
Systemy diagnostyki
laboratoryjnej
Tekst z wykładów i nie tylko
by
MC_OMEN

Woltamperometria - definicja
Woltamperometria – technika analityczna, której podstawą jest pomiar zależności natężenie prądu – potencjał
elektryczny w układzie elektrody pracującej i odniesienia zanurzonych w roztworze badanym zawierającym oznaczaną
substancję i elektrolit podstawowy. Elektroda porównawcza (odniesienia) jest niepolaryzowana (np. elektroda
kalomelowa), natomiast elektroda pracująca jest polaryzowaną obojętną.
Woltamperometria to badania procesów elektrodowych polegające na wymuszaniu pożądanego kierunku przebiegu
procesu elektrodowego poprzez polaryzację elektrody wskaźnikowej zewnętrznym potencjałem. Pomiar jest realizowany
w warunkach dynamicznych, mierzonym sygnałem jest natężenie prądu rejestrowane w funkcji zmian napięcia
polaryzującego elektrodę (woltamperometria, polarografia) lub w funkcji czasu bądź zmiany składu próbki, dla E=const,
(chronoamperometria, amperometria).
Warunki
• procesy będące podstawą pomiaru przebiegają na elektrodzie pracującej, która ma niewielką powierzchnię i jest
elektrodą polaryzowalną. Niewielka powierzchnia zapewnia, że mierzone prądy są małe a ilośd analitu ulegającego
redukcji jest tak mała, że nawet wielokrotne powtarzanie pomiaru nie powoduje zauważalnej zmiany jego stężenia.
• druga elektroda w układzie to odwracalna i niepolaryzowalna elektroda odniesienia o powierzchni na tyle dużej by
przepływający przez nią prąd nie powodował zmiany jej pote ncjału. Jeżeli nie jest to możliwe do spełnienia stosowany
jest dodatkowy układ elektroniczny – potencjostat i trzecia elektroda pomocnicza przez którą przepływa prąd.
• potencjał elektrody pracującej jest zmieniany w trakcie pomiaru zgodnie z programem wy nikającym ze stosowanej
techniki a mierzony prąd jest rejestrowany w postaci zależności od przyłożonego napięcia tworząc krzywą o kształcie fali
lub piku nazywaną woltamogramem lub polarogramem.
• w roztworze badanym oprócz substancji oznaczanej – depolaryzatora obecny jest celowo dodany elektrolit
podstawowy zapewniający przewodnictwo roztworu.
• jeżeli elektroda pracująca polaryzowana jest napięciem ujemnym (na elektrodzie zachodzi proces redukcji) z roztworu
musi byd usunięty tlen ponieważ jego obecnośd zakłóca pomiar.
Elektrody
Elektroda pracująca, na której przebiegają procesy będące podstawą pomiaru, jest elektrodą polaryzowalną. Oznacza to,
że w nieobecności depolaryzatora przyjmuje potencjał zewnętrznego źródła napięcia. W zależności od znaku
przyłożonego napięcia może byd katodą (potencjał ujemny) lub anodą (potencjał dodatni). Najbardziej zbliżona do
elektrody idealnie polaryzowalnej jest elektroda rtęciowa. Wynika to z prawie idealnie gładkiej i czystej powierzchni
rtęci, która jest w temperaturze pokojowej cieczą. Zakres polaryzacji elektrody ograniczony jest od strony potencjałów
ujemnych rozkładem elektrolitu podstawowego (wydzielaniem wodoru) a od strony potencjałów dodatnich
elektrochemicznym rozpuszczaniem materiału elektrody.
Elektroda odniesienia powinna byd niepolaryzowalna tzn. jej potencjał nie powinien ulegad zmianie przy przepływie
prądu. W woltamperometrii wykorzystywane są głównie trzy elektrody odniesienia - chlorosrebrowa, kalomelowa i
siarczanowa.
Elektroda pomocnicza przyjmuje prąd płynący przez elektrodę pracującą. Wykonywana jest zwykle z metalu
szlachetnego (najczęściej platyny) lub węgla szklistego. Na elektrodzie pomocniczej przebiega także reakcja
elektrochemiczna, a jej produkty mogą zanieczyścid roztwór badany.
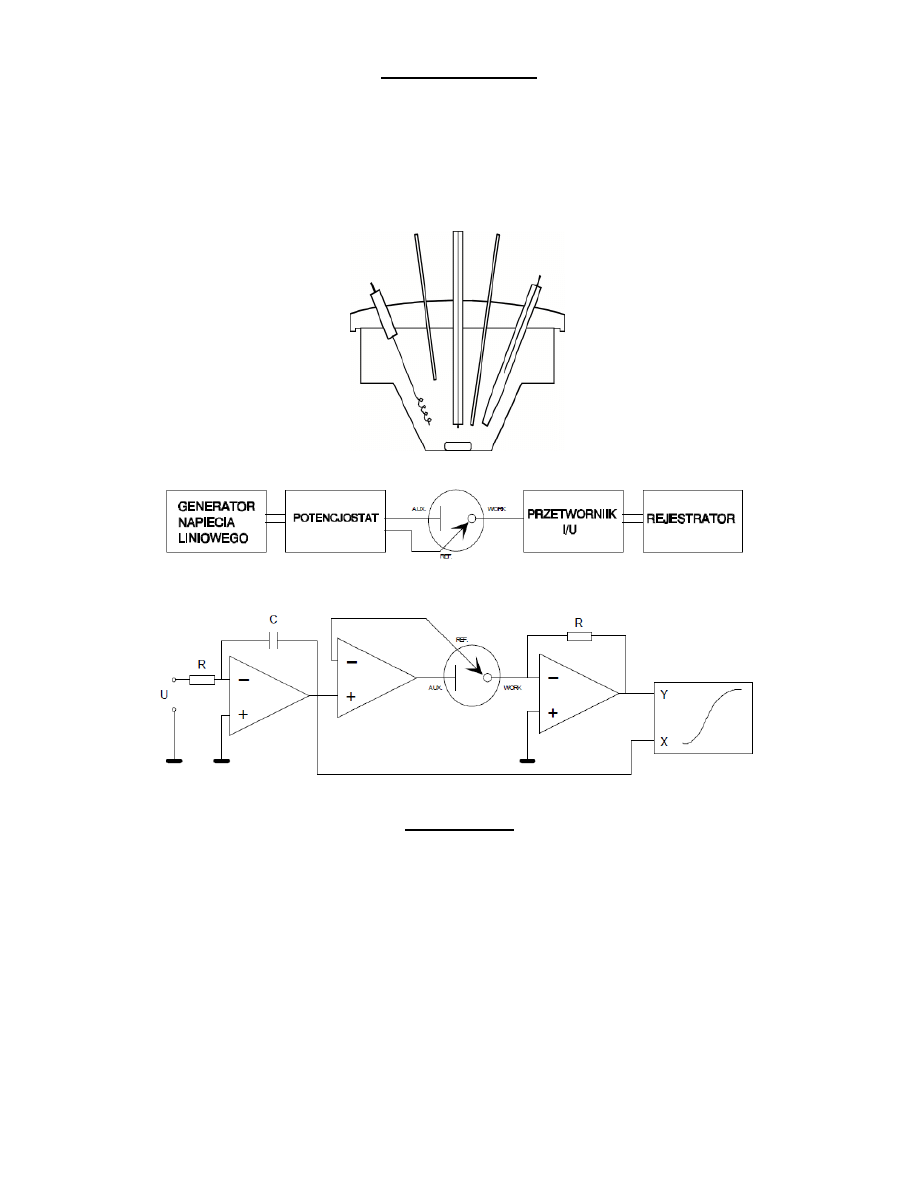
Układ pomiarowy
3- elektrody - pracująca (wskaźnikowa), odniesienia, pomocnicza
Mieszadło - najczęściej magnetyczne. Wykorzystywane jest do intensyfikacji transportu depolaryzatora do
elektrody przy zatężaniu w woltamperometrii stripingowej oraz do szybkiego wymieszania roztworu po dodatku
wzorca.
Doprowadzenia gazu obojętnego - doprowadzają gaz służący do usuwania tlenu (Ar, N2).
Polarografia
Polarografia – częśd woltamperometrii, elektrochemiczna metoda analityczna polegająca na przyłożeniu liniowo
wzrastającego potencjału elektrycznego do kroplowej elektrody rtęciowej będącej elektrodą pracującą z cyklicznie
odnawianą się w trakcie pomiaru powierzchnią i rejestracji natęże nia prądu płynącego przez nią. Wartośd natężenia
prądu jest proporcjonalna do stężenia obecnej w roztworze substancji ulegającej utlenieniu lub redukcji. Krzywa
zależności natężenia prądu od liniowo rosnącego potencjału, rejestrowana za pomocą aparatu zwan ego polarografem, w
postaci tzw. krzywej polarograficznej, pozwala zidentyfikowad substancję badaną i określid jej stężenie.
Rtęd jako materiał elektrodowy pomimo licznych wad ma również szereg niepodważalnych zalet, takich jak np. rtęd jest
metalem szlachetnym i zachowuje się obojętnie względem większości elektrolitów, wykazuje wysokie nadnapięcie
wydzielania wodoru, tworzy amalgamaty z większością metali a wytwarzaną elektrodę cechuje wyjątkowa gładkośd i
dobre zdefiniowanie powierzchni. Rtęd jest chętnie wykorzystywana do budowy różnego typu elektrod stacjonarnych.
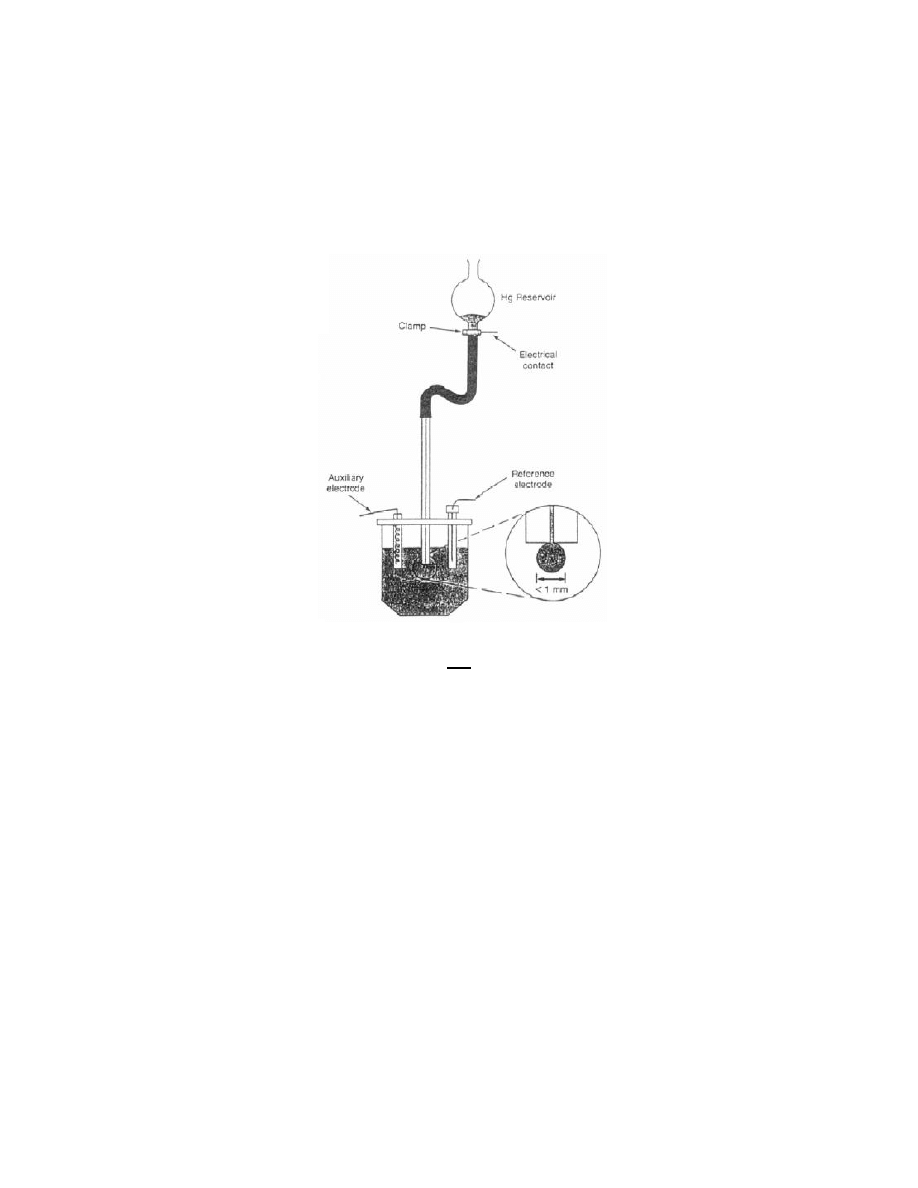
Wspólną cechą charakterystyczną dla wszystkich rodzajów elektrod kroplowych jest bardzo niski stosunek powierzchni
rtęci do jej całkowitej objętości i łatwośd obrywania się kropli. Tych wad pozbawione są tzw. cienkowarstwowe
(błonkowe) elektrody rtęciowe BER. Pojęcie cienkowarstwowa elektroda rtęciowa obejmuje swoim znaczeniem
wszystkie znane i stosowane rodzaje elektrod rtęciowych, które są wytwarzane w wyniku mechanicznego bądź
elektrolitycznego nakładania warstewki rtęci, o grubości od kilkudziesięciu nanometrów do kilku mikrometrów, na stałe,
metaliczne (Pt, Au, Ag, Ir), amalgamatowe lub węglowe (Glassy Carbon, GC) podłoże, dowolnego kształtu i powierzchni.
Zaletą elektrod błonkowych jest korzystny stosunek powierzchni elektrody do objętości Hg, istotny na etapie zatężania i
późniejszego utleniania nagromadzonych amalgamatów, wadą zła odtwarzalnośd powierzchni.
Hg
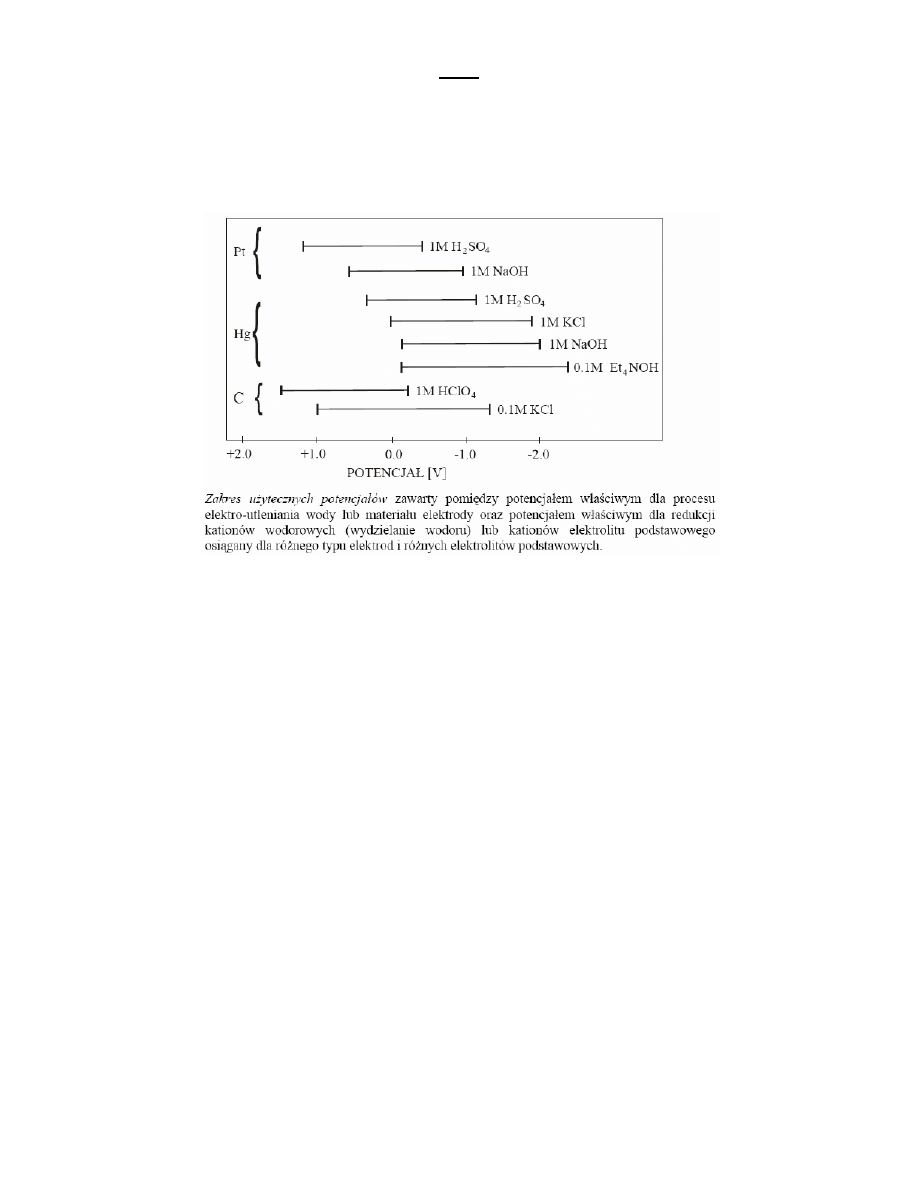
Wszystkie elektrody rtęciowa cechuje mały zakres w stosowaniu dla potencjałów dodatnich; rtęd nie może byd
stosowana przy potencjale większym niż +0.4V względem nasyconej elektrody kalomelowej NEK, ponieważ ulega
anodowemu rozpuszczeniu. W obecności anionów – tworzących z Hg2 2+ lub Hg2+ sole trudno rozpuszczalne albo
kompleksy –rtęd łatwiej ulega anodowemu rozpuszczeniu, odpowiednie potencjały wynoszą w obecności Cl- (0.0V), OH-
(-0.2V), CN- (-0.6V). Zakres stosowania rtęci dla potencjałów ujemnych z uwagi na wysokie nadnapięcie wydzielania
wodoru jest znacznie większy i jest ograniczony z reguły rozkładem elektrolitu podstawowego. Jeżeli elektrolitem
podstawowym są sole sodu i potasu wynosi on w roztworze obojętnym lub alkalicznym –1.8V, dla soli litu –2.0V i dla soli
czteroalkiloamoniowych –2.6V. W roztworach niewodnych zakres ten może byd obniżony nawet do –3V, co pozwala na
polarograficzne oznaczanie jonów metali alkalicznych.
MFE
Obecnie najbardziej popularną elektrodą rtęciową jest błonkowa elektroda rtęciowa (mercury film electrode - MFE).
Powstaje ona przez elektrochemiczne lub mechaniczne osadzenie rtęci na powierzchni elektrody stałej – najczęściej
złotej, srebrnej lub z węgla szklistego. Elektrody te charakteryzują się korzystnym stosunkiem powierzchni do objętości
umożliwiającym polepszenie czułości i rozdzielczości oznaczeo metodą woltamperometrii stripingowej.
W woltamperometrii i polarografii sygnałem jest prąd związany z redukcją lub utlenianiem oznaczanej substancji
(depolaryzatora) na elektrodzie pracującej. Proces ten traktowany jako całośd podlega prawom elektrolizy. Natomiast
chwilowa wielkośd przepływającego prądu zależy od szeregu czynników. Zakładając, że potencjał elektrody jest
wystarczający do zajścia reakcji elektrodowej wyróżnid można trzy stadia procesu:
doprowadzenie depolaryzatora do powierzchni elektrody (transport)
właściwa reakcja elektrodowa polegająca na przeniesieniu n elektronów z elektrody do depolaryzatora
(redukcja) lub z depolaryzatora do elektrody (utlenienie)
produkt reakcji, w zależności od jego postaci chemicznej może byd odtransportowany od powierzchni elektrody
(do głębi roztworu lub do wnętrza elektrody rtęciowej) bądź pozostad na powierzchni elektrody
O wielkości prądu decyduje etap najwolniejszy. W warunkach pomiaru woltamperometrycznego nie obserwuje się
ustalenia prądu, krzywa ma charakter piku, którego wielkośd jest także ograniczona najwolniejszym procesem. W
typowych warunkach prowadzenia pomiaru woltamperometrycznego, transport substratu do powierzchni elektrody
następuje na drodze dyfuzji i ten proces jest najwolniejszy. Dlatego tak ograniczony prąd nazywamy prądem dyfuzyjnym.
Jeżeli w roztworze elektrolitu zanurzymy dwie elektrody i doprowadzimy do nich napięcie, to jony depolaryzatora mogą
dopływad do elektrody wskutek dyfuzji, która jest wynikiem różnicy stężeo depolaryzatora w pobliżu elektrody i w głębi
roztworu, wskutek migracji, tj. uporządkowanego ruchu jonów do elektrody o odpowiednim znaku oraz konwekcji
wywołanej np. mieszaniem roztworu. Całkowity mierzony prąd będzie sumą natężeo prądów składowych
Gdzie
– prąd dyfuzyjny graniczny
– prąd migracyjny graniczny
– prąd wywołany konwekcją (mieszaniem) roztworu
– prąd szczątkowy
Eliminacja składowych migracyjnych prądu faradajowskiego ma duże znaczenie praktyczne i pozwala uzyskiwad liniowe
zależności natężenia prądu granicznego lub prądu piku od stężenia depolaryzatora. W migracyjnym transporcie ładunku
uczestniczą wszystkie jony obecne w elektrolicie podstawowym, a ilośd ładunku przenoszonego przez poszczególne
rodzaje jonów jest proporcjonalna do stosunku ich stężeo. W transporcie tym mogą uczestniczyd również jony
depolaryzatora, co przyczynia się do zwiększenia natężenia rejestrowanego prądu i może wydawad się zjawiskiem
korzystnym. Należy jednak pamiętad, że wkład transportu migracyjnego depolaryzatora jest zależny od stężeo wszystkich
jonów. Konsekwencją tego mogą byd znaczne zmiany składowej migracyjnej prądu faradajowskiego towarzyszące
zmianom składu elektrolitu podstawowego, nawet wtedy gdy stężenie depolaryzatora pozostaje stałe. Aby
wyeliminowad prąd migracyjny z prądu granicznego, stosuje się elektrolit podstawowy o stężeniu co najmniej 1000 razy
większym od stężenia oznaczanego depolaryzatora, który przyjmuje na siebie całe przewodnictwo. W ten sposób udział
jonów depolaryzatora w migracyjnym transporcie ładunku staje się pomijalnie mały, a migrujące jony elektrolitu
podstawowego nie ulegają rozładowaniu na elektrodzie pracującej i nie przyczyniają się do zmian natężenia prądu.
Elektrolit podstawowy
Elektrolit podstawowy powinien spełniad następujące wymagania:
powinien zapewniad wystarczająco szeroki zakres potencjałów umożliwiający oznaczenie obecnych w próbce
depolaryzatorów
nie powinien tworzyd z depolaryzatorami soli nierozpuszczalnych
depolaryzatory powinny występowad w nim w zdefiniowanej i jednorodnej formie chemicznej
nie powinien powodowad kontaminacji badanej próbki Spośród często stosowanych elektrolitów podstawowych
wymienid można KCl, KNO3, HCl, NaOH oraz bufory amonowy, fosforanowy, octanowy i wiele innych.
Polarogramy
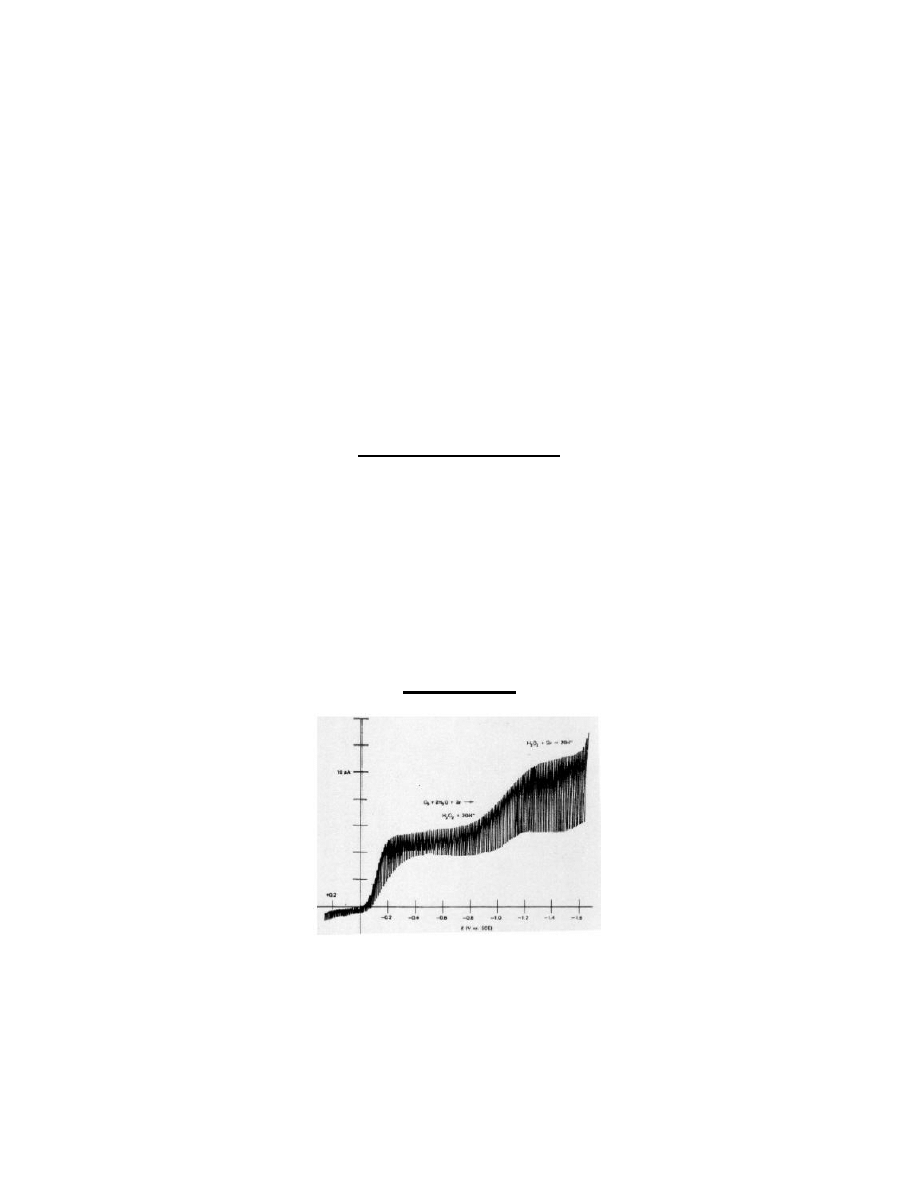
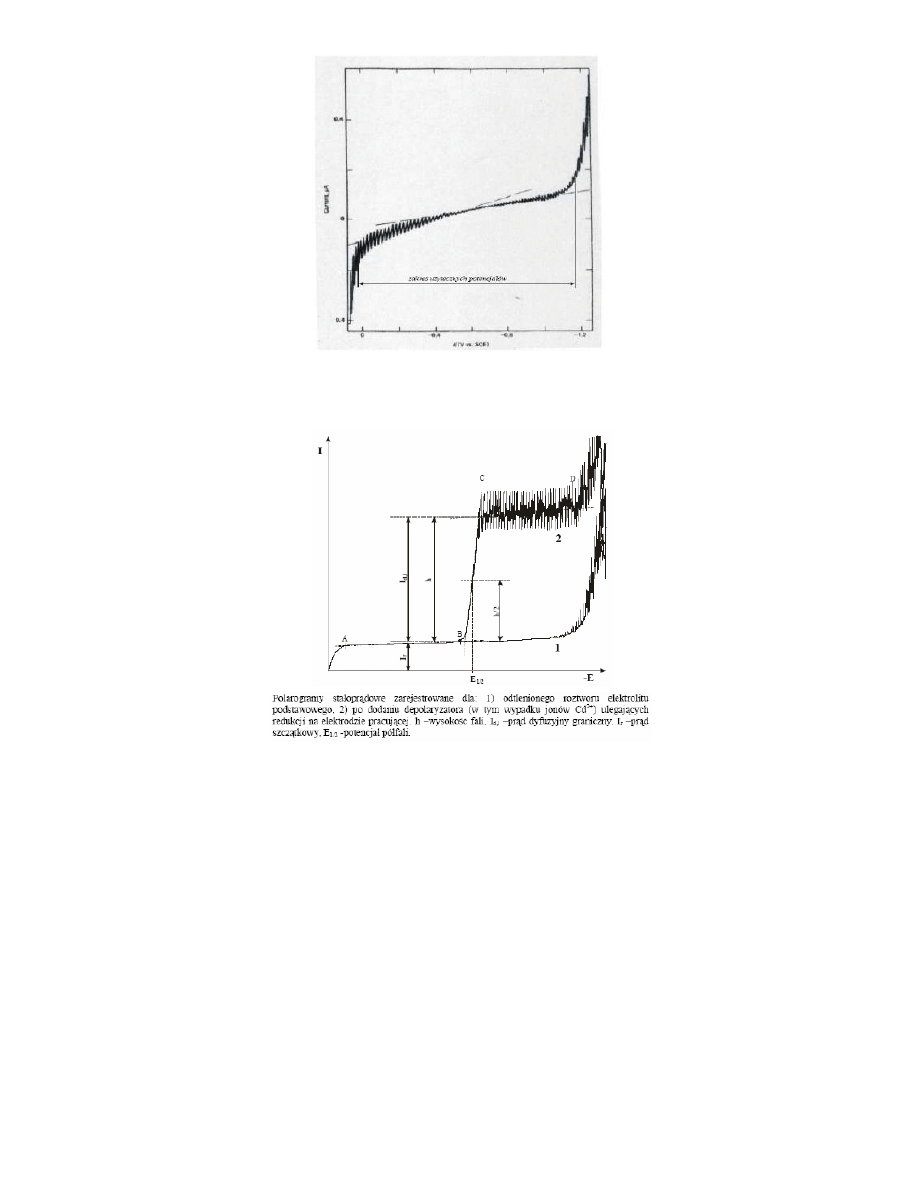
Polarogram stałoprądowy zarejestrowany dla ultraczystego roztworu 0,1M KNO3 przed usunięciem z roztworu
rozpuszczonego w nim tlenu.
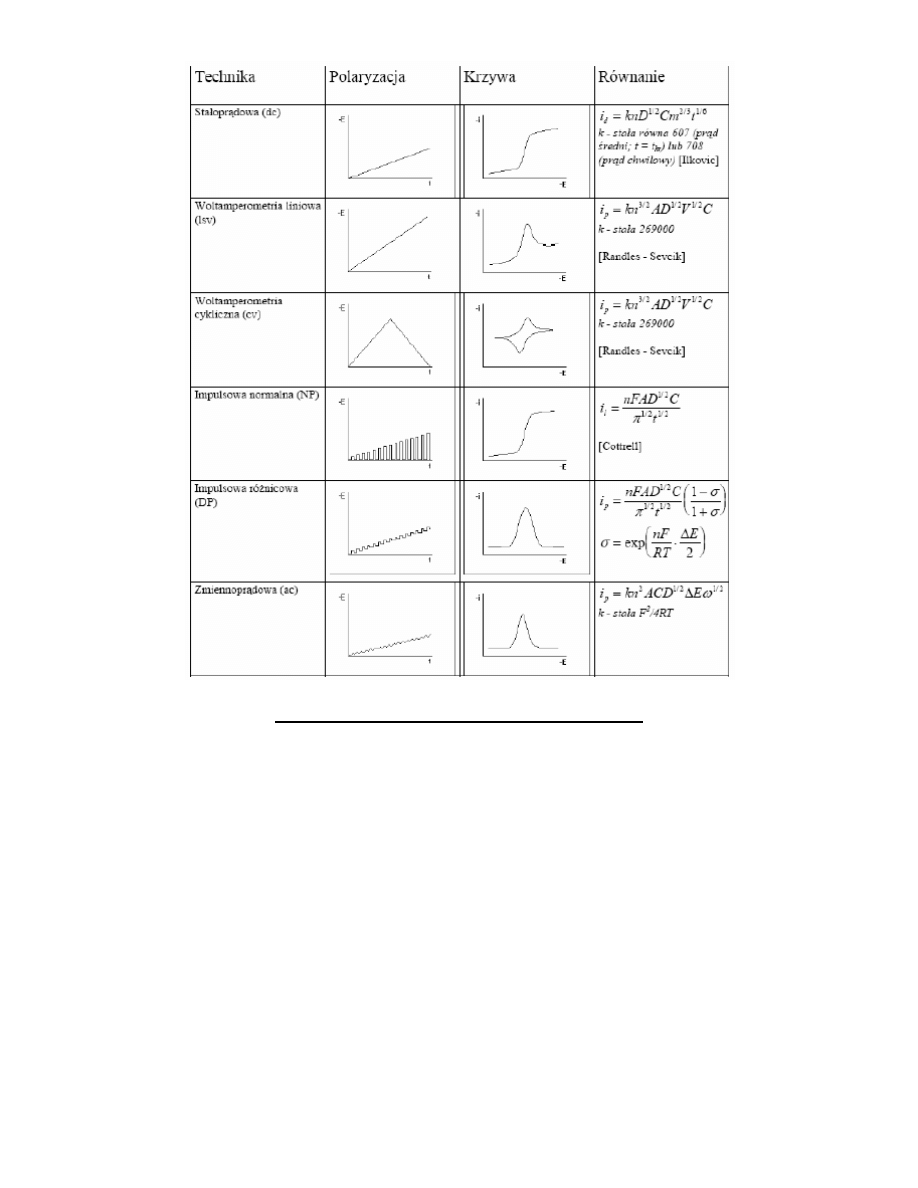
Polarogram stałoprądowy zarejestrowany dla ultraczystego roztworu 0,1M KNO3 po usunięciem rozpuszczonego w nim
tlenu, z zaznaczonym zakresem użytecznych potencjałów
Anodowa woltamperometria stripingowa
Anodowa woltamperometria stripingowa (Anodic Stripping Voltammetry - ASV), to technika, w której proces akumulacji
realizowany jest poprzez elektrochemiczną redukcję oznaczanej substancji na powierzchni stacjonarnej elektrody
pracującej. Stosowany potencjał akumulacji, o 200¸300mV bardziej ujemny aniżeli potencjał półfali w klasycznej
polarografii stałoprądowej, zapewnia metodzie zadowalającą powtarzalnośd granicznego prądu elektrolizy, a tym samym
odtwarzalnośd warunków zatężania.
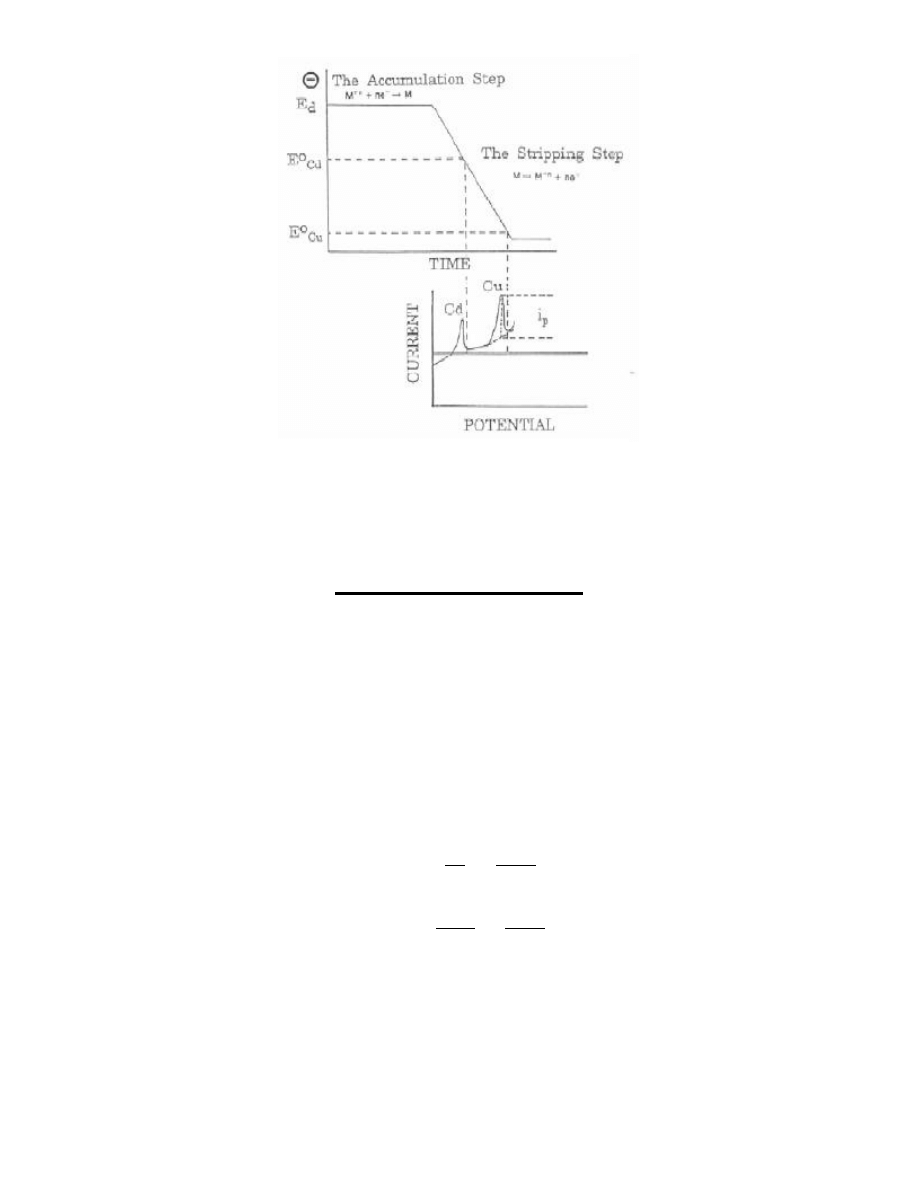
W zależności od chemicznego charakteru wydzielanego metalu oraz materiału z jakiego wykonano elektrodę, ulega on
rozpuszczeniu w rtęci, tworząc amalgamat lub wyłącznie cienką warstewkę metalu na powierzchni elektrody. Po
zakooczeniu procesu akumulacji elektroda jest polaryzowana w kierunku dodatniego potencjału i przy odpowiedniej jego
wartości atomy wydzielonego uprzednio metalu są utleniane i przechodzą z elektrody do roztworu.
Metody potencjometryczne
Metody potencjometryczne sprowadzają się do pomiaru siły elektromotorycznej SEM ogniwa złożonego z dwóch
półogniw (elektrod) zanurzonych w badanym roztworze. Mierzona siła elektromotoryczna zależy od stężenia w
roztworze oznaczanego składnika. Zasadniczym elementem układu pomiarowego służącego do oznaczeo
potencjometrycznych jest elektroda zwana wskaźnikową oraz elektroda odniesienia. Na granicy faz (powierzchnia
elektrody – roztwór) ustala się równowaga, którą ogólnie można przedstawid w następującej postaci
Potencjał elektrody E zależy m.in. od właściwości chemicznych składników roztworu oraz wartości stężeo składników
reakcji elektrodowych zarówno w postaci zredukowanej jak też utlenionej. Opisuje go równanie Nernsta:
(
[ ]
[ ]
)
(
[ ]
[ ]
)
- standardowy potencjał elektrodowy
– stała gazowa
– temperatura w skali Kelwina
– liczba elektrodów biorących udział w reakcji elektrodowej
– stała Faradaja
[ ] – stężenie molowe formy utlenionej
[ ] – stężenie molowe formy zredukowanej
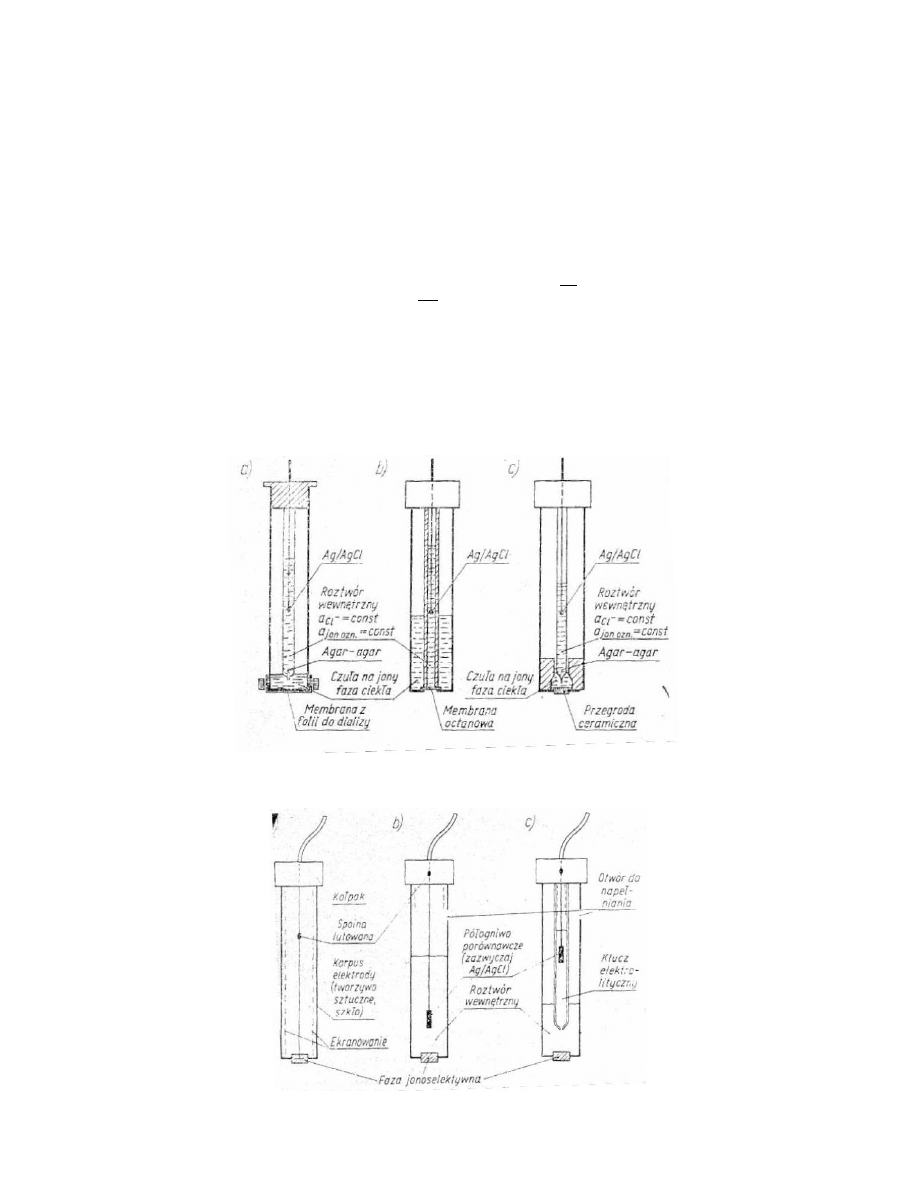
Pod pojęciem elektrod jonoselektywnych rozumiemy przyrządy, których zasadniczym elementem jest membrana
wykonana z materiału, na powierzchni którego w roztworze ustala się równowaga pomiędzy materiałem membrany a
oznaczanymi jonami znajdującymi się w roztworze. Równaniem opisującym potencjał elektrody jonoselektywnej jest
równanie Nikolskiego:
(
∑
)
– aktywnośd jonu oznaczanego,
– aktywnośd jonu przeszkadzającego,
– ładunek jonu oznaczanego
– ładunek jonu przeszkadzjącego,
– współczynnik selektywności
Przykłady konstrukcji elektrod z ciekłą membraną.
Przykłady konstrukcji elektrod ze stałą membraną.
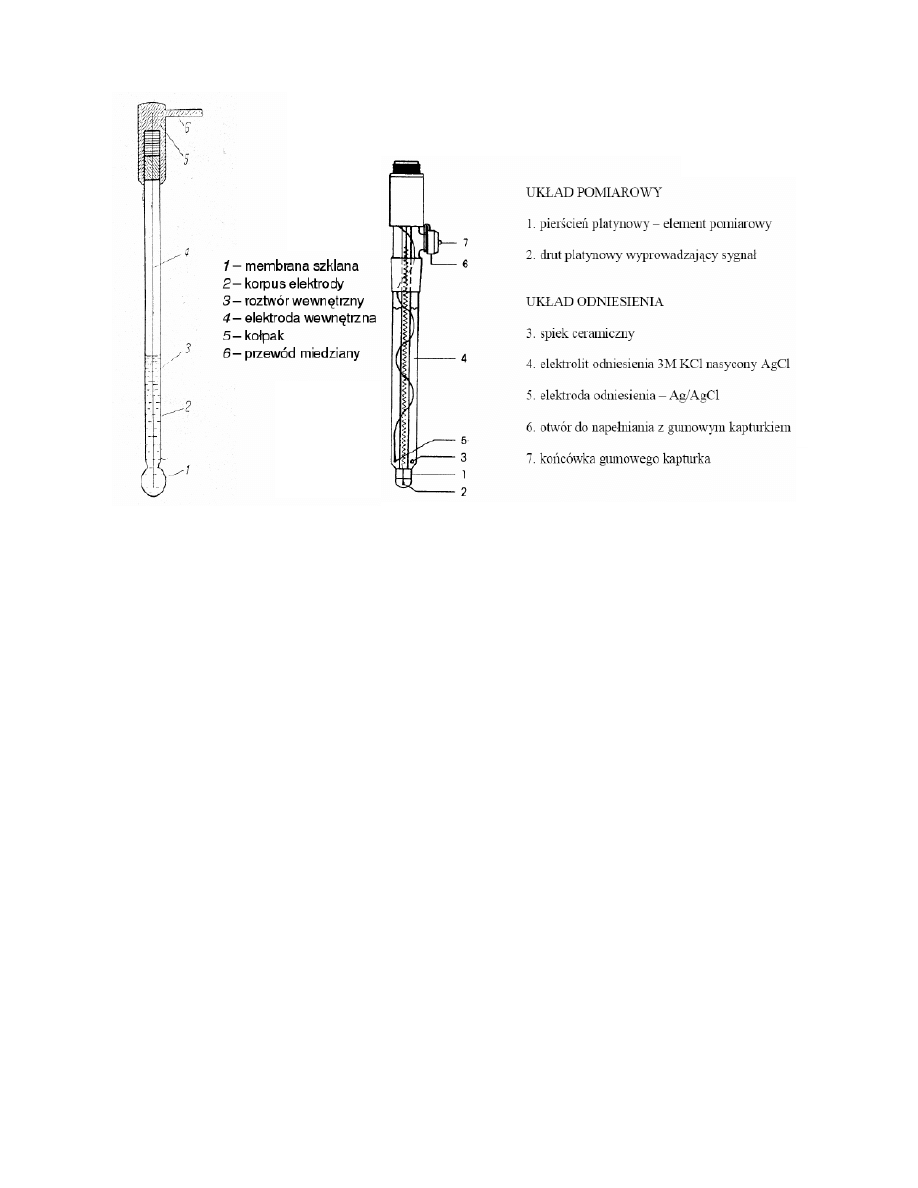
Jako elektroda porównawcza największe znaczenie teoretyczne ma normalna elektroda wodorowa (NEW). Jest to
blaszka platynowa pokryta czernią platynową, omywana wodorem pod ciśnieniem 760 mm Hg i zanurzona w roztworze
kwasu solnego o aktywności równej 1. Reakcje zachodzące na tej elektrodzie można przedstawid równaniem
analogicznym do równania opisującego procesy zachodzące na elektrodach metalowych.
Nie jest ona jednak wygodna w użyciu i w praktyce stosuje się najczęściej nasyconą elektrodę kalomelową (NEK) i
chlorosrebrową.
Elektroda kalomelowa
Elektrodę kalomelową stanowi drut platynowy będący w kontakcie z rtęcią metaliczną pokrytą warstwą chlorku rtęci(I)
Hg2Cl2 (kalomelu), zanurzoną w nasyconym roztworze chlorku potasu. Półogniwo takie można zapisad: Hg,
Hg2Cl2(s)|nas. KCl. Przemiany zachodzące na elektrodzie można przedstawid równaniem:
Elektroda chlorosrebrowa
Elektrodę chlorosrebrową stanowi drut srebrny pokryty warstewką chlorku srebra zanurzony w roztworze zawierającym
jony Cl-, pochodzące z chlorku potasu lub kwasu solnego. Schemat takiego półogniwa można zapisad: Ag, AgCl(s)|KCl.
Przemiany zachodzące na elektrodzie można przedstawid równaniem:

Podział metod potencjometrycznych
1. Metoda krzywej kalibracji
2. Metoda dodatku wzorca
3. Metoda dodatku próbki do wzorca
4. Miareczkowanie potencjometryczne
Metoda krzywej kalibracji
Metoda krzywej kalibracji – polega na sporządzeniu serii wzorców o określonym stężeniu i pomiarze siły
elektromotorycznej ogniwa pomiarowego. Na podstawie otrzymanych wyników sporządza się wykres zależności E (SEM)
jako funkcji log ci, bądź E jako funkcji log a. Następnie wyznaczamy wartośd E w próbce badanej i odczytujemy wartośd
stężenia z krzywej kalibracyjnej.
Metoda dodatku wzorca
Metoda dodatku wzorca – polega na pomiarze SEM ogniwa złożonego z elektrody czułej na oznaczany typ jonu oraz
elektrody odniesienia. Najpierw dokonujemy pomiaru w ściśle określonej objętości roztworu, którego stężenia nie
znamy, następnie dodajemy ściśle określoną objętośd oznaczanego jonu o znanym stężeniu czyli wzorca. Metodę tę
stosujemy gdy stężenie próbki jest znacznie mniejsze niż stężenie wzorca.
Metoda dodatku próbki do wzorca
Metoda dodatku próbki do wzorca – stosuje się gdy stężenie próbki jest znacznie większe od stężenie wzorca. Najpierw
dokonuje się pomiaru E ściśle określonej objętości roztworu wzorca a następnie dodaje ściśle określoną objętośd
roztworu próbki.
Miareczkowanie potencjometryczne
Miareczkowanie potencjometryczne – polega na pomiarze E ogniwa zbudowanego z elektrody wskaźnikowej oraz
elektrody odniesienia o stałym potencjale. E tak zbudowanego ogniwa zmienia się wraz ze zmianą stężenia jonu
biorącego udział w reakcji z odczynnikiem miareczkującym. Siłę elektromotoryczną mierzy się po dodaniu każdej porcji
odczynnika miareczkującego a następnie wykreśla krzywą E = f(V). Z tak otrzymanej krzywej wyznacza się graficznie
punkt przegięcia na krzywej miareczkowania
Konduktometria
Konduktometria - pomiar przewodnictwa elektrycznego (lub rezystancji) słupa cieczy znajdującego się pomiędzy dwoma
elektrodami. W przypadku konduktometrii staramy się uniknąd jakichkolwiek reakcji elektrodowych i dlatego do
pomiarów stosuje się prąd zmienny.
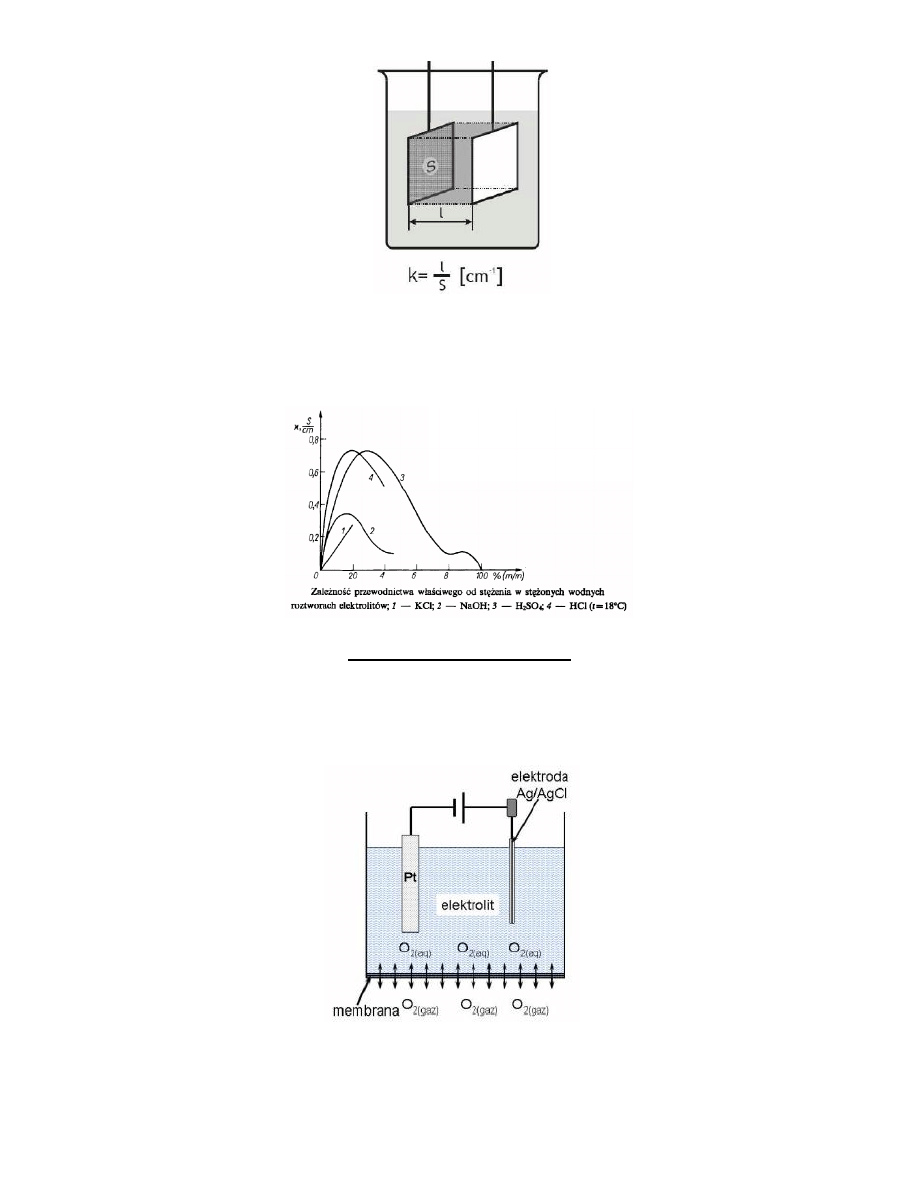
Przewodnictwo roztworu rośnie wraz ze wzrostem stężenia tylko do pewnego momentu, po czym zaczyna spadad – co
związane jest z niecałkowitą dysocjacją elektrolitów i wzrastającym oddziaływaniem międzyjonowym, zmniejszającym
ruchliwośd jonów
Elektroda tlenowa Clarka
Elektroda tlenowa Clarka jest amperometrycznym sensorem do oznaczania tlenu. Klasyczna tlenowa elektroda Clarka
zawiera ciekły elektrolit oraz dwie elektrody. Anoda jest elektroda chlorosrebrowa, a katoda metal szlachetny platyna
lub złoto.
Katoda znajduje się w szklanej otoczce izolującej. Anoda posiada dużą powierzchnię, aby zapewnid sensorowi dobra
stabilnośd podczas pracy i zabezpieczyd przed zmianami stężenia elektrolitu. Elektrody zanurzone są w roztworze
elektrolitu. Zazwyczaj elektrolitem jest to 0,1 mM roztwór chlorku potasu (KCl). Całośd przykryta jest półprzepuszczalna
membrana. Jako materiał membran w takich czujnikach stosuje się różne polimery: teflon, polietylen, polimery
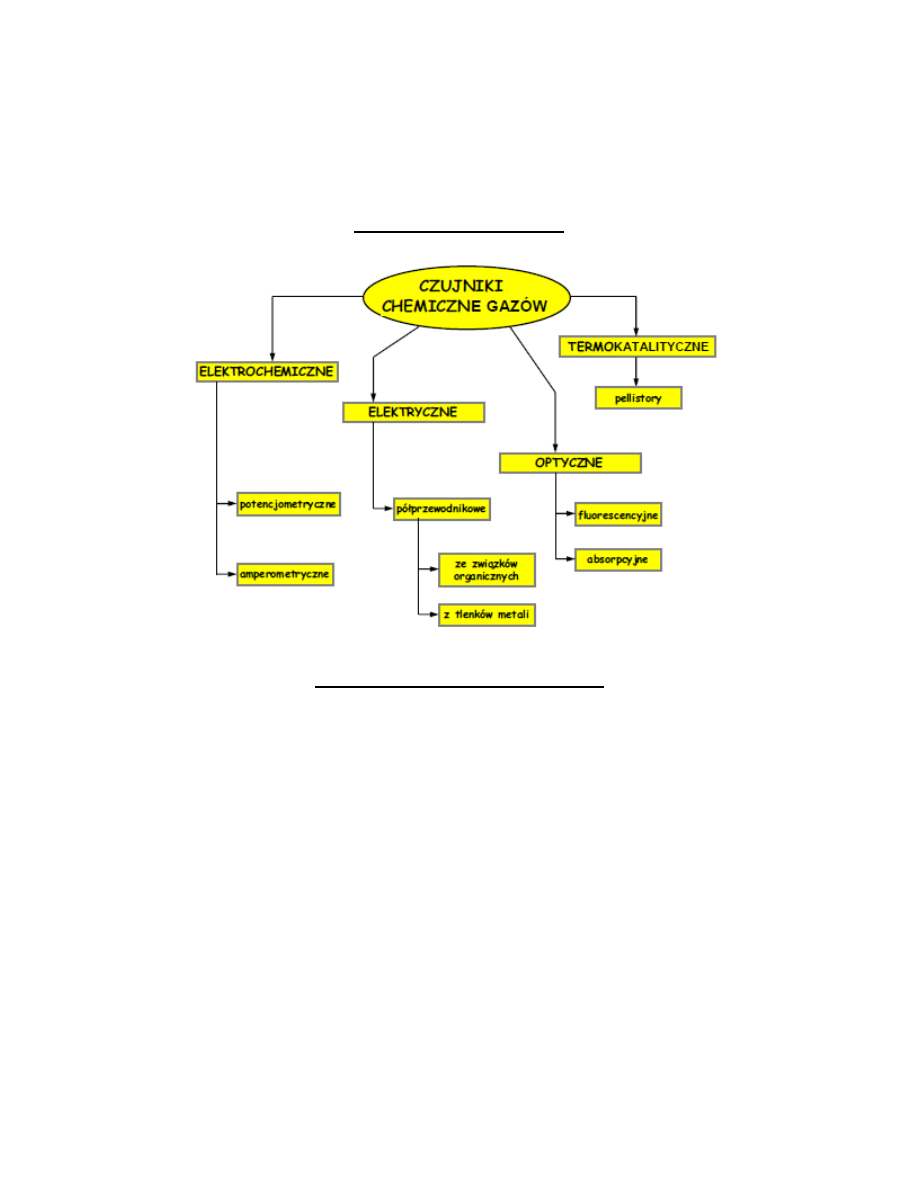
silikonowe przez które tlen łatwo przenika, w przeciwieostwie do innych gazowych składników próbki. Membrany te nie
przepuszczają zanieczyszczeo i redukowalnych jonów. Stosowane napięcie polaryzacji w elektrodzie Clarka jest stałe i
wynosi zazwyczaj 0,7 V lub 0,8 V względem elektrody Ag/AgCl. Na anodzie zachodzi proces utleniania srebra, które
natychmiast w chodzi w reakcje z jonami chlorkowymi znajdującymi się w roztworze i powstaje trudno rozpuszczalny
chlorek srebra. Tlen dyfunduje przez membranę do elektrolitu wewnętrznego elektrody i dalej do katody gdzie ulega
reakcji redukcji.
Czujniki stężenia gazów
Półprzewodnikowe czujniki gazu
Dzielimy na :
- oparte na przewodnictwie barierowym
- oparte na zjawiskach zachodzących na defektach sieci
Chemisorpcja - adsorpcja chemiczna, polegająca na tworzeniu się silnych wiązao chemicznych między adsorbentem i
adsorbatem.
Chemisorpcja przyczynia się nie tylko do zmian koncentracji elektronów lub dziur w materiale czułym chemicznie
sensora. Zjawisko to wpływa również na ruchliwośd nośników ładunku elektrycznego. Zależnośd wymienionych wielkości
od chemisorpcji oznacza, że zaadsorbowane gazy mogą zmienid przewodnictwo elektryczne kryształów półprzewodnika,
z których składa się element receptorowo przetwornikowy sensora. Wielkością charakteryzującą grubośd warstwy
podwójnej, w obrębie której dochodzi do ekranowania ładunku powierzchniowego, jest długośd Debye’a LD.
Na podstawie badao stwierdzono dwie ważne prawidłowości:
– przewodnictwo elektryczne porowatego elementu receptorowo-przetwornikowego zależy w decydującym stopniu od
niezmieniającej się pod wpływem gazów rezystancji wewnętrznej półprzewodnika, gdy średnica d jego ziaren jest
większa od podwójnej długości Debye’a 2LD;
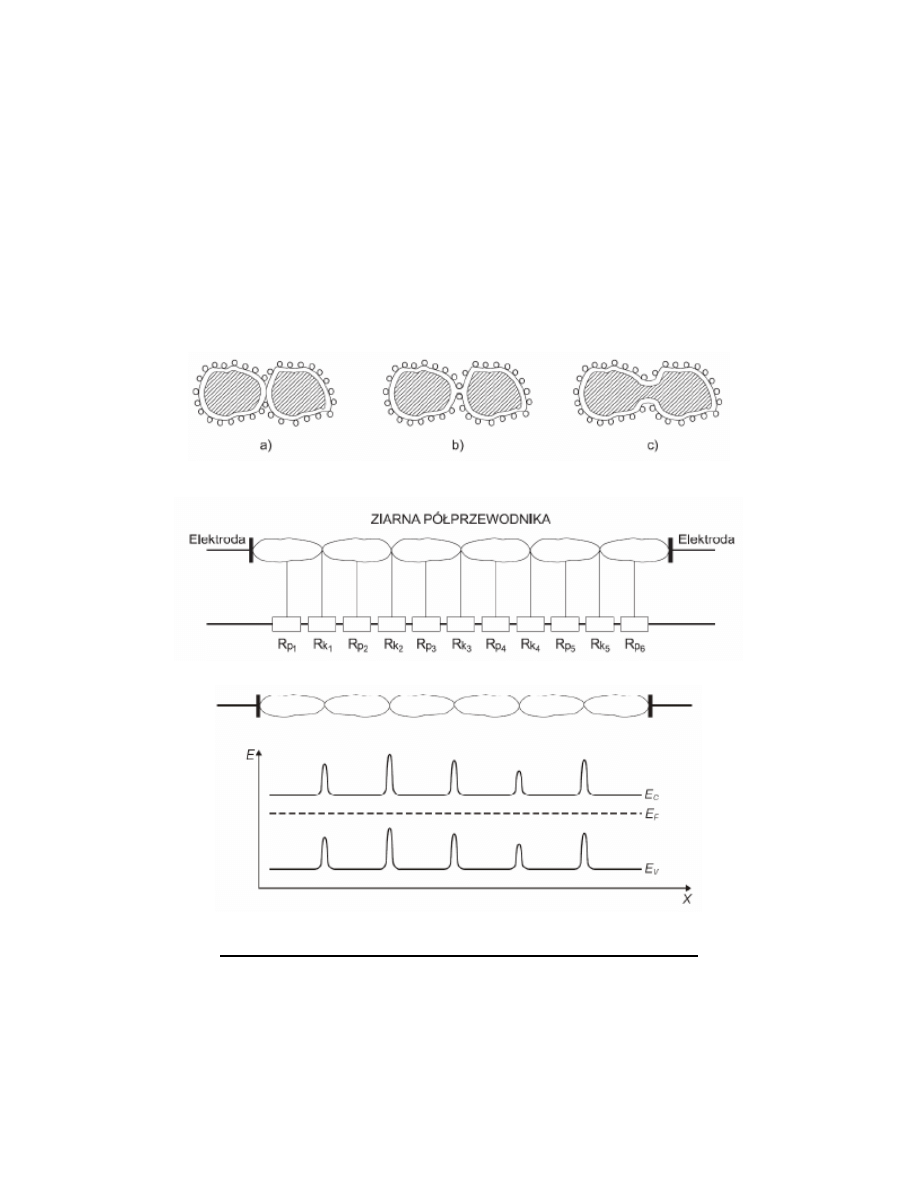
– wpływ otoczenia gazowego na natężenie prądu płynącego przez sensor staje się istotne, kiedy wymienione wielkości są
porównywalne.
Właściwości pomiarowe elementu receptorowo-przetwornikowego sensora związane są również z wielkością połączeo,
jakie występują między ziarnami materiału czułego chemicznie. W półprzewodnikach stosowanych w rezystancyjnych
czujnikach gazów przyjmują one postad:
– kontaktów o niewielkiej powierzchni (x < LD);
– cienkich szyjek (x ≅ LD);
– grubych szyjek (x > LD).
LD i x oznaczają odpowiednio długośd Debye’a i wymiar poprzeczny kontaktu.
Oparte na zjawiskach zachodzących na defektach sieci
defekty te są silnymi centrami adsorpcyjnymi i dlatego odgrywają ważną rolę w chemisorpcji. Ich migracja powoduje
degradację powierzchni między ziarnami półprzewodnika. Ponadto omawiane defekty uczestniczą w zjawiskach
elektronowych zarówno na powierzchni, jak i wewnątrz kryształu półprzewodnika. Występujące na granicy faz wakansje
tlenowe reagują z zaadsorbowanymi cząsteczkami tlenu. Między tymi defektami i gazem tworzy się stan równowagi
termodynamicznej.
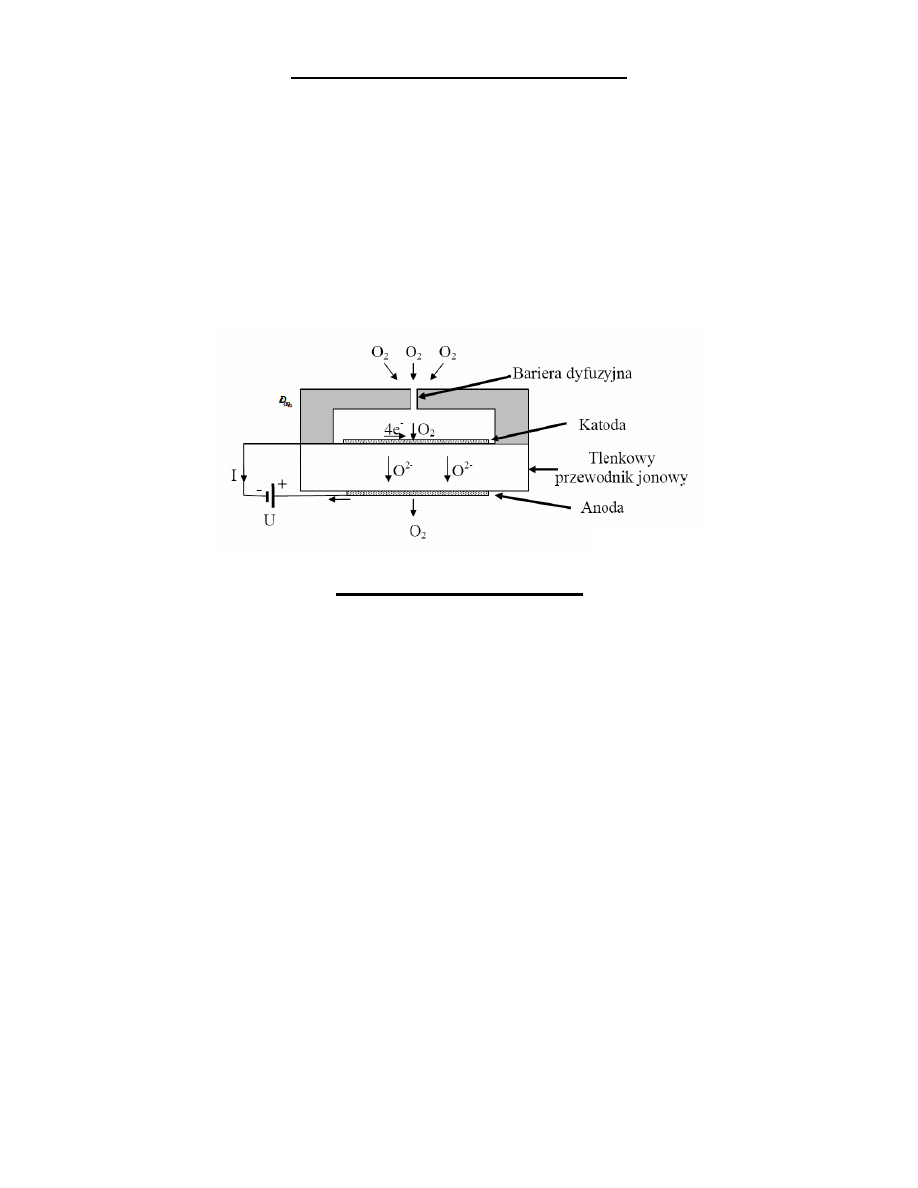
Czujniki gazów ze stałym elektrolitem
• czujniki amperometryczne
• czujniki potencjometryczne Nernsta
• czujniki potencjometryczne z mieszanym
potencjałem
• czujniki elektrokatalityczne
Czujniki amperometryczne
Czujniki potencjometryczne
Warstwa podwójna
• samorzutne przechodzenie elektronów lub jonów jednej fazy do drugiej. W wyniku tego procesu jedna faza wykazuje
nadmiar, a druga niedomiar ładunku elektrycznego określonego znaku. Powstaje pole elektryczne .
• wybiórcza adsorpcja jednego rodzaju jonów, powodująca nagromadzenie się ładunku jednego znaku w sąsiedztwie
granicy faz. Dla zachowania elektroobojętności całego układu powstaje warstwa rozmytego ładunku we wnętrzu tej
samej fazy.
• adsorpcja polarnych cząsteczek rozpuszczalnika lub cząsteczek substancji rozpuszczonej. Cząsteczki te ulegają wówczas
orientacji na granicy faz.
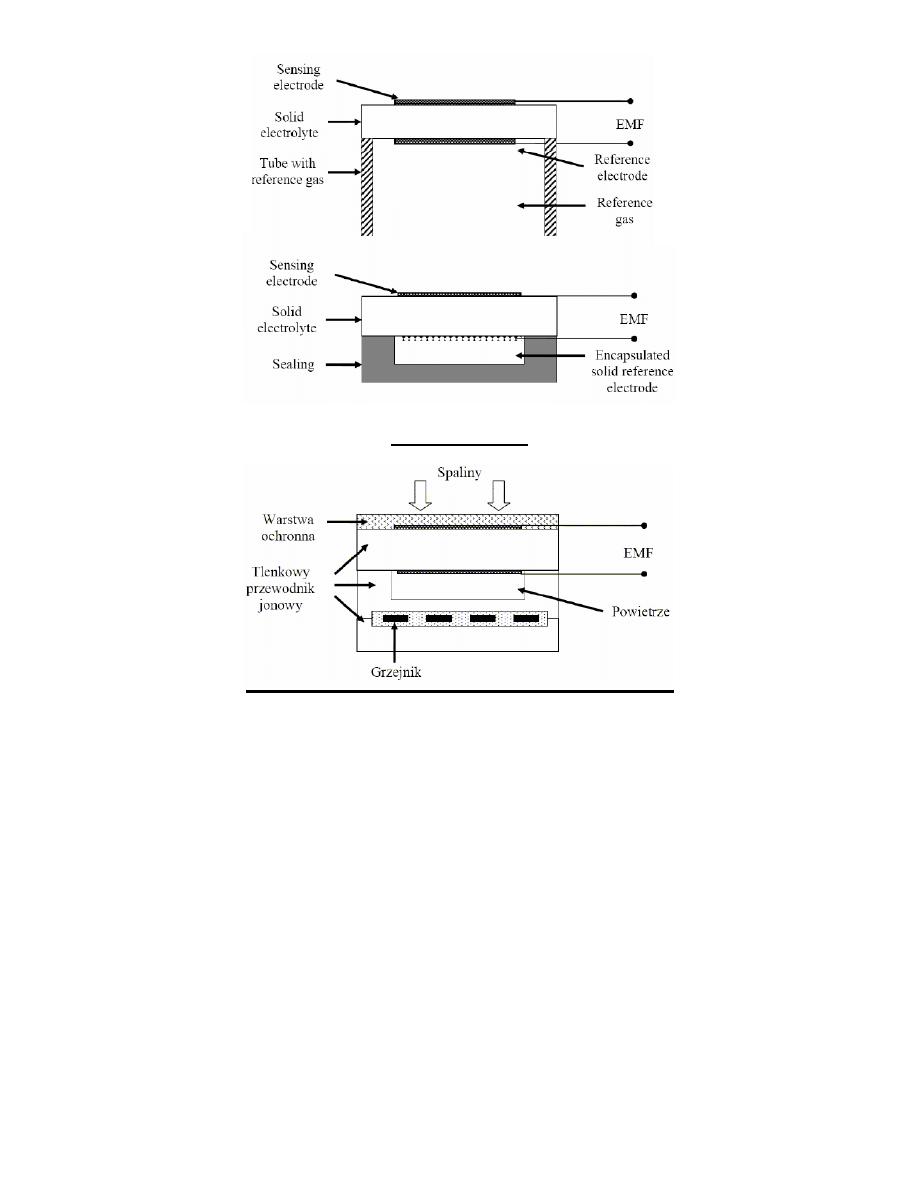
Czujnik Lambda
Działanie sondy lambda opiera się na prawie sformułowanym przez Walthera Nernsta (równanie Nernsta). Sond a
lambda, czyli ogniwo galwaniczne zbudowane z materiału ceramicznego – dwutlenku cyrkonu, na którym napylone są po
obu jego stronach cienkie warstwy platyny, zostało opracowane pod koniec lat 60 XX wieku w Robert Bosch GmbH przez
dr. Güntera Baumana. Sondę umieszcza się w układzie wydechowym, tak, że jedna strona urządzenia styka się z
gorącymi gazami spalinowymi, osiągającymi w tym miejscu około 300 °C, a druga strona styka się z powietrzem z
otoczenia.
Spiek ceramiczny dwutlenku cyrkonu w temperaturze powyżej 300 °C staje się swoistym elektrolitem zdolnym do
przewodnictwa jonowego. W celu utrzymania odpowiedniej temperatury, niektóre sondy mają grzałkę. Warstwy platyny
na jego powierzchni pełnią rolę elektrod, przy czym ich potencjał elektryczny jest funk cją stężenia tlenu w gazach, z
którymi się stykają. Przy dużej różnicy stężenia tlenu po obu stronach ogniwa generuje ono siłę elektromotoryczną
dochodzącą do 1 V.
Napięcie generowane przez ogniwo jest przekazywane do modułu sterującego składem mieszanki p aliwowo-powietrznej
w silniku. Moduł ten dostosowuje na bieżąco skład mieszanki, tak aby w określonych warunkach obciążenia silnika,
rodzaju paliwa i warunków atmosferycznych zapewnid jak najdoskonalszą pracę silnika i minimalizowad emisję tlenku
węgla.
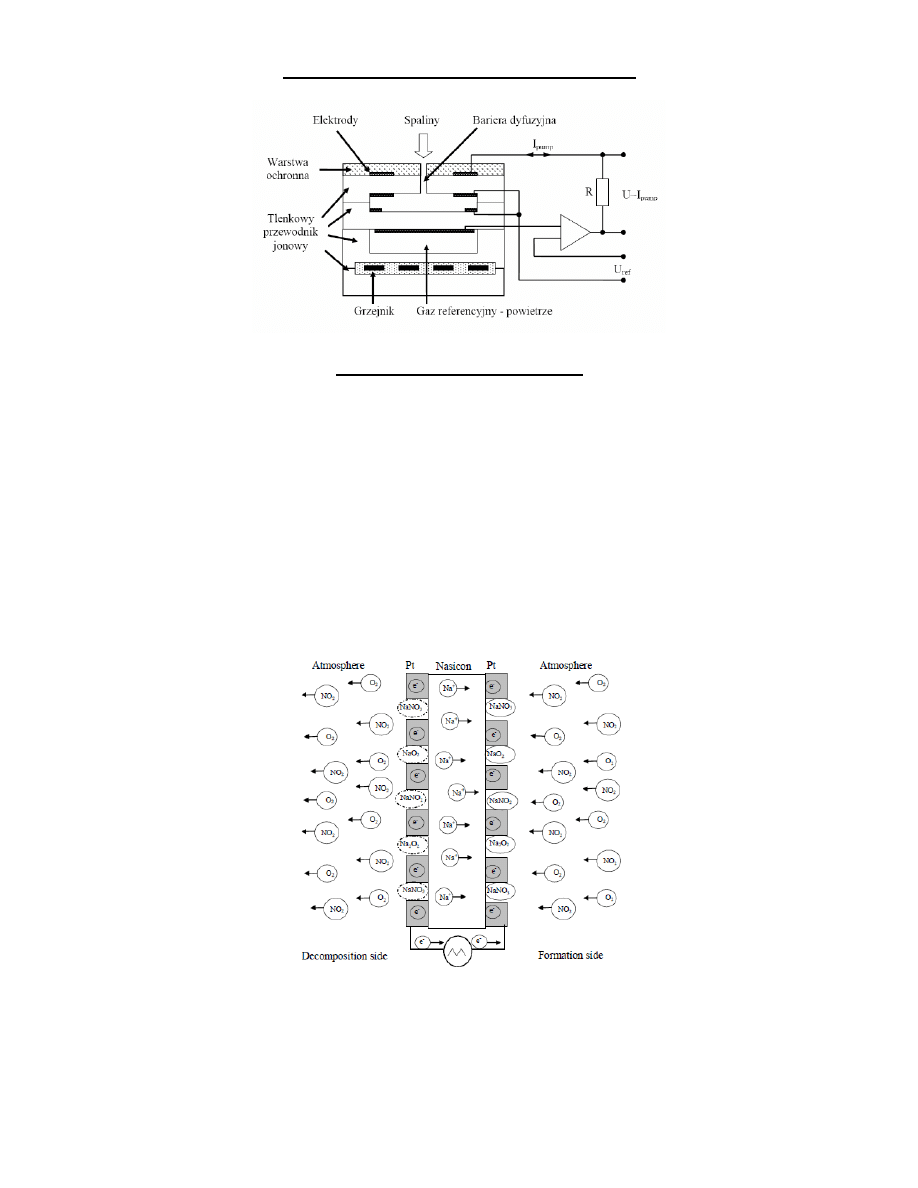
Szerokopasmowy czujnik stężenia tlenu
Czujniki elektrokatalityczne
Elektrochemiczne czujniki na bazie elektrolitów stałych dzieli się zwykle na czujniki potencjometryczne i czujniki
amperometryczne, w zależności od tego czy elektrycznym sygnałem wyjściowym jest odpowiednio napięcie czy prąd.
Wadą tych czujników, bardzo trudną do usunięcia, jest brak selektywności - sygnał wyjściowy czujników jest zależny nie
tylko od stężenia mierzonego analitu, a także zwykle od wielu czynników interferujących.
Elektrokatalityczne czujniki gazów przypisane są przez niektórych autorów do grupy czujników amperometrycznych ze
względu na fakt, że mierzona jest w nich odpowiedź prądowa czujników. Inni autorzy uznają odrębnośd czujników
wykorzystujących kinetykę reakcji chemicznej jako drugą, obok czujników z ograniczeniem prądu, podgrupę czujników
amperometrycznych. Wydaje się jednak, że czujniki te tworzą odrębną podgrupę czujników elektrochemicznych, ze
względu na fakt pracy daleko od stanu równowagi termodynamicznej i brak bariery dyfuzyjnej, typowych dla
amperometrycznych czujników na bazie elektrolitów stałych, oraz specyficzny mechanizm działania.
Zasada pracy czujnika związana jest z tworzeniem i dekompozycją warstwy chemicznej na jego powierzchni oraz
oddziaływania tej warstwy z otaczającym czujnik gazem. Przy pobudzaniu czujnika liniowo narastającym napięciem, na
ujemnie spolaryzowanej elektrodzie czujnika (strona tworzenia) tworzy się związek chemiczny, którego ilośd zależy od
stężenia gazu otaczającego czujnik. Jednocześnie, na dodatnio spolaryzowanej elektrodzie czujnika (strona rozkładu)
następuje rozkład istniejącej tam warstwy chemicznej. Odwrócenie polaryzacji czujnika powoduje, że tworzenie i rozkład
warstwy będzie występowad na przeciwnych elektrodach.
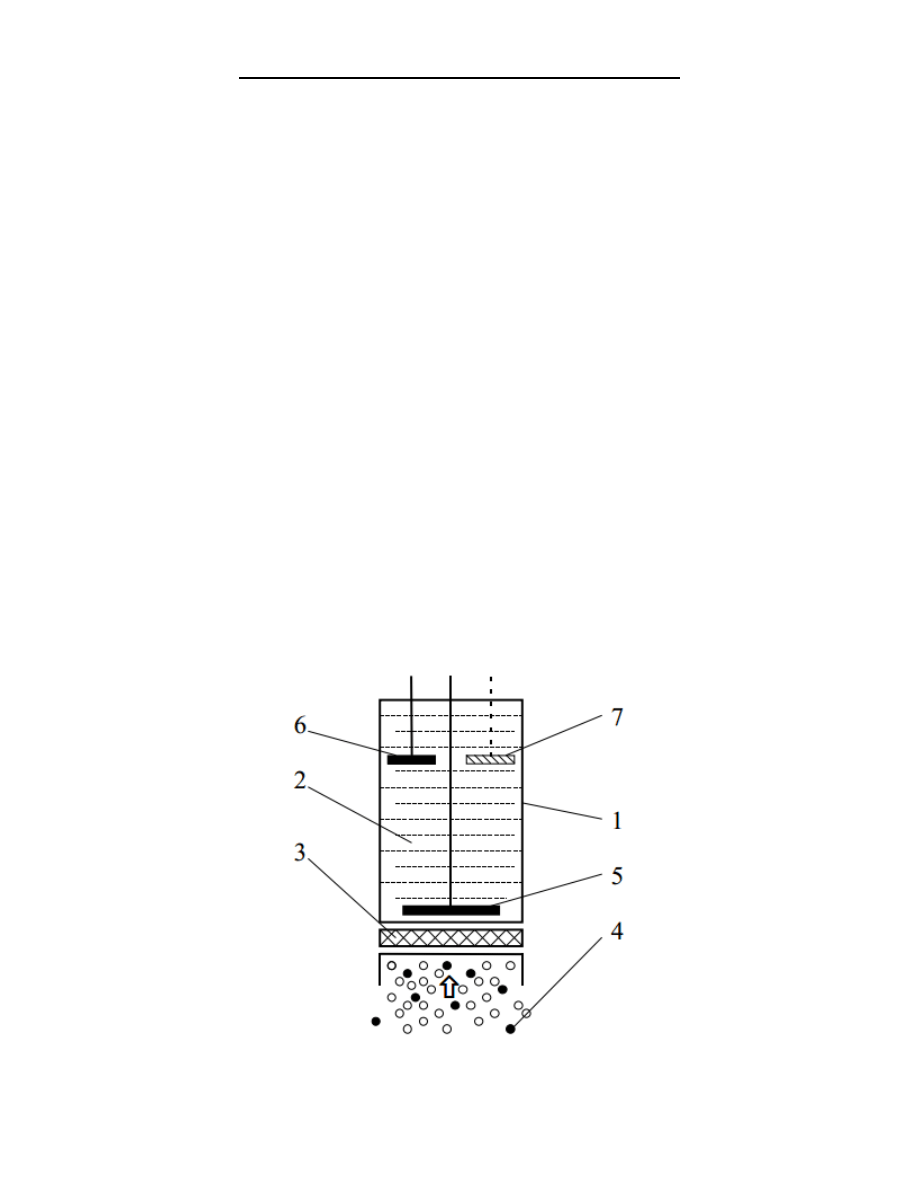
Czujniki amperometryczne z ciekłym elektrolitem
Elektrochemiczne czujniki gazowe są urządzeniami umożliwiającymi uzyskanie ilościowych informacji dotyczących stężeo
różnych gazów. Są przy tym wystarczająco czułe, w miarę selektywne a przy tym posiadają niewielkie ro zmiary i są
względnie tanie. Są one szeroko stosowane w urządzeniach do monitoringu osobistego powietrza atmosferycznego w
miejscu pracy a także do kontroli emisji toksycznych gazów z instalacji przemysłowych czy silników samochodowych.
Większośd w tej grupie czujników stanowią czujniki amperometryczne, których sygnałem jest prąd elektryczny będący
skutkiem elektrochemicznego utleniania lub redukcji cząsteczek oznaczanego gazu na powierzchni jednej z elektrod
czujnika. W budowie czujników amperometrycznych możemy wyróżnid kilka podstawowych elementów:
elektrodę wskaźnikową (sensing, indicating or working electrode), która usytuowana jest w łatwo osiągalnym
przez cząsteczki oznaczanego gazu miejscu czujnika, na powierzchni tej elektrody przebiega elektrochem iczna
reakcja redukcji lub utlenienia cząsteczek oznaczanego gazu,
przeciwelektrodę (counter electrode) ulokowaną w takim miejscu czujnika dokąd dostęp oznaczanego gazu jest
utrudniony,
trzecią elektrodę pełniącą rolę elektrody odniesienia (reference electrode) - względem tej elektrody, w
niektórych wersjach czujników amperometrycznych, ustalany jest optymalny potencjał elektrody wskaźnikowej
(roboczej),
stężony roztwór dobrze jonowo przewodzącego elektrolitu umieszczony wewnątrz obudowy czujnika,
barierę dyfuzyjną kontrolującą proces dyfuzji cząsteczek gazu do powierzchni elektrody wskaźnikowej. Zwykle
jest to małej średnicy kapilara lub przepuszczalna dla gazów membrana z folii polimerowej,
niskooporowy zewnętrzny układ elektryczny łączący elektrody w dwuelektrodowej wersji czujnika i
umożliwiający pomiar płynącego pomiędzy elektrodami prądu elektrycznego,
w wersji trójelektrodowej układ elektryczny w postaci tzw. potencjostatu, umożliwiający precyzyjną kontrolę
wartości potencjału elektrody pracującej względem elektrody odniesienia a jednocześnie pomiar sygnału
czujnika (prądu) w obwodzie przeciwelektrody
Budowa czujnika amperometrycznego: 1-obudowa, 2- roztwór elektrolitu, 3-membrana, 4- cząsteczki oznaczanego gazu,
5- elektroda wskaźnikowa (pracująca), 6- przeciwelektroda, 7- elektroda odniesienia (nie we wszystkich wersjach

czujników).
Spektrometria masowa
Spektrometria masowa jest nowoczesną techniką analityczną pozwalającą na dokładny pomiar masy. Od czasu
pierwszego doświadczenia, opisanego przez J.J.Thomsona w 1912 roku, MS otworzyła nowe możliwości w naukach
biologicznych, stając się wygodnym narzędziem, dzięki któremu możemy uzyskad szereg istotnych informacji o badanych
substancjach. Wysokorozdzielcze aparaty umożliwiają wyznaczenie masy cząsteczkowej z dokładnością do 4 miejsca po
przecinku dla związków o masie do ok. 1000 Da, (w przypadku cyklotronowego rezonansu jądrowego z transformacją
Fouriera (FTICR) rozdzielczośd dla pików o m/z rzędu 1000 sięga nawet miliona) co umożliwia określenie składu
elementarnego próbki z rozdzielczością na poziomie jednego elektronu.
Spektrometria masowa jest metodą badania substancji przy pomocy widma mas atomów i cząsteczek wchodzących w jej
skład. Istota metody polega na tym, że zjonizowane atomy lub cząsteczki substancji są rozdzielane ze względu na
wartośd stosunku m/q (m-masa, q-ładunek jonu) i rejestrowane oddzielnie za pomocą spektrometru masowego. Z
otrzymanego widma mas wyznacza się wartości mas oraz względną zawartośd składników badanej substancji.
Początkowo zadaniem spektrometrii masowej było badanie składu izotopowego pierwiastków oraz precyzyjne
wyznaczanie mas atomów. Obecnie znalazła szerokie zastosowanie jako ogólna metoda analityczna, stosowana w fizyce
doświadczalnej, chemii, biologii molekularnej, technice czy też w ochronie środowiska.
MS jako metoda sekwencjonowania białek umożliwia identyfikację modyfikacji posttranslacyjnych, czy analizę związków
endogennych występujących w bardzo niskich stężeniach. Technikami uzupełniającymi dla MS są elektroforeza kapilarna
(CZE) oraz wysokosprawna chromatografia cieczowa (HPLC). Stosowanie tych metod sprzężonych ze spektrometrem
dostarcza dodatkowych informacji i umożliwia identyfikację związków na jeszcze niższym poziomie stężeo. MS znajduje
również zastosowanie w dziedzinach takich jak ochrona środowiska, kontrola antydopingowa, farmakologia, diagnostyka
medyczna, biotechnologii czy ostatnio dynamicznie się rozwijającej proteomice czyli badaniach nad identyfikacją
proteomu. (PROTEin complement of the genOME - białkowa składowa kodowana przez genom).
Spektrometr masowy – instrument pozwalający na pomiar stosunku masy do ładunku (m/z) analizowanych substancji.
Rozdzielczośd spektrometru – wartośd liczbowa informująca o możliwości rozróżnienia na widmie masowym pików o
zbliżonych masach. W przypadku pojedynczego piku wartośd określająca dokładnośd oznaczenia masy cząsteczkowej
(atomowej) substancji analizowanej. Jeśli spektrometr masowy w danym momencie analizy posiada rozdzielczośd
R=1000 istnieje możliwośd rozróżnienia pików o m/z=1000 oraz m/z=1001. Dla izolowanego piku rozdzielczośd definiuje
jego szerokośd połówkową, tzn. dla R=1000 i piku o m/z=1000 stosunek jego wysokości do szerokości w 0,5 wysokości
wynosi co najmniej 10 (H/L0,5h>=10)
Zasada działania spektrometru masowego - Podstawą działania każdego spektrometru, jest jonizacja cząsteczek badanej
substancji, co umożliwia przyspieszenie jej w polu elektrycznym w próżni. Heterogeniczny strumieo jonów (dodatnich
lub ujemnych) zostaje rozdzielony na szereg składowych, zależnie od stosunku masy do ładunku (m/z). W przypadku
jonów naładowanych dodatnio, masa próbki mierzona w spektrometrze jest powiększona o masę protonu lub protonów
przyłączonych do cząsteczki analizowanej substancji. Dla jonów naładowanych ujemnie masa próbki pomniejszona jest o
masę protonu lub protonów oderwanych od cząsteczki analizowanej substancji. Jonizacja elektronami (EI) jest tu
wyjątkiem i metoda ta powoduje jedynie wybicie elektronu bez przyłączania protonu. Istnieje możliwośd oznaczenia m/z
substancji nie jonizujących się poprzez dołączenie (reakcja chemiczna lub oddziaływanie fizyczne) podstawnika
obdarzonego ładunkiem lub podlegającego jonizacji (tzw. derywatyzacja) lub poprzez utworzenie adduktów np. z sodem
lub potasem.
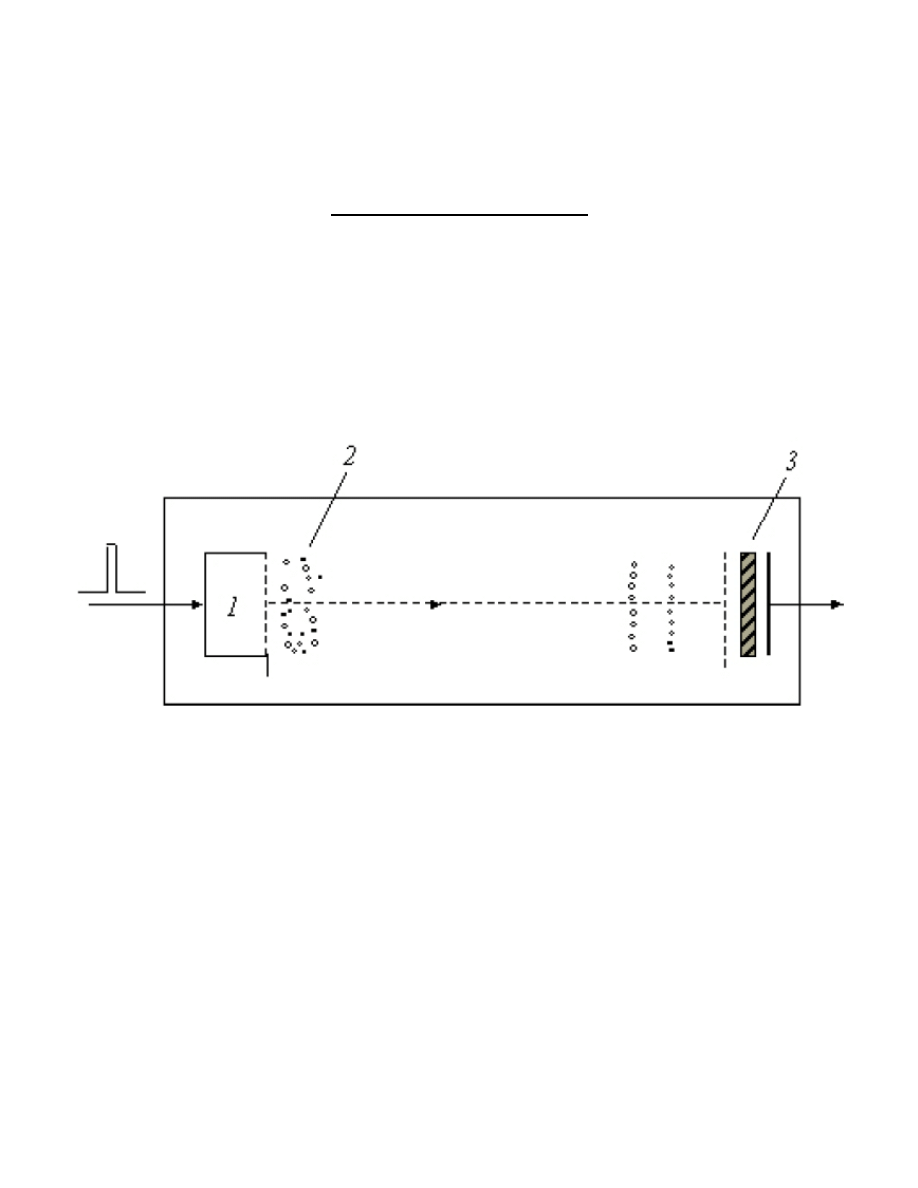
Budowa spektrometru masowego
Każdy spektrometr masowy składa się z pewnych niezbędnych podzespołów, niezależnych od typu instrumentu czy
sposobu jego wykorzystania. Konstrukcję spektrometru przedstawiono za pomocą ogólnego schematu blokowego.
układ wprowadzania próbki → źródło jonów → analizator jonów → detektor jonów → analiza danych
Spektrometry czasu przelotu
W analizatorach czasu przelotu (time-of-flight, TOF) wykorzystana jest zależnośd czasu przelotu jonów od ich stosunku
masy do ładunku. Przypuśdmy, że jony po wyjściu ze źródła są przyspieszane w polu elektrycznym za pomocą różnicy
potencjałów Us i po przebyciu drogi d docierają do detektora.
Rozdzielczośd masowa spektrometru zdefiniowana jest jako wartośd największej masy, przy której możliwe jest
rozdzielenie jonów różniących się o masę jednostkową
Schemat liniowego (jednosegmentowego) spektrometru typu czasu przelotu (TOF). 1 - źródło jonów, 2 - pakiet jonów w
pobliżu źródła, 3 - kanałowy detektor elektronów wtórnych
Rozdzielczośd spektrometru TOF zależy od szeregu czynników:
1) niedokładnośd ogniskowania wynikająca ze skooczonej (niezerowej) szerokości obszaru, w którym są formowane
jony,
2) poszerzenie szerokości pakietu jonów z powodu rozrzutu kątów trajektorii jonów,
3) odchylenie od prostopadłości źródła jonów w stosunku do osi,
4) efektywna głębokośd płytki detektora kanałowego,
5) efekt niejednakowych prędkości początkowych jonów wychodzących ze źródła,
6) niedoskonale prostokątny kształt sygnału wyzwalającego jony w źródle,
7) poszerzenie impulsu prądu w przedwzmacniaczu,
8) wpływ resztkowych pól elektrycznych w obszarze siatek,
9) wpływ ładunku przestrzennego wytworzonego przez jony,
10) czas trwania impulsu jonizacyjnego.
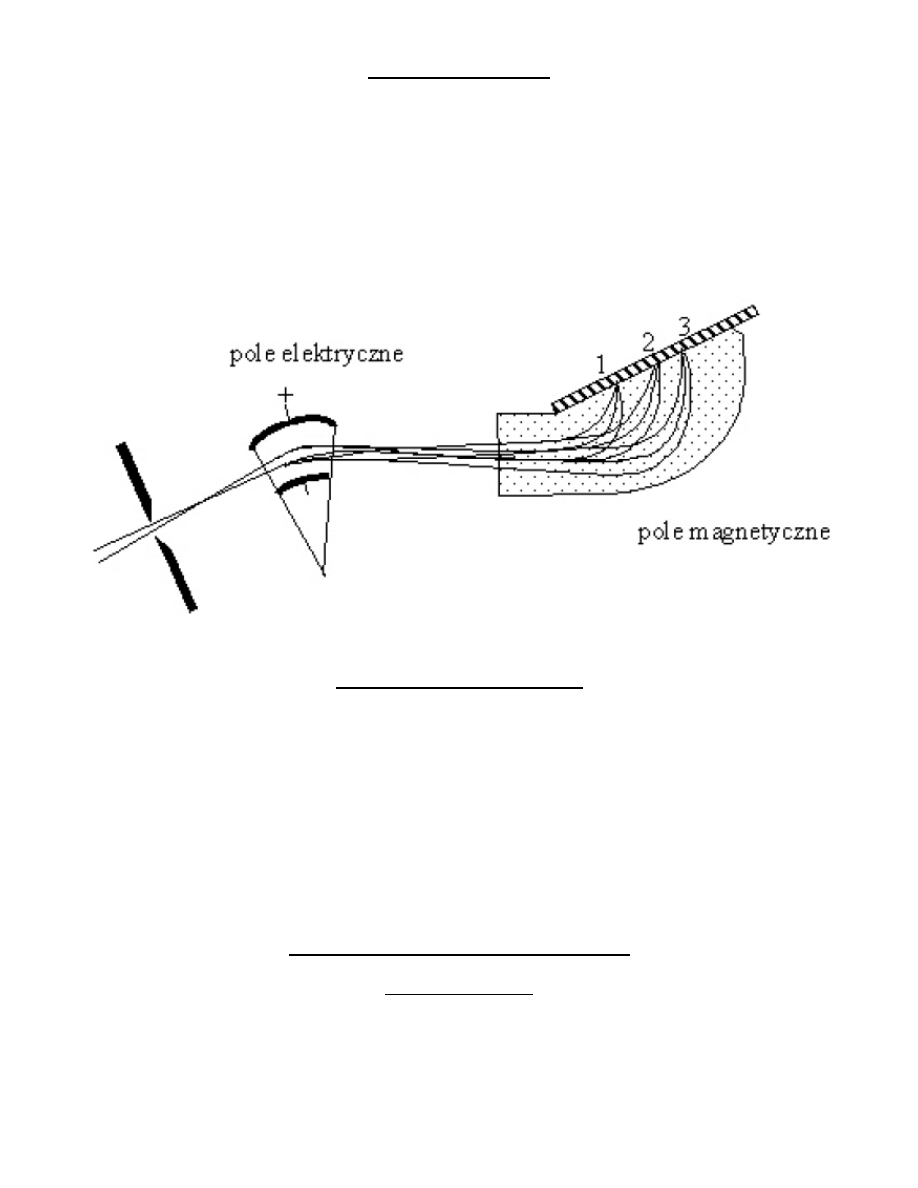
Spektrometr Astona
Ulepszoną wersję spektrografu mas Thomsona zastosował w 1919 roku Aston, który zamiast równoległych względem
siebie pól elektrycznych i magnetycznych zastosował pola wzajemnie prostopadłe. Pole elektryczne rozszczepia wiązkę
jonów według stosunku m/e, ale także według różnych prędkości. Dobierając odpowiednie natężenia pól można
spowodowad aby pole magnetyczne kierowało wszystkie cząstki o różnych prędkościach do jednego określonego punktu
w przestrzeni, pozostawiając jednocześnie rozdzielone wiązki cząstek o różnym e/m. Dzięki temu spektrograf posiada
wyższą transmisję dla jonów dzięki czemu uzyskuje się większą zdolnośd rozdzielczą.
Układ wprowadzania próbki
Poszczególne układy różnią się zależnie od stanu skupienia analizowanej próbki jak również metody jonizacji
zastosowanej do analizy:
stan stały – sondy z probówką, płytki (jonizacja typu EI, MALDI)
stan ciekły – zawory wstrzykowe, pompy strzykawkowe, systemy HPLC, FPLC, systemy elektroforezy kapilarnej (jonizacja
typu ESI, MALDI)
stan gazowy – układy chromatografii GC, komory próżniowe, systemy strzykawek gazoszczelnych (jonizacja typu EI, CI,
ICP)
Źródła jonów - metody jonizacji próbki
Electron impact (EI)
Historycznie pierwsze i najbardziej rozpowszechnione źródło jonów. Jego integralnym elementem jest katoda (filament).
Po przyłożeniu napięcia emituje elektrony o ściśle określonej energii, które zderzając się z cząsteczkami próbki wybijają
elektron/elektrony z ich orbit walencyjnych. Najczęściej stosowany zakres analizy: 10-1000 Da. Cząsteczki o wyższych
masach ulegają łatwo dekompozycji przy stosowaniu jonizacji typu EI. Należy wówczas rozważyd użycie innej techniki
jonizacji. Najczęściej analizowane próbki: niskocząsteczkowe, nieorganiczne, organiczne w postaci stałej. Zastosowania:

potwierdzanie masy cząsteczkowej substancji po syntezie, ochrona środowiska (analiza niskocząsteczkowych
zanieczyszczeo), nadzór prawidłowości procesów technologicznych, kontrola antydopingowa. Postad wyników: piki
obserwowane na widmie masowym posiadają zazwyczaj ładunek +1. W przypadku niskorozdzielczej analizy typu EI masa
cząsteczkowa próbki jest zazwyczaj równa m/z.
Jonizacja chemiczna (CI)
Metoda jonizacji “łagodniejsza” w porównaniu do EI, tzn. daje możliwośd uzyskania jonu molekularnego o większej
intensywności w porównaniu do jonów fragmentacyjnych niż w metodzie EI. Jonizacja substancji następuje na skutek
zderzeo z tzw. jonami pierwotnymi występującymi w źródle jonów (najczęściej są to jony gazów obojętnych, metanu,
izobutanu, amoniaku). Aby zderzenia pomiędzy analitem i jonami pierwotnymi zachodziły wystarczająco często, w źródle
jonów typu CI należy wytworzyd ciśnienie około 60 Pa. Najczęściej stosowany zakres analizy: 10 - 3000 Da. Najczęściej
analizowane próbki: niskocząsteczkowe nieorganiczne i organiczne w postaci stałej. Zastosowania: detekcja w
chromatografii gazowej, farmakologia, kontrola antydopingowa, ochrona środowiska (np. analiza TCDD, PCB). Postad
wyników: jonizacja następuje zazwyczaj poprzez przyłączenie protonu (-ów) do analizowanych cząsteczek co należy
uwzględnid podczas obliczania rzeczywistej masy analitu (dla z=+1 m/z = M-1).
Electrospray (ESI)
Electrospray to jedna z nowszych metod jonizacji próbki w spektrometrii masowej. Próbka ulega jonizacji pod ciśnieniem
atmosferycznym przy użyciu napięcia 2-5kV. Podstawowe zalety metody: minimalna fragmentacja próbki podczas
jonizacji, wysoka czułośd oznaczeo, kompatybilnośd z met. chromatograficznymi (HPLC) oraz elektroforetycznymi (CE),
możliwośd analizy dużych cząsteczek (do ok. 80 000 Da). Cechą charakterystyczną widm masowych ESI są serie pików
odpowiadających kolejnym, wielokrotnie naładowanym, protonowanym jonom *M+zH+z+, [M+(z+1)H](z+1)+, itd.
Powyższe zjawisko umożliwia analizowanie mas znacznie większych niż nominalny zakres pomiarowy analizatora przy
zachowaniu stosunkowo wysokiej rozdzielczości, np. związek o masie 5000 Da po przyłączeniu 5 protonów będzie
obserwowany przy m/z=1001 ([5000 + 5]/5=1001). Najczęściej stosowany zakres analizy: 50 – 80 000 Da. Najczęściej
analizowane próbki: średnio- i wysokocząsteczowe substancje organiczne, peptydy, białka, polimery, kwasy nukleinowe
w postaci ciekłej. Zastosowania: biochemia, biotechnologia, farmakologia, neurochemia, medycyna sądowa, detekcja w
chromatografii cieczowej. Postad wyników: serie pików o różnej ilości przyłączonych/oderwanych protonów. M = (m-
nz)/nz dla z+.
MALDI (desorpcja/jonizacja laserowa w matrycy)
W metodzie MALDI analizowaną substancję jonizuje się po jej uprzednim zmieszaniu z roztworem matrycy (małe
cząsteczki organiczne silnie absorbujące promieniowanie przy stosowanej długości fali lasera). Po odparowaniu
rozpuszczalnika próbkę naświetla się impulsami lasera, co powoduje wzbudzenie elektronów w matrycy. Jony,
utworzone przez przeniesienie protonu między wzbudzoną matrycą a analizowaną substancją, ulegają następnie
desorpcji. Jedną z zalet metody jest możliwośd analizowania substancji o masach nawet do 1 000 000 Da.
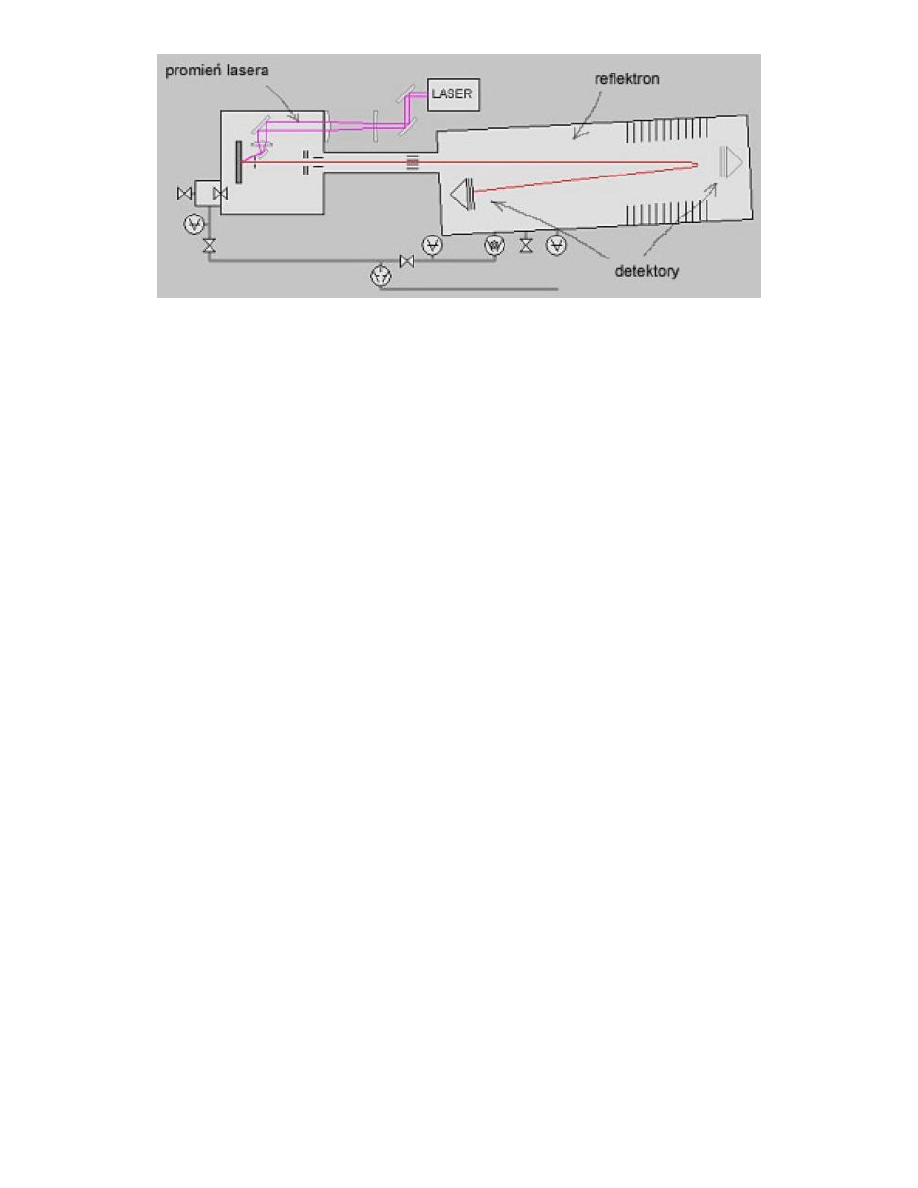
Jonizacja próbki metodą MALDI jest techniką jonizacji, rozwijaną w spektrometrii mas od połowy lat 80–tych. Akronim
MALDI pochodzi od Matrix Assisted Laser Desorption and Ionisation — laserowa desorpcja i jonizacja próbki
wspomagana matrycą. Wykorzystuje ona ideę matrycy pośredniczącej w przekazywaniu energii do badanej substancji,
ułatwiającej jonizację próbki i umożliwiającej badanie substancji nielotnych, wielkocząsteczkowych, polarnych.
Matrycami stosowanymi w technice MALDI są substancje, które:
Dobrze absorbują promieniowanie z zakresu UV
Łatwo sublimują
Po desorpcji dostarczają dużych ilości jonów (protonów) potrzebnych do jonizacji badanej substancji
Analizowaną substancję rozpuszcza się w dowolnym, lotnym rozpuszczalniku. Następnie roztwór próbki miesza się z
roztworem matrycy (użytym w nadmiarze), ewentualnie dodając niewielkie ilości substancji ułatwiających powstawanie
jonów (np. kwas trifluorooctowy, sole litowców i miedziowców).
Mieszaninę, w ilości ok. 1 ml, nanosi się na płytkę ze stali nierdzewnej (slider) i pozwala odparowad rozpuszczalnikowi w
strumieniu powietrza. Celem jest doprowadzenie do wspólnego, jednorodnego wy-krystalizowania mieszaniny badanej
substancji i matrycy. Ponieważ jakośd widma uwarunkowana jest równomiernym rozproszeniem badanej substancji w
materiale matrycy, jest to etap niezwykle istotny dla jakości widma.
Po dokładnym wysuszeniu próbkę wprowadza się do komory pomiarowej aparatu i usuwa powietrze (średnia droga
swobodna jonów ~1 m wymaga próżni 10–6 –10–7 Torr). Po uzyskaniu dostatecznej próżni wykonuje się właściwy
pomiar. W aparacie wykorzystuje się laser gazowy N2 , pracujący impulsowo w zakresie nadfioletu (l = 337 nm).
Skoncentrowany na powierzchni o średnicy 80–100 mm impuls laserowy trwający ok. 3 ns wywołuje ciąg reakcji:
Absorpcja promieniowania głównie przez materiał matrycy.
Odparowanie próbki na głębokośd 2–3 l, czyli 0,5–1 mm i wyrzucenie strumienia gazów prostopadle do jej
powierzchni.
Dysocjacja termiczna matrycy.
Tworzenie jonów (głównie H+, Na+, K+).
Reakcje jonów z badaną substancją i matrycą:
o dysocjacja termiczna z utworzeniem pary kation–anion
o oderwanie elektronu
o oderwanie bądź przyłączenie protonu
o przyłączenie kationu bądź anionu
Wytworzone w ten sposób jony są przyspieszane w polu elektrycznym i kierowane do detektora.
Całkowity czas trwania wymienionych procesów nie przekracza kilku nanosekund.
Rodzaje jonów generowanych w technice MALDI: Dodatnie: [M+H]+ [M+Na]+, [M+K]+, [M+Ag]+[M+ n Matryca]+ [M–
–OH]+ Ujemne: [M–H]– generowane zwłaszcza w przypadku peptydów, polikwasów.
W przypadku próbek biologicznych o wzajemnej proporcji jonów typu *M+H++ w stosunku do *M+Na++, *M+K++, [M+Ag]+
… decyduje zawartośd tych kationów w próbce. Natomiast polimery generują przede wszystkim jony stabilizowane
kationami metali (przeważnie sodu). Jony–addukty zawierające cząsteczkę matrycy, lub cząstkę powstałą w wyniku jej
rozpadu, powstają na ogół z małą wydajnością.
Podsumowanie MALDI :
Zakres analizy: 500-1 000 000 Da
Najczęściej analizowane próbki: średnio- i wysokocząsteczowe substancje organiczne, peptydy, białka, polimery, kwasy
nukleinowe
Zastosowania: biochemia, biotechnologia, farmakologia, neurochemia, immunologia
Postad wyników: zazwyczaj jonizacja +1, rzadziej +2, możliwośd obserwacji niekowalencyjnych kompleksów
Połączenie wysokosprawnej chromatografii cieczowej ze spektrometrią masową
Połączenie wysokociśnieniowej chromatografii cieczowej (HPLC) ze spektrometrią masową łączy zalety obu technik.
Podstawowe zalety:
zwiększona czułośd analizy (próbka zagęszczona na kolumnie chromatograficznej, unikanie strat związanych z
przenoszeniem próbki),
duża oszczędnośd analizowanego materiału,
bezpośrednia analiza skomplikowanych mieszanin,
niewielkie zużycie eluentów ze względu na specyfikę stosowanych kolumn chromatograficznych.
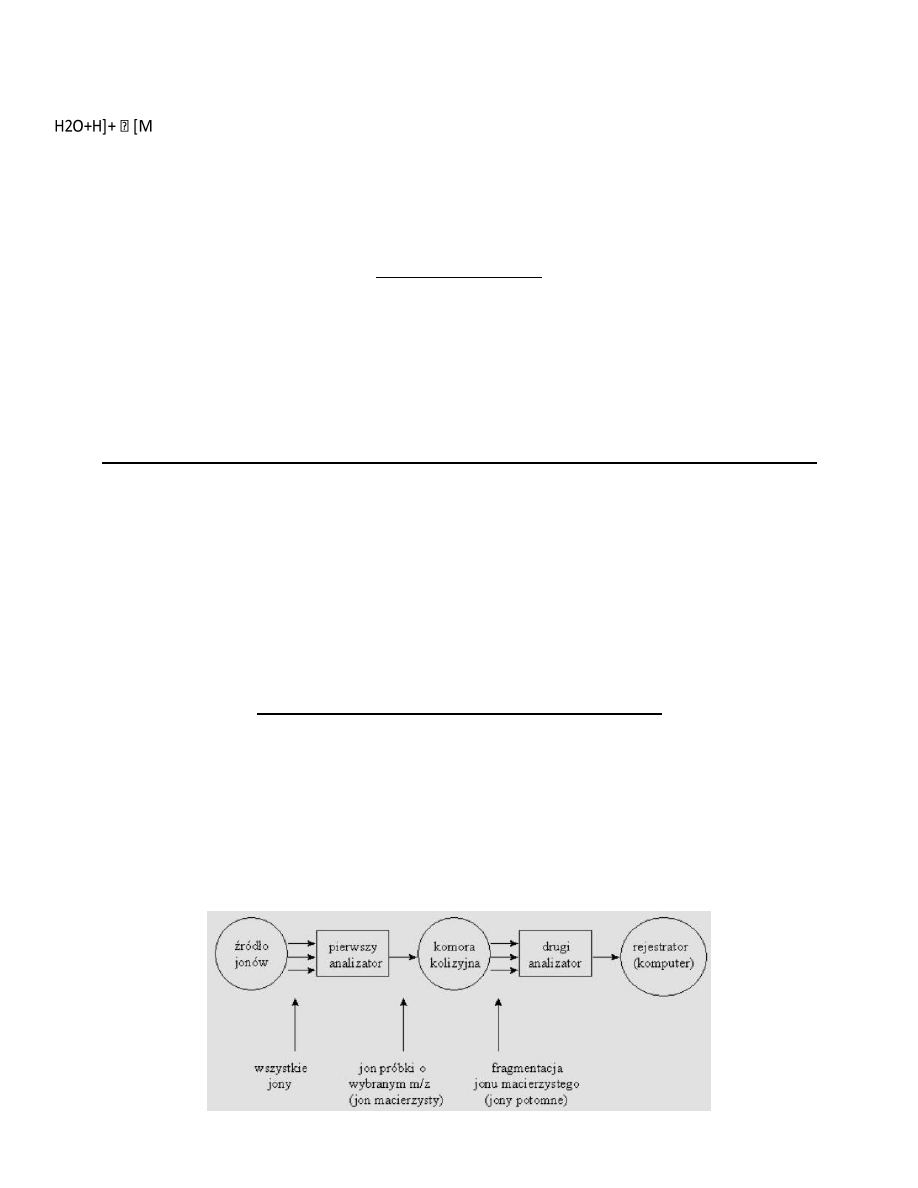
Tandemowa spektrometria masowa (MS/MS)
Pozwala na określanie struktury badanego związku na podstawie jonów fragmentacyjnych, które są charakterystyczne
dla danego związku lub grupy związków. W praktyce metoda MS/MS polega na tym, iż z widma masowego selekcjonuje
się wybrany jon (najczęściej jon molekularny). Jon ten poddawany jest kolizjom (następuje jego zderzenie z
wprowadzonym gazem obojętnym). Na skutek zderzeo jon macierzysty (parent ion) rozpada się na jony fragmentacyjne
(daughter ions). Do przeprowadzania eksperymentów MS/MS znakomicie nadają się spektrometry wyposażone w
pułapkę jonów pełniącą rolę analizatora i detektora. Za pomocą tego typu instrumentów można przeprowadzad analizy
MSn (gdzie n = 2,3 ....ok. 9), co wydatnie zwiększa możliwośd identyfikacji próbek.

Podstawowe defnicje
Jon molekularny – jon obdarzony ładunkiem (ładunkami) powstający w wyniku fragmentacji próbki w źródle jonów
Jon fragmentacyjny – jon powstały w wyniku spontanicznej fragmentacji substancji (np. podczas jonizacji metodą EI) lub
uzyskany techniką tandemowej spektrometrii masowej. Dostarcza informacji o strukturze substancji analizowanej.
Addukt - jon powstały poprzez przyłączenie do analizowanej substancji np. jonu sodowego
Proteom - PROTEin complement of the genOME (ogół białek kodowanych przez genom)
Genom - całośd informacji genetycznej komórki
Pułapka jonowa - typ analizatora stosowany w spektrometrii masowej, często umożliwiający sekwencyjną fragmentację
MSn
Mapa peptydowa – zbiór peptydów powstały w wyniku proteolizy białka z zastosowaniem enzymów o ściśle
zdefiniowanej swoistości. Jest jego „odciskiem palca” (fingerprint) i służy do identyfikacji białek.
Dalton - jednostka masy, dokładnie odpowiada 1,0000 na skali mas atomowych
Dekonwolucja - uzyskanie rzeczywistej masy substancji z widma pików wielokrotnie zjonizowanych
Matryca - niskocząsteczkowe związki organiczne absorbujące promieniowanie lasera.
Derywatyzacja - przeprowadzenie trudno lotnych związków w ich lotne i trwałe pochodne (dla celów GC i GC/MS)
EI (Electron Impact) - jonizacja elektronami
MALDI (Matrix-Assisted Laser Desorption/Ionization) - jonizacja laserem wspomagana matrycą
FTICR (Fourier Transform Ion Cyclotron Resonance) – cyklotronowy rezonans jonowy z transformacją Fouriera
ESI (Electrospray Ionization) - jonizacja przez rozpylanie w polu elektrycznym
HPLC (High Performance Liquid Chromatography) – wysokosprawna chromatografia cieczowa)
MS/MS (Tandem Mass Spectrometry) - tandemowa spektrometria masowa
TOF (Time of Flight Analyser) - analizator czasu przelotu
PSD (Post Source Decay) - rozpad poza źródłem jonów m/z - stosunek wartości masy do liczby ładunków
DIOS (Desorption/Ionization on Porous Silicon) - desorpcja/jonizacja na porowatym krzemie
ICP (Inductively Coupled Plasma) - jonizacja plazmą wzbudzoną indukcyjnie
Wyszukiwarka
Podobne podstrony:
Opracowanie Sciaga MC OMEN
Opracowanie pytań MC OMEN 2
Opracowanie pytań MC OMEN 3
Opracowanie projektu MC OMEN
Opracowanie pytań MC OMEN
Opracowanie pytań MC OMEN
Opracowanie Sciaga MC OMEN
Opracowanie pytań MC OMEN 2
Opracowanie pytań MC OMEN 2
Opracowanie pytań MC OMEN
Opracowanie wykładów biofyzka 1 MC OMEN
Opracowanie wykładów biofyzka 2 MC OMEN
Opracowanie wykładów biofyzka 3 MC OMEN
więcej podobnych podstron