
CHOROBY
JEDNOGENOWE
CZŁOWIEKA II.
CHOROBY
WIELOCZYNNIKOWE.
Joanna Skrzypek
GS 46
(III rok lekarski)

C
HOROBY JEDNOGENOWE SPRZĘŻONE Z
CHROMOSOMEM
X

W zaburzeniach sprzężonych z chromosomem X
zmutowany gen występuje na chromosomie X.
U kobiet występują dwa chromosomy X (X
mat
i X
pat
) –
jeden z tych chromosomów jest nieaktywny w każdej
komórce organizmu.
Hipoteza Lyon: jeden chromosom X w każdej
komórce jest losowo inaktywowany we wczesnej fazie
rozwoju embrionalnego kobiet. Dzięki temu kobiety,
posiadające dwie kopie chromosomu X, wytwarzają
produkty genów związanych z chromosomem X w
ilościach podobnych do mężczyzn – kompensacja
dawki.

W wyniku losowej inaktywacji jednego
chromosomu X w każdej komórce, wszystkie
prawidłowo rozwijające się kobiety mają dwie
odrębne populacje komórek (jedna – aktywny
chromosom X od ojca, druga- aktywny
chromosom X od matki). Kobiety są „mozaikami”
wobec tego chromosomu.
Wynika z tego, że kobiety mogą być nosicielami
zaburzeń sprzężonych z chromosomem X
(zarówno dominujące, jak i recesywne).

W przypadku mężczyzn występuje tylko jeden
chromosom X- mężczyzna posiada tylko jedną
kopię genu sprzężonego z tym chromosomem (jest
hemizygotą).
W przeciwieństwie do kobiet, u mężczyzn
chromosom X pozostaje czynny W KAŻDEJ
KOMÓRCE ORGANIZMU (każdy allel danego
genu –prawidłowy czy zmutowany, ulega
ekspresji).
Mężczyźni nie wykazują mozaikowatości tych
genów!
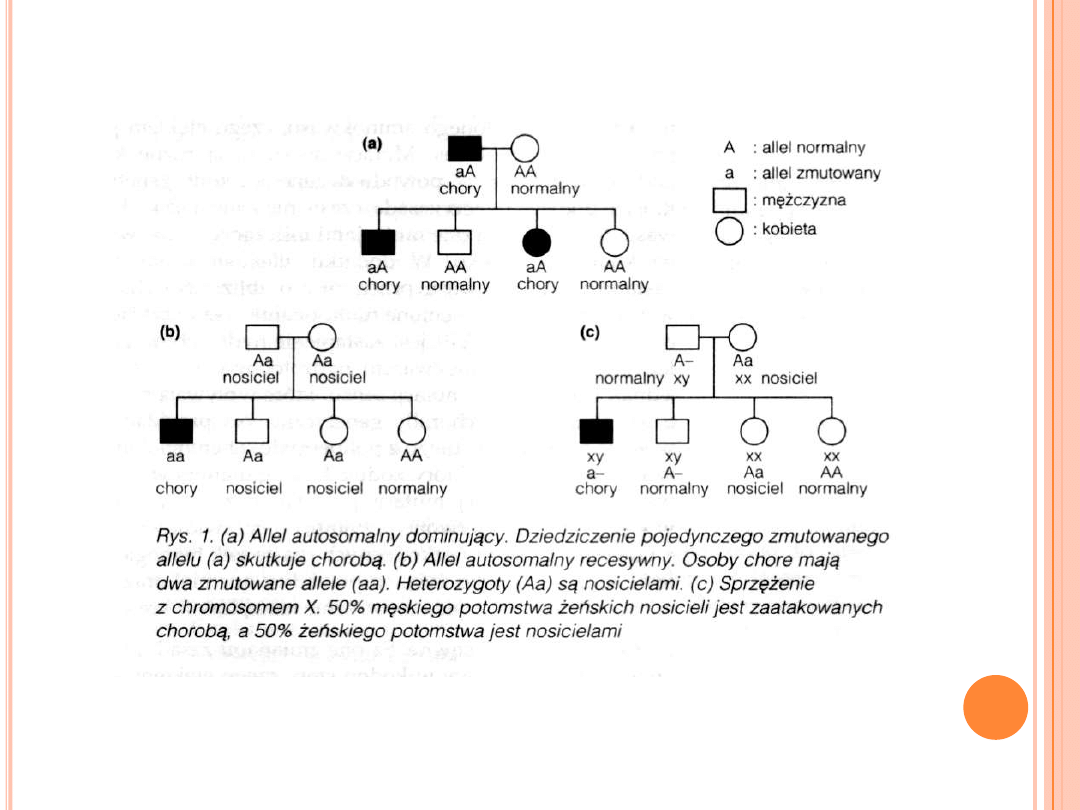

C
HOROBY DZIEDZICZONE
DOMINUJĄCO SPRZĘŻONE Z
CHROMOSOMEM
X
Cechy dziedziczenia:
chory mężczyzna ma wyłącznie chore córki i
wyłącznie zdrowych synów (niemożliwe jest
dziedziczenie z ojca na syna)
chore kobiety heterozygoty przekazują cechę 50%
swego potomstwa (niezależnie od płci)
chore kobiety homozygoty przekazują cechę
wszystkim swoim dzieciom.

Z
ESPÓŁ ŁAMLIWEGO CHROMOSOMU
X
(
ZESPÓŁ
M
ARTINA
-B
ELLA
)
Zespół łamliwego chromosomu X jest najczęstszą
przyczyną upośledzenia umysłowego związanego
z chromosomem X. Ponadto jest drugą co do
częstości przyczyną upośledzenia umysłowego u
chłopców ( 1- zespół Downa)
Częstość występowania:
M - 1:2500
K – 1:2000

Z
ESPÓŁ ŁAMLIWEGO CHROMOSOMU
X
(
ZESPÓŁ
M
ARTINA
-B
ELLA
)
Za chorobę odpowiedzialna jest mutacja genu
FMRI znajdującego się w Xq27.3. Koduje on
białko FMRP (białko wiążące RNA).
Rejon genu nie ulegający translacji zawiera
powtórzenia CGG. U osób zdrowych liczba tych
powtórzeń wynosi od 6 do 50; u nosicieli (tzw.
premutacja) – od 50-200; natomiast u osób
chorych występuje ponad 200 powtórzeń CGG.

Z
ESPÓŁ ŁAMLIWEGO CHROMOSOMU
X
(
ZESPÓŁ
M
ARTINA
-B
ELLA
)
W dziedziczeniu tej choroby występuje tzw.
paradoks Shermana, z którego wynika, że:
- córki nosiciela nigdy nie są chore,
-synowie nosiciela mogą zachorować.
*Premutacje mają skłonność do powiększania się
w kolejnych pokoleniach – im większa jest
premutacja, tym większe prawdopodobieństwo
wystąpienia pełnej mutacji w kolejnym
pokoleniu.

Z
ESPÓŁ ŁAMLIWEGO CHROMOSOMU
X (
ZESPÓŁ
M
ARTINA
-B
ELLA
)
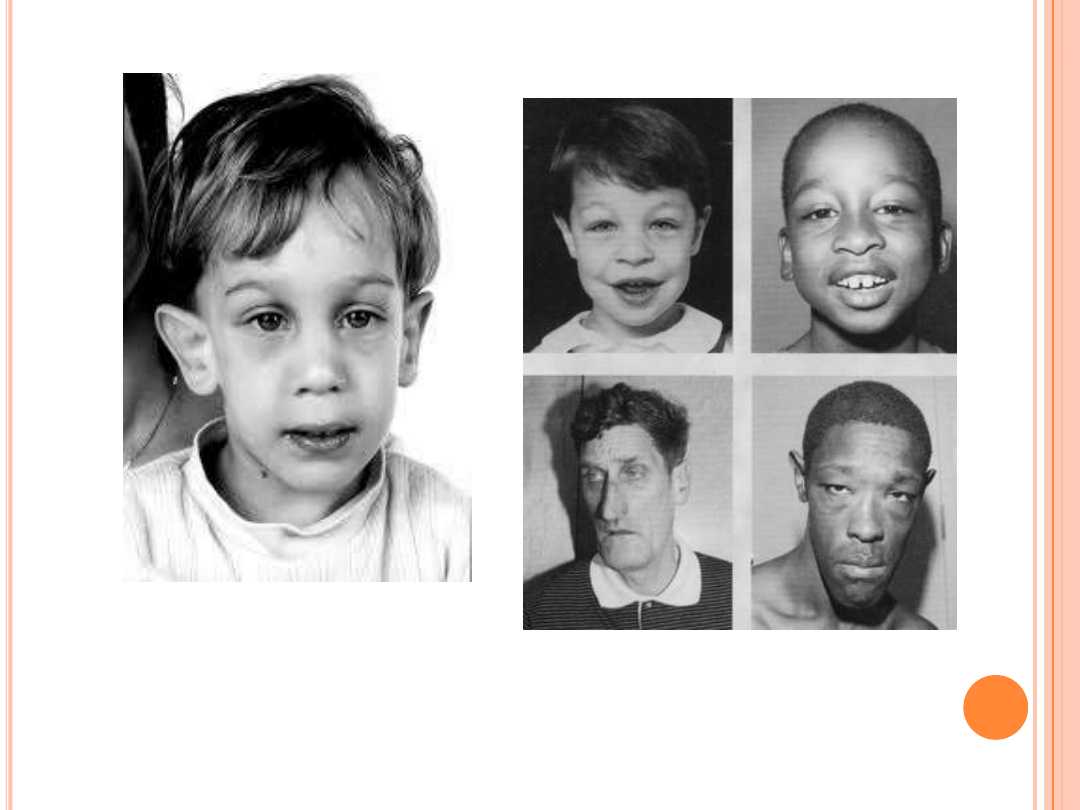
-niska waga urodzeniowa,
mały obwód głowy
-zwiększona objętość jąder
-duże małżowiny uszne
-hipotonia mięśniowa
*u dzieci: objawy autyzmu,
zaburzenia mowy, opóźnienie
rozwoju psychoruchowego
-deformacje twarzoczaszki
(twarz podłużna, wydatne guzy
czołowe, duże i odstające
małżowiny uszne, duża żuchwa,
wysokie podniebienie,
hipoteloryzm)
-charakterystyczne
bladoniebieskie tęczówki
-makroorchidyzm
-zniekształcenie kręgosłupa,
szerokie i krótkie palce
-napady padaczki
-postępująca encefalopatia
Cechy fenotypowe
niemowląt męskich:
Cechy fenotypowe
dorosłych mężczyzn:

Z
ESPÓŁ
R
ETTA
Zespół Retta jest genetycznie uwarunkowanym,
postępującym zaburzeniem neurorozwojowym, jedna z
najczęstszych przyczyn upośledzenia umysłowego u
dziewczynek.
Częstość występowania- 1:10000-15000
Jest wynikiem mutacji genu MEC2 zlokalizowanego na
Xq20 (mutacja ta odpowiedzialna za ok. 80% zachorowań).
Gen ten koduje białko MeCP2 (represor transkrypcji).
Inne przyczyny: mutacja genu CDKL5 na Xp22, kodujące
białkową kinazę serynowo-treoninową 9, odpowiedzialną za
fosforylację białka MeCP2; translokacja pomiędzy 1 a 7
chromosomem, utrata funkcji genu Netrin G1.
*ok. 70% mutacji powstaje de novo!

Z
ESPÓŁ
R
ETTA
Istotny wpływ na stopień nasilenia i zakres
zmian klinicznych w tej chorobie ma położenie
mutacji w obrębie genu, typ mutacji oraz sposób
inaktywacji chromosomu X.
Wyróżniamy mutacje:
-w części kodującej domenę MBP
-w części kodującej domenę TRP
-uszkadzające domenę NLS (najcięższa postać).

Z
ESPÓŁ
R
ETTA
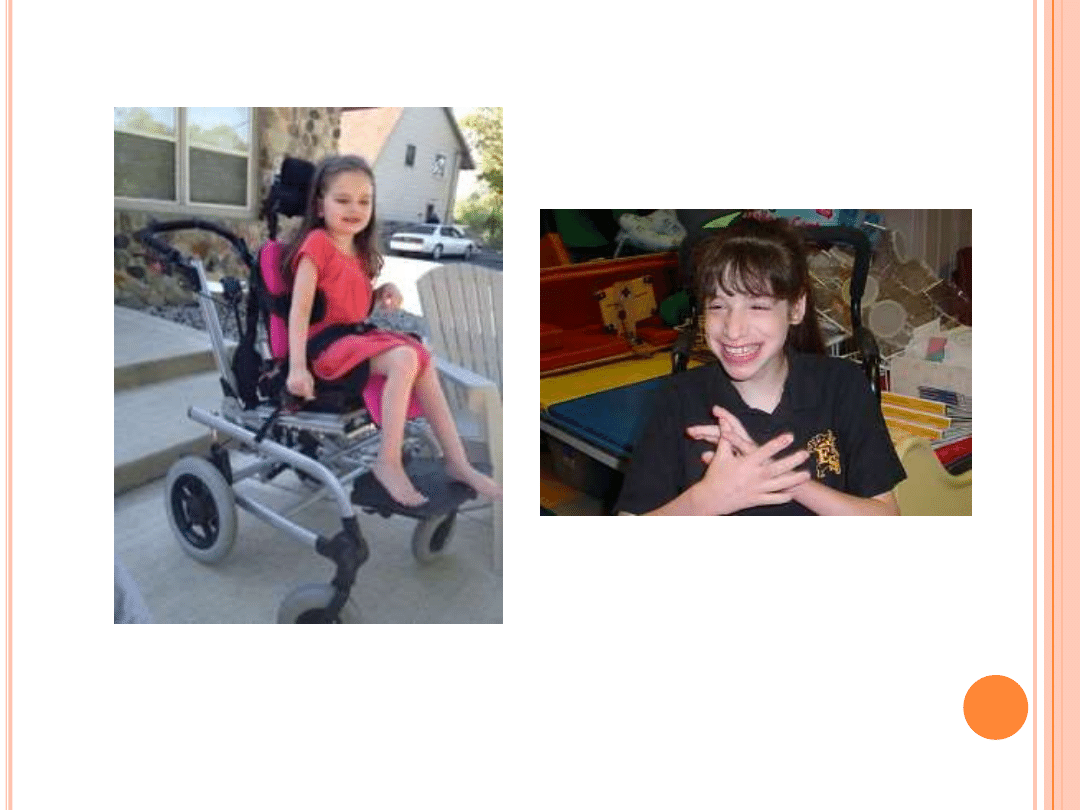
Pierwsze zaburzenia pojawiają się ok. 16-18 mż
(po prawidłowym okresie wzrostu i rozwoju).
Polegają na REGRESJI, czyli dziecko przestaje
siedzieć, chodzić, traci zdolności manualne.
Spada przyrost obwodu główki (małogłowie).
Dochodzi do zahamowania rozwoju mowy.
Pojawiają się ruchy stereotypowe rąk(klaskanie,
ugniatanie, uderzanie w głowę).
Inne objawy: zaburzenia snu, napady pobudzenia,
zachowania autystyczne, drgawki, drżenia,
zaburzenia oddychania (bezdech/hiperwentylacja).
POSTĘPUJĄCE UPOŚLEDZENIE RUCHOWE!!

Z
ESPÓŁ
R
ETTA
Mutacja genu MEC2 jest zazwyczaj letalna u płci
męskiej. Niewielki odsetek (ok. 1,5%) okazuje się
nieletalna (spowodowane tylko częściową utratą
białka MeCP2).

C
HOROBY DZIEDZICZONE RECESYWNIE
SPRZĘŻONE Z CHROMOSOMEM
X
Cechy dziedziczenia:
choroba występuje znacznie częściej u mężczyzn
niż u kobiet
kobiety chorują tylko w układzie
homozygotycznym; u kobiet heterozygot
(nosicieli) choroba nie występuje
istnieje 50% prawdopodobieństwo, że
heterozygotyczne kobiety przekażą zmutowany
gen zarówno synom, jak i córkom
chory mężczyzna nigdy nie przekazuje choroby
synom, a wszystkie córki chorego mężczyzny są
nosicielkami genu

H
EMOFILIA
A
Hemofilia A jest chorobą spowodowaną brakiem lub
niedoborem VIII czynnika krzepnięcia krwi (czynnika
antyhemolitycznego).
Gen kodujący VIII czynnik krzepnięcia F8C, którego
mutacja determinuje chorobę, znajduje się na Xq28.
1/5 przypadków choroby powstaje w wyniku mutacji
de novo.
Częstość występowania:
1:10000-1:20000

H
EMOFILIA
A
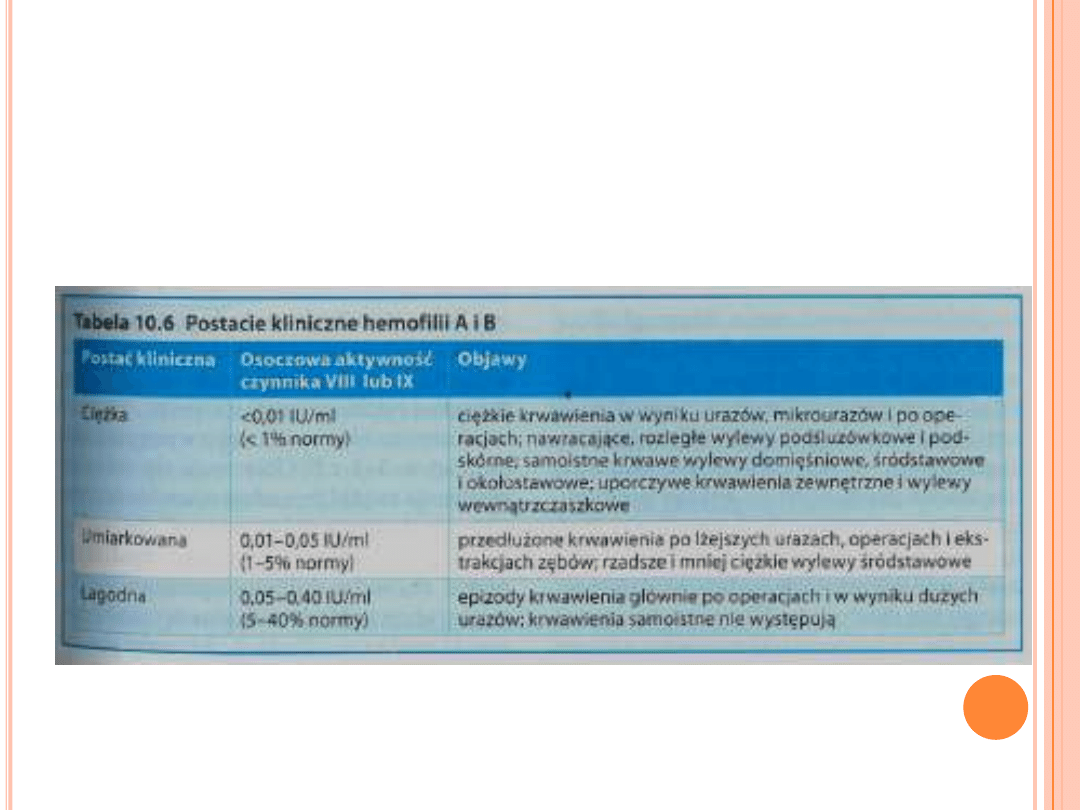
Wyróżniamy 3 postacie hemofilii A:
a) postać łagodna (poziom VIII czynnika krzepnięcia
wynosi od 5-25% normy)- dochodzi do krwawień
pooperacyjnych lub po cięższych urazach;
b) postać umiarkowana (poziom VIII czynnika
krzepnięcia wynosi od 1-5% normy)- dochodzi do
krwawień po lekkich urazach;
c) postać ciężka (poziom VIII czynnika krzepnięcia
wynosi poniżej 1% normy) – dochodzi do częstych
epizodów krwawień, nawet do kilku razy w miesiącu.
* Około 50% chorych na hemofilię A są to osoby z
postacią ciężką choroby.

H
EMOFILIA
A
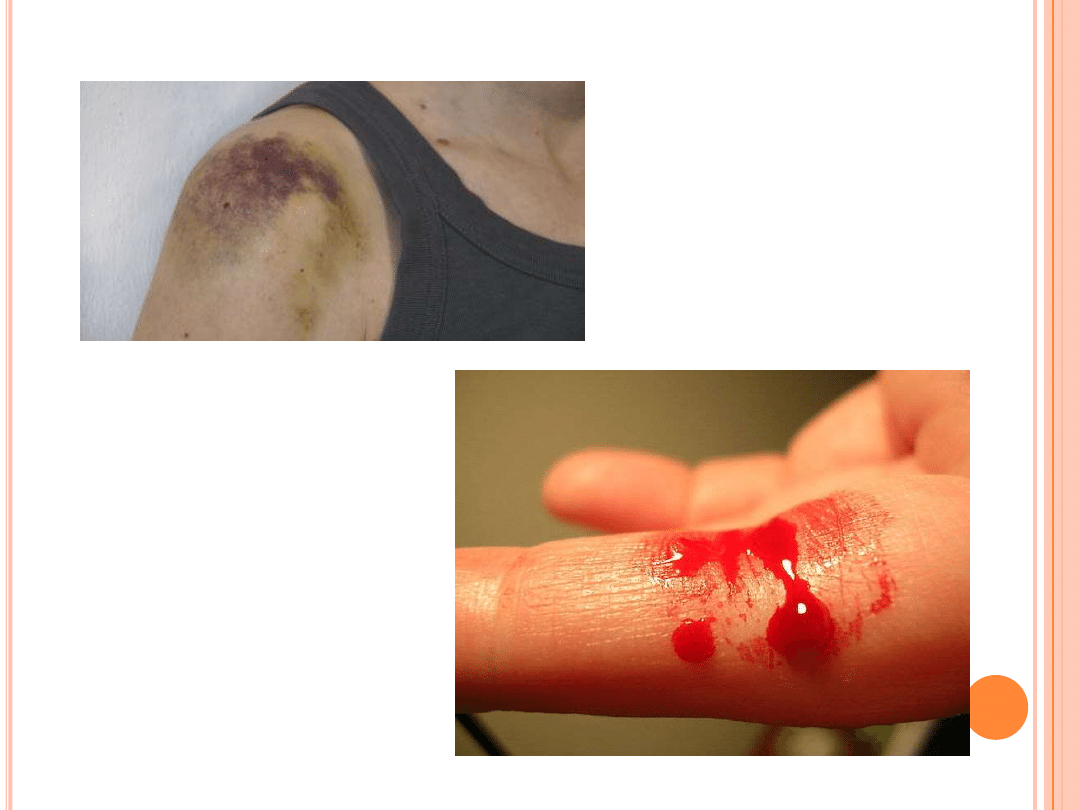
Objawy
(są zależne od stopnia niedoboru
czynnika VIII):
-siniaki (krwawienie do tkanki podskórnej)
-wylewy krwi do stawów (skokowy, biodrowy,
kolanowy..)
-krwawienie wewnątrzczaszkowe
-wylewy do mięśni
-wylewy do OUN
Należy podkreślić, że w przypadku hemofilii aktywność
płytek krwi jest prawidłowa!

H
EMOFILIA
B (
CHOROBA
C
HRISTMASA
)
Hemofilia B jest chorobą polegającą na
niedoborze lub braku IX czynnika krzepnięcia
krwi.
Gen F9 kodujący IX czynnik krzepnięcia, którego
mutacja determinuje chorobę, znajduje się na
Xq27.1-27.2.
Częstość występowania:
1:30000
*Chorują wyłącznie mężczyźni!
H
EMOFILIA
B (
CHOROBA
C
HRISTMASA
)
Klinicznie hemofilia B nie różni się od hemofilii
A.

D
YSTROFIE MIĘŚNIOWE
Jest najczęstszą i
najcięższą postacią
dystrofii mięśniowych.
Częstość
występowania –
1:3600 (żywych
urodzeń chłopców)
Jest alleliczną
odmianą DMD.
Lżejszy przebieg niż
DMD.
Częstość
występowania –
1:30000
Dystrofia mięśniowa
Duchenne’a (DMD)
Dystrofia mieśniowa
Beckera (BMD)

D
YSTROFIE MIĘŚNIOWE
Przyczyną wystąpienia dystrofii mięśniowych
sprzężonych z chromosomem X jest mutacja genu
dystrofina znajdującego się na Xp21.2.
Około 30% mutacji powstaje de novo. Ponad 60%
stanowią delecje, mogą również wystąpić mutacje
punktowe i duplikacje.
W przypadku DMD delecje i duplikacje mogą
zmieniać ramkę odczytu w genie, wskutek czego
następuje zanik dystrofiny w komórkach
mięśniowych. W BMD mutacje uszkadzające gen
zwykle nie powodują zmiany ramki odczytu, a
dystrofina występuje w błonie komórkowej w
postaci poprzerywanej warstwy.

D
YSTROFIE MIĘŚNIOWE
-
DYSTROFINA
Funkcją dystrofiny jest zachowanie strukturalnej
integralności sarkolemy, jej stabilizację w czasie
skurczu i rozkurczu oraz przenoszenie sił w
aparacie skurczowym. Chroni ona komórki
mięśniowe przed nekrozą, ponad to pośredniczy w
komunikacji międzykomórkowej.
Brak dystrofiny powoduje niestabilność
kompleksu dystrofino-glikoproteinowego, co
prowadzi do postępującej martwicy włókien
mięśniowych i zastępowanie ich przez tkankę
łączną i tłuszczową.

D
YSTROFIE MIĘŚNIOWE
-
OBJAWY
Pierwsze objawy choroby w DMD pojawiają się około 3-4
r.ż. i obejmują osłabienie mięśni manifestujące się
trudnościami przy wstawaniu, wchodzeniu po schodach,
kaczkowaty chód.
-symetryczny zanik obręczy biodrowej, a następnie
barkowej
-pseudohipertrofia mięśni łydek
-w surowicy: wysoki poziom kinazy kreatyniny (praktycznie
od urodzenia) i aldolazy dehydrogenazy mleczanowej
-około 10-14 r.ż. przestaje chodzić
-osłabienie mięśnia sercowego (nasilenie objaw
niewydolności krążeniowo-oddechowej)
-może pojawić się lekkie opóźnienie rozwoju umysłowego.

D
YSTROFIE MIĘŚNIOWE
-
OBJAWY
Przebieg BMD jest znacznie łagodniejszy niż
DMD i ma wolniejszy postęp.
Objawy pojawiają się około 11 r.ż. i są
różnorodne. Jednym ze stałych objawów jest
rzekomy przerost mięsni łydek.
Średnia długość życia osoby z BMD niewiele różni
się od średniej długości życia osoby zdrowej.
Natomiast średnia długość życia osoby z DMD
wynosi około 25 lat (śmierć na skutek
niewydolności krążeniowo-oddechowej)

CHOROBY
WIELOCZYNNIKOWE
Rodzinne występowanie chorób wieku dorosłego.
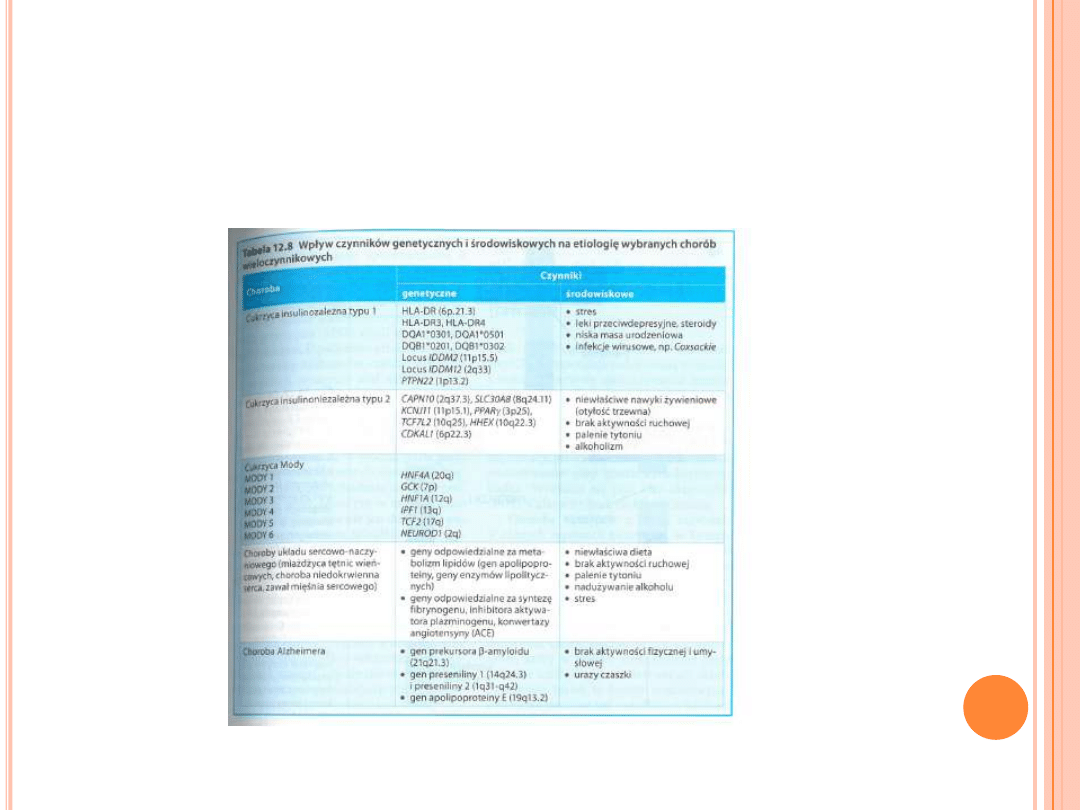
D
ZIEDZICZENIE WIELOCZYNNIKOWE

D
ZIEDZICZENIE WIELOCZYNNIKOWE
-
NADCIŚNIENIE
Nadciśnienie występuje u ok. 15% populacji
większości krajów rozwiniętych. Jest kluczowym
czynnikiem w chorobach serca, nerek, w udarach.
Badania wykazały, że rodzinne dziedziczenie
ciśnienia krwi (skurczowego, jak i rozkurczowego)
wynosi ok. 20-40%. Stąd można wnioskować, że
czynniki środowiskowe muszą również być
istotnymi przyczynami zróżnicowania ciśnienie
krwi wśród populacji ludzkiej.

D
ZIEDZICZENIE WIELOCZYNNIKOWE
-
NADCIŚNIENIE
Najważniejsze czynniki środowiskowe:
-przyjmowanie zwiększonej ilości sodu
-mała aktywność fizyczna
-napięcie psychofizyczne
-otyłość
( na otyłość mają wpływ zarówno geny jak i
środowisko)\

D
ZIEDZICZENIE WIELOCZYNNIKOWE
-
NADCIŚNIENIE
Wyodrębniono wiele genów polimorficznych
związanych z pierwotnym nadciśnieniem tętniczym-
geny odpowiedzialne za syntezę:
-angiotensynogenu (AGT); 1q42-43
-konwertazy angiotensyny (ACE); 17q23
-receptora angiotensyny typu 1 (AT1R); 3q21-25
-przedsionkowego peptydu natriuretycznego (ANP)
Niewielki odsetek nadciśnienia wynika z rzadko
występujących zaburzeń jednogenowych, np. zespół
Cushinga, zespół Liddle’a. Wykryto około 10 genów
odpowiedzialnych za te choroby. Wszystkie z nim
mają wpływ na resorpcję wody i soli w nerkach.

D
ZIEDZICZENIE WIELOCZYNNIKOWE
-
MIAŻDŻYCA
Potencjalnie rozwojowi miażdżycy (jak również
chorobie niedokrwiennej serca) sprzyjają geny:
-metabolizmu lipidów (gen apolipoproteiny)
-czynników zapalenia (geny cytokin, czynnika TNF-α
-czynników wazoaktywnych (gen ANA, BNP, CNP)
-układu RAA (gen konwertazy angiotensyny, gen
syntezy aldosteronu)
-układu krzepnięcia i trombolizy (gen inhibitora
aktywatora plazminogenu, gen fibrynogenu)

D
ZIEDZICZENIE WIELOCZYNNIKOWE
-
MIAŻDŻYCA
Ponadto do rozwoju miażdżycy predysponują pewne
czynniki środowiskowe:
-nieodpowiednia dieta
-brak aktywności fizycznej
-otyłość
-palenie tytoniu
-nieprawidłowe stężenia frakcji cholesterolu i
trójglicerydów w osoczu
Środowiskowe czynniki ryzyka mogą być w
dużym stopniu modyfikowane przez samego
chorego!

D
ZIEDZICZENIE
WIELOCZYNNIKOWE
–
CHOROBY
UKŁADU SERCOWO
-
NACZYNIOWEGO
W celu określenia roli czynników genetycznych w
etiopatogenezie chorób układu sercowo-naczyniowego
uwzględnia się wywiad rodzinny oraz testy genetyczne.
Za pomocą obecnie stosowanych testów można
potwierdzić lub ustalić rozpoznania jednogenowe.

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
1
Cukrzyca typu 1 jest przewlekłym procesem
prowadzącym do niszczenia komórek β wysp trzustki.
Proces ten jest zależny zarówno od czynników
genetycznych jak i środowiskowych.
W populacji ogólnej ryzyko wystąpienia cukrzycy typu
1 wynosi 0,2 – 0,4%.
Postacie cukrzycy typu 1:
a) typ 1A- postać autoimmunologiczna
-uwarunkowana poligenowo
-uwarunkowana jednogenowo
b) typ 1B – postać idiopatyczna
c) postać wtórna (np. polekowa, w chorobie
alkoholowej)

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
1
Do rozwoju cukrzycy typu 1 przyczynia się wiele
genów na różnych chromosomach.
Duże znaczenie mają pewne regiony w genomie
określane jako IDDM1, IDDM2, IDDM3…
W regiony IDDM1 znajdują się geny kodujące
antygeny układu HLA (I i II). W patogenezie
cukrzycy znaczenie mają polimorficzne geny
kodujące cząsteczki HLA-DR i HLA-DQ (II).
U około 95% osób z cukrzycą występują antygeny
HLA-DR3 lub HLA-DR4.
Heterozygoty (posiadają HLA-DR3 i HLA-DR4)
mają większe ryzyko zachorowania!

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
1
W regionie IDDM2 znajduje się gen kodujący
insulinę. Region 5’ tego genu posiada zmienną liczbę
powtórzeń tandemowych – VNTR. Im większa liczba
powtórzeń, tym mniejsze ryzyko wystąpienia
cukrzycy.
IDDM12 (2q33) – gen kodujący białko CTLA-4,
odpowiedzialne za działanie cytotoksyczne wobec
autoreaktywnych limfocytów T.
PTPN22 (1p13.2)- gen kodujący limfocytarną
fosfatazę tyrozynową, odpowiedzialną za eliminację
autoreaktywnych limfocytów T. Gdy poziom białka
jest obniżony, wówczas mamy zmniejszone działanie
destrukcyjne wobec autoreaktywnych limfocytów.

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
1
Czynniki środowiskowe sprzyjające wystąpieniu
cukrzycy:
-niewłaściwe odżywianie
-infekcje wirusowe
-niska masa urodzeniowa
-nadużywanie alkoholu
- leki przeciwdepresyjne, leki steroidowe

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
2
W rozwoju cukrzycy typu 2 znaczenie mają czynniki
genetyczne, środowiskowe, ale również epigenetyczne
(tj. metylacja DNA< modyfikacja histonów..)
Podobnie jak cukrzyca typu 1 może być
uwarunkowana monogenowo lub poligenowo.
Mutacje w obrębie genów wpływają na:
-sekrecję insuliny
-obniżenie wrażliwości na insulinę
-homeostazę glukozy
-regulacje szlaków metabolicznych węglowodanów i
lipidów

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
2
Ryzyko wystąpienia cukrzycy typu 2:
1 rodzic chory - 40%
2 rodziców chorych- 70%
Czynniki środowiskowe sprzyjające wystąpieniu
cukrzycy 2:
-siedzący tryb życia
-mała aktywność fizyczna
-otyłość, szczególnie brzuszna
-alkohol
-palenie tytoniu
-złe nawyki żywieniowe

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
2
Geny wpływające na rozwój cukrzycy typu 2:
1)
Gen SLC30A8 (zaburzenia sekrecji insuliny)
2)
Gen CDKL1 (zaburzenia sekrecji insuliny)
3)
Gen KCNJ11 (zaburzenia sekrecji insuliny)
4)
Gen CAPN10 (zmniejszenie wrażliwości na
insulinę)

D
ZIEDZICZENIE WIELOCZYNNIKOWE
–
CUKRZYCA TYPU
2
Do cukrzycy typu 2 należy podgrupa tzw. Cukrzycy MODY
(„młodzieńcza cukrzyca dorosłych”), ujawniająca się
najczęściej przed 25 rż. Dotyczy poniżej 5% pacjentów, u
których zdiagnozowano cukrzycę typu 2.
Kryteria diagnostyczne:
a) hiperglikemia u 1-2 członków rodziny przed 25 rż
b) autosomalny dominujący tor dziedziczenia w co najmniej
3 pokoleniach
c) brak konieczności leczenia insuliną po 5 latach od
rozpoznania lub brak zwiększonego stężenia peptydu C
nawet u pacjentów leczonych insuliną
d) prawidłowy poziom insuliny, ale niewystarczający w
stosunku do hiperglikemii
e) rzadko występuje nadwaga/otyłość

C
HOROBA
A
LZHEIMERA
Choroba Alzheimera charakteryzuje się
postępująca demencją i tratą pamięci oraz
tworzeniem się blaszek amyloidowych i
neurofibrylarnych ziarnistości w mózgu
(zwłaszcza kora i hipokamp!).
Śmierć po około 5-10 lat od pierwszych objawów.
Ryzyko pojawienie się choroby podwaja się u
osób, które mają chorego krewnego pierwszego
stopnia.

C
HOROBA
A
LZHEIMERA
Choroba ta może być dziedziczona zarówno
autosomalnie dominująco (ok. 10-15%, choroba o
wczesnym początku, objawiająca się przed 65 rż),
jak również wielogenowo.
W chorobie o wczesnym początku za chorobę
odpowiedzialne są mutacje w jednym z trzech
genów:
1) presenilina 1 (chromosom 14)
2) presenilina 2 (chromosom 1)
3) gen prekursora białka amyloidu (chromosom
21)

C
HOROBA
A
LZHEIMERA
W chorobie o późnym początku istotnym
czynnikiem ryzyka jest zróżnicowanie alleliczne
w locus apolipoproteiny E (APOE). Gen kodujący
APOE znajduje się na ramieniu długim
chromosomu 19 (19q13.2).
Wiele badań wskazuje na to, że allel APOE4
wywołuje zwiększenie ryzyka wystąpienia
choroby, podczas gdy allel APOE2 ma działanie
ochronne.

C
HOROBA
A
LZHEIMERA
Objawy choroby:
-postępujące otępienie umysłowe
-zaburzenia pamięci i orientacji
-zaburzenia zachowania
-depresja
-konfabulacja
Czynniki sprzyjające wczesnemu objawieniu się
choroby to przede wszystkim brak aktywności
ruchowej i umysłowej.

S
CHIZOFRENIA
Schizofrenia jest poważnym zaburzeniem
emocjonalnym, charakteryzującym się
urojeniami, halucynacjami, ucieczką od
rzeczywistości, dziwnym, obojętnym lub
niewłaściwym zachowaniem.
Ryzyko wystąpienia choroby w populacji: ok. 1%
Ryzyko wystąpienia choroby u potomstwa
chorego: 8-10%

S
CHIZOFRENIA

S
CHIZOFRENIA
Jak dotąd nie zlokalizowano i nie
sklonowano genów przyczyniających się do
wystąpienia schizofrenii.

Dziękuję za uwagę
Wyszukiwarka
Podobne podstrony:
4 Ch jednogenowe cz 1 Ładosz
Koncert Skrzypcowy D Dur Op 35 Cz 3 Finał II Skrzypce
Symfonia F dur KV130 Cz 1 Allegro II Skrzypce
2 Ch chromosomowe cz 1 Stec
Ch zw krŕg cz 2 do wydruku
ch stawow o podl imm cz II
ch stawow o podl imm psow i kotow cz. II
2 Ch chromosomowe cz 1 Puzio
Geometria w praktyce, cz 2 ?ch czterospadowy i kopertowy
4 Ch jednogenowe autos dominujące Skowronek
3 Ch chromosomowe cz 2 Rabiej
Ch zw krŕg cz 1 do wydruku
Geometria w praktyce, cz 1 ?ch pulpitowy i dwuspadowy
ch stawow o podl imm psow i kotow cz I
więcej podobnych podstron