
1
ZNAKOWANIE KWASÓW NUKLEINOWYCH
W pracach biologii molekularnej często zachodzi konieczność używania znakowanych cząsteczek
kwasów nukleinowych, czyli takich, które zawierają wbudowany radioaktywny izotop lub znacznik
nieradioaktywny, np. biotynę. Znakowany DNA lub RNA wykorzystywany jest w takich technikach jak
testy hybrydyzacji, primer extension, foot-printing, EMSA, RNA protection, sekwencjonowanie i wiele
innych.
1. Najczęściej wykorzystywanymi znacznikami kwasów nukleinowych są radioaktywne izotopy
32
P,
35
S,
3
H (Tab.1) oraz związki nieradioaktywne, takie jak: biotyna, digoxigenina, fluoresceina, rodamina,
kumaryna. Wybór znacznika zależy głównie od czułości i rozdzielczości testu, do którego zostanie użyty
znakowany DNA a także od trwałość znacznika oraz bezpieczeństwa pracy.
a. Czułość oznacza najmniejszą ilość kwasu nukleinowego, jaką można zidentyfikować z wykorzystaniem
danego znacznika. Najczulszym ze stosowanych obecnie znaczników jest izotop
32
P (1,71 MeV), który
umożliwia detekcję nawet 10fg DNA. Mniejszą czułość reakcji można natomiast uzyskać wykorzystując
izotop
35
S (0,167 MeV), używany do niedawna powszechnie do reakcji sekwencjonowania. Parametry
pośrednie wykazuje natomiast izotop
33
P (0,249 MeV). Szybki postęp w technikach znakowania
nieradioaktywnego powoduje jednak, że znaczniki takie biotyna i digoxigenina umożliwiają detekcję
kwasów nukleinowych z czułością porównywalną do znaczników radioaktywnych.
b. Rozdzielczość oznacza najmniejszą odległość pomiędzy dwoma sygnałami, jaką można uzyskać dla
danego znacznika. Izotopy radioaktywne o wysokiej aktywności promieniowania (wysoka czułość
detekcji), np.
32
P, charakteryzują się niższą rozdzielczością niż izotopy o mniejszej aktywności, np.
35
S.
Najwyższą rozdzielczość uzyskuje się jednak przy wykorzystaniu technik znakowania
nieradioaktywnego, które pozwalają na lokalizację kw. nukleinowych in situ, czyli w preparatach
mikroskopowych.
c. Trwałość znacznika. Izotopy radioaktywne, ze względu na okres półrozpadu, charakteryzują się
ograniczonym okresem trwania, np.
32
P - 14 dni,
33
P - 25 dni,
35
S – 87 dni. W przeciwieństwie do nich,
zestawy do znakowania nieradioaktywnego, przechowywane we właściwych warunkach, nie ulegają tak
szybkiemu starzeniu i mogą być wykorzystane w stosunkowo długim okresie czasu.
d. Bezpieczeństwo pracy. Izotopy radioaktywne używane w biologii molekularnej emitują różne typy
promieniowania, tj.
α, β lub γ. Izotopy wykorzystywane powszechnie do znakowania kwasów
nukleinowych, np.
32
P,
33
P i
35
S emitują promieniowanie
β, które może być potencjalnie niebezpieczne
dla zdrowia. Ponieważ jednak stosunkowo łatwo ulega ono ekranowaniu przez takie materiały jak: ołów,
który stosowany jest do przechowywania prób, czy pleksia używana do budowy ekranów ochronnych,
praca z nim jest bezpieczna przy zastosowaniu odpowiednich środków ostrożności. Ponieważ znaczniki
nieradioaktywne nie stanowią żadnego zagrożenia dla zdrowia i środowiska są coraz bardziej popularne
w pracach biologii molekularnej.
e. Nukleotydy zawierające znaczniki. Większość technik znakowania wykonuje się syntetyzując nową nić
DNA na wybranej matrycy, z wykorzystaniem nukleotydów dNTP, zawierających określony znacznik.
Ponieważ podczas wbudowywania nukteotydów do DNA następuje odłączenie reszt fosforanowych z
pozycji
β i γ, do znakowania radioaktywnego wykorzystuje się izotopy obecne w pozycji αdNTP ,
np.
α[
32
P]dCTP,
α[
35
S]dATP. Znaczniki nieradioaktywne, np. biotyna i digoxigenina, przyłączone są do
C5 UTP. Znakowane cząsteczki RNA można uzyskać dzięki syntezie RNA in vitro z wykorzystaniem
nukleotydów typu
α rNTP, np. α[
32
P]UTP. Znakowanie z wykorzystaniem kinazy polinukleotydowej
wykonuje się z wykorzystaniem nukleotydów zawierających izotop w pozycji
γ, np. γ[
32
P]ATP.
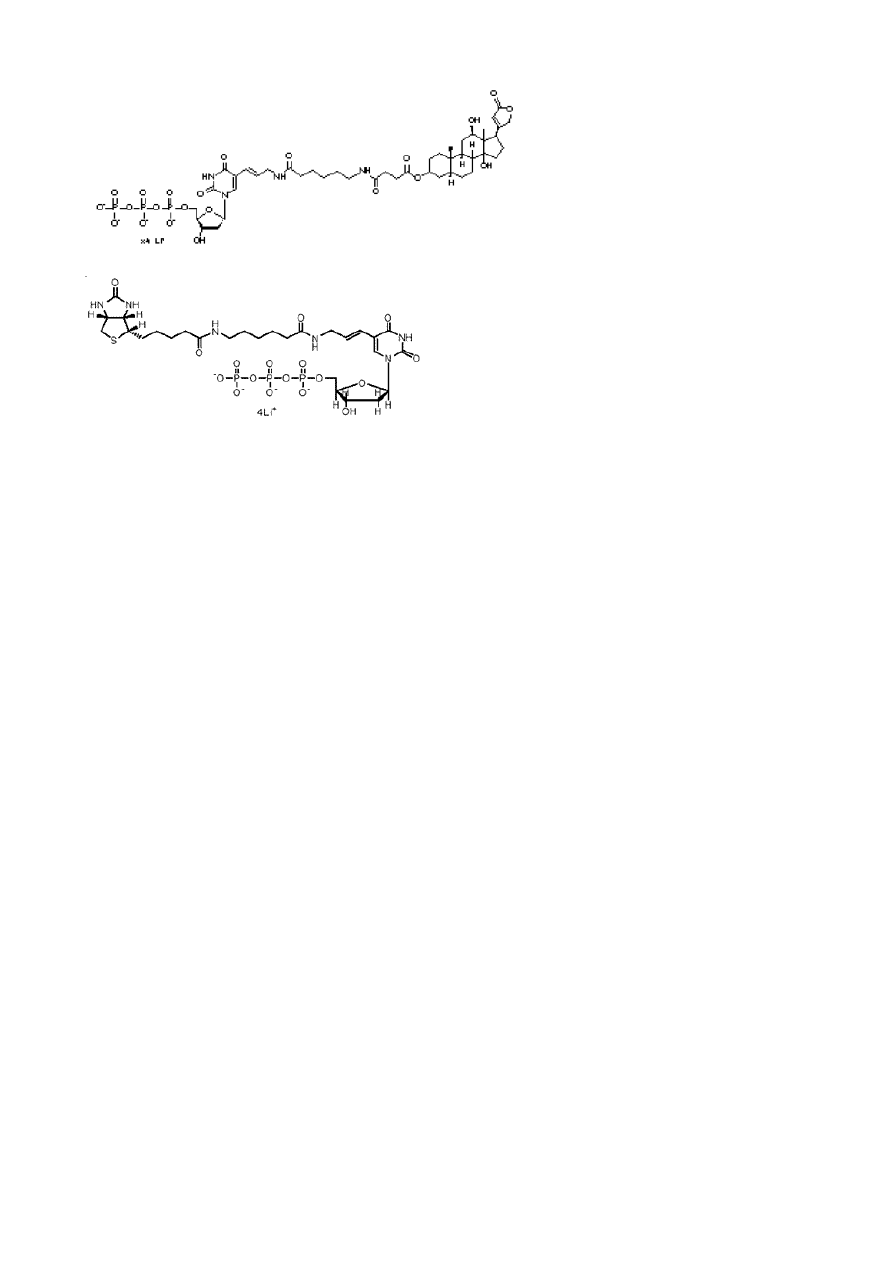
2
Struktura DIG-dUTP.
Biotin-11-dUTP. Liczba podana przed nazwą
nukleotydu oznacza długość łańcucha oddzielającego
zasadę od biotyny
2. Enzymy używane do znakowania. Znakowanie wykonuje się zazwyczaj w warunkach in vitro z
wykorzystaniem enzymów dostępnych komercyjnie.
a. Większość technik znakowania wykorzystuje zdolności różnych polimeraz do wbudowywania
nukleotydów zawierających znacznik do syntetyzowanej nici kwasu nukleinowego. Do znakowania
cząsteczek DNA używa się w tym celu polimerazy I, podjednostki Klenova polimerazy I, Taq
polimerazy, odwrotnej transkryptazy itp. Znakowane cząsteczki RNA można natomiast uzyskać dzięki
wykorzystaniu polimerazy RNA, np. polimerazy RNA T7 lub SP6. Enzymy te mogą wprowadzać do nici
DNA lub RNA nukleotydy zawierające zarówno znaczniki radioaktywne jak i nieradioaktywne.
b. Niektóre techniki znakowania opierają się na zdolności enzymów do modyfikacji cząsteczek kwasów
nukleinowych, np. fosforylacji 5’ końca nici DNA w wykorzystaniem kinazy polinukleotydowej.
c. W eksperymentach, w których badamy syntezę kw. nukleinowych w warunkach in-vivo, np. technika
„run-on”, znakowanie kwasów nukleinowych następuje w wyniku aktywności enzymów występujące w
badanym materiale biologicznym i znaczników podanych z zewnątrz.
3. Podstawowe techniki znakowania
a. Nick translation – do znakowania zostaje podany DNA w postaci dwuniciowej. DNAaza I
(endonukleaza) wykonuje nacięcia w różnych pozycjach obu nici DNA, generując wolne 3’ końce.
Następnie polimeraza I, dzięki aktywności 5’
→3’ polimerazy, dobudowuje do końców 3’ nową nić DNA
ze znakowanymi nukleotydami. Równocześnie, dzięki aktywności 5’
→3’ exonukleazy, polimeraza ta
hydrolizuje starą nić DNA.
b. Metoda hexamerowa (ang. random primer) – do znakowania podawany jest DNA w postaci
jednoniciowej np. po denaturacji termicznej. Do reakcji dodaje się ponadto mieszaninę
sześcionukleotydowych oligonukleotydów o zróżnicowanej sekwencji. Cząsteczki te po przyłączeniu się
do różnych miejsc na obu niciach DNA rozpoznawane są przez fragment Klenowa polimerazy I, który
dobudowuje komplementarną nić od pozycji startera, włączając nukleotydy ze znacznikiem.
c. Kinazowanie – w metodzie tej znakuje się końce 5’ nici DNA pozbawione grupy fosforanowej. Enzym,
kinaza polinukleotydowa, przenosi radioaktywną grupę fosforanową z
γ[
32
P]ATP na koniec 5’ cząsteczek
DNA.
d. Synteza „gorącego” transkryptu (RNA). Technika ta umożliwia otrzymanie wyznakownych
radioaktywnie cząsteczek RNA, komplementarnych do nici kodującej lub matrycowej wklonowanego
fragmentu DNA. Dzięki obecności w wektorze promotorów dla polimeraz RNA, takich jak SP6, T7 i T3,
wklonowany fragment DNA można poddać transkrypcji in vitro, prowadzącej do powstania cząsteczki
RNA z wbudowanymi nukleotydami
α[
32
P]rNTP, zawierającymi radioaktywny izotop.
4. Oczyszczanie kwasów nukleinowych po znakowaniu. Ponieważ po reakcji znakowania znaczna część
nukleotydów ze znacznikiem pozostaje w roztworze, muszą one zostać usunięte przed następnym etapem
prac. Do oczyszczania znakowanego DNA stosować można wiele komercyjnie dostępnych systemów
3
przeznaczonych do oczyszczania DNA po reakcjach enzymatycznych, jeżeli umożliwiają one oddzielenie
DNA od wolnych nukleotydów. Ponadto DNA można oczyszczać następującymi metodami:
a. wytrącanie etanolem
b. rozdział chromatograficzny na kolumnie z sephadexem G-50.
c. elucja z żelu po rozdziale elektroforetycznym
5. Detekcja znaczników.
a. Znaczniki radioaktywne. Identyfikacja DNA znakowanego radioaktywnie opiera się na detekcji
promieniowania
β. Promieniowanie to można zlokalizować wykonując ekspozycję sygnału na błonie
radiologicznej lub przy wykorzystaniu sprzętu do pomiaru promieniowani radioaktywnego, takiego jak
licznik scyntylacyjny. Aparaty typu Phosphoimager łączą natomiast obie te funkcje, gdyż umożliwiają
zarówno lokalizację sygnału na membranie (skanowanie) jak i pomiar jego intensywności.
b. Znaczniki nieradioaktywne. Obecność znaczników nieradioaktywnych można zidentyfikować na
podstawie ich aktywności własnej (bezpośrednie) lub aktywności enzymów połączonych ze znacznikiem
(pośrednie).
− bezpośrednie. Związki wbudowane do DNA, takie jak rodamina, kumaryna czy fluoresceina,
charakteryzuje się zdolnością do emisji światła o określonej długości fali w świetle UV. Ich detekcja
odbywa się przy pomocy mikroskopu fluorescencyjnego.
− pośrednie. Czulsze metody detekcji znaczników nieradioaktywnych oparte są na technikach
enzymatycznych. Znaczniki takie jak biotyna, digoxigenina oraz floresceina rozpoznawane są przez
specyficzne przeciwciała (ew. streptavidynę w przypadku biotyny) sprzężone z enzymami takimi jak
alkaliczna fosfataza, peroksydaza. Enzymy te po przyłączeniu się do znacznika katalizują reakcje, które
prowadzą do luminescencji lub powstania barwnych produktów.
6. Wybrane właściwości izotopów radioaktywnych używanych w pracach eksperymentalnych biologii
molekularnej.
izotop okres
półrozpadu energia
rozpadu promieniowanie
32
P
14,3 dni
1,71 MeV
β
35
S
87 dni
0,167 MeV
β
3
H
12,3 lat
0,018 MeV
β
14
C
5730 lat
0,156 MeV
β
125
I
60 dni
0,035 MeV
γ
a. Jednostki aktywności promieniowania.
– jednostką promieniowania w układzie SI jest Bq (becquerele). 1 Bq = 1dps (1 rozpad na sekundę).
Jednostki pochodne to GBq, MBq, kBq.
– ponadto wciąż u użyciu są jednostki Ci (curie). 1Ci = 3,7 x 10
10
Bq (37GBq) (1Gbq = 0,027Ci).
Jednostkami pochodnymi dla Ci są
μCi, mCi.
b. Aktywność specyficzna. Ilość rozpadów radioaktywnych na jednostkę masy lub objętości, np. Ci/g,
mCi/mmol, dpm/
μg.
c. Rodzaje promieniowania emitowane przez izotopy radioaktywne:
α - jądra helu
4
2
He,
β - elektrony, γ -
fotony (fale elektromagnetyczne).
TEST HYBRYDYZACJI
Testy hybrydyzacji wykonuje się w celu identyfikacji określonych sekwencji DNA lub RNA w
badanym materiale genetycznym, który zazwyczaj umieszczony jest na membranie nitrocelulozowej lub
nylonowej (patrz ćw. membrany hybrydyzacyjne).
1. Prehybrydyzacja. Ponieważ membrany hybrydyzacyjne wykazują duże powinowactwo do kwasów
nukleinowych, powierzchnia nie związana z badanym materiałem genetycznym powinna zostać
zablokowana w celu uniknięcia wiązania do niej sondy. W tym celu membranę należy inkubować w
4
roztworze prehybrydyzacyjnym, zawierającym takie składniki blokujące takie jak BSA lub DNA Salmon
sperm.
2. Hybrydyzacja. Warunki, w jakich należy prowadzić hybrydyzację powinny zostać dostosowane do
stopnia homologii, jaka istnieje pomiędzy sondą i kwasami nukleinowymi umieszczonymi na
membranie. Jeżeli celem eksperymentu jest identyfikacja sekwencji o najwyższym stopni homologii
należy zastosować rygorystyczne warunki hybrydyzacji. Jeżeli natomiast sonda nie posiada pełnej
homologii do badanego DNA, jak to ma miejsce w przypadku poszukiwania homologów znanego genu
w genomie innego gatunku, warunki hybrydyzacji powinny być mniej rygorystyczne. O możliwości
wiązania dwóch cząsteczek DNA o ograniczonej homologii decyduje głównie temperatura i stężenie soli
w środowisku reakcji. Wzrost temperatury zwiększa rygorystyczność warunków hybrydyzacji, czyli
zaniża możliwość powstawania wiązań niespecyficznych. Podobny efekt można również uzyskać
dodając formamid do roztwory hybrydyzacyjnego. Rygorystyczność reakcji wzrasta również w wyniku
zmniejszania stężenie soli w buforze.
3. Odmywanie. Celem odmywania jest usunięcie z powierzchni filtra sondy, która nie związała się z
badanym DNA.
4. Ekspozycja. Membrany po hybrydyzacji eksponuje się w na błony radiologiczne, lub poddaje się
skanowaniu przy zastosowaniu odpowiedniego czytnika radioaktywności (ang. Phosphoimager).
Sprzęt i oddczynniki
-
odczynniki do znakowania
-
pasterówka
-
wata szklana
-
sephadex G50 Fine
-
Dextran blue
-
statyw ochronny
-
statyw do scyntylatora
-
naczynia scyntylacyjne
-
odczynniki do buf.
hybrydyzacyjnego (tab.1)
-
50x Denhardt (1% PVP,
1%Ficoll 400, 1% BSA)
-
łaźnia wodna
-
naczynie do hybrydyzacji
-
kaseta do ekspozycji RTG

5
Postępowanie :
Uwaga. Prace z radioaktywnością prowadzimy w rękawiczkach, za osłoną z pleksi.
I
. Przygotowanie sondy hybrydyzacyjnej.
1. Preparat DNA (ok. 20 ng/20
μl) zdenaturować we wrzącej łaźni wodnej przez 5 minut a następnie umieścić
preparat w lodzie.
2. Do zdenaturowanego DNA dodać następujące odczynniki:
− ssDNA
20
μl
− H
2
O (do 50
μl reakcji)
8
μl
− bufor
(10
x
st.)
5
μl
− mieszanina heksamerów
5
μl
− nukleotydy nieradioaktywne
5
μl
− fragment Klenowa polimerazy I (1u/μl)
2
μl
−
32
P
αdCTP (10μCi/μl)
5
μl
Całość dokładnie wymieszać i krótko zwirować.
3. Mieszaninę inkubować w temp. +37
o
C przez 20 min.
4. Oczyszczanie wyznakowanej sondy przez chromatografię na kolumnie z Sephadexem G-50 fine.
a)
przygotowanie
kolumny
-
umieścić w przewężeniu pasterówki niewielką ilość waty szklanej
-
pasterówkę wypełnić Sephadexem G-50 fine
-
kolumnę przemyć 3 obj. buforu TE
b) oczyszczanie wyznakowanej sondy
- do mieszaniny reakcyjnej dodać 5
μl barwnika Dekstran blue
-
nałożyć preparat na kolumnę
-
zbierać frakcje po 5 kropli do probówek Eppendorfa
-
dokonać pomiaru radioaktywności zebranych prób
-
połączyć frakcje zawierające DNA
II. Prehybrydyzacja i hybrydyzacja.
1. Przygotować mieszaninę, która posłuży zarówno do prehybrydyzacji jak i hybrydyzacji. Na 1cm
2
powierzchni filtra
należy przygotować 0,45 ml roztworu, z czego do prehybrydyzacji należy stosować ilość 0,3 ml/cm
2
membrany,
natomiast do hybrydyzacji 0,15 ml/cm
2
.
składniki stężenie wyjściowe stężenie końcowe
SSC 20X
5X
formamid 100%
50%
salmon sperm DNA
10mg/ml
50
μg/ml
EDTA
0,5 M
1 mM
SDS 20%
0,1%
Denhardt sol.
50X
1X
H
2
O
do
objętości końcowej
2. Umieścić membranę w naczyniu hybrydyzacyjnym i zalać go roztworem prehybrydyzacyjnym.
3. Prehybrydyzację prowadzić w temp. +42
o
C przez 2 godz. w piecu hybrydyzacyjnym
4. Sondę zdenaturować we wrzącej łaźni wodnej przez 5 min. a następnie schłodzić w lodzie.
5. Usunąć z naczynia hybrydyzacyjnego roztwór prehybrydyzacyjny a na jego miejsce umieścić mieszaninę
hybrydyzacyjną.
6. Dodać zdenaturowaną, radioaktywną sondę.
7. Hybrydyzację prowadzić w temp. +42
o
C przez 18 godz.
III. Aby odmyć membranę po hybrydyzacji należy ją płukać w roztworach zgodnie z poniższymi warunkami:
1. 1 x 5 minut - 2xSSC, 0,1%SDS
temp.pokojowa.
2. 3 x 20 minut - 2xSSC, 0,1%SDS
temp.pokojowa.
3. 1 x 60 minut - 0,1xSSC, 0,1%SDS
42
o
C
4. 1 x 10 minut - 2xSSC, 0,1%SDS
temp.pokojowa.
IV. Wykonać ekspozycję mambrany na błonę radiologiczną.
Wyszukiwarka
Podobne podstrony:
hybrydyz id 207415 Nieznany
002 Analiza AMI Wyklad r1 id 59 Nieznany (2)
Naped hybrydowy id 313570 Nieznany
Montaz znakow pionowych id 3075 Nieznany
Analityk kredytowy 241302 id 59 Nieznany (2)
analiza czestotliwosciowa id 59 Nieznany (2)
59 01 032 036 id 41760 Nieznany (2)
59 4 id 41741 Nieznany (2)
59 id 41735 Nieznany
Anamnesis71 3b str 59 63 id 62 Nieznany (2)
59 3 id 41740 Nieznany (2)
59 01 032 036 id 41760 Nieznany (2)
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
więcej podobnych podstron