WYKŁADY Z CHEMII 2009/2010
Wykładowca: dr hab. inż. Magdalena Hasik
Opracowała: Karolina Jaworska
Spis treści
............................................................................................
............................................................................................
............................................................................................
............................................................................................
.........................................................................................
.........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
..........................................................................................
........................................................................................
........................................................................................
........................................................................................
........................................................................................
........................................................................................
........................................................................................
........................................................................................
........................................................................................
Wykład 1
(Postaci węgla w przyrodzie; właściwości węgla; klasy zw. organicznych; właściwości zw. organicznych)
Wykład 2
(Hybrydyzacja orbitali węgla; hybrydyzacje: sp
3
,
sp
2
, sp; konsekwencje hybrydyzacji; sposoby
przedstawiania związków organicznych; tworzenie wzorów szkieletowych; wzory projekcyjne)
Wykład 3
(Polaryzacja wiązań kowalencyjnych; znaczenie polaryzacji; efekt indukcyjny, efekt mezomeryczny;
struktury rezonansowe; izomeria zw. organicznych)
Wykład 4
(Rodzaje izomerii przykłady stereoizomerii, konfiguracji, cząstki chiralne)
Wykład 5
(Reakcje związków, reagenty reakcji org.; kwasy i zasady Lewisa; mechanizmy reakcji organicznych;
alkany-nazewnictwo, budowa, reakcje)
Wykład 6
(Mechanizmy reakcji alkanów, cykloalkany-nazewnictwo, budowa i wł. chemiczne; alkeny-nazewnictwo,
budowa, wł. chemiczne; addycja elektrofilowa)
Wykład 7
(Addycja HZ do alkenów; reguła Markownikowa; przegrupowywanie karbokationów; inne reakcje
alkenów: ozonoliza i polimeryzacja; alkadieny; alkiny-nazewnictwo i budowa)
1

Wykład 8
(Własności alkinów; areny: rodzaje, historia, budowa, reguła Hűckla; nazewnictwo; własności)
Wykład 9
(Mechanizm reakcji S
E
; mechanizm nitrowania i sulfonowania; podstawniki w pierścieniach
aromatycznych: podstawniki aktywujące, dezaktywujące, fluorowce jako podstawniki;
halogenopochodne węglowodorów: rodzaje i własności
Wykład 10
(Mechanizm i cechy reakcji S
N1
i S
N2
; mechanizm reakcji eliminacji; reguła Zajcewa; halogenki arylów,
związki metaloorganiczne; halogenopochodne alkenów)
Wykład 11
(Alkohole: nazewnictwo, podział, właściwości; fenole: budowa i własności; związki karbonylowe:
aldehydy, ketony, właściwości fizyczne)
Wykład 12
(Aldehydy i ketony, kwasy karboksylowe, aminy)
Wykład 13
(Biocząsteczki: lipidy, sacharydy, aminokwasy)
2

WYKŁAD 1 (01.03.2010)
1
.
Dawniej chemia organiczna
była chemią związków pochodzących od
organizmów żywych i przez nie produkowanych. W 1828 r. Friedrich Wöhler przeprowadził
eksperyment w wyniku którego został sztucznie zsyntetyzowany pierwszy związek
organiczny- mocznik.
NH
4
+
-
[ O-C≡N ]
Temp
NH
2
-C-NH
2
O
(cyjanian amonu)
(mocznik)
Zapoczątkowało to serie udanych prób syntezowania innych związków
organicznych. Jednym z największych odkrywców był Rober B. Woodward, który
zsyntetyzował m. in. chininę, chlorofil, strychninę, cholesterol i kortyzol.
Definicję chemii organicznej zmieniono wtedy na: chemia organiczna to chemia związków
węgla.
2.
Postaci węgla w przyrodzie:
- kopalniany - 60%-98% węgla pierwiastkowego
•
•
torf (~60%)
•
•
w. brunatny (65% - 78%)
•
•
w. kamienny (78% - 92%)
•
•
antracyt (~96%)
•
•
szungit (~98%)
- grafit
- diament
-fulereny (i nanorurki węglowe)
3.
Związki węgla:
-nieorganiczne
•
•
tlenki i ditlenki ( CO ; CO
2
)
•
•
węglany ( CO
3
2-
)
•
•
cyjanki (M-CN
<M=metal>
)
-organiczne
•
•
białka, kwasy nukleinowe
•
•
oleje opałowe, benzyna, oleje napędowe
•
•
barwniki, leki, papier
4.
Specyficzne własności węgla wpływające na liczebność i różnorodność zw. org.
–
–
Atomy węgla mogą łączyć się ze sobą, tworząc łańcuchy proste, rozgałęzione
lub pierścienie
–
–
Atomy węgla w związkach mogą być połączone wiązaniami pojedynczymi
(związki nasycone) lub wielokrotnymi (związki nienasycone): podwójnymi lub
potrójnymi
–
–
Atomy węgla mogą tworzyć wiązania pojedyncze lub wielokrotne z atomami
innych pierwiastków (tzw. heteroatomami): O, N, F, Cl, Br, I
3
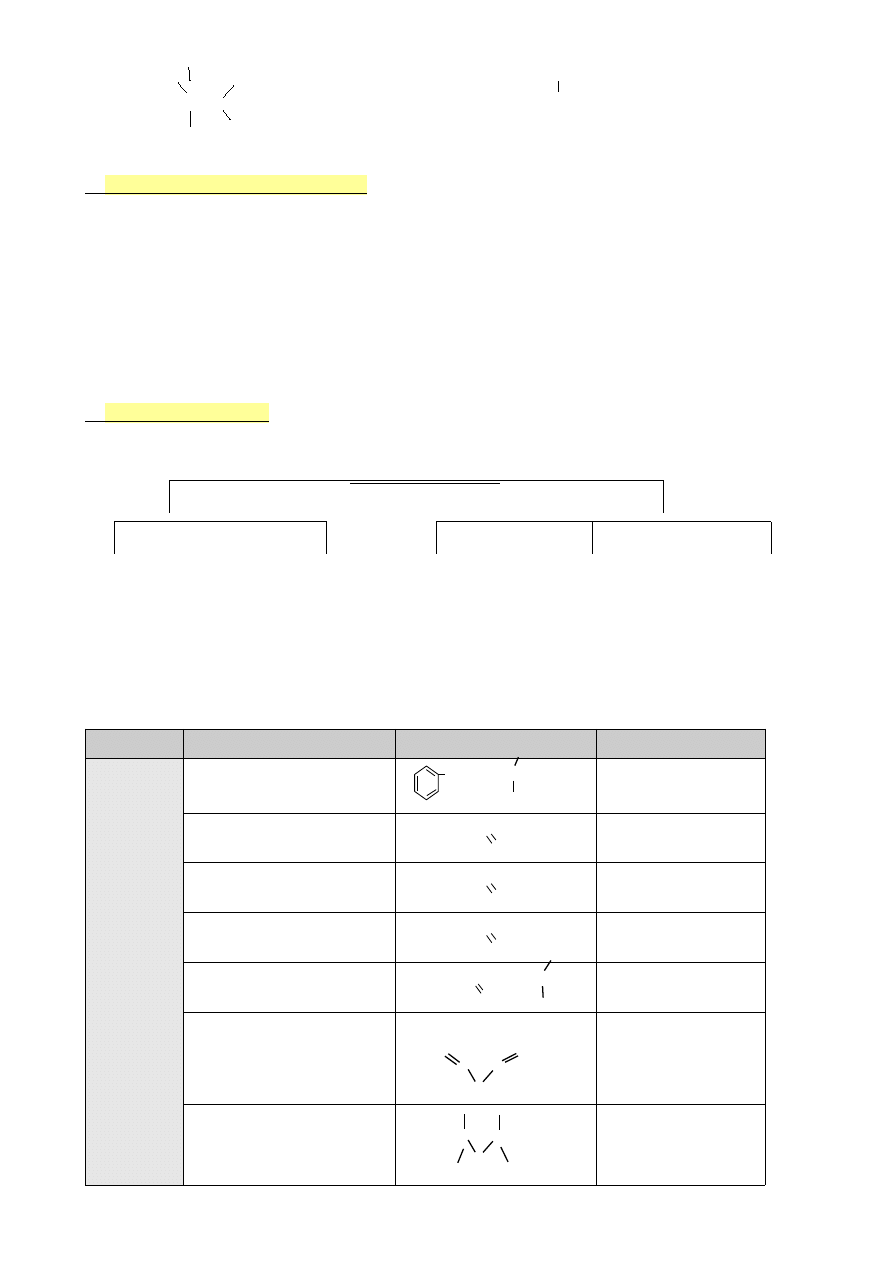
—C=O
gr. karbonylowa
—C≡N
nitryle
—C—N
gr. aminowa
—C=N—
iminy
5.
Klasy związków organicznych:
(zawierają pierwiastki)
A)
Węglowodory
→ C, H
B)
Halogenopochodne węglowodorów
→ C, H, X
C)
Etery, alkohole, fenole, zw. karbonylowe
→ C, H, O
D)
Aminy, iminy, nitryle, zw. azowe
→ C, H, N
E)
Amidy, zw. nitrowe
→ C, H, O, N
F)
Chlorki kwasowe
→ C, H, O, Cl
6.
Związki organiczne
WĘGLOWODORY
ŁAŃCUCHOWE
PIERŚCIENIOWE
NASYCONE
NIENASYCONE
NASYCONE NIENASYCONE
AROMATYCZNE
Halogenopochodne węglowodorów posiadają taki sam podział jak węglowodory.
Mogą być jednopodstawnikowe lub wielopodstawnikowe.
Pierwiastki
Związki
Grupa
Nazwa grupy
C, H, O
Alkohole, Fenole
OH
,
—C—OH
gr. hydroksylowa
Keton
—C—
O
gr. karbonylowa
Aldehydy
—C—H
O
gr. aldehydowa
Kwasy karboksylowe
—C—OH
O
gr. karboksylowa
Estry
—C—O—C—
O
gr. estrowa
Bezwodniki kwasowe
—C(O)OC(O) —
O O
—C C —
O
—
Etery
—C C —
O
—
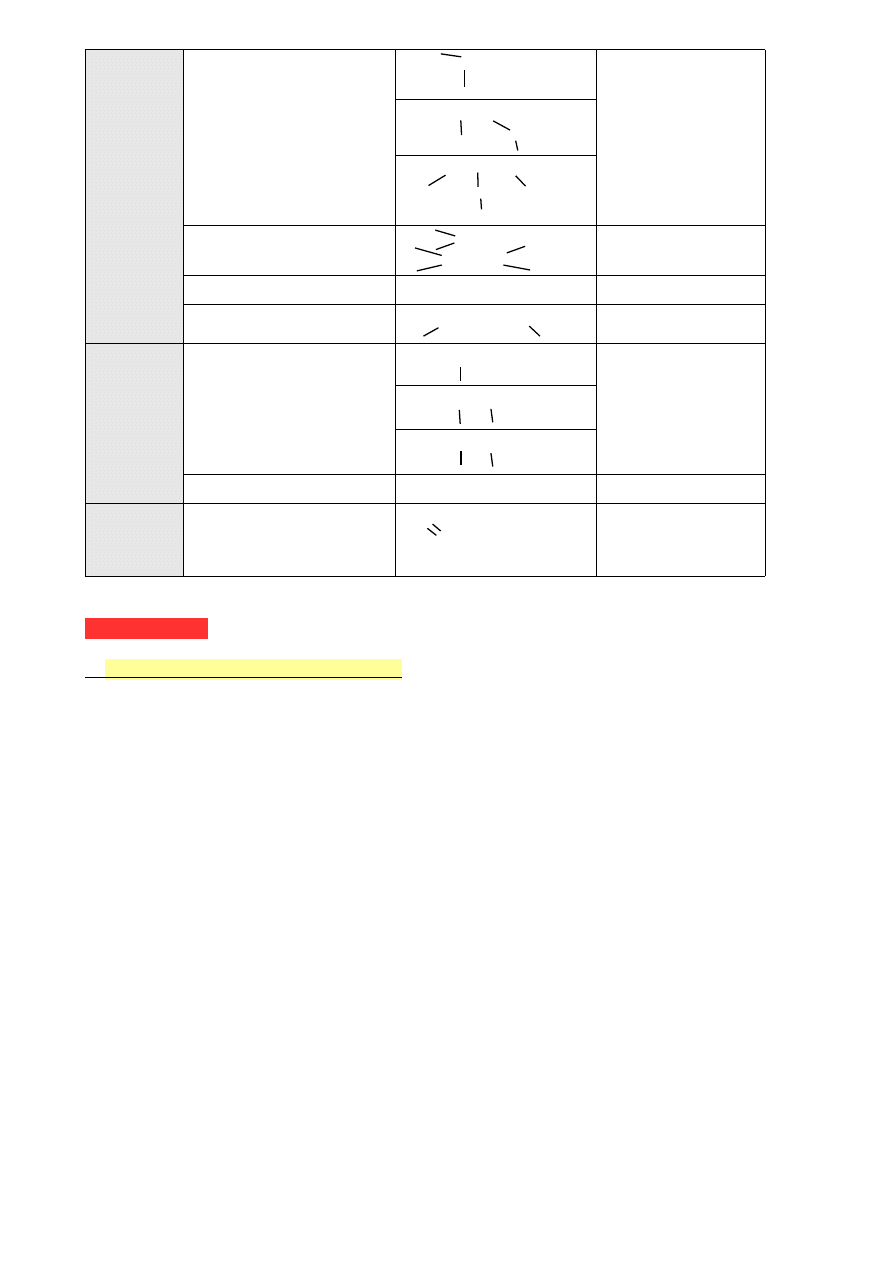
C, H, N
Aminy
A) —C—NH
2
gr. aminowa
A) a. pierwszorzędowa
B) a. drugorzędowa
C) a. trzeciorzędowa
B)
—C—NH
—C—
C) —C—N—C—
—C—
Iminy
C=N—
C=N—C—
—
Nitryle
—C≡N
gr. nitrylowa
Związki azowe
—C—N=N—C—
—
C, H, O, N Amidy
A) O=C
—NH
2
gr. amidowa
A) a. pierwszorzędowy
B) a. drugorzędowy
C) a. trzeciorzędowy
B) O=C
—NH
C) O=C—N—
Związki nitrowe
—NO
2
gr. nitrowa
C, H, O, Cl Chlorki kwasowe
— C—Cl ,
O
— C(O)Cl
—
OSTATECZNIE- chemia organiczna to chemia węglowodorów i jego pochodnych
7.
Własności związków organicznych
–
–
atomy w cząstkach połączone wiązaniami kowalencyjnymi
–
–
występują we wszystkich stanach skupienia
–
–
ciała stałe – substancje krystaliczne o niezbyt wysokich temperaturach topnienia
ciecze – o niezbyt wysokich temperaturach wrzenia
–
–
rozkład termiczny zw. organicznych występuje w temperaturach < 1000°C
–
–
właściwości termiczne (temperatury przemian fazowych i inne własności fizyczne)
uwarunkowane oddziaływaniami międzycząsteczkowymi
–
–
reaktywność chemiczna uwarunkowana obecnością określonych grup funkcyjnych
5

WYKŁAD 2 (08.03.2010)
1.
Hybrydyzacja orbitali atomowych węgla w związkach organicznych i jej wpływ na
geometrię cząsteczek organicznych
C:
•
•
6 elektronów na 2 powłokach
•
•
stan podstawowy
- węgiel nieaktywny
- C: 1s
2
2s
2
2p
x
1
2p
y
1
2p
z
0
→ el. walencyjne
•
•
stan wzburzony
- *C: 1s
2
2s
1
2p
x
1
2p
y
1
2p
z
1
→ el. walencyjne
W cząsteczkach związków organicznych orbitale atomowe węgla są zhybrydyzowane
( wymieszane )
+ =
s
p
sp
2.
Hybrydyzacja
A) 2s
1
2p
x
1
2p
y
1
2p
z
1
sp
3
B) 2s
1
2p
x
1
2p
y
1
2p
z
0
sp
2
C) 2s
1
2p
x
1
2p
y
0
2p
z
0
sp
A)
sp
3
•
•
4 równocenne zhybrydyzowane orbitale
•
•
109,5°- kąt między wiązaniami
•
•
budowa tetraedryczna
•
•
występują we wszystkich związkach organicznych zawierających pojedyncze
wiązania między atomami węgla
•
•
występuje też w cząsteczkach: NH
3
, H
2
O
Metan- CH
4
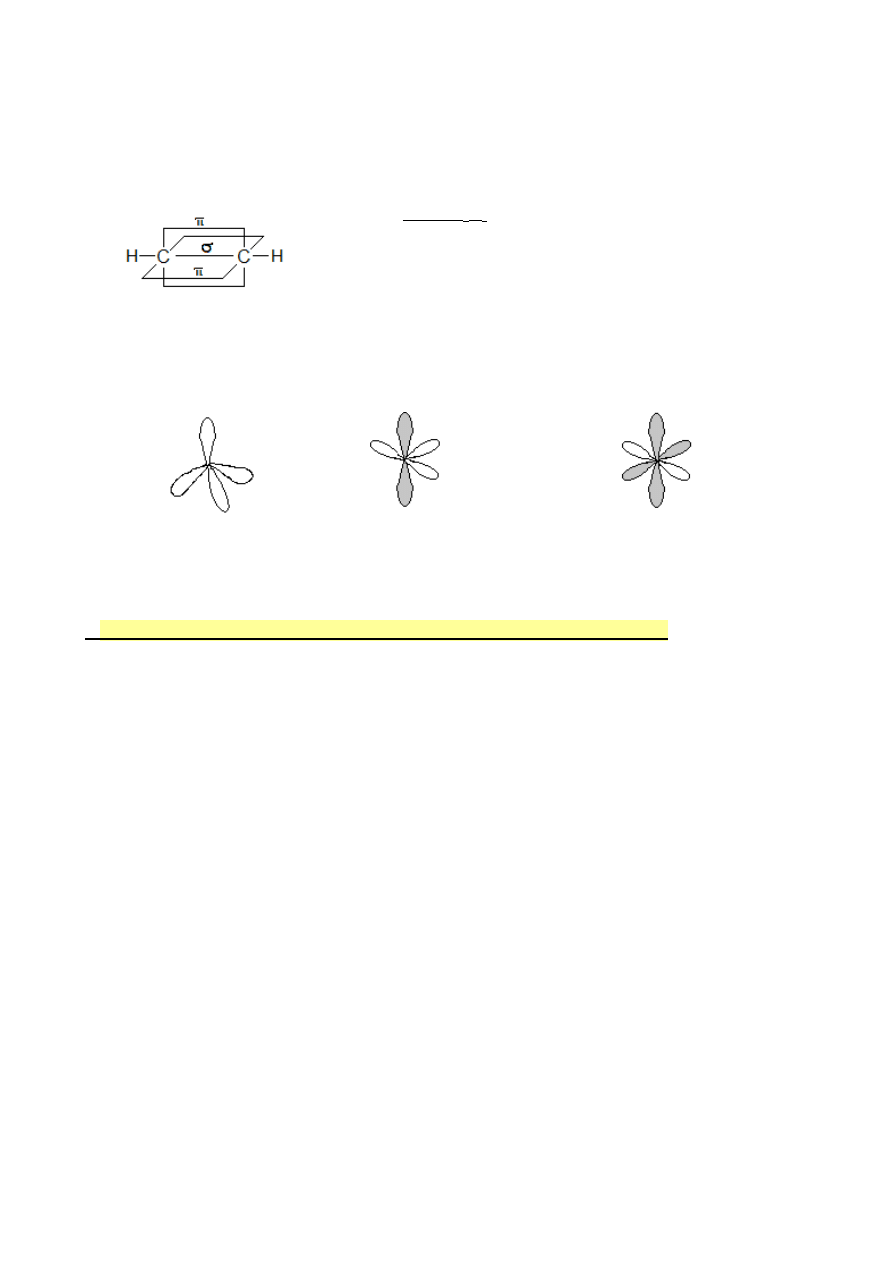
Wiązania pojedyncze C-H i C-C to wiązania σ
( czołowe nakładanie się orbitali sp
3
-s lub sp
3
-sp
3
)
B)
sp
2
•
•
120° - kat między wiązaniami
•
•
trójkąt- budowa płaska
•
•
występuje we wszystkich związkach organicznych zawierających at. węgla
związane wiązaniami podwójnymi także w związkach aromatycznych
Eten- C
2
H
4
Wiązanie podwójne węgiel – węgiel to
jedno wiązanie σ i jedno wiązanie
π
6
C)
sp
•
•
dwa równocenne wiązania
•
•
180
° - kąt między wiązaniami
•
•
cząstka liniowa (digonalna)
•
•
wszystkie związki organiczne zawierające at. C związane wiązaniami
potrójnymi
Etyn- C
2
H
2
Wiązanie potrójne składa się z jednego
wiązania σ i dwóch wiązaniań
π
Hybrydyzacje:
sp
3
sp
2
sp
[na szaro zaznaczone orbitale typu
π ]
3.
Konsekwencje hybrydyzacji atomów C w związkach organicznych
A) Geometria cząsteczek
B) Długość wiązań węgiel-węgiel:
–
–
pojedyncze: 0,153 nm
–
–
podwójne: 0,134 nm
–
–
potrójne: 0,121 nm
Skrócenie wiązania związane jest ze zwiększeniem udziału orbitalu s w
zhybrydyzowanym orbitalu tworzącym wiązanie.
C) Możliwość (lub brak możliwości) obrotu atomów wokół wiązań węgiel-węgiel
(alkany- TAK, alkeny, alkiny- NIE)
Konsekwencją obrotu lub jego braku wokół wiązań C-C jest występowanie
związków organicznych w postaci różnego rodzaju izomerów.
7
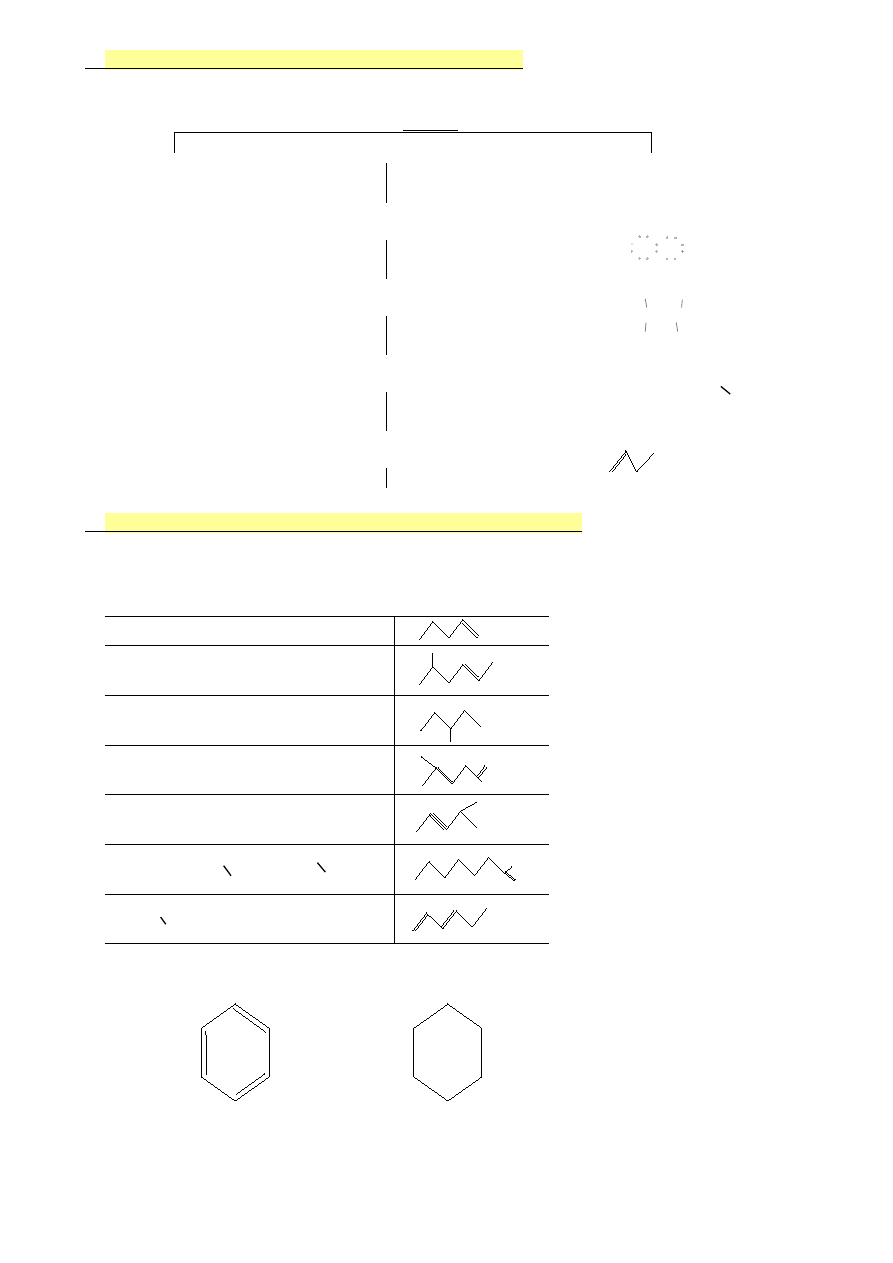
4.
Sposoby przedstawiania związków organicznych
WZÓR
sumaryczny
strukturalny
H H
–
–
CH
4
metan
- wzór Lewisa
H C C H
–
–
C
2
H
6
etan
H H
–
–
C
2
H
4
O
2
kwas etanowy
H H
–
–
C
2
H
6
O
etanol
- wzór kreskowy
H
‒C—C‒H
lub
eter dimetylowy
H H
- wzór półstrukturalny
CH
3
—
CH
2
—
C=O
OH
- wzór grupowy
CH
3
CH
3
- wzór szkieletowy
5.
Zasady tworzenia szkieletowych wzorów strukturalnych
A) Atomy węgla na ogół nie uwidaczniane we wzorze
B) Atomy wodoru nie uwidaczniane we wzorze
CH
3
CH
2
CH
2
CHO
O
(CH
3
)
2
CHCH
2
CH=CHCH
3
CH
3
CH
2
CH(CH
3
)CH(OH)CH
3
OH
(CH
3
)
2
C=CHCH
2
COOH
O
OH
CH
3
C≡CCH(CH
3
)
2
(CH
3
)
2
CHCH
2
CHCH
2
CH
2
C=O
NH
2
OH
NH
2
OH
O
CH
2
=CCH=CHCH
2
CH
3
Cl
Cl
Benzen
Cykloheksan
8
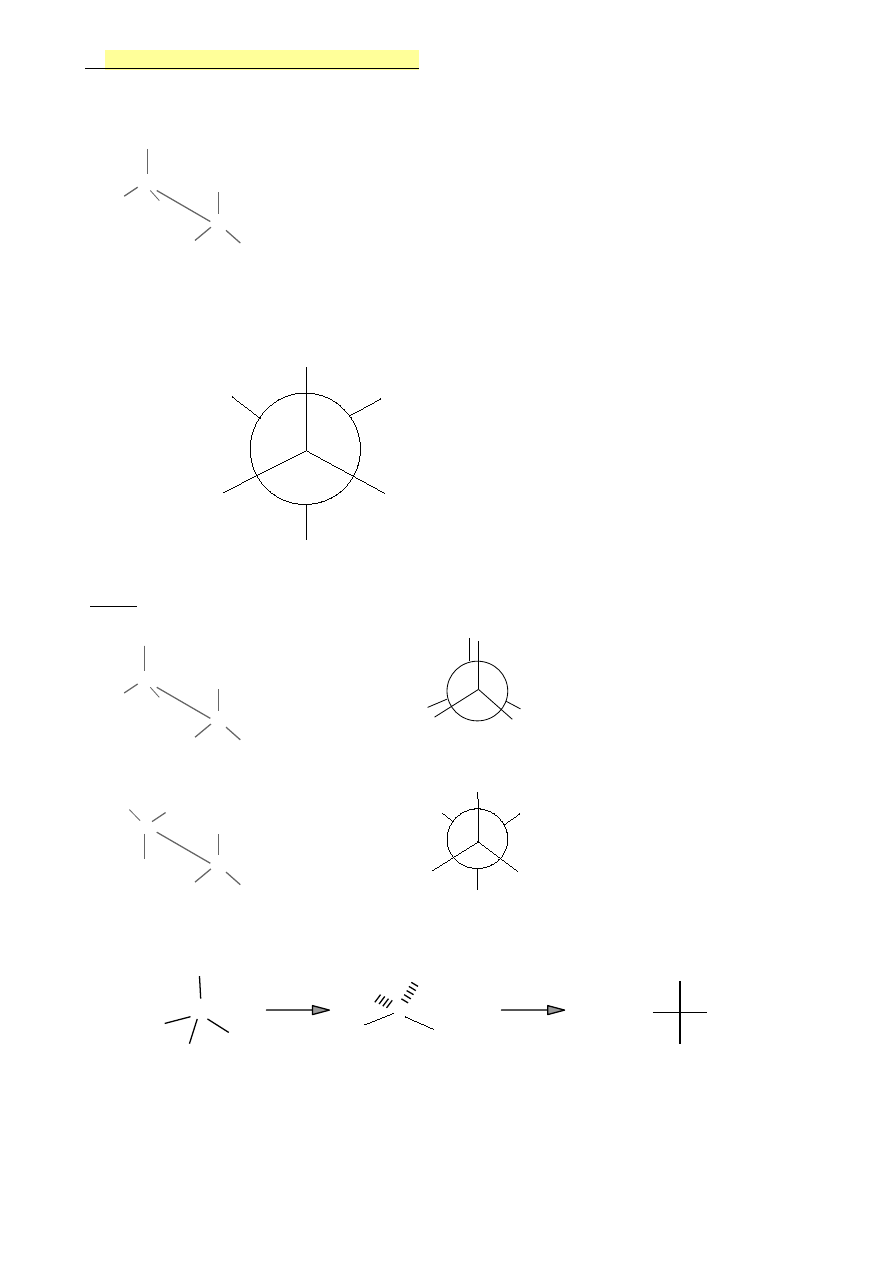
6.
Związki nasycone-wzory projekcyjne
Projekcja konikowa (kozłowa)- patrzymy na cząsteczkę ( i wiązanie C-C) z ukosa
H
C
H
H H
Etan
C
H H
Projekcja Newmana- patrzymy na cząsteczkę wzdłuż wiązania C-C
H
H
H
H
H
H
Butan
CH
3
CH
CH
C
CH
3
H H
H
H
C
H
H
H H
CH
3
H H
H
H
C
CH
3
CH
3
C
H
H
H H
CH
3
Projekcja Fishera- stosowane są do przedstawiania cząsteczek czynnych optycznie
CH
3
CH
3
CH
3
HO
C
C
H
2
N Cl
OH
Cl
NH
2
Cl
NH
2
OH
Wzór Fishera
Wzory projekcyjne pokazują budowę przestrzenną !
9

WYKŁAD 3 (15.03.2010)
1.
CH
3
CH
3
CH
3
—CH
2
—
CH
—CH—CH
3
CH
3
CH
3
H
H
C
H
H
CH(CH
3
)
2
H
C
H
CH(CH
2
)
3
CH
3
CH
3
ZADANIE
Przedstawić w projekcji kozłowej i Newmana poniższe związki ze względu na
zaznaczone wiązania.
•
•
ClCH
2
—CH
—
CH
3
Cl
CH
3
CH
3
•
•
CH
3
—C
—
C—CH
3
CH
3
CH
3
•
•
ClCH
2
—
CH
2
—CH
3
•
•
ClCH
2
—CH
2
—
CH
2
—CH
2
Cl
2.
Polaryzacja wiązań kowalencyjnych w związkach organicznych
Wynika z elektroujemności między atomem węgla a atomem związanego z nim
pierwiastka.
H
H H
↓
σ
-
σ
+
σ
-
σ
+
A)
H→ C ←H
metan
B)
H‒C‒C←MgBr
bromek etylomagnezu
↑
H
H H
H
H
σ
+
σ
-
σ
+
σ
-
C)
H‒C→Cl
chlorometan
D)
H‒C‒C≡N
acetylonitryl
H
H
-przesunięcie elektronów π.
*To, co po prawej stronie węgla w układzie okresowym jest bardziej elektroujemne więc
strzałka skierowana do tego. Jeżeli różnica elektroujemności jest mała, to polaryzacja jest
słaba.
10

ZADANIE
Ustalić polaryzację wiązań dla podanych cząstek.
σ
+
σ
-
σ
-
σ
+
σ
+
σ
-
A) H
3
C‒HC=O
B) H
7
C
3
‒CH
2
←Li
C) H
3
C→NO
2
propanon
n-butylolit
nitrometan
(keton dimetylowy)
3.
Znaczenie polaryzacji wiązań w cząsteczkach organicznych
•
•
Polaryzacja wiązań często decyduje o kierunku przebiegu reakcji chemicznej np.
Reakcje związków karbonylowych z odczynnikiem Grignarda, w wyniku których
powstają alkohole:
σ
-
σ
+
σ
-
σ
+
C=O + RMgX
H
2
0
‒C‒OH + Mg
2+
+ X
-
R
‒C‒Mg X
R
•
•
Polaryzacja wiązań wpływa na kwasowość podstawowych kwasów karbonylowych
X ← CH
2
← COOH
wodór wiązany słabiej niż halogen
Im podstawnik bardziej elektrododatni, tym moc kwasu mniejsza
Im podstawnik bardziej elektroujemny, tym moc kwasu większa
Z- bardziej elektroujemny od węgla –
podstawnik elektronoakceptorowy
H
H H
H H H
σ
+
σ
-
σ
+
σ
-
σ
+
σ
-
H‒C→Z
,
H‒C→C→Z ,
H‒C→C→C→Z
-I
H
H H
H H H
Y- mniej elektroujemny od węgla –
podstawnik elektronodonorowy
H
H H
H H H
σ
-
σ
+
σ
-
σ
+
σ
-
σ
+
H‒C←Y
,
H‒C←C←Y , H‒C←C←C←Y
+I
H
H H
H H H
Przesunięcie elektronów wiązań
σ
C‒C w cząsteczce w kierunku atomu bardziej
elektroujemnego podstawnika nazywamy EFEKTEM INDUKCYJNYM (I).
11
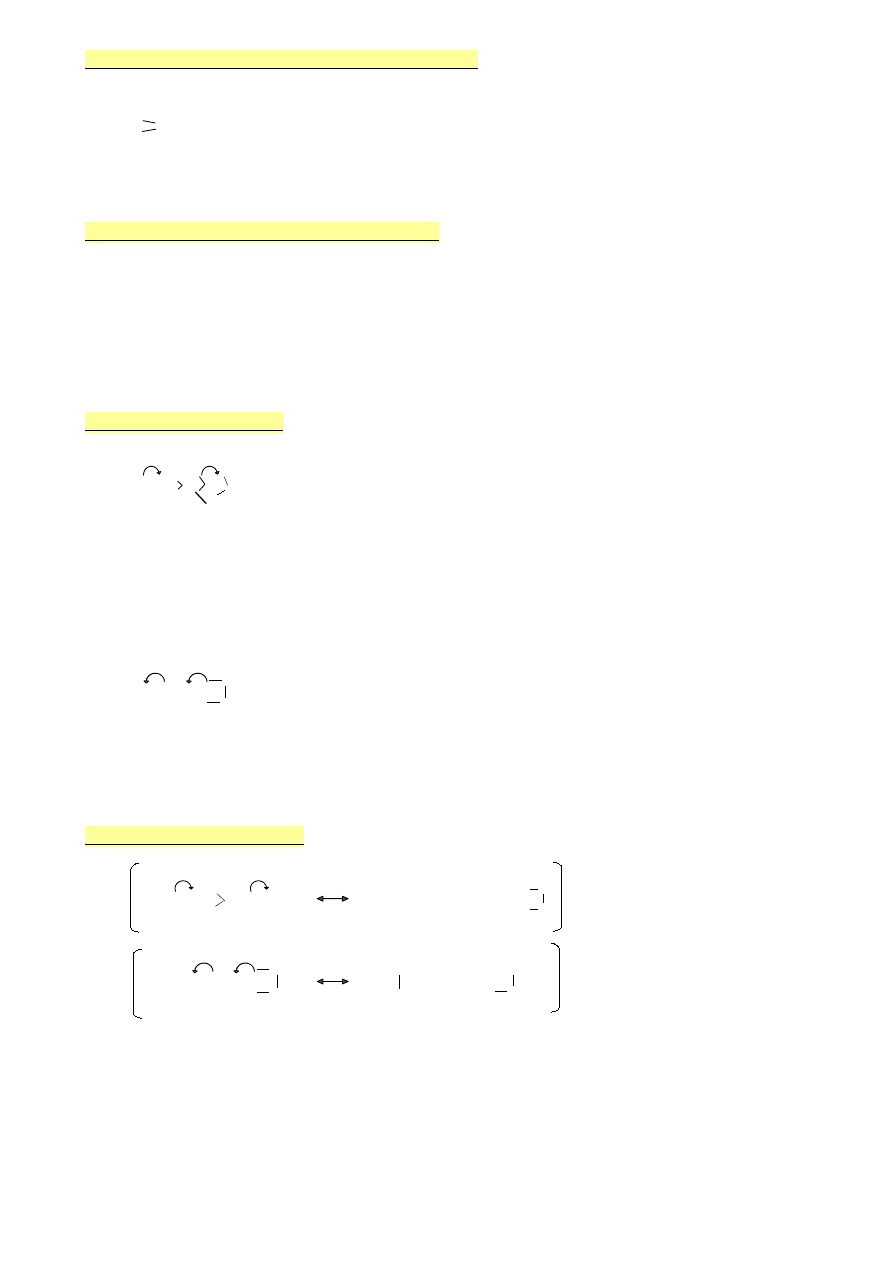
4.
Podstawniki elektronoakceptorowe
(efekt -I)
•
•
F; Cl; Br; I
•
•
C=O; ‒CHO; ‒COOH; ‒COOR; ‒C(O)Cl; ‒C(O)NH
2
•
•
‒OH; ‒OR
•
•
CN; NO
2
; ‒NH
2
; ‒NHR; ‒NR
2
Podstawniki elektronodonorowe
(efekt +I)
•
•
grupy alkilowe, tj. C
n
H
2n+1
(słaby efekt)
•
•
atomy metali (Mg, Li)
Poza efektem indukcyjnym w związkach organicznych występuje też efekt mezomeryczny
(rezonansowy)
5.
Efekt mezomeryczny
σ
+
σ
+
σ
-
A) CH
2
=CH‒C=O
- propenal ( akroleina, aldehyd akrylowy)
H
-I
=CH‒CH= - sprzężony układ wiązań podwójnych
-M
( sprzężenie π – π )
Ponieważ elektrony π są bardziej mobilne, to efekt M jest dużo silniejszy niż efekt I.
σ
-
σ
+
B) CH
2
=CH—Cl
- chloroeten (chlorek winylu)
-I
-układ sprzężony p- π ( wiązanie σ rozdziela wiązanie π
+M
i wolne elektrony orbitalu p)
Efekty +M i -M mają istotny wpływ na reakcje zwłaszcza związków aromatycznych
6.
Struktury rezonansowe
+
-
CH
2
=CH—HC=O
CH
2
—CH=HC—O
propenal
-M
-
+
CH
2
=CH—Cl
CH
2
—CH=Cl
chlorek winylu +M
Prawdziwa struktura cząsteczki jest czymś pomiędzy strukturami rezonansowymi
cząsteczka to hybryda struktur rezonansowych. Im więcej struktur rezonansowych tym
⇒
cząsteczka ma mniejszą energię.
12
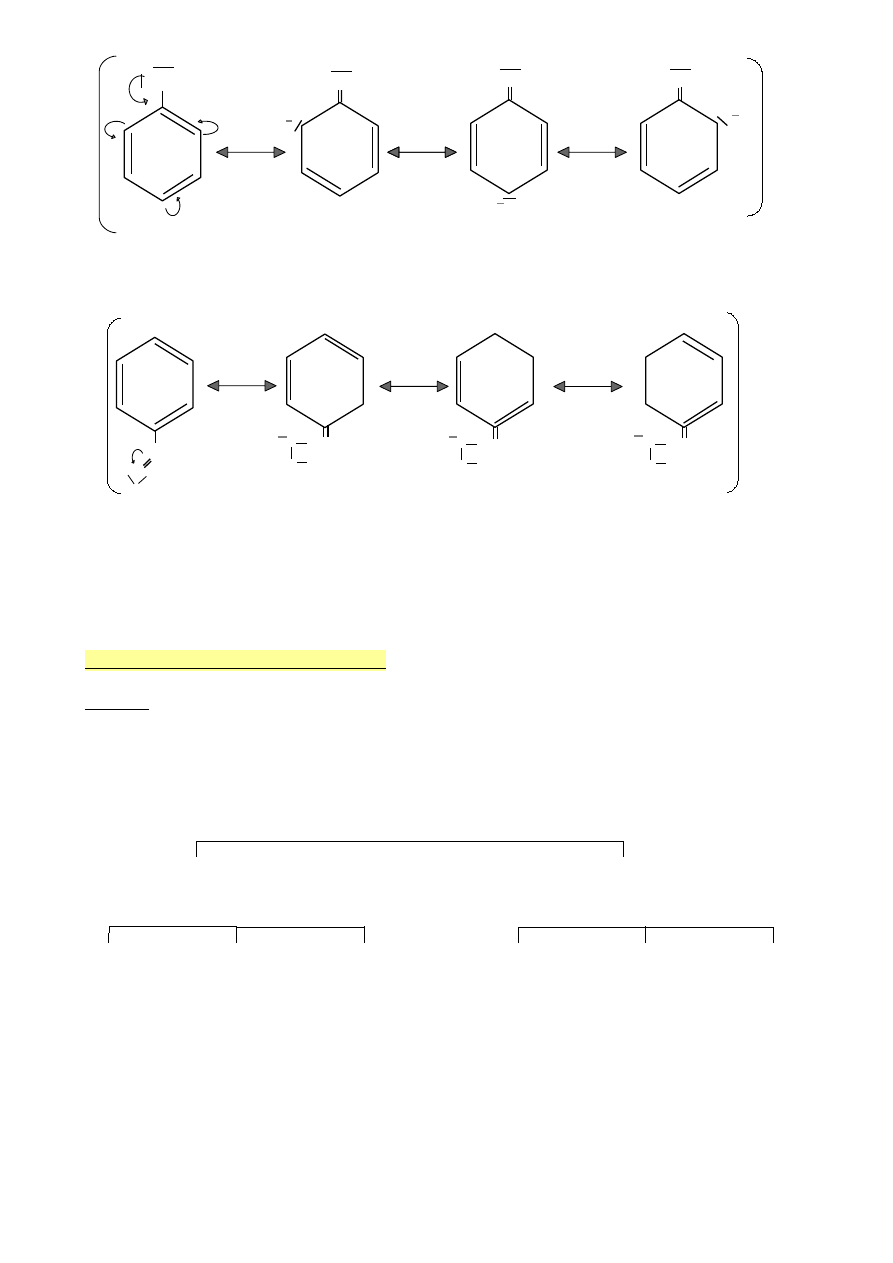
OH
+ OH
+ OH
+ OH
Hydroksybenzen ( fenol )
Efekt +M ( sprzężenie p - π )
+
+
+
C‒H
O‒C‒H
O‒C‒H
O‒C‒H
O
Aldehyd benzoesowy
Efekt -M (sprzężenie π – π )
7.
Izomeria związków organicznych
Izomery- cząsteczki o tym samym wzorze sumarycznym, ale różniące się sposobem
rozmieszczenia atomów w cząsteczce. Różnią się one właściwościami fizycznymi
a czasem i chemicznymi.
IZOMERIA
KONSTYTUCYJNA
PRZESTRZENNA
różna kolejność i sposób
( stereoizomeryczna )
powiązania atomów
różna budowa przestrzenna
łańcucha
położenia
budowy
konformacja geometryczna enancjomeria
(szkieletu) (podstawienia)
(funkcyjna)
(cis-trans ; E-Z)
(optyczna)
13
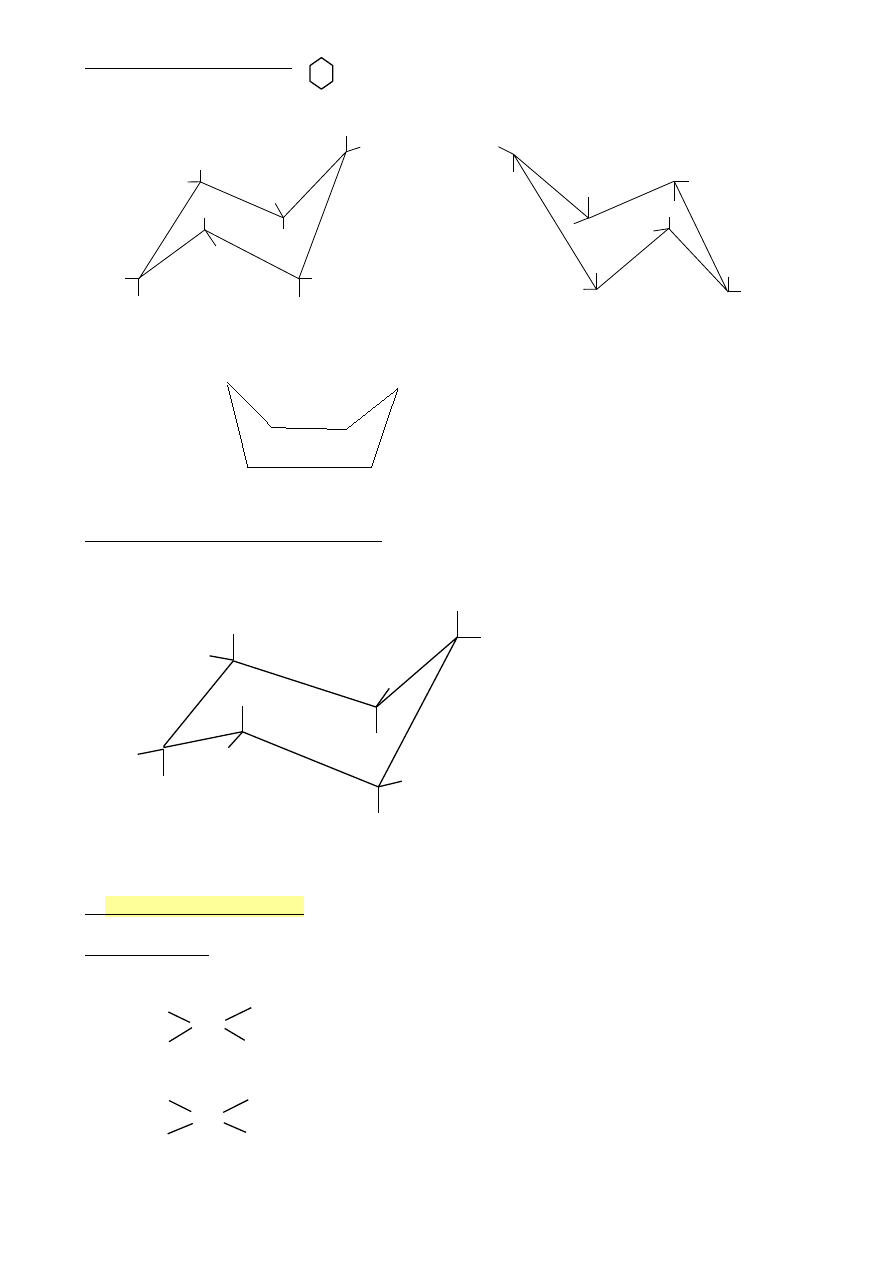
Izomerie konstytucyjne
Izomeria łańcucha
CH
3
CH
2
CH
2
CH
2
CH
3
n- pentan
CH
3
CH
3
CH
2
CH
2
CH
CH
3
2-metylobutan
cykloheksan
CH
3
1-metylopentan
Izomeria położenia
CH
3
CH
2
CH
3
|
OH
1-propanol
CH
3
CH
2
CH
3
|
OH
2-propanol
Br
Br
o-dibromobenzen
Br
Br
m-dibromobenzen
Br
Br
p-dibromobenzen
CH
3=
CHCH
2
CH
3
but-1-en
CH
3
CH
=
CHCH
3
but-2-en
Izomeria budowy
CH
3
CH
2
OH
etanol
CH
3
-O-CH
3
eter dimetylowy
CH
3
-C-CH
3
||
O
propanon
CH
3
CH
2
C=O
|
H
propanal
CH
3
CH=CHCH
3
but-2-en
cyklobutan
ZADANIE
Napisać i określić typ izomerii i nazwać związki
A) C
7
H
16
(9 izomerów)
B)
COOH
(3 izomery)
NH
2
C) C
4
H
6
(2 izomery)
D) C
4
H
8
O
(2 izomery)
14
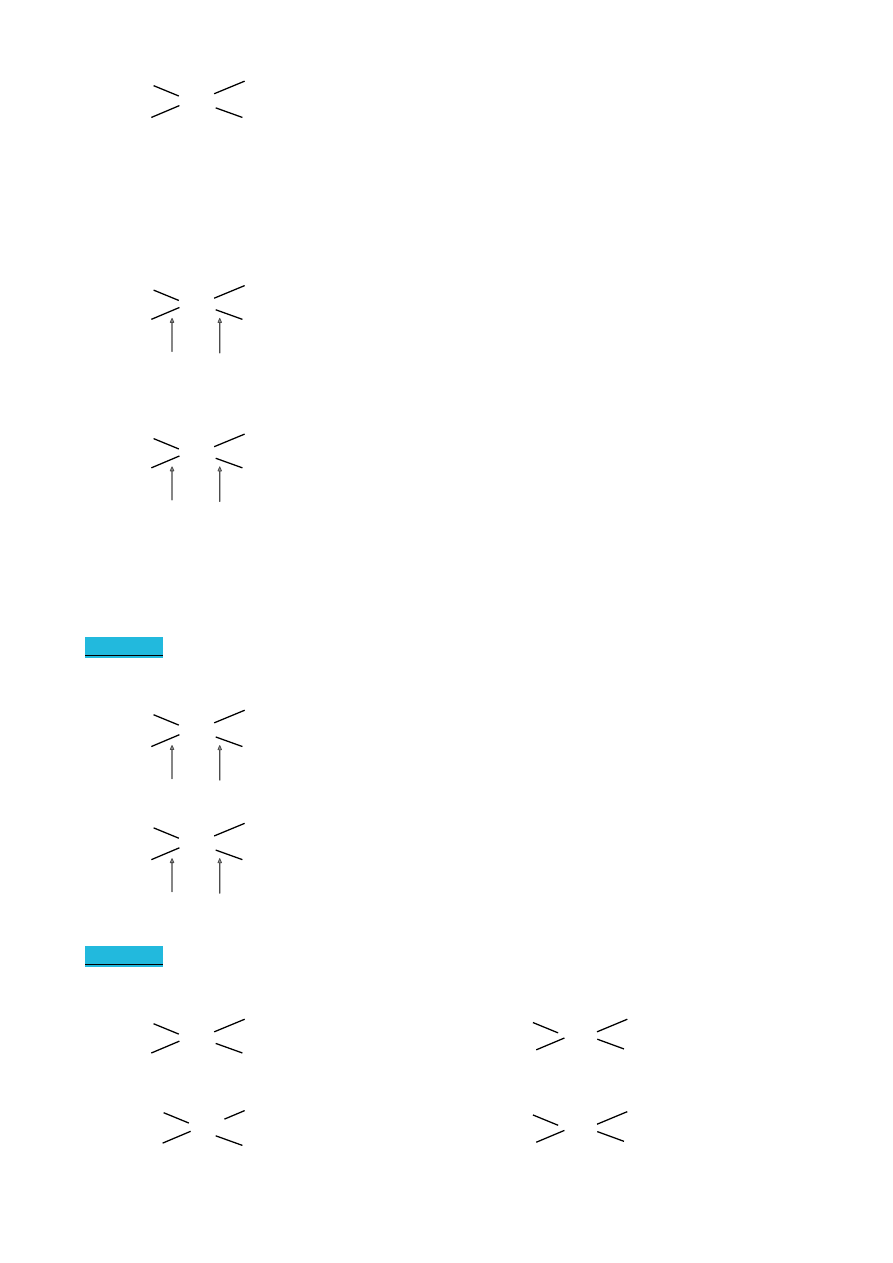
WYKŁAD 4 (22.03.2010)
1.
Stereoizomeria
- budowa przestrzenna związków organicznych. Są to izomery różniące
się rozmieszczeniem atomów w przestrzeni
Konformacja
- w alkanach i cykloalkanach oraz ich pochodnych
Geometryczna
- występuje w alkenach i cykloalkenach
Optyczna
- występuje w cząsteczkach zawierających niesymetryczny
(chiralny) atom węgla
2.
Izomeria konformacyjna
- związana z rotacją wokół wiązań węgiel-węgiel ( związki
łańcuchowe) lub naprężeniem kątowym albo torsyjnym ( związki
cykliczne)
Konformery etanu
H
H
H
C
H
H H
H
H
naprzeciwległy
C
H
H
(synperiplanarny)
H H
H
H H
H
H
C
H
naprzemianległy
(antyperiplanarny)
H
C
H
H
H H
H
Konformacja naprzemianległa jest bardziej korzystna energetycznie
Konformery butanu
CH
3
CH
3
H CH
3
H
H
H
CH
3
H
H
H
H
H
CH
3
H
H
CH
3
H
naprzemianległy
synklinarny
naprzeciwległy
(antyperiplanarny, anti)
(gauche)
(synperiplanarny)
-wzrost energii
15
Konformery cykloheksanu
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
konformacja krzesłowa
konformacja łódkowa
Jak rysować konformację krzesłową?
a
a
e
e
e
a
a
- pozycje aksjalne (osiowe)
e- pozycje ekwatorialne (równikowe)
a
e
e
a
e
a
Podstawniki w pierścieniu cykloheksanu zawsze zajmują pozycje ekwatorialne.
3.
Izomeria geometryczna
-polega na różnym położeniu podstawników względem
płaszczyzny wiązania π
Izomery butenu
CH
3
CH
3
C=C
izomer cis
(cis-but-2-en)
H
H
CH
3
H
C=C
izomer trans (trans-but-2-en
)
H
CH
3
16
H
CH
3
C=C
brak izomerii geometrycznej
H
CH
3
Dla alkenów z 3 podstawnikami obowiązuje
reguła E-Z
Z ( zusammen) - razem
E ( entgegen) - naprzeciw
CH
3
Br
C=C
izomer Z ( Z-1-bromo-1chloroprop-1-en)
H
Cl
C>H Br>Cl
CH
3
Cl
C=C
izomer E ( E-1-bromo-1chloroprop-1-en)
H
Br
C>H Br>Cl
Wartość podstawników określamy na podstawie ich „starszeństwa” (ustalane za
pomocą liczby atomowej). Jeżeli starsze podstawniki znajdą się w jednej płaszczyźnie, to
powstaje izomer Z, w przeciwnym razie izomer E.
Przykłady
A)
H
COOH
C=C
izomer E
Cl
CH
3
(tu porównujemy dalsze atomy podstawników)
Cl>H O>H
B)
H
Cl
C=C
izomer Z
Cl
Br
C>H Br>Cl
ZADANIE
Określ rodzaj izomerii geometrycznej w poniższych cząsteczkach
A) CH
3
CH
3
B)
Br
CH
2
CH
3
C=C
Z
C=C
E
Br
Cl
CH
3
CH
2
CH
2
Br
C) H
3
CO
CH
2
CH
3
D)
Cl
NO
2
C=C
E
C=C
Z
CH
3
Cl
CH
3
CH
2
Br
17
Związki cykliczne
CH
3
CH
3
CH
3
CH
3
cis-dimetylocyklopropan
trans-dimetylocyklopropan
4.
Izomeria optyczna
– charakterystyczna dla związków nasyconych
cząsteczka chiralna- cząsteczka, której struktura nie ma płaszczyzny symetrii ani środka
symetrii. Atom węgla jest chiralny, gdy jest połączony z czterema
różnymi podstawnikami.
CH
3
*C- chiralny atom
(centrum asymetrii,centrum chiralne)
*C
Cl
OH
NH
2
*
A)
CH
3
-CH-CH
2
CH
3
NH
2
Cl
*
B)
BrCH
2
-CH-CH-CH
3
*
OH
C)
*
OH
Izomery te wykazują czynność optyczną tzn. skręcają płaszczyznę polaryzacji światła
źródło światła
polaryzator
światło
próbka
analizator
spolaryzowane
18

-prawoskrętne (+) -
zgodne z ruchami wskazówek zegara
-lewoskrętne (-)
-
przeciwne do ruchu wskazówek zegara
Enancjomery- stereoizomery, których cząsteczki mają się do siebie jak przedmiot i jego
lustrzane odbicie. Każdy z enancjomerów skręca płaszczyznę polaryzacji o
ten sam kąt, ale w odwrotną stronę.
(racemat- mieszanina złożona z takiej samej ilości cząsteczek
prawoskrętnych i lewoskrętnych)
2 węgle asymetryczne
A)
COOH
B)
COOH
H—C—NH
2
NH
2
—C—H
H—C—OH
OH—C—H
CH
3
CH
3
enancjomery
C)
COOH
D)
COOH
H—C—NH
2
NH
2
—C—H
OH—C—H
H—C—OH
CH
3
CH
3
enancjomery
A,C
A,D
diastereoizomery- mają różne właściwości chemiczne i fizyczne
B,C
B,D
Wzory projekcyjne Fishera
CH
3
OH
CH
3
CH
3
C
C
NH
2
Cl
Cl
NH
2
Cl
OH NH
2
OH
Enancjomery:
CH
3
CH
3
NH
2
Cl
NH
2
Cl
OH
OH
19
5.
Konfiguracja
- określony sposób rozmieszczenia podstawników względem wybranego
fragmentu cząsteczki
- względna (D,L)
- absolutna (R,S)
Aldehyd glicerynowy
CHO
CHO
H
OH
OH
H
CH
2
OH
CH
2
OH
D(+)
L(-)
Żeby określić konfigurację trzeba przekształcić w taki sposób, żeby podstawniki
leżały tak, jak w aldehydzie glicerynowym (choć nie zawsze możliwe).
Konfiguracja absolutna
1. Określić starszeństwo podstawników
2. Ułożyć tak, by najmłodszy podstawnik był jak najdalej
CH
3
C
Cl
OH
NH
2
3. Rozpatrujemy kolejność pozostałych trzech poruszając się od najstarszego do
najmłodszego
CH
3
C
Cl
obrót w lewo
⇒ S
OH
NH
2
R (rectus) - prawy
S (sinister) - lewy
20
WYKŁAD 5 (29.03.2010)
1.
Reakcje związków organicznych
A) Reakcje podstawienia -
SUBSTYTUCJI
(S)
–C–Y + Z → –C–Z +Y
B) Reakcje przyłączania –
ADDYCJI
(A)
C=C + ZY → –C–C–
| |
Z Y
C) Reakcje
ELIMINACJI
(E)
–C–C– → C=C + ZY
| |
Z Y
D) Reakcje
przegrupowania
CH
3
CH
2
H
CH
3
H
C=C
kwas
C=C
H
H
H
CH
3
but-1-en
but-2-en
Reorganizacja prowadzi do powstawania izomerów.
Przykłady reakcji organicznych
•
•
CH
3
CH
2
Br + NaCN → CH
3
CH
2
CN +NaBr
(S)
•
•
CH
3
CH
3
+ Cl
2
→
CH
3
CH
2
Cl + HCl
(S)
•
•
CH
3
CH
2
OH → CH
2
=CH
2
+H
2
O
(E)
•
•
CH
3
OH + HBr → CH
3
Br +H
2
O
(S)
•
•
CH
2
=CH
2
+H
2
→ CH
3
CH
3
(A)
•
•
CH
2
=CH
2
+HCl
→ CH
3
CH
2
Cl
(A)
2.
Reagenty biorące udział w reakcjach organicznych
A)
WOLNE RODNIKI
- elektrycznie obojętne, zawierające nieparzystą liczbę elektronów
walencyjnych (pojedynczy niesparowany elektron)
A• •B
→ A• + B•
homolityczny
wolne
rozpad wiązania
rodniki
21
B)
CZĄSTKI ELEKTROFILOWE
(elektrofile)
I NUKLEOFILOWE
(nukleofile)
•
•
elektrofile- cząstka z niedoborem elektronów; jon lub cząstka elektrycznie obojętna
•
•
nukleofile- cząstka z nadmiarem elektronów; jon lub cząstka elektrycznie obojętna
A ••B
→
A
+
+
B
-
••
heterolityczny
elektrofil nukleofil
rozpad wiązania
Rozpad homolityczny
- wiązania słabo spolaryzowane, niespolaryzowane
Rozpad heterolityczny
- wiązania spolaryzowane
Elektrofile:
kationy (np. H
+
), kwasy Lewisa (np. AlCl
3
, BF
3
)
Nukleofile:
aniony (np. OH
-
), zasady Lewisa (np. H
2
O, NH
3
)
3.
Kwasy i zasady Lewisa
A) Kwasy (elektrycznie obojętne)
Cl
Al Cl
- dziura elektronowa- niedobór elektronów ( brakuje 2 )
Cl
F
B F
- 3 elektrony walencyjne, fluor wnosi kolejne 3
- do 8 brakuje 2 elektronów
F
B) Zasady (elektrycznie obojętne)
H
N H
H O H
H
el. niewiążące
2 wolne pary elektronów
22
4.
Nowe pojęcia
–
–
równania sumaryczne reakcji
–
–
machanizm reakcji
A) CH
2
=CH
2
+ HCl → CH
3
-CH
2
Cl
R.S.
-pokazuje jakie są produkty i substraty reakcji
+ n
B) CH
2
=CH
2
+ HCl → CH
3
-CH
2
+ Cl → CH
3
-CH
2
Cl
eten
karbokation
chloroetan
M.R.
-pokazuje drogę reakcji, czyli jak z substratów powstają produkty reakcji
(ilość etapów, produkty przejściowe)
Mechanizm reakcji może przebiegać:
•
•
wolnorodnikowo – powstają wolne rodniki i biorą udział we wszystkich etapach
•
•
elektrofilowo – reakcję inicjują elektrofile, we wszystkich etapach przejściowych
występują elektrofile
•
•
nukleofilowo – reakcję inicjują elektrofile; występują we wszystkich etapach
Oznaczenia reakcji organicznych
S
R
–
substytucja wolnorodnikowa
S
E
- substytucja elektrofilowa
S
N
- substytucja nukleofilowa
A
E
- addycja elektrofilowa
A
R
- addycja wolnorodnikowa
E
- eliminacja
Przykład
Reakcja etenu z HCl
CH
2
=CH
2
+ HCl → CH
3
-CH
2
Cl
← addycja
+ n
1. CH2=CH2 + HCl → CH3-CH2 + Cl
← inicjowana przez H
+
(elektrofil)
+ n
2. CH
3
-CH
2
+ Cl → CH
3
-CH
2
Cl
← z etenu powstaje karbokation
(elektrofil), który bierze udział w obu
etapach reakcji.
23
BUDOWA ZWIĄZKÓW ORGANICZNYCH
ALKANY
5.
Alkany
•
•
wzór ogólny CnH2n+2
•
•
grupa homologiczna- ‒CH
2
‒
•
•
Podstawniki węglowodorowe- alkile
CH
3
metyl (metylen)
CH
3
‒CH
3
etyl (etylen)
CH
4
metan
CH
3
‒CH
3
etan
CH
3
‒CH
2
‒CH
3
propan
CH
3
‒CH
2
‒CH
2
‒CH
3
butan
CH
3
‒CH
2
‒CH
2
‒CH
2
‒CH
3
pentan
6.
Nazewnictwo węglowodorów
A) Podstawa nazwy- najdłuższy łańcuch węglowodorowy
B) Nazywamy podstawniki
C) Numerujemy łańcuch tak, żeby numery atomów węgli, przy których znajdują się
podstawniki była najmniejsza (suma cyfr określających podstawniki ma być jak
najmniejsza)
D) W przypadku kilku podstawników wymienia się je w kolejności alfabetycznej
E) Gdy jest kilka takich samych podstawników, ich nazwę poprzedza się przedrostkiem
di-, tri-, tetra-, penta-,itd.; oraz numerami węgli przy których te podstawniki
występują.
Przykładowe nazwy alkanów
CH
2
‒CH
3
|
•
•
CH
3
‒CH
2
‒CH‒CH‒CH
3
3-etylo-2-metylopentan
|
CH
3
CH
2
‒CH
3
CH
3
|
|
•
•
CH
3
‒CH
2
‒CH‒CH‒CH
2
‒CH‒CH
3
5-etylo-2-metylo-4-propyloheptan
|
CH
2
‒CH
2
‒CH
3
CH
3
CH
3
|
|
•
•
CH
3
‒CH‒CH
2
‒CH
2
‒CH‒CH‒CH
2
‒CH
3
3-etylo-4,7-dimetylooktan
|
CH
2
CH
3
24
7.
Budowa alkanów
•
•
wyłącznie wiązania pojedyncze (σ)
•
•
orbitale atomowe węgla w alkanach mają hybrydyzację sp
3
(węgiel tetraedryczny)
•
•
alkany mogą tworzyć izomery konstytucyjne
Izomery konstytucyjne alkanów
C
4
H
10
- 2 izomery
C
5
H
12
- 3 izomery
C
6
H
14
- 5 izomerów
C
7
H
16
- 9 izomerów
C
10
H
22
- 75 izomerów
C
20
H
42
- 366319 izomerów
ZADANIE
Narysować izomery heksanu i heptanu i nazwać je.
8.
Rzędowość atomów węgla w alkanach
H
A)
H‒C‒C‒
I rzędowy (1°) - związany z jednym atomem węgla
H
H
B)
H‒C‒C‒
II rzędowy (2°) - związany z dwoma atomami węgla
‒C‒
H
C)
‒C‒C‒C‒
III rzędowy (3°) - związany z trzema atomami węgla
‒C‒
‒C‒
D)
‒C‒C‒C‒
IV rzędowy (4°) - wszystkie wiązania z innymi atomami węgla
‒C‒
ZADANIE
Określić rzędowość wszystkich węgli
•
•
CH
3
‒CH
2
‒CH
2
‒CH
3
•
•
CH
3
‒CH‒CH
2
‒CH
3
CH
3
|
|
CH
2
‒CH
3
•
•
CH
3
‒CH‒CH
2
‒CH
3
25
9.
Występowanie w przyrodzie i własności fizyczne alkanów
•
•
naturalne źródła: gaz ziemny (głównie metan)
ropa naftowa ( mieszanka alkanów do C
40
)
•
•
właściwości fizyczne:
◦
◦
stan skupienia C
1
-C
4
– gazy (bez zapachu)
do C
17
– ciecze (zapach benzynopodobny)
od C
18
– ciała stałe ( bez zapachu)
◦
◦
temp. Wrzenia i gęstość izomerów rozgałęzionych niższa
niż odpowiadających im węglowodorów prostych
◦
◦
rozpuszczalność: praktycznie nierozpuszczalne w wodzie,
za to bardzo dobrze rozpuszczalne w eterze
10.
Reakcje
–
–
spalania (utlenianie tlenem z powietrza)
–
–
halogenowania (z fluorowcami)
Spalanie
A) Spalanie całkowite
CH
4
+ 2O
2
→ CO
2
+ 2H
2
O + 890 kJ/mol
B) Półspalanie
2CH
4
+ 3O
2
→ 2CO
2
+ 4H
2
O + 607 kJ/mol
C) Spalanie niecałkowite
CH
4
+ O
2
→ C + H
2
O + E
Spalanie alkanów to ważna praktycznie reakcja- silnie egzotermiczna reakcja
ciepło
wydzielone
Zastosowanie:
- kuchenki gazowe
- piecyki gazowe
- benzyna (ropa)
liczba atomów węgla
w cząsteczce
Halogenowanie
H‒C‒ + X
2
h
ν (ΔT)
X‒C‒ + HX
Na ogół powstaje mieszanina produktów
26
WYKŁAD 6 (12.04.2010)
Przykład - chlorowanie metanu
4CH
4
+ 10 Cl
2
h
ν
CH
3
Cl + CH
2
Cl
2
+ CHCl
3
+ CCl
4
+ 10HCl
chlorometan dichlorometan trichlorometan tetrachlorometan
1.
Mechanizm reakcji halogenowania
Każda reakcja S
R
to reakcja łańcuchowa i przebiega w trzech elementarnych etapach.
Mechanizm reakcji halogenowania alkanów (S
R
) na przykładzie chlorowania metanu:
A)
I etap – inicjowanie – powstawanie wolnych rodników
Cl
2
h
ν
2 Cl*
B)
II etap – propagacja – wzrost łańcucha
CH
4
+ Cl* → CH
3
* + HCl
CH
3
* + Cl
2
→ CH
3
Cl + Cl*
C)
III etap – terminacja – zakończenie reakcji
CH
3
* + Cl* → CH
3
Cl
CH
3
* + CH
3
* → CH
3
CH
3
Cl* + Cl* → Cl
2
2.
Monohalogenowanie wyższych alkanów
•
•
chlorowanie (temp. reakcji – 25 °C)
Cl
CH
3
CH
2
CH
2
CH
3
+ Cl
2
h
ν
CH
3
CH
2
CH
2
CH
2
Cl + CH
3
CH
2
CHCH
3
n-butan
1-chlorobutan (28%) 2-chlorobutan (72%)
•
•
bromowanie (temp. reakcji – 127 °C)
Br
CH
3
CH
2
CH
2
CH
3
+ Br
2
h
ν
CH
3
CH
2
CH
2
CH
2
Br + CH
3
CH
2
CHCH
3
n-butan
1-bromobutan (2%)
2-bromobutan (98%)
Wnioski:
–
–
reaktywność bromu jest mniejsza niż reaktywność chloru
–
–
brom jest bardziej selektywny niż chlor
Reaktywność = łatwość tworzenia wolnych rodników
III rz > II rz > I rz > CH
4
ZADANIE
Napisać mechanizm chlorowania i bromowania n-butanu i przekonać się, że
III rzędowe halogenki powstają z II rzędowych wolnych rodników, a I rzędowe
halogenki z I rzędowych rodników.
27

3.
Przyczyny, dla których S
R
jest reakcją charakterystyczną alkanów
Wynika z ich budowy:
•
•
związki nasycone
⇒ substytucja, eliminacja
(S)
⇑
•
•
słaba polaryzacja C–H
⇒ związki mało polarne
⇒ homolityczny rozpad wiązań prowadzący (R)
do powstania wolnych rodników
CYKLOALKANY
4.
Cykloalkany
•
•
węglowodory nasycone o budowie pierścieniowej
•
•
wzór ogólny (CH
2
)
n
•
•
wzory strukturalne
cyklopropan
cyklobutan
cyklopentan
cykloheksan
itd.
•
•
Miarą trwałości jest energia zgromadzona przez pierścień
(CH
2
)
n
+ 3/2 nO
2
→ nCO
2
+ nH
2
O + Q
697,5 kJ
686,6 kJ
664,4 kJ
659,0 kJ
najmniej trwały
najtrwalszy
5.
Budowa cykloalkanów i nazewnictwo
•
•
atomy węgla o hybrydyzacji sp
3
→ kąt między wiązaniami pominien wynosić 109°C
•
•
cykloalkany nie są płaskimi cząstkami ( wyjątek – cyklopropan)
•
•
jeżeli w pierścieniu występują podstawniki, to zajmują one pozycje ekwatorialne
•
•
nazewnictwo jak w alkanach + przedrostek "cyklo" przed rdzeniem nazwy
•
•
jeżeli cykloalkan zawiera podstawniki alkilowe, to o rdzeniu nazwy decyduje liczba
atomów węgla w cykloalkanie i w podstawniku
CH
3
CH
2
CH
2
CH
2
CH
3
metylocyklopentan
1-cyklopropylobutan
•
•
naturalnie w przyrodzie występują jako produkty rafinacji. Cyklo propan i cyklobutan
nie występują naturalnie
28
6.
Właściwości chemiczne cykloalkanów
•
•
główna reakcja- S
R
+ Cl
2
hν
+ HCl
Cl
•
•
Br
2
300 °C
Br + HBr
•
•
3 i 4- członowe cykloalkany- nietrwałe i mocno reaktywne
Ni, H
2
, 80°C →
CH
3
–CH
2
–CH
3
propan
Cl
2
, FeCl
3
→
CH
2
Cl–CH
2
–CH
2
Cl
1,3-dichloropropan
stęż. H
2
SO
4
→
CH
3
–CH
2
–CH
2
OH
alkohol n-propylowy
ALKENY
7.
Alekny
•
•
wiązanie podwójne C=C
•
•
wzór ogólny C
n
H
2n
•
•
grupa homologiczna –CH
2
–
•
•
końcówka- alken
CH
2
=CH
2
eten (etylen)
CH
2
=CH–CH
3
propen (propylen)
CH
2
=CH–CH
2
–CH
3
buten (butylen)
CH
3
–CH=CH–CH
3
8.
Nazewnictwo i budowa
A)
Rdzeń nazwy- najdłuższy łańcuch zawierający wiązanie podwójne
B)
Numeracja atomów węgla tak, by wiązanie podwójne miało jak najniższy lokant
C)
Wymieniamy podstawniki w kolejności alfabetycznej
D)
Jeżeli wiązanie podwójne jest w środku cząsteczki,to zaczynamy numerację od
strony podstawników
E)
Jeżeli kilka wiązań podwójnych to nazywamy je: dieny, trieny, tetraeny itd.
CH
3
CH
2
=C–CH=CH
2
2-metylobuta-1,3-dien
CH
2
CH
3
CH
2
=C–CH–C=CH–CH=CH–CH
3
3-etylo-3,4-dimetylookta-1,4,6-trien
CH
3
CH
3
29
Budowa
•
•
hybrydyzacja C=C → sp
2
•
•
wiązania podwójne utworzone z wiązania σ i wiązania π
•
•
izomeria konstytucyjna i geometryczna
•
•
grupy pochodzące od alkenów:
CH
2
=CH–
gr. winylowa
CH
2
=CH–CH
2
–
gr. allilowa
ZADANIE
Nazwać alkeny
CH
3
CH
3
•
•
CH
2
=CH–CH–C–CH
3
CH
3
•
•
CH
3
–CH
2
–CH=C–CH
2
–CH
3
CH
3
•
•
CH
3
–CH=CH–CH–CH=CH–CH–CH
3
CH
2
CH
2
CH
3
CH
2
CH
3
9.
Występowianie i właściwości
•
•
jedynym naturalnie występującym alkenem jest eten, który tworzy się i wydziela
podczas dojrzewania owoców
Właściwości fizyczne
•
•
n
2-4
- gazy
5-17 - ciecze
18≤
- ciała stałe
•
•
nierozpuszczalne w wodzie
•
•
dobrze rozpuszczalne w benzynie, eterze dimetylowym, chloroformie
Właściwości chemiczne
•
•
energia wiązań:
σ 400 kJ/mol
trwalsze
π 280 kJ/mol
mało trwałe
A
E
alkeny są podatne na addycję
•
•
są nukleofilami:
podatne na atak reagentów elektrofilowych
30

10.
Addycja elektrofilowa
–
–
symetryczne cząstki
uwodornienie (+kat)
R–CH=CH
2
+ H
2
kat
R–CH
2
–CH
3
bromowanie
R–CH=CH
2
+ Br
2
R–CHBr–CH
2
Br
–
–
niesymetryczne cząstki
R–CH=CH
2
+ HCl = R–CHCl–CH
3
–
–
inne podstawniki
Cl
2
H–OH, H–Br, H–I, H–HSO
4
Produkty
HX
→
RCHX–CH
3
fluorowcoalkany
RCH=CH +
H
2
O
→
CH
3
–CHR
alkohole
OH
H
2
SO
4
→
R–CH–CH
3
wodorosiarczany
alkilów
OSO
3
H
R
1
CH=CHR
2
+ Br
2
→
R
1
CH–CHR
2
cząsteczka symetryczna
Br Br
R
1
CH=CHR
2
+ HCl
→
R
1
CH–CHR
2
+ R
1
CH–CHR
2
cząsteczka niesymetryczna
Cl H
H Cl
Kierunek addycji do niesymetrycznych alkenów jest zgodne z
regułą Markownikowa
RCH=CH
2
+ HCl →
RCHCl–CH
3
Wodór przyłącza się do tego atomu węgla przy wiązaniu podwójnym, który jest już
związany z większą liczbą atomów wodoru. W obecności nadtlenów reakcja ta przebiega
wbrew regule Markownikowa (
efekt nadtlenkowy
)
RCH=CH
2
+ HBr
ROOR
RCH
2
–CH
2
Br
*ROOR- nadtlenek
31
WYKŁAD 7 (19.04.2010)
1.
Mechanizm przyłączania HZ do alkenów
HZ= HCl, HBr (śr. Polarne), HI, H
2
O, H
2
SO
4
2-etapowa addycja elektrofilowe (A
E
):
1)
C=C + H • •Z → —C—C— + :Z
-
etap wolny
H
+
2) —C—C— + :Z
-
→ —C —C —
etap szybki
H
+
H Z
Trwałość karbokationów
III rz > II rz > I rz
Szereg trwałości karbokationów jest związany z polaryzacją wiązania C—H
( przesunięcie el
-
tworzących wiązanie w kierunku berdziej elektroujemnego atomu węgla).
2.
Wytłumaczenie reguły Markownikowa w oparciu o A
E
RCH=CH
2
+ HZ
RCH—CH
2
+ Z
-
RCH—CH
2
KK II rz
+
H
Z
H
RCH—CH
2
+ Z
-
KK I rz
H +
Nowa definicja
: Przyłączenie HZ do alkenów przebiega w taki sposób, że z dwóch
karbokationów, które mogą powstać w pierwszym etapie powstaje ten o wyższej
rzędowości.
3.
Przegrupowanie karbokationów w czasie reakcji A
E
Karbokationy dążą do jak najwyższej rzędowości
CH
3
A)
CH
3
—CH—CH=CH
2
HCl
CH
3
—CH—CH—CH
3
+ CH—C—CH
2
—CH
3
CH
3
CH
3
Cl
Cl
3-metylobut-1-en
2-chloro-3-metylobutan
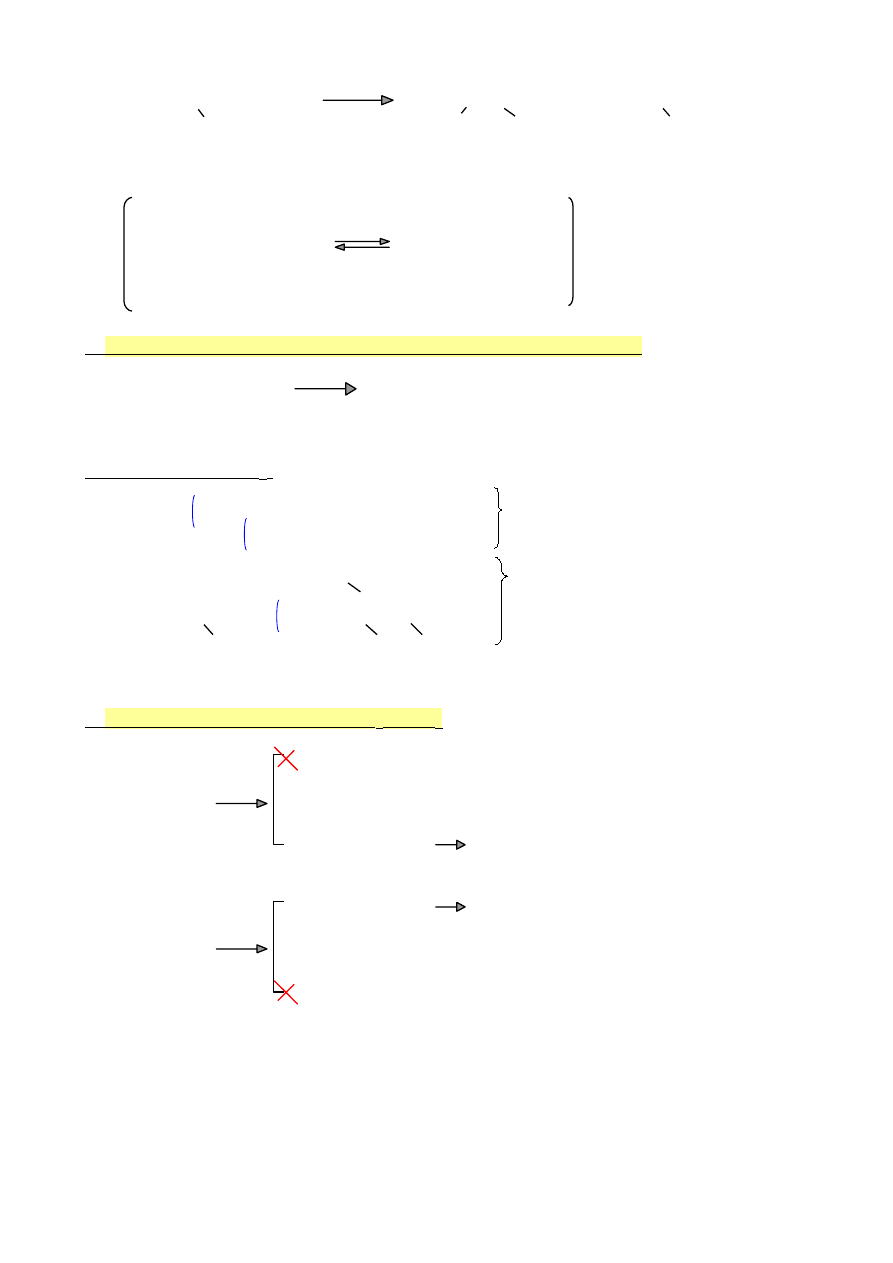
2-chloro-2-metylobutan
Powstanie 2 produktów w reakcji związane jest z przegrupowaniem przejściowego
karbokationu.
CH
3
CH
3
|
|
CH
3
—CH—CH—CH
3
CH
3
—C—CH
2
—CH
3
+
+
KK II rz
KK III rz
Polaryzacja polega na migracji np. jonu wodorkowego
32
CH
3
CH
3
CH
3
CH
3
|
|
| |
B)
CH
3
—CH—CH=CH
2
HCl
CH
3
—C—CH—CH
3
+ CH
3
—C—CH—CH
3
CH
3
CH
Cl
Cl
3,3-dimetylobut-1-en
2-chloro-3,3-dimetylobutan
2-chloro-2,3-dimetylobutan
Przegrupowanie karbokationu polega tu na migracji grupy metylowej
+
+
CH—C—CH—CH
3
CH
3
—C—CH—CH
3
|
| |
CH
3
CH
3
CH
3
4.
Mechanizm przyłączania do alkenó Hbr w obecności nadtlenku
RCH=CH
2
+ HBr
ROOR
RCH
2
—CH
2
Br
Reakcja zachodzi wbrew regule Markownikowa
Addycja rodnikowa (A
R
)
A)
R—O• •O—R → 2 R—O
Inicjacja
R—O• + H• •Br → R—OH + Br•
B)
Br• + RCH=CH
2
→ RCH—CH
2
Wzrost łańcucha
Br
RCH—CH
2
+ H• •Br → RCH—CH
2
+ Br•
Br
H Br
C)
Łączenie się dowolnych 2 wolnych rodników
5.
Przyłączanie Hbr do alkenów: A
E
vs A
R
A
E
CH
3
—CH
2
—CH
2
KK I rz
+
CH
3
—CH=CH
2
+ HBr
CH
3
—CH—CH
3
Br-
CH
3
—CHBr—CH
3
KK II rz
+
A
R
CH
3
—CH—CH
2
HBr
CH
3
—CH
2
—CH
2
Br
WR II rz
•
CH
3
—CH=CH
2
+ Br •
CH
3
—CHBr—CH
2
WR I rz
•
Wolne rodniki nie ulegają przegrupowaniu
33
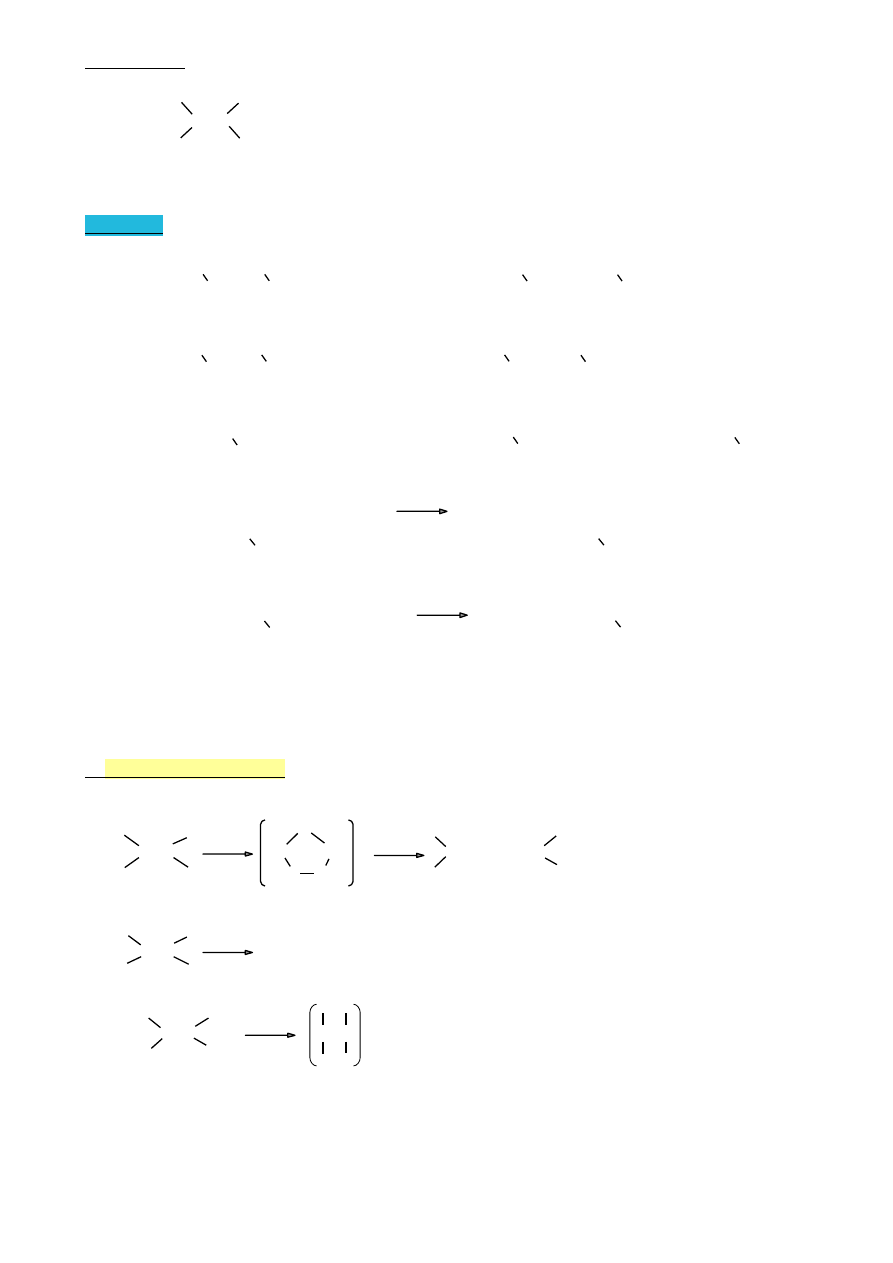
Bromowanie- ważna reakcja analityczna,pozwalająca wykryć wiązanie wielokrotne w
cząsteczce
|
|
C=C + Br
2
/ CCl
4
→ Br—C—C—Br
|
|
czerwony
bezbarwny
ZADANIE
Uzupełnij przykłady addycji elektrofilowej do alkenów
1) CH
3
‒CH
2
‒C=CH‒CH‒CH
3
+ Br
2
→ CH
3
‒CH
2
‒CBr‒CHBr‒CH‒CH
3
CH
3
CH
3
CH
3
CH
3
2,4-dimetyloheks-2-en
3,4-dibromo-2,4-dimetyloheksan
2) CH
3
‒CH
2
‒C=CH‒CH‒CH
3
+ HI → CH
3
‒CH
2
‒CI‒CH
2
‒CH‒CH
3
CH
3
CH
3
CH
3
CH
3
2,4-dimetyloheks-2-en
4-jodo-2,4-dimetyloheksan
3) CH
3
‒CH=CH‒CH‒CH
3
+ HCl → CH‒CHCl‒CH
2
‒CH‒CH
3
*
+
CH
3
‒CH
2
‒CHCl‒CH‒CH
3
C
2
H
5
C
2
H
5
C
2
H
5
4-metyloheks-2-en
2-chloro-4-metyloheksan
3-chloro-4-metyloheksan
*
powstanie chętnej, bo wtedy panuje mniejszy "tłok" w cząsteczce
4) CH
3
‒CH
2
‒CH=C‒CH
2
‒CH
3
+ HBr
śr. polarne
CH
3
‒CH
2
‒CH
2
‒CBr‒CH
2
‒CH
3
CH
3
CH
3
3-metyloheks-3-en
3-bromo-3-metyloheksan
5)
CH
3
‒CH
2
‒CH=C‒CH
2
‒CH
3
+ HBr
ROOH
CH
3
‒CH
2
‒CHBr‒CH‒CH
2
‒CH
3
CH
3
CH
3
3-metyloheks-3-en
4-bromo-3-metyloheksan
Napisać mechanizmy reakcji z przykładów 2-5
6.
Inne reakcje alkenów
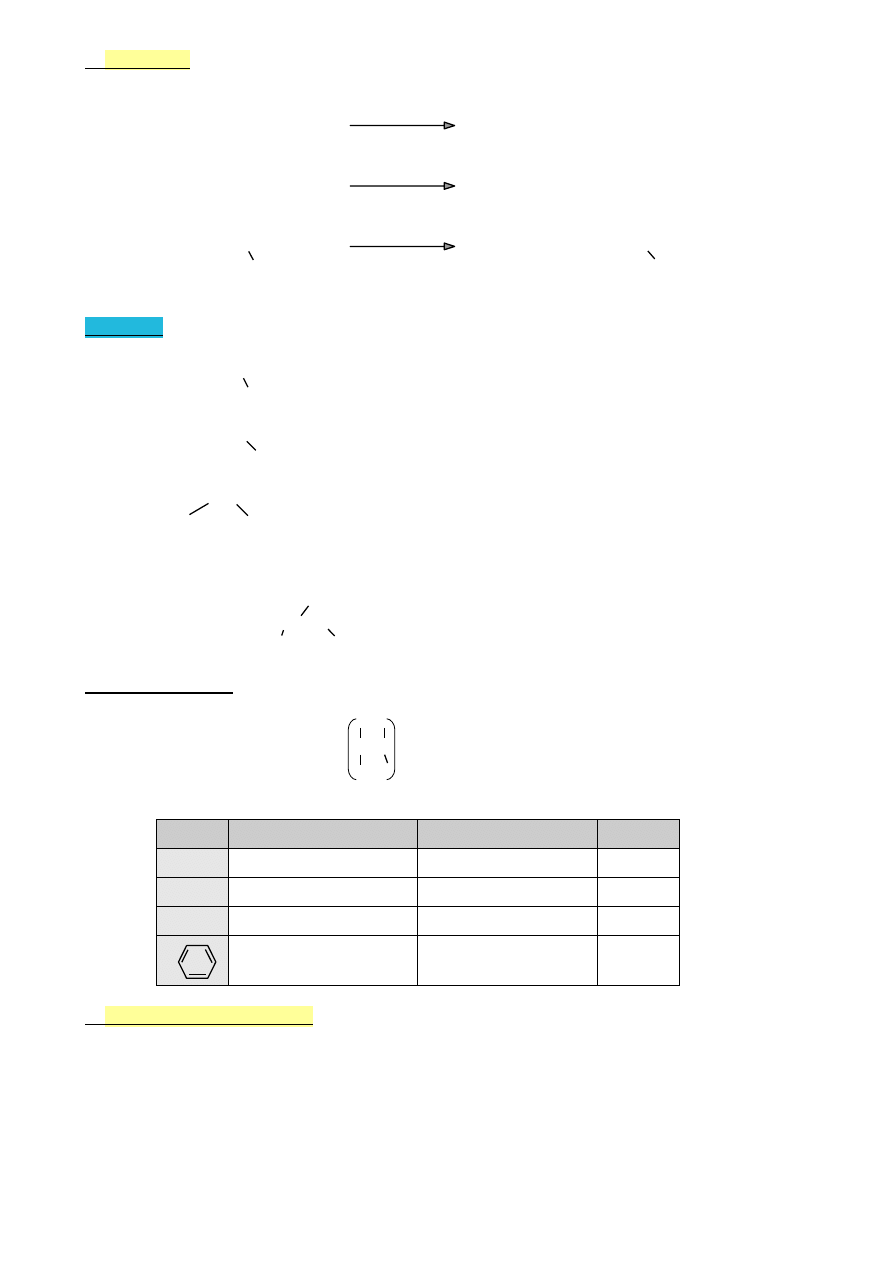
A)
Ozonoliza
O
C=C
O
3
C C
H
2
O, Zn
C=O + O=C
O O
Aldehydy i ketony
B)
Utlenianie
C=C
KMnO
4
kwasy, ketony, CO
2
C)
Polimeryzacja
n C=C
‒C‒C‒
n
Polimer
34
7.
Ozonoliza
Sposób na wykrywanie położenia wiązania C=C w cząsteczce
•
•
CH
3
CH
2
CH
2
CH=CHCH
3
1) O
3
2) H
2
O / Zn
CH
3
CH
2
CH
2
CH=O + O=CHCH
3
Aldehydy
•
•
CH
3
CH
2
CH=CHCH
2
CH
3
1) O
3
2) H
2
O / Zn
CH
3
CH
2
CH=O + O=CHCH
2
CH
3
Aldehyd
•
•
CH
3
CH
2
CH=CCH
3
1) O
3
2) H
2
O / Zn
CH
3
CH
2
CH=O + O=CCH
3
CH
3
CH
3
Aldehyd i keton
ZADANIE
A) Napisać produkty reakcji ozonolizy podanych alkenów
•
•
CH
3
CH=CHCHCH
3
C
2
H
5
•
•
CH
3
CH
2
CH=CCH
2
CH
3
CH
3
•
•
CH
3
CH
2
C=CCH
2
CH
3
C
2
H
5
CH
3
B) Napisać jaki alken był substratem reakcji ozonolizy w wyniku której
otrzymano podane nieżej produkty:
CH
2
CH
3
CH
2
=O + O=CCHC=O + O=CHCH=O + O=CHCH
3
CH
3
CH
3
Polimery winylowe- produkty polimeryzacji monomerów winylowych
n ‒CH
2
=CHX → ‒C‒C‒
x n
monomer winylowy
polimer winylowy
X
Nazwa monomeru
Nazwa polimeru
skrót
H
Eten (etylen)
Polietylen
/PE/
CH
3
Propen (propylen)
Polipropylen
/PP/
Cl
Chlorek winylu
Polichlorek winylu
/PCW/
Winylobenzen (styren) Polistyren (styropian) /PS/
8.
Podsumowanie alkenów
•
•
reaktywność uwarunkowana wiązaniem C=C
•
•
najważniejszą reakcją jest addycja elektrofilowa
•
•
w efekcie nadtlenkowym zachodzi reakcja A
R
•
•
największe znaczenie praktyczne ma reakcja polimeryzacji
•
•
reakcja bromowania pozwala na wykrycie wiązania podwójnego, a ozonoliza
określa ich położenie
35
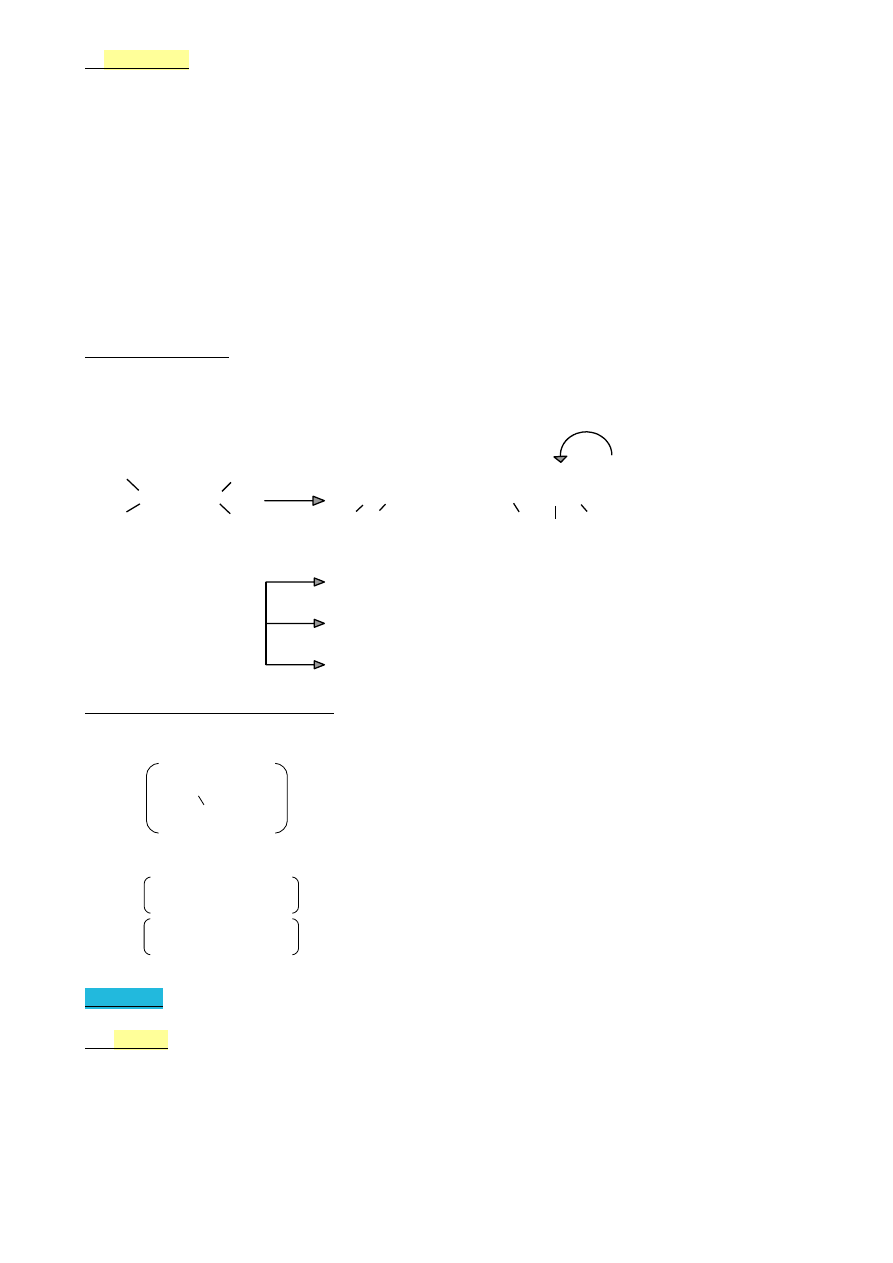
9.
Alkadieny
| | | |
A) ‒C=C‒C‒C=C‒
dien izolowany
(wiązanie izolowane)
| | | |
| | |
B) ‒C=C‒C=C‒C‒
dien sprzężony
(wiązanie sprzężone)
| | |
| | | |
C) ‒C‒C=C=C‒C‒
dien skumulowany
(wiązanie skumulowane)
|
|
Własności dienów
Reakcja charakterystyczna – addycja dp wiązania C=C:
•
•
dieny izolowane – każde wiązanie reaguje niezależnie
•
•
dieny sprzężone – addycja 1,2 i 1,4
produkt główny
|
| | | |
| | |
C=C‒C=C
+YZ
‒C‒C‒C=C‒ + ‒C‒C=C‒C‒
|
Y Z
Y Z
Br
2
CH
2
Br‒CH=CH‒CH
2
Br + CH
2
Br‒CHBr‒CH=CH
2
CH
2
=CH‒CH=CH
2
HCl
CH
3
‒CH=CH‒CH
2
Cl + CH
3
‒CHCl‒CH=CH
2
H
2
, kat
CH
3
‒CH=CH‒CH
3
+ CH
3
‒CH
2
‒CH=CH
2
Polimery dimerów sprzężonych
•
•
kauczuk naturalny – cis-polizopren
‒CH
2
‒C=CH‒CH
2
‒
polimer izoprenu
CH
3
n
•
•
kauczuki syntetyczne
‒CH
2
‒CH=CH‒CH
2
‒
n
polibutadien
(polimer buta 1-3 dienu)
‒CH
2
‒CCl=CH‒CH‒
n
polichloropropen
(polimer chloropropenu)
ZADANIE
Narysować monomery w/w kauczuków
10.
Alkiny
•
•
wiązanie potrójne węgiel-węgiel
•
•
gr. homologiczna -CH
2
•
•
wzór ogólny C
n
H
2n-2
36
11.
Nazewnictwo
•
•
końcówka yn lub in ( po spółgłoskach ch, f, g, k, l )
•
•
konstruowanie nazw jak w alkenach
•
•
enyny- zawierają wiązanie podwójne i potrójne. Początek cząsteczki
jak najbliżej wiązania wielokrotnego, a jeżeli to możliwe, to wiązania
podwójnego
CH≡CH
etyn
CH≡C‒CH
3
propyn
CH≡C‒CH
2
‒CH
3
butyn
CH
3
‒CH
2
‒C≡C‒CH
2
‒CH
3
heks-3-yn
Przykłady
CH
3
‒CH
2
‒CH‒CH
2
‒C≡C‒CH
2
‒CH
3
6-metylookt-3-yn
CH
3
CH
3
CH
3
CH
3
‒CH
2
‒C‒C≡C‒CH‒CH
3
2,5,5-trimetylohept-3-yn
CH
3
CH
3
CH
3
‒CH
2
‒C‒C≡C‒CH
2
‒CH
2
‒CH
3
3,3-dimetylookt-3-yn
CH
3
CH≡C‒C≡CH
but-1,3-diyn
CH≡C‒CH
2
‒CH‒CH=CH‒CH
3
4-etylohept-5-en-1-yn
C
2
H
5
CH≡C‒CH
2
‒CH
2
‒CH=CH
2
heks-1-en-5-yn
12.
Budowa
•
•
hybrydyzacja sp
•
•
wiązanie potrójne złożone jest z 1 wiązania σ i dwóch wiązań π
•
•
izomeria konstytucyjna (łańcucha lub położenia wiązania)
•
•
brak izomerii przestrzennej
37
WYKŁAD 8 (26.04.2010)
1.
Właściwości
Fizyczne
•
•
etyn i propyn- gazy; wyższe alkiny- ciecze
•
•
gęstość, temperatury wrzenia i topienia rosną ze wzrostu łańcucha
•
•
rozgałęzione alkiny mają w.w. Parametry niższe w porównaniu z cząstkami
liniowymi
•
•
acetylen (etyn)- gaz termochemicznie nietrwały, bardzo łatwo wybucha od iskry
podczas sprężania
Chemiczne
•
•
najbardziej charakterystyczne – reakcje addycji (A
E
) do wiązania potrójnego.
Ogólny przebieg takich reakcji
Y
Z
‒C≡C‒ + YZ → ‒C=C‒
YZ
‒C‒C‒
Y Z
Y Z
•
•
alkiny terminalne (zawierające atom H przy wiązaniu potrójnym) mają własności
słabych kwasów i reagują z bardzo silnymi zasadami
‒C≡C‒H + zasada → ‒C≡C:
-
C-H kwas
karbokation
A) Reakcje przyłączenia do alkanów
H H
H
2
, kat
‒C=C‒
H
2
, kat
‒C‒C‒
H H
H H
X X
X
2
‒C=C‒
X
2
‒C‒C‒
X X
X X
‒C≡C‒ +
X H
HX
‒C=C‒
HX
‒C‒C‒
X H
X H
H
H
2
O, H+, Hg, SO
4
‒C=C‒
‒C‒C‒
H OH
enol
H O
B) Reakcje alkinów jako kwasów
Alkiny- bardzo słabe kwasy, reagują z bardzo mocnymi zasadami
H‒C≡C‒H + Na
NH
3
ciekły
H‒C≡C:
-
Na
+
acetylen
acetylenek sodu
CH
3
‒CH‒C≡C‒H + NaNH
2
eter
CH
3
‒CH‒C≡C:
-
Na
+
+ NH
3
CH
3
CH
3
3-metylobut-1-yn
izopropyloacetylenek sodu
38
C) Polimeryzacja
n H‒C≡C‒H
kat
—CH=CH—
n
Poliacetylen (Pac)
•
•
pierwszy przewodzący polimer
•
•
najprostszy
przedstawiciel
polimerów
sprzężonych
(skoniugowanych)
Za odkrycie własności poliacetylenu 3 uczonych: Alan J. Heeger, Alan G. MacDiarmid
i Hideki Hirakawa otrzymało Nagrodę Nobla w 2000r.
WĘGLOWODORY AROMATYCZNE (ARENY)
2.
Areny
Węglowodory pierścieniowe (cykliczne) zawierają w cząsteczce atomy węgla o
hybrydyzacji sp
2
- jak alkeny. Wykazują jednak zupełnie inne własności chemiczne niż
alkeny.
Rodzaje związków aromatycznych
•
•
benzen i jego analogi
CH
3
CH
3
CH
CH
3
CH
3
CH
3
benzen
toluen
kumen
o-ksylen
(metylobenzen) (izopropylobenzen) (1,2-dimetylobenzen)
•
•
związki policykliczne (wielopierścieniowe)
•
•
zawierające pojedyncze pierścienie benzenowe
CH
2
CH=CH
difenylometan
difenyloeten
gr. fenylowa
CH
trifenylometan
39
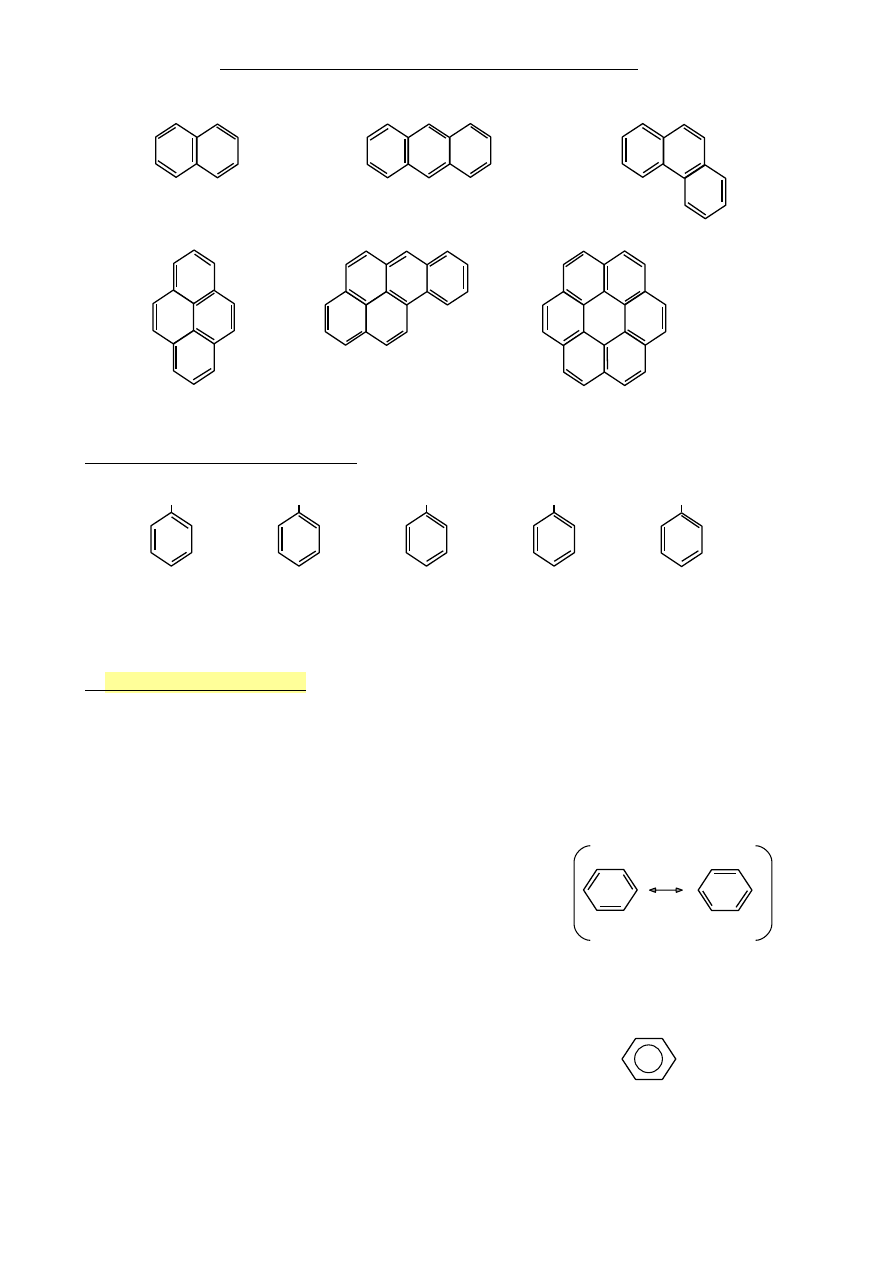
•
•
zawierające skondensowane pierścienie benzenowe
naftalen
antracen
fenantren
piren
benzopiren
koronen
Inne przykłady analogów benzenu
OH
NH
2
CH=CH
2
CHO
COOH
fenol
anilina
styren
Aldehyd
Kwas
(hydroksybenzen) (aminobenzen) (nirylobenzen)
benzoesowy
3.
Historia i wzór benzenu
•
•
1825 – Michael Faraday odkrył benzen w skroplonym gazie świetlnym
•
•
1834 – Eilhardt Mitscherlich określił wzór C
6
H
6
•
•
1865 – August Kekulé – zaproponował strukturę benzenu
•
•
Benzen to hybryda dwóch struktur rezonansowych
•
•
Benzen to sprzężony cykloheksatrien
•
•
Struktury rezonansowe
(tautomeryczne/mezomeryczne)
Wzór alternatywny (już nie zalecany)
Kółko w środku oznacza 6 elektronów
zdelokalizowanych π
40
4.
Budowa benzenu
•
•
Płaski pierścień sześciowęglowy, kąt między wiązaniami 120°
•
•
Cząstki stabilizowane rezonansem
-354 kJ/mol
H
H
-236 kJ/mol
-204 kJ/mol
-118 kJ/mol
H
H
H
H
oczekiwane rzeczywiste
ciepło uwodarniania
do cykloheksanu
E=150 kJ/mol – energia rezonansu
•
•
niska energia – bardzo trwała cząsteczka
•
•
6 elektronów π →
Reguła Hűckla
(w związkach aromatycznych liczba elektronów
wynosi 4n + 2 (n ∊ Z {0,1,2...})
n = 1
4n + 2 = 6
jest aromatyczny
•
•
Związki cykliczne
•
•
Ze względu na zdelokalizowane elektrony π, cząsteczki są bardzo trwałe
•
•
Ponieważ są bardzo trwałe, nie ulegają reakcji addycji, tylko substytucji
•
•
Im większa liczba struktur rezonansowych, tym energia cząsteczki niższa
5.
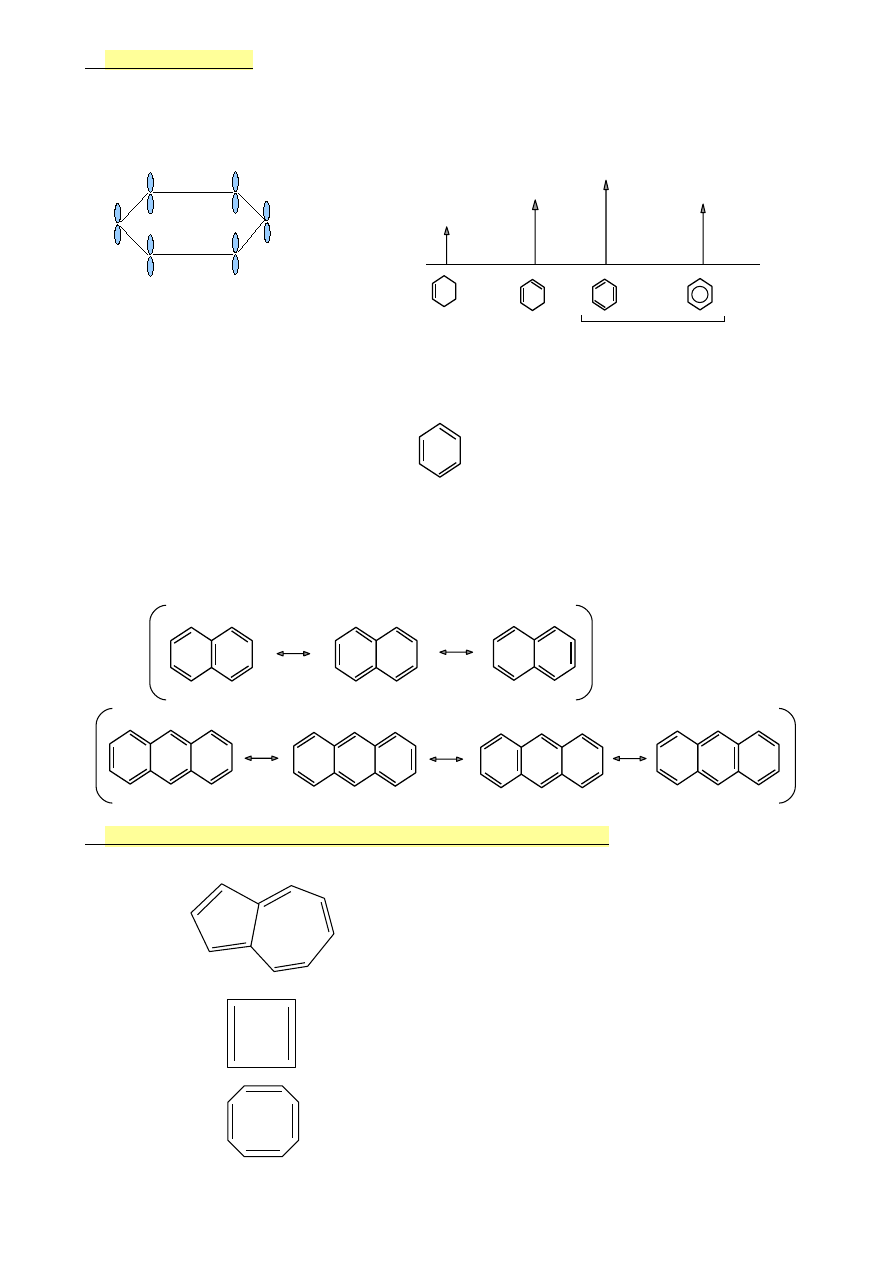
Zastosowanie reguły Hűckla do określania aromatyczności
•
•
azulen
10el π (4n+2)
TAK
•
•
cyklobutadien
4el π
NIE
•
•
cyklooktatetraen
8el π
NIE
41
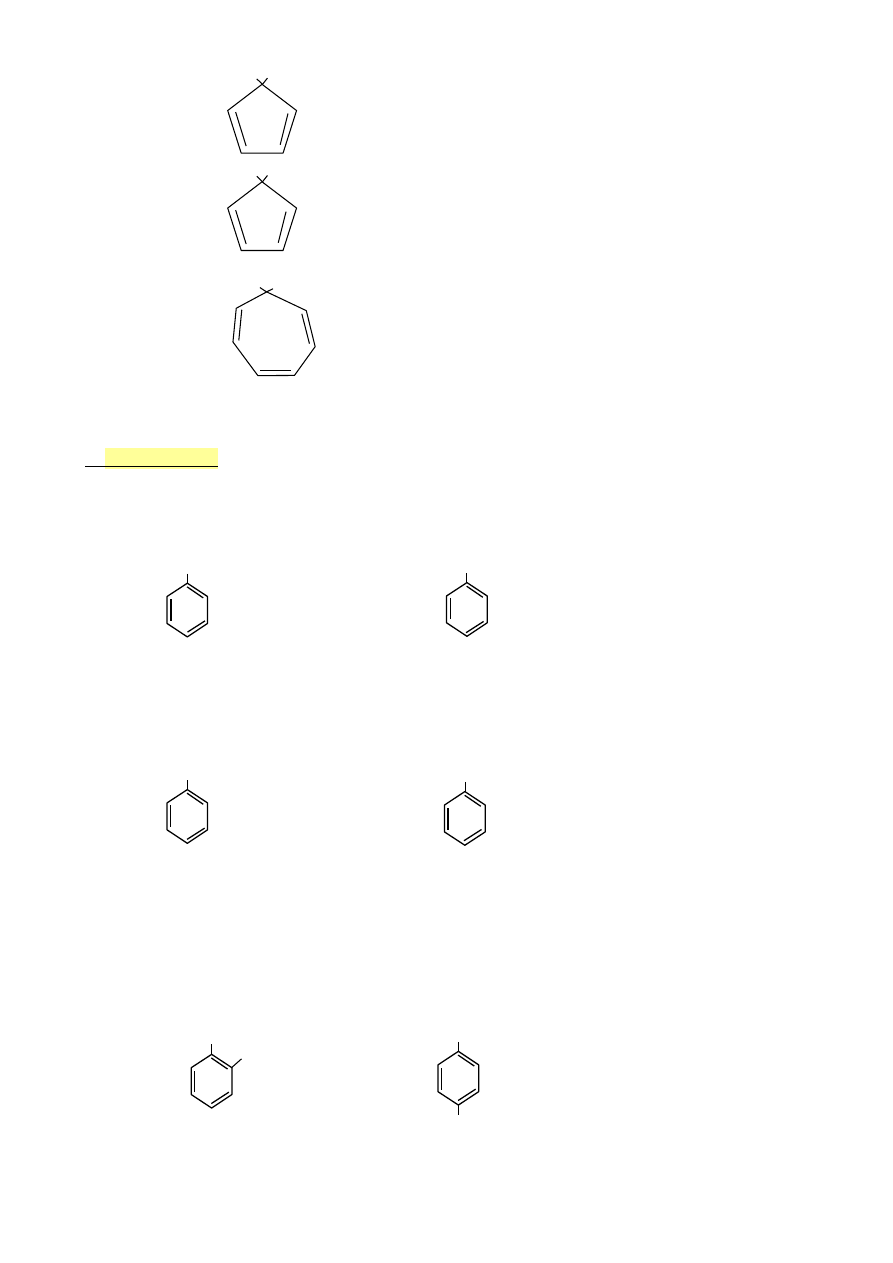
H H
•
•
cyklopentadien
4el π
NIE
H :|
•
•
anion cykloheptadienylowy 6el π TAK
H +
•
•
kation cykloheptatienylony 6el π TAK
6.
Nazewnictwo
•
•
stosuje się wiele potocznych, zwyczajowych nazw (anilina, fenol, toluen)
•
•
nazewnictwo systematyczne
NH
2
OH
aminobenzen
hydroksybenzen
•
•
w przypadku monoalkilowych pochodnych benzenu, podstawa nazwy zależy od
długości grupy alkilowej, np.
CH
2
-CH
2
-CH
3
CH
3
-CH-CH
2
-CH
2
-CH
2
-CH
2
-CH
3
propylobenzen
2-fenyloheptan
Jeżeli liczba atomów węgla w podstawniku jest równa liczbie atomów węgla w pierścieniu,
to ważniejszy jest pierścień
•
•
nazwy dipodstawionych związków konstruujemy przez dodanie przedrostków
położenia: -orto ; -meta ; -para. Podstawniki wymienia się w kolejności
alfabetycznej.
Cl
Cl
Cl
CH
3
o-dichlorobenzen
1-chloro-4-metylobenzen
42
X
ORTO
META
PARA
*
Klasyfikacja podstawników pierścienia może być oparta na kryterium
elektroujemności podstawnika względem elektroujemności pierścienia aromatycznego:
Podstawniki I rodzaju – ich elektroujemność jest mniejsza niż elektroujemność
pierścienia i w związku z tym część ładunku ujemnego podstawnika zostaje przeniesiona
do pierścienia aromatycznego.
◦
◦
Podstawniki I rodzaju: -CH
3
-OH -SH -Cl (X) -NH
2
◦
◦
Podstawniki te kierują następne podstawniki w pozycje o- i p-.
Podstawniki II rodzaju – ich elektroujemność jest większa niż elektroujemności
pierścienia i w związku z tym część ładunku ujemnego pierścienia aromatycznego zostaje
przeniesiona do podstawnika.
◦
◦
Podstawniki II rodzaju: -COOH -NO
2
-SO
3
H -CHO -COOR
◦
◦
Podstawniki te kierują następne podstawniki w pozycję m-.
Dokładniejszy opis podstawników w wykładzie 9
Przykłady
NO
2
OH
Cl
Cl
Cl
Cl
COOH
NO
2
2-chloro-1,4-dinitrobenzen
2,6-dichlorofenol
kwas m-chlorobenzoesowy
7.
Numeracja atomów węgla w aromatycznych węglowodorach policyklicznych
•
•
jest narzucona
8 1
8 9
1
7
2
7
2
6
3
6
3
5 4
5 10 4
•
•
można też określać położenie za pomocą liter greckich
α
α
α γ α
β
β
β
β
β
β
β
β
α
α
α γ α
43
Przykłady
NO
2
Cl
CH
3
C
2
H
5
2-metylonaftalen
9-nitroantracen
1-chloro-6-etylonaftalen
8.
Nazwy grup aromatycznych
fenyl
o-fenylen
m-fenylen
p-fenylen
CH
2
-
CH
C
benzyl
benzyliden
benzylidyn
9.
Właściwości fizyczne
•
•
związki o zróżnicowanych własnościach fizycznych
•
•
ciecze i ciała stałe
•
•
ciecze (zwłaszcza toluen i ksyleny) szeroko stosowane jako rozpuszczalniki (toluen
jest ŁATOWOPALNY!)
•
•
benzen (też ciecz) nie jest stosowany jako rozpuszczalnik, gdyż ma własności
rakotwórcze
10.
Właściwości chemiczne – związane z budową
•
•
delokalizacja elektronów π w pierścieniu aromatycznym
•
•
pierścień trwały, trudno ulega rozpadowi, tzn. Reakjca przebiegają z zachowaniem
pierścienia/
•
•
związki aromatyczne to nukleofile, więc podatne są na atak reagentów
elektrofilowych
Reakcja podstawienie elektrofilowego w związkach aromatycznych
X
2
/ FeX
3
ArX
chloro/bromobenzen
halogenowanie
+ HX
H
2
SO
4
/ SO
3
ArSO
3
H
kwas benzenosulfonowy
sulfonowanie
+ H
2
O
ArH +
HNO
3
/H
2
SO
4
ArNO
2
nitrobenzen
nitrowanie
+ H
2
O
RCl / AlCl
3
ArR
alkilobenzen
alkilowanie
+ HCl
Friedla-Craftsa
RC(=O)Cl / AlCl
3
ArCR
acetylobenzen
acylowanie
O
+ HCl
Friedla-Craftsa
44
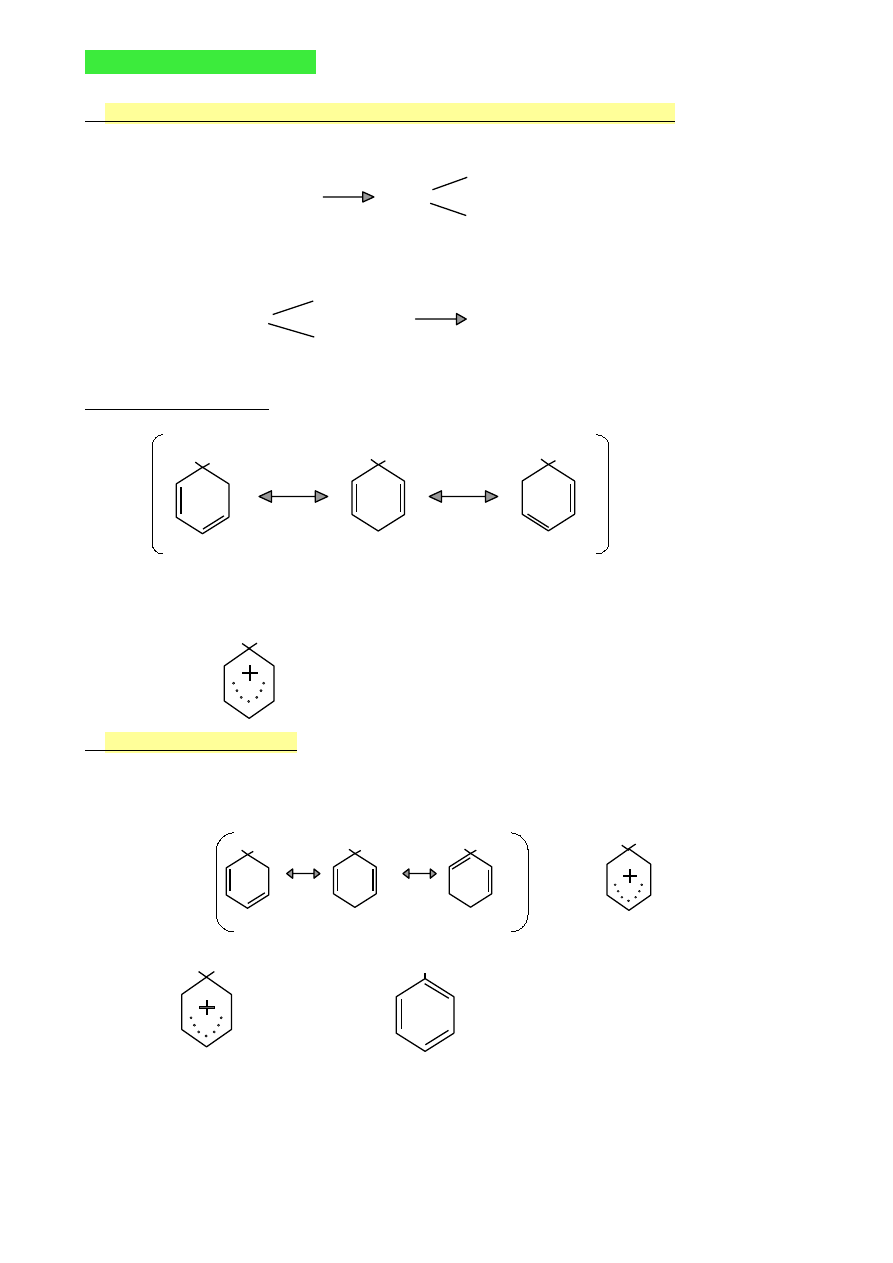
WYKŁAD 9 (10.05.2010)
1.
Ogólny mechanizm reakcji elektrofilowej związków aromatycznych
Reakcje dwuetapowe
H
I etap
Ar + E
+
+
Ar
wolny etap
E
elektrofil
karbokation
H
II etap
+
Ar
+ : Z
-
ArE + HZ
szybki etap
E
nukleofil
Struktury rezonansowe
(na przykładzie benzenu)
H E
H E
H E
H
+
H
+
H
+
Inny zapis struktury karbokationu
H E
2.
Mechanizm nitrowania
1. HONO
2
+ 2H
2
SO
4
↔ H
3
O
+
+ 2HSO
4
-
+ NO
2
+
jon nitroniowy
H NO
2
H NO
2
H NO
2
2. NO
2
+
+
=
H NO
2
NO
2
3.
+ HSO
4
-
→
+ H
2
SO
4
nitrobenzen
elektrofil- NO
2
+
45
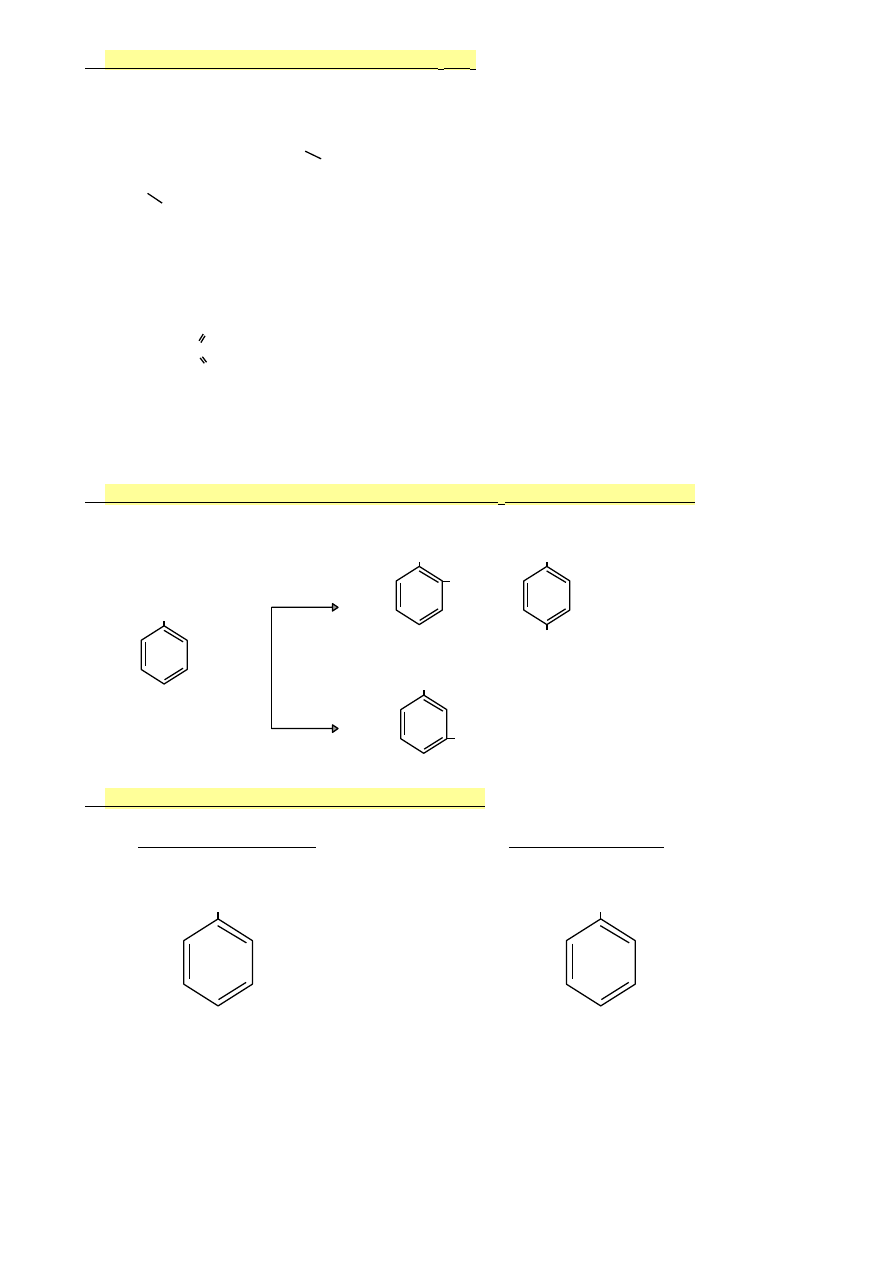
3.
Mechanizm sulfonowania za pomocą H
2
SO
4
1.
2H
2
SO
4
↔ H
3
O
+
+ HSO
4
-
+ SO
3
2.
SO
3
+ C
6
H
6
↔ + C
6
H
5
‒H
SO
3
-
3.
C
6
H
5
‒H + HSO
4
-
↔ C
6
H
5
SO
3
-
+ H
2
SO
4
SO
3
-
4.
C
6
H
5
SO
3
-
+ H
3
O
+
↔ C
6
H
5
SO
3
H + H
2
O
(równowaga przesunięta w lewo)
O
elektrofil
S=O
O
Gdy sulfonowanie przebiega za pomocą oleum (roztworu SO
3
w stężonym H
2
SO
4
),
to elektrofilem jest HSO
3
+
i uprzywilejowane jest tworzenie się kwasu sulfonowego.
4.
Kierujący wpływ podstawników w reakcjach S
E
pochodnych benzenu
Y
Y
E
szybko
p. aktywujący
Y
( I rodzaju )
E
+ E
Y
wolno
p. dezaktywujący
E
( II rodzaju )
Y- podstawnik kierujący
5.
Podstawniki w pierścieniach aromatycznych
Elektronoakceptorowe
Elektronodonorowe
(przyciągają elektrony)
(odpychają elektrony)
Y
σ-
Z
σ+
σ+
σ-
Wyciągają elektrony z pierścienia,
Podają elektrony na pierścień, powodują,
powodują, że staje się on słabszym
że staje się on mocniejszym nukleofilem
nukleofilem tj. Jest mniej podatny na
tj. Podatne na reakcje z elektrofilami
reakcje z elektrofilami
46
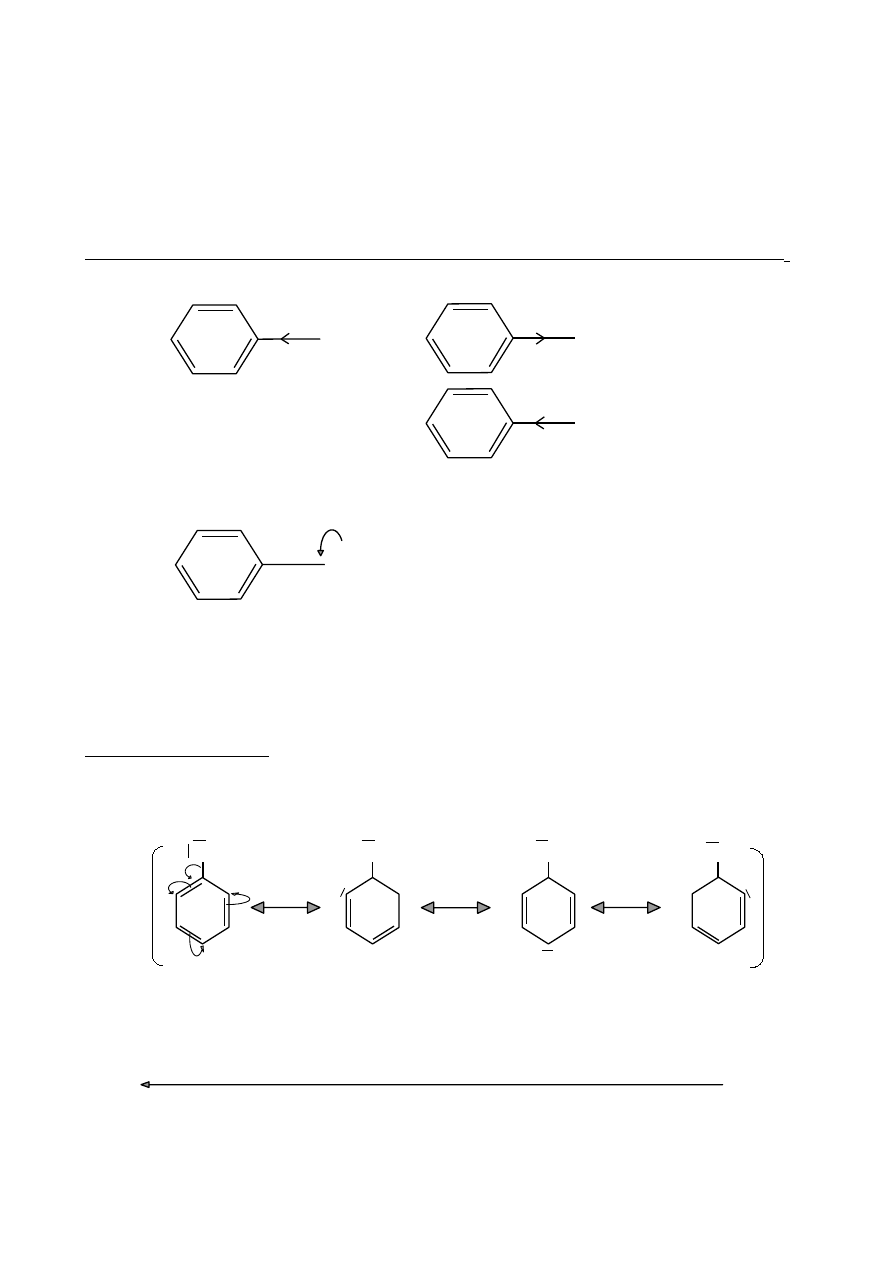
PODSTAWNIKI AKTYWUJĄCE
- kierują w pozycje orto- i para-
•
•
silnie aktywujące –grupy aminowe
-NH
2
-NHR -NR
2
– grupa hydroksylowa
-OH
•
•
średnio aktywujące – grupy alkokrylowe
-OR (np. -OCH
3
)
oraz
-OAr (np. -OC
6
H
5
)
•
•
słabo aktywujące – grupy alkilowe
-R (np. -CH
3
)
– grupy arylowe
-Ar (np. -C
6
H
5
)
Efekty elektronowe podstawników aktywujących pierścienie aromatyczne w reakcjach S
E
σ-
σ+
CH
3
σ+
σ-
OH
+ I
- I
σ-
σ+
NH
2
(efekt mezomeryczny związany ze sprzężeniem p-π)
••
σ-
Y
σ+
+ M
Y: O→ (OH, OR)
X→ halogen
N→ (NH
2
, NHR, NR
2
)
W przypadku atomów O, N związanych z pierścieniem aromatycznym, efekt
mezomeryczny jest silniejszy od efektu indukcyjnego.
Struktury rezonansowe
Hydroksybenzen (fenol)
Efekt +M (sprzężenie p-π)
OH
+
OH
+
OH
+
OH
-
-
-
PODSTAWNIKI DEZAKTYWUJĄCE
– kierują w pozycję meta–
‒N(CH
3
)
3
+
> ‒NO
2
> ‒CN > ‒SO
3
H > ‒COR > ‒COOH > ‒COOR > ‒CHO
moc dezaktywacji
47
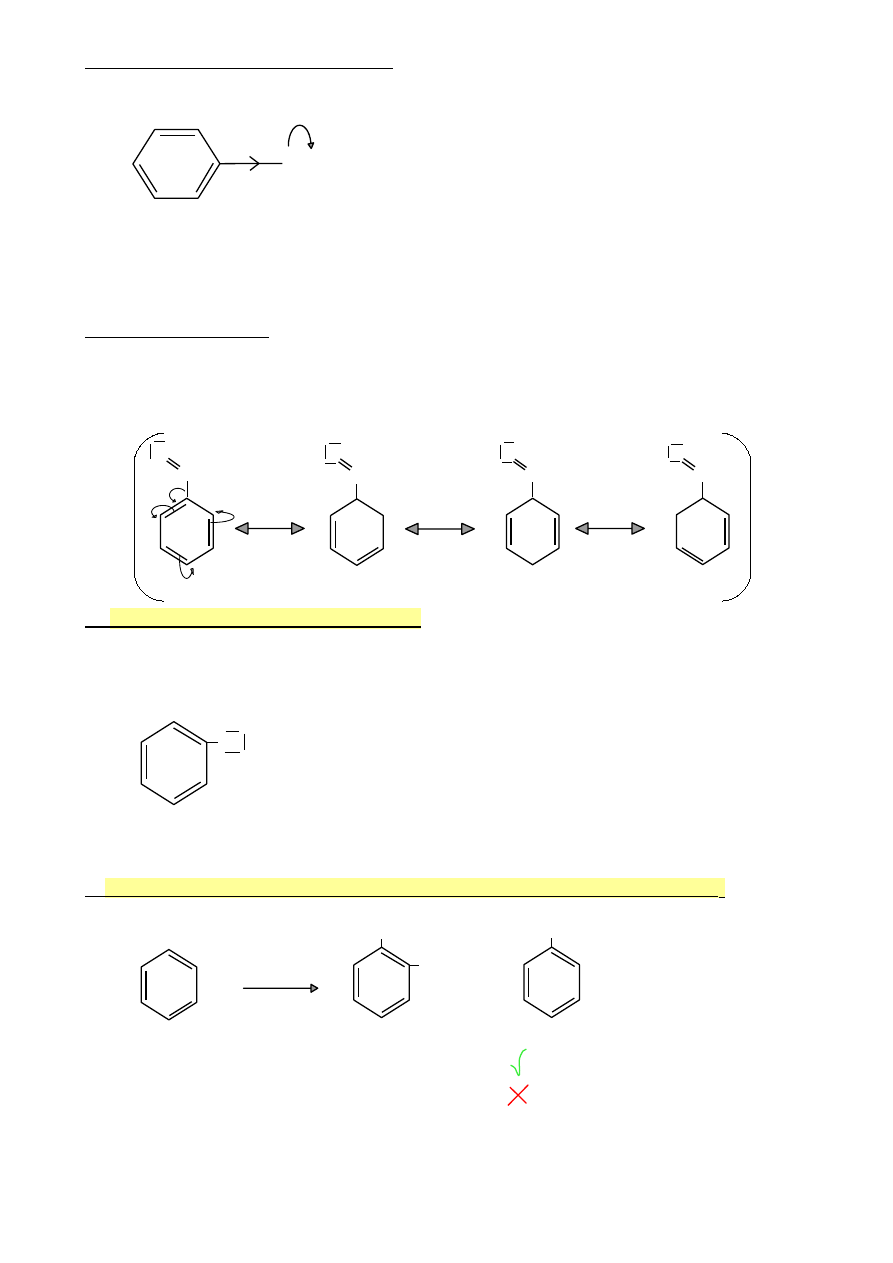
Efekty elektronowe tych podstawników
σ+
Y=Z
σ-
-M
Y=Z : NO
2
, SO
3
H, gr. karbonylowa, gr. nitrylowa
M: efekt mezomeryczny związany ze sprzężeniem π-π
Struktury rezonansowe
Aldehyd benzoesowy
Efekt -M (sprzężenie π-π)
O
O
-
O
-
O
-
C‒H
C‒H
C‒H
C‒H
+
+
+
6.
Atomy fluorowców jako podstawniki
Szczególna grupa podstawników: (‒F, ‒Cl, ‒Br, ‒I ) dezaktywują pierścień w reakcjach S
E
,
ale kierują następny podstawnik w pozycje o- i p-
X
-I, +M
X=F, Cl, Br, I
W przypadku fluorowców efekt indukcyjny i mezomeryczny są porównywalne
7.
Praktyczne znaczenie kierującego wpływu podstawników w reakcjach S
E
CH
3
CH
3
A)
?
NO
2
NO
2
2 możliwości:
–
–
najpierw alkilowanie, potem nitrowanie
–
–
najpierw nitrowanie, potem alkilowanie
Mieszaninę o- i p- nitrotoluenu z benzenem można otrzymać tylko najpierw alkilując, a
potem nitrując pierścień.
48
B)
Dipodstawienie pochodne benzenu
Y
Y
Y
Z
Z
Z
O pozycji wprowadzenia do pierścienia aromatycznego kolejnego podstawnika w wyniku
reakcji S
E
decyduje:
•
•
podstawnik aktywujący, gdy jeden podstawnik jest aktywujący a drugi
dezaktywujący
•
•
podstawnik silniej aktywujący, gdy oba podstawniki są podstawnikami
aktywującymi
8.
Reakcje S
R
związków aromatycznych- podsumowanie
•
•
reakcję zapoczątkowują cząstki ubogie w elektrony tj. Czynniki elektrofilowe:
kationy, kwasy Lewisa
•
•
podstawniki znajdujące się w pierścieniu aromatycznym decydują o miejscu
podstawienia kolejnych
Przykłady – zadania
A)
?
NO
2
B)
Br
?
C)
H
2
SO
4
/ SO
3
?
D)
C
2
H
5
Cl / AlCl
3
?
O CH
3
E)
?
C‒CH
F)
CH
3
H
2
SO
4
?
CH
3
NO
2
CH
3
Br
G)
H
2
SO
4
?
H)
CH
3
Cl / Al Cl
3
?
CH
3
A)
HNO
3
/ H
2
SO
4
E)
CH
3
‒CH‒C‒Cl / AlCl
3
B)
Br
2
/ FeBr
3
CH
3
O
C)
HSO
3
F)
SO
3
H
SO
3
H
CH
3
CH
3
D)
C
2
H
5
G)
CH
3
SO
3
H
CH
3
NO
2
H)
SO
3
H CH
3
NO
2
CH
3
Br
49
9.
Reaktywność związków aromatycznych w S
E
A)
CH
3
NO
2
II
I
III
B)
CHO
CH
3
OCH
3
III
II
I
C)
CH
3
CH
3
CH
3
II
I
III
10.
Węglowodory- podsumowanie
WĘGLOWODORY
TYP REAKCJI
Alkany, cykloalkany
S
R
Alkeny, alkiny, cykloalkeny
A
E
(A
R
)
Areny
S
E
HALOGENOPOCHODNE WĘGLOWODORÓW
11.
Halogenopochodne węglowodorów – związki organiczne
, w których przynajmniej
jeden z atomów wodoru został podstawiony przez atom fluorowca. Mogą być:
•
•
fluoro-, chloro-, bromo-, jednopochodne węglowodorów
•
•
mono- lub wielopodstawione (di-, tri-, tetra-, ...)
•
•
związki, które mają wszystkie wodory podstawione halogenkami nazywamy
perhalogenopochodnymi
np. CF
2
=CF
2
(polimer to teflon)
12.
Halogenopochodne alkanów
•
•
wzór ogólny: R‒X
•
•
nazewnictwo obejmuje zasady nazewnictwa alkanów
•
•
nazwy zwyczajowe np. halogenek metylu zamiast halogenometanu
•
•
rzędowość halogenków alkilów
‒C‒
‒C‒
‒C‒C‒C‒
‒C‒C‒
‒C‒X
X
X
III rz
II rz
I rz
50

13.
Własności
Fizyczne
•
•
wyższe temperatury wrzenia halogenoalkanów niż alkanów o tej samej liczbie
atomów węgla z powodu większej masy cząsteczkowej
•
•
nierozpuszczalne w wodzie, ale rozpuszczalne w rozpuszczalnikach organicznych
•
•
ich gęstość wzrasta ze wzrostem liczby atomowej ( monohalogenopochodne)
Chemiczne
•
•
związki nasycone
–
substytucja, eliminacja
•
•
wiązanie spolaryzowane –
węgiel jest elektrofilowy,
S
N
więc podatny na atak
E
reagentów nukleofilowych
* Eliminacja też zachodzi nukleofilowo, ale nie zaznacza się tego w skrócie
51
WYKŁAD 10 (24.05.2010)
1.
Mechanizm reakcji S
N1
Reakcja 2-etapowa:
•
•
1 etap: - rozpad R ‒ X na jony pod wpływem rozpuszczalnika
R ‒ X → R
+
+ X
-
WOLNO
•
•
2 etap: - reakcja karbokationu z nukleofilu
R
+
+ Nu
-
→ RNu
SZYBKO
σ
-
σ
-
R – X + Nu
-
→ [Nu ‒ R ‒ X] R ‒ Nu + X
-
stan przejściowy
Szybkość reakcji określa etap wolniejszy, czyli V
SN
1
=k[R ‒ X]
Jeżeli reakcja jest 1-etapowa, wtedy jej szybkość zależy od stężania obydwu substratów
v = k [R ‒ X] [Nu
-
]
Stereochemia reakcji S
N1
1 etap
R
1
R
1
C
→
C
+
+ X
-
R
3
X
sp
2
R
2
R
2
R
3
tetraedyczny
płaski karbokation
R ‒ X czynny optycznie
mieszanina racemiczna
nieczynna optycznie
+ Nu
-
+ Nu
-
R
1
R1
R
1
C +
→
C
+
C
R
2
R
3
Nu
Nu
R
3
R
3
R
2
R
2
cz. optyczna 50%
cz. optyczna 50%
52
Każdy enancjomer skręca płaszczyznę polaryzacji światła – ponieważ efekty się znoszą to
enancjomery nie są czynne optycznie (racemat).
W wyniku reakcji S
N
1 czynny optycznie R ‒ X przekształca się w nieczynną optycznie
mieszaninę racemiczną.
2.
Stereochemia reakcji S
N2
R
1
σ
-
R
1
R
+ Nu
-
Nu
C
→
C
→
C
R
3
X
R
3
X
Nu
R
3
R
2
R
2
R
2
czynny optycznie
stan przejściowy
czynny optycznie
R ‒ X (+)
(pięciocentryczny)
R ‒ Nu (-)
W wyniku reakcji S
N
2, czynny optycznie R ‒ X (halogenek alkilu) przekształca się w
czynny optycznie R ‒ Nu
3.
Kiedy reakcja biegnie wg. S
N1
a
kiedy wg. S
N2
?
•
•
Im wyższa rzędowość alkanu, tym łatwiej powstają karbokationy
•
•
Wg S
N
1
chętniej reagują III – rzędowe halogenki alkilów
•
•
Wg S
N
2
chętniej reagują I – rzędowe halogenki alkilów
•
•
Reaktywność h.a. w reakcjach S
N
1
i S
N
2
III rz.
II rz.
I rz.
S
N
1
S
N
1
S
N
2
S
N
2
R ‒ I > R ‒ Br > R ‒ Cl > R ‒ F
wzrost mocy (energii) wiązania R ‒ X
Najtrudniej ulegają reakcji fluoropochodne
53
4.
Cechy reakcji S
N
1
i S
N
2
SN
1
SN
2
2 – etapowa
1 – etapowa
r. I rzędu, jej szybkość
r. II rzędu, szybkość zależy
zależy od stężenia
od stężenia R ‒ X i Nu
-
R ‒ X w środowisku
reakcji
z czynnego optycznie
z czynnego optycznie
substratu powstaje
substratu powstaje
nieczynna optycznie
czynny optycznie produkt
mieszanina racemiczna
Przykłady
NaOH aq
CH
3
‒CH‒CH
3
CH
3
CHCH
3
|
|
Cl
OH
2-chloropropan
propan-2-ol
Dodatek zasady przyspiesza reakcję. (Szybkość zależy od stężenia nukleofilu).
Ponieważ halogenek jest drugorzędny, nie wiadomo by było, który mechanizm działa.
Ponieważ jednak zasada przyspiesza reakcję, jest to S
N2
.
SN2 – Reakcja 1 – etapowa
v = k [(CH
3
)
2
CHCl] [NaOH]
CH
3
σ
-
| σ
-
CH
3
CH CH
3
+ OH
-
→ HO HC Cl →
CH
3
CH CH
3
+ Cl
-
|
|
|
Cl
CH
3
OH
stan przejściowy
odwrócenie konfiguracji
CH
3
CH
3
CH
3
σ
-
atak
OH
C →
C
→
C
+
Cl
-
σ
-
H
Cl
H
Cl
OH
H
CH
3
CH
3
CH
3
+ OH
-
nie ma czynności optycznej
- nie ma asymetrycznego węgla!
54
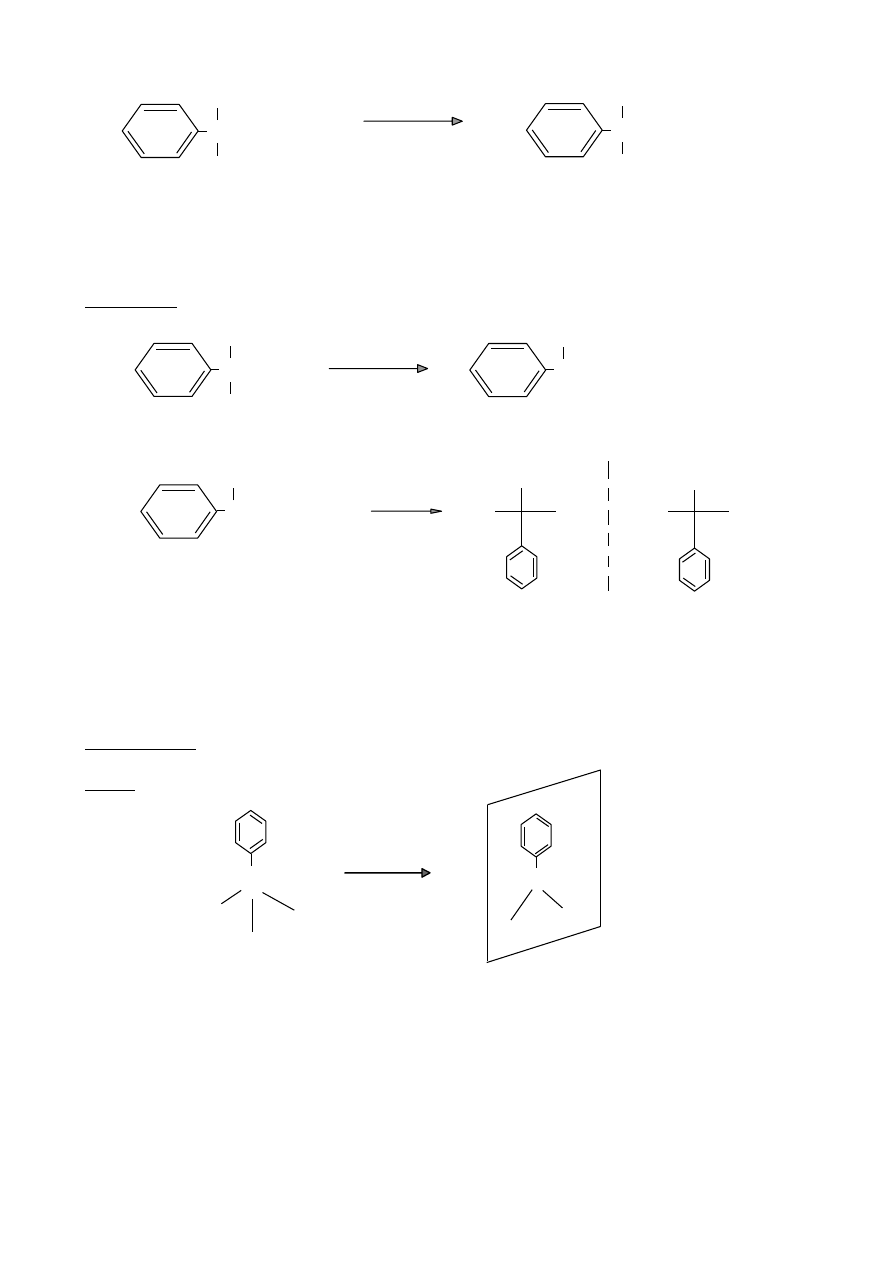
C
CH
3
+NH
3
CCH
2
CH
3
-HBr
CCH
2
CH
3
Br
NH
2
2-bromo-2-fenylobutan
powstaje grupa aminowa
Bromek aminu jest III-rzędowy, więc zachodzi mechanizm S
N1
, produktem jest mieszanina
racemiczna, nieczynna optycznie.
Mechanizm
1) C
CH
3
CCH
2
CH
3
CCH
2
CH
3
+ Br
-
+
Br
ETAP WOLNY
2)
CH
3
CH
2
CH
3
CH
2
CH
3
CCH
2
CH
3
+ 2NH
3
H
3
C
NH
2
+ H
2
N
CH
3
+
-HBr
v = k[(CH
3
)(CH
2
CH
3
)PhCBr]
Równomolowa mieszanina enancjomerów,
nieczynna optycznie
Stereochemia
1 etap
sp
3
sp
2
C*
C
+
+ Br
-
H
3
C
Br
C
2
H
5
CH
3
C
2
H
5
tetraedyczny
płaski karbokation
bromek alkilu
(czynny optycznie)
55
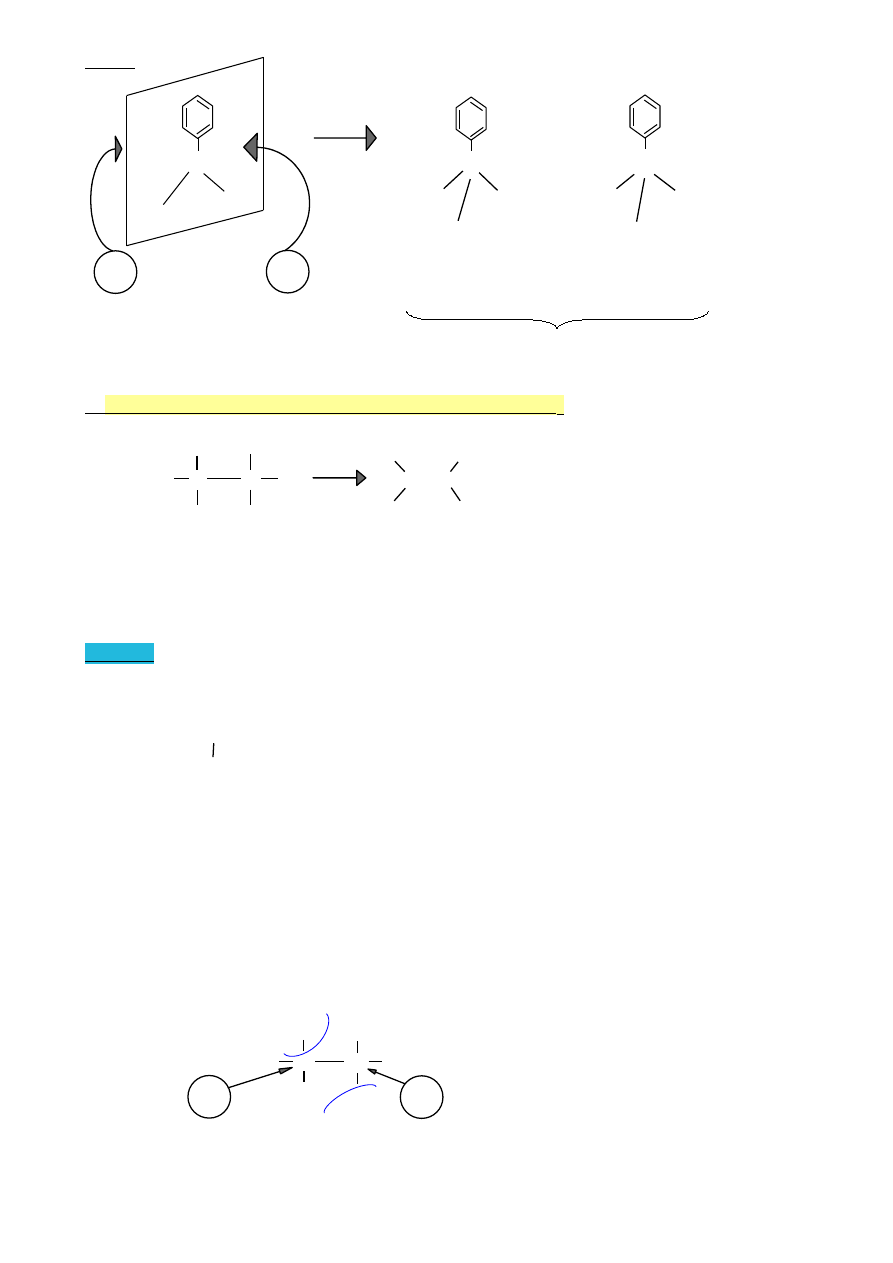
2 etap
sp
3
C
+
C
+ C
C
2
H
5
H
3
C
NH
2
H
2
N
CH
3
CH
3
C
2
H
5
C
2
H
5
+NH
3
+NH
3
czynny optycznie
czynny optycznie
50%
50%
płaski karbokation
mieszanina racemiczna
nieczynna optycznie
5.
Eliminacja – reakcja konkurencyjna w stosunku do S
N
X
C
C
C = C + HX
H
Dwa rodzaje eliminacji w R-X
1. E
1
– konkurencyjna w stosunku do S
N
1
2. E
2
– konkurencyjna w stosunku do S
N
2
Przykład
Orientacja eliminacji – reguła Zajcewa
CH
3
‒CH
2
‒CH‒CH
3
→ CH
3
‒CH=CH‒CH
3
+ CH
3
‒CH
2
‒CH=CH
2
Br
2-bromobutan
but-2-en
but-1-en
(2-buten)
81%
19%
Reguła Zajcewa
W reakcji eliminacji atom wodoru odrywa się od tego atomu węgla, który ma mniej atomów
wodoru.
Substytucja a eliminacja
odłącza się
X
C
C
+ Nu
-
H
+ Nu
-
substytucja
odłącza się
eliminacja
56

Przykład
CH3CH2ONa
CH
3
CH
2
CH
2
Br
CH
3
CH=CH
2
+HBr
propen
Pierwszorzędowy halogenek SN
⇒
2
E
⇒
2
CH
3
CH
3
NaOH
CH
3
CCH
2
CH
3
CH
3
C=CHCH
3
+HCl
Cl
Zgodnie z r. Zajcewa
2-metylobut-2-en
Trzeciorzędowy halogenek E
⇒
1
NH
3
CH
3
CHCH
2
CH
3
CH
3
CH=CHCH
3
+HCl
Cl
but-2-en
Drugorzędowy halogenek E
⇒
1
lub E
2
(zależy od środowiska reakcji)
HALOGENKI ARYLÓW
6.
Halogenki arylów
Wzór ogólny
Ar – X
Halogenopochodne węglowodorów aromatycznych, atom halogenu związany z atomem
węgla pierścienia tj. z atomem węgla o hybrydyzacji sp
2
Np.
Cl
Cl
Cl
Cl
Br
I
o-bromochlorobenzen
m-jodochlorobenzen
Halogenopochodne benzylowe nie są halogenopochodnymi arylów
CH
2
I
jodek benzylu
57

Właściwości fizyczne
Nie rozpuszczają się w wodzie, rozpuszczalne w rozpuszczalnikach organicznach.
Właściwości chemiczne
Związane ściśle z budową, tj. sposobem związania atomu halogenu z pierścieniem
aromatycznym:
•
•
Struktury rezonansowe
•
•
Efekt mezomeryczny
SN w halogenkach arylów zachodzi łatwo w pozycjach orto- i para-
Cl
OCH
3
O
2
N
NO
2
O
2
N
NO
2
CH
3
O
-
+Cl
-
NO
2
NO
2
ZWIĄZKI METALOORGANICZNE
7.
Związki metaloorganiczne
Halogenki alkilu i arylu reagują z metalami tworząc związki metaloorganiczne, tj.
zawierające wiązanie C-M (M – metal), np.
C
4
H
9
Br+2Li
C
4
H
9
Li+LiBr
bromek butylu
butylolit
C
4
H
9
Br+Mg
C
4
H
9
MgBr
bromek butylu
bromek butylomagnezowy
związki metaloorganiczne – nukleofile
Reagują z wieloma elektrofilami i są bardzo przydatne w syntezie organicznej.
σ
-
σ
+
C
M
Odczynnik Grignarda
O
||
‒C‒
RMgX
alkohole, kwasy, aldehydy, ketony
związki B-, P-, Sn- organiczne
58
RX
ArX
halogenki
halogenki
alkilów
arylów
atom halogenku związany
atom halogenku związany
z tetraedrycznym
bezpośrednio z atomem
atomem węgla
węgla pierścienia aromatycznego
łatwo ulegają
nie ulegają reakcjom
reakcjom S
N
i E
S
N
ani E
tworzą związki metaloorganiczne
HALOGENOPOCHODNE ALKENÓW
8.
Halogenopochodne alkenów
σ
+
σ
-
CH
2
=CH‒CH
2
‒
CH
2
=CH‒CH
2
X
grupa alkinowa
halogenek alkilu
- I
budową podobne do halogenków alkilowych
halogenki alkilu reaktywne jak RX
σ
-
σ
+
CH
2
=CH‒
CH
2
=CH N
grupa winylowa
halogenek winylu
- I
podobna do halogenków arylowych
+ M
59
WYKŁAD 11 (31.05.2010)
ALKOHOLE
1.
Alkohole
•
•
wzór ogólny R-OH
•
•
alkohole mogą być
•
•
monowodorotlenowe lub wielowodorotlenowe (poliole)
•
•
I-rzędowe, II-rzędowe, III-rzędowe
‒C‒OH
I-rzędowe
‒C‒C‒C‒
II-rzędowe
OH
C
‒C‒C‒C‒
III-rzędowe
OH
Jako pochodne węglowodorów
ALKOHOLE
łańcuchowe
pierścieniowe
nasycone
nienasycone
nasycone
nienasycone
Alkohole, w których gr. -OH znajduje się w pozycji winylowej ( >C=C‒OH ) to enole:
nietrwałe związki przegrupowujące się do związku karbonylowego. Związki w których
grupa -OH jest bezpośrednio przy pierścieniu aromatycznym to fenole, odrębna grupa
związków.
2.
Nazewnictwo:
•
•
końcówka „-ol”
•
•
łańcuch numeruje się tak, żeby grupa hydroksylowa miała jak
najniższy lokant
OH
CH
3
‒C‒CH
2
‒CH
2
‒CH
3
2-metylopentan-2-ol
CH
3
Nazwy zwyczajowe
CH
3
OH
alkohol benzylowy
→
fenylometanol
CH
2
=CH‒CH
2
OH
alkohol allilowy
→
prop-2-en-1-ol
CH
CH
3
‒C‒CH
3
alkohol tert-butylowy
→
2-metylopropan-2-ol
OH
60

CH
2
‒CH
2
glikol etylenowy
→
etano-1,2-diol
OH OH
CH
2
‒CH‒CH
2
gliceryna, glicerol
→
propano-1,2,3-triol
OH OH
OH
3.
Właściwości fizyczne
R‒OH
słabo polarna
polarna
część lipofilowa,
część fipofobowa,
hydrofobowa
hydrofilowa
Właściwości fizyczne alkoholi uwarunkowane są wpływem grup alkilowej i
hydroksylowej. Im dłuższy łańcuch alkilowy, tym bardziej hydrofobowe właściwości, im
więcej grup -OH, tym bardziej hydrofilowa cząstka.
Heksanol (6-C 1-OH)
cząstka nierozpuszczalna w wodzie
Glukoza (6-C 5-OH)
bardzo dobrze rozpuszczalna w wodzie
O właściwościach fizycznych decydują też wiązania wodorowe:
A) Między cząstkami alkoholi
R—O‒H
O—R
ciecz zasocjowana – cząsteczki alkoholu tworzą
H
dimery, trimery, oligomery
R—O
H
Ta własność powoduje wysokie temperatury wrzenia
B) Między cząsteczkami wody i alkoholu
H‒O H‒O H R
Ta własność pozwala na dużą
H
rozpuszczalność w wodzie niższych
O—R R O‒H O‒H
alkoholi
4.
Właściwości chemiczne
•
•
związki bardzo aktywne, więc są używane jako odczynniki w syntezie organicznej
•
•
alkohole są amfoteryczne:
•
•
jako kwas – dysocjacja wiązania -OH
•
•
jako zasada – protonowanie atomu tlenu grupy OH z
utworzeniem kationu alkilooksoniowego
•
•
Alkohole (prócz III-rzędowych) mogą ulegać reakcjom utleniania
61
A)
Właściwości kwasowe – tworzenie alkoholanów
R—O‒H + Z
-
RO
-
+ Z‒H
hetorolityczny
zasada
anion
rozpad wiązania
alkoholanowy
Równowaga przesunięta jest w prawo, gdy użyta zasada jest silniejsza od anionu
alkoholanowego np. anion amidowy:
R—O‒H + Na
+
NH
2
-
RO
-
Na
+
+ NH
3
amidek sodu
alkoholan sodu
Alkoholany – zasady silniejsze od wodorotlenków. Alkoholany sodu i potasu otrzymuje się
w reakcji alkoholu z metalem (Na, K)
R—O‒H + Na
+
RO
-
Na
+
+ H
+
Kwasowość alkoholi maleje wraz ze wzrostem długości łańcucha węglowego i
wzrostem rzędowości (związane z efektem indukcyjnym).
B)
Właściwości zasadowe – reakcje z halogenowodorami (S
N
)
R—O‒H + HX
RX + H
2
O
HX= HCl, HBr, HI
halogenek alkilu
Przykłady
CH
3
CH
3
CH
3
‒C‒OH + HCl CH
3
‒C‒Cl + H
2
O
CH
3
CH
3
tert-butanol
chlorek tert-butylu
CH
3
OH + HI
CH
3
I + H
2
O
metanol
jodometan
Reakcje mogą zachodzić zgodnie z mechanizmem S
N1
lub S
N2
62
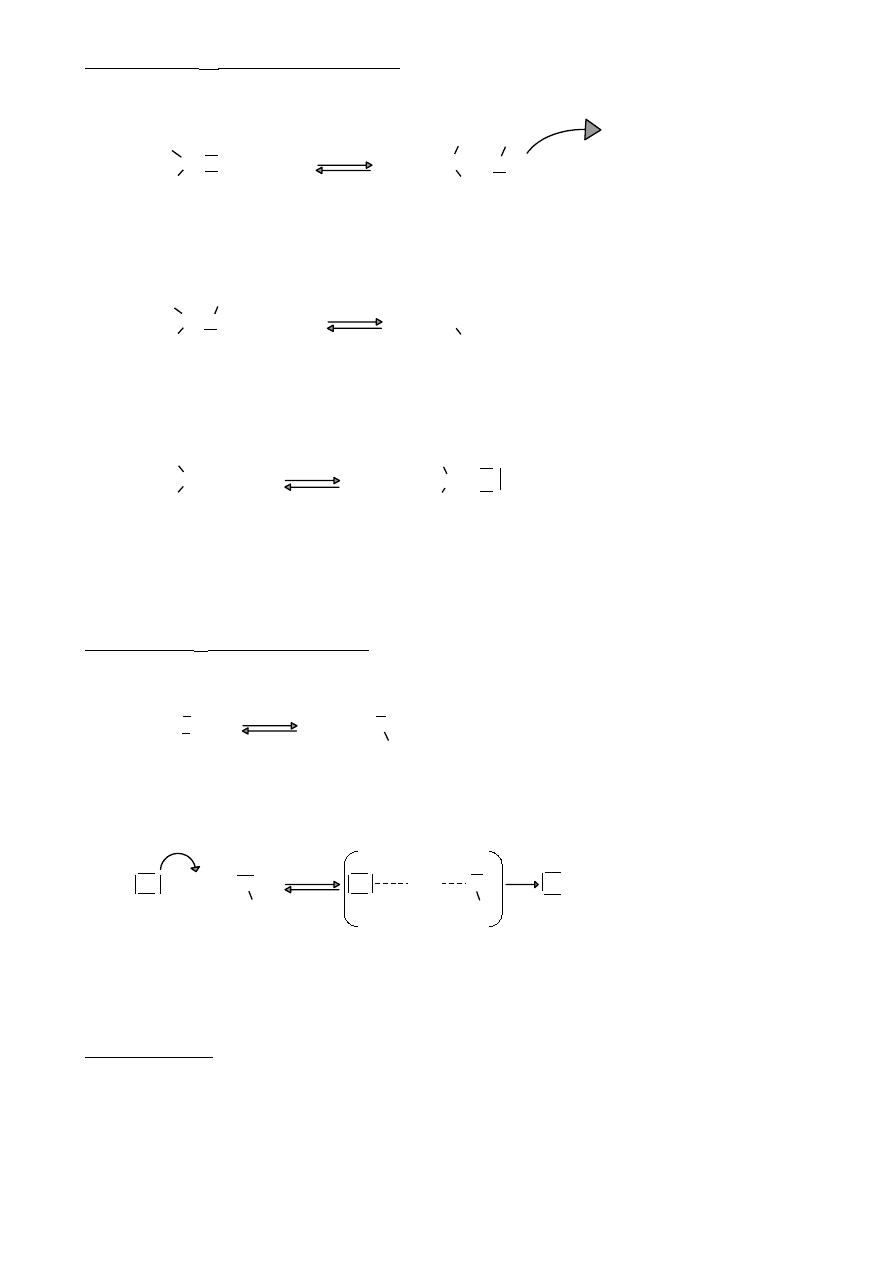
Mechanizm S
N1
(np. tert-butanol z HCl
)
1. Protonowanie alkoholu
grupa OH jest grupą
CH
3
CH
3
H
„źle” opuszczającą
CH
3
‒C‒OH +
H+
CH
3
‒C‒—OH
CH
3
CH
3
+
ETAP SZYBKI
tert-butanol
kation tert-butylooksoniowy
2. Wytworzenie karbokationu
CH
3
H
CH
3
CH
3
‒C‒OH + HCl CH
3
‒C
+
+ H
2
O
ETAP WOLNY
CH
3
+
CH
3
kation tert-butylooksoniowy
karbokation
3. Reakcja karbokationu z nukleofilem
CH
3
CH
3
CH
3
‒C
+
CH
3
‒C ‒ Cl
ETAP SZYBKI
CH
3
CH
3
karbokation
chlorek tert-butylu
szybkość S
N1
: v = k [(CH
3
)
3
H
2
O
+
] = k' [(CH
3
)
3
OH] ⇒
v = k [ROH]
Mechanizm S
N2
(np. metanol i HBr)
1. Protonowanie alkoholu
+
CH
3
‒OH
H+
CH
3
‒OH
ETAP SZYBKI
H
metanol
kation metylooksoniowy
2. Reakcja kationu metylooksoniowego z nukleofilem (Br
-
)
Br
-
CH
3
‒
+
OH
Br
-
CH
3
+
OH Br‒CH
3
+ H
2
O
H
H
ETAP WOLNY
Ogólna szybkość S
N2
: v=k [ROH][HX]
Podsumowanie:
Ponieważ grupa OH jest złą grupą opuszczającą, reakcja musi zacząć
się od protonowania alkoholu.
63
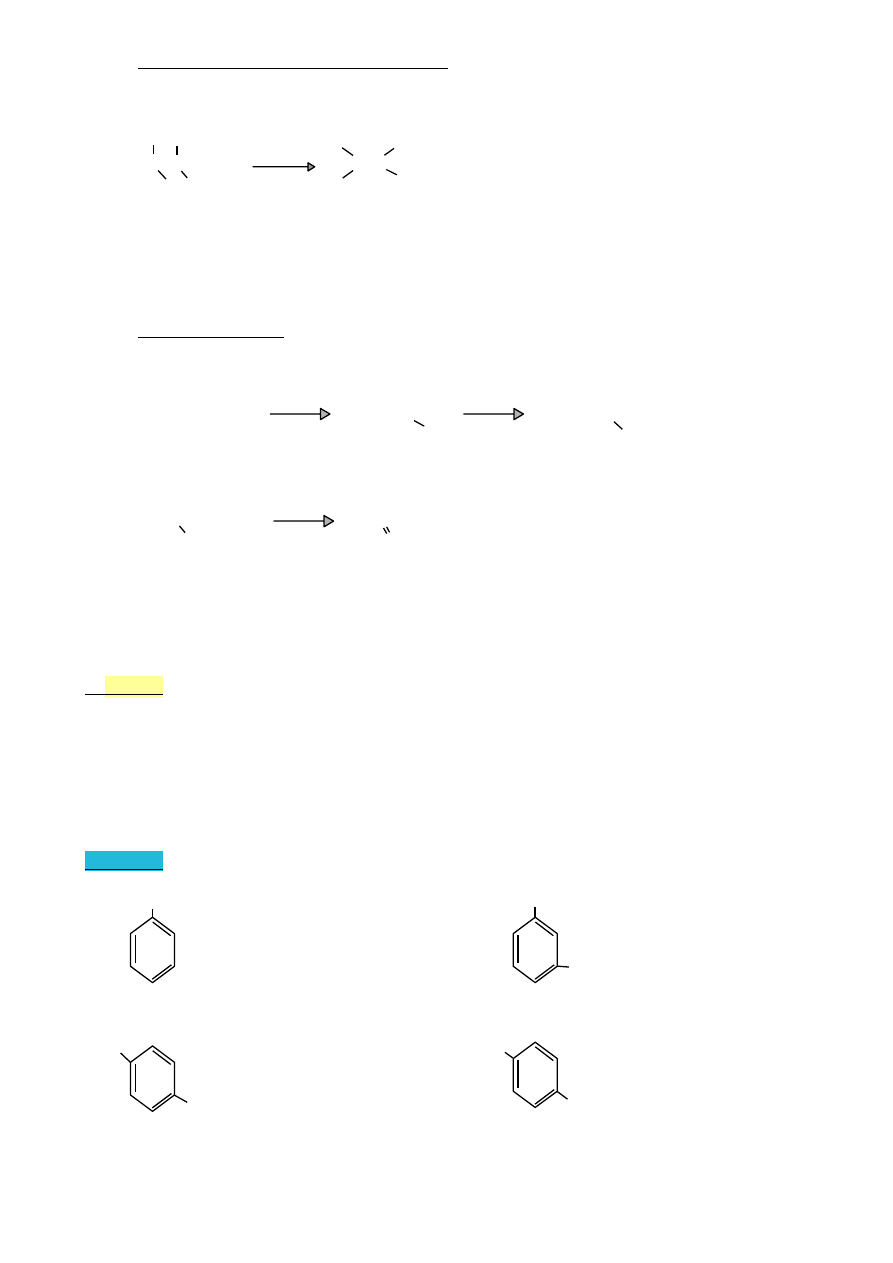
C)
Dehydratacja alkoholi- eliminacja wody
Reakcja konkurencyjna w stosunku do S
N
, odwrotna do uwadniania alkenów
‒C‒C‒
H
2
SO
4
, ΔT
C=C + H
2
O
H OH
Reakcje przebiegają według mechanizmu E
1
, rzadziej E
2
Obowiązuje
reguła Zajcewa
: wodór odłącza się od tego atomu węgla, który
połączony jest z mniejszą liczbą atomów wodoru.
D)
Utlenianie alkoholi
•
•
Alkohole I-rzędowe ulegają utlenieniu do aldehydów i kwasów
CH
3
CH
2
CH
2
OH
KMnO
4
CH
3
CH
2
C=O
KMnO
4
CH
3
CH
2
C=O
H
OH
•
•
Alkohole II-rzędowe ulegają utlenieniu do ketonów
CH
3
CHCH
2
CH
3
K
2
Cr
2
O
7
CH
3
CHCH
2
CH
3
OH
O
•
•
Alkohole III-rzędowe nie ulegają utlenieniu (w bardzo drastycznych warunkach
utlenianie prowadzi do degradacji cząsteczki)
FENOLE
5.
Fenole
•
•
wzór ogólny Ar‒OH
•
•
grupa -OH bezpośrednio związana z pierścieniem aromatycznym
•
•
Przeciwny kierunek polaryzacji wiązania C‒OH w fenolach niż w alkoholach
powoduje, że fenole w porównaniu do alkoholi są:
◦
◦
silniejszymi kwasami
◦
◦
słabszymi zasadami (atom tlenu grupy -OH fenoli trudniej ulega protonowaniu
Przykłady
OH
OH
fenol
m-krezol
(hydroksybenzen)
CH
3
(3-metylofenol)
HO
HO
p-hydroksyanilina
hydrochinon
(p-aminofenol)
(benzeno-1,3-diol)
NH
2
OH
64
COOH
OH
HO
OH
kwas salicylowy
katechol
(kwas o-hydroksybenzoesowy)
benzeno-1,2-diol
OH
OH
OH
rezorcynol
pirogalol
benzeno-1,3-ol
benzeno-1,2,3-triol
HO
OH
6.
Właściwości fizyczne fenoli
Na właściwości fizyczne fenoli wpływają wiązania wodorowe:
•
•
między cząsteczkami
H‒O H‒O
•
•
wewnątrz cząsteczki
H
O
H
N
H
•
•
między cząsteczkami wody a fenolu
H‒O H‒O H‒O
H
Większość prostych fenoli to ciała stałe o niskiej temperaturze topnienia i wysokiej
temperaturze wrzenia; średnio lub słabo rozpuszczalne w wodzie i żrące.
7.
Właściwości chemiczne fenoli
A) Reakcje grupy hydroksylowej
B) Podstawienia elektrofilowego grupy aromatycznej
65
•
•
Właściwości kwasowe grupy hydroksylowej
◦
◦
dysocjacja elektrolityczna fenoli w wodzie
OH
O
-
+ H
2
O ↔
+ H
3
O
+
anion fenolanowy
fenol – kwas mocniejszy od wody
◦
◦
reakcje z wodorotlenkami metali alkalicznych
OH
O
-
Na
+
•
•
+ NaOH ↔
+ H
2
O
•
•
Właściwości kwasowe fenoli modyfikowane są przez podstawniki w pierścieniu:
elektronoakceptorowe zwiększają moc kwasu, elektronodonorowe-zmiejszają
np.
OH
NO
2
NO
2
kwas pikrynowy (2,4,6-trinitrofenol)
- moc porównywalna z mocą silnych
kwasów nieorganicznych
NO
2
•
•
Utlenianie fenoli (łatwiejsze niż alkoholi)
OH
O
Na
2
Cr
2
O
7
/ H
+
OH
O
hydrochinon
chinon
66
•
•
Reakcje podstawienia elektrofilowego
S
E
zachodzi bardzo łatwo (-OH podstawnikiem aktywującym). Jeszcze łatwiej
reaguje anion fenolanowy. Np. reakcje Kolbego
OH
O
-
Na
+
OH
H
O
NaOH
1) C=O 2) H
2
O / H
+
ZWIĄZKI KARBONYLOWE
8.
Związki karbonylowe
Zawierają w cząsteczkach grupę karbonylową
C=O
Do związków karbonylowych zalicza się:
–
–
aldehydy
–
–
ketony
–
–
kwasy karboksylowe i ich pochodne
9.
Aldehydy i ketony
Aldehydy mogą być:
Ketony mogą być:
alifatyczne
alifatyczno-alifatyczne
R
R
C=O
C=O
H
R
1
H
alifatyczno-aromatyczne
C=O
H
R
C=O
aromatyczne
Ar
H
aromatyczno-aromatyczne
C=O
Ar
Ar
C=O
Ar
grupa aldehydowa
-CHO (nie -COH) !
67
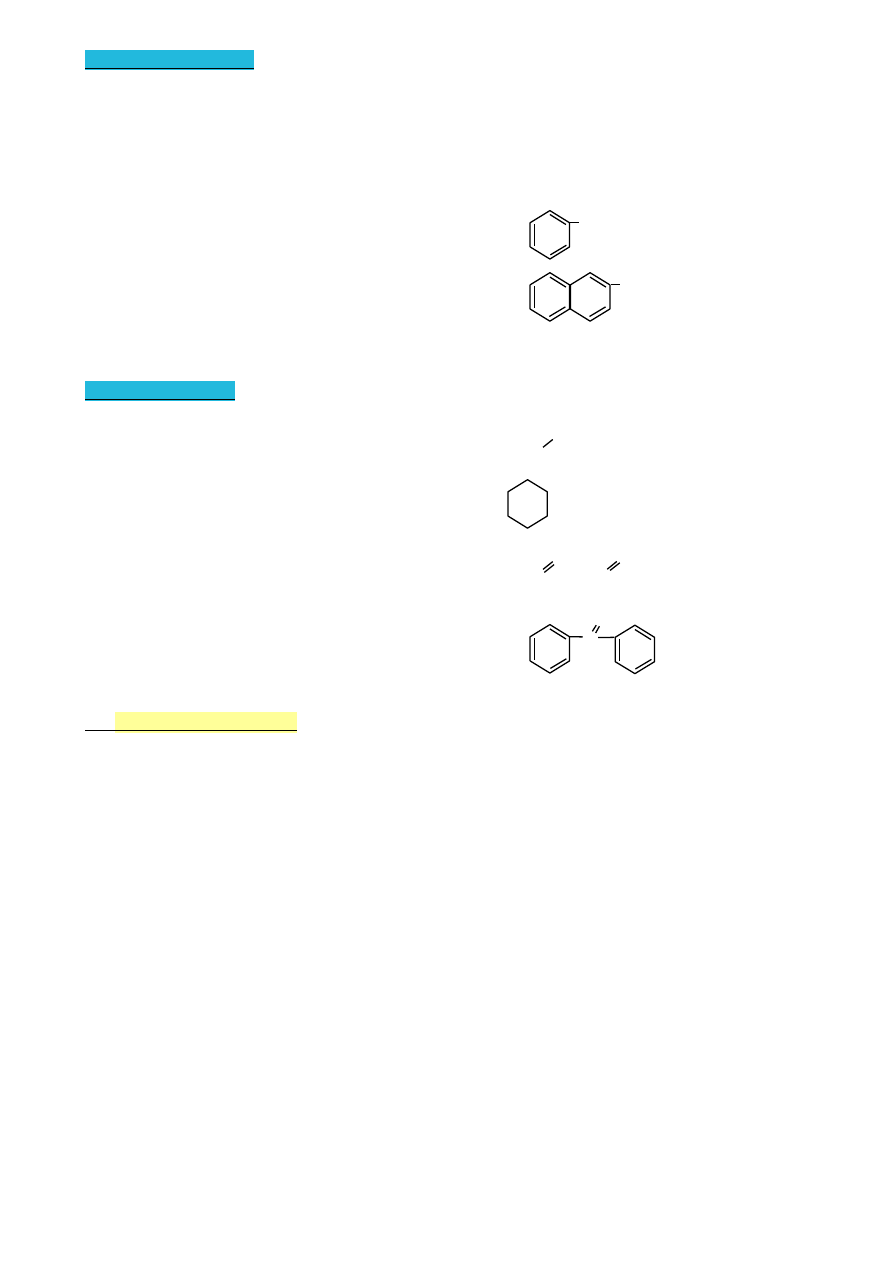
Przykłady aldehydów
metanal
(aldehyd mrówkowy)
HCHO
etanal
(aldehyd octowy)
CH
3
‒CHO
propanal
(aldehyd propionowy)
CH
3
‒CH
2
‒CHO
butanal
(aldehyd masłowy)
CH
3
‒CH
2
‒CH
2
‒CHO
pentanodial
CHO‒CH
2
‒CH
2
‒CH
2
‒CHO
benzaldehyd
(aldehyd benzoesowy)
CHO
aldehyd 1-naftoesowy
CHO
Przykłady ketonów
propanon
(k. dimetylowy, aceton)
CH
3
‒C=O
CH
3
cykloheksanon
=O
pentano-2,4-dion
(acetyloaceton)
CH
3
‒C‒CH
2
‒C‒CH
3
O O
O
benzofenon
(keton difenylowy)
C
10.
Właściwości fizyczne
•
•
większość aldehydów i ketonów alifatycznych to ciecze w warunkach normalnych;
wyjątkiem jest gazowy formaldehyd
•
•
temperatura wrzenia wyższa niż w alkanach o zbliżonej masie ale niższa niż
odpowiednich alkoholi. Mogą się tworzyć wiązania wodorowe
•
•
wiązania wodorowe między atomami tlenu grupy karbonylowej a atomami wodoru
wody powodują, że niższe aldehydy i ketony rozpuszczają się w wodzie
•
•
większość aldehydów i ketonów rozpuszcza się w polarnych rozpuszczalnikach
organicznych (alkohole i halogenki)
68
WYKŁAD 12 (07.06.2010)
1.
Nazewnictwo aldehydów i ketonów
•
•
aldehydy- końcówka -al
•
•
ketony- końcówka -on lub keton np. etylowo-metylowy – podstawniki wymieniamy
w kolejności alfabetycznej
•
•
aldehydy – numerujemy łańcuch tak, by pierwszy węgiel był przy grupie
aldehydowej
•
•
ketony – numerujemy tak, by węgiel z grupą karbonylową miał jak najniższy lokant
2.
Właściwości chemiczne
Związane z obecnością grupy karbonylowej :
sp
2
C=O wiązanie podwójne, słabe →
reakcje addycji
A
N
σ+
σ-
C = O atom węgla elektrofilowy →
reakcje z nukleofilami
addycja nukleofilowa do grup karbonylowej
Dwie możliwości w zależności od rodzaju nukleofila
σ-
O
O
-
OH
||
|
H
+
|
σ+
C
+ Nu
-
C
C
Nu
Nu
HCN (cyjanowodór), ROH (alkohole), RMgX (odczynniki Grignarda)
σ-
O
O
-
OH
Nu
||
|
H
+
- H
2
O
||
σ+
C
+
-
NuH
C
C
C
NuH
NuH
NH
3
(amoniak), RNH
2
(aminy), NH
2
OH (hydroksyloamina)
69
Przykłady A
N
do grupy karbonylowej
HCN
R
OH
C
cyjanohydryna
R'
CN
R”OH
R
OH
C
hemiacetal
O
R'
OR”
(hemiketal)
||
2R”OH
R
OR”
C
C
acetal (ketal)
R'
OR”
R
R'
NH
3
R
aldehyd
C = NH
imina
keton
R'
R”NH
2
R
C = NR”
alkiloimina
R'
NH
2
OH
R
C = NOH
oksym
R'
R”MgX
R
OH
C
alkohol
R'
R”
3.
Inne reakcje aldehydów i ketonów
A)
Utlenianie
R‒CHO
[O]
R‒COOH
[O] = KMnO
4
, Na
2
Cr
2
O
7
, [Ag(NH
3
)]
+
, O
2
aldehyd
kwas karboksylowy
Ketony nie ulegają utlenianiu
B)
Próba Tollensa (lustra srebrnego) - pozwala rozróżnić aldehyd od ketonów:
RCHO + [Ag(NH
3
)
2
]
+
RCOO
-
+ Ag
aldehyd bezbarwny roztwór
lustro srebrne
np.
CH
3
CHO + 2[Ag(NH
3
)
2
]
+
2Ag + CH
3
COO
-
+ 4NH
3
+ 2 H
2
O
aldehyd octowy
C)
Redukcja
R‒CHO
[H]
R‒CH
2
‒OH
[H] = NaBH
4
, LiAlH
4
, H
2
/kat (Ni, Pd, Pt)
O
O
R‒C‒R'
[H]
R‒CH‒R'
70

4.
Aldehydy i ketony podsumowanie
•
•
aldehydy i ketony to bardzo reaktywne związki. Najbardziej charakterystyczne dla
nich są reakcje addycji nukleofilowej do grupy karbonylowej (A
N
)
•
•
Aldehydy ulegają utlenianiu do kwasów karboksylowych, ketony nie ulegają
utlenianiu
•
•
Aldehydy i ketony ulegają redukcji do alkoholi
•
•
Szczególne właściwości związków karbonylowych zawierających α atomy wodoru
KWASY KARBOKSYLOWE
5.
Kwasy karboksylowe
•
•
Wzory ogólne
O
O
O
R‒C‒OH
Ar‒C‒OH
H‒C‒OH
szczególny przypadek
•
•
nazewnictwo
W kwasach karboksylowych ( podobnie jak w aldehydach ) atom węgla
grupy karbonylowej zawsze oznaczamy numerem 1
Przykłady kwasów
HCOOH
kwas metanowy
(mrówkowy)
CH
3
COOH
kwas etanowy
(octowy)
CH
3
CH
2
COOH
kwas propanowy
(propionowy)
CH
3
CH
2
CH
2
COOH
kwas butanowy
(masłowy)
CH
2
COOH
kwas fenylooctowy
COOH
kwas benzoesowy
COOH
kwas 1-naftoesowy
6.
Właściwości
Fizyczne:
•
•
na te właściwości wpływają wiązania wodorowe
•
•
wiązania wodorowe tworzą się między cząsteczkami kwasu → stosunkowo
wysokie temperatury wrzenia kwasów
•
•
wiązania wodorowe między cząsteczkami kwasu i wody: dobra
rozpuszczalność w wodzie niższych kwasów karboksylowych
71

Chemiczne
•
•
reakcje grupy karboksylowej (rozerwanie wiązania O-H, wiązania C-O)
•
•
reakcje zachodzące z udziałem atomów wodoru związanych z atomem
węgla α względem grupy karboksylowej, tj. Atomem węgla sąsiadującym z
grupą karboksylową – atomów wodoru α
7.
Reakcje grupy karboksylowej
A)
Dysocjacja grupy karboksylowej (rozpad wiązania O-H)
Kwasy karboksylowe – kwasy mocniejsze od fenoli, alkoholi, wody, ale słabsze od
kwasów nieorganicznych. W wodzie dysocjują:
R–COOH + H
2
O ↔ R– COO
-
+ H
3
O
+
anion karboksylanowy
Anion karboksylanowy stabilny dzięki rozproszeniu ładunku między obydwa atomy tlenu
B)
Kwasy karboksylowe reagują z metalami, wodorotlenkami, i tlenkami metali oraz z
aminokwasem tworząc sole:
2 RCOOH + Zn → (RCOO
-
)
2
Zn
2+
+ H
2
RCOOH + NaOH → RCOO
-
Na
+
+ H
2
RCOOH + NH
3
→ RCOO
-
NH
4
+
Przykłady
HCOOH + NaOH → HCOONa + H
2
O
kwas mrówkowy
mrówczan sodu
CH
3
COOH + NH
3
→ CH
3
COONH
4
+ H
2
O
kwas octowy
octan amonu
COOH + NaOH →
COONa + H
2
O
kwas benzoesowy
benzoesan sodu
Z soli pod działaniem mocniejszego kwasu powstają kwasy organiczne
RCOO
-
Na
+
+ HCl → RCOOH + NaCl
Przykłady
COONa + HCl →
COOH + NaCl
benzoesan sodu
kwas benzoesowy
CH
3
COONH
4
+ HCl → CH
3
COOH + NH
4
Cl
octan amonu
kwas octowy
72
C)
Podstawienie grupy hydrofilowej (rozpad wiązania C-O)
Pochodne kwasów karboksylowych (poza nitrylami) zawierają grupę acylową a po
hydrolizie tworzą kwasy.
O
O
O
R—C–X
R—C–OR'
R—C–NH
2
R—C≡N
halogenek
ester
amid
nitryl
kwasowy
O O
O
O
|| ||
||
||
R–C–NH–C–R
R–C—O—C–R
imid
bezwodnik
kwasowy
Otrzymywanie chlorków kwasowych
O
O
||
||
R–C–OH
+
SOCl
2
→ R–C–Cl + SO
2
+ HCl
chlorek tionylu
Otrzymywanie estrów
O
O
||
||
R–C–OH
+
R'OH → R–C–OR' + H
2
O
kwas karboksylowy
alkohol
ester
Otrzymywanie amidów
O
O
O
||
||
||
R–C–OH + NH
3
→ R–C–O
-
NH
4
+
P
4
O
10
R–C–NH
2
+ H
2
O
sól amonowa
amid
kwasu
Przykład podstawienia na acylowym atomie węgla
O
O
||
||
CH
3
CCl + (CH
3
)
3
COH
pirymidyna
CH
3
CO(CH
3
)
3
+ HCl
chlorek
tert-butanol
octan tert-butylu
Reakcje kwasów z udziałem α atomów H
Halogenowanie kwasów karboksylowych
α
R–CH
2
–COOH
PX
3
R–CH–COOH
|
X
73

Kwasy zawierające atom halogenu – mocniejsze niż niepodstawione:
X—CH
2
–COOH
wodór słabiej związany
X—CH
2
–COO
-
stabilizacja anionu karbosylanowego
Moc kwasów wzrasta wraz z ilością podstawianych halogenków
8.
Kwasy karboksylowe – podsumowanie
•
•
Kwasy karboksylowe są słabymi kwasami, ale mocniejszymi od fenoli, alkoholi,
wody.
•
•
Moc kwasów karboksylowych zwiększa się, gdy przy atomie węgla α względem
grupy karboksylowej znajdują się podstawniki elektronoakceptorowe
•
•
Kwasy karboksylowe tworzą szereg pochodnych. Zalicza się do nich: chlorki
kwasowe, estry, amidy, bezwodniki, estry i nitryle.
AMINY
9.
Aminy
Organiczne pochodne amoniaku, w których jeden, dwa lub trzy atomy wodoru zastąpiono
grupami alifatycznymi lub aromatycznymi.
NH
3
amoniak
R–NH
2
amina I rzędowa
R–NH–R'
amina II rzędowa
R–N–R'
amina III rzędowa
|
R”
R'
|
czwartorzędowa sól amoniowa
R–N
+
–R'' X
-
|
R'''
10.
Nazwy i przykłady amin
CH
3
NH
2
metyloamina
C
5
H
11
NH
2
pentyloamina
CH
3
CHC
3
H
7
2-pentyloamina
|
NH
2
•
•
Aminy alifatyczne drugo- i trzecio- rzędowe
:
N – podstawione pochodne amin pierwszorzędowych
CH
3
NHC
2
H
5
N-metyoetyloamina
74
•
•
aminy symetryczne
– przedrostki di- tri- ...
•
•
aminy aromatyczne
– nazwy tworzone jak pochodne aniliny
NH
2
CH
3
2,4-dimetytyloanilina
CH
3
11.
Właściwości amin
•
•
Obecność wolnej pary elektronowej na atomie azotu wpływa w decydujący sposób
na właściwości amin
•
•
Większość amin to ciecze o nieprzyjemnym zapachu
•
•
Aminy I i II rz. Mogą tworzyć wiązania wodorowe, które są słabsze niż w alkoholach
– temperatura wrzenia amin niższe niż alkoholi
•
•
Dzięki wiązaniom wodorowym z cząsteczkami wody niższe aminy ( do 6. at. C )
rozpuszczają się w wodzie
Wł. chemiczne:
•
•
Ze względu na obecność wolnej pary elektronowej na atomie azotu aminy mają
właściwości zasadowe ( zasady Lewisa )
•
•
Aminy to zasady mocniejsze niż woda:
R–NH
2
+ H
2
O ↔ R– NH
3
+
+ OH
-
kation amoniowy
•
•
Z kwasami aminy tworzą sole amoniowe:
RNH
2
+ HCl ↔ RNH
3
+
Cl
-
RNHR' + HCl ↔ RNH
2
R'
+
Cl
-
•
•
Aminę można otrzymać z soli działając mocniejszą zasadą, np.:
RNH
3
+
Cl
-
RNH R' + NaOH ↔ RNH
2
+ NaCl + H
2
O
Przykładowe sole amin i ich nazwy
CH
3
NH
2
+ HCl
↔
CH
3
NH
3
Cl
metyloamina
chlorek metyloamoniowy
NH
2
+ H
2
SO
4
↔
NH
3
OSO
3
H
anilina
wodorosiarczan fenyloamoniowy/ anilinowy
NHCH
3
+ CH
3
COOH ↔
NH
2
‒OCCH
3
| ||
CH
3
O
75

WYKŁAD 13 (14.06.2010)
BIOCZĄSTECZKI
1.
Lipidy
LIPIDY
tłuszcze
steroidy
woski
prostaglandyny
fosfolipidy
terpentoidy
↓
↓
estry
nie są estrami
(ulegają hydrolizy)
(nie ulegają hydrolizie)
2.
Tłuszcze (glicerydy)
Źródło energii w organizmach żywych roślinnych i zwierzęcych gromadzone jako rezerwa
pokarmowa.
Budowa chemiczna
Estry różnych kwasów karboksylowych i glicerolu:
O
O
CH
2
‒OH
CH
2
‒O‒C‒R
CH
2
‒O‒C‒R
1
|
|
O
|
O
CH ‒OH
CH ‒O‒C‒R
CH ‒O‒C‒R
2
|
|
O
|
O
CH
2
‒OH
CH
2
‒O‒C‒R
CH
2
‒O‒C‒R
3
glicerol
Tłuszcze to triestry ( triglicerydy)
Kwasy tłuszczowe mają proste łańcuchy węglowodorowe ( nasycone lub nienasycone) i
parzystą liczbę atomów węgla w cząsteczkach (głównie C
12-20
)
Podstawowe kwasy tłuszczowe
•
•
nasycone
◦
◦
CH
3
(CH
2
)
10
COOH (C
11
H
23
COOH)
– kwas dodekanowy /laurynowy/
◦
◦
CH
3
(CH
2
)
14
COOH (C
15
H
31
COOH) – kwas heksadekanowy /palmitynowy/
◦
◦
CH
3
(CH
2
)
16
COOH (C
17
H
35
COOH) – kwas oktadekanowy /stearynowy/
76

•
•
nienasycone (zawsze izomery Z)
◦
◦
CH
3
(CH
2
)
7
(CH
2
)
7
COOH
C=C
– kwas oleinowy (C
17
H
33
COOH)
H
H
◦
◦
CH
3
(CH
2
)
4
CH
2
(CH
2
)
7
COOH
– kwas linolowy (C
17
H
31
COOH)
C=C C=C
H H H H
◦
◦
CH
3
CH
2
CH
2
CH
2
(CH
2
)
7
COOH
C=C C=C C=C
– kwas linolenowy(C
17
H
29
COOH)
H H H H H H
Jeżeli występują wiązania podwójne, to cząsteczka się zagina na tym wiązaniu, co
powoduje zmniejszenie oddziaływań między łańcuchami kwasów tłuszczowych w
cząsteczce tłuszczu. Objawia się to stanem ciekłym w temperaturze pokojowej i niskimi
temperaturami topnienia.
3.
Reakcje tłuszczów
A) Uwodornianie tłuszczów nienasyconych – utwardzanie
O
O
CH
2
‒O‒C‒C
17
H
33
CH
2
‒O‒C‒C
17
H
35
|
O
|
O
CH
‒O‒C‒C
17
H
33
3H
2
, kat. Ni
CH
‒O‒C‒C
17
H
35
|
O
|
O
CH
2
‒O‒C‒C
17
H
33
CH
2
‒O‒C‒C
17
H
35
trioleinian glicerolu
B) Zmydlanie
O
O
CH
2
‒O‒C‒R
1
CH
2
‒OH
KO‒C‒R
1
|
O
|
O
CH
‒O‒C‒R
2
+ KOH →
CH
2
‒OH +
KO‒C‒R
1
|
O
|
O
CH
2
‒O‒C‒R
3
CH
2
‒OH
KO‒C‒R
1
Mydła sodowe – stałe
Mydła potasowe – ciekłe
Mydła – najstarsze detergenty
Detergenty- środki powierzchniowo czynne
COO
-
Na
+
gr. lipofilowa (hydrofobowa)
gr. lipofobowa (hydrofilowa)
FAZA ORGANICZNA
FAZA WODNA
77

Detergenty zmniejszają napięcie powierzchniowe wody, wzrasta wtedy zdolność
tworzenia piany przez roztwór i wzrasta zwilżalność powierzchni. Wadą mydeł jest to, że
sole wapnia i magnezu kwasów tłuszczowych są nierozpuszczalne w wodzie. W trwałej
wodzie tworzą się osady.
2 C
17
H
35
COONa + Mg
2+
→ (C
17
H
35
COO)
2
Mg↓ + 2Na
+
2 C
17
H
35
COONa + Ca
2+
→ (C
17
H
35
COO)
2
Ca↓ + 2Na
+
SACHARYDY
4.
Sacharydy
Materiał zapasowy i źródło energii ( rośliny- skrobia, zwierzęta-glikogen). Są to związki
polifunkcyjne: zawierają 2 grupy funkcyjne:
-OH i -CHO
-OH i -C=O
hydroksyaldehydy
hydroksyketony
ALDOZY
KETOZY
Większość sacharydów ma wzór ogólny (CH
2
O)
2
, dlatego zostały nazwane
węglowodanami (XIX wiek).
SACHARYDY
MONOSACHARYDY DISACHARYDY
OLIGOSACHARYDY
POLISACHARYDY
(cukry proste)
2 cząstki cukrów
3 – kilkunastu cząsteczek
kilkanaście do
prostych
cukrów prostych
kilkuset
monosacharydów
Monosacharydy – cząsteczki, które w wyniku hydrolizy nie tworzą cząsteczek prostych
cukrów ( w przeciwieństwie do innych)
W zależności od liczby atomów węgla w cząsteczce monosacharydy dzielą si na:
–
–
triozy – 3 atomy C
–
–
tetrozy – 4 atomy C
–
–
pentozy – 5 atomów C
–
–
heksozy – 6 atomów C
Aldehyd glicerynowy – najważniejsza trioza. Stanowi wzorzec do określania
konfiguracji względem asymetrycznego atomu węgla ( D, L )
O
C‒H
H‒
*
C‒OH
CH
2
OH
aldehyd glicerynowy
aldotrioza
78

Heksozy (wszystkie w konfiguracji D)
D-glukoza
D-galaktoza
D-fruktoza
(A-H)
(A-H)
(K-H)
O
O
C‒H
C‒H
CH
2
OH
|
|
|
H‒
*
C‒OH
H‒
*
C‒OH
C=O
|
|
|
HO‒
*
C‒H
HO‒
*
C‒H
HO‒
*
C‒H
|
|
|
H‒
*
C‒OH
HO‒
*
C‒H
H‒
*
C‒OH
|
|
|
H‒
*
C‒OH
H‒
*
C‒OH
H‒
*
C‒OH
|
|
|
CH
2
OH
CH
2
OH
CH
2
OH
* A-H aldoheksoza
A-K ketoheksoza
Glukoza – powstaje w procesie fotosyntezy
6CO
2
+ 6H
2
O
hν , chlorofil
C
6
H
12
O
6
+ 6O
2
5.
Wzory łańcuchowe
O
C‒H
CHO
CHO
CHO
|
H‒*C‒OH
H OH
OH
|
HO‒*C‒H
HO
H
HO
|
→
H‒*C‒OH
H OH
OH
|
H‒*C‒OH
H OH
OH
|
CH
2
OH
CH
2
OH
CH
2
OH
CH
2
OH
projekcyjne
U sacharydów możliwa jest wewnątrzcząsteczkowa reakcja addycji grupy ‒OH do grupy
karbonylowej (A
N
)
O
R
OH
||
R”OH
C
hemiacetal
C
R'
OR”
hemiketal
R
R'
R
OR”
2R”OH
C
acetal
R'
OR”
ketal
79
6.
Struktury pierścieniowe monosacharydów- projekcje Hawortha
Glukoza
7
CH
2
OH
O
H
6
O
H
1
C‒H
H
2
|
4
OH
H
1
H‒
*
C‒OH
OH
3
2
OH
3
|
HO‒
*
C‒H
H
OH
4
|
α-D-glukopiranoza
H‒
*
C‒OH
powstaje acetalowy (hemi-
5
|
acetalowy) atom węga
H‒
*
C‒OH
(asymetryczny!)
6
|
CH
2
OH
6
CH
2
OH
D-glukoza
H
5
O
OH
H
4
OH
H
1
O
OH
3
2
H
H
OH
β-D-glukopiranoza
piran
O pozycji α i β decyduje położenie -OH
Fruktoza
H
CH
2
OH
H
O
CH
2
OH
|
H
C=O
H
OH
|
OH
OH
HO‒C‒H
OH
H
|
H‒C‒OH
α-D-fruktopiranoza
|
H‒C‒OH
|
H
C
2
HOH
H
O
OH
H
H
OH
OH
CH
2
OH
OH
H
β-D-fruktopiranoza
80
CH
2
OH
|
C=O
HOCH
2
O
CH
2
OH
|
OH‒C‒H
|
H H
OH
OH
H‒C‒OH
|
H‒C‒OH
OH
H
|
CH
2
‒OH
α-D-fruktofuranoza
O
furan
7.
Reakcje monosacharydów
Tworzenie glikozydów – reakcja monosacharydów z alkoholem. Są to acetale (lub
hemiacetale) Część pierścienia nazywamy glikonem, a grupę OR
– aglikonem. Związki te mają duże znaczenie biologiczne: fenole
są usuwane z organizmu w postaci glikozydów.
CH
2
OH
CH
2
OH
CH
2
OH
H
O
H
H
O
H
H
O
OCH
3
OH H H
CH
3
OH / H +
OHH H
+
OH H H
OH
OH
OH
O‒CH
3
OH
H
H OH
H OH
H OH
α-D-glukopiramidozyd metylowy
β-D-glukopiramidozyd metylowy
8.
Disacharydy
Rolę alkoholu pełni cząstka drugiego cukru
Sacharoza – owoce, trzcina cukrowa
Laktoza
– mleko
Maltoza
– hydroliza skrobi
Celobioza
– hydroliza celulozy
Hydroliza disacharydów prowadzi do wytworzenia cukrów prostych z których powstały.
Przykłady oligo- i polisacharydów
Cyklodekstryny – oligosacharydy składające się z 6-8 cząsteczek glukozy otrzymuje się
je w wyniku hydrolizy skrobi.
Polisacharydy:
skrobia
glikogen
chityna
81
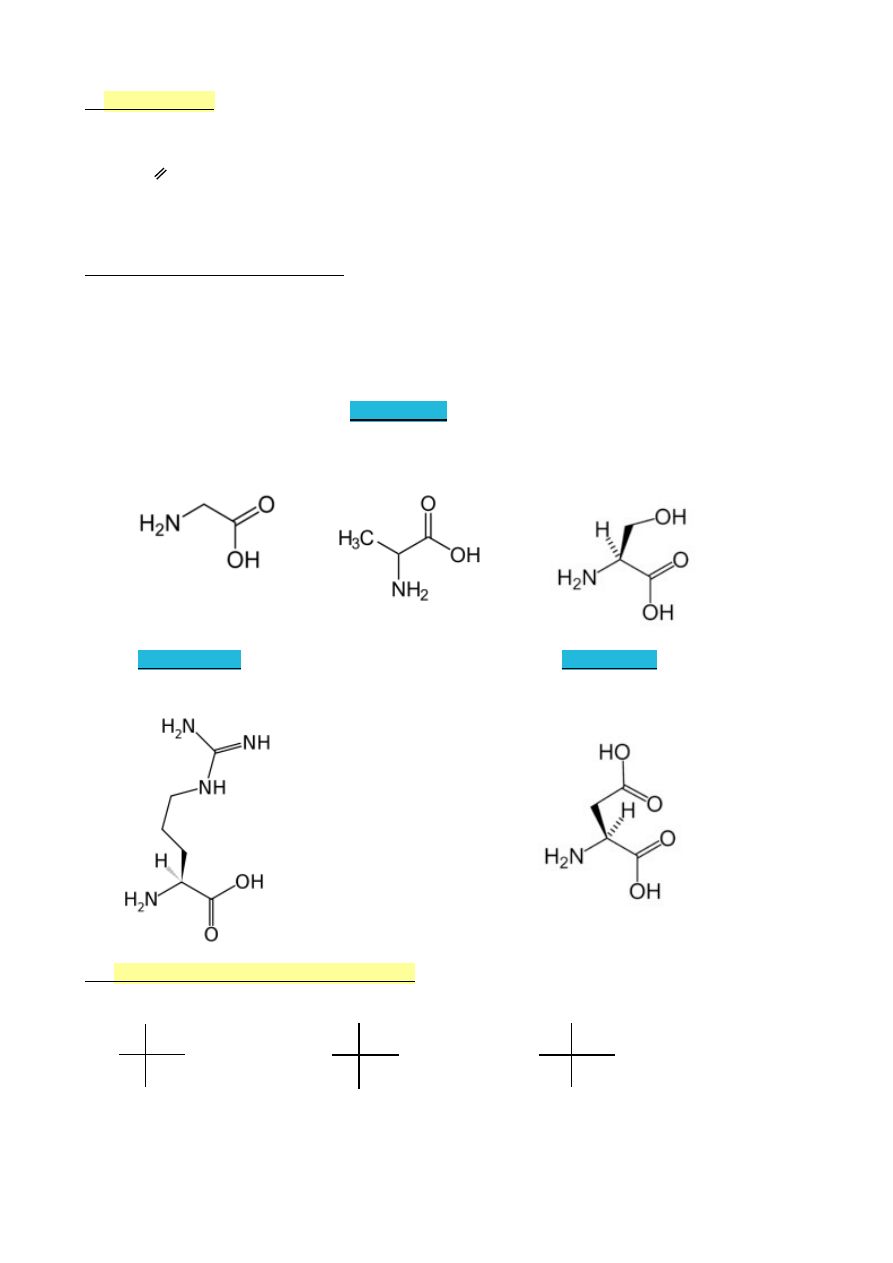
AMINOKWASY
9.
Aminokwasy
Związki zawierające w cząsteczkach grupy funkcyjne : -NH
2
i -COOH
O
R‒CH‒C‒OH
Aminokwasy wchodzące w skład białek (aminokwasy białkowe)
|
są w pozycji α
NH
2
Aminokwasy białkowe mogą być:
Endogenne – wytwarzane przez organizm
Egzogenne – muszą być dostarczane z pokarmem
Aminokwasy podstawowe (20) – egzogenne aminokwasy, niezbędne do życia m.in.
OBOJĘTNE
Glicyna (Gly, G)
Alanina (Ala, A)
Seryna (Ser, S)
ZASADOWE
KWASOWE
Arginina (Arg, R)
kwas asparginowy (Asp, D)
10.
Aminokwasy- izomeria (izomery L)
COOH
COOH
CHO
NH
2
H
NH
2
H
OH
H
CH
3
CH
2
OH
CH
2
OH
L- alanina
L-seryna
ald. L-glicerynowy
82

11.
Budowa aminokwasów a właściwości fizyczne
Grupa aminowa – zasadowa, a karboksylowa – kwasowa. Może zajść reakcja
wewnątrzcząsteczkowa:
O
O
R–CH–C–OH
↔
R–CH–C–O
-
|
|
NH
2
NH
3
+
sól wewnętrzna,
jon obojnaczy, zwitterjon
Aminokwasy jak sole – są substancjami krystalicznymi o wysokich temperaturach
topnienia, rozpuszczalne w wodzie, nierozpuszczalne w węglowodorach.
12.
Właściwości chemiczne
•
•
Aminokwasy- związki amfoteryczne: reagują z kwasami i zasadami
•
•
Cząsteczki kwasów mogą łączyć się ze sobą tworząc peptydy. Mogą być dipeptydy
, tripeptydy, oligoeptydy, polipeptydy. Wysokie polipeptydy to białka.
A)
Reakcja w środowisku kwaśnym
O
O
R–CH–C–O
-
+ H
3
O
+
↔
R–CH–C–OH + H
2
O
|
|
NH
3
+
NH
3
+
jon hydroniowy
kation amonowy
B)
Reakcje w środowisku zasadowym
O
O
R–CH–C–O
-
+ OH
-
↔
R–CH–C–O
-
+ H
2
O
|
|
NH
3
+
NH
2
jon hydroksylowy
anion karboksylowy
C)
Reakcje z kwasami i zasadami nieorganicznymi
O
O
R–CH–C–OH + HCl
↔
R–CH–C–OH + H
2
O
|
|
NH
2
NH
3
Cl
O
O
R–CH–C–OH + NaOH ↔
R–CH–C–ONa + H
2
O
|
|
NH
2
NH
2
83
ZADANIE
Rozpisać powyższe reakcje dla alaniny
13.
Punkt izoelektryczny
pH roztworu, przy którym w roztworze aminokwas istnieje jako jon obojnaczy
O
O
O
R–CH–C–O
-
OH-
R–CH–C–O
-
H+
R–CH–C–OH
|
|
|
NH
2
NH
3
+
NH
3
+
W punkcie izoelektrycznym roztwór aminokwasu, nie przewodzi prądu
elektrycznego. Każdy aminokwas charakteryzuje się określony wartością punktu
izoelektrycznego.
14.
Tworzenie peptydów
Amidy
O
O
O
R–C–OH + NH
3
→ R–C–O
-
NH
4
+
P
4
O
10
R–C–NH
2
+ H
2
O
sól amonowa kwasu
amid
R–C–OH + RNH
2
→ R–C–NHR + H
2
O
alkiloamid
A)
Tworzenie dipeptydów z dwóch cząsteczek tego samego aminokwasu:
O
O
NH
2
–CH
2
–C–OH
+ NH
2
–CH
2
–C–OH
O
||
O
NH
2
–CH
2
–C–N–CH
2
–C–OH
+ H
2
O
|
H
wiązanie peptydowe
B)
Jeżeli reagują aminokwasy A i B to istnieją 2 możliwości:
•
•
reakcja grupy aminowej aminokwasu A z grupą karbonylową aminokwasu B
•
•
reakcja grupy aminowej aminokwasu B z grupą karbonylową aminokwasu A
Powstają 2 różne izomeryczne względem siebie peptydy
O
O
O
O
NH
2
–CH
2
–C–N–CH–C–OH
NH
2
–CH–C–N–CH
2
–C–OH
H CH
3
CH
3
H
84

Tripeptydy: Produkty kondensacji aminokwasów. W przypadku 3 różnych aminokwasów
możliwych jest 6 różnych tripeptydów. Wykazują one odrębne właściwości
fizyczne i chemiczne.
PEPTYDY
oligo-
poli-
białka
(2-10 aminokwasów)
(11-100 aminokwasów)
(kilkaset- kilka tysięcy
aminokwasów)
Oddziaływania między łańcuchami aminokwasów:
•
•
mostki siarczkowe
•
•
oddziaływania wodorowe
•
•
oddziaływania jonowe
•
•
oddziaływania hydrofobowe
15.
Struktury białek
–
–
struktura I rzędowa - sekwencja i rodzaj aminokwasów w łańcuchu
polipeptydów
–
–
struktura II rzędowa – konfiguracja łańcucha peptydowego
(α-helisa i β-harmonijka)
–
–
struktura III rzędowa
–
–
struktura IV rzędowa
Denaturacja białek- degradacja w II, III- i IV-rzędowej strukturze białka, które prowadzą
do utraty aktywności biologicznej lub innej indywidualnej cechy charakterystycznej przy
zachowaniu jego struktury pierwszorzędowej .
85
Wyszukiwarka
Podobne podstrony:
chemia organiczna wykład 6
dyd k3a r, chemia, 0, httpzcho.ch.pw.edu.pldydaktyk.html, Technologia Chemiczna, Chemia Organiczna -
Instrukcja do prób barwnych I (alkohole, Studia, Biotechnologia, Chemia, Chemia organiczna, Wykłady
Chemia organiczna wykłady
dyd e2b, chemia, 0, httpzcho.ch.pw.edu.pldydaktyk.html, Technologia Chemiczna, Chemia Organiczna - w
Ćwiczenia – węglowodory alifatyczne, Studia, Biotechnologia, Chemia, Chemia organiczna, Wykłady II
Chemia organiczna wykład 8
UZ1 - chemia - wyklady-1, II semestr, Chemia budowlana, Wykłady, Całość
dyd k2a r, chemia, 0, httpzcho.ch.pw.edu.pldydaktyk.html, Technologia Chemiczna, Chemia Organiczna -
CHEMIA ORGANICZNA REAKCJE sciaga 111, Technologia chemiczna, 3 semestr, Chemia organiczna, wykłady
Instrukcja do zmydlania tłuszczów, Studia, Biotechnologia, Chemia, Chemia organiczna, Wykłady II
dyd kzb, chemia, 0, httpzcho.ch.pw.edu.pldydaktyk.html, Technologia Chemiczna, Chemia Organiczna - w
dyd kza r, chemia, 0, httpzcho.ch.pw.edu.pldydaktyk.html, Technologia Chemiczna, Chemia Organiczna -
dyd e1c, chemia, 0, httpzcho.ch.pw.edu.pldydaktyk.html, Technologia Chemiczna, Chemia Organiczna - w
Chemia organiczna wykład 14
więcej podobnych podstron