
WYBRANE ZAGADNIENIA Z
GENETYKI CHORÓB TKANKI
ŁĄCZNEJ

Wrodzona podatność na chorobę tkanki łącznej ma
znaczenie dla wystąpienia, rozwoju i ciężkości jej
przebiegu
Badania populacyjne, w tym badania nad rodzinnym
występowaniem chorób autoimmunologicznych,
wykazały związek tych chorób
z układem HLA
W przeciwieństwie do wielu chorób uwarunkowanych
genetycznie przyczyną chorób tkanki łącznej nie jest
defekt 1 genu, lecz
dysfunkcja wzajemnej relacji wielu
genow
WPROWADZENIE

Toczeń rumieniowaty układowy (łac. lupus
erythematosus systemicus, ang. systemic lupus
erythematosus, SLE) – choroba
autoimmunologiczna
,
rozwijająca się na tle złożonych i niejasnych zaburzeń
układu odpornościowego, doprowadzający do
procesu zapalnego wielu tkanek i narządów.
TOCZEŃ RUMIENIOWATY
UKŁADOWY

Zachorowalność szacuje się na 40–50 przypadków na
100000,
10-‐krotnie częściej chorują kobiety
i szczyt
zachorowań występuje w wieku 16–55 lat.
Choroba charakteryzuje się bardzo zróżnicowanym
nasileniem i przebiegiem naturalnym.
Przez dłuższy czas mogą dominować objawy z
jednego narządu (co może być niewystarczające do
pewnego rozpoznania SLE), mogą pojawiać się
częściowe remisje i zaostrzenia. W miarę trwania
choroby mogą pojawiać się objawy z kolejnych
narządów, jednak
objawy uprzednio występujące nie
ulegają wycofaniu.
TOCZEŃ RUMIENIOWATY
UKŁADOWY

Genetyczne badania epidemiologiczne dowiodły
silnego TRU z wrodzoną podatnością na chorobę
Stwierdza się 100 razy większe ryzyko występowania
TRU u kolejnych członków rodziny w porównaniu z
ryzykiem stwierdzanym w populacji ogólnej
TOCZEŃ RUMIENIOWATY
UKŁADOWY

Podostry toczeń skórny
Toczeń indukowany lekami
Toczeń noworodków
Toczeń z zespołem Sjögrena
Toczeń z zespołem antyfosfolipidowy
TOCZEŃ RUMIENIOWATY
UKŁADOWY-‐ PODGRUPY

Kryteria Amerykańskiego Towarzystwa Reumatologicznego:
Zmiany skórne typu rumienia (często w kształcie motyla na
twarzy),
nigdy nie przekraczają bruzd nosowo-‐wargowych
.
Zmiany skórne rumieniowo-‐bliznowaciejące-‐ rumień krążkowy
(typu DLE).
Nadwrażliwość na światło słoneczne.
Nadżerki w jamie ustnej.
Zapalenie lub ból stawów – dotyczące co najmniej dwóch
stawów, bez zmian w obrazie RTG.
Zapalenie błon surowiczych – opłucnej (pleuritis) lub osierdzia
(pericarditis), stwierdzone w wywiadzie lub w chwili badania.
Zmiany w nerkach – utrzymująca się proteinuria (białkomocz)
powyżej 0,5 g/dobę lub obecność wałeczków nerkowych w
moczu.
TOCZEŃ RUMIENIOWATY
UKŁADOWY

Zaburzenia neuropsychiatryczne – szeroki wachlarz,
najczęściej napady drgawek lub psychoza (po wykluczeniu
przyczyn polekowych, metabolicznych, mocznicy).
Zaburzenia hematologiczne – niedokrwistość
hemolityczna z retikulocytozą lub limfopenia (poniżej 1500
w 1 mm³) lub leukopenia (poniżej 4000 w 1 mm³) lub
trombocytopenia (poniżej 100 000 w 1 mm³)
Zaburzenia immunologiczne – obecność
komórek LE lub
przeciwciał przeciw dsDNA (natywne DNA), lub
przeciwciał anty-‐Sm, lub fałszywie dodatnie serologiczne
odczyny kiłowe przy ujemnym teście na immobilizację
krętków
.
Przeciwciała przeciwjądrowe (ANA) – w mianie nie niższym
niż 80 (zwykle ANA>160)
, badane metodą
immunofluorescencji lub inna odpowiednia jeśli nie
stosowano leków powodujących lekowe zespoły LE.
TOCZEŃ RUMIENIOWATY
UKŁADOWY

Za pewnym rozpoznaniem tocznia przemawia
spełnienie co najmniej
czterech spośród 11 kryteriów
,
przy czym kryteria mogą być spełnione w chwili
badania lub w wywiadzie. Spełnienie dwóch lub
trzech kryteriów pozwala na rozpoznanie choroby
toczniopodobnej.
U ok
. 5%
chorych nie wykrywa się obecności
przeciwciał przeciwjądrowych. Rozpoznaje się wtedy
toczeń seronegatywny
.
Nie można rozpoznać SLE, jeżeli chory nie spełnia
żadnego z kryteriów immunologicznych choroby, tzn.
kryteriów 10 lub 11.
TOCZEŃ RUMIENIOWATY
UKŁADOWY

objaw Raynauda
przerzedzenie włosów (allopecia)
obniżenie stężenia dopełniacza
obecność kompleksów immunologicznych w skórze
pozornie nie zmienionej
hipergammaglobulinemia
charakterystyczne zmiany histologiczne z
odkładaniem się kompleksów immunologicznych w
nerkach (biopsja
TOCZEŃ RUMIENIOWATY UKŁADOWY –
Kryteria pomocnicze rozpoznania
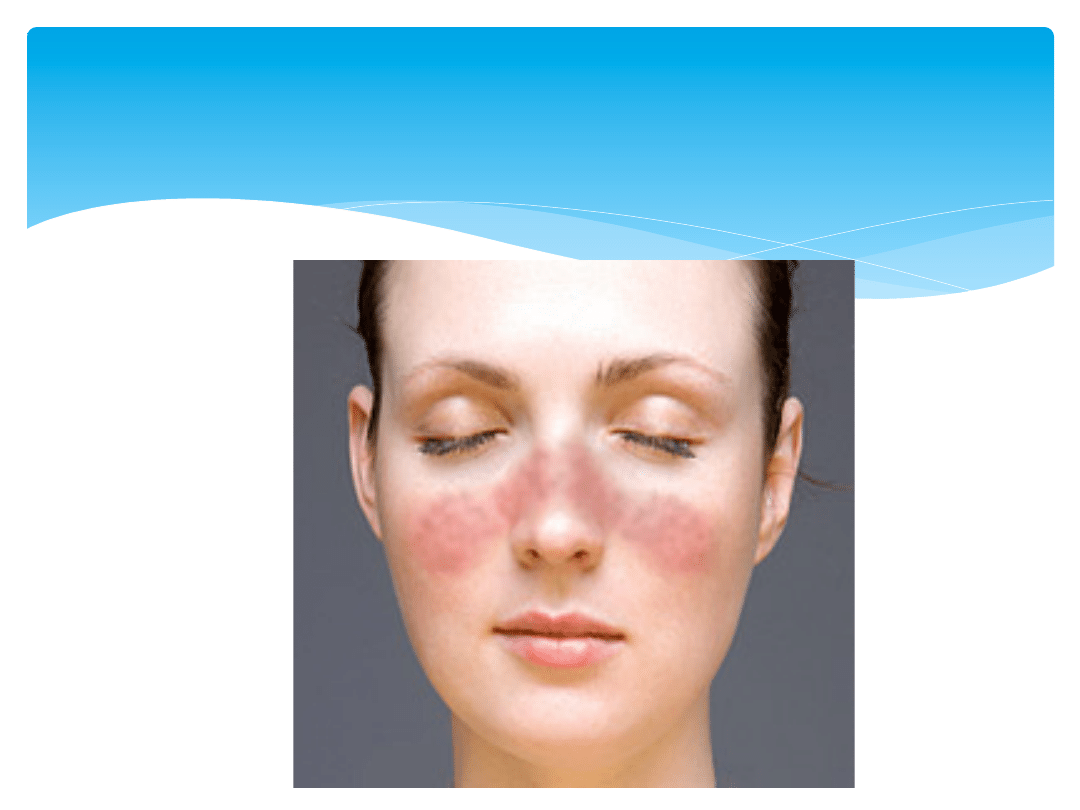
Rumień na twarzy młodej pacjentki z toczniem
rumieniowatym układowym

Skóra, błony śluzowe, narząd wzroku: LBT, badanie okulistyczne,
badanie laryngologiczne
Układ ruchu: RTG stawów.
Układ oddechowy: RTG klatki piersiowej, dodatkowo: HRCT
klatki piersiowej, scyntygrafię płuc, testy czynnościowe płuc,
badanie płynu opłucnowego (ogólne lub bakteriologiczne)
Układ sercowo-‐naczyniowy: EKG, echokardiogram, dodatkowo:
przepływy naczyniowe.
Układ wydalniczy: ogólne badanie moczu i posiew moczu,
proteinuria dobowa, kreatynina z klirensem i mocznik, USG jamy
brzusznej, dodatkowo: biopsja nerki.
Układ nerwowy: badanie neurologiczne, badanie psychiatryczne,
badanie psychologiczne, EEG, tomografia komputerowa głowy,
MRI głowy/kręgosłupa, EMG
TOCZEŃ RUMIENIOWATY UKŁADOWY –
postępowanie diagnostyczne

Stosuje się leczenie objawowe, najczęściej
farmakologiczne, oparte o leki immunosupresyjne i
przeciwmalaryczne.
Zwiększenie spożycia omega-‐3 wielonienasyconych
kwasów tłuszczowych może mieć korzystny wpływ
na objawy choroby
TOCZEŃ TRZEWNY UKŁADOWY -‐
leczenie

Na toczeń rumieniowaty układowy choruje m.in.
amerykański raper Trick Daddy i brytyjski wokalista
Seal

Twardzina, sklerodermia (łac. scleroderma) – rzadka,
przewlekła choroba charakteryzującą się
stwardnieniem skóry i tkanek w wyniku nadmiernego
gromadzenia kolagenu. Choroba ta jest
spowodowana
występowaniem przeciwciał przeciw
topoizomerazie oraz centromerom (w CREST).
Częściej występuje w rodzinach, w których choroba
już cześniej się pojawiała
TWARDZINA

Występuje w dwóch postaciach
-‐
ograniczonej (skóra palców, przedramion i twarzy) i
-‐
układowej (zmiany w skórze, układzie naczyniowym,
mięśniowym, kostnym i w narządach wewnętrznych).
TWARDZINA

Zmiany skórne charakteryzują się twardymi, wyraźnie
odgraniczonymi ogniskami barwy porcelanowej.
Początkowo są one otoczone obwódką barwy
fiołkowej, a następnie ulegają przebarwieniu i
zanikowi.
TWARDZINA

Łagodną postacią twardziny jest zespół CREST (od
Calcinosis -‐ ogniskowych wapnień, występowania
objawu Raynauda, zaburzeń przełykowych –
Esophageal dysmotility, Sclerodactylia i
Teleangiectasia).
Z czasem dochodzi do zwiększenia ilości i pogrubienia
wiązek kolagenu, zaniku odczynu zapalnego i zaniku
przydatków skórnych "zatopionych" w kolagenie
TWARDZINA

Dermatomyositis (zapalenie skórno-‐mięśniowe, DM) –
jest odmianą zapalenia wielomięśniowego, w której
zmiany dotyczą głównie mięśni obręczy barkowej i
biodrowej. Występuje dwa razy częściej u kobiet niż u
mężczyzn. Czasami dochodzi do wystąpienia zmian
narządowych z zajęciem przełyku (trudności w
połykaniu – dysfagia) i pozostałych mięśni gładkich
przewodu pokarmowego, mięśnia serca
(myocarditis), mięśni oddechowych. W przypadku
zajęcia mięśni oddechowych, może dochodzić do
niewydolności oddechowej, która w ciężkich
przypadkach może doprowadzić do śmierci. U 20-‐40%
chorych dochodzi do śródmiąższowej choroby płuc,
która może doprowadzić do włóknienia płuc.
ZAPALENIE SKÓRNO-‐MIĘŚNIOWE

Objawy i przebieg
Postępujące, symetryczne osłabienie siły mięśniowej,
podwyższony poziom enzymów mięśniowych
(aldolazy i kinazy kreatynowej), zmiany w
elektromiogramie, zmiany w biopsji mięśnia (zmiany
zapalno-‐zwyrodnieniowe).
ZAPALENIE SKÓRNO-‐MIĘŚNIOWE

Towarzyszą temu zmiany skórne zlokalizowane
najczęściej w obrębie dłoni (rumienie, grudki nad
drobnymi stawami – objaw Gottrona), wybroczyny,
rumienie w obrębie wałów paznokciowych,
hyperkeratoza na dłoniach („ręce mechanika”) oraz
zmiany rumieniowe w obrębie twarzy z nasilonym
obrzękiem i rumieniem w okolicy oczodołów
(„rzekome okulary”, "heliotrop").
Przy długo trwającej chorobie, częściej u dzieci,
pojawiają się złogi wapnia w tkance podskórnej.
ZAPALENIE SKÓRNO-‐MIĘŚNIOWE

Dermatomyositis jest rewelatorem nowotworowym
(w około 20-‐50% towarzyszy nowotworom narządów
wewnętrznych), zwykle u pacjentów, u których
choroba pojawiła się po 60. roku życia.
U dzieci DM nie jest związane z chorobą
nowotworową.
ZAPALENIE SKÓRNO-‐MIĘŚNIOWE

Polimorfizm genów klasy III HLA (C2, C4A, C4B)
Nosicielstwo allela T1858 PTPN22
Polimorfizm genu CTLA4 kodującego antygen
związany z cytotoksycznym limfocytem T
Polimorfizmy genów kodujących cytokiny, TNF-‐alfa,
TNF-‐beta, IL-‐10, IL1A
Polimorfizm promotora genu MCP1 (monocyte
chemotactic protein 1)
Polimorfizm geny
fibrylaryny (FBN1) – związek tylko z
TU
POLIMORFIZMY GENETYCZNE A PREDYSPOZYCJA DO
TRU, TWARDZINY UKŁADOWEJ, ZAPALENIA
SKÓRNO-‐MIĘŚNIOWEGO I WIELOMIĘŚNIOWEGO

Allele klasy II ukladu HLA są mocniej związane z
autoprzeciwciałami niż podatnością na chorobę lub jej
obrazem klinicznym
Związek przeciwciał u chorych na TRU, TU, DM i PM z
określonym i genami wskazuje na znaczne
zróżnicowanie w zależnosci od badanej rasy i kraju, w
którym badanie przeprowadzono
Opisano związek przeciwciał przeciw topoizomerazie
I, ACA, PM-‐Scl, przeciwciał przeciw fibrylarynie, U1-‐
RNP z niektórymi genami
POLIMORFIZM GENETYCZNY A AUTOPRZECIWCIAŁA U
CHORYCH NA TU, TWARDZINĘ UKŁADOWĄ, ZAPALENIE
SKÓRNO-‐MIĘŚNIOWE I WIELOMIĘŚNIOWE

W niektórych badaniach wykazano związek między
genami i zmianami narządowymi u chorych na TU
Zwrócono także uwagę na powiązanie pewnych
zmian narządowych w zapaleniu mięśni z obecnością
genow. Chorzy z przeciwciałam antysyntetazowymi
mieli istotnie częściej zapalenie stawów, gorączkę, ,
śródmiąższowe zapalenie płuc i allel HLA-‐DRw52
Allel A (-‐308) TNFA zwiazany jest z wapnieniem
tkanek w młodzińczym DM i przewlekłym zapaleniu
mięśni
POLIMORFIZM GENETYCZNY A ZMIANY
NARZĄDOWE U CHORYCH NA TU, ZAPALENIE
WIELOMIĘŚNIOWE I SKÓRNO-‐MIĘŚNIOWE
Wyszukiwarka
Podobne podstrony:
DO TEL ! WYBRANE ZAGADNIENIA Z GENETYKI CHOR B TKANKI CZNEJ
streszczenie- postawy, Psychologia, Psychologia I rok, semestr zimowy, Wprowadzenie do psychologii-
DO TEL! 6 Dziedziczenie zwiaza Nieznany
LD NM , Elektrotechnika-materiały do szkoły, wybrane zagadnienia z teorii obwodów Szymański
9617-opracowanie epok od starożytności do renesansu wybrane zagadnienia, OPRACOWANIA LEKTUR , STRESZ
Zakres tematyczny kolokwium z wybranych zagadnień, Psychologia, Psychologia I rok, semestr zimowy, W
Wprowadznie do psychologii wybrane zagadnienia- opracowanie zagadnień, Psychologia, Psychologia I ro
Ściąga 4.3, Elektrotechnika-materiały do szkoły, wybrane zagadnienia z teorii obwodów Szymański
Wprowadzenie do psychologii-wybrane zagadnienia do egzaminu 2013-2014, Psychologia, Psychologia I r
DO TEL! 5= Genetyka nadci nieni Nieznany
Pytania Wybrane zagadnienia do egz Asys jak, Wybrane zagadnienia do egzaminu „Asystent SZJ&quo
DO TEL! 4= Genetyka chorEb uk a Nieznany
WYBRANE ZAGADNIENIA ROZWOJU ZAWODNIKÓW DO 14 ROKU ŻYCIA, Tenis ziemny
10942 wybrane zagadnienia z geografii powtórka do egzaminu
( ) Wybrane zagadnienia z farma Nieznany (2)
Wybrane zagadnienia ochrony przeciwporażeniowej w instalacjach elektrycznych do 1 kV
WYBRANE ZAGADNIENIA DO MAPY FIZYCZNEJ EUROPY, Geografia
5 wybrane zagadnienia z wytrzy Nieznany (2)
więcej podobnych podstron