagromadzenie
komórek w stadium metafazowym poprzez
wprowadzenie analogu alkaloidu roślinnego (kolchicyny)
który zatrzymuje podziały w stadium metafazy w którym
morfologia chromosomów jest najbardziej czytelna.

Badanie cytogenetyczne
3. Zakończenie hodowli i sporządzenie preparatów
chromosomowych
. W tym celu komórki poddawane są:
•
szokowi hipotonicznemu (w celu rozproszenia chromosomów w
komórce)
•
utrwalaniu
•
nakrapianiu na szkiełko podstawowe
4. Barwienie
chromosomów z zastosowaniem jednej lub
kilku technik prążkowych.
5. Analiza
chromosomów w mikroskopie świetlnym
(1000x), wykonanie dokumentacji
fotograficznej oraz sporządzenie
kariotypu.

Badanie cytogenetyczne
•
Hodowla limfocytów:
•
wymaga czynników mitotycznych, najczęściej uzyskiwanej z fasoli
fitohemaglutininy
(PHA) która pobudza je do podziałów.
•
Makrometoda:
•
wymaga 5-10 ml krwi i hodowla limf. izolowanych przez wirowanie lub
sedymentację
•
Mikrometoda (prostsza):
•
0,3-
0,5 ml pełnej krwi
•
czas hodowli zwykle 72h
•
wady: brak możliwości bezpośredniego powtórnego badania lub
dalszych badań z tych samych komórek.
Wynika to z ograniczonych zdolności limf. T do wzrostu in vitro.
•
można jednak „unieśmiertelnić” hodowlę wirusem EBV.

Badanie cytogenetyczne
•
Hodowla fibroblastów:
•
zwykle tylko w określonych przypadkach (zwykle gdy wynik z
limfocytów nie odpowiada obrazowi klinicznemu – co może się
zdarzyć przy mozaikowatości komórkowej)
•
Fibroblasty uzyskuje się przez pobranie niewielkiego wycinka
skóry, który poddawany jest działaniu trypsyny lub kolagenazy w
celu uzyskania zawiesiny komórek.
•
Komórki rosną in vitro przytwierdzone do naczynia
•
zaleta: długotrwała możliwość wielokrotnego przeprowadzania
analizy kariogramu
. Mogą być także przechowywane w ciekłym
azocie i wykorzystane po latach
•
wada: długi czas potrzebny do uzyskania odpowiedniej ilości
dzielących się komórek

Badanie cytogenetyczne
•
Hodowla
amniocytów i kom. trofoblastu:
•
amniocyty
uzyskuje się przez amniopunkcję wczesną (12-14 tydz.
ciąży) lub późną (15-17); stanowią one heterogenną populację
komórek spośród których tylko część ma zdolność wzrostu in vitro
•
amniocyty
tak jak fibroblasty przyrastają do dna naczynia
•
z chwilą zaobserwowania odpowiedniej ilości metafaz, kończy się
hodowlę i sporządza preparaty; zwykle trwa to 10-21 dni
•
komórki trofoblastu pobiera się z kosmówki za pomocą biopsji przez
powłoki brzuszne między 8 a 11. tygodniem ciąży. Preparaty można
uzyskać bezpośrednio z materiału lub hodowli (trwającej podobnie
długo co hodowla amniocytów).
•
konieczne jest
oczyszczenie komórek kosmówki od domieszki
tkanki matczynej czyli doczesnej (które w hodowli rosną szybciej).
•
preparat bezpośredni jest możliwy do uzyskania dzięki wysokiej
aktywności mitotycznej kosmówki in vivo
•
ryzyko wiąże się z możliwością stwierdzenia aberracji
występującej w kosmówce a nieobecnej u płodu

Badanie cytogenetyczne
•
Hodowla z komórek nowotworowych szpiku:
•
materiałem może być preparat biopsyjny (nakłucie mostka, talerza kości
biodrowej u dzieci) lub z krwi obwodowej pacjenta
•
możemy badać kariotyp bezpośrednio po pobraniu lub po krótkotrwałej hodowli
in vitro (24-48h)
•
ogólnie stosowaną zasadą jest dążenie do przedstawienia wyników na
podstawie analizy 20 metafaz
•
Hodowla z litych guzów nowotworowych:
•
komórki pochodzące z płynów wysiękowych jamy otrzewnej lub opłucnej lub
fragmentów tkanki guzów
•
źródłem komórek metafazowych są krótkotrwałe hodowle in vitro
•
tkanki pobrane umieszczamy w dowolnym płynie hodowlanym z antybiotykiem
i niekiedy środkiem mikostatycznym a następnie rozdrobione umieszczamy w
pożywce z dodatkiem kolagenazy.
•
po uzyskaniu zawiesiny i założeniu hodowli in vitro, cytogenetyk obserwuje
hodowle oczekując na uzyskanie odpowiedniej ilości podziałów mitotycznych
•
w przypadku „zanieczyszczenia” próbek prawidłowymi komórkami np.
nabłonkowymi, stosuje się metodę sedymentacji, specjalne podłoża oraz
pożywki stymulujące wzrost specyficznych komórek.

Badanie cytogenetyczne
•
Uzyskane chromosomy metafazowe barwi się
technikami prążkowymi.
•
Sporządzenie kariotypu polega na ułożeniu
chromosomów wg obowiązujących reguł klasyfikacji ISCN
(ang. międzynarodowy system nazewnictwa
cytogenetycznego) z 1995r.
•
Za kryteria klasyfikacji przyjęto:
•
wielkość chromosomów
•
położenie centromeru
•
rozmieszczenie prążków w chromosomach
•
Chromosomy autosomalne podzielono na 7 grup i do
dwóch z nich przypisano chromosomy płci.

Grupa
Para
A
1,2,3
B
4,5
C
6,7,8,9,10,11,12, X
D
13,14,15
E
16,17,18
F
19,20
G
21, 22, Y
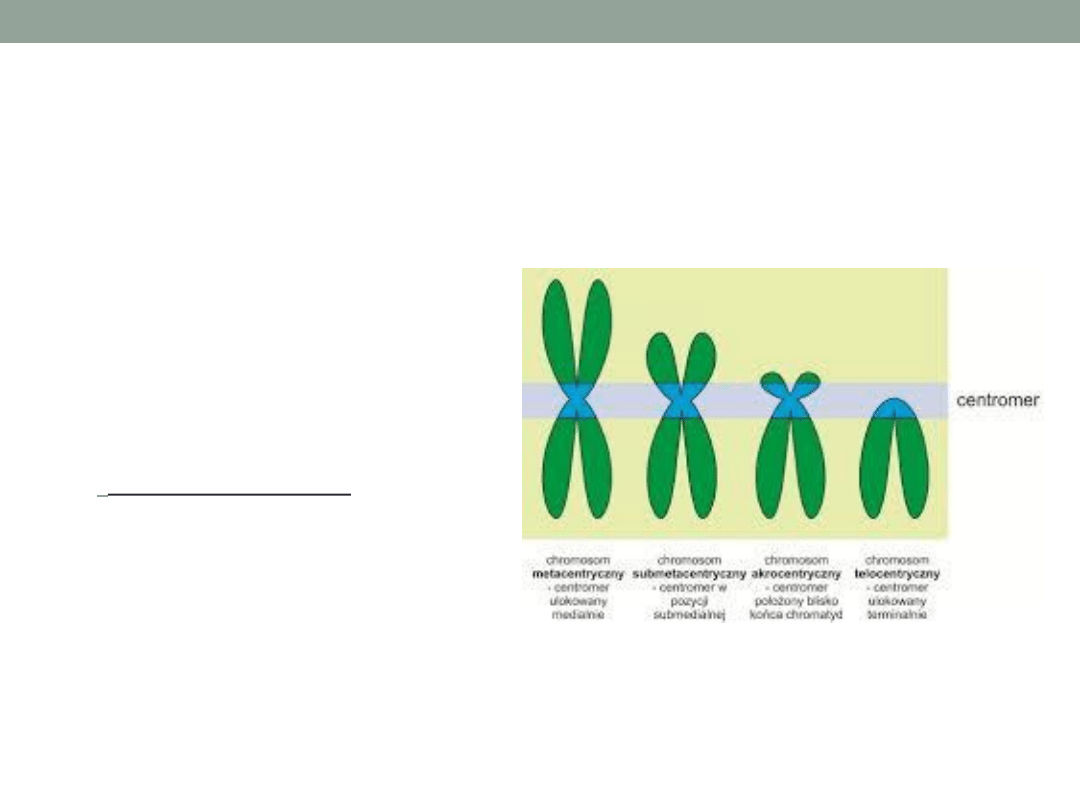
Badanie cytogenetyczne
•
Chromosomy dzielimy
na typy:
•
metacentryczne
•
submetacentryczne
•
akrocentryczne
•
telocentryczne

Grupa Cechy
A
1,3
– duże metacentryczne
2
– submetacen.
B
4,5
– duże submetacen.
C
6,7,8,9,10,11,12, X
średnie submetacen.
D
13,14,15 -
duże
akrocen
. z możliwymi
satelitami
E
16,17,18
–małe meta- i
submetacentryczne
F
19,20
– najmniejsze
metacentryczne
G
21, 22- akrocentryczne + sat
Y
– akroc. nie posiada satelit

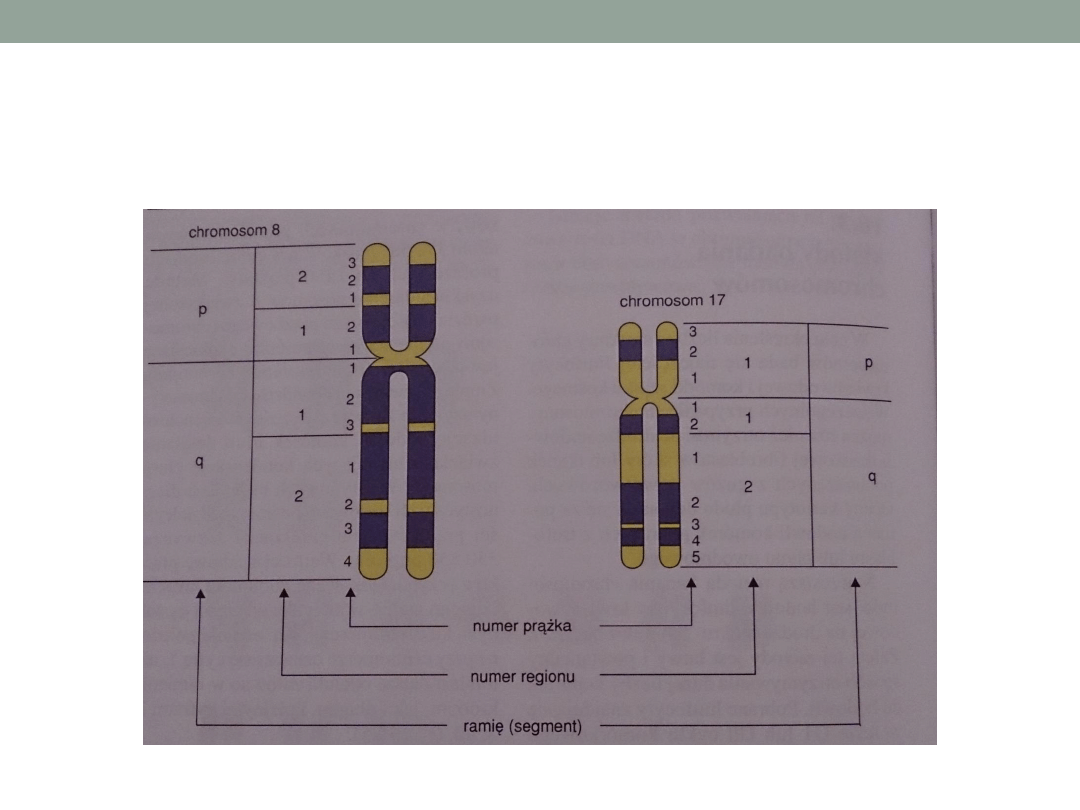
Badanie cytogenetyczne
– analiza
prążkowa
•
Każdy chromosom ma specyficzny dla siebie wzór
prążkowy różniący się wielkością i intensywnością
zabarwienia.
•
W haploidalnym zestawie metafazowym można wyróżnić
ok. 400 prążków a na wcześniejszych stadiach podziału
– w prometafazie lub profazie, zależnie od kondensacji
500-
1250 prążków. Metoda uzyskiwania chromosomów o
zwiększonej rozdzielczości (HRT) opiera się na
synchronizacji hodowli lub działanie czynników
hamujących kondensację.
•
Wielkość najmniejszej zauważalnej aberracji przy
rozdzielczości na poziomie 400 prążków odpowiada 7-10
x 10
6
pz
a przy rozdzielczości 850 prążków wynosi 2-5 x
10
6
co jest jednak w praktyce nieosiągalne.

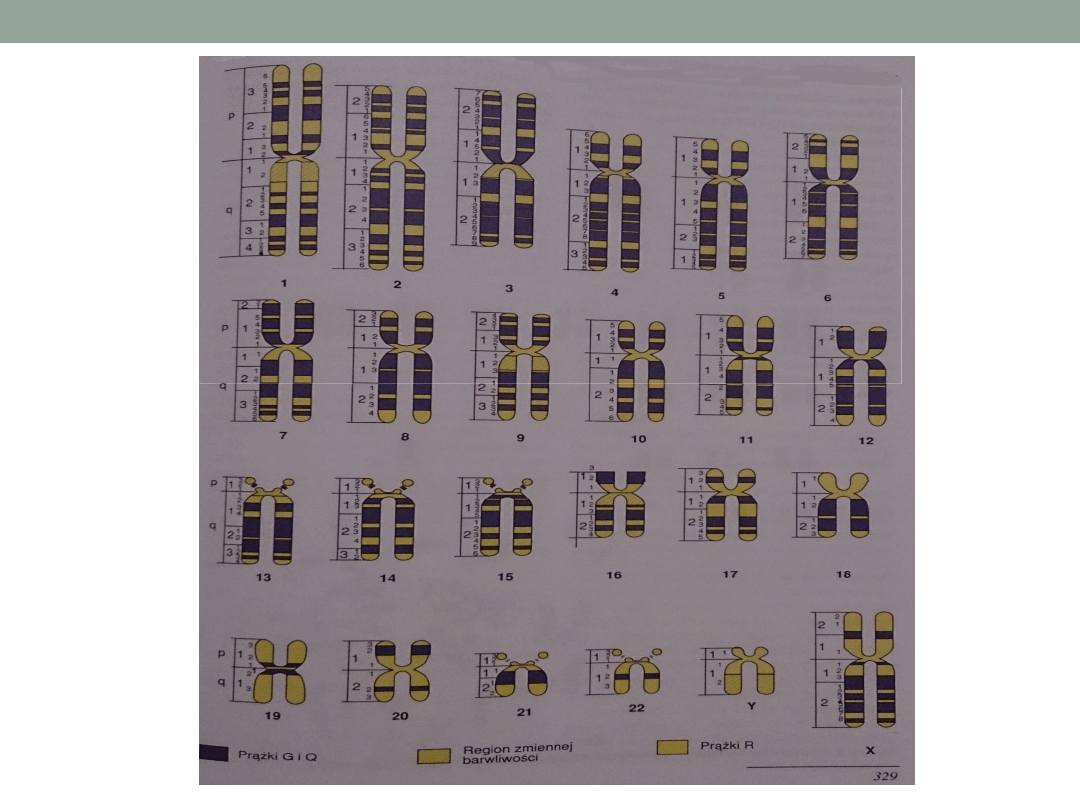
Metody barwienia
chromosomów i
zastosowanie
•
Obraz prążkowy umożliwiający rozpoznanie każdego z 46
chromosomów jest otrzymywany za pomocą trzech
podstawowych technik barwienia.
Są to techniki uzyskiwania
prążków określanych symbolami: G (od barwnika Giemsy),
Q (ang. quinacrine) oraz R (ang. reverse).
•
Obraz ten stanowi odbicie ich strukturalnego i funkcjonalnego
zróżnicowania.
•
ciemne prążki G odpowiadają regionom o dużej zawartości par AT
(późno replikującym, zawierającym nieliczne aktywne geny); różnią się
także zawartością i składem towarzyszących białek (bogate w
wiązania S-S)
•
jasne prążki odpowiadają regionom wcześnie replikującej i aktywnej
transkrypcyjnie euchromatyny
•
wzór prążków G, poza nielicznymi przypadkami, odpowiada obrazowi
prążków Q

Metody barwienia
chromosomów i
zastosowanie
•
Prążki G powstają standardowo przez trawienie
chromosomów trypsyną a następnie barwienie
odczynnikiem Giemsy (GTG) lub Wrighta (GTW)
•
Prążki Q powstają w wyniku barwienia chromosomów
pochodnymi akrydyny (QFQ) mającymi zdolność
interkalacji nici DNA i wykazującymi powinowactwo do
pewnych zasad; prążki Q silnie fluoryzujące odpowiadają
rejonom bogatym w pary A-T a miejsca wygaszenia
parom G-C
•
Wzór prążków R stanowi odwrotność prążków G, a może
być uzyskiwany różnymi metodami (RHG, RBA, RBG) a
używany jest do ujawnienia drobnych aberracji
niedostrzegalnych w barwieniu G lub Q

Metody barwienia chromosomów i
zastosowanie
•
Stwierdzenie jakiejkolwiek nieprawidłowości wzoru
prążkowego, sugerującej aberrację chromosomu, wymaga
zwykle dodatkowych
barwień i badań w celu charakterystyki i
identyfikacji aberracji; zwykle to:
•
technika wybarwiania heterochromatyny centromerowej (
prążki C)
•
srebrzenie organizatorów jąderka (AgNOR)
•
barwienie
distamycyną A/DAPI
•
autoradiografia
•
cytometria
przepływowa
•
hybrydyzacja in situ (ISH)
•
Jednym z elementów analizy chromosomowej jest także
identyfikacja aktywnego i nieaktywnego chr.
X, szczególnie w
analizie translokacji między X ojcowskim i autosomami;
badanie ma znaczenie w ustalaniu związku między fenotypem
a aberracją chromosomową. Identyfikacja polega na słabszej
intensywność fluorescencji nieaktywnego X.
Metody barwienia chromosomów i
zastosowanie

Identyfikacja aberracji
– dodatkowe
barwienia
•
barwienie organizatorów jąderkowych AgNOR:
•
NOR są zlokalizowane w nitkach satelitonośnych chromosomów
akrocentrycznych
a zawierają geny rybosomalne dla 18S i 28S
rRNA.
•
Organizatory przejawiające aktywną transkrypcję rRNA barwią się
selektywnie azotanem srebra
•
ponadto stosowana jest do identyfikacji chromosomów
markerowych, które często są utworzone z ramion krótkich
chromosomów akrocentrycznych

Identyfikacja aberracji
– dodatkowe
barwienia
•
barwienie
distamycyną A/DAPI:
•
DAPI jest to barwnik o powinowactwie do par A-T a distamycyna to
antybiotyk
•
daje obraz prążkowy podobny do wzoru prążkowego Q
•
uzyskany obraz jest bardziej kontrastowy
•
silnie fluoryzują regiony heterochromatyny konstytutywnej
chromosomów 1,9,15p,16 i Yq
•
służy przede wszystkim do identyfikacji chromosomów
markerowych pochodzących z 15

Identyfikacja aberracji
– dodatkowe
barwienia
•
autoradiografia
•
najczęściej wykorzystuje się tryt lub trytowaną aktynomycynę D
(H3-AMD)
•
zdolność AMD do wiązania się z DNA umożliwia stosunkowo
precyzyjną analizę rozmieszczenia rejonów G-C lub A-T

Identyfikacja aberracji
– dodatkowe
barwienia
•
cytometria
przepływowa:
•
wykorzystuje się pomiar ugięcia i rozproszenia światła oraz
wzbudzenia fluorescencji w zawiesinie komórkowej
•
można ocenić ploidalność komórki na podstawie pomiaru
intensywności fluorescencji w specjalnym aparacie – cytometrze
przepływowym
•
sporządza się wykresy zawartości DNA w poszczególnych
populacjach komórek czyli histogramy DNA.
•
Z liczbą chromosomó wiąże się ilość DNA w jądrze komórkowym a
wynosi ona 2n dla prawidłowej komórki diploidalnej nie będącej w
trakcie podziału. W przypadku zaburzeń genomu zawartość DNA
zmienia się.
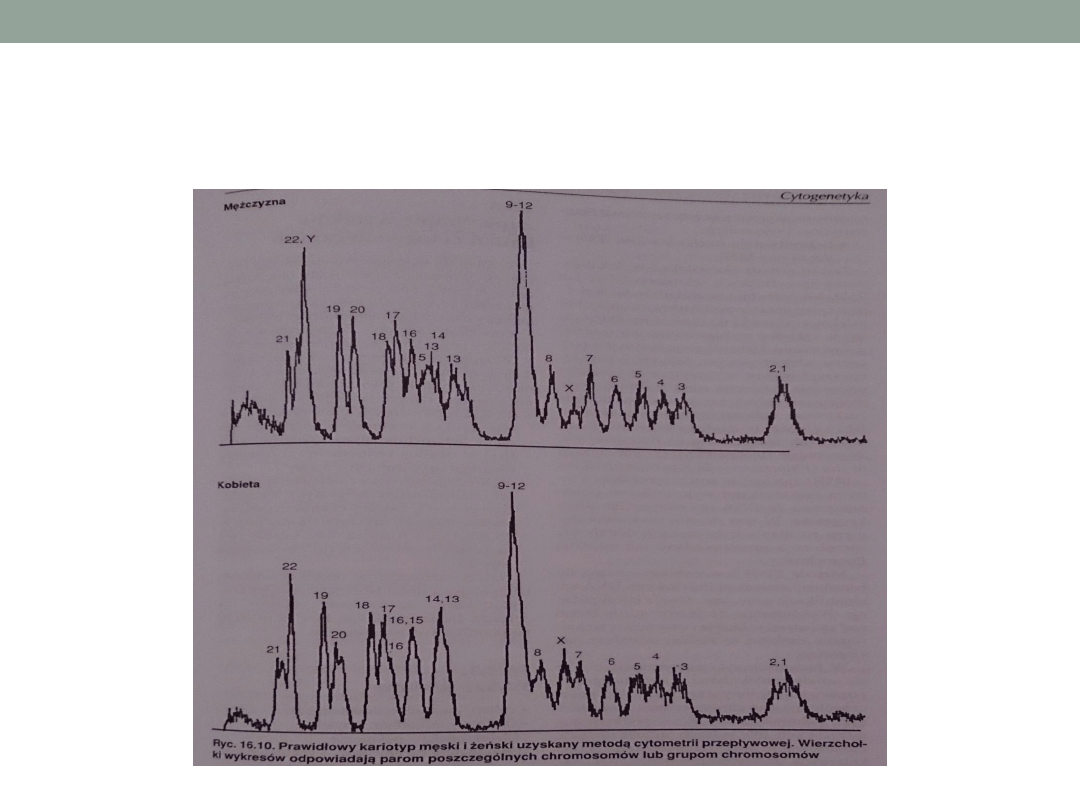
Identyfikacja aberracji
– dodatkowe
barwienia

Identyfikacja aberracji
– dodatkowe
barwienia
•
hybrydyzacja in situ (ISH):
•
pozwala na lokalizację specyficznych sekwencji
DNA lub RNA w materiale biologicznym na
szkiełku podstawowym lub w zawiesinie.
•
jest to proces wieloetapowy wymagający
przygotowania sond molekularnych znakowanych
radioaktywnie lub fluorochromem.
•
W metodzie fluorescencyjnej hybrydyzacji in situ
(FISH) stosuje się sondy sprzężone innymi
cząsteczkami wykazującymi powinowactwo do
DNA (np. biotyna, digoksygenina).
•
FISH jest stosowana do badań nad zmianami
ilościowymi DNA np. amplifikacje, delecje
•
metoda ta pozwala wykrywać sekwencji nie tylko
w stanie metafazowym chromosomów ale także w
interfazie

Metoda
barwienia
Zastosowanie do identyfikacji
Prążki Q
Wszystkich chromosomów. wariantów Y, satelitów i
regionów centromerowych chr. akrocentrycznych
Prążki G
Wszystkich chromosomów i ich aberracji
Prążki R
Wszystkich chromosomów i ich aberracji a szczególnie tych
w dystalnych
częściach chromosomu
Prążki C
Heterochromatyny
okołocentromerowej oraz wariantów
heterochromatyny konstytutywnej chr. 1,9,16, Y
AgNOR
Organizatrów jąderka na krótkich ramionach chromosomów
akrocentrycznych 13,14,14,21,22 i ich wariantów
DA/DAPI
Heterochromatyny
okołocentromerowej chromosomów
1,9,19, Yq
ale najczęściej chromosomów markerowych z
ramionami 15q
RBA
(replikacyjne
prążki R)
Nieaktywnego, późno replikującego się chromosomu X

Zapisywanie wyników badań
cytogenetycznych
•
Według obowiązującego systemu zapis kariotypu człowieka
zawiera liczbę określającą ilość chromosomów w kom.
diploidalnej, przecinek, po przecinku litery oznaczające
chromosomy płciowe.
Prawidłowy kariotyp żeński: 46,XX i męski: 46,XY
•
W zapisie
prawidłowego kariotypu uwzględnia się też
obecność większej niż w granicach normy ilość
heterochromatyny konstytutywnej oraz dużych satelitów, np.:
•
46,XX,1qh+ oznacza kobietę z obecnością heterochr. konstytutuwnej
(h+) w okolicy centromerowej
ramienia długiego chrom. 1
•
46,XY,9qh-
oznacza mężczyznę z ubytkiem heterochr. konstytutywnej
w ok. centromerowej
ramienia długiego chr. 9
•
Należy zaznaczyć, że obecnosć większej lub mniejszej ilości
heterochromatyny konstytutywnej oraz wielkość satelitów nie
oznacza patologii a jest to cecha polimorfizmu chromosomów.

Zapisywanie wyników badań
cytogenetycznych -
skróty
+ (plus)
– nadmiar, dodatek
- (minus)
– niedobór, ubytek
: -
pęknięcie
:: -
pęknięcie i połączenie
, (przecinek)
– rozdziela liczbę chromosomów i
opisu aberracji
; -
rozdziela zapisy dotyczące różnych chromosomów
. (kropka)
– oddziela zapis subprążków i prążków
→ - od do (zakres regionu chromosomu)
( )
– strukturalnie zmienione chromosomy lub miejsca
pęknięć za skrótem typu aberracji
/ -
oddziela różne linie w mozaikach

Zapisywanie wyników badań
cytogenetycznych -
skróty
cen
– centromer
del
– delecja
der
– chromosom pochodny
dic
– chromosom dicentryczny
dup
– duplikacja
h
– heterochromatyna konstytutywna
i
– izochromosom
ins
– insercja
inv
– inwersja
kpz
– kilo par zasad
mar
– chromosom markerowy
mat
– pochodzenie matczyne
pat
– pochodzenie ojcowskie
NOR
– organizator jąderkowy
p
– ramię krótkie
q
– ramię długie
PAR
– region pseudoaktywny
r
– chromosom pierścieniowy
s
– satelity
t
– translokacja
tel
– telomer
ter
– koniec chromosomu
UDP
– uniparentalna disomia
UDHD
– uniparentalna heterodisomia
UPID
– uniparentalna izodisomia

Przykłady

Bibliografia
•
„Diagnostyka laboratoryjna” Journal of Laboratory
Diagnostics 2010 • Volume 46 • Number 1 • 81-86
•
„Podstawy genetyki dla studentów i lekarzy” – pod
redakcją Gerarda Drewy i Tomasza Ferenca wyd. II,
Wrocław 2003
•
„Biologia molekularna. Elementy genetyki klinicznej.”
Jerzy Bal, wyd. PWN, Warszawa 2013a
Wyszukiwarka
Podobne podstrony:
2 Ch chromosomowe cz 1 Puzio
3 Ch chromosomowe cz 2 Rabiej
5 Ch jednogenowe cz 2 Ch wieloczynnikowe Skrzypek
4 Ch jednogenowe cz 1 Ładosz
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Genetyka
Ch zw krŕg cz 2 do wydruku
ch stawow o podl imm cz II
ch stawow o podl imm psow i kotow cz. II
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Lekarski I rok ŚUM, biologia
Geometria w praktyce, cz 2 ?ch czterospadowy i kopertowy
Ch zw krŕg cz 1 do wydruku
więcej podobnych podstron