
- 1 -
Automated NMR structure calculation
P
ETER
G
ÜNTERT
Tatsuo Miyazawa Memorial Program, RIKEN Genomic Sciences Center, 1-7-22 Suehiro,
Tsurumi, Yokohama 230-0045, Japan
e mail: guentert@gsc.riken.jp
telephone: +81-45-503-9345
fax: +81-45-503-9343
© 2005 Peter Güntert. All Rights reserved.
Contents
1 Introduction .......................................................................................................................................... 2
2 Principles of automated NOE assignment ............................................................................................ 3
2.1 Chemical shift assignment ............................................................................................................. 3
2.2 Requirements on input data ........................................................................................................... 4
2.3 Ambiguity of chemical shift-based NOE assignment.................................................................... 7
2.4 Ambiguity of structure-based NOE assignment ............................................................................ 8
2.5 Network-anchoring ........................................................................................................................ 8
2.6 Ambiguous distance restraints ..................................................................................................... 11
2.7 Partial NOE assignment............................................................................................................... 11
2.8 Calibration of distance restraints ................................................................................................. 12
2.9 Constraint combination................................................................................................................ 13
2.10 Removal of erroneous restraints by violation analysis .............................................................. 15
2.11 Error-tolerant target function ..................................................................................................... 15
2.12 Refinement in explicit solvent ................................................................................................... 16
2.13 Quality control ........................................................................................................................... 17
2.14 Troubleshooting......................................................................................................................... 19
3 Implementations of automated NOESY assignment .......................................................................... 20
3.1 Semiautomatic methods ............................................................................................................... 20
3.2 The NOAH algorithm .................................................................................................................. 21
3.3 The ARIA algorithm.................................................................................................................... 21
3.4 The CANDID algorithm .............................................................................................................. 23
3.5 CYANA ....................................................................................................................................... 23
3.6 The AUTOSTRUCTURE algorithm ........................................................................................... 26
3.7 The KNOWNOE algorithm ......................................................................................................... 27
4 Assignment-free structure calculation ................................................................................................ 27

- 2 -
1 Introduction
NMR protein structure determination has remained until recently a laborious undertaking that
occupied a trained spectroscopist over several months for each new protein structure. It has
been recognized that many of the time-consuming interactive steps carried out by an expert
during the process of spectral analysis could be accomplished by automated, computational
methods (Moseley and Montelione, 1999; Altieri and Byrd, 2004; Baran et al., 2004;
Gronwald and Kalbitzer, 2004), and many approaches have already been proposed in order to
automate parts of NMR protein structure determination. Today automated methods for NMR
structure calculation are playing a more and more prominent role and will most likely
supersede the conventional manual approaches to solving three-dimensional protein structures
in solution. This chapter gives an introduction to the current state of automated NMR
structure calculation.
So far, all de novo NMR protein structure determinations have followed the “classic way”
(Wüthrich, 1986) that proceeds through the successive steps of sample preparation, NMR
measurements, NMR data processing, peak picking, chemical-shift assignment, NOESY
assignment and collection of other conformational restraints, structure calculation, and
structure refinement. Section 2 is devoted to basic aspects of the principles and problems of
automated NOESY assignment and structure calculation, including questions of reliability,
quality control and troubleshooting. Section 3 presents a selection of various specific
implementations of automated NOESY assignment and structure calculation for which either
the literature bears testimony of widespread use or that embody concepts of particular interest
and future potential. Alternatives to the classic approach that bypass the potentially
cumbersome chemical shift and NOESY assignment steps have been proposed, and will be
discussed in Section 4.
For consistency and simplicity, the following conventions will be used: An interaction
between two or more atoms is manifested by a signal in a multidimensional spectrum. A peak
refers to an entry in a peak list that has been derived from an experimental spectrum by peak
picking. A peak may or may not represent a signal, and there may be signals that are not
represented by a peak. Chemical shift assignment is the process and the result of attributing a
specific chemical shift value to an atom. Peak assignment is the process and the result of

- 3 -
identifying in each spectral dimension the atom(s) that are involved in the signal represented
by the peak. NOESY assignment is peak assignment in NOESY spectra.
2 Principles of automated NOE assignment
Because of resonance and peak overlap it is in practice not straightforward to obtain a
comprehensive set of distance restraints from a NOESY spectrum. NOESY assignment
instead becomes an iterative process in which preliminary structures, calculated from limited
numbers of distance restraints, serve to reduce the ambiguity of the cross peak assignments.
In addition to this problem, considerable difficulties may arise from spectral artifacts and
noise, and from the absence of expected signals because of fast relaxation. These inevitable
shortcomings of NMR data collection are the main reason why until recently laborious
interactive procedures have dominated 3D protein structure determinations. Automated
procedures follow the same general scheme as the interactive approach but do not require
manual intervention during the assignment/structure calculation cycles (Figs. 1 and 2). Two
main obstacles have to be overcome by an automated method starting without any prior
knowledge of the structure: First, based on chemical shifts alone the number of NOESY cross
peaks with unique assignment based on chemical shifts is in general not sufficient to define
the fold of the protein. Therefore, automated methods should have the ability to make use also
of NOESY cross peaks that cannot yet be assigned unambiguously. Second, the automated
program must be able to cope with erroneously or inaccurately picked peaks and with the
incompleteness of the chemical shift assignment of typical experimental data sets. An
automated procedure needs devices to substitute the intuitive decisions made by an
experienced spectroscopist in dealing with imperfect experimental NMR data.
2.1 Chemical shift assignment
In de novo three-dimensional structure determinations of proteins by NMR, the key
conformational data are upper distance limits derived from nuclear Overhauser effects
(NOEs) (Solomon, 1955; Macura and Ernst, 1980; Kumar et al., 1980; Neuhaus and
Williamson, 1989). In order to extract distance restraints from a NOESY spectrum, its cross
peaks have to be assigned, i.e. the pairs of interacting hydrogen atoms have to be identified.
The assignment of NOESY cross peaks requires as a prerequisite the knowledge of the
chemical shifts of the spins from which NOEs are arising. There have been many attempts to

- 4 -
automate this chemical shift assignment step that conventionally has to precede the collection
of conformational restraints and the structure calculation. These methods have been reviewed
recently (Moseley and Montelione, 1999; Altieri and Byrd, 2004; Baran et al., 2004;
Gronwald and Kalbitzer, 2004), and will not be discussed in detail here. Some automated
approaches (Friedrichs et al., 1994; Hare and Prestegard, 1994; Olson and Markley, 1994;
Buchler et al. 1996; Li and Sanctuary, 1997a; Lukin et al., 1997; Zimmerman et al., 1997;
Leutner et al., 1998; Atreya et al., 2000; Bailey-Kellog et al., 2000; Güntert et al., 2000;
Bhavesh et al., 2001; Moseley et al., 2001; Tian et al., 2001; Andrec and Levy, 2002;
Chatterjee et al., 2002; Coggins and Zhou, 2003) target the question of assigning the
backbone and, possibly,
β chemical shifts, usually on the basis of triple resonance
experiments that delineate the protein backbone through one- and two-bond scalar couplings.
Other algorithms (Chin et al., 1992; Xu et al., 1993, 1994; Oschkinat and Croft, 1994; Bartels
et al., 1996, 1997; Choy et al., 1997; Croft et al., 1997; Li and Sanctuary, 1997b; Gronwald et
al., 1998; Pristovšek et al., 2002; Hitchens et al., 2003) are concerned with the more
demanding problem of complete assignment of the amino acid backbone and side-chain
chemical shifts. In most cases, these algorithms require peak lists from a specific set of NMR
spectra as input, and produce lists of chemical shifts of varying completeness and correctness,
depending on the quality and information content of the input data and the capabilities of the
algorithm.
2.2 Requirements on input data
A limiting factor for the application of automated NOE assignment methods is that they rely
on the availability of an essentially complete list of chemical shifts from the preceding
sequence-specific resonance assignment. At present, chemical shift assignment remains
largely the domain of interactive or semi-automated methods, despite of the aforementioned
promising attempts towards automation. Experience shows that in general the majority of the
chemical shifts can be assigned readily whereas others pose difficulties that may require a
disproportionate amount of the spectroscopist’s time. Hence, NMR structure determination
would be speeded up significantly if NOE assignment and structure calculation could be
based on incomplete lists of assigned chemical shifts, provided that the reliability and
robustness of the NMR method for protein structure determination is not compromised.

- 5 -
The influence of incomplete chemical shift assignments on the reliability of NMR structures
obtained by automated NOESY cross peak assignment has been investigated in detail (Jee and
Güntert, 2003) using the program CYANA for combined automated NOESY assignment with
the CANDID algorithm (Herrmann et al., 2002a; see Section 3.4) and torsion angle dynamics-
based structure calculations (Güntert et al., 1997). Various degrees of completeness of the
chemical shift assignment were simulated by randomly omitting entries from the experimental
1
H chemical shift lists that had been used for the earlier, conventional structure
determinations of two proteins. Overall, the results showed that for reliable automated
NOESY assignment with the CYANA program, and, presumably, other NOE assignment
algorithms based on the same principles, around 90% completeness of the chemical shift
assignments for the backbone amide and non-labile protons is required. Furthermore, the
input data must be self-consistent in the sense that the peak lists are faithful representations of
the NOESY spectra and that the positions of the NOESY cross peaks fit the chemical shift
lists within the specified error ranges. The chemical shift tolerances should not significantly
exceed 0.02 ppm for
1
H when working with homonuclear [
1
H,
1
H]-NOESY spectra, or 0.03
ppm when working with heteronuclear-resolved 3D or 4D NOESY spectra, and 0.6 ppm for
15
N and
13
C shifts (Herrmann et al., 2002a). The algorithm was more tolerant against the lack
of chemical shift assignments when using data from a uniformly
13
C- and
15
N-labelled protein
than in the case of homonuclear data for a much smaller protein. This is due to the availability
of
13
C and
15
N chemical shifts that allow resolving many
1
H chemical shift degeneracies such
that the probability of accidental, erroneous NOE assignments is decreased compared to the
case of homonuclear data. In certain cases the lack of a small number of “essential” chemical
shifts can lead to a significant deviation of the structure. For example, the lack of aromatic
chemical shifts was in general found to be more harmful to the outcome of a structure
calculation than that of a similar number of other protons, presumably because aromatic
protons tend to be located in the hydrophobic core of the protein where they give rise to a
higher-than-average number of NOEs. With exclusively homonuclear data significant
deviations from the reference structure of more than 2 Å were sometimes observed already at
the omission of 20% of the aromatic chemical shifts, which corresponds to an overall
omission ratio of less than 2% of all assigned
1
H chemical shifts. On the other hand, in
practice the algorithm might be expected to tolerate a slightly higher degree of
incompleteness in the chemical shift assignments than the simulations of Jee and Güntert
(2003) suggested if most missing assignments are of “unimportant” chemical shifts that are

- 6 -
involved in few NOEs only. This is usually the case because the chemical shifts of protons
that are involved in many NOEs are intrinsically easier to assign than those exhibiting only
few NOEs.
CYANA uses network-anchoring and constraint combination, two devices that have been
designed and shown to be effective in minimizing the impact of incomplete and/or erroneous
pieces of input data (see Sections 2.5 and 2.9). Chemical shift assignment-based automated
NOE assignment without network-anchoring and constraint combination can be more
susceptible to the deleterious effects from missing chemical shift assignments or artifacts in
the input data.
Instead of using an invariable, fixed list of user-supplied chemical shift assignments,
programs may try to find additional chemical shift assignments during automated NOESY
assignment and the structure calculation. Such methods have been proposed and applied when
a preliminary structure was available (Hare and Wagner; 1999): Starting from nearly
complete chemical shift assignments for the backbone and for 348 side-chain protons of the
28 kDa single-chain T cell receptor protein, the chemical shifts of 40 additional side-chain
protons could be found by a combination of chemical shift prediction with the program
SHIFTS (Ösapay and Case, 1991; Sitkoff and Case, 1997) and NOE assignment with ARIA
(Nilges et al., 1997).
In contrast to the susceptibility against missing chemical shift assignments, automated
structure calculation with the CYANA program was found to be tolerant with respect to
incomplete NOESY peak picking (Jee and Güntert, 2003). The algorithm tolerated the
omission of up to 50% of the NOESY cross peaks that were used for the conventional
structure determinations with only a moderate decrease in the precision and accuracy of the
resulting structure. Even when half of the NOESY peaks were omitted from the experimental
input peak lists from 3D NOESY spectra, RMSD values to the reference structure remained in
the region of 2 Å. Similar behavior was observed when only homonuclear data was available,
albeit with a somewhat more pronounced dependence on the omission rate and RMSD bias
values occasionally exceeding 2 Å in runs with 30% NOESY peak omission ratio. These
findings suggest that it is better to strive for correctness than for ultimate completeness of the
input NOESY peak lists.

- 7 -
2.3 Ambiguity of chemical shift-based NOE assignment
Because of the limited accuracy of experimentally determined chemical shift values and peak
positions many NOESY cross peaks cannot be attributed to a single, unique spin pair but have
an ambiguous NOE assignment comprising multiple spin pairs. A simple mathematical model
of the NOESY assignment process by chemical shift matching gives insight into this problem
(Mumenthaler et al., 1997). It assumes a protein with n hydrogen atoms, for which complete
and correct chemical shift assignments are available, and N cross peaks picked in a 2D
[
1
H,
1
H]-NOESY spectrum with an accuracy of the peak position of
∆ω, i.e. the position of the
picked peak differs from the resonance frequency of the underlying signal by no more than
∆ω in both spectral dimensions. Under the simplifying assumption of a uniform distribution
of the proton chemical shifts over a spectral width
∆Ω, the chemical shift of a given proton
falls within an interval of half-width
∆ω about a given peak position with probability
∆Ω
∆
=
ω
2
p
. Peaks with unique chemical shift-based assignment have in both spectral
dimensions exactly 1 out of all n proton shifts inside the tolerance range
∆ω from the peak
position. Their expected number is
∆Ω
∆
−
−
−
=
≈
−
=
/
4
2
2
2
unique
)
1
(
ω
n
np
n
Ne
Ne
p
N
N
. (1)
N
unique
decreases exponentially with increasing size of the protein (n) and increasing chemical
shift tolerance range (
∆ω). For a typical small protein with, for instance, n = 500 proton
chemical shifts within a range of
∆Ω = 10 ppm and chemical shift accuracies of ∆ω = 0.01,
0.02 or 0.03 ppm, respectively, Eq. 1 predicts that only 14%, 1.8% or 0.25% of the NOEs can
be assigned unambiguously based solely on chemical shift information, which is generally
insufficient to calculate a preliminary three-dimensional structure. For peak lists obtained
from
13
C- or
15
N-resolved 3D [
1
H,
1
H]-NOESY spectra, the ambiguity in one of the proton
dimensions can usually be resolved by reference to the hetero-spin, so that Eq. 1 is replaced
by
∆Ω
∆
−
−
=
≈
/
2
unique
ω
n
np
Ne
Ne
N
. (2)
With regard to assignment ambiguity, 3D NOESY spectra are thus equivalent to homonuclear
NOESY spectra from a protein of half the size or with twice the accuracy in the determination
of the chemical shifts and peak positions.

- 8 -
The influence of chemical shift tolerances on NMR structure calculations using ARIA
protocols for assigning NOE data has been assessed systematically by Fossi et al. (2005).
2.4 Ambiguity of structure-based NOE assignment
Once available, a preliminary three-dimensional structure may be used to resolve ambiguous
NOE assignments. The ambiguity is resolved if only one out of all chemical shift-based
assignment possibilities corresponds to an interatomic distance shorter than the maximal
NOE-observable distance, d
max
. Assuming that the hydrogen atoms are evenly distributed
within a sphere of radius R that represents the protein, the probability q that two given
hydrogen atoms are closer to each other than d
max
can be estimated by the ratio between the
volumes of two spheres with radii d
max
and R, respectively:
3
max
)
/
(
R
d
q
=
. Using d
max
= 5 Å,
one obtains q ≈ 4% for a nearly spherical protein with a radius of about 15 Å. Thus, under
ideal conditions about 96% of the peaks with two assignment possibilities can be assigned
uniquely by reference to the protein structure. Even by reference to a perfectly refined
structure, however, it is impossible to resolve all assignment ambiguities, since the
probability q will always be larger than 0.
2.5 Network-anchoring
Network-anchoring (Herrmann et al., 2002a) exploits the observation that the correctly
assigned restraints form a self-consistent subset in any network of distance restraints that is
sufficiently dense for the determination of a protein 3D structure. In contrast, the erroneously
assigned restraints are randomly distributed in space, generally contradicting each other.
Network-anchoring evaluates the self-consistency of NOE assignments independent from any
previous knowledge on the 3D protein structure and can thus compensate for the absence of
3D structural information at the outset of a de novo structure determination (Fig. 3). Network-
anchoring is important for finding a well-defined, essentially correct structure already in the
first cycle of the structure calculation and is a major factor for the robustness of automated
NOESY assignment with the program CYANA (Herrmann et al., 2002a; Güntert, 2004). The
requirement that each NOE assignment must be embedded in the network of all other
assignments makes network-anchoring a sensitive approach for detecting erroneous restraints.
These may also include “lonely” restraints that artificially constrain unstructured parts of the
protein. Since such lonely restraints do not lead to systematic restraint violations during the

- 9 -
structure calculation, they could not be detected and eliminated by 3D structure-based peak
filters.
In the CANDID algorithm, the network-anchoring score N
αβ
for a given initial assignment of
a NOESY cross peak to an atom pair (
α,β) is calculated by searching all atoms γ in the same
or in the neighboring residues of either
α or β that are connected simultaneously to both
atoms
α and β (Herrmann et al., 2002a). The connection may either be an initial assignment
of another peak (in the same or in another peak list) or the fact that the covalent structure
implies that the corresponding distance must be short enough to give rise to an observable
NOE. Each such indirect path contributes to the total network-anchoring score for the
assignment (
α,β) an amount given by the product of the generalized volume contributions
(Herrmann et al., 2002a) of its two parts,
α→γ and γ→β. N
αβ
has an intuitive meaning as the
number of indirect connections between the atoms
α and β through a third atom γ, weighted
by their respective generalized volume contributions.
In the program CYANA, network-anchoring is implemented in the probabilistic NOE
assignment algorithm. The program calculates the probability P
network
that a given initial
assignment to an atom pair (
α,β) corresponds to a distance d
αβ
shorter than the upper distance
bound u derived from the NOESY cross peak volume. The network-anchoring based
probability is computed from individual probabilities, P
1
, P
2
,… , defined below, that represent
different possible ways to confirm that the assignment (
α,β) corresponds to a short enough
distance:
L
)
1
)(
1
(
1
2
1
network
P
P
P
−
−
−
=
. (3)
P
network
is always larger than the individual probabilities, P
1
, P
2
,… Therefore, network-
anchoring requires that some (not necessarily all) individual probabilities are high. The
individual probabilities include the following cases:
(a) The a priori probability that two atoms in a protein of radius R are closer than the upper
limit u is
3
1
)
/
(
)
(
R
u
u
d
P
=
≤
αβ
. (4)
(b) The covalent structure may imply that the distance d
αβ
is shorter than an upper bound, c:
(
)
1
,
)
/
(
min
)
(
3
2
c
u
u
d
P
=
≤
αβ
. (5)

- 10 -
This applies to short-range assignments.
(c) Another NOE, e.g. a symmetry-related peak, exists with probability P’ of having the same
assignment, (
α,β),
(
)
1
,
)
'
/
(
min
)
'
(
'
)
(
3
3
u
u
u
d
P
u
d
P
⋅
≤
=
≤
αβ
αβ
.
(6)
)
'
(
'
u
d
P
≤
αβ
is the probability that the assignment (
α,β) is correct for symmetry-related peak
with upper distance bound u’.
(d) Two NOEs exist that connect atoms
α and β through a third atom, γ:
)
,
;
(
)
(
)
(
)
(
4
βγ
αγ
βγ
βγ
αγ
αγ
αβ
u
u
u
f
u
d
P
u
d
P
u
d
P
⋅
≤
⋅
≤
=
≤
,
(7)
)
(
αγ
αγ
u
d
P
≤
and )
(
βγ
βγ
u
d
P
≤
denote the probabilities that the assignments (
α,γ) and (β,γ)
of the two “indirect” NOEs with upper distance bounds u
αγ
and u
βγ
are correct. The function f
is a geometric factor that describes the probability for the distance d
αβ
to be shorter than the
upper bound u, given that the two distances d
αγ
and d
βγ
are shorter than u
αγ
and u
βγ
,
respectively. One of the two NOEs can be replaced by a covalently constrained distance. In
this case the NOE-derived upper bound is replaced by the one implied by the covalent
structure and the corresponding probability is set to 1.
(e) The atoms
α and β are close in the covalent structure to atoms α’ and β’, respectively, that
are connected by an NOE:
)
,
,
;
(
)
(
)
(
'
'
'
'
'
'
'
'
5
β
α
ββ
αα
β
α
β
α
αβ
u
c
c
u
g
u
d
P
u
d
P
⋅
≤
=
≤
.
(8)
)
(
'
'
'
'
β
α
β
α
u
d
P
≤
and g are defined as the analogous quantities in Eq. 7. c
αα
’
and c
ββ
’
are the
upper bounds derived from the covalent structure for the distances d
αα
’
and d
ββ
’
.
The overall network-anchoring probability can include in the product of Eq. 3 multiple terms
of types (c)-(e) that reflect multiple indirect paths. The calculation of the network-anchoring
probability is recursive in the sense that its calculation for a given peak requires the
knowledge of the probabilities from other peaks, which in turn involve the corresponding
network-anchoring probabilities. Therefore, the calculation of these quantities is iterated until
convergence. Note that the peaks from all peak lists contribute simultaneously to network-
anchored assignment.

- 11 -
2.6 Ambiguous distance restraints
Ambiguous distance restraints (Nilges, 1993, 1995) are an important and powerful concept
for the handling of ambiguities in NOESY cross peak assignments. When using ambiguous
distance restraints, each NOESY cross peak is treated as the superposition of the signals from
each of its multiple assignments, using relative weights proportional to the inverse sixth
power of the corresponding interatomic distance. A NOESY cross peak with a unique
assignment possibility gives rise to an upper bound b on the distance d(
α,β) between two
hydrogen atoms,
α and β. A NOESY cross peak with n > 1 assignment possibilities can be
seen as the superposition of n degenerate signals and interpreted as an ambiguous distance
restraint,
b
d
≤ , with
6
/
1
1
6
−
=
−
⎟
⎠
⎞
⎜
⎝
⎛
=
∑
n
k
k
d
d
. (9)
Each of the distances d
k
= d(
α
k
,
β
k
) in the sum of Eq. 9 corresponds to one assignment
possibility to a pair of hydrogen atoms,
α
k
and
β
k
. Because the “r
-6
-summed distance” d is
always shorter than any of the individual distances d
k
, an ambiguous distance restraint is
never falsified by including incorrect assignment possibilities, as long as the correct
assignment is present.
2.7 Partial NOE assignment
Despite of the property of ambiguous distance restraints that additional, even wrong
assignment possibilities added to an ambiguous distance restraint that contains one or several
correct assignments do not render the restraint incompatible with the correct structure, it is
important to keep the ambiguity of NOE assignments small in order to obtain a well-defined
structure. This is because additional assignment possibilities “dilute” the information
contained in an ambiguous distance restraint and make it more difficult for the structure
calculation algorithm to find the correct structure.
To this end, the “volume contribution”, i.e. the relative contribution C
k
of each assignment
possibility k to the total peak intensity, is estimated from the three-dimensional structure from
the previous cycle by (Nilges et al., 1997)

- 12 -
6
−
⎟
⎟
⎠
⎞
⎜
⎜
⎝
⎛
=
d
d
C
k
k
, (10)
where
L
denotes the average over the individual conformers of the structure bundle.
Alternatively, when spin diffusion is taken into account by a relaxation matrix treatment, the
volume contributions C
k
are obtained from the back-calculated NOE intensities (Linge et al.,
2004a). In either case, the volume contributions are normalized such that the sum over all
contributions to a given peak equals 1. A partial assignment is then achieved by ordering the
contributions by decreasing size, and discarding the smallest contributions such that
p
C
p
N
k
k
>
∑
=1
, (11)
where p is the “assignment cutoff” and N
p
the number of contributions to the peak necessary
to account for a fraction of the peak volume larger than p (Nilges et al., 1997). For instance,
in the ARIA algorithm the parameter p is decreased from cycle to cycle and typically takes
the values 1.0, 0.9999, 0.999, 0.99, 0.98, 0.96, 0.93, 0.9, 0.8 in cycles 0 to 8, respectively
(Linge et al., 2001). To give an intuitive meaning to the assignment cutoff p, a cross peak
with two assignments may be considered (Nilges and O’Donoghue, 1998): If the shorter of
the two distances is 2.5 Å, a value p = 0.999 will exclude a second distance of 7.9 Å, a value
p = 0.95 a second distance of 4.1 Å, and a value p = 0.8 a second distance of 3.3 Å. If the
shorter distance is 4 Å, the corresponding minimal excluded distances are 12.6, 6.6 and 5.2 Å,
respectively.
2.8 Calibration of distance restraints
Under the assumption of isolated spin pairs in a rigid molecule, the target distances d
NOE
can
be obtained from the cross peak volume V by a simple calibration function,
6
/
1
NOE
)
(
−
= CV
d
.
The calibration constant C can be set by the user or determined automatically, for example by
setting
∑
−
=
NOEs
6
/V
d
C
, where the sum runs over all NOEs with a corresponding average
distance d smaller than a cutoff of typically 6 Å (Linge et al., 2001). In the ARIA
algorithm, an upper bound
2
NOE
NOE
d
d
u
ε
+
=
and a lower bound
2
NOE
NOE
d
d
l
ε
−
=
(typically
ε
= 0.125 Å
−1
) are derived from each target distance d
NOE
(Linge et al., 2001). Most other
algorithms apply only an upper bound. Alternatively, spin diffusion effects (Kalk and

- 13 -
Berendsen, 1980) can be taken into account by a relaxation matrix approach based on the
simulation of the NOE spectrum rather than the direct use of the individual distances d
(Linge et al., 2004a). A fast matrix squaring scheme performs the potentially time-consuming
relaxation matrix analysis efficiently, and the deviation of the calculated NOE from the value
resulting from the isolated spin pair approximation is used to derive a correction factor for the
target distance. In this way, severe cases of spin diffusion can be detected and corrected
within the framework of the automated algorithm.
2.9 Constraint combination
In NMR structure determinations of biological macromolecules spurious distance restraints
may arise from misinterpretation of noise and spectral artifacts. This situation is particularly
critical at the outset of a structure determination, before the availability of a preliminary
structure for 3D structure-based filtering of restraint assignments. Constraint combination
(Herrmann et al., 2002a) aims at minimizing the impact of such imperfections on the resulting
structure at the expense of a temporary loss of information. It is typically applied in the first
two cycles of automated NOESY assignment with the program CYANA and consists of
generating distance restraints with combined assignments from different, in general unrelated,
cross peaks (Fig. 4). The basic property of ambiguous distance restraints—that the restraint
will be fulfilled by the correct structure whenever at least one of its assignments is correct,
regardless of the presence of additional, erroneous assignments—then implies that such
combined restraints have a lower probability of being erroneous than the corresponding
original restraints, provided that the fraction of erroneous original restraints is smaller than
50%.
Two basic modes of constraint combination are “2→1” combination of all assignments of two
long-range peaks each into a single restraint and “4→4” pairwise combination of the
assignments of four long- range peaks into four restraints (Herrmann et al., 2002a). Let A, B,
C, D denote the sets of assignments of four peaks. Then, 2→1 combination replaces two
restraints with assignment sets A and B, respectively, by a single ambiguous restraint with
assignment set A
∪ B, the union of sets A and B. 4→4 pairwise combination replaces four
restraints with assignments A, B, C and D by four combined ambiguous restraints with
assignment sets A
∪ B, A ∪ C, A ∪ D and B ∪ C, respectively. In both cases constraint
combination is not applied to the short-range peaks, because in case of error their effect on

- 14 -
the global fold of a protein is minimal (Nabuurs et al., 2003). The number of long-range
restraints is cut in half by 2→1 combination but stays constant upon 4→4 pairwise
combination. The latter approach thus preserves more of the original structural information,
and can furthermore take into account that certain peaks and their assignments are more
reliable than others, because the peaks with assignment sets A, B, C, D are used 3, 2, 2, 1
times, respectively, to form combined restraints. To this end, the peaks included in constraint
combination are sorted according to their total residue-wise network-anchoring score
(Herrmann et al., 2002a) and 4→4 combination is performed by selecting the assignments A,
B, C, D from the first, second, third, and fourth quarter of the sorted list.
The effect of constraint combination on the expected number of erroneous distance restraints
in the case of 2→1 combination can be estimated quantitatively by assuming an original data
set containing N long-range peaks, and a uniform probability p
<< 1 that a long-range peak
would lead to an erroneous restraint. By 2→1 constraint combination, these are replaced by
N/2 restraints that are erroneous with probability p
2
. In the case of 4→4 combination, it may
be assumed that the same N long-range peaks can be classified into four equally large classes
with probabilities
αp, p, p,
p
)
2
(
α
−
respectively, that they would lead to erroneous restraints.
The overall probability for an input restraint to be erroneous is again p. The parameter
α,
1
0
≤
≤
α
, expresses how much “safer” the peaks in the first class are compared to those in
the two middle classes, and in the fourth, “unsafe” class. After 4→4 combination, there are
still N long-range restraints but with an overall error probability of
2
2
)
4
/
)
1
(
(
p
α
α
−
+
, which
is smaller than the probability p
2
obtained by simple 2→1 combination provided that the
classification into more and less safe classes was successful (
α < 1). For instance, 4→4
combination will transform an input data set of 900 correct and 100 erroneous long-range
cross peaks (i.e., N = 1000, p = 0.1) that can be split into four classes with
α = 0.5 into a new
set of approximately 993 correct and 7 erroneous combined restraints. Alternatively, 2→1
combination will yield under these conditions approximately 495 correct and 5 erroneous
combined restraints. Unless the number of erroneous restraints is high, 4→4 combination is
thus preferable over 2→1 combination in the first two NOESY assignment and structure
calculation cycles.
The upper distance bound b for a combined restraint is formed from the two upper distance
bounds b
1
and b
2
of the original restraints either as the r
-6
-sum,
6
/
1
6
2
6
1
)
(
−
−
−
+
=
b
b
b
, or as the
maximum,
)
,
max(
2
1
b
b
b
=
. The first choice minimizes the loss of information if two already

- 15 -
correct restraints are combined, whereas the second choice avoids the introduction of too
small an upper bound if a correct and an erroneous restraint are combined.
2.10 Removal of erroneous restraints by violation analysis
Experimental peak lists can in practice not be assumed to be completely free of errors,
especially in the early stages of a structure determination or if they originate from automatic
peak picking. In addition, if the chemical shift assignment is incomplete, even the most
carefully prepared peak list will contain peaks that cannot be assigned correctly, namely those
involving unassigned spins, because most automated NOE assignment algorithms do not
attempt to extend or modify the chemical shift assignments provided by the user. When
building a three-dimensional structure from NOE data, most erroneous distance restraints will
be inconsistent with each other and with the correct ones. The erroneous restraints can
therefore, in principle, be detected by analyzing the violations of restraints with respect to the
bundle of three-dimensional structures from the previous cycle of calculation. The problem is
to distinguish violations arising from incorrect restraints from those of correct restraints that
appear as a result of insufficient convergence of the structure calculation algorithm, or as an
indirect effect of structural distortions caused by other erroneous restraints. Violations due to
incorrect restraints can be expected to occur in the majority of conformers rather than
sporadically. Therefore, a violation analysis can be performed by counting the conformers in
which a given restraint is violated by more than a cutoff that is decreased gradually from an
initial large value of 1–2 Å in the second cycle to about 0.1 Å in the final cycle of the
automated structure calculation. If this is the case for a given restraint in more than, say, 50%
of all conformers, several options are possible (Mumenthaler and Braun, 1995; Linge et al.,
2001; Herrmann et al., 2002a): The peak may either be reported as a problem but still used
without change, or the upper distance bound may be increased, or the restraint may be
removed from the input for the structure calculation in the current cycle. Obviously, this kind
of violation analysis can be applied only after a first preliminary structure has been obtained.
2.11 Error-tolerant target function
In order to reduce distortions in the structures resulting from erroneous distance restraints that
passed undetected through the violation analysis, the contribution to the target function from a
severely violated restraint should be limited (Mumenthaler and Braun, 1995). For instance,

- 16 -
ARIA uses in the structure calculation with CNS a target function with a linear asymptote for
large violations which limits the maximal force exerted by a violated distance restraint. The
target function for a single distance restraint is (Nilges and O’Donoghue, 1998):
⎪
⎪
⎪
⎩
⎪⎪
⎪
⎨
⎧
+
≥
−
+
−
−
+
−
+
<
<
−
≤
≤
<
−
=
.
if
)
(
)
2
(
)
2
3
(
;
if
)
(
;
if
0
;
if
)
(
)
(
2
2
2
a
u
d
u
d
u
d
a
a
a
a
a
u
d
u
u
d
u
d
l
l
d
l
d
d
f
γ
γ
γ
(12)
Here,
d
denotes the r
-6
-summed distance of Eq. 9, l and u are the lower and upper distance
bounds,
γ is the slope of the asymptotic potential, and a is the violation at which the potential
switches from harmonic to asymptotic behavior.
The use of NOE pseudo-potential energy function that is linear in the size of the restraint
violation for each individual assignment possibility has been proposed by Kuszewski et al.
(2004). In this approach, a violated restraint for a given assignment results in a force of
constant magnitude, independent of the size of the restraint violation.
As an alternative, implemented in the program CYANA, the idea of ambiguous distance
restraints can be extended in order to confine the actual contribution of a strongly violated
restraint to the target function in an intuitive way to a certain maximum value, v
max
, regardless
of the actual size of the large violation. When violation confinement is active, the effective
distance, d of Eq. 9, to be compared with the upper distance bound, b, is calculated as
6
/
1
1
6
6
max
)
(
−
=
−
−
⎟
⎠
⎞
⎜
⎝
⎛
+
+
=
∑
n
k
k
d
v
b
d
. (13)
The basic property of ambiguous distance restraints implies that
max
v
b
d
+
<
and thus
confines the apparent distance restraint violation to less than v
max
.
2.12 Refinement in explicit solvent
Strongly simplified, “soft” force fields are generally used for the de novo calculation of NMR
structures. There are two reasons for this: Computational efficiency and the need to allow for
a reasonably smooth folding pathway of the polypeptide chain from a random initial structure
to the native conformation. This pathway should not be obstructed by high energy barriers as
they occur if steep, divergent potentials such as the Lennard-Jones potential of standard

- 17 -
classical molecular dynamics force fields are used. The stiffness incurred by potentials that
impede the interpenetration of parts of the molecule during the initial stages of the simulated
annealing procedure would result in most conformers being trapped far from the native
structure in local minima with unfavorable energies.
However, since the physical reality of the non-bonded attractive and repulsive interactions is
only crudely approximated in this way, the resulting structures have often appeared to be of
low quality when submitted to structure validation programs that put much emphasis on such
features as the appearance of the Ramachandran plot, staggered rotamers of side-chain torsion
angles, covalent and hydrogen bond geometry, and electrostatic interactions. To remedy this
situation, a short molecular dynamics trajectory in explicit solvent (Allen and Tildesley, 1987;
Leach, 2001) may be used to refine the final structure in ARIA (Linge et al. 2004b). It could
be shown that a thin layer of solvent molecules around the protein is sufficient to obtain a
significant improvement in validation parameters over unrefined structures, while maintaining
reasonable computational efficiency (Linge et al., 2004b; Spronk et al., 2002).
2.13 Quality control
A variety of methods and criteria for the validation of NMR protein structures have been
proposed or are in use (Spronk et al., 2004), and their importance has recently been assessed
by a large-scale effort to recalculate NMR solution structures for which the experimental
restraints have been deposited in the Protein Data Bank (Nederveen et al., 2005).
Final structures from an automatic algorithm that have a low RMSD within the bundle of
conformers but differ significantly from the “correct” structure are problematic because,
without knowledge of a reference structure, they may appear at first glance as good, well-
defined solutions. In a conventional structure calculation based on manual NOESY
assignment, incomplete or inconsistent input data will be manifested by large RMSD and/or
target function values of the final structure bundle, which will prompt the spectroscopist to
correct and/or complete the input data for a next round of structure calculation. The test
calculations of Jee and Güntert (2003) showed that for structure calculation with automated
NOE assignment neither the RMSD value of the final structure nor the final target function
value are suitable indicators to discriminate between correct and biased results. Other criteria
are needed to evaluate the outcome.

- 18 -
On the basis of the initial experience with the CANDID algorithm, guidelines for successful
runs were proposed (Herrmann et al., 2002a). These comprised six criteria that should be met
simultaneously: (1) average CYANA target function value of cycle 1 below 250 Å, (2)
average final CYANA target function value below 10 Å
2
, (3) less than 20% unassigned
NOEs, (4) less than 20% discarded long-range NOEs, (5) RMSD value in cycle 1 below 3 Å,
and (6) RMSD between the mean structures of the first and last cycle below 3 Å. Criterion (4)
refers to the percentage of NOEs discarded by the CANDID algorithm among all NOEs with
assignments exclusively between atoms separated by 4 or more residues along the
polypeptide sequence. Criteria (3) and (4) impose a limit on the number of NOEs that are not
used to generate distance restraints for the final structure calculation, and thus measure the
completeness with which the picked NOE cross peaks can be explained by the resulting
structure.
The validity of the original guidelines as sufficient conditions for successful CYANA runs
was confirmed by the fact that all the structure calculations in the systematic study of Jee and
Güntert (2003) with an RMSD bias (Güntert, 1998) to the reference structure of more than 2
Å violated one or several of the six criteria. On the other hand, these test calculations revealed
a certain redundancy among the six original criteria. Provided that the input peak lists do not
deliberately misinterpret the underlying NOESY spectra (to which the algorithm has no direct
access), the aforementioned criteria of Herrmann et al. (2002a) can be replaced by only two
conditions for successful structure calculation with automated NOESY assignment: Less than
25% of the long-range NOEs have been discarded by the automated NOESY assignment
algorithm for the final structure calculation, and the backbone RMSD to the mean coordinates
for the structure bundle of the first cycle does not exceed 3 Å.
The percentage of discarded long-range NOEs cannot be calculated readily outside the
program that generates the NOE assignments, because it requires knowledge of the possible
assignments also for the NOESY cross peaks that were excluded from the generation of
conformational restraints. In this case, an overall percentage of unused cross peaks of less
than 15 % can be used as an alternative criterion that is straightforward to evaluate from the
final assigned output peak lists, in which unused cross peaks remain unassigned. Among these
two alternatives, the percentage of discarded long-range NOEs is a slightly more sensitive
indicator of the accuracy of the final structure than the overall percentage of unused cross
peaks because the latter includes also peaks with short-range assignment or with no

- 19 -
assignment possibility at all that are expected to have little distorting effect on the resulting
structure.
The ability of the program to find a well-defined structure in the initial cycle of NOE
assignment and structure calculation, as measured by the RMSD within the structure bundle
in cycle 1, is an important factor that strongly influences the accuracy of the final structure.
This can be understood by considering the iterative nature of the automated NOE assignment
algorithm, in which each cycle except cycle 1 is dependent on the structure obtained in the
preceding cycle. A low precision of the structure from cycle 1 may hinder convergence to a
well-defined final structure, or, more dangerously, opens the possibility of a structural drift in
later cycles towards a precise but inaccurate final structure.
2.14 Troubleshooting
If the output of a structure calculation based on automated NOESY assignment does not
fulfill the aforementioned guidelines, the structure will in many cases still be essentially
correct, but should not be accepted without further validation. The normal approach in this
case is to improve the quality of the input chemical shift and peak lists, and to perform a new
complete structure calculation, until the criteria are met. Usually, this can be achieved
efficiently because the output from an unsuccessful run, even though the structure cannot be
trusted, clearly points out problems in the input, e.g. peaks that cannot be assigned and might
therefore be artifacts or indications of erroneous or missing sequence-specific assignments.
The program CYANA provides for each peak informational output that greatly facilitates this
task: the list of its chemical shift-based assignment possibilities, the assignment(s) finally
chosen, and the reasons why an assignment is chosen or not, or why a peak is not used at all.
In addition, even when the criteria of the previous section are met already, a higher precision
and accuracy of the structure might still be achieved by further improving the input data. A
completely refined input data set should contain well below 5% of peaks that cannot be
assigned and used by the program.

- 20 -
3 Implementations of automated NOESY assignment
3.1 Semiautomatic methods
Semiautomatic NOESY assignment methods relieve the spectroscopist from the burden of
checking the two straightforward criteria for NOESY assignments, i.e. the agreement of
chemical shifts and the compatibility with a preliminary structure, while entrusting the
assignment decisions to the spectroscopist who may have additional relevant information at
his disposal. Such approaches (e.g. Güntert et al., 1993; Meadows et al., 1994; Duggan et al.,
2001) use the chemical shifts and a model or preliminary structure to provide the user with
the list of possible assignments for each cross peak. The user decides interactively about the
assignment and/or temporary removal of individual NOESY cross peaks, possibly taking into
account supplementary information such as line shapes or secondary structure, and performs a
structure calculation with the resulting input. In general, several cycles of NOESY assignment
and structure calculation are required to obtain a high-quality structure.
A prototype of this semiautomatic approach is the program ASNO (Güntert et al., 1993). The
input for ASNO consists of a list of the proton chemical shifts, a peak list containing the
chemical shift coordinates of the cross peaks in the NOESY spectrum, and a bundle of
conformers calculated using a previous, in general preliminary set of input of NOE distance
restraints. Alternatively, the structural input can consist of the crystal structure of the protein
under investigation or originate from a homologous protein. In that case care must be
exercised to rule out possible bias by the imported reference data. In addition, the user
specifies the maximally allowed chemical shift differences between corresponding cross peak
coordinates and proton chemical shift values to be used for chemical shift-based assignments,
the maximal proton-proton distance d
max
in the structure that may give rise to an observable
NOE, and the minimal number of conformers for which a given proton–proton distance must
be shorter than d
max
for an acceptable NOE assignment. For each NOESY cross peak ASNO
first determines the set of all possible chemical shift-based assignments. These are then
checked against the corresponding
1
H–
1
H distances in the group of preliminary conformers
and retained only if the distance between the two protons is shorter than d
max
in at least the
required number of conformers. After several rounds of structure calculation, NOE
assignment with ASNO, and interactive checking and refinement of the assignments, a final,
high-quality structure is obtained.

- 21 -
The program SANE (Structure Assisted NOE Evaluation) (Duggan et al., 2001) is an
alternative protocol in which ambiguous distance restraints are generated for cross peaks with
multiple possible assignments. The user is directly involved in violation analysis after each
round of structure calculation. Throughout the structure determination the user provides input
that can help to circumvent erroneous local structures and reduce the number of iterations
required to reach acceptable structures. Like ASNO, the SANE program includes a distance
filter that is based on an initial search model structure, which may be an X-ray structure, an
ensemble of solution structures, or even a homology-modeled structure. To minimize the
problem of multiple possible assignments SANE makes use of a suite of filters that take into
account existing partial assignments, the average distance between protons in one or more
structures, relative NOE contributions calculated from the structures, and the expected
secondary structure in order to iterate to an accurately assigned NOE cross peak list,
including both unambiguous and ambiguous NOEs for the structure calculation.
3.2 The NOAH algorithm
In a first approach and proof of feasibility of automated NOESY assignment, the programs
DIANA (Güntert et al., 1991) and DYANA (Güntert et al., 1997) were supplemented with the
automated NOESY assignment routine NOAH (Mumenthaler and Braun, 1995; Mumenthaler
et al., 1997). In NOAH, the multiple assignment problem is treated by temporarily ignoring
cross peaks with too many (typically, more than two) assignment possibilities and instead
generating independent distance restraints for each of the assignment possibilities of the
remaining, low-ambiguity cross peaks, where one has to accept that part of these distance
restraints may be incorrect. In order to reduce the impact of these incorrect restraints on the
structure, an error-tolerant target function is used. NOAH requires high accuracy of the input
chemical shifts and peak positions. It makes use of the fact that only a set of correct
assignments can form a self-consistent network, and convergence towards the correct
structure has been achieved for several proteins (Mumenthaler and Braun, 1995;
Mumenthaler et al., 1997; Xu et al., 1999; 2001; Oezguen et al., 2002).
3.3 The ARIA algorithm
The widely used automated NOESY assignment procedure ARIA (Nilges et al., 1997; Nilges
and O’Donoghue, 1998; Linge et al., 2001, 2003) has been interfaced initially with the

- 22 -
program XPLOR (Brünger, 1992) and later with the program CNS (Brünger et al., 1998) for
the structure calculation. ARIA introduced many new concepts, most importantly the use of
ambiguous distance restraints (Nilges, 1993, 1995; see Section 2.6) for handling ambiguities
in the initial, chemical shift-based NOESY cross peak assignments. Prior to the introduction
of ambiguous distance restraints, in general only unambiguously assigned NOEs could be
used as distance restraints in the structure calculation. Since the majority of NOEs cannot be
assigned unambiguously from chemical shift information alone, this lack of a general way to
directly include ambiguous data into the structure calculation considerably hampered the
performance of automatic NOESY assignment algorithms.
ARIA starts from lists of peaks and chemical shifts in the formats of the common spectral
analysis programs ANSIG (Kraulis 1989; Helgstrand et al., 2000), NMRView (Johnson and
Blevins, 1994), PIPP (Garrett et al., 1991) or XEASY (Bartels et al., 1995) and proceeds in
cycles of NOE assignment and structure calculation. Constraints on dihedral angles, J-
couplings, residual dipolar couplings, disulfide bridges and hydrogen bonds can be used in
addition, if available. In each cycle, ARIA calibrates and assigns the NOESY spectra, merges
the restraint lists from different spectra, and calculates a bundle of (typically 20) conformers
with the program CNS. Normally, an extended “template” structure is used in the initial cycle
0. In all later cycles, NOE assignment, calibration and violation analysis are based on the
average
1
H-
1
H distances d calculated from the (typically 7 out of 20) lowest energy
conformers from the previous cycle.
The ARIA algorithm is particularly efficient for improving and completing the NOESY
assignment once a correct preliminary polypeptide fold is available. To obtain a correct fold
in the initial phase of a de novo structure determination when the powerful structure-based
filters for the elimination of erroneous cross peak assignments cannot be active yet, it can be
of help if the user supplies a limited number of already assigned long-range distance
restraints. ARIA has been used in the NMR structure determinations of many proteins (Linge
et al., 2001, 2003). Similar algorithms that also relies on ambiguous distance restraints and
the program XPLOR for the structure calculation has been implemented (Gilquin et al., 1999;
Savarin et al., 2001; Kuszewski et al., 2004).

- 23 -
3.4 The CANDID algorithm
The CANDID algorithm (Herrmann et al., 2002a) in the programs DYANA (Güntert et al.,
1997) and CYANA version 1.0 (Güntert, 2004) combines features from NOAH and ARIA,
such as the use of three-dimensional structure-based filters and ambiguous distance restraints,
with the new concepts of network-anchoring and constraint combination that further enable
an efficient and reliable search for the correct fold in the initial cycle of de novo NMR
structure determinations. Automated structure calculation with CYANA proceeds in iterative
cycles of NOE assignment followed by structure calculation. Between subsequent cycles,
information is transferred exclusively through the intermediary three-dimensional structures,
in that the molecular structure obtained in a given cycle is used to guide the NOE assignments
in the following cycle. Otherwise, the same input data are used for all cycles, that is, the
amino acid sequence of the protein, one or several chemical shift lists, and one or several lists
containing the positions and volumes of cross peaks in 2D, 3D or 4D NOESY spectra. The
assignment of NOEs with CANDID is based on the concept of “generalized volume
contributions” (Herrmann et al., 2002a). The original, “physical” volume contribution of a
given assignment to the total intensity of a peak (Eq. 10) is generalized in CANDID by
factors that reflect the covalent structure of the protein, the presence of transposed peaks, and
network-anchoring.
The CANDID method has been evaluated in test calculations (Herrmann et al., 2002a, b; Jee
and Güntert, 2003) and used in many de novo structure determinations, including four
variants of the human prion protein (Calzolai et al., 2001; Zahn et al., 2003), two distinct
forms of the pheromone binding protein from Bombyx mori (Horst et al., 2001; Lee et al.,
2002), the calreticulin P-domain (Ellgard et al., 2001, 2002), the class I human ubiquitin-
conjugating enzyme 2b (Miura et al., 2002), the heme chaperone CcmE (Enggist et al., 2002)
(Fig. 2), the nucleotide-binding domain of Na,K-ATPase (Hilge et al., 2003).
3.5 The CYANA algorithm
A new, probabilistic automated NOE assignment algorithm has been implemented in program
CYANA, version 2.0. Input chemical shift lists can be in the formats of XEASY (Bartels et
al., 1995) or the BioMagResBank (Doreleijers et al., 2003). NOESY peak lists can be
prepared either using interactive spectrum analysis programs such as XEASY, NMRView
(Johnson and Blevins, 1994), ANSIG (Kraulis 1989; Helgstrand et al., 2000), or automated

- 24 -
peak picking methods such as AUTOPSY (Koradi et al., 1998) or ATNOS (Herrmann et al.,
2002b) that allow to start the NOE assignment and structure calculation process directly from
the NOESY spectra. The input may further include previously assigned NOE upper distance
restraints or other previously assigned conformational restraints. These will not be modified
during automated NOE assignment but used for the CYANA structure calculation. An
automated CYANA structure calculation typically comprises seven cycles (Figs. 1 and 2),
each of which consists of the following steps:
1. Read experimental input data: Amino acid sequence; chemical shift list from sequence-
specific resonance assignment; list(s) of NOESY cross peak positions and volumes; and,
optionally, conformational restraints from other sources for use in addition to the input
from automated NOE assignment.
2. Calibrate distance bounds. From the NOESY peak volumes or intensities upper distance
bounds are derived.
3. Create initial assignment list. For each NOESY cross peak, one or several initial
assignments are determined based on chemical shift agreement within a user-defined
tolerance range.
4. Filter initial assignments. For each initial assignment of a NOESY cross peak an overall
probability for its correctness is calculated as the product of three probabilities that reflect
(a) the agreement between the values of the chemical shift list and the peak position, (b)
self-consistency within the entire NOE network (see Section 2.5), and, if available (i.e. in
cycles 2, 3,...), (c) the compatibility with the three-dimensional structure from the
preceding cycle (Fig. 3). Initial assignments with overall probability below a given
threshold are discarded.
5. Create distance restraints. Distance restraints are created for all cross peaks with at least
one assignment with overall probability above the threshold. Peaks with a single accepted
assignment yield unambiguous distance restraints, those with more than one accepted
assignment result in ambiguous distance restraints.
6. Constraint combination. In cycles 1 and 2 groups of (2 or) 4, a priori unrelated long-range
distance restraints are combined into new virtual distance restraints that carry each the
assignments from two of the original restraints (see Section 2.9).
7. Structure calculation. Using simulated annealing (Kirkpatrick et al., 1983) driven by
torsion angle dynamics (Jain et al., 1993; Güntert et al., 1997) a 3D structure of the protein
is calculated that is added to the input for the following cycle. Distance restraints from

- 25 -
NOEs with multiple assignments and those resulting from constraint combination are
introduced as ambiguous distance restraints into the structure calculation.
8. Return to Step 1.
In the first cycle, the structure-independent NOE self-consistency check has a dominant
impact on the filtering of individual assignment possibilities (step 4) and entire distance
restraints (step 5), since structure-based criteria cannot be applied yet. The second and
subsequent cycles differ from the first cycle by the use of an additional probability for NOE
assignments and cross peaks that exploit the protein 3D structure from the preceding cycle.
Since the precision of the structure determination normally improves with each subsequent
cycle, the criteria for accepting assignments (step 4) are tightened in more advanced cycles of
the structure calculation.
The output from a cycle includes a listing of all NOESY cross peak assignments, comments
about individual assignment decisions that can help to recognize potential artifacts in the
input data, and a three-dimensional structure in the form of a bundle of conformers. A final
structure calculation is performed with unique assigned distance restraints only, in order to
allow their direct use in subsequent refinement and analysis programs that cannot handle
ambiguous distance restraints.
A complete automated CYANA structure calculation requires the calculation of 7 x 100
individual conformers, and hence a substantial amount of computation. Because of the
efficiency of the CYANA torsion angle dynamics algorithm (Jain et al., 1993; Güntert et al.,
1997) it is nevertheless possible to perform a complete automated structure calculation with
CYANA in short time. For instance, the computation time for the calculation of one
conformer of the 136-residue heme chaperone protein CcmE on the basis of 2453 NOE upper
distance bounds and 56 torsion angle restraints (Enggist et al., 2002) using 10000 torsion
angle dynamics steps on a single processor is less than one minute on modern hardware:
Linux PC, Pentium IV, 3.06 GHz:
29 s
Linux PC, Pentium IV, 1.8 GHz:
42 s
Compaq Alpha Server GS 320:
23 s
Silicon Graphics, R16000, 700 MHz:
39 s
Silicon Graphics, R12000, 400 MHz:
59 s
Time-consuming structure calculations are most efficiently performed in parallel. Since an
NMR structure calculation always involves the computation of a group of conformers, it is
highly efficient and straightforward with CYANA to run calculations of multiple conformers

- 26 -
in parallel, for example on clusters of Linux computers using the Message Passing Interface
MPI for interprocess communication (Gropp et al., 1996) or on shared-memory
multiprocessor systems. Nearly ideal speedup, i.e., an overall computation time almost
inversely proportional to the number of processors, can be achieved with CYANA (Güntert et
al., 1997).
The CYANA algorithm has been used for a large number of the NMR protein structures
determined by the RIKEN Structural Genomics/Proteomics Initiative, and elsewhere. These
structure determinations have confirmed that network-anchored assignment and restraint
combination enable reliable, truly automated NOESY assignment and structure calculation
without prior knowledge about NOESY assignments or the three-dimensional structure.
NOESY assignments and the corresponding distance restraints for these de novo structure
determinations were made using CYANA, confining interactive work to the stage of the
preparation of the input chemical shift and peak lists. If used sensibly, automated NOESY
assignment with CYANA has no disadvantage compared to the conventional, interactive
approach but is a lot faster, and more objective. Network-anchored assignment and constraint
combination render automated NOE assignment with CYANA stable also in the presence of
the imperfections typical for experimental NMR data sets. Using CYANA, the evaluation of
NOESY spectra is no longer the time-limiting step in protein structure determination by
NMR.
3.6 The AUTOSTRUCTURE algorithm
An approach that uses rules for assignments similar to the ones used by an expert to generate
an initial protein fold has been implemented in the program AUTOSTRUCTURE and applied
to protein structure determination (Huang et al., 2003, 2005; Greenfield et al., 2001; Moseley
and Montelione, 1999). AUTOSTRUCTURE is aimed to identify iteratively self-consistent
NOE contact patterns, without using any 3D structure model, and to delineate secondary
structures, including alignments between
β-strands, based upon a combined pattern analysis
of secondary structure-specific NOE contacts, chemical shifts, scalar coupling constants, and
slow amide proton exchange data. The software generates conformational restraints, e.g.
distance, dihedral angle and hydrogen bond restraints, automatically and submits parallel
structure calculations with the program DYANA (Güntert et al., 1997). The resulting

- 27 -
structure is then refined automatically by iterative cycles of self-consistent assignment of
NOESY cross peaks and regeneration of the protein structure with the program DYANA.
3.7 The KNOWNOE algorithm
The program KNOWNOE (Gronwald et al., 2002) presents a “knowledge-based” approach to
the problem of automated assignment of NOESY spectra that is, in principle, devised to work
directly with the experimental spectra without interference of an expert. Its central part is a
“knowledge-driven Bayesian algorithm” for resolving ambiguities in the NOE assignments.
NOE cross peak volume probability distributions were derived for various classes of proton-
proton contacts by a statistical analysis of the corresponding interatomic distances in more
than 300 protein NMR structures. For a given cross peak with n possible assignments
n
A
A
,
,
1
K
, the conditional probabilities P(Ak, a|V) that an assignment Ak is responsible for at
least a fraction a of the cross peak volume V can then be calculated from the volume
probability distributions using Bayes’ theorem. Peaks with one assignment Ak with a
probability P(Ak, a|V
0
) higher than a cutoff, typically in the range 0.8 to 0.9, are transiently
considered as unambiguously assigned. Note that no preliminary structure is needed to
achieve this discrimination that yields a higher number of unambiguous assignments as would
be possible based on chemical shifts alone (see Section 2.4). With this list of unambiguously
assigned peaks a set of structures is calculated. These structures are used as input for a next
cycle in which only assignments are accepted that correspond to distances shorter than a
threshold d
max
, which is decreased from cycle to cycle until
5
max
=
d
Å, the assumed
detection limit for NOEs. Since this algorithm essentially relies on the unambiguously
assigned NOEs in order to calculate the intermediate structures (only for the final structure
calculation some ambiguous distance restraint are used), it requires, like NOAH (see Section
3.2), a high accuracy of the chemical shifts of typically 0.01 ppm. The program KNOWNOE
was tested successfully on 2D NOESY spectra of the 66 amino acid cold shock protein from
Thermotoga maritima for which automated NOESY assignment resulted in a structure of
comparable quality to the one obtained from manual data evaluation (Gronwald et al., 2002).
4 Assignment-free structure calculation
It is almost universally assumed that a protein structure determination by NMR requires the
sequence-specific resonance assignments (Wüthrich, 1986). However, the chemical shift

- 28 -
assignment by itself has no biological relevance. It is required only as an intermediate step in
the interpretation of the NMR spectra. Several attempts have been made to devise a strategy
for NMR protein structure determination that circumvents the tedious chemical shift
assignment step. There is a loose analogy between these approaches and the direct phasing
methods in X-ray crystallography (Drenth, 1994). Although until today no de novo NMR
protein structure determination has been accomplished without prior chemical shift
assignment, an introduction into the concepts assignment-free NMR structure calculation is
warranted because recent progress in this field may open the avenue to an alternative strategy
of NMR structure determination.
The underlying idea of assignment-free NMR structure calculation methods is to exploit the
fact that NOESY spectra provide distance information even in the absence of any chemical
shift assignments. This proton-proton distance information can be exploited to calculate a
spatial proton distribution. Since there is no association with the covalent structure at this
point, the protons of the protein are treated as a gas of unconnected particles. Provided that
the emerging proton distribution is sufficiently clear, a model can then be built into the proton
density in a manner analogous to X-ray crystallography in which the structural model is
constructed into the electron density.
This general idea was first tested by Malliavin et al. (1992) with simulated NOEs between
backbone amide protons of lysozyme. From simulations with synthetic NOE data for BPTI
and combining metric matrix distance geometry with graph theoretical approaches to identify
secondary structure elements and, eventually, sequence-specific assignments, Oshiro and
Kuntz (1993) concluded that “this approach is only useful with excellent quality stereo-
resolved data”.
By then the most thorough attempt at simultaneous protein structure determination and
sequence-specific assignment of
13
C and
15
N-separated NOE data using “a novel real-space
ab initio approach” came with Per Kraulis’ ANSRS algorithm (Kraulis, 1994). The input data
are a list of NOESY cross peaks including knowledge of the chemical shifts of the
13
C or
15
N
atoms covalently bound to the protons that make the NOE (i.e., a 4D NOESY peak list), and a
complete but unassigned list of the chemical shifts of all detectable
1
H-
13
C and
1
H-
15
N
moieties. The ANSRS algorithm then proceeds in three stages. First,
1
H spin 3D real-space
structures are calculated using dynamical simulated annealing. Second, a list for each residue
type of plausible
1
H spin combinations with probability scores is generated in a recursive
combinatorial search with spatial restraints. Finally, the sequence-specific assignment and a

- 29 -
low-resolution 3D structure are obtained by Monte Carlo simulated annealing. With simulated
data for two small proteins of 32 and 58 residues the resulting average 3D real-space
1
H spin
structures were within less than 2 Å RMSD from the previously known 3D structure, and the
ANSRS procedure was able to determine the sequence-specific assignments for more than
95% of the spins. Despite these encouraging figures, the ANSRS program has not become a
routine tool for NMR structure determination, presumably because the requirements on the
quality of the input data are still formidable from the experimental point of view, and because
the algorithm has no facilities to deal with overlap among
1
H-X chemical shift pairs.
Atkinson and Saudek proposed an interesting algorithm for direct fitting of structure and
chemical shift data to NMR spectra (Atkinson and Saudek, 1997). Optimization of four
variables per atom, three Cartesian coordinates and the chemical shift value, directly against
the NOESY spectrum, rather than peak lists, by simulated annealing was shown to succeed in
finding sets of coordinates (i.e. structures) and chemical shifts that match the reference
configuration, albeit only in the case of a peptide fragment with six atoms. Subsequently, the
same authors realized that the direct determination of protein structures by NMR without
chemical shift assignment is not restricted to using only NOESY spectra, but can incorporate,
in a natural way, data from the same set of heteronuclear and dipolar coupling experiments as
normally used in the conventional approach (Atkinson and Saudek, 2002). NOEs are again
interpreted as distances between unassigned and unconnected atoms, while cross peaks in all
other spectra are also interpreted as distances instead of being used for assignment purposes.
For example, a
15
N-
1
H HSQC peak yields a distance equal to the N-H bond length between
the two corresponding atoms, and the HNCA spectrum yields, for each N-H pair, four
distances to the two adjacent C
α
atoms. RMSD values to the crystal structure below 2 Å were
obtained when using simulated peak lists for the protein ubiquitin with no prior assignment of
any spectral resonance or cross peak, but every hydrogen atom in the structure was labeled by
both its own chemical shift and that of the attached heavy atom.
The most recent approach to NMR structure determination without chemical shift assignment
is the CLOUDS protocol of Grishaev and Llinás (2002a, b). For the first time, the feasibility
of the assignment-free structure determination concept could be demonstrated using
experimental data rather than simulated data sets. The CLOUDS method relies on precise and
abundant inter-proton distance restraints calculated via a relaxation matrix analysis of sets of
experimental NOESY cross peaks (Madrid et al., 1991). A gas of unassigned, unconnected

- 30 -
hydrogen atoms is condensed into a structured proton distribution (cloud) via a molecular
dynamics simulated annealing scheme in which the inter-nuclear distances and van der Waals
repulsive terms are the only active restraints. Proton densities are generated by combining a
large number of such clouds, each computed from a different trajectory. After filtering by
reference to the cloud closest to the mean, a minimal dispersion proton density (“family of
clouds”) is identified that affords a quasi-continuous hydrogen-only probability distribution
and conveys immediate information on the shape of the protein. The NMR-generated proton
density provides a template to which the molecule has to be fitted to derive the structure. The
primary structure is threaded through the unassigned proton density by a Bayesian approach,
for which the probabilities of sequential connectivity hypotheses are inferred from likelihoods
of H
N
-H
N
, H
N
-H
α
, and H
α
-H
α
interatomic distances as well as
1
H NMR chemical shifts, both
derived from public databases. Once the polypeptide sequence is identified, directionality
becomes established, and the N and C termini are recognized. Side chain hydrogen atoms are
found by a similar procedure. The folded structure is then obtained via a direct molecular
dynamics embedding into mirror image-related representations of the proton density and
selected according to a lowest energy criterion.
The feasibility of the method was tested with experimental NMR data measured for two
globular proteins of 60 and 83 residues, for which excellent unambiguously identified
homonuclear NOESY peak lists were available from the previous, conventional structure
determinations. At the outset of a de novo structure determination it may not be
straightforward to produce a NOESY peak list of such completeness and quality. In
particular, it was assumed that the NOEs can be identified unambiguously, i.e. that it is
known with certainty whether any two NOESY peaks involve the same proton or not. The
resulting structures deviated by 1.0–1.4 Å RMSD for the backbone heavy atoms from the
previously reported X-ray and NMR structures (Grishaev and Llinás, 2002b). These results
show that assignment-free NMR structure calculation can successfully generate 3D protein
structures from experimental data.
As for all NMR spectrum analysis, resonance overlap presents a major difficulty also in
applying “no assignment” strategies. Indeed, if two resonances from nuclei that are far apart
in the structure have identical chemical shifts but distinct sets of neighbors they would be
represented by a single atom with one set of neighbors, leading to a gross distortion of the
calculated structure. In that respect, the use of heteronuclear-edited NOESY spectra

- 31 -
drastically reduces the likelihood of overlap. At present, a full de novo protein structure
determination by the assignment-free approach has not been reported yet, and it remains to be
seen whether the assignment-free approach will be able to provide the reliability and the
structure quality of the conventional method.
References
Abe H, Braun W, Noguti T, Gō N (1984) Rapid calculation of first and second derivatives of conformational energy with
respect to dihedral angles in proteins. General recurrent equations. Computers & Chemistry 8:239–247
Allen MP, Tildesley, DJ (1987) Computer Simulation of Liquids. Clarendon Press, Oxford
Altieri AS, Byrd RA (2004) Automation of NMR structure determination of proteins. Curr. Opin. Struct. Biol. 14: 547–553.
Andrec M, Levy RM (2002) Protein sequential resonance assignments by combinatorial enumeration using 13Ca chemical
shifts and their (i, i
−1) sequential connectivities. J. Biomol. NMR 23:263–270
Atkinson RW, Saudek V (1997) Direct fitting of structure and chemical shift to NMR spectra. J. Chem. Soc. Faraday Trans.
93:3319–3323
Atkinson RW, Saudek V (2002) The direct determination of protein structure by NMR without assignment. FEBS Lett.
510:1–4
Atreya HS, Sahu SC, Chary KVR, Govil G (2000) A tracked approach for automated NMR assignments in proteins
(TATAPRO). J. Biomol. NMR 17:125–136
Bailey-Kellogg C, Widge A, Kelley JJ, Berardi MJ, Bushweller JH, Donald BR (2000) The NOESY JIGSAW: Automated
protein secondary structure and main-chain assignment from sparse, unassigned NMR data. J. Comp. Biol. 7:537–558
Baran MC, Huang YJ, Moseley HNB, Montelione GT (2004) Automated analysis of protein NMR assignments and
structures. Chem. Rev. 104: 3541-3555
Bartels C, Xia, TH, Billeter M, Güntert P, Wüthrich K (1995) The program XEASY for computer-supported NMR-spectral
analysis of biological macromolecules. J. Biomol. NMR 6:1–10
Bartels C, Billeter M, Güntert P, Wüthrich K (1996) Automated sequence-specific NMR assignment of homologous proteins
using the program GARANT. J. Biomol. NMR 7:207–213
Bartels C, Güntert P, Billeter M, Wüthrich K (1997) GARANT-A general algorithm for resonance assignment of
multidimensional nuclear magnetic resonance spectra. J. Comp. Chem. 18:139–149
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984). Molecular dynamics with coupling to an
external bath. J. Chem. Phys. 81:3684–3690
Bhavesh NS, Panchal SC, Hosur RV (2001) An efficient high-throughput resonance assignment procedure for structural
genomics and protein folding research by NMR. Biochemistry 40:14727–14735
Brünger AT (1992) X-PLOR version 3.1. A system for X-ray crystallography and NMR. Yale University Press, New Haven
Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu
NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: A new software suite for
macromolecular structure determination. Acta Crystallogr. D 54: 905–921
Calzolai L, Lysek DA, Güntert P, von Schroetter C, Riek R, Zahn R, Wüthrich K (2000) NMR structures of three single-
residue variants of the human prion protein. Proc. Natl. Acad. Sci. USA 97:8340–8345
Buchler, NEG, Zuiderweg ERP, Wang H, Goldstein RA (1997) Protein heteronuclear NMR assignments using mean-field
simulated annealing. J. Magn. Reson. 126:34–42

- 32 -
Chatterjee A, Bhavesh NS, Panchal SC, Hosur RV (2002) A novel protocol based on HN(C)N for rapid assignment in (
15
N,
13
C) labeled proteins: implications to structural genomics, Biochem. Biophys. Res. Commun. 293:427–432
Chin Y, Hwang JF, Chen TB, Soo VW (1992) RUBIDIUM, a program for computer-aided assignment of 2-dimensional
NMR-spectra of polypeptides. J. Chem. Inf. Comput. Sci. 32:183–187
Choy WY, Sanctuary BC, Zhu G (1997) Using neural network predicted secondary structure information in automatic
protein NMR assignment. J. Chem. Inf. Comput. Sci 37:1086–1094
Coggins BE, Zhou P (2003) PACES: Protein sequential assignment by computer-assisted exhaustive search. J. Biomol. NMR
26:93–111
Croft D, Kemmink J, Neidig KP, Oschkinat H (1997) Tools for the automated assignment of high-resolution three-
dimensional protein NMR spectra based on pattern recognition techniques. J. Biomol. NMR 10:207–219
Doreleijers JF, Mading S, Maziuk D, Sojourner K, Yin L, Zhu J, Markley JL, Ulrich EL (2003) BioMagResBank database
with sets of experimental NMR constraints corresponding to the structures of over 1400 biomolecules deposited in the
Protein Data Bank. J. Biomol. NMR 26:139–146
Drenth J (1994) Principles of protein X-ray crystallography. Springer, New York
Duggan BM, Legge GB, Dyson HJ, Wright PE (2001) SANE (Structure Assisted NOE Evaluation): An automated model-
based approach for NOE assignment. J. Biomol. NMR 19:321–329
Ellgaard L, Riek R, Herrmann T, Güntert P, Braun D, Helenius A, Wüthrich K (2001) NMR structure of the calreticulin P-
domain. Proc. Natl. Acad. Sci. USA 98:3133–3138
Ellgaard L, Bettendorff P, Braun D, Herrmann T, Fiorito F, Jelesarov I, Herrmann T, Güntert P, Helenius A, Wüthrich K
(2002) NMR structures of 36 and 73-residue fragments of the calreticulin P-domain. J. Mol. Biol. 322, 773–784
Enggist E, Thöny-Meyer L, Güntert P, Pervushin K (2002) NMR structure of the heme chaperone CcmE reveals a novel
functional motif. Structure 10:1551–1557
Friedrichs MS, Mueller L, Wittekind M (1994) An automated procedure for the assignment of protein
1
HN,
15
N,
13
C
α
,
1
H
α
,
13
C
β
and
1
H
β
resonances. J. Biomol. NMR 4:703–726
Fossi M, Linge J, Labudde D, Leitner D, Nilges M, Oschkinat H (2005) Influence of chemical shift tolerances on NMR
structure calculations using ARIA protocols for assigning NOE data. J. Biomol. NMR 31:21–34
Garrett DS, Powers R, Gronenborn AM, Clore GM (1991) A common-sense approach to peak picking in 2-dimensional, 3-
dimensional, and 4-dimensional spectra using automatic computer-analysis of contour diagrams. J. Magn. Reson. 95:214–
220
Gilquin B, Lecoq A, Desné F, Guenneugues M, Zinn-Justin S, Ménez A (1999) Conformational and functional variability
supported by the BPTI fold: Solution structure of the Ca2+ channel blocker calcicludine. Proteins 34:520–532
Greenfield NJ, Huang YJ, Palm T, Swapna GVT, Monleon D, Montelione GT, Hitchcock-DeGregori SE (2001) Solution
NMR structure and folding dynamics of the N terminus of a rat non-muscle alpha-tropomyosin in an engineered chimeric
protein. J. Mol. Biol. 312:833–847
Grishaev A, Llinás M (2002a) CLOUDS, a protocol for deriving a molecular proton density via NMR. Proc. Natl. Acad. Sci.
USA 99:6707–6712
Grishaev A, Llinás M (2002b) Protein structure elucidation from NMR proton densities. Proc. Natl. Acad. Sci. USA
99:6713–6718
Gronwald W, Kalbitzer HR (2004) Automated structure determination of proteins by NMR spectroscopy. Prog. NMR
Spectrosc. 44:33–96.
Gronwald W, Willard L, Jellard T, Boyko RE, Rajarathnam K, Wishart DS, Sonnichsen FD, Sykes BD (1998) CAMRA:
Chemical shift based computer aided protein NMR assignments. J. Biomol. NMR 12:395–405

- 33 -
Gronwald W, Moussa S, Elsner R, Jung A, Ganslmeier B, Trenner J, Kremer W, Neidig KP, Kalbitzer HR (2002) Automated
assignment of NOESY NMR spectra using a knowledge based method (KNOWNOE). J. Biomol. NMR 23:271–287
Gropp W, Lusk E, Doss N, Skjellum, A (1996) A high-performance, portable implementation of the MPI message passing
interface standard. Parallel Computing 22:789–828
Güntert P (2004) Automated NMR protein structure calculation with CYANA. Meth. Mol. Biol. 278:353–378
Güntert P (2003) Automated NMR protein structure calculation. Prog. NMR Spectrosc. 43:105–125
Güntert P (1998) Structure calculation of biological macromolecules from NMR data. Q. Rev. Biophys. 31:145–237
Güntert P, Braun W, Wüthrich K (1991) Efficient computation of three-dimensional protein structures in solution from
nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA.
J. Mol. Biol. 217:517–530
Güntert P, Berndt KD, Wüthrich K (1993) The program ASNO for computer-supported collection of NOE upper distance
constraints as input for protein structure determination. J. Biomol. NMR 3:601–606
Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program
DYANA. J. Mol. Biol. 273:283–298
Güntert P, Salzmann M, Braun D, Wüthrich K (2000) Sequence-specific NMR assignment of proteins by global fragment
mapping with the program MAPPER. J. Biomol. NMR 18:129–137.
Hare BJ, Prestegard JH (1994) Application of neural networks to automated assignment of NMR structures of proteins. J.
Biomol. NMR 4:35–46
Hare BJ, Wagner G (1999) Application of automated NOE assignment to three-dimensional structure refinement of a 28 kDa
single-chain T cell receptor. J. Biomol. NMR 15:103–113
Helgstrand M, Kraulis P, Allard P, Härd T (2000) ANSIG for Windows: An interactive computer program for semiautomatic
assignment of protein NMR spectra J. Biomol. NMR 18:329–336
Herrmann T, Güntert P, Wüthrich K (2002a) Protein NMR structure determination with automated NOE assignment using
the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319:209–227
Herrmann T, Güntert P, Wüthrich K (2002b) Protein NMR structure determination with automated NOE-identification in the
NOESY spectra using the new software ATNOS. J. Biomol. NMR 24:171–189
Hilge M, Siegal G, Vuister GW, Güntert P, Gloor SM, Abrahams JP (2003) ATP-induced conformational changes of the
nucleotide binding domain of Na,K-ATPase. Nat. Struct. Biol. 10:468–474
Hitchens TK, Lukin JA, Zhan YP, McCallum SA, Rule GS (2003) MONTE: An automated Monte Carlo based approach to
nuclear magnetic resonance assignment of proteins. J. Biomol. NMR 25:1–9
Horst R, Damberger F, Luginbühl P, Güntert P, Peng G, Nikonova L, Leal WS, Wüthrich K (2001) NMR structure reveals
intramolecular regulation mechanism for pheromone binding and release. Proc. Natl. Acad. Sci. USA 98:14374–14379
Huang YJ, Swapna GVT, Rajan PK, Ke H, Xia B, Shukla K, Inouye M, Montelione GT (2003) Solution NMR structure of
ribosome-binding factor A (RbfA), a cold-shock adaptation protein from Escherichia coli. J. Mol. Biol. 327:521-536
Huang YJ, Moseley HNB, Baran MC, Arrowsmith C, Powers R, Tejero R, Szyperski T, Montelione GT (2005) An
integrated platform for automated analysis of protein NMR structures. Meth. Enzymol. 394: 111-141
Jain A, Vaidehi N, Rodriguez G (1993) A fast recursive algorithm for molecular dynamics simulation. J. Comp. Phys.
106:258–268
Jee JG, Güntert P (2003) Influence of the completeness of chemical shift assignments on NMR structures obtained with
automated NOE assignment. J. Struct. Funct. Genom. 4:179-189
Johnson BA, Blevins RA (1994) NMR View - a computer program for the visualization and analysis of NMR data. J.
Biomol. NMR 4:603–614
Kalk A, Berendsen HJC (1976) Proton magnetic-relaxation and spin diffusion in proteins J Magn. Reson. 24:343–366

- 34 -
Kirkpatrick S, Gelatt Jr CD, Vecchi, MP (1983) Optimization by simulated annealing. Science 220:671–680
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J.
Mol. Graph. 14:51–55
Koradi R, Billeter M, Engeli M, Güntert P, Wüthrich K (1998) Towards fully automatic peak picking and integration of
biomolecular NMR spectra. J. Magn. Reson. 135:288–297
Kraulis PJ (1989) ANSIG - a program for the assignment of protein 1H 2D NMR spectra by interactive computer graphics. J.
Magn. Reson. 24:627–633
Kraulis PJ (1994) Protein 3-dimensional structure determination and sequence-specific assignment of 13C-separated and
15N-separated NOE data—a novel real-space ab-initio approach. J. Mol. Biol. 243:696–718
Kumar A, Ernst RR, Wüthrich K (1980) A two-dimensional nuclear overhauser enhancement (2D NOE) experiment for the
elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res.
Commun. 95:1–6
Kuszewski J, Schwieters CD, Garrett DS, Byrd RA, Tjandra N, Clore GM (2004) Completely automated, highly error-
tolerant macromolecular structure determination from multidimensional nuclear overhauser enhancement spectra and
chemical shift assignments. J. Am. Chem. Soc. 126:6258–6273
Leach AR (2001) Molecular modeling. Principles and applications. 2
nd
edition. Prentice Hall, Harlow, UK
Lee D, Damberger FD, Peng G, Horst R, Güntert P, Nikonova L, Leal WS, Wüthrich K (2002) NMR structure of the
unliganded Bombyx mori pheromone-binding protein at physiological pH. FEBS Lett. 531:314–318
Leutner M, Gschwind RM, Liermann J, Schwarz C, Gemmecker G, Kessler H (1998) Automated backbone assignment of
labeled proteins using the threshold accepting algorithm. J. Biomol. NMR 11:31–43
Li KB, Sanctuary BC (1997a) Automated resonance assignment of proteins using heteronuclear 3D NMR. 1.Backbone spin
systems extraction and creation of polypeptides. J. Chem Inf. Comput. Sci. 37:359–366
Li KB, Sanctuary BC (1997b) Automated resonance assignment of proteins using heteronuclear 3D NMR. 2. Side chain and
sequence-specific assignment. J. Chem Inf. Comput. Sci. 37:467–477
Linge JP, O’Donoghue SI, Nilges M (2001) Automated assignment of ambiguous nuclear Overhauser effects with ARIA.
Meth. Enzymol. 339:71–90
Linge JP, Habeck M, Rieping W, Nilges M (2003) ARIA: automated NOE assignment and NMR structure calculation.
Bioinformatics 19:315–316
Linge JP, Habeck M, Rieping W, Nilges M (2004a) Correction of spin diffusion during iterative automated NOE assignment.
J. Magn. Reson. 167:334–342
Linge JP, Williams MA, Spronk CAEM, Bonvin AMJJ, Nilges M (2004b) Refinement of protein structures in explicit
solvent. Proteins 50:496–506
Lukin JA, Gove AP, Talukdar SN, Ho C (1997) Automated probabilistic method for assigning backbone resonances of
(
13
C,
15
N)-labeled proteins. J. Biomol. NMR 9:151–166
Macura, S, Ernst, RR (1980) Elucidation of cross relaxation in liquids by 2D NMR spectroscopy. Mol. Phys. 41:95–117
Madrid M, Llinás E, Llinás M (1991) Model-independent refinement of interproton distances generated from 1H-NMR
Overhauser intensities. J. Magn. Reson. 93:329–346
Malliavin TE, Rouh A, Delsuc M, Lallemand JY (1992) Approche directe de la détermination de structures moléculaires à
partir de l’effet Overhauser nucléaire. Compt. Rend. Acad. Sci. Serie II 315:635–659
Meadows RP, Olejniczak ET, Fesik SW (1994) A computer-based protocol for semiautomated assignments and 3D structure
determination of proteins. J. Biomol. NMR 4:79–96
Miura T, Klaus W, Ross A, Güntert P, Senn H (2002) The NMR structure of the class I human ubiquitin-conjugating enzyme
2b. J. Biomol. NMR, 22:89–92

- 35 -
Moseley, HNB, Montelione, GT (1999) Automated analysis of NMR assignments and structures for proteins. Curr. Op.
Struct. Biol. 9:635–642
Moseley, HNB, Monleon D, Montelione, GT (2001) Automatic determination of protein backbone resonance assignments
from triple resonance nuclear magnetic resonance data. Meth. Enzymol. 339:91–107
Mumenthaler C, Braun W (1995) Automated assignment of simulated and experimental NOESY spectra of proteins by
feedback filtering and self-correcting distance geometry. J. Mol. Biol. 254:465–480
Mumenthaler C, Güntert P, Braun W, Wüthrich K (1997) Automated procedure for combined assignment of NOESY spectra
and three-dimensional protein structure determination. J. Biomol. NMR 10:351–362
Nabuurs SB, Spronk CAEM, Krieger E, Maassen H, Vriend G, Vuister GW (2003) Quantitative evaluation of experimental
NMR restraints. J. Am. Chem. Soc. 125:12026–12034.
Nederveen AJ, Doreleijers JF, Vranken W, Miller Z, Spronk CAEM, Nabuurs, SB, Güntert P, Livny M, Markley JL, Nilges
M, Ulrich EL, Kaptein R, Bonvin AMJJ (2005) RECOORD: a REcalculated COORdinates Database of 500+ proteins from
the PDB using restraints from the BioMagResBank. Proteins 59:662–672.
Neuhaus D, Williamson MP (1989) The nuclear Overhauser effect in structural and conformational analysis. VCH,
Weinheim
Nilges M (1993) A calculation strategy for the structure determination of symmetric dimers by
1
H NMR. Proteins 17:297–
309
Nilges M (1995) Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous
NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 245:645–660
Nilges M, O’Donoghue SI (1998) Ambiguous NOEs and automated NOE assignment. Prog. NMR Spectrosc. 32:107–139
Nilges M, Macias M, O’Donoghue SI, Oschkinat H (1997) Automated NOESY interpretation with ambiguous distance
constraints: The refined NMR solution structure of the pleckstrin homology domain from
β-spectrin. J. Mol. Biol. 269:408–
422
Oezguen N, Adamian L, Xu Y, Rajarathnam K, Braun W (2002) Automated assignment and 3D structure calculations using
combinations of 2D homonuclear and 3D heteronuclear NMR spectra. J. Biomol. NMR 22:249–263
Olson Jr JB, Markley JL (1994) Evaluation of an algorithm for the automated sequential assignment of protein backbone
resonances - a demonstration of the connectivity tracing assignment tools (CONTRAST) software package. J. Biomol. NMR
4:385–410
Ösapay K, Case DA (1991) A new analysis of proton chemical shifts in proteins. J. Am. Chem. Soc. 113:9436–9444
Oshiro CM, Kuntz ID (1993) Application of distance geometry to the proton assignment problem. Biopolymers 33:107–115
Oschkinat H, Croft D (1994) Automated assignment of multidimensional nuclear-magnetic-resonance spectra. Meth.
Enzymol. 239:308–318
Pristovšek P, Rüterjans H, Jerala R (2002) Semiautomatic sequence-specific assignment of proteins based on the tertiary
structure—the program st2nmr. J. Comput. Chem. 23:335–340
Savarin P, Zinn-Justin S, Gilquin B (2001) Variability in automated assignment of NOESY spectra and three-dimensional
structure determination: A test case on three small disulfide-bonded proteins. J. Biomol. NMR 19:49–62
Sitkoff D, Case DA (1997) Density functional calculations of proton chemical shifts in model peptides. J. Am. Chem. Soc.
119:12262–12273
Solomon I (1955) Relaxation processes in a system of two spins. Phys. Rev. 99:559–565
Spronk CAEM, Nabuurs SB, Krieger E, Vriend G, Vuister GW (2004) Validation of protein structures derived by NMR
spectroscopy. Prog. NMR Spectrosc. 45:315–347
Spronk CAEM, Linge JP, Hilbers CW, Vuister GW (2002) Improving the quality of protein structures derived by NMR
spectroscopy. J. Biomol. NMR 22: 281–289

- 36 -
Tian F, Valafar H, Prestegard JH (2001) A dipolar coupling based strategy for simultaneous resonance assignment and
structure determination of protein backbones. J. Am. Chem. Soc. 123:11791–11796
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Xu J, Strauss SK, Sanctuary BC, Trimble L (1993) Automation of protein 2D proton NMR assignment by means of fuzzy
mathematics and graph-theory. J. Chem. Inf. Comput. Sci. 33:668–682
Xu J, Strauss SK, Sanctuary BC, Trimble L (1994) Use of fuzzy mathematics for complete automated assignment of peptide
1
H 2D NMR-spectra. J. Magn. Reson. B 103:53–58
Xu Y, Wu J, Gorenstein D, Braun W (1999) Automated 2D NOESY assignment and structure calculation of
crambin(S22/I25) with the self-correcting distance geometry based NOAH/DIAMOD programs. J. Magn. Reson. 136:76–85
Xu Y, Jablonsky MJ, Jackson PL, Braun W, Krishna NR (2001) Automated 2D NOESY assignment and structure calculation
of crambin(S22/I25) with the self-correcting distance geometry based NOAH/DIAMOD programs. J. Magn. Reson. 148:35–
46
Zahn R, Güntert P, von Schroetter C, Wüthrich K (2003) NMR structure of a human prion protein with two disulfide bridges.
J. Mol. Biol. 326:225–234
Zimmerman DE, Kulikowski CA, Huang YP, Feng WQ, Tashiro M, Shimotakahara S, Chien CY, Powers R, Montelione GT
(1997) Automated analysis of protein NMR assignments using methods from artificial intelligence. J. Mol. Biol. 269:592–
610
Figure captions
Fig. 1 General scheme of automated combined NOESY assignment and structure calculation.
Fig. 2 Structures of the heme chaperone CcmE (Enggist et al., 2002) obtained with the program CYANA in
seven consecutive cycles of combined automated NOESY assignment and structure calculation using torsion
angle dynamics. The backbones of the 10 conformers with lowest target function value in each cycle were
drawn with the program MOLMOL (Koradi et al., 1996).
Fig. 3 Three conditions that must be fulfilled by a valid assignment of a NOESY cross peak to two protons A
and B in the automated NOESY assignment with CYANA: (a) Agreement between chemical shifts and the peak
position, (b) network-anchoring, and (c) spatial proximity in a (preliminary) structure.
Fig. 4 Schematic illustration of the effect of constraint combination (Herrmann et al., 2002a) in the case of two
distance restraints, a correct one connecting atoms A and B, and a wrong one between atoms C and D. A
structure calculation that uses these two restraints as individual restraints that have to be satisfied simultaneously
will, instead of finding the correct structure (a), result in a distorted conformation (b), whereas a combined
restraint that will be fulfilled already if one of the two distances is sufficiently short leads to an almost
undistorted solution (c).
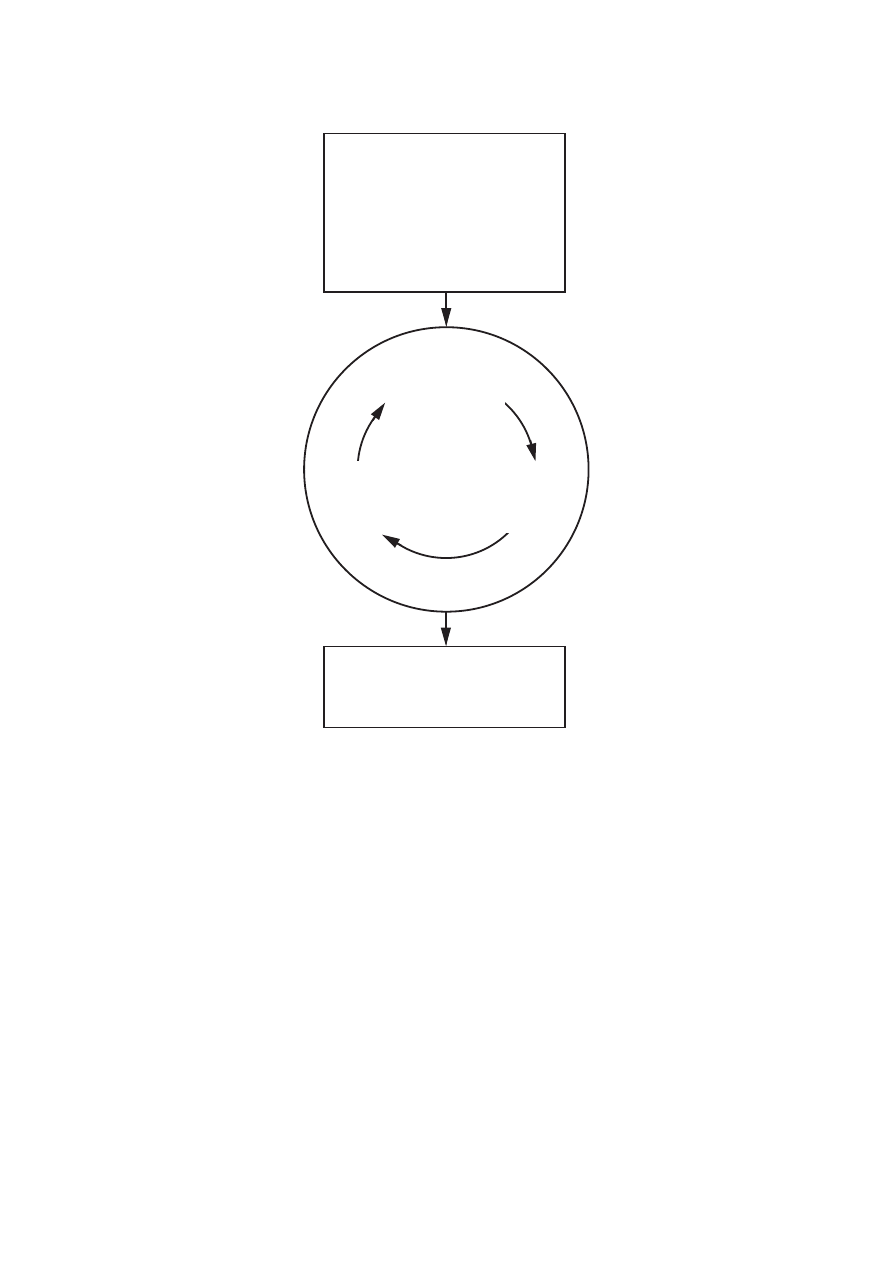
Find NOE
assignments
Evaluate NOE
assignments
Structure
calculation
Amino acid sequence
Sequence-specific
NOESY cross peak
positions and volumes
NOE assignments
3D Structure
assignments
Figure 1 (Güntert)
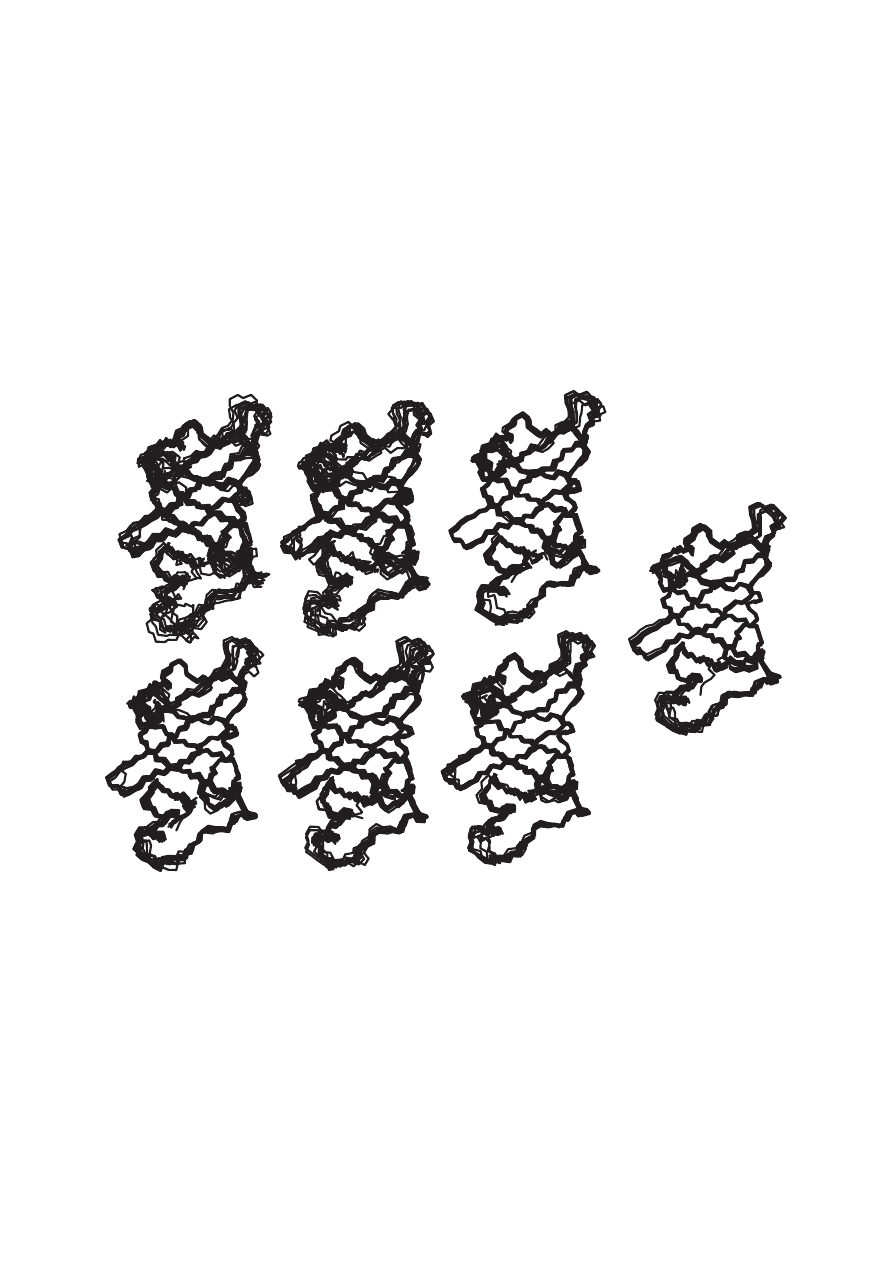
Cycle 1
Cycle 7
Cycle 6
Cycle 5
Cycle 4
Cycle 3
Cycle 2
Final structure
Figure 2 (Güntert)
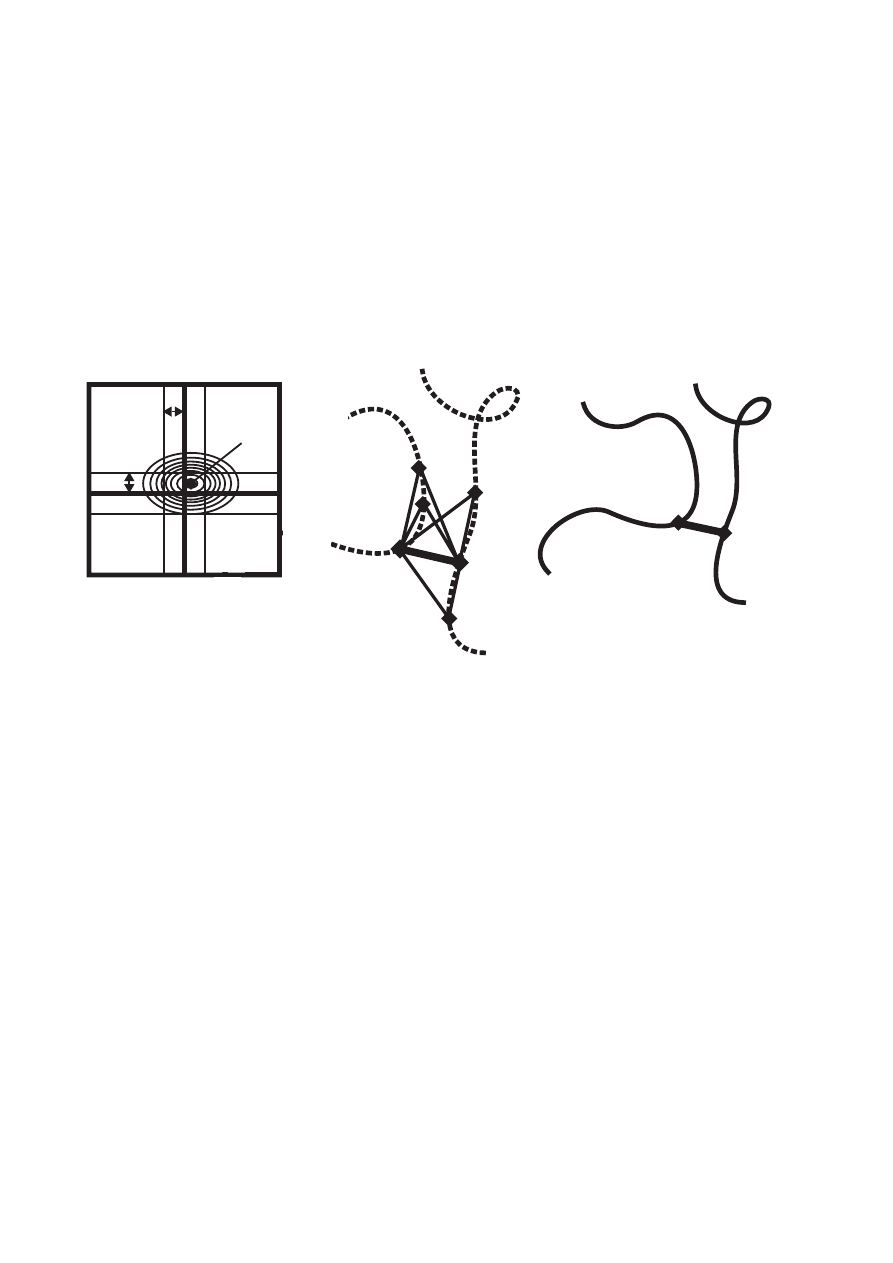
∆ω
Peak at
(
ω
1
,
ω
2
)
ω
A
ω
A
w
B
d
AB
< d
max
atom A
atom B
|ω
1
− ω
A
| < ∆ω |ω
2
− ω
B
| < ∆ω
ω
B
∆ω
(a)
(c)
(b)
A
B
Figure 3 (Güntert)
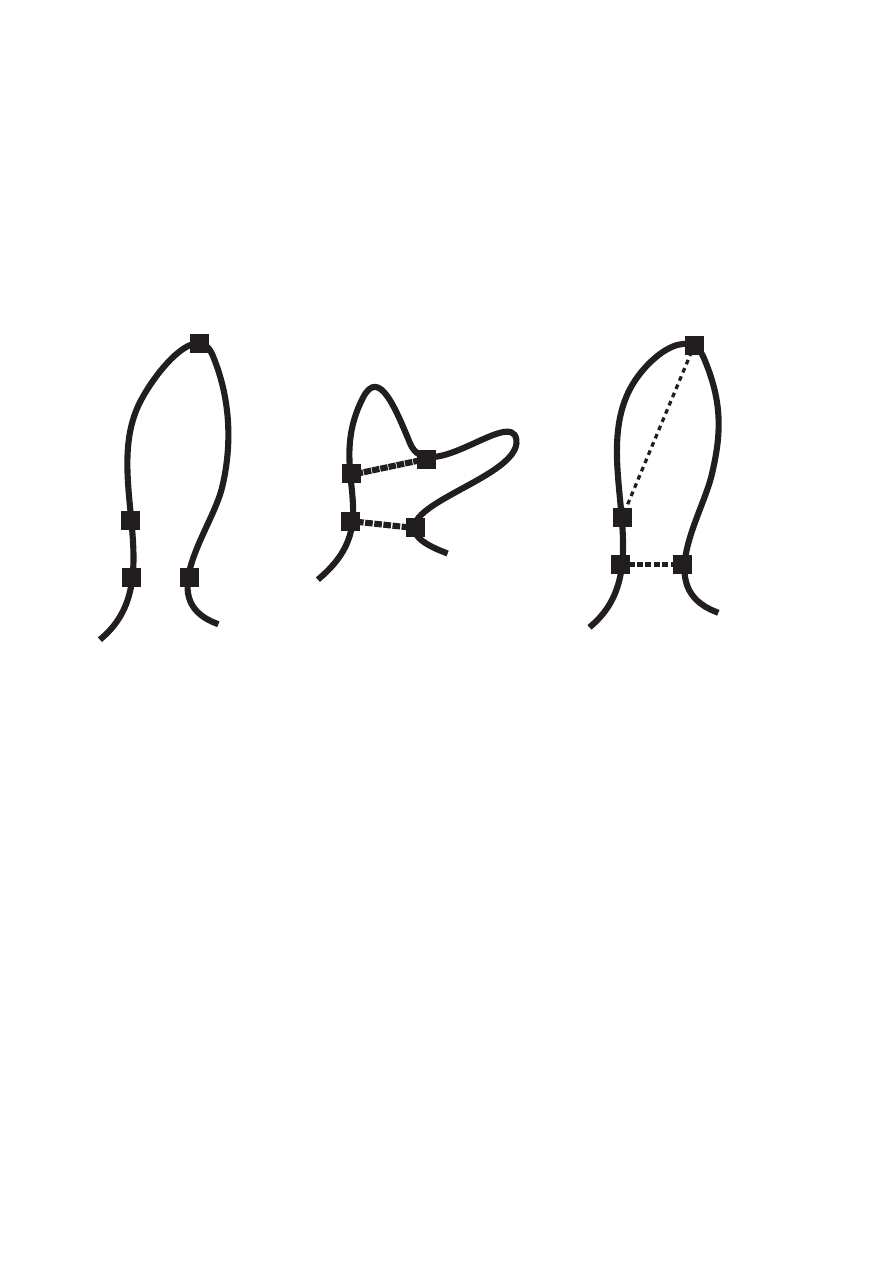
A
B
C
D
Correct
A
B
C
D
A
B
C
D
Combined
Individual
constraint
constraints
structure
A–B (correct)
C–D (wrong)
(unknown)
(a)
(c)
(b)
(ambiguous)
Figure 4 (Güntert)
Wyszukiwarka
Podobne podstrony:
lecture 3 Structural constraints, 3D structure calculation
Eurocode 6 Part 3 1996 2006 Design of Masonry Structures Simplified Calculation Methods for Maso
Spektroskopia NMR
FESTO Podstawy automatyzacji
Automatyka (wyk 3i4) Przel zawory reg
Automatyzacja w KiC (w 2) Obiekty reg
Structures sp11
AutomatykaII 18
196 Capital structure Intro lecture 1id 18514 ppt
Widmo NMR
4 Plant Structure, Growth and Development, before ppt
Automatyka wykład 8
WEEK 8 Earthing Calculations
Wybrane elementy automatyki instalacyjnej
12 Podstawy automatyki Układy sterowania logicznego
Automatyka dynamiakPAOo 2
Automatyka okrętowa – praca kontrolna 2
więcej podobnych podstron