
146
Nr 4–6
Zawał serca
WIADOMOŚCI LEKARSKIE 2008, LXI, 4–6
Małgorzata Z. Lisik, Aleksander L. Sieroń
NIEPEŁNOSPRAWNOŚĆ INTELEKTUALNA
SPRZĘŻONA Z CHROMOSOMEM X – SCHEMAT POSTĘPOWANIA
Z Katedry i Zakładu Biologii Ogólnej, Molekularnej i Genetyki
Śląskiego Uniwersytetu Medycznego w Katowicach
Niepełnosprawność intelektualna (NI) stanowi poważny problem medyczny i społeczny. Dotyczy 2–3% populacji. Ustalenie etiologii NI
ma ogromne znaczenie dla prognozy możliwości wsparcia oraz poradnictwa genetycznego. Szacuje się, że 25–35% przypadków ma podłoże ge-
netyczne. W NI uwarunkowanej czynnikami genetycznymi 25–30% przypadków ma związek z mutacjami w genach zlokalizowanych w chro-
mosomie X (tzw. niepełnosprawność intelektualna sprzężona z chromosomem X – X-linked mental retardation – XLMR). Niepełnosprawność
intelektualna sprzężona z chromosomem X stanowi heterogenną grupę zaburzeń. Historycznie na podstawie obrazu klinicznego podzielono ją
na 2 grupy: specyficzną oraz niespecyficzną. Z biegiem czasu podział ten staje się coraz mniej przejrzysty, a spektrum objawów klinicznych
bardzo szerokie. Obie postacie opisano dla kilku genów. Mutacje w genach XLMR stwierdzono w niewielkim odsetku rodzin. Połowa chorych
z XLMR może mieć mutacje w jednym z genów zlokalizowanych w chromosomie X. Dostępne metody diagnostyczne nie pozwalają jednak
na poszukiwanie mutacji we wszystkich znanych genach. W trakcie udzielania porady genetycznej rodzinie obciążonej NI musimy opierać
się na danych empirycznych dotyczących ryzyka powtórzenia choroby u kolejnych dzieci danej pary. [Wiad Lek 2008; 61(4–6): 146–153]
Słowa kluczowe: niepełnosprawność intelektualna (NI), sprzężenie z chromosomem X, poradnictwo genetyczne.
Niepełnosprawność intelektualna (NI) jest jednym
z najczęściej obserwowanych zaburzeń neuropsychia-
trycznych u dzieci i dorosłych. Częstość występowania
u młodych osób szacuje się na 1–3%. Stanowi ona
główną przyczynę skierowań do diagnostyki w prak-
tyce pediatrycznej, neurologii dziecięcej oraz genetyce
klinicznej. Bardzo często pomimo intensywnych badań
nie udaje się ustalić etiologii, pozostawiając rodzinę
bez dokładnego poradnictwa genetycznego oraz dia-
gnostyki prenatalnej. Niepełnosprawność intelektualna
to powstały przed 18 rokiem życia istotnie niższy od
przeciętnego (co najmniej 2 odchylenia standardowe
– standard deviation – SD) ogólny poziom funkcjonowa-
nia intelektualnego, występujący łącznie z trudnościami
w zakresie przystosowania. W definicji użyto terminu
„niepełnosprawność intelektualna”, który jest tożsamy
z wcześniejszym określeniem „upośledzenie umysło-
we”. Ze względu na pejoratywny i piętnujący charakter
drugiego pojęcia, zalecane jest używanie określenia
„niepełnosprawność intelektualna” [1].
Definicja NI według DSM-IV z 1994 r. obejmuje
3 kryteria: 1) istotnie niższy ogólny poziom funkcjono-
wania intelektualnego; 2) współwystępowanie znacznych
ograniczeń w zakresie przystosowania w przynajmniej
2 obszarach spośród następujących: porozumiewanie się,
troska o siebie, życie domowe, sprawność społeczno-
-interpersonalna, korzystanie ze środków zabezpieczenia
społecznego, kierowanie sobą, troska o bezpieczeństwo,
troska o zdrowie, zdolności szkolne, sposoby organi-
zowania czasu wolnego; 3) początek tego stanu musi
wystąpić przed 18 rokiem życia.
Poziom rozwoju umysłowego określa się za pomocą
ilorazu inteligencji (II) wartości liczbowej testu psycho-
metrycznego, którego celem jest obiektywny pomiar
inteligencji kognitywnej, polegającej na umiejętności
kojarzenia informacji i operacji na symbolach. Wartość
ta nie jest absolutną miarą inteligencji, lecz jest to po-
miar zawsze relatywny. Wartość II 100 oznacza średnią
inteligencję kognitywną w grupie wiekowej danej oso-
by. Zazwyczaj test konstruuje się tak, aby otrzymany
wynik dla całej grupy wiekowej układał się w typową
krzywą Gaussa o kształcie dzwonu, a średni rozrzut (σ)
statystyczny wyników wynosił 15. W praktyce oznacza
to, że: a) wynik powyżej 115 wskazuje na inteligencję
wybitną, b) 85–115 na inteligencję przeciętną, c) poniżej
85 na inteligencję niską, d) 70–85 na inteligencję niższą
niż przeciętna, czyli dolną granicę normy [1,2].
Termin „niepełnosprawność intelektualna” nie jest
diagnozą, lecz objawem. Nie informuje o etiologii, pro-
gnozie czy specyficznym leczeniu. Odnosi się do stanu
klinicznego i dotyczy funkcjonowania intelektualnego
oraz socjalnego. Niepełnosprawność intelektualna sta-
nowi bardzo heterogenną grupę zaburzeń i niestety w
większości przypadków (20–50%) jej etiologia pozo-
staje nieznana. W celu ułatwienia klinicznego badania
NI przyjęto różne podziały. Wcześniejsze doniesienia
wyróżniały NI „patologiczną”, czyli ciężką, oraz „ro-
dzinną”, zwykle łagodną. Niepełnosprawność inte-
lektualną można także podzielić ze względu na czas
wystąpienia na prenatalną, okołoporodową oraz post-
natalną [3]. Klasyczny podział opiera się na wartości II,
wyłaniając 4 grupy: NI lekkiego stopnia (II 50–70, 2–3
SD), NI umiarkowanego stopnia (II 35–49, 3–4 SD),
NI znacznego stopnia (II 20–34, 4–5 SD) oraz NI głę-
bokiego stopnia (II < 20, > 5 SD) [2]. Lekka postać NI
występuje 7 razy częściej niż postacie umiarkowana lub

147
Nr 4–6
ciężka. Bardziej praktyczny podział NI ze względu na
wartość II obejmuje 2 grupy: lekkiego stopnia, gdy II
jest wyższy od 50, oraz umiarkowanego do znacznego
stopnia, gdy II jest niższy od 50.
Szanse ustalenia etiologii NI są większe u osób
z ciężką postacią tego zaburzenia. Przyczyny NI mogą
być genetyczne lub środowiskowe, wrodzone (aberracje
chromosomowe, działanie czynników teratogennych
na płód) lub nabyte (infekcje ośrodkowego układu
nerwowego, urazy głowy) [4]. Niepełnosprawność in-
telektualną można także podzielić na specyficzną, gdy
towarzyszą jej cechy dysmorficzne lub wady narządów
wewnętrznych, oraz niespecyficzną, gdy jedynym
objawem jest niepełnosprawność intelektualna [5].
Zdecydowana większość (90%) przypadków NI należy
do grupy lekkiego stopnia (II > 50), jedynie 10% sta-
nowią chorzy z NI umiarkowanego i znacznego stopnia
(II < 50). Przyczynę zaburzeń udaje się ustalić jedynie
u około 50% osób z NI od umiarkowanego do znacznego
stopnia oraz u znacznie mniejszego odsetka osób z lekką
postacią NI [3].
Przeszukanie bazy danych OMIM (On-Line Men-
delian Inheritance in Man; http://www3.ncbi.nml.
nih.gov/omim/) w czerwcu 2007 r. za pomocą słowa
kluczowego „niepełnosprawność intelektualna” ujaw-
niło 1418 dokumentów. Penrose [6] w 1938 r. po raz
pierwszy zaobserwował, że NI znamiennie częściej
dotyczy mężczyzn niż kobiet; według niego, stosunek
ten wynosił 1,3:1. Podobne badania prowadzone przez
liczne grupy badawcze w USA, Kanadzie, Australii i Eu-
ropie potwierdziły tę obserwację, wskazując na większą
o 30% liczbę osób płci męskiej z NI. Lehrke [7] w 1974 r.
przedstawił koncepcję, że wyjaśnieniem obserwowanej
przewagi może być sprzężenie z chromosomem X.
Obserwacje te oraz opisy licznych dużych rodzin z NI
u wielu członków danej rodziny, wskazujących na
dziedziczenie sprzężone z chromosomem X, dało
podstawę do wysunięcia hipotezy, że geny sprzężone
z chromosomem X odgrywają ważną rolę w etiologii
NI. Przypuszcza się, że obserwowana przewaga męż-
czyzn może być następstwem niemożności kompensacji
patogennych mutacji w chromosomie X w stanie hemi-
zygotycznym u mężczyzny. Inne wytłumaczenie tego
zjawiska może stanowić obserwowany zazwyczaj brak
objawów klinicznych u kobiet nosicielek z 2 chromo-
somami X, z których jeden ulega lionizacji. Choroby
sprzężone z chromosomem X są następstwem mutacji
w genach zlokalizowanych w chromosomie X i dotyczą
głównie mężczyzn. Kobiety nosicielki nieprawidłowego
genu zwykle nie wykazują cech choroby lub cechy te
są u nich bardzo subtelnie wyrażone. Szacuje się, że
monogenowa, sprzężona z chromosomem X niepełno-
sprawność intelektualna (X-linked mental retardation
– XLMR) dotyczy 10% mężczyzn z tym zaburzeniem,
czyli 1/550 mężczyzn jest nosicielem nieprawidłowego
genu w chromosomie X. Obecnie trudno oszacować
proporcje specyficznych postaci NI sprzężonych z chro-
mosomem X, lecz na podstawie badań klinicznych
oraz biorąc pod uwagę zespół łamliwego chromosomu X
(Fra X) mogą one stanowić 30–40%. Wyniki badań mo-
lekularnych wskazują, że częstość mutacji w XLMR
wynosi 8–14%. Szacowana na tej podstawie częstość
występowania XLMR oceniana jest na 0,9–1,4/1000
mężczyzn [8].
Dotychczas opisano 143 specyficzne postacie NI
sprzężonej z chromosomem X (MRXS) i zmapowano
dla nich 48 genów. Udało się także sklonować 59 genów
oraz opisano 88 rodzin z niespecyficzną postacią NI
Niepełnosprawność intelektualna
OMIM
Nazwa białka
Locus
Gen
300034
300096
300104
300142
300157
300189
300206
300267
300286
300336
300427
300499
300573
300576
300585
309548
312173
314995
314998
Angiotensin rec.2
Tetraspanin
RABGDIA
P21 Act. Kinase 3
Fatty acid-CoA ligase 4
Discs large homolog 3
IL1 receptor accessory protein
αPIX
Kruppel-like Factor 8
Neuroligin 3
Neuroligin 4
FTSJ homolog 1
zinc-finger 674
zinc-finger, DHHC-type containing 15
zinc-finger 673
AFF2 protein
Ribosomal protein L10
zinc-finger 41
zinc-finger 81
Xq23
Xq11.4
Xq28
Xq23
Xq23
Xq13.1
Xp21.1
Xq26.3
Xp11.21
Xq13
Xp22.3
Xq11.23
Xp11.3
Xq13.3
Xp11.3
Xq28
Xq28
Xp11.3
Xp11.23
AGTR2
TM4SF2
GDI1
PAK3
FACL4 (ACSL4)
DLG3
IL1RAPL
ARHGEF6
KLF8/ZNF741
NLGN3
NLGN4
FTSJ1
ZNF674
ZDHHC15
ZNF673
FMR2
RLP10
ZNF41
ZNF81
Tabela I. Dziewiętnaście sklonowanych genów odpowiedzialnych za niespecyficzną postać niepełnosprawności
intelektualnej (MRX)
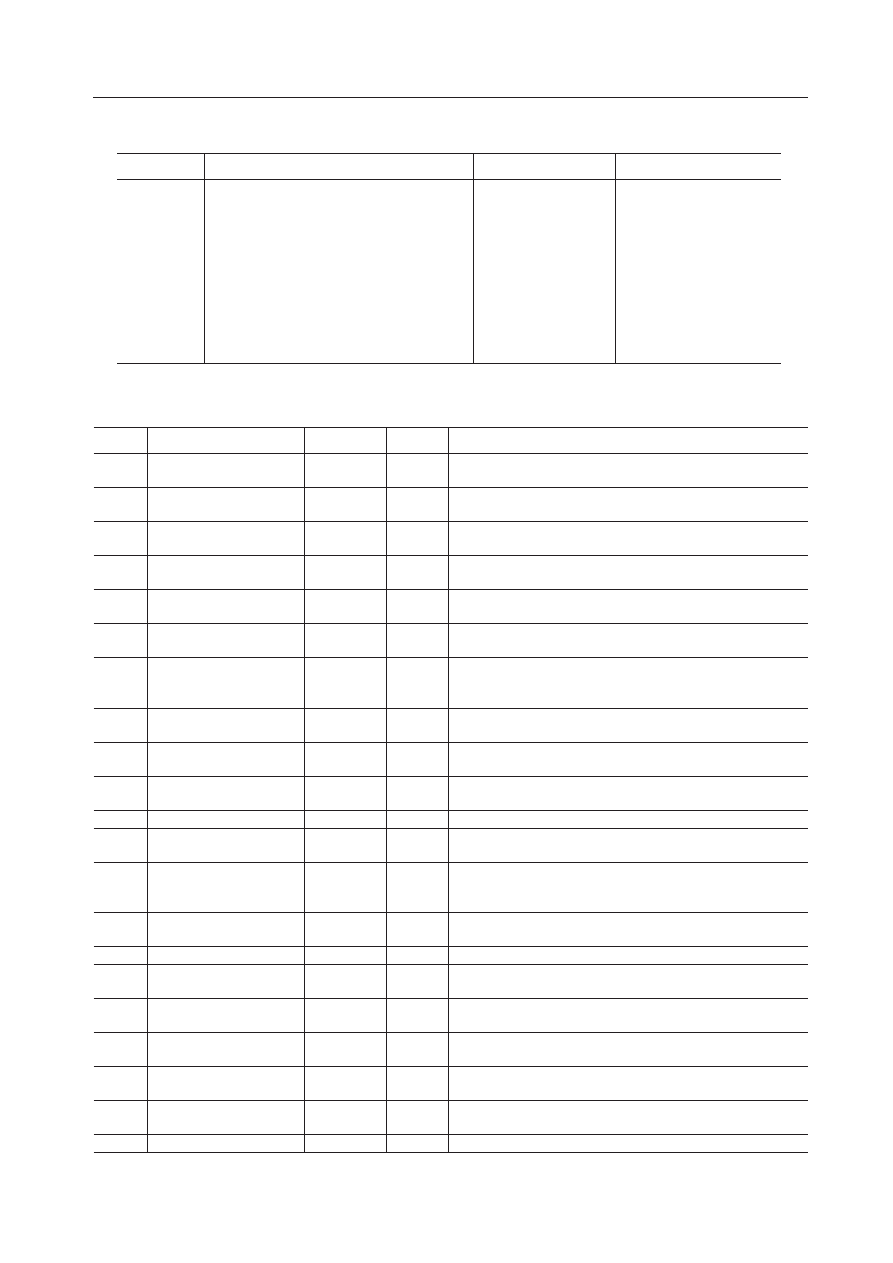
148
Nr 4–6
M.Z. Lisik, A.L. Sieroń
OMIM
Schorzenie
Locus
Gen
300005
300032
300036
300075
300127
300382
300463
300523
300630
305400
314690
zespół Retta
ATR-X
niedobór transportera kreatyniny
zespół Coffina i Lowry’ego
ataksja móżdżkowa
zespół Westa/drgawki niemowlęce
zespół Sutherlanda i Haana/MRXS3
zespół Allana, Herndona i Dudleya
zespół Turnera
zespół Aarskoga i Scotta
JARID1C-related XLMR
Xq28
Xq13.2
Xq28
Xp22.1
Xq12
Xp22.1
Xp11.23
Xq13.2
Xp22.2
Xp11.22
Xp11.22
MECP2
ATRX (XNP)
SLC6A8
RSK2 (RPS6KA3)
OPHN1
ARX
PQBP1
ALC16A2/MCT8
AP1S2
FGD1
JARID1C/SMCX
OMIM
Schorzenie
Gen
Locus
Opis
305400
zespół Aarskoga
i Scotta
FGD1
Xp11.2
niskorosłość, hiperteloryzm, moszna szalowa,
nadmierna wiotkość stawów
301040
zespół ATRX
ATRX (XNP) Xq13.3 małogłowie, pogrubiałe rysy twarzy, nieprawidłowości szkieletu
oraz genitalii, inkluzje HbH
301900
zespół Borjesona,
Forsmanna i Lehmanna
PHF6
Xq26.2
otyłość, hipogonadyzm, okrągła twarz, wąskie szpary
powiekowe, padaczka
300354
zespół Cabezasa
CUL4B
Xq24
niskorosłość, otyłość, hipogonadyzm, wydatna dolna warga,
zanik mięśni
300524
zespół Cantagrela
KIAA2022
Xq13.2
niskorosłość, małogłowie, krótka rynienka podnosowa,
znacznego stopnia NI, spastyczne porażenie czterokończynowe
303600
zespół Coffina
i Lowry’ego
RSK2
(RPS6KA3)
Xp22.1
pogrubiałe rysy twarzy, zwężające się palce, nieprawidłowości
szkieletu
300900
zespół Cornelii de Lange,
sprzężony
z chromosomem X
SMC1A/
/SMC1L1
Xp11.22 dysmorfia twarzy, niedobór wzrostu z zaburzeniami karmienia,
małe dłonie
305000
wrodzona dyskeratoza
DKC1
Xq28
siatkowata pigmentacja skóry, dystrofia paznokci, leukoplakia
śluzówek
305450
FG
MED12
Xq13.1 wielkogłowie, agenezja ciała modzelowatego, nieprawidłowości
układu pokarmowego, głuchota
300321
FGS-2
FLNA
Xq28
wydatne czoło, obniżone napięcie mięśniowe, przemieszczony
do przodu odbyt
FGS-like
UPF3B
Xq24
300624
fragile X syndrome
FMR1
Xq27.3
wielkogłowie, podłużna twarz, duże małżowiny uszne,
powiększenie jąder
305600
zespół Goltza/
/ogniskowa hipoplazja
skóry
PORCN
Xp11.23
ogniskowy niedorozwój skóry, krótkie palce, brak palców,
dodatkowe palce z syndaktylią, małoocze
300472
zespół Grahama
IGBP1
Xq13
niskorosłość, agenezja ciała modzelowatego, ubytek tęczówki,
nisko osadzone małżowiny uszne, głuchota
307030
hiperglicerolemia
GK
Xp21.2
gliceroluria, słaby przyrost wzrostu, zez zbieżny, osteoporoza
309900
choroba Huntera
IDS
Xq28
pogrubiałe rysy twarzy, dysostoza wielu stawów, niskorosłość,
powiększenie wątroby i śledziony
308300
incontinentia pigmenti/
/IP-2
IKBKG/
/NEMO
Xq28
nietrzymanie barwnika, nieprawidłowości uzębienia,
nieprawidłowości tęczówki
300534
JARID1C-related XLMR
JARID1C/
/SMCX
Xp11.2
niskorosłość, powoli postępująca spastyczna paraplegia,
niedorozwój szczęki
300319
Jun
NXF5
Xq22.1
niskorosłość, skośne dolne ustawienie szpar powiekowych,
małżowiny uszne
309000
zespół Lowe’a/oczno-
-mózgowo-nerkowy
OCRL1
Xq25
wodoocze, zaćma, krzywica witamino-D-oporna,
wielkogłowie XLMR
BRWD3
Xq21.1
wielkogłowie
Tabela II. Geny odpowiedzialne za specyficzną (MRXS) oraz niespecyficzną postać niepełnosprawności intelektualnej
(MRX); http://xlmr.interfree.it
Tabela III. Lista sklonowanych genów odpowiedzialnych za specyficzną postać niepełnosprawności intelektualnej (MRXS);
http://xlmr.interfree.it

149
Nr 4–6
sprzężonej z chromosomem X (MRX; http://xlmr.inter-
free.it; Greenwood Medical Center). Niestety mutację
w poznanych genach można stwierdzić tylko u niewiel-
kiego odsetka rodzin obciążonych niepełnosprawnością
intelektualną XLMR oraz u jeszcze mniejszego odsetka
chorych w przypadku sporadycznej postaci NI. Na pod-
stawie danych można przedstawić koncepcję, że mutacje
ponad 100 genów zlokalizowanych w chromosomie X
mogą się wiązać z NI. Liczba genów XLMR przekracza
5–10-krotnie szacowaną pierwotnie liczbę [9].
Niepełnosprawność intelektualną sprzężoną z chro-
mosomem X można podzielić na 3 grupy: 1) zespół
łamliwego chromosomu X, FRAXA (fragile X syn-
drome type A), który stanowi najczęstszą przyczynę NI;
szacuje się, iż występuje u 1/4000 mężczyzn oraz
u 1/8000 kobiet; 2) MRXS, rozpoznawalną klinicznie
ze względu na towarzyszące jej specyficzne nieprawi-
dłowości fizyczne, neurologiczne oraz metaboliczne;
3) MRX, w której głównym objawem klinicznym jest
niepełnosprawność intelektualna [10]. Użyteczne wy-
daje się wyróżnienie w MRXS 4 podgrup, takich jak:
a) zespoły malformacji, obejmujące liczne wady roz-
wojowe, b) choroby nerwowo-mięśniowe, obejmujące
układ nerwowy i/lub mięśnie, c) choroby metaboliczne
oraz d) choroby dominujące [11]. Chociaż podział
XLMR na postacie specyficzną oraz niespecyficzną
pozostaje użyteczny dla celów klinicznych, ostatnie
badania zależności między fenotypem i genotypem
oraz szczegółowa analiza kliniczna chorych wskazują
na zanikanie granic między tymi postaciami. Niektóre
postacie wcześniej kwalifikowane jako niespecyficzne
obecnie zaliczane są do postaci specyficznych. Przy-
kładem może być Fra X. Początkowo był on uważany
za niespecyficzną postać NI, jednak po kompleksowej
analizie klinicznej został zakwalifikowany jako po-
stać specyficzna. Niemniej jednak podział ten może
okazać się pomocny w kwalifikowaniu pacjentów do
badań molekularnych. Mutacje w niektórych genach
odpowiadają zarówno za postać specyficzną, jak i
niespecyficzną NI. Mutacje w genie ARX są przyczyną
MRXS w postaci zespołu Westa, zespołu Partingtona,
lissencefalii (gładkomózgowia) oraz MRX, w której
jedyny objaw choroby stanowi niepełnosprawność
intelektualna [12,13].
Niepełnosprawność intelektualna
ATRX – zespół alfa-talasemia/niepełnosprawność intelektualna (X-linked alpha-thalassaemia mental retardation syndrome).
Marfanoid II
ZDHHC9
Xq25
obniżone napięcie mięśniowe, cechy marfanoidalne, opóźniony
rozwój mowy
300166
zespół oczno-
-twarzowo-sercowo-
-zębowy
BCOR
Xp11.4
małoocze, zaćma, powiększenie korzonków nerwowych, ubytki
w przegrodzie międzykomorowej
309801
MCOPS7/MIDAS
HCCS
Xp22.2
małoocze, niedorozwój skóry, sclerocornea
309400
choroba Menkesa
ATP7A
Xp21.1
niedobór wzrostu, skręcone włosy,
ogniskowa degeneracja móżdżku i mózgu
300123
MRGH
SOX3
Xq27.1
izolowany niedobór hormonu wzrostu, niskorosłość,
małe siodło tureckie
302350
zespół Nance’a
i Horana
NHS
Xp22.13
zaćma, mała rogówka, stożkowate siekacze,
dodatkowe zęby
312180
Nascimento
UBE2A
Xq24
uogólniony hirsutyzm, zrośnięcie brwi, duże usta, otyłość,
niskorosłość, padaczka
300000
zespół Opitza-G/BBB
MID1
Xp22.2
hiperteloryzm, nieprawidłowości linii środkowej,
wada serca, spodziectwo
311200
zespół ustno-twarzowo-
-palcowy 1
OFD1
Xp22.2
środkowy rozszczep twarzy, guzki języka, syndaktylia
311300
zespół uszno-
-podniebienno-palcowy 1
FLNA
Xq28
niskorosłość, utrata słuchu, rozszczep podniebienia, szerokie
kciuki i paluch, syndaktylia
309500
zespół Renpenninga
PQBP1
Xp11.3
małogłowie, niskorosłość
300263 zespół Sideriusa i Hamela
PHF8
Xp11.22
rozszczep wargi i podniebienia, szeroki czubek nosa,
duże dłonie
312870
zespół Simpsona,
Golabiego i Behmela
GPC3
Xq26.2
makrosomia, pogrubiałe rysy twarzy, dodatkowe palce,
dodatkowe brodawki sutkowe, wada serca
309583
zespół Snydera
i Robinsona
SMS
Xp22.11
wielkogłowie, długa szczupła twarz, wysoko wysklepione
podniebienie/rozszczep podniebienia, asteniczna budowa ciała
300434
zespół Stocco dos Santos
KIAA1202/
/SHROOM4 Xp11.22
niskorosłość, obustronne zwichnięcie stawów biodrowych,
zanik kory, padaczka
300630
zespół Turnera
AP1S2
Xp22.2
znacznego stopnia obniżenie napięcia mięśniowego,
niskorosłość, wysokie czoło, mały podbródek
314390
zespół VACTERL
z wodogłowiem
FANCB
Xp22.2
anomalie kręgów, odbytu, krtani, przełyku, nerek,
kości promieniowych, wodogłowie

150
Nr 4–6
Kamieniem milowym w rozumieniu patofizjologii
NI okazało się poznanie genu FMR1. Od tego momentu
wiele laboratoriów zaczęło badać funkcję kodowanego
białka oraz patogenezę Fra X. Białko kodowane przez
gen FMR1, FMRP (fragile X mental retardation pro-
tein), posiada domeny wiążące mRNA, bierze udział
w rozwoju dendrytów oraz funkcji synaps. Dużo mniej
wiadomo o funkcji innych genów, które ulegają mutacji
w MRX. Taki stan wiedzy częściowo jest następstwem
stosunkowo niedawnego zidentyfikowania tych genów.
Geny poznane kilka lat temu są funkcjonalnie związane
z tworzeniem i dekonstrukcją cytoszkieletu aktyny oraz
kontrolą wzrostu neurytów [13].
Wyniki projektu mapowania ludzkiego genomu oraz
badania zwierząt laboratoryjnych, u których przerwano
ciągłość genu, pozwoliły na określenie specyficznych
zmian wewnątrzkomórkowych w przypadku mutacji
genu oraz wpływu braku białka na zaburzenia funkcji
poznawczych, pozwalając na ustalenie mechanizmów
komórkowych. Remodelowanie synapsy, zmiany
w kształcie wypustek oraz gęstości dendrytów leżą u pod-
staw wielu funkcji mózgu, takich jak nauka i pamięć.
Liczne białka kodowane przez geny, których mutacje
prowadzą do XLMR, aktywują szlaki sygnałów regulu-
jące morfologię wypustek dendrytów, uwalnianie neuro-
transmiterów, wzrost aksonów oraz cytoszkieletu.
Obecna teoria sugeruje, że NI jest następstwem
zaburzeń w strukturze i funkcji synaps. Pierwszym z ge-
nów związanych z XLMR był FMR2, którego mutacje
powodują powstanie miejsca łamliwego (fragile X
syndrome type E – FRAXE) w miejscu Xq28. Następ-
nie zidentyfikowano kolejne geny związane z XLMR:
GDI1, OPHN1, PAK3, RPS6KA3, IL1RAPL1, TM4SF2,
ARHGEF6, MECP2, FACL4, ARX. Mutacje w każdym
z tych genów są stosunkowo rzadkie, odpowiadają za
mniej niż 1% przypadków XLMR. Zidentyfikowano
21 genów XLMR, w których mutacje stwierdzono u 24
z 82 rodzin z MRX [13]. Białka kodowane przez geny
związane z XLMR można ze względu na ich funkcję
podzielić na kilka grup: 1) białka regulatory lub efektory
RHO GTP-azy (OPHN1, RhoGEF, PAK3, ARHGEF6,
FGD1); 2) białka biorące udział w szlakach transdukcji
sygnałów pochodzenia zewnątrzkomórkowego z po-
wierzchni komórki do aktyny cytoszkieletu komórki
i jądra, niezbędne dla wzrostu aksonów i/lub ustalenia
i stabilizacji połączeń nerwowych; 3) białka kontrolujące
ekspresję genów poprzez modulacje struktury chroma-
tyny (MeCP2, NP, CDKL5, RSK2, ZNF41, ZNF81);
4) białka biorące udział w tworzeniu synaps (NLGN4,
SYN1, DLG3, GDI1); 5) białka regulatory transkrypcji
(ARX, ZNF41, JARD). Inne postacie XLMR są następ-
stwem zaburzeń takich fundamentalnych procesów, jak:
składanie RNA (PQBP1), translacja (FTSJ1), degradacja
białek (MID1, UBE2A), metabolizm energii (SLC6A8)
lub defekty metaboliczne (SMS, ACSL4) [13,14,15].
Dane genetyczne w połączeniu z badaniami funkcjo-
nalnymi wskazują, że zaburzenia regulacji subtelnych
mechanizmów zarządzających aktywnością synaptyczną
oraz plastycznością synaps mogą być uznane za jeden
z podstawowych procesów biorących udział w patoge-
nezie różnych postaci autosomalnych oraz sprzężonych
z chromosomem X niepełnosprawności intelektualnej.
Kandel i wsp. [16] opisali proces przechowywania
w pamięci i uczenia się jako dialog między genami oraz
synapsami. Zaproponowali oni model pamięci krót-
koterminowej, która jest następstwem bezpośrednich
biochemicznych zmian synaptycznych polegających
m.in. na aktywacji CaMKII, wzroście aktywności re-
ceptora glutaminianowego AMPA. Według ich modelu,
pamięć długoterminowa wymaga transkrypcji i trans-
lacji nowych białek, wzmacniających siłę oraz liczbę
aktywnych synaps [17]. Można więc spekulować, że
w przypadku NI nieodpowiednie pobudzenie odpowiedzi
synaptycznej przez bodźce czuciowo-motoryczne może
być następstwem zaburzeń w regulowaniu kaskady
sygnałów transkrypcyjnych regulujących ekspresję
czynników kluczowych dla morfogenezy, aktywności
i plastyczności synaps [16].
W świetle obecnej wiedzy można zaproponować
hipotezę „opartą na synapsie” dla kilku postaci NI.
Zaburzenia funkcji białek kodowanych przez geny
objęte szerokim spektrum deficytów poznawczych
– od łagodnej postaci NI, z występowaniem lub bez
cech autystycznych i zaburzeń zachowania, do ciężkiej
NI – mogą prowadzić poprzez zmiany specyficznych
w regulacji swoistych szlaków i procesów komórkowych
do defektów w strukturze i/lub funkcji synaps oraz sieci
neuronalnej, a w konsekwencji do hamowania zdolności
mózgu do przetwarzania informacji [15,16]. Wydaje się,
że w niektórych postaciach NI białka potrzebne w okresie
postnatalnym (w czasie aktywnej nauki) oraz powsta-
jące deficyty są subtelne i do pewnego stopnia można
im zapobiegać lub leczyć je w przypadku wczesnej
diagnozy i odpowiedniej terapii. Terapia behawioralna
oraz poznawcza może pomóc pacjentom z NI wyko-
rzystać w pełni tkwiący w nich potencjał. W przypadku
większości chorych cechy kliniczne nie są wystarczająco
specyficzne, aby ukierunkować badanie na poszukiwanie
określonej mutacji właściwego genu. Analiza mutacji
wymaga zastosowania tańszych metod. Obserwujemy
intensywny rozwój nowych genomowych technologii
sekwencjonowania, które są coraz szybsze i tańsze, np.
wykorzystanie mikromacierzy DNA czy masowego
sekwencjonowania z wykorzystaniem strategii shotgun.
W następnych dekadach nowe technologie umożliwią
sekwencjonowanie wielu, jeżeli nie wszystkich genów
XLMR w jednej próbie [16,17].
Odkrycie mutacji w genie ARX w dużej liczbie
rodzin zarówno ze specyficzną, jak i niespecyficzną
postacią NI wiązało się z nadzieją, że poznano ważny
M.Z. Lisik, A.L. Sieroń

151
Nr 4–6
gen, który będzie można łatwo badać, a powtarzalne
mutacje w tym genie będą wykrywane u większości
rodzin z NI. Szczególnie ważne było odkrycie, że
7 z 9 mutacji polegało na powieleniu lub duplikacji jed-
nego z 2 ciągów wieloalaninowych. Najczęstszą mutację,
duplikację 24 par zasad (zwaną dup24), prowadzącą do
powielenia ciągu alanin z 12 do 20, opisano dla rodziny
z różnymi fenotypami jej członków. Powtórna ocena
kliniczna badanej rodziny wykryła wewnątrzrodzinne
zróżnicowanie ekspresji objawów choroby oraz ich hete-
rogenności. Badania przeprowadzone przez Europejskie
Konsorcjum XLMR wykazały, że mutacja w genie ARX
występowała w 8,1% rodzin z NI, a mutacja duplikacja
24 u 6,6% z nich [18,19]. Ocenia się, że 50% genów ule-
ga ekspresji w mózgu. Liczba genów zlokalizowanych
w autosomach prawdopodobnie stanowi wielokrot-
ność liczby genów sprzężonych z chromosomem X.
Identyfikacja postaci autosomalnej recesywnej będzie
zdecydowanie trudniejsza, ponieważ będzie się objawiać
głównie w postaci sporadycznej. Większość z licznych
autosomalnych recesywnych genów odpowiedzialnych
za NI pozostaje niezidentyfikowana. Autosomalny
recesywny typ dziedziczenia niespecyficznej postaci
NI często pozostaje nierozpoznany. Mapowanie oraz
identyfikacja sprawczych genów będzie w dużej mierze
uzależniona od dostępności rodzin spokrewnionych. Do
tej pory opisano mutacje w 3 genach, które występowały
w pojedynczych bardzo licznych spokrewnionych rodzi-
nach. Są to: PRSS12 – neurotrypsyna (MIM#249500),
CRBN – cerebron (MIM#607417) oraz CC2D1A (coiled-
-coil and C2 domain containing 1A; MIM#608441)
[20,21,22].
Konsensus opracowany w czasie konferencji eksper-
tów, sponsorowanej przez American College of Medical
Genetics (wrzesień 1995 r.), obejmował schemat postę-
powania w NI [23]. Pierwszym krokiem w przypadku
dziecka lub dorosłego z NI powinien być ukierunkowany
wywiad medyczny oraz badanie fizykalne. Wywiad
powinien dotyczyć występowania w rodzinie chorób
neurologicznych oraz NI, pokrewieństwa między rodzi-
cami, poziomu wykształcenia rodziców, szczegółowego
przebiegu ciąży, w tym narażenia na działanie toksyn,
leków, infekcji, przebieg porodu oraz okresu okołopo-
rodowego ze zwróceniem uwagi na urodzeniową masę
i długość ciała oraz obwód głowy, testy noworodkowe
w kierunku fenyloketonurii oraz niedoczynności tarczy-
cy, parametry rozwoju psychoruchowego (wiek, w jakim
dziecko siedziało, chodziło, mówiło) oraz parametry
rozwoju fizycznego (wzrost, masa ciała oraz obwód gło-
wy). Następnie należy sporządzić rodowód, obejmujący
co najmniej 3 pokolenia. Badanie fizykalne powinno
obejmować pomiar obwodu głowy i umieszczenie go
w siatkach centylowych dla grupy wiekowej osoby
badanej, szczegółowe oględziny skóry, jeśli to możliwe
z użyciem lampy Wooda, badanie neurologiczne z oceną
zachowania oraz badanie w kierunku wad wrodzonych.
Należy pamiętać, że cechy te mogą być bardzo sub-
telnie wyrażone. Użyteczny może się okazać przegląd
zdjęć oraz nagrań wideo, które są szczególnie cenne
w przypadku zaburzeń ruchowych oraz zachowania.
W badaniu fizykalnym powinno się dokonać oglądu całej
sylwetki pacjenta, ze zwróceniem szczególnej uwagi na
ocenę cech dysmorficznych twarzy, co często pomaga
w postawieniu diagnozy klinicznej. Często zachodzi
konieczność wykonania badań audiologicznych, okuli-
stycznych oraz psychometrycznych. Nieprawidłowości
w badaniu fizykalnym powinny zostać udokumentowane
szczegółowymi pomiarami, opisem, a także dokumen-
tacją fotograficzną [24,25,26].
Testy diagnostyczne
Analiza chromosomów powinna stanowić główny
element procesu diagnostycznego w przypadku NI.
U każdego dziecka z NI o nieustalonej etiologii wymaga-
ne jest wykonanie kariotypu o rozdzielczości minimum
500 prążków. W przypadku podejrzenia specyficznego
zespołu mikrodelecji po wykluczeniu aberracji chromo-
somowej należy wykonać badanie FISH (fluorescent in
situ hybridization – FISH) z zastosowaniem specyficznej
sondy lub sond telomerowych. U pacjentów z asyme-
trią i/lub zmianami pigmentacji skóry należy wykonać
biopsję skóry w celu określenia kariotypu z fibroblastów
skóry, aby wykluczyć mozaikowatość somatyczną.
Badanie w kierunku Fra X należy rozważyć zarówno
u kobiet, jak i u mężczyzn z niewyjaśnioną NI, szcze-
gólnie w przypadku dodatniego wywiadu rodzinnego,
zaburzeń zachowania i braku dużych wad strukturalnych.
Badania neuroobrazujące należy wykonać w przypadku
objawów neurologicznych oraz nieprawidłowego obwo-
du głowy (małogłowie, wielkogłowie). W większości
przypadków metodę z wyboru powinien stanowić
rezonans magnetyczny mózgu. Testy metaboliczne
powinno się wykonać w przypadku cech klinicznych
oraz fizykalnych wskazujących na ten typ zaburzenia.
Selektywny skrining metaboliczny jest wskazany w róż-
nych sytuacjach klinicznych od hipotonii u noworodków
do postępującego pogrubienia rysów twarzy i regresu
w rozwoju psychomotorycznym. Badania takie powinny
obejmować ocenę równowagi kwasowo-zasadowej,
analizę aminokwasów oraz kwasów organicznych
w osoczu i moczu, analizę enzymów lizosomalnych,
analizę karnityny w osoczu i moczu, bardzo długie kwasy
tłuszczowe w osoczu i inne [23,24,25].
W przypadku NI należy objąć poradnictwem ge-
netycznym całą rodzinę. W trakcie udzielania porady
genetycznej powstaje pytanie dotyczące ryzyka po-
wtórnego wystąpienia NI u kolejnych dzieci danej pary
małżeńskiej. Postawienie diagnozy na podstawie wyni-
ku badania molekularnego, wskazującego na mutacje
w konkretnym genie, pozwala na udzielenie precyzyjnej
porady genetycznej oraz wykonanie diagnostyki prena-
talnej w czasie następnej ciąży. Niestety jest to możliwe
Niepełnosprawność intelektualna

152
Nr 4–6
u mniejszości rodzin z NI. W przypadku gdy etiologia
NI pozostaje nieznana, istnieje konieczność korzystania
z empirycznych danych dotyczących ryzyka powtórzenia
NI u kolejnych dzieci. Większość danych opublikowano
w latach 1971–1987, i chociaż obserwowane ryzyko
powtórzenia choroby 2–12% wciąż jest ważne, postęp
w dziedzinie genetyki molekularnej, w tym jakość ana-
lizy chromosomów, możliwość analizy molekularnej
genu FMR1, poprawa w ocenie klinicznej przypadków
postaci specyficznych, stawia pytanie o prawdziwość
niektórych z tych danych. Przeprowadzone ostatnio
badania populacyjne w Atlancie (USA) obejmowały
dzieci urodzone w latach 1981–1991. Oszacowane na
ich podstawie ryzyko powtórzenia w przypadku izolo-
wanej postaci NI wynosiło 8,4%, w przypadku postaci
łagodnej 7,1%, w postaci ciężkiej 4,7% [27]. Turner
i Partington [29] wykorzystując szczegółowe badanie
kliniczne oraz dostępne metody genetyki molekularnej
oraz cytogenetyki zaobserwowali ryzyko powtórzenia NI
dla rodzeństwa probanda 1:7,5 dla par braci oraz 1:20
dla par sióstr [30].
Niepełnosprawność intelektualna sprzężona z chro-
mosomem X stanowi bardzo heterogenną grupę zabu-
rzeń ze względu na liczbę genów odpowiedzialnych za
XLMR, która szacowana jest na około 100–130 [9].
Stwarza to znaczne trudności w stworzeniu specyficz-
nego protokołu ułatwiającego identyfikację mutacji
w genach zlokalizowanych w chromosomie X. Do-
świadczenie wskazuje, że dokładne badanie kliniczne ze
szczególnym zwróceniem uwagi na cechy dysmorficzne
pozwalające zakwalifikować chorych do właściwej gru-
py specyficznej/niespecyficznej postaci NI powinno być
pierwszym krokiem w diagnostyce różnicowej chłopca
z opóźnieniem rozwoju lub mężczyzny niepełnospraw-
nego intelektualnie. Rola chromosomu X w funkcjach
poznawczych jest intensywnie badana od lat 70. XX
wieku. Identyfikacja części genów pozwoliła na badanie
ich roli w rozwoju neuronów oraz procesów poznaw-
czych poprzez rozszerzenie badań molekularnych do
badania białek na poziomie komórkowym, wiązanych
ze wzrostem neuronów, tworzeniem dendrytów oraz
synaps. Istnieje potrzeba poprawy używanych obecnie
testów diagnostycznych oraz tworzenia nowych testów
psychodiagnostycznych, mających ocenić różne aspekty
procesów poznawczych pacjentów z NI. Korelacje mię-
dzy fenotypem i genotypem oraz badanie zachowania
powinny umożliwić spojrzenie na mechanizmy biorące
udział w poznaniu, nauce, zachowaniu, rozwoju mózgu.
Szczegółowe badanie kliniczne pozwala na ustalenie
etiologii NI w 50–70% przypadków.
Postępowanie w przypadku niepełnosprawności
intelektualnej
1. Analiza danych z wywiadu rodzinnego oraz ich
graficzne przedstawienie w postaci drzewa genealo-
gicznego, obejmującego co najmniej 3 pokolenia.
2. Analiza przebiegu ciąży oraz okresu okołoporodo-
wego.
3. Badanie fizykalne powinno polegać na dokładnych
oględzinach całej sylwetki pacjenta, ze szczególnym
zwróceniem uwagi na występowanie małych anoma-
lii, zmian zabarwienia skóry. Należy także wykonać
podstawowe pomiary antropometryczne, obejmujące
wzrost, masę ciała oraz obwód głowy, i porównać
uzyskane wyniki z danymi dla dzieci zdrowych
w odpowiednim wieku.
4. Bardzo ważny element stanowi badanie dysmorfo-
logiczne oraz dokumentacja fotograficzna. Szcze-
gółowe badanie fizykalne przeprowadzone przez
doświadczonego specjalistę pozostaje podstawą
w przypadku ustalania etiologii NI.
5. Badanie neurologiczne ze szczególnym zwróceniem
uwagi na występowanie zaburzeń zachowania.
6. Badanie cytogenetyczne o wysokiej rozdzielczości
(> 550 prążków).
7. W przypadku niemożności wykonania molekular-
nego kariotypowania należy wykonać co najmniej
badanie FISH w kierunku aberracji subtelomerowych
chromosomów.
8. W przypadku prawidłowego kariotypu należy wy-
konać badanie molekularne w kierunku Fra X jako
najczęstszej rodzinnej postaci NI z częstością 1/4000
urodzeń.
9. W przypadku stwierdzenia odchyleń od stanu pra-
widłowego w badaniu neurologicznym oraz niepra-
widłowego obwodu głowy należy wykonać badania
neuroradiologiczne oraz neuroobrazowanie. Obra-
zowanie mózgu za pomocą rezonansu magnetycz-
nego jest bardziej czułe w porównaniu z tomografią
komputerową. Problem może stanowić konieczność
znieczulenia ogólnego w celu neuroobrazowania
u wielu pacjentów niepełnosprawnych intelektual-
nie, szczególnie u młodszych. Znieczulenie ogólne
jest działaniem inwazyjnym i niesie ze sobą pewne
ryzyko powikłań.
10. Badania metaboliczne w kierunku wrodzonych
błędów metabolizmu powinny być selektywnie celo-
wane na podstawie danych z wywiadu oraz badania
fizykalnego [25].
Podsumowanie
Chociaż rozumienie mechanizmów molekularnych
funkcji białek w NI jest coraz lepsze, ich zastosowanie
w terapii jest odległe w czasie. Procesy patofizjologiczne
w ludzkim mózgu prowadzące do NI są bardzo złożone,
dodatkowo zaczynają się w bardzo wczesnym okresie
rozwoju płodowego. Identyfikacja genów pozwoliła na
badania funkcji ich produktów białkowych w rozwoju
neuronów oraz procesów poznawczych. Precyzyjna rola
wielu genów pozostaje nieznana. Jej poznanie pozwoli
na fundamentalnie nowe spojrzenie na mechanizm
funkcjonowania mózgu, odkrycie krzyżowych szlaków
M.Z. Lisik, A.L. Sieroń

153
Nr 4–6
Piśmiennictwo
[1] WHO. The ICD-10 classification of mental and behavioral disorders. WHO: 1992. [2] American Psychiatric Association. Diagnostic and statistical manual
of mental disorders DSM-IV: American Psychiatric Association: 1994. [3] Vasconelos MM. Mental retardation. J Pediatr 2004; 80(Suppl. 2): 71–82. [4] Win-
nepenninckx B, Rooms L, Kooy RF. Mental retardation: a review of the genetic causes. Br J Dev Disabil 2003; 49: 29–44. [5] Chelly J, Khelfaoui M, Francis F,
Cherif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet 2006; 14: 701–713. [6] Penrose LS. A clinical and genetic study
of 1 280 cases of mental defect. Medical Research Council Special Report Series 1938: 229. [7] Lehrke RG. X-linked mental retardation and verbal disability.
Birth Defects Orig Artic Ser 1974; 10: 1–100. [8] Kerr B, Turner G, Mulley J, Gedeon A, Partington M. Non-specific mental retardation. J Med Genet 1991; 28:
378–382. [9] Ropers HH, Hoeltzenbein M, Kalscheuer V, Yntema H, Hamel B, Fryns JP, Chelly J, Partington M, Gecz J, Moraine C. Nonsyndromic X-linked
mental retardation: where are the missing mutations? Trends Genet 2003; 19: 316–320. [10] Frints SG, Froyen G, Marynen P, Fryns JP. X-linked mental retarda-
tion: vanishing boundaries between non-specific (MRX) and syndromic (MRXS) forms. Clin Genet 2002; 62: 423–432.
[11] Stevenson RE. Splitting and lumping in the nosology of XLMR. Am J Med Genet 2000; 97: 174–182. [12] Kleefstra T, Hamel BC. X-linked mental retar-
dation: further lumping, splitting and emerging phenotypes. Clin Genet 2005; 67: 451–467. [13] Chiurazzi P, Tabolacci E, Neri G. X-linked mental retardation
(XLMR): from clinical conditions to cloned genes. Crit Rev Clin Lab Sci 2004; 41: 117–158. [14] Raymond LF, Tarpey P. The genetics of mental retardation.
Hum Mol Genet 2006; 15: 110–116. [15] Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet 2005; 6: 46–57. [16] Kandel ER. Nerve cells and
behavior. W: Principles of Neural Science. 4
th
ed. Red. Kandel ER, Schwartz JH, Jessel TM. McGraw-Hill. New York 2000, 19–35. [17] Chechlacz M, Gleeson
JG. Is mental retardation a defect of synapse structure and function? Pediatr Neurol 2003; 29: 11–17. [18] Mandel JL, Chelly J. Monogenic X-linked mental
retardation: is it as frequent as currently estimated? The paradox of the ARX (Aristaless X) mutations. Eur J Hum Genet 2004; 12: 689–693. [19] Gecz J, Clooster-
man D, Partington M. ARX: gene for all seasons. Curr Opin Genet Dev 2006; 16: 308–316. [20] Molinari F, Rio M, Meskenaite V, Encha-Razavi F, Auge J,
Bacq D, Briault S, Vekemans M, Munnich A, Attie-Bitach T i wsp. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation.
Science 2002; 298: 1779–1781.
[21] Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation.
Neurology 2004; 63: 1927–1931. [22] Basel-Vanagaite L, Attia R, Yahav M, Ferland RJ, Anteki L, Walsh CA, Olender T, Straussberg R, Magal N, Taub E i wsp.
The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J Med Genet 2006; 43:
203–210. [23] Curry JC, Stevenson RE, Aughton D, Byrne J, Carey JC, Cassidy S, Cunniff C, Graham JM Jr, Jones MC, Kaback MM i wsp. Evaluation of mental
retardation: recommendations of consensus conference. Am J Med Genet 1997; 72: 468–477. [24] Moeschler JB, Shevell M; American Academy of Pediatrics
Committee on Genetics. Clinical genetic evaluation of the child with mental retardation or developmental delay. Pediatrics 2006; 117: 2304–2316. [25] van Kar-
nebeek CD, Jansweijer MC, Leenders AG, Offringa M, Hennekam RC. Diagnostic investigations in individuals with mental retardation: a systematic literature
review of their usefulness. Eur J Hum Genet 2005; 13: 6–25. [26] Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, Zenker M, Huffmeier U, Thiel C,
Ruschendorf F i wsp. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet
A 2006; 140: 2063–2074. [27] Raymond FL. X-linked mental retardation: a clinical guide. J Med Genet 2006; 43: 193–200. [28] Van Naarden Braun K, Autry A,
Boyle C. A population-based study of the recurrence of developmental disabilities – Metropolitan Atlanta Developmental Disabilities Surveillance Program,
1991-94. Paediatr Perinat Epidemiol 2005; 19: 69–79. [29] Turner G, Partington M. Recurrence risks in undiagnosed mental retardation. J Med Genet 2000: 37:
45–47. [30] Crow YJ, Tolmie JL. Recurrence risks in mental retardation. J Med Genet 1998; 35: 177–182.
Adres autorów: Małgorzata Lisik, Katedra i Zakład Biologii Ogólnej, Molekularnej i Genetyki SUM, ul. Medyków 18, 40-752 Katowice,
e-mail: mlisik@slam.katowice.pl
dla jego prawidłowej funkcji oraz rozszerzenie wiedzy
na temat patofizjologii XLMR. Będzie także stanowić
podstawę do przyszłych badań neurobiologicznych,
pozwalających na szersze spojrzenie na rozwój intelek-
tualny i motoryczny. Badania molekularne i kliniczne
są kluczowe dla poznania szlaków biorących udział
w powstaniu NI i będą stanowić fundament przyszłej
interwencji terapeutycznej.
M.Z. Lisik, A.L. Sieroń
X-LINKED MENTAL RETARDATION – TREATMENT SCHEME
Summary
Mental retardation is a serious medical and social problem. The prevalence of mental retardation is estimated at 2–3%. Establishing
the cause of mental retardation is extremely important for prognosis, management, and genetic counseling. It is postulated that 25–35% of
mental retardation cases may be of genetic background. Among the genetic causes 25–30% are probably result of mutations located in the
X chromosome (X-linked mental retardation – XLMR). X-linked mental retardation is a heterogeneous set of conditions responsible for
a large proportion of inherited mental retardation. More than 200 XLMR conditions and 45 cloned genes are listed in catalogue available on
the Internet. Traditionally, based on clinical presentation, XLMR conditions were divided into specific and nonspecific forms or syndromic
and nonsyndromic. The distinction between specific and non-specific forms of XLMR is gradually becoming less clear and spectrum of phe-
notypic variability is very large as both syndromic and nonsyndromic forms have been described for several of the XLMR genes. Mutations
in patients suffering from X-linked mental retardation genes have been found only in a relatively limited number of cases. Up to 50% of the
patients from XLMR families might have mutations in one of the known genes implicated in XLMR so far. However, current methods are
generally too expensive or too unreliable to justify mutation screening of all known XLMR genes in diagnostic testing. Thus it is necessary
to use empirical data of recurrence risk in genetic counseling of the family with mental retardation.
Key words: mental retardation, X-linked, genetic counseling.
Niepełnosprawność intelektualna
Wyszukiwarka
Podobne podstrony:
Zastosowanie wybranych metod w pracy z dzieckiem z niepełnosprawnością intelektualną i sprzężoną nie
Seksualnosc dzieci niepelnosprawnych intelektualnie
Poczucie tożsamości seksualnej u osób niepełnosprawnych intelektualnie
Metodyka pracy?ukacyjno terapeutycznej z uczniem z niepełnosprawnością intelektualną
zal[1]. nr 7 Karta poziomu funkcjonowania ucznia, Pedagogika niepełnosprawnych intelektualnie
Techniki plastyczne w terapii dzieci o specjalnych potrzebach edukacyjnych, Niepełnosprawność intele
UPOŚLEDZENIE UMYSŁOWE W STOPNIU UMIARKOWANYM, Pedagogika dziecka z niepełnosprawnością intelektualną
Niepełnosprawność intelektualna w stopniu głębokim, Oligofrenopedagogika, NIEPEŁNOSPRAWNOŚĆ
Pedagogika Specjalna P L i T Z, Metodyka pracy z dziećmi niepełnosprawnymi intelektualnie
godnosc osoby niepoelnosprawnej (1), Niepełnosprawność intelektualna(1)
Młodzi dorośli z niepełnosprawnością intelektualną kontekst społeczny
FUNKCJONOWANIE OSÓB NIEPEŁNOSPRAWNYCH INTELEKTUALNIE W SPOŁECZEŃSTWIE POLSKIM orzginl licencjata
Niepełnosprawni intelektualnie
DokZal 2608 idzal. nr 1 do uchwaly nr 495, Niepełnosprawność, Niepełnosprawność intelektualna, WTZ
Metodyka nauczania i wychowania osób z lekką niepełnosprawnością intelektualną - wykłady, Metody nau
maciol1, Metodyka nauczania zintegrowanego i wychowania dziecka z lekką niepełnosprawnością intelekt
Jak posługiwać się pieniędzmi tekst w formie dla dziecka z niepełnosprawnością intelektualnax
więcej podobnych podstron