MIKROBIOLOGIA
SKRYPT DO ĆWICZEŃ
WERSJA DUŻA
przygotowany przez zespół pracowników
Zakładu Genetyki Bakterii
INSTYTUT MIKROBIOLOGII
WYDZIAŁ BIOLOGII
UNIWERSYTET WARSZAWSKI
2012

2
SPIS TREŚCI
I. REGULAMIN PRACOWNI
3
II. ĆWICZENIA
Ćwiczenie 1.
4
Przygotowanie i jałowienie szkła i podłoży
Podstawowe techniki mikrobiologiczne
Ćwiczenie 2.
9
Izolowanie drobnoustrojów z różnych środowisk naturalnych
Izolowanie czystych kultur
Ćwiczenie 3.
14
Wzrost hodowli bakteryjnej
Określanie liczebności mikroorganizmów
Ćwiczenie 4.
18
Barwienie bakterii
Formy morfologiczne bakterii
Ćwiczenie 5.
20
Barwienie bakterii cd.
Budowa komórki bakteryjnej
Ćwiczenie 6.
22
Metabolizm bakterii – źródła węgla, azotu i energii
Ćwiczenie 7.
28
Metabolizm bakterii – procesy oddechowe i fermentacje
Ćwiczenie 8.
33
Koniugacja u bakterii
Ćwiczenie 9.
38
Analiza mikrobiologiczna wody
Oznaczanie bakterii grupy coli
Ćwiczenie 10.
41
Podstawowe techniki pracy z bakteriofagami
III. ADDENDUM
44
1. Opis kolonii bakteryjnych
2. Komora Thomy
IV. KRÓTKA CHARAKTERYSTYKA BAKTERII STOSOWANYCH NA
mm
ĆWICZENIACH
45
V. SŁOWNICZEK POLSKO-ANGIELSKI
49
(terminy mikrobiologiczne)
VI. ZALECANA LITERATURA
51

3
I. Regulamin pracowni
1. W pracowni obowiązuje noszenie fartucha ochronnego (najlepiej białego).
2. Wszystkie odczynniki i ich roztwory, a także podłoża i hodowle mikroorganizmów muszą
być czytelnie oznakowane.
3. Na stołach laboratoryjnych mogą znajdować się wyłącznie materiały potrzebne do
wykonywania ćwiczenia.
4. Należy zachować daleko idącą ostrożność przy pracy z materiałem mikrobiologicznym, a
w szczególności:
a) stosować się do zasad pracy jałowej (zachować szczególną ostrożność przy pracy
w bezpośrednim sąsiedztwie płomienia palnika);
b) wstawiać hodowle bakteryjne w probówkach do statywów (nie wolno ich kłaść na
stole);
c) opisywać każdą posianą próbę (rodzaj posiewu, inicjały osoby wykonującej
posiew, itp.);
d) po skończeniu posiewów wyżarzać ezy, opalać głaszczki i zawsze wstawiać je do
statywów, a zużyte pipety wkładać do specjalnych pojemników.
5. W pracowni obowiązuje szczególna ostrożność przy pracy z odczynnikami chemicznymi
(do pipetowania ich roztworów należy używać WYŁĄCZNIE pompek lub gruszek);
6. W pracowni zabronione jest spożywanie posiłków, a także picie napojów.
7. Po zakończeniu pracy należy:
a) posprzątać stół laboratoryjny;
b) niepotrzebny już sprzęt odłożyć na miejsce, pozostawić w porządku mikroskopy;
8. Niepotrzebne hodowle drobnoustrojów oraz szkło zużyte w trakcie pracy należy odłożyć do
specjalnie przygotowanych pojemników, po uprzednim usunięciu wszelkich napisów, i
zanieść do zmywalni.
9. Nie wolno wynosić z pracowni żadnych hodowli bakteryjnych, preparatów i odczynników
bez pozwolenia prowadzącego zajęcia.
10. Po zakończeniu pracy należy sprawdzić, czy został wyłączony gaz oraz aparatura nie
przeznaczona do pracy ciągłej, a przed wyjściem z sali – umyć ręce.
11. W razie pojawienia się jakichkolwiek problemów należy niezwłocznie zgłosić się do
osoby prowadzącej zajęcia lub do opiekuna.

4
Ćwiczenie 1
Temat: Przygotowanie i jałowienie szkła i podłoży
rrr
Podstawowe techniki mikrobiologiczne
I. WPROWADZENIE
1. Podłoża
Obiektem badań w mikrobiologii są mikroskopijnej wielkości organizmy występujące
powszechnie we wszystkich środowiskach naturalnych. Badanie tych organizmów w naturalnych
warunkach bytowania jest jednak z wielu względów bardzo trudne, a często niemożliwe.
Poznanie morfologii, fizjologii, wymagań pokarmowych i środowiskowych drobnoustrojów jest
możliwe dzięki stworzeniu sztucznych środowisk do ich hodowli – tzw. podłoży (pożywek)
mikrobiologicznych. Umożliwiły one hodowanie drobnoustrojów w warunkach laboratoryjnych i
uzyskanie czystych kultur – tzn. hodowli stanowiących potomstwo jednego osobnika i będących
materiałem do badań mikrobiologicznych.
Podłoża mikrobiologiczne są to mieszaniny odpowiednio dobranych składników
odżywczych, dostarczających hodowanym na nich organizmom niezbędnych pierwiastków
chemicznych oraz źródła energii. Każde podłoże musi mieć odpowiednią dla danego gatunku
wartość odżywczą, odpowiednie pH, rH i ciśnienie osmotyczne. Ważne jest również określenie,
do jakiego celu ma służyć dane podłoże (np. czy chodzi nam o uzyskanie hodowli, o
wyselekcjonowanie jakiegoś określonego gatunku, o zróżnicowanie gatunków występujących w
mieszaninie, itd.).
Zależnie od potrzeby możemy zastosować podłoże:
(i)
minimalne – zawiera tylko te składniki, które są niezbędne do wzrostu
mikroorganizmu;
(ii)
pełne – zapewnia optymalny wzrost danego mikroorganizmu;
(iii)
selekcyjne – umożliwia wyłącznie wzrost mikroorganizmów o określonych
cechach;
(iv)
różnicujące – pozwala na zróżnicowanie mikroorganizmów względem określonej
cechy na podstawie morfologii kolonii;
(v)
syntetyczne – charakteryzuje się ściśle określonym składem jakościowym
i ilościowym;
(vi)
złożone – zawiera ekstrakty drożdżowe, autolizaty drożdżowe, peptony, ekstrakty
mięsne, itp., a więc jego skład jakościowy i ilościowy nie jest ściśle określony.
Podłoża wzbogacone stosuje się do hodowli drobnoustrojów, które wymagają dodatkowych
substancji odżywczych, występujących w naturalnych produktach. Podłoże (np. bulion
odżywczy) wzbogaca się, dodając do niego krew, surowicę, wyciąg glebowy, mleko, żółtko jaj,
namok siana, soki warzywne, itp. Podłoża mineralne nie zawierają związków organicznych i
stosuje się je do hodowania różnego rodzaju mikroorganizmów autotroficznych.
Ze względu na konsystencję dzielimy podłoża na: (i) płynne, (ii) półpłynne i (iii) stałe.
Podłoża zestala się najczęściej agarem, który jest uzyskiwany z krasnorostów morskich. Podłoża
zestalone można wykorzystywać w postaci słupków, skosów i na płytkach Petriego.
2. Zasady przygotowywania podłoży
Przygotowując podłoża należy:
a) używać szkła odpowiednio wymytego i wypłukanego;
b) przestrzegać ściśle przepisów przygotowania pożywek (odważać dokładnie składniki,
zwracać uwagę na kolejność ich dodawania i na uwodnienie soli);

5
c) używać tylko wody destylowanej;
d) ustawiać odpowiednie pH podłoża, pamiętając o tym, że po jałowieniu w autoklawie pH
może się obniżyć;
e) do jałowienia rozlewać podłoże do kolb do objętości nie większej niż 3/4 naczynia, aby nie
wykipiało w czasie jałowienia w autoklawie;
f) kolby zatykać korkami z waty, zabezpieczać korki przed zamoczeniem folią aluminiową i
odpowiednio podpisywać;
g) stosować możliwie najniższą temperaturę do jałowienia.
3. Sposoby sterylizacji
Jednym z koniecznych warunków, jaki muszą spełniać wszystkie podłoża, jest ich sterylność, co
oznacza, że muszą być pozbawione wszelkich organizmów – zarówno ich form wegetatywnych
jak i przetrwalnikowych, a także wirusów. Proces sterylizacji można przeprowadzić dwoma
sposobami:
1. przez zabicie drobnoustrojów i ich form przetrwalnikowych, a także inaktywację
wirusów, w danym środowisku w wyniku działania:
(a) wysokiej temperatury – suszarka, autoklaw, aparat Kocha;
(b) promieniowania UV,
X;
(c) związków chemicznych, np. tlenek etylenu lub propylenu;
2. przez usunięcie drobnoustrojów i ich form przetrwalnikowych z danego środowiska
(filtracja).
Wybór metody sterylizacji zależy od rodzaju sterylizowanego podłoża (środowiska), a także od
wyposażenia laboratorium; należy wybrać metodę nie niszczącą podłoża, a skuteczną, możliwie
szybką i tanią. Trzeba też pamiętać, że w pracowni mikrobiologicznej sterylizujemy wszystko,
czym moglibyśmy zakazić hodowlę badanych drobnoustrojów.
Jałowienie szkła w suszarce
W suszarce jałowimy przede wszystkim szkło (odpowiednio zabezpieczone i zapakowane) i te
przedmioty, które nie ulegną zniszczeniu w tak wysokiej temperaturze. Sterylizację
przeprowadza się w 160
o
C przez 2 godziny lub rzadziej w 180
o
C przez 1 godzinę (nie wolno
przekraczać temp. 180
o
C, gdyż grozi to zwęgleniem papieru i waty). Zabite zostają wszystkie
mikroorganizmy, w tym ich formy przetrwalnikowe, i wirusy.
Jałowienie w autoklawie
Temperatura 100
o
C nie zabija form przetrwalnikowych i niektórych wirusów. Wyższą
temperaturę wrzenia wody można osiągnąć po zastosowaniu nadciśnienia. W autoklawie –
hermetycznie zamkniętym kotle – stosując nadciśnienie 1 atm, uzyskuje się atmosferę nasyconej
pary wodnej o temp. 121
o
C. W tej temperaturze wszystkie mikroorganizmy i ich przetrwalniki
zostają zabite w ciągu około 30 min. Czas trwania sterylizacji w autoklawie zależeć będzie od
rodzaju jałowionego materiału i jego objętości. W autoklawie jałowi się:
a) podłoża (oprócz tych, które rozkładają się w tej temperaturze), np. bulion i agar
odżywczy,
b) sól fizjologiczną, roztwory soli, bufory,
c) wodę destylowaną,
d) narzędzia chirurgiczne, opatrunki, itp.
W autoklawie nie sterylizuje się stężonych roztworów cukrów oraz substancji łatwo
hydrolizujących, np. witamin, aminokwasów, puryn, pirymidyn, mocznika.
6
Jałowienie w aparacie Kocha (tyndalizacja)
W aparacie Kocha sterylizuje się substancje, które ulegają rozkładowi w temp. powyżej 100
o
C.
Tyndalizacja polega na trzykrotnym ogrzewaniu jałowionego podłoża w temp. 100
o
C przez 30
min, co 24 godz. W czasie pierwszego ogrzewania zabite zostają formy wegetatywne, a
przetrwalniki są aktywowane do kiełkowania, w wyniku działania wysokiej temperatury i
obecności pewnych związków organicznych. Proces ten jest możliwy, dzięki pozostawieniu
jałowionego materiału w temp. pokojowej przez około 24 godz. Następne ogrzewanie zabija
kiełkujące przetrwalniki (które utraciły ciepłooporność). Trzecie ogrzewanie zabija ewentualne
formy endospory, których kiełkowanie było opóźnione.
Ponieważ metoda tyndalizacji wykorzystuje zjawisko kiełkowania przetrwalników,
możemy ją stosować tylko do jałowienia wodnych roztworów substancji umożliwiających ten
proces, a więc zawierających określone związki organiczne. W ten sposób jałowi się stężone
roztwory cukrów, witamin, aminokwasów, puryn, pirymidyn, itp.
Jałowienie przez filtrację
Filtracja pozwala na jałowienie płynów, które ulegają rozkładowi pod wpływem ciepła (np.
roztwór mocznika, surowica), a także gazów. Polega ona na przepuszczaniu jałowionego płynu
przez filtr o określonej wielkości porów przy zastosowaniu nad- lub podciśnienia. Filtr
zatrzymuje bakterie na zasadzie mechanicznej i/lub fizyko-chemicznej. Filtry i oprawki do
filtrów należy przed użyciem wyjałowić (na ogół w autoklawie); sterylne musi też być naczynie,
do którego filtrujemy jałowy płyn. Do jałowienia niewielkich ilości płynu bardzo wygodne są,
dostępne w sprzedaży, wysterylizowane jednorazowe zestawy filtracyjne. Trzeba pamiętać, że
filtracja nie usuwa wirusów.
II. CZĘŚĆ PRAKTYCZNA
1. Przygotowanie bulionu odżywczego, agaru odżywczego oraz składników podłoża Davisa
i ich jałowienie
1.1. Bulion odżywczy
Skład:
bulion w proszku
15 g
woda destylowana
1000 ml
a) Bulion w proszku wsypać do kolby, wlać wodę, rozpuścić mieszając. Sprawdzić pH
podłoża przy pomocy papierka wskaźnikowego – powinno wynosić 7,2 - 7,4.
b) Część bulionu rozlać po około 200 ml do 2 kolb o pojemności 300 ml. Kolby zatkać
korkami z waty i zabezpieczyć przed zamoknięciem folią aluminiową.
c) Jedną kolbę jałowić w autoklawie przy nadciśnieniu 1 atm przez 30 min, drugą pozostawić
bez jałowienia.
d) Pozostały bulion zużyć do przygotowania agaru odżywczego.
1.2 Agar odżywczy
Skład:
bulion odżywczy
200 ml
agar-agar w proszku
4 g
a) Do kolby o pojemności 300 ml odważyć 4 g sproszkowanego agaru i wlać 200 ml
przygotowanego bulionu odżywczego (nie mieszać!).
b) Kolbę zatkać korkiem z waty, zabezpieczyć folią aluminiową.

7
c) Jałowić w autoklawie przy nadciśnieniu 1 atm przez 30 min.
Każdy zespół studentów przygotowuje sobie 200 ml agaru odżywczego na następne
ćwiczenia.
1.3. Podłoże Davisa płynne i stałe
Podłoże Davisa przygotowuje się przez zmieszanie, oddzielnie przygotowanych i
wyjałowionych, następujących składników:
a) 150 ml wody destylowanej lub 150 ml agaroidu (woda dest. + agar)
(zależnie od konsystencji podłoża)
b) 40 ml soli Davisa
c) 4 ml 20 % glukozy
Przygotowanie składników podłoża:
ad. a)
- Do 2 kolb o pojemności 300 ml wlać po 150 ml wody destylowanej.
- Do jednej z nich niczego nie dodawać - będzie służyła do przygotowania podłoża płynnego.
- Do drugiej odważyć 4 g sproszkowanego agaru (nie mieszać!). Otrzymamy w ten sposób
agaroid.
- Obie kolby zatkać korkami z waty, zabezpieczyć folią aluminiową.
- Jałowić w autoklawie przy nadciśnieniu 1 atm przez 30 min.
ad. b)
- Do kolby o pojemności 200 ml wlać około 90 ml wody destylowanej.
- Odważyć wymienione niżej sole i wsypać je do kolby z wodą i wymieszać:
K
2
HPO
4
3,5 g
KH
2
PO
4
1,0 g
MgSO
4
x7H2O
0,05 g
(NH
4
)
2
SO
4
0,05 g
cytrynian sodu
0,25 g
- Przelać roztwór do cylindra i uzupełnić wodą dest. do 100 ml, wymieszać.
- Rozlać roztwór soli po 40 ml do kolbek à 100 ml.
- Jałowić w autoklawie przy nadciśnieniu 1 atm przez 30 min.
ad. c)
- W kolbie o pojemności 100 ml przygotować 50 ml 20 % roztworu glukozy w wodzie
destylowanej.
- Rozlać do 2 probówek po około 5 ml, a resztę zostawić w kolbce.
- Jedną probówkę pozostawić bez jałowienia.
- Drugą probówkę wyjałowić w autoklawie przy nadciśnieniu 1 atm przez 30 min.
- Kolbkę z glukozą wyjałowić w aparacie Kocha (tyndalizacja).
Zaobserwować zmianę barwy roztworu po wyjałowieniu w autoklawie i ewentualne zmiany w
niejałowionym roztworze glukozy.
2. Technika pracy sterylnej
a) Dezynfekcja stołów laboratoryjnych za pomocą sterinolu lub alkoholu etylowego.
b) Praca w bezpośrednim sąsiedztwie płomienia palnika.
c) Sterylizacja ez (wyżarzanie w płomieniu palnika) i głaszczek (opalanie w etanolu).

8
2.1. Praktyczne zastosowanie poznanych technik i metod posiewu bakterii
a) Przestrzegając zasad pracy sterylnej rozlać do 3 probówek po 2 ml jałowego bulionu.
Po tygodniu sprawdzić jałowość bulionu we wszystkich 4 probówkach (również w tej, z której
pobierany był bulion).
b) Otrzymaną hodowlę bakteryjną wysiać ezą, według wskazówek prowadzącego zajęcia, na
płytkę z agarem odżywczym – posiewem redukcyjnym (p. rysunek 1).
Rys. 1. Posiew redukcyjny. 1
–
początek linii posiewu; 2 i 3
–
miejsca, w których przerywa się posiew
i opala ezę w celu usunięcia nadmiaru materiału.
III. ZAGADNIENIA DO OPRACOWANIA
1. Skład podłoży mikrobiologicznych i ich przygotowanie.
2. Kryteria podziału podłoży mikrobiologicznych.
3. Sterylizacja i sposoby jej przeprowadzania.
4. Pasteryzacja, dezynfekcja i zastosowanie tych procesów w praktyce.
5. Metody przedłużania trwałości żywności.

9
Ćwiczenie 2
Temat: Izolowanie drobnoustrojów z różnych środowisk naturalnych
Izolowanie czystych kultur
I. WPROWADZENIE
Woda, gleba i organizmy żywe są środowiskami dogodnymi dla wzrostu różnych
mikroorganizmów. Mikroorganizmy znajdują się również w powietrzu, które nie jest jednak
środowiskiem sprzyjającym ich rozwojowi. To właśnie mikroorganizmy wytyczają granice
biosfery, a tak szerokie rozprzestrzenienie w przyrodzie zawdzięczają następującym cechom: 1)
małe rozmiary; 2) krótki czas generacji; 3) różnorodność metaboliczna (zdolność do
wykorzystania wielu źródeł węgla, azotu, energii, różnych końcowych akceptorów elektronów);
4) zdolność adaptacji do zmieniających się warunków środowiska; 5) zdolność do życia w
warunkach ekstremalnych (dotyczy to temperatury, pH, potencjału oksydo-redukcyjnego,
ciśnienia osmotycznego, ciśnienia hydrostatycznego i bardzo niskich stężeń substancji
pokarmowych); 6) wytwarzanie form przetrwalnikowych.
Na ogół w danym środowisku występują różne mikroorganizmy. Jeśli chcemy
wyizolować określony gatunek, musimy zastosować odpowiednie podłoża i warunki hodowli,
hamujące wzrost innych mikroorganizmów i prowadzące do selektywnego namnażania
mikroorganizmu poszukiwanego. Jest to tzw. hodowla wzbogacająca. Jeśli spodziewamy się, że
w danym środowisku jest mało komórek poszukiwanego mikroorganizmu, hodowlę
wzbogacającą należy założyć na podłożu płynnym, a dopiero po uzyskaniu wzrostu, przesiać
mikroorganizmy na podłoże stałe. Z wyrosłych kolonii możemy następnie założyć czyste
kultury, a po ich identyfikacji, uzyskać czystą kulturę poszukiwanego przez nas gatunku.
1. Izolowanie czystych kultur
Metody izolowania czystych kultur dzielimy na bezpośrednie i pośrednie. W metodach
bezpośrednich
pobieramy
pojedynczą
komórkę
mikroorganizmu
(np.
stosując
mikromanipulator) i przenosimy do jałowego podłoża płynnego. W metodach pośrednich
izolujemy pojedyncze kolonie na podłożu stałym. Zakładając, że pojedyncza kolonia stanowi
potomstwo pojedynczej komórki, pobieramy z niej materiał i przenosimy na podłoże stałe,
wykonując posiew redukcyjny. Pojedyncze kolonie możemy uzyskać dzięki posiewowi:
1) powierzchniowemu na podłoże stałe w płytce Petriego, wykonanemu za pomocą:
- ezy – posiew redukcyjny (metoda suchych rozcieńczeń);
- pipety – metoda rozcieńczeń; zwykle wysiewa się 0,1 ml i rozprowadza po
powierzchni płytki głaszczką;
2) wgłębnemu, w którym zawiesinę mikroorganizmów wprowadza się na dno pustej
szalki Petriego, a następnie zalewa upłynnionym i odpowiednio schłodzonym
podłożem stałym (zwykle wysiewa się 1 ml).
Jeśli w badanej próbie jest mało mikroorganizmów, możemy ją przefiltrować, a filtr umieścić na
odpowiednim podłożu (p. punkt 3, str. 15).
2. Podstawowe pojęcia mikrobiologiczne
Hodowla to podłoże z namnożonymi mikroorganizmami. Hodowle można prowadzić na
podłożu płynnym bądź stałym. Hodowle mogą być jednogatunkowe (gdy na podłożu rośnie
jeden gatunek bakterii) lub mieszane (gdy rosną w nich przynajmniej dwa gatunki).
Kolonia to widoczne gołym okiem skupisko drobnoustrojów na podłożu stałym. Na ogół kolonia
powstaje w wyniku podziałów pojedynczej komórki.
10
Czystą kulturą nazywamy hodowlę, w której bakterie stanowią potomstwo jednej, pierwotnie
wyosobnionej komórki bakteryjnej.
Szczepy to różne klony należące do tego samego gatunku, a wyprowadzone z poszczególnych
czystych kultur, izolowanych niezależnie od siebie.
II. CZĘŚĆ PRAKTYCZNA
1. Przygotowanie podłoży
a) Rozlać na płytki Petriego upłynniony agar odżywczy (przygotowany na poprzednich
ćwiczeniach).
b) Przygotować podłoże stałe Davisa poprzez zmieszanie:
- 150 ml agaroidu
- 40 ml soli Davisa
- 4 ml 20 % glukozy
c) Po zastygnięciu podłoży, płytki wysuszyć, w celu odparowania części wody.
d) Przed dokonaniem posiewów płytki należy odpowiednio podpisać flamastrem na denku,
uwzględniając rodzaj posiewanej próbki, rozcieńczenie, inicjały osoby wykonującej posiew.
2. Izolowanie drobnoustrojów z powietrza metodą sedymentacyjną Kocha i określanie
ich liczby
a) Płytki z agarem odżywczym postawić w miejscu, gdzie wykonany będzie posiew.
b) Zdjąć wieczko i wystawić pożywkę na działanie powietrza przez 5, 10 lub 15 minut
zależnie od przewidywanego skażenia powietrza. Po ekspozycji płytki zakryć.
c) Po kilkudniowej inkubacji w temperaturze pokojowej policzyć kolonie mikroorganizmów, a
następnie obliczyć ich liczbę (X) w 1 m
3
powietrza według zamieszczonego niżej wzoru
(według założenia Omeliańskiego na 1 m
2
podłoża osiada w ciągu 5 minut tyle
mikroorganizmów, ile znajduje się 1 m
3
powietrza):
n x 5
X = ---------
gdzie: n – uśredniona liczba kolonii na płytce
p x t
p – powierzchnia płytki w m
2
t – czas otworzenia płytki w min
Tabela 1. Badanie czystości mikrobiologicznej powietrza
Miejsce badania czystości
powietrza
Liczba drobnoustrojów
w 1 m
3
powietrza
Powietrze atmosferyczne uważamy za:
- nie zanieczyszczone, jeśli ogólna liczba bakterii w 1 m
3
wynosi mniej niż 1000;
- średnio zanieczyszczone, jeśli ogólna liczba bakterii w 1 m
3
wynosi od 1000 do 3000;
- silnie zanieczyszczone, jeśli ogólna liczba bakterii w 1 m
3
wynosi więcej niż 3000.
Dopuszczalny stopień mikrobiologicznego zanieczyszczenia powietrza atmosferycznego wynosi 3000
mikroorganizmów w 1 m
3
.
11
Dopuszczalna liczba mikroorganizmów w 1 m
3
powietrza pomieszczeń użytkowych wynosi:
- pomieszczenia służby zdrowia
sala operacyjna - 100
sala opatrunkowa - 150
sala z chorymi – 1000
- pomieszczenia domów mieszkalnych
kuchnia i jadalnia – 2000
pokój do przyjęć – 1500
sypialnia – 1000
- pomieszczenia szkolne
sale wykładowe – 1500
sale do ćwiczeń – 2000
sale gimnastyczne – 3000
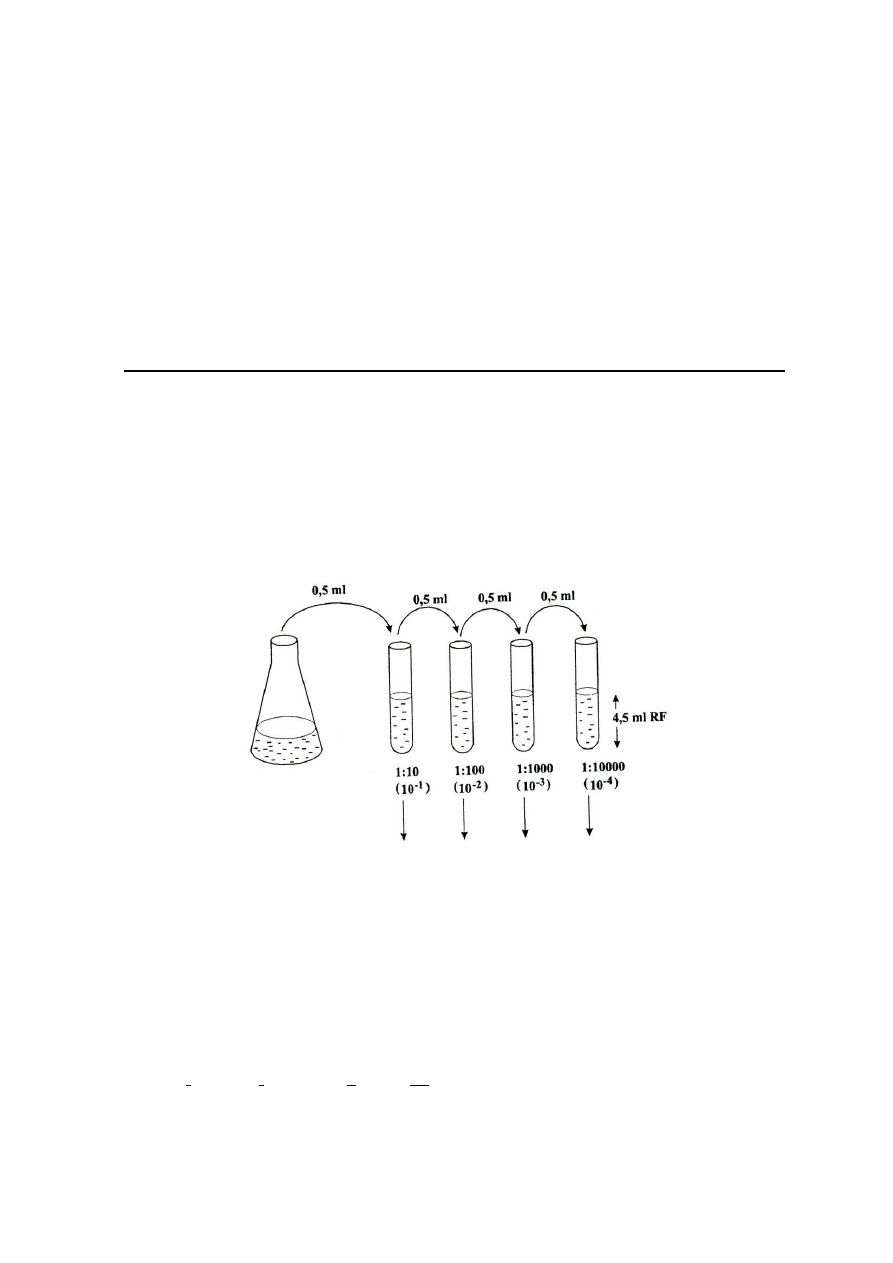
3. Izolowanie mikroorganizmów z naturalnego zbiornika wodnego i określanie ich liczby
a) Rozcieńczyć wodę ze zbiornika (10
-1
- 10
-4
) wg schematu przedstawionego na Rys. 2.
- Wlać do 4 probówek po 4,5 ml soli fizjologicznej.
- Do pierwszej probówki odmierzyć pipetą (à 1 ml) 0,5 ml badanej wody. Pipetę odłożyć, a
zawartość probówki dobrze wymieszać. W ten sposób rozcieńczyliśmy badaną wodę
dziesięciokrotnie (10
-1
).
- Przenieść 0,5 ml rozcieńczenia 10
-1
do następnej probówki z solą fizjologiczną i
wymieszać. Uzyskujemy stukrotne rozcieńczenia badanej wody (10
-2
).
- Postępując analogicznie otrzymujemy rozcieńczenia 10
-3
i 10
-4
.
Rys. 2. Schemat rozcieńczania hodowli bakteryjnej.
b) Wysiać po 0,1 ml każdego rozcieńczenia na płytki z: (i) agarem odżywczym w 2
powtórzeniach; (ii) na płytki z podłożem Davisa (po jednej płytce na każde rozcieńczenie).
c) Po kilkudniowej inkubacji w temperaturze pokojowej zaobserwować zróżnicowaną
morfologię kolonii mikroorganizmów.
d) Następnie policzyć kolonie na płytkach. Do liczenia należy wziąć te płytki, na których
wyrosło od 30 do 300 kolonii. Określić liczbę mikroorganizmów zdolnych do wytworzenia
oo
kolonii (jednostek tworzących kolonie, jtk) w 1 ml badanej wody (X) wg wzoru:
X = a x b x 10 [jtk/ml]
gdzie: a – średnia liczba bakterii na płytkach
b – odwrotność wysianego rozcieńczenia
10 – przeliczenie na 1 ml
wysiew po 0,1ml z każdej probówki na dwie płytki
z agarem odżywczym i jedną z podłożem Davisa
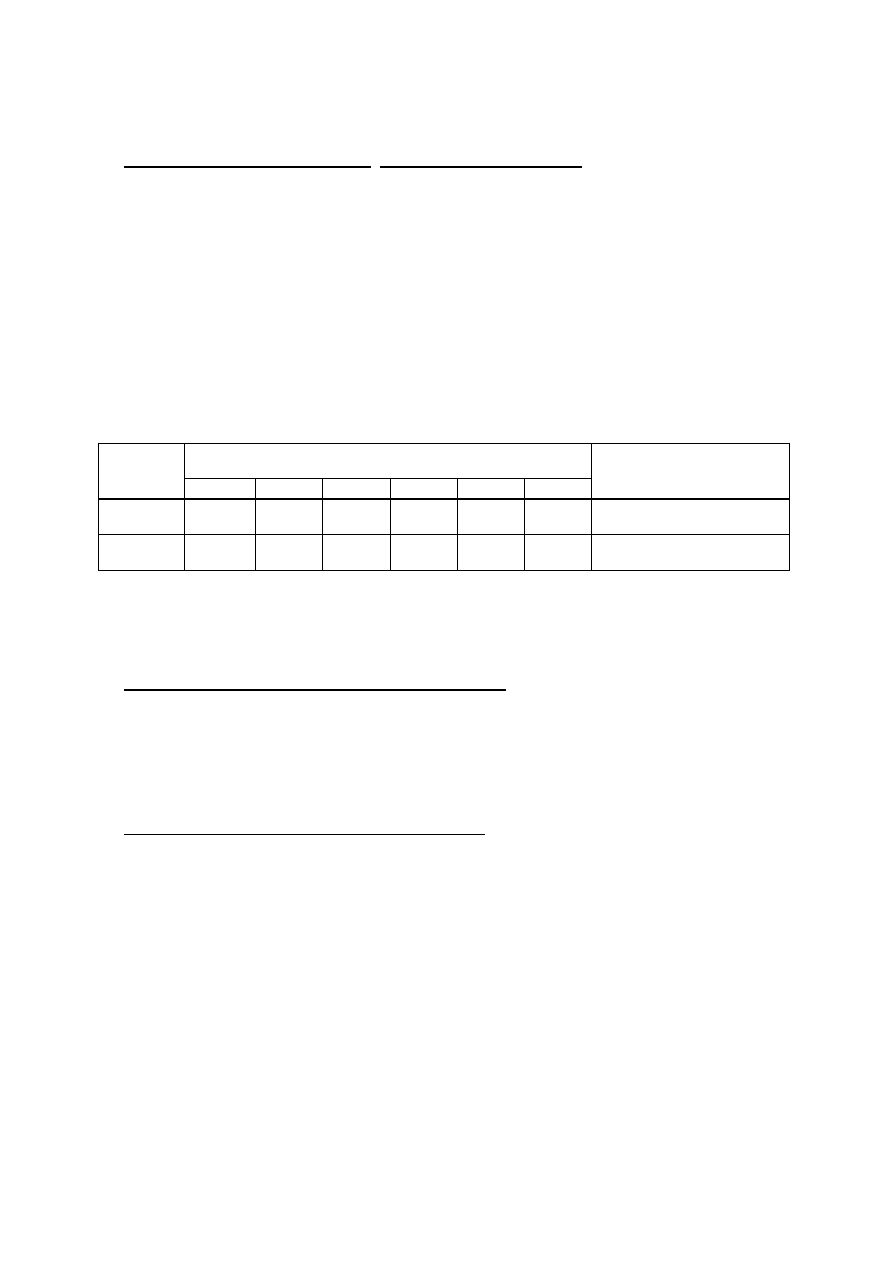
12
Wyniki zapisać w tabeli 2.
4. Izolowanie mikroorganizmów z gleby i określanie ich liczby
a) Przygotować roztwór glebowy. W tym celu odważyć 2 g gleby i wsypać do kolby
zawierającej 18 ml soli fizjologicznej i całość wytrząsać kilka minut w celu wymycia
mikroorganizmów z cząstek gleby. Poczekać, aż cząstki stałe opadną na dno.
b) Przygotować kolejne rozcieńczenia gleby (10
-1
- 10
-6
) tak jak w punkcie 3a, traktując
roztwór glebowy jako rozcieńczenie 10
-1
. Wysiać na dwie płytki z agarem odżywczym po
0,1 ml każdego z rozcieńczeń od 10
-2
do 10
-6
.
c) Po kilkudniowej inkubacji w temperaturze pokojowej zaobserwować zróżnicowaną
morfologię kolonii mikroorganizmów.
d) Policzyć kolonie, a następnie określić liczbę jednostek (mikroorganizmów) zdolnych do
wytworzenia kolonii (jtk) w 1 g gleby, stosując wzór z punktu 3d). Wyniki zapisać w tabeli 2.
Tabela 2. Określenie liczby bakterii w wodzie i glebie
Badana
próbka
Średnia liczba kolonii bakterii na agarze odżywczym
na rozcieńczeniu:
Liczba jtk* w 1 ml wody
lub w 1 g gleby
10
-1
10
-2
10
-3
10
-4
10
-5
10
-6
Woda
X
X
Gleba
X
X
* jtk = jednostka tworząca kolonię (cfu, ang. colony forming unit)
e) Porównać: (i) liczebność bakterii w badanej wodzie i glebie i (ii) liczby jtk/ml uzyskane na
podłożu Davisa i na agarze odżywczym.
.
5. Izolowanie mikroorganizmów z innych środowisk
a) Na jednej płytce z agarem odżywczym wykonać posiew dowolny, np. kaszlnąć, dotknąć
palcem brudnym i przetartym etanolem, dotknąć jakimś przedmiotem, zrobić posiew
materiału pobranego jałową wymazówką, itp.
b) Po inkubacji w temperaturze pokojowej obserwować wzrost bakterii.
6. Charakterystyka wybranej kolonii bakteryjnej
a) Wybrać jedną z kolonii bakteryjnych i opisać w zeszycie jej morfologię (patrz
III.1 Addendum) uwzględniając:
- kształt i brzeg kolonii;
- wielkość (średnicę) w mm;
- wzniesienie;
- barwę kolonii i jej otoczenia (barwnik może dyfundować do podłoża);
- typ wzrostu (na powierzchni lub częściowo wrośnięta w podłoże);
- przejrzystość (przejrzysta, nieprzejrzysta, opalizująca);
- powierzchnię (gładka, lśniąca, matowa, pomarszczona, pofałdowana, krzaczkowata,
koncentrycznie pierścieniowata);
- strukturę (np. ziarnista, włóknista, skórzasta, krucha, ciągnąca się – strukturę bada się za
pomocą ezy).

13
Należy przy tym pamiętać, że morfologia kolonii zależy nie tylko od rodzaju
mikroorganizmu, ale również od składu podłoża i warunków hodowli. Hodowla danego
mikroorganizmu może charakteryzować się też swoistym zapachem.
b) Wybraną kolonię bakteryjną (z dowolnej płytki) posiać metodą posiewu redukcyjnego na agar
odżywczy (tak jak pokazano na rysunku 1, str. 8). Płytkę inkubować w temperaturze
pokojowej. Następnie sprawdzić, czy uzyskana hodowla jest jednorodna i czy pojedyncze
kolonie mają taką samą morfologię jak pobrana kolonia wyjściowa. Jeśli tak, to udało się
uzyskać czystą kulturę. Posiew redukcyjny należy powtórzyć, pobierając ponownie materiał z
pojedynczej kolonii. Należy pamiętać, że mikroorganizmy stanowiące zakażenia mogą
charakteryzować się wolniejszym wzrostem, a tym samym ich kolonie mogą pojawić się na
płytce w późniejszym czasie.
III. ZAGADNIENIA DO OPRACOWANIA
1. Przyczyny szerokiego rozpowszechnienia mikroorganizmów w przyrodzie.
2. Metody izolowania mikroorganizmów z różnych środowisk naturalnych.
3. Metody izolowania czystych kultur – bezpośrednie i pośrednie.
4. Podstawowe pojęcia mikrobiologiczne – hodowla, czysta kultura, kolonia, szczep, klon.
14
Ćwiczenie 3
Temat: Wzrost hodowli bakteryjnej
Określanie liczebności mikroorganizmów
I. WPROWADZENIE
1. Hodowle mikroorganizmów
Po wsianiu bakterii na odpowiednie podłoże płynne i inkubacji w optymalnych dla danego
gatunku warunkach następuje, po pewnym okresie adaptacji, intensywny przyrost liczby bakterii
w hodowli. Jednakże skład podłoża takiej hodowli podlega ciągłym zmianom; ubożeje ono w
składniki pokarmowe, natomiast nagromadzają się w nim stopniowo metabolity, wykazujące
często działanie toksyczne. Tego typu hodowle noszą nazwę hodowli okresowych (Rys. 3A). W
hodowlach tego typu obserwuje się kilka faz wzrostu. (i) Faza zastoju (adaptacyjna), następuje
po wsianiu bakterii do nowego podłoża. Bakterie przystosowują się do nowego środowiska i ich
liczba nie rośnie. (ii) Faza młodości fizjologicznej następuje pod koniec fazy zastoju, komórki
rosną i zaczynają się dzielić, więc ich liczba w hodowli zaczyna powoli rosnąć. (iii) Faza
wzrostu wykładniczego – wszystkie komórki są w dobrej kondycji fizjologicznej, rosną i dzielą
się w stałych odstępach czasu, równych czasowi podwojenia (czasowi generacji). W
zamkniętym systemie wzrost nie może trwać wiecznie. W hodowli rośnie stężenie szkodliwych
produktów przemiany materii, a stężenie związków odżywczych maleje, w konsekwencji
częstość podziałów komórek bakteryjnych zmniejsza się i rozpoczyna się (iv) faza stacjonarna,
która składa się z kilku etapów: faza zwolnionego wzrostu, faza równowagi (w tej fazie liczba
żywych bakterii nie zmienia się), faza zamierania (liczba żywych komórek stale się zmniejsza,
zaś rośnie liczba komórek martwych).
Jeżeli chcemy utrzymać hodowlę w fazie intensywnego wzrostu, musimy ciągle
uzupełniać zużywane z podłoża składniki pokarmowe i odprowadzać z hodowli nagromadzające
się metabolity i nadmiar komórek. W takiej hodowli ciągłej (Rys. 3B) faza wzrostu
wykładniczego może trwać przez czas nieograniczony, jeśli nie dopuści się do pojawienia sie
warunków obniżających szybkość wzrostu.
Jeszcze inny rodzaj hodowli uzyskamy, jeśli wszystkie bakterie będą na tym samym
etapie cyklu komórkowego, a więc będą się równocześnie dzieliły. Taka synchroniczna
hodowla (Rys. 3C) jest odbiciem zachowania się pojedynczej komórki i przez to może być
przydatna w badaniach dotyczących fizjologii bakterii.
Rys. 3. Krzywe wzrostu bakterii. (A) hodowla okresowa, (B) hodowla ciągła, (C) hodowla
i
synchroniczna.
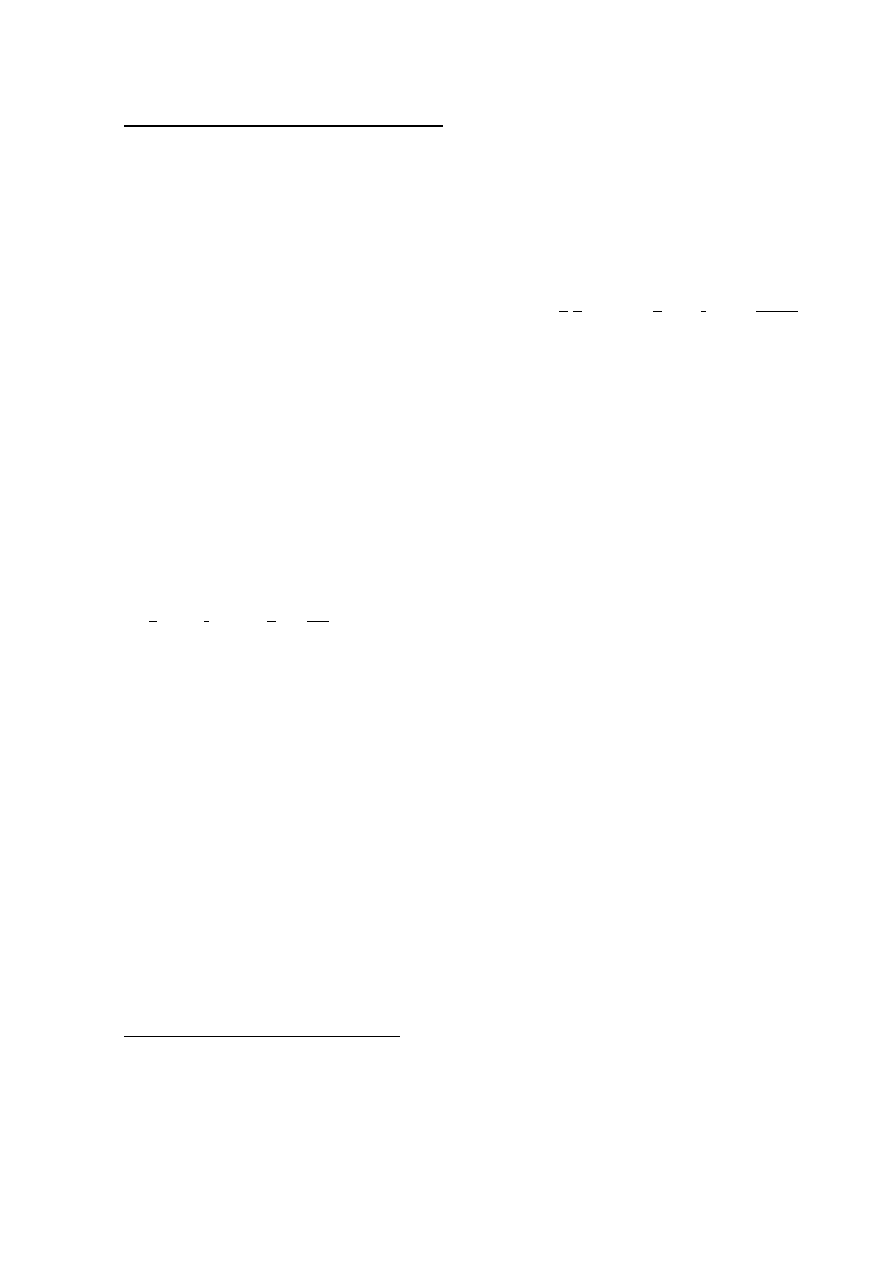
15
2. Określanie liczebności mikroorganizmów
Liczebność mikroorganizmów można badać, stosując metody bezpośrednie i pośrednie. W
metodach bezpośrednich najczęściej liczymy mikroorganizmy pod mikroskopem, np. w
komorze Thomy (p. str. 44), Halbera lub innej. Mikroorganizmy są liczone pod niedużym
powiększeniem (obiektyw 40x), a więc można w ten sposób określić tylko liczbę
drobnoustrojów odpowiednio dużych (powyżej 4 µm średnicy). Ponadto ich liczba powinna
przekraczać 10
7
komórek/ml.
Jeśli mikroorganizmów jest bardzo mało, należy przefiltrować odpowiednią objętość
badanej wody przez specjalny czarny filtr membranowy, a następnie wybarwić osadzone na
nim komórki, stosując 4,6-diamidyno-2-fenyloindol (ang. 4,6-diamidino-2-phenylindole,
DAPI),
który wiąże się z cząsteczkami dwuniciowego DNA. Filtr ogląda się w mikroskopie
epifluorescencyjnym (z bocznym światłem UV). Komórki oświetlone światłem o długości np.
365 nm (ultrafiolet) emitują światło o długości fali 420 nm (jasnoniebieskie). Drobnoustroje
liczy się w polach siatki narysowanej na okularze. Znając wielkość pól, powierzchnię filtra i
objętość przesączonej próby określa się liczbę drobnoustrojów. Metoda ta jest szybka i bardzo
dokładna.
Metody pośrednie polegają na odpowiednim, ilościowym wysiewie badanego materiału
na podłoże stałe i uzyskaniu tylu kolonii, aby (i) można je było policzyć, (ii) błąd statystyczny
był jak najmniejszy (zwykle liczy się kolonie na tych płytkach, gdzie jest ich ~ 30-300). Zakłada
się, że każda kolonia wyrasta z podziałów pojedynczej komórki mikroorganizmu. Znając liczbę
wyrosłych kolonii, objętość wysianej próbki i ewentualne rozcieńczenie materiału, można
oszacować liczbę żywych mikroorganizmów, zdolnych do wytworzenia kolonii (ang. viable
count) przypadających na 1 mililitr czy 1 g badanego środowiska. W metodzie tej oznaczamy w
rzeczywistości nie liczbę żywych komórek, lecz liczbę jednostek tworzących kolonie (jtk/ml)
(ang. colony forming unit, cfu). Dokładność metody płytkowej jest zadawalająca w przypadku
określania gęstości bakterii w hodowli określonego gatunku o znanych wymaganiach
pokarmowych. Metodę tę stosuje się np. w mikrobiologii żywności, w mikrobiologii medycznej,
w analizie sanitarnej wody, gdzie ważne jest dla nas poznanie liczby żywych bakterii.
W metodach pośrednich stosuje się różne techniki wysiewu, pamiętając o tym, by wysiew robić
co najmniej w dwóch powtórzeniach.
1) Posiew powierzchniowy kolejnych rozcieńczeń na płytki
Materiał zawierający mikroorganizmy odpowiednio się rozcieńcza i wysiewa po 0,1 ml
kolejnych rozcieńczeń na odpowiednie podłoża.
2) Posiew wgłębny (metoda płytek lanych)
Jej zaletą jest to, że można wysiać więcej niż 0,1 ml zawiesiny (zwykle 1 ml), ale
trzeba pamiętać, że mikroorganizmy będą musiały przez krótki okres czasu znieść
temperaturę 45-50
o
C (p. punkt I.2., str. 9).
3) Metoda filtrów membranowych
Jeśli szacuje się, że w badanym środowisku (np. wodzie) liczba drobnoustrojów będzie
mała, określoną objętość wody przesącza się przez filtr membranowy. Filtr z
osadzonymi bakteriami nakłada się jałową pęsetą na płytkę Petriego z odpowiednim
podłożem. Mikroorganizmy rosną i dzielą się, wytwarzając kolonie na filtrze. Po
inkubacji liczy się wyrosłe kolonie.
3. Przechowywanie mikroorganizmów
Mikroorganizmy można, przez krótki okres czasu (tzn. od kilku dni do kilku tygodni, w
zależności od drobnoustroju), przechowywać w lodówce na płytkach Petriego, dbając o to, by
podłoże nie wyschło. Lepiej w tych warunkach przechowywać hodowle założone na skosach, a
jeszcze lepiej na słupkach w podłożu półpłynnym. Dłuższe przechowywanie wymaga
zamrożenia hodowli po dodaniu do niej jałowego glicerolu (stężenie końcowe 15 %).

16
Zastosowanie temperatury -20
o
C pozwala na przechowywanie kilkumiesięczne, natomiast w
temperaturze -70
o
C mikroorganizmy mogą przetrwać wiele lat. Hodowle mikroorganizmów
można też przechowywać bardzo długo w stanie zliofilizowanym.
II. CZĘŚĆ PRAKTYCZNA
Szczepy bakteryjne:
- Escherichia coli (dziki szczep)
Podłoża i roztwory:
- bulion odżywczy
- podłoże Davisa (płynne)
- agar odżywczy
- roztwór fizjologiczny (0,85 % NaCl)
1. Badanie wzrostu bakterii w hodowli okresowej
a) Przygotować nocne hodowle szczepu (i) w bulionie odżywczym oraz (ii) w minimalnym
podłożu Davisa.
b) Każdą z hodowli rozcieńczyć odpowiednim, świeżym podłożem w stosunku 1 : 20.
c) Zmierzyć gęstość optyczną (ang. optical density, OD
600
) każdej z rozcieńczonych hodowli –
nie powinna być wyższa niż 0,1.
d) Każdą z rozcieńczonych hodowli podzielić na 3 części:
- jedną inkubować w warunkach statycznych w 37
o
C;
- drugą – z napowietrzaniem również w 37
o
C;
- trzecią pozostawić w temp. pokojowej bez napowietrzania.
e) Inkubację prowadzić przez 3 godziny. Co godzinę pobierać próbki hodowli i oznaczać ich
OD
600
.
f) Na podstawie uzyskanych wyników przygotować wykres zależności OD
600
każdej z
prowadzonych hodowli od czasu. Porównać uzyskane wyniki.
2. Określanie liczby bakterii w hodowli metodą pośrednią
a) Upłynnić agar odżywczy, rozlać na szalki Petriego; po zakrzepnięciu agaru płytki wysuszyć.
b) Otrzymaną hodowlę bulionową E. coli rozcieńczyć w roztworze soli fizjologicznej do
wartości 10
-6
(milion razy).
c) Z rozcieńczeń 10
-5
i 10
-6
wysiać po 0,1 ml na szalki z agarem odżywczym – każde
rozcieńczenie na dwie szalki.
d) Inkubować w temp. 37
o
C przez 24 godz. Policzyć kolonie otrzymane na szalkach.
e) Obliczyć liczbę bakterii zdolnych do utworzenia kolonii w 1 ml wyjściowej hodowli (x) wg
wzoru:
x = a x b x 10 [jtk/ml]
gdzie: a – średnia liczba kolonii na płytce
b – odwrotność wysianego rozcieńczenia
10 – przelicznik na 1 ml

17
III. ZAGADNIENIA DO OPRACOWANIA
1. Hodowle bakteryjne – kryteria podziału.
2. Warunki prowadzenia hodowli.
3. Fazy wzrostu w hodowli okresowej.
4. Typy wzrostu bakterii na różnych podłożach.
5. Określanie liczebności mikroorganizmów – metody bezpośrednie i pośrednie.
6. Sposoby przechowywania mikroorganizmów.
18
Ćwiczenie 4
Temat: Barwienie bakterii
Formy morfologiczne bakterii
I. WPROWADZENIE
1. Barwienia
Komórki bakteryjne są zwykle bardzo małe i charakteryzują się małą gęstością – słabo załamują
i pochłaniają światło, dlatego też trudno jest odróżnić je od podłoża. Przed oglądaniem w
mikroskopie, najczęściej się je wybarwia, stosując różne metody, w zależności od rodzaju
bakterii oraz celu, jaki chcemy osiągnąć. W tym celu należy przygotować rozmaz bakterii na
szkiełku podstawowym, wysuszyć go, a następnie w odpowiedni sposób utrwalić przed
barwieniem. W barwieniu prostym stosujemy tylko jeden barwnik, natomiast w złożonym – co
najmniej dwa barwniki, a często również różne bejce (zaprawy) i odbarwiacze. Przykładem
barwienia złożonego jest barwienie Grama, które jest podstawą klasyfikacji i identyfikacji
bakterii. W metodzie tej stosujemy dwa barwniki (fiolet krystaliczny i safraninę), płyn Lugola
jako bejcę i etanol jako odbarwiacz. Z powodu różnic w budowie ściany komórkowej bakterie
gramujemne barwią się na różowo, zaś gramdodatnie na fioletowo.
Oprócz barwienia pozytywnego, w którym oglądamy wybarwione bakterie na
bezbarwnym tle, istnieją też barwienia negatywne, w których „wybarwia się” tło (czyli szkiełko
podstawowe) za pomocą tuszu bądź nigrozyny. W ten sposób uwidacznia się otoczki bakteryjne,
które trudno barwią się metodami pozytywnymi.
2. Kształt komórek bakteryjnych
Podstawowe kształty bakterii to kula (ziarniaki), walec (pałeczki i laseczki) i skręcony walec
(przecinkowce, krętki i śrubowce). Istnieją też bakterie o kształtach nieregularnych, np.
maczugowce. Bakterie mogą tworzyć charakterystyczne układy komórek, takie jak: dwoinki,
pakiety, grona (takie układy tworzą ziarniaki), a także łańcuszki (ziarniaki i laseczki).
II. CZĘŚĆ PRAKTYCZNA
1. Barwienie proste preparatów bakteryjnych
Szczepy bakteryjne:
- pałeczki: Escherichia coli, Proteus vulgaris, Pseudomonas fluorescens
- laseczki: Bacillus subtilis, B. megaterium
- ziarniaki: Micrococcus luteus, Staphylococcus epidermidis, Enterococcus faecalis
Barwniki:
- fiolet krystaliczny (barwić 1 minutę)
- błękit metylenowy (barwić 5 minut)
a) Przygotowanie preparatu
- Zrobić rozmaz na szkiełku podstawowym.
- Wysuszyć go w temperaturze pokojowej.
- Utrwalić, przeprowadzając ostrożnie szkiełko trzykrotnie przez płomień palnika. Przed
barwieniem poczekać, aż szkiełko ostygnie.

19
b) Barwienie preparatu
- Zalać cały preparat wybranym barwnikiem.
- Po określonym czasie zlać barwnik i przemywać szkiełko wodą wodociągową aż do
momentu, gdy woda spływająca z preparatu będzie bezbarwna.
- Delikatnie usunąć wodę ze szkiełka za pomocą bibuły.
- Po całkowitym wyschnięciu preparatu oglądać go pod mikroskopem, najpierw pod małym
powiększeniem, a następnie stosując obiektyw immersyjny.
- Narysować wszystkie oglądane formy morfologiczne bakterii i tworzone przez te bakterie
układy komórek.
2. Barwienie złożone metodą Grama
Szczepy bakteryjne:
- Escherichia coli i Bacillus megaterium
- Proteus vulgaris i Bacillus megaterium
- Proteus vulgaris i Micrococcus luteus
Roztwory stosowane do barwienia:
- fiolet krystaliczny
- safranina
- płyn Lugola
- 95 % etanol
a) Przygotowanie preparatu
- Na szkiełku podstawowym zmieszać hodowle bakterii gramujemnej (np. E. coli) i
gramdodatniej (np. Bacillus megaterium).
- Zrobić rozmaz, wysuszyć i utrwalić jak w punkcie 1a).
b) Barwienie metodą Grama
- Preparat barwić fioletem krystalicznym przez 2 minuty.
- Zlać fiolet, wypłukać szkiełko płynem Lugola i zalać je płynem Lugola na 2 minuty.
- Spłukać preparat wodą, osuszyć bibułą i zalać na 30 sekund 95 % etanolem.
- Spłukać wodą, osuszyć bibułą i dobarwiać safraniną przez 5 minut.
- Spłukać wodą, osuszyć i oglądać.
- Narysować obraz widziany w mikroskopie, uwzględniając kolor, na jaki wybarwiły się
komórki poszczególnych bakterii.
III. ZAGADNIENIA DO OPRACOWANIA
1. Podstawowe kształty komórek bakteryjnych i tworzone przez nie układy.
2. Technika sporządzania preparatów mikroskopowych – wykonanie rozmazu, cel i sposoby
utrwalania preparatów.
3. Podział metod barwienia: barwienia proste i złożone; pozytywne i negatywne.
4. Zasada barwienia metodą Grama.
5. Bakterie gramujemne i gramdodatnie.
6. Technika mikroskopowania (w tym zastosowanie immersji).

20
Ćwiczenie 5
Temat: Barwienie bakterii cd.
Budowa komórki bakteryjnej
I. WPROWADZENIE
Strukturami występującymi w każdej komórce bakteryjnej są: nukleoid, błona cytoplazmatyczna
i rybosomy. Poza tym, większość bakterii ma ścianę komórkową, a niektóre - otoczki i rzęski.
Ściana komórkowa zawiera warstwę mureiny (peptydoglikanu), która u typowych bakterii
gramdodatnich jest gruba i silnie usieciowana, zaś u gramujemnych – cienka, słabo usieciowana
i pokryta błoną zewnętrzną. Stosując bejce (zaprawy), można pogrubić ścianę komórkową i
rzęski, a następnie je wybarwić, dzięki czemu stają się widoczne w zwykłym mikroskopie
optycznym. Dzięki rzęskom bakterie poruszają się, co można zobaczyć pod mikroskopem w
tzw. kropli wiszącej, stosując szkiełko z łezką. Ruch bakterii można też wykazać, stosując
posiew na słupek z podłożem półpłynnym, a w przypadku bakterii zdolnych do wzrostu
„rozpełzliwego” (ang. swarming) – punktowy posiew na środek niewysuszonej płytki.
Otoczki pełnią rolę: (i) ochronną (przed wysychaniem, fagocytozą, niektórymi
bakteriofagami i pewnymi związkami chemicznymi), a także (ii) w adhezji do podłoża stałego,
co jest ważne przy kolonizacji środowiska przez mikroorganizmy. Obecność otoczek można
wykazać, stosując złożone barwienie negatywno-pozytywne.
Pewne gatunki bakterii (np. laseczki) wytwarzają formy przetrwalnikowe, zwane
endosporami, które umożliwiają im przetrwanie w niekorzystnych warunkach środowiska.
Endospory są niewrażliwe na wysoką temperaturę (niektóre mogą przeżyć ogrzewanie w 100
o
C)
i działanie toksycznych związków chemicznych. Można je uwidocznić, dzięki barwieniu
złożonemu, w którym stosuje się zieleń malachitową (na gorąco), wodę jako odbarwiacz i
safraninę.
II. CZĘŚĆ PRAKTYCZNA
Szczepy bakteryjne:
Barwniki, utrwalacze, zaprawy i inne:
Bacillus megaterium
wodne roztwory safraniny (0,01 %; 0,2 %; 0,5 %)
Bacillus subtilis
wodny roztwór zieleni malachitowej (5 %)
Micrococcus luteus
roztwór taniny (10 %)
Proteus vulgaris
tusz chiński
alkohol metylowy
1. Barwienie ściany komórkowej
a) Przygotować rozmaz z nocnej hodowli Bacillus megaterium.
b) Po wysuszeniu preparat utrwalać przez 1 min alkoholem metylowym.
c) Zlać alkohol, przepłukać preparat roztworem taniny (bejca) i zalać roztworem taniny
na 30 min.
d) Spłukać preparat wodą.
e) Barwić 30-60 sek. 0,01 % roztworem safraniny.
f) Obejrzeć pod mikroskopem najpierw pod małym powiększeniem, potem pod immersją.
Narysować obraz mikroskopowy i opisać.

21
2. Barwienie przetrwalników metodą Schaeffer-Fultona
a) Wykonać rozmazy z 24- i 48-godzinnej hodowli B. megaterium lub B. subtilis.
b) Preparat wysuszyć i utrwalić w płomieniu palnika.
c) Barwić zielenią malachitową przez około 8 min, podgrzewając preparat, co pewien czas,
płomieniem palnika, aż do ukazania się pary.
d) Spłukać wodą.
e) Barwić 0,5 % safraniną przez 4 min.
f) Spłukać wodą i osuszyć.
g) Preparat obejrzeć pod immersją. Przetrwalniki powinny być wybarwione na zielono, a
wnętrze komórki na różowo.
3. Barwienie otoczek metodą Burriego-Ginsa
a) Na odtłuszczone szkiełko podstawowe nanieść kroplę płynnej hodowli Micrococcus luteus
(lub B. megaterium) lub zrobić zawiesinę bakterii z hodowli stałej i nanieść kroplę na
szkiełko.
b) Dodać kroplę tuszu i zmieszać z hodowlą.
c) Drugim szkiełkiem podstawowym rozprowadzić zawiesinę po powierzchni szkiełka tak,
aby powstała jednorodna, ciemna (lecz nie czarna) warstwa.
d) Rozmaz wysuszyć na powietrzu, ostrożnie utrwalić w płomieniu palnika.
e) Barwić 0,2 % safraniną przez około 15 sek.
f) Spłukać barwnik, preparat osuszyć.
g) Obejrzeć pod mikroskopem. Tło powinno być ciemne, bakterie zabarwione na różowo, zaś
otoczki są niewybarwione.
4. Wykazanie zdolności bakterii do ruchu
a) Z płynnej hodowli Proteus vulgaris przygotować preparat w postaci tzw. kropli wiszącej:
- na szkiełko nakrywkowe nanieść maleńką kroplę płynnej hodowli szczepu;
- za pomocą wazeliny nałożonej na rogach szkiełka przykleić je nad łezką szkiełka
podstawowego.
b) Nastawić pod mikroskopem obraz brzegu kropli na małym powiększeniu, a następnie
zmienić obiektyw na immersyjny. Obserwować ruch własny bakterii.
III. ZAGADNIENIA DO OPRACOWANIA
1. Budowa i funkcja bakteryjnych struktur komórkowych: ściana komórkowa (w tym błona
zewnętrzna bakterii gramujemnych), otoczki, rzęski.
2. Zastosowanie złożonych metod barwienia do uwidocznienia niektórych struktur
komórkowych (otoczka, ściana komórkowa, endospory).
3. Metody określania zdolności bakterii do ruchu.
22
Ćwiczenie 6
Temat: Metabolizm bakterii - źródła węgla, azotu i energii
I. WPROWADZENIE
Każdy mikroorganizm musi mieć zapewnione do wzrostu: (i) źródła pierwiastków biogennych
(węgla, azotu, siarki i fosforu), (ii) odpowiednie kationy (Na
+
, K
+
, Mg
2+
, itd), (iii)
mikroelementy oraz (iv) źródło energii.
Typ pokarmowy bakterii wskazuje zwykle na: (i) główny proces, za pomocą którego
bakteria zdobywa energię (fototrofy wykorzystują energię świetlną, a chemotrofy –
chemiczną), (ii) donory elektronów niezbędne do redukcji NAD lub NADP (litotrofy
wykorzystują związki nieorganiczne, a organotrofy – organiczne) i wreszcie (iii) na główne
źródło węgla (autotrofy wykorzystują dwutlenek węgla, a heterotrofy – związki organiczne).
Chemolitoautotrofy to mikroorganizmy, które wykorzystują pierwiastki chemiczne bądź
związki nieorganiczne jako źródło energii, a także donor elektronów. Głównym źródłem węgla
jest dla nich dwutlenek węgla, więc potrafią one rosnąć na podłożach mineralnych. Należą do
nich np. bakterie nitryfikacyjne, które uzyskują energię z utleniania amoniaku (nitrozobakterie) i
azotynów (nitrobakterie), a także bezbarwne bakterie siarkowe, które wykorzystują do tego celu
siarkę i jej związki nieorganiczne. Ten typ pokarmowy występuje wyłącznie u
mikroorganizmów prokariotycznych (bakterie i archeony)
Chemoorganoheterotrofy to mikroorganizmy, dla których związki organiczne są
źródłem węgla, energii i elektronów. Każdy związek organiczny pochodzenia naturalnego może
być wykorzystany do tego celu przez pewną grupę prokariotów. Pierwsze etapy rozkładu
związków wielkocząsteczkowych zachodzą na zewnątrz komórek dzięki wydzielanym przez
drobnoustroje ektoenzymom (amylazy, lipazy, proteazy, nukleazy).
Prokarioty potrafią wykorzystać jako źródło azotu nie tylko związki nieorganiczne (jony
NH
4
+
, NO
3
-
) i organiczne (np. aminokwasy), ale również azot cząsteczkowy. Zdolność do
wiązania azotu cząsteczkowego jest cechą występującą u wielu różnych bakterii (zarówno
wolnożyjących jak i symbiotycznych, autotroficznych jak i heterotroficznych, tlenowych i
beztlenowych), a także u niektórych archeonów. Nitrogenaza jest kompleksem enzymatycznym,
który umożliwia zredukowanie cząsteczki N
2
do NH
3
, gdy brakuje w środowisku innych,
dostępnych form azotu.
Niektóre bakterie potrafią syntetyzować wszystkie związki budujące ich komórki z: (i)
jednego, prostego związku węgla (nieorganicznego lub organicznego, w zależności od tego, czy
są to autotrofy czy heterotrofy) i (ii) soli mineralnych. Nazywamy je prototrofami. Inne, zwane
auksotrofami, nie potrafią syntetyzować pewnych związków organicznych, które muszą tym
samym znajdować się w ich podłożu. Związki te, zwane czynnikami wzrostowymi, to
aminokwasy, witamy, zasady purynowe i pirymidynowe i inne.
II. CZĘŚĆ PRAKTYCZNA
1. Przygotowanie podłoży
a) Podłoże AB
Zmieszać ze sobą:
50 ml soli A (Na
2
HPO
4
, KH
2
PO
4
, pH 8,4)
50 ml soli B (NH
4
Cl, MgSO
4
, Na
2
S
2
O
3
, pH 7,0)
2 ml roztworu czerwieni fenolowej
23
2 ml soli Tuovinena (wersenian sodu, ZnSO
4
, CaCl
2
, MnCl
2
,
FeSO
4
, (NH
4
)
6
Mo
7
O
24
, CuSO
4
, CoCl
2
, pH 6,0)
100 ml 4 % agaroidu
b) Agar odżywczy
Skład: wyciąg mięsny, ekstrakt drożdżowy, pepton, NaCl, woda, agar-agar.
c) Podłoże dla nitryfikatorów
Do 2 kolb à 300 ml wlać po 50 ml wody wodociągowej i dodać do każdej z nich po 2 ml
następujących roztworów: 6 % (NH
4
)
2
SO
4
; 0,6 % MgSO
4
; 0,1 % FeSO
4
; 0,3 % K
2
HPO
4
;
0,6 % NaCl; 20 % CaCO
3
.
d) Agar odżywczy ze skrobią (1 %)
e) Agar odżywczy z mlekiem
Do upłynnionego agaru odżywczego dodać 8 ml jałowego, odtłuszczonego mleka.
f) Agar odżywczy z tłuszczem (3 %)
Do upłynnionego agaru odżywczego dodać 6 g wrzącej margaryny.
g) Podłoże EMB z laktozą
Zmieszać: 190 ml upłynnionego podłoża (o składzie: ekstrakt drożdżowy, hydrolizat kazeiny,
K
2
HPO
4
, NaCl, woda, agar-agar)
2 ml wodnego roztworu 4 % eozyny
2 ml wodnego roztworu 0,65 % błękitu metylenowego
10 ml 20 % roztworu laktozy.
h) Żelatyna odżywcza: woda, żelatyna, pepton.
i) Podłoże Davisa dla prototrofa
Zmieszać ze sobą: 40 ml soli Davisa (K
2
HPO
4
, KH
2
PO
4
, MgSO
4
, (NH
4
)
2
SO
4
,
cytrynian sodu, woda)
4 ml 20 % glukozy
150 ml agaroidu
j) Podłoże Davisa dla auksotrofa
Do podłoża Davisa, sporządzonego jak wyżej, dodać 2 ml hydrolizatu kazeiny i 2 ml
roztworu tiaminy.
k) Podłoże dla bakterii wiążących azot
Skład: woda, mannitol, K
2
HPO
4
, KH
2
PO
4
, MgSO
4
, NaCl, CaCO
3
, ślady MnSO
4
,
FeCl
2
, Na
2
MoO
4
.
l) Podłoże Davisa z różnymi źródłami azotu
Zmieszać ze sobą:
40 ml bezazotowych soli Davisa
4 ml 20 % glukozy
2 ml roztworu zawierającego źródło azotu
150 ml agaroidu
Źródła azotu to: 10 % roztwór NH
4
Cl

24
10 % roztwór KNO
3
20 % roztwór hydrolizatu kazeiny
2. Źródła węgla i energii
a) Określenie typu pokarmowego badanych szczepów
Szczepy bakteryjne:
Podłoża:
Bacillus subtilis
podłoże mineralne AB
Halothiobacillus neapolitanus
agar odżywczy
Paracoccus versutus
Micrococcus luteus
- Płytkę z podłożem AB i z agarem odżywczym podzielić na 4 sektory i odpowiednio
podpisać.
- Każdy ze szczepów wysiać rysą na oddzielnych sektorach obu podłoży.
- Po inkubacji w temperaturze pokojowej określić typ pokarmowy badanych bakterii na
podstawie uzyskanych wyników..
Zmiana barwy podłoża AB z czerwonej na żółtą świadczy o jego zakwaszeniu.
b) Wykazanie obecności bakterii nitryfikacyjnych w glebie
Materiał badany
Podłoże
gleba
podłoże mineralne z punktu 1c)
- Podłoże przygotowane w punkcie 1c) rozlać do 2 kolbek à 100 ml.
- Do jednej z nich wprowadzić grudkę gleby. Drugą zostawić niezaszczepioną, jako kontrolę.
Po 1 i 2 tygodniach inkubacji w temperaturze pokojowej zbadać, czy w hodowli pojawiły się
azotyny. Do jednej probówki pobrać około 2 ml pożywki z kolby zaszczepionej glebą, a do
drugiej z kolby kontrolnej. Do obu probówek dodać po kilka kropli mieszaniny -
naftyloaminy z kwasem sulfanilowym lub wsypać odrobinę odczynnika firmy Merck do
wykrywania azotynów. Pojawienie się różowego zabarwienia świadczy o obecności azotynów
w próbie.
c) Badanie zdolności bakterii heterotroficznych do wykorzystywania różnych organicznych
źródeł węgla
Szczepy bakteryjne:
Podłoża:
Escherichia coli dzika (Lac
+
)
agar odżywczy ze skrobią
Escherichia coli mutant (Lac
-
)
agar odżywczy z mlekiem
Bacillus subtilis
agar odżywczy z tłuszczem
Pseudomonas fluorescens
EMB z laktozą
żelatyna odżywcza
- Płytki z pożywkami podzielić na pół, odpowiednio podpisać i zrobić posiewy:
- na agar odżywczy z mlekiem i ze skrobią posiać rysą B. subtilis i dziką E. coli;
- na agar odżywczy z tłuszczem posiać rysą E. coli i P. fluorescens;
- na podłoże EMB z laktozą posiać posiewem redukcyjnym dziką E. coli (Lac
+
) i
mutanta E. coli (Lac
-
) lub Proteus vulgaris (Lac
-
);
- Na jeden słupek z żelatyną odżywczą wsiać igłą B. subtilis, a na drugi E. coli.

25
- Płytki z podłożem EMB inkubować w 37
o
C przez noc (jeśli nie mogą być obejrzane po 24
godz., wstawić do lodówki).
- Pozostałe posiewy inkubować w temperaturze pokojowej przez kilka dni.
Odczyt posiewów po inkubacji:
- Płytki z agarem odżywczym i skrobią należy zalać płynem Lugola. Brak fioletowego
zabarwienia wokół strefy wzrostu bakterii świadczy o rozkładzie skrobi.
- Na agarze odżywczym z mlekiem obserwuje się przejrzyste strefy wokół linii wzrostu
szczepów hydrolizujących kazeinę.
- Płytki z agarem odżywczym i tłuszczem należy zalać 20 % roztworem CuSO
4
. Pojawienie się
szmaragdowego zabarwienia wokół linii wzrostu świadczy o rozkładzie tłuszczu.
- Na podłożu EMB kolonie bakterii zużywających cukier są fioletowe z metalicznym
połyskiem, a kolonie bakterii niezdolnych do zużycia danego cukru są różowe.
d) Prototrofy i auksotrofy
Szczepy bakteryjne:
Podłoża:
E. coli dzika
podłoże Davisa
E. coli AB1157 thr leu pro his arg thi
podłoże Davisa z hydrolizatem
E. coli 108 pro met thy
kazeiny i tiaminą
agar odżywczy
- Płytkę z agarem odżywczym, podłożem Davisa oraz podłożem Davisa uzupełnionym
hydrolizatem kazeiny i tiaminą podzielić na trzy sektory i odpowiednio podpisać.
- Na jednym sektorze wszystkich trzech pożywek posiać wężykiem dziką E. coli, a na dwóch
pozostałych oba mutanty auksotroficzne.
- Po inkubacji zinterpretować otrzymane wyniki.
3. Źródła azotu
a) Izolowanie z gleby bakterii wiążących azot cząsteczkowy
- Z kolbki zawierającej podłoże bezazotowe (punkt 1k, str.23) odlać do probówki z nakrętką
około 10 ml podłoża.
- Do probówki dodać „szczyptę” sacharozy.
- Probówkę i kolbę zaszczepić grudką gleby. Probówkę szczelnie zakręcić. Inkubować w
temperaturze pokojowej.
- Po tygodniu zaobserwować wzrost Azotobacter sp. w postaci błonki na powierzchni
pożywki.
- Pobrać próbkę z błonki na dwa szkiełka podstawowe.
- Jedno przykryć szkiełkiem nakrywkowym i obserwować przyżyciowo komórki
Azotobacter sp. oraz jego cysty, widoczne jako okrągłe twory, dość silnie załamujące
światło.
- Na drugim wykonać preparat tuszowy (ewentualnie dobarwiony błękitem metylenowym).
Obserwować komórki z otoczkami.
- Wyjałowiona ezą pobrać materiał z kożucha i zrobić posiew redukcyjny na stałe podłoże dla
bakterii wiążących N
2
. Po inkubacji obserwować pojawienie się śluzowatych kolonii
Azotobacter spp.

26
- Jeśli w probówce z pożywką bezazotową namnożył się Clostridium pasteurianum, z dna
probówki powinny odrywać się pęcherzyki gazu, a po odkręceniu nakrętki czuć jest
zapach kwasu masłowego.
- Z dennej części probówki pobrać pipetą pasterowską nieco hodowli, nanieść kroplę na
szkiełko podstawowe i zmieszać z równą objętością płynu Lugola. Przykryć szkiełkiem
nakrywkowym.
- Obserwować w mikroskopie laseczki z zabarwionym na fioletowo materiałem zapasowym
(granulozą) wypełniającym komórki.
b) Wykorzystywanie różnych innych źródeł azotu
- Trzy płytki z podłożem Davisa z trzema różnymi źródłami azotu – NH
4
Cl, KNO
3
i
hydrolizatem kazeiny – podzielić na dwie części.
- Na jednej połowie posiać rysą E. coli, a na drugiej – B. subtilis.
- Po inkubacji zaobserwować, jakie źródła azotu może wykorzystywać każda z badanych
bakterii.
III. ZAGADNIENIA DO OPRACOWANIA
1. Źródła pierwiastków biogennych i energii.
2. Typy pokarmowe bakterii.
3. Prototrofy i auksotrofy; czynniki wzrostowe.
4. Różnorodność źródeł węgla i azotu wykorzystywanych przez bakterie.
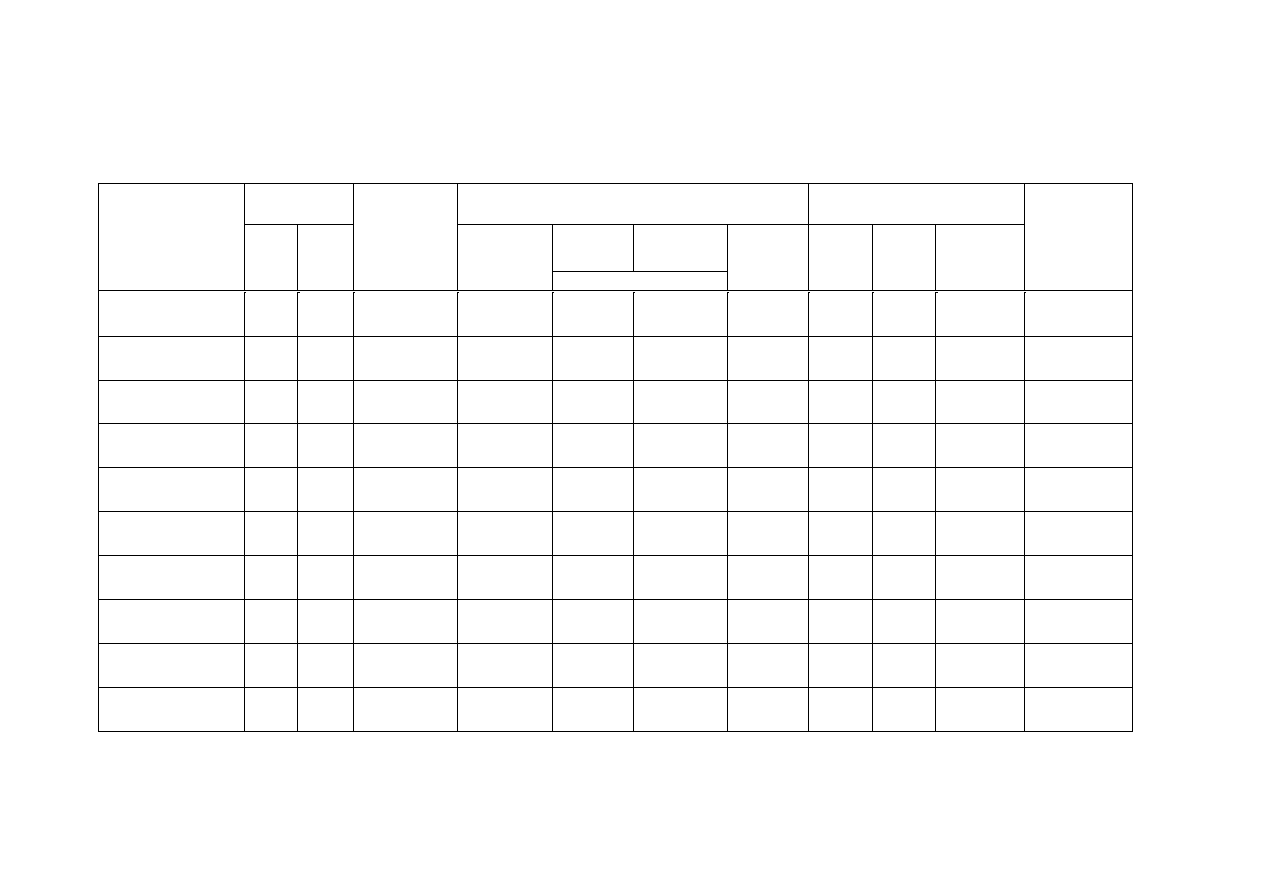
27
Tabela 3. Właściwości metaboliczne badanych bakterii (źródło węgla, azotu i energii)
BAKTERIA
Wzrost na
podłożu
Typ
pokarmowy
Rozkład związków wielkocząsteczkowych
Źródła azotu na podłożu
Davisa z
Wymagania
pokarmowe
AO
AB
skrobia
(amylazy)
kazeina
żelatyna
wzrost/
upłynnienie
tłuszcz
(lipazy)
NH
4
Cl
KNO
3
hydrolizat
kazeiny
proteazy
Escherichia
coli
Micrococcus
luteus
Serratia
marcescens
Pseudomonas
stutzeri
Pseudomonas
fluorescens
Staphylococcus
epidermidis
Bacillus
subtilis
Enterococcus
faecalis
Paracoccus
versutus
Halothiobacilus
neapolitanus

28
Ćwiczenie 7
Temat: Metabolizm bakterii - procesy oddechowe i fermentacje
I. WPROWADZENIE
Chemotrofy uzyskują energię z utleniania jakichś pierwiastków bądź związków chemicznych.
Proces utleniania zachodzący z udziałem łańcucha transportu elektronów i egzogennego
końcowego akceptora elektronów nazywamy oddychaniem. W tym procesie ATP powstaje
w wyniku fosforylacji oksydacyjnej. Ilość uwalnianej energii jest tym większa, im dłuższy
jest łańcuch transportu elektronów, dlatego też najbardziej wydajne energetycznie jest
oddychanie tlenowe, w którym końcowym akceptorem jest tlen. Organizmy zdolne do
wykorzystania tlenu w oddychaniu nazywamy tlenowcami. Wśród tlenowców wyróżniamy
(i) tlenowce bezwzględne – nie potrafią wykorzystać innych niż tlen akceptorów elektronów,
(ii) tlenowce warunkowe – potrafią wykorzystać inne niż tlen akceptory i (iii) mikroaerofile
– wykorzystują tlen, ale rosną wówczas, gdy jego stężenie jest niższe niż w powietrzu (od 1
do 15 %, w zależności od mikroorganizmu).
Istnieje duża grupa prokariotów, które potrafią oddychać beztlenowo, wykorzystując
inne niż tlen końcowe akceptory elektronów. Mogą to być pierwiastki chemiczne (np. siarka),
utlenione związki nieorganiczne (np. azotany, siarczany), a także pewne związki organiczne
(np. fumaran). Do oddychania beztlenowego zdolne są nie tylko beztlenowce, ale także
niektóre tlenowce warunkowe, np. bakterie denitryfikacyjne. Bakterie te, gdy w środowisku
brakuje tlenu, a występują w nim azotany, przerzucają się z oddychania tlenowego na
oddychanie azotanowe. Do beztlenowców zdolnych do uzyskiwania energii w procesie
oddychania należą np. bakterie redukujące siarczany, które wykorzystują siarczany jako
końcowy akceptor elektronów.
Wśród
beztlenowców
możemy
wyróżnić
grupę
(i)
beztlenowców
aerotolerancyjnych, które, choć nie potrafią wykorzystać tlenu jako akceptora elektronów,
mogą przeżyć pewien okres w warunkach tlenowych i (ii) beztlenowców bezwzględnych,
którym szkodzi wszelka ekspozycja na działanie tlenu, więc ich hodowle trzeba prowadzić w
specjalnych urządzeniach zwanych anaerostatami.
Prokarioty mogą też uzyskiwać energię z utleniania związków organicznych, bez
udziału łańcucha transportu elektronów, z wykorzystaniem endogennych akceptorów
elektronów. Taki sposób uzyskiwania energii nazywamy fermentacją. Do fermentacji
zdolne są niektóre warunkowe tlenowce (np. E. coli) i niektóre beztlenowce (np. Clostridium
spp.). W metabolizmie fermentacyjnym ATP powstaje w wyniku fosforylacji substratowej.
Nazwy fermentacji wywodzą się od najbardziej charakterystycznego produktu końcowego,
którym jest związek (lub związki) organiczny.
II. CZĘŚĆ PRAKTYCZNA
Szczepy bakteryjne:
Podłoża i roztwory:
Escherichia coli
bulion odżywczy
Bacillus subtilis
bulion odżywczy z KNO
3
(0,1 %)
Pseudomonas fluorescens
mleko z lakmusem
Pseudomonas stutzeri
20 % roztwór glukozy
Enterococcus faecalis
0,3 % roztwór błękitu metylenowego
Paracoccus versutus
woda utleniona
Staphylococcus epidermidis
jałowa parafina
Micrococcus luteus
Serratia marcescens

29
1. Określenie stosunku bakterii do tlenu
a) Przygotowane w probówkach podłoża zaszczepić jedną kroplą nocnej hodowli bulionowej
bakterii wg wskazówek prowadzącego zajecia:
- bulion odżywczy z dodatkiem 0,5 % glukozy – wysoki słup podłoża, ogrzanego
przed posiewem we wrzącej łaźni wodnej szybko schłodzonego w lodzie;
- bulion odżywczy – niski słup podłoża.
b) Po posiewie – wysoki słup podłoża należy zalać jałową parafiną, aby uniemożliwić dostęp
powietrza. Zaszczepione podłoża inkubować w 30 lub 37
o
C (w zależności od bakterii)
przez 24 godz.
c) Obserwować wzrost lub jego brak w poszczególnych probówkach. Wyniki zamieścić w
tabeli 4. Określić stosunek badanych bakterii do tlenu.
2. Wykazanie obecności katalazy w wybranych szczepach bakterii
Nocne hodowle szczepów bakterii na płytkach z agarem odżywczym zalać wodą utlenioną.
Wydzielanie się pęcherzyków gazu świadczy o obecności katalazy – enzymu rozkładającego
nadtlenek wodoru na wodę i tlen.
3. Oddychanie azotanowe (w tym denitryfikacja)
a) Probówki zawierające bulion odżywczy z KNO
3
(wysoki słup pożywki i rurka Durhama)
zaszczepić bakteriami wg wskazówek prowadzącego zajęcia; jedną probówkę pozostawić
nieszczepioną jako kontrolę.
b) Po inkubacji obserwować obecność gazu w rurkach Durhama, sprawdzić obecność jonów
NO
2
-
w hodowli (za pomocą odpowiedniego odczynnika firmy Merck).
c) Na podstawie uzyskanych wyników określić, który ze szczepów jest zdolny do
oddychania azotanowego. Wyniki zamieścić w tabeli 4.
4. Fermentacja i peptonizacja mleka
a) Do probówek zawierających odtłuszczone, jałowe mleko z lakmusem wsiać kolejno:
Enterococcus faecalis, E. coli, B. subtilis, czwartą probówkę pozostawić nie zaszczepioną.
b) Inkubować 24 godz w 37
o
C, zaobserwować zmiany w podłożu: zakwaszenie lub
alkalizację, powstanie skrzepu kazeiny, hydrolizę kazeiny, redukcję lakmusu.
c) Na podstawie zaobserwowanych zmian określić zdolność badanych szczepów do
fermentacji lub peptonizacji mleka. Wyniki zamieścić w tabeli 4.
Mleko odtłuszczone zawiera laktozę, kazeinę, sole mineralne i witaminy – jest więc
znakomitą pożywką, na której może rozwijać się ogromna liczba gatunków bakterii.
Jeśli bakterie fermentują laktozę, wówczas powstaje kwas mlekowy w takiej ilości, że
wytrąca się kazeina jako tzw. skrzep kwaśny. Następuje zmiana barwy lakmusu na różową.
Wskutek wytworzenia się warunków prawie beztlenowych w dolnej części słupa pożywki
następuje redukcja lakmusu, który pełni rolę końcowego akceptora elektronów.
Bakterie, które nie fermentują laktozy mogą wykazywać zdolność do peptonizacji
kazeiny po jej uprzednim wytrąceniu przez podpuszczkę (tzw. skrzep słodki). W tym
przypadku obserwuje się alkalizację mleka (zmiana barwy lakmusu na niebieską), a następnie
w miarę upływu czasu – upłynnienie skrzepu.
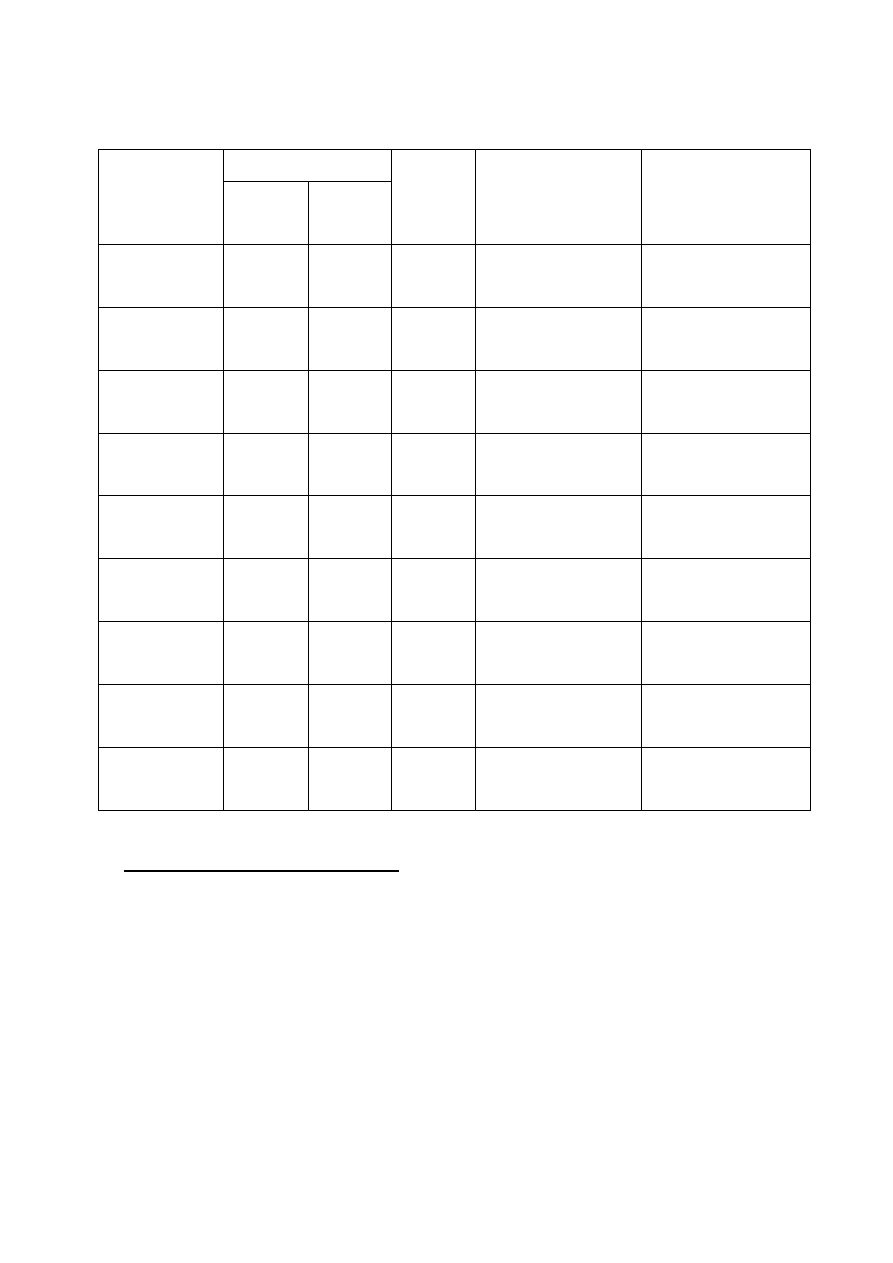
30
Tabela 4. Właściwości metaboliczne badanych bakterii (procesy oddechowe i fermentacje)
5. Test redukcji błękitu metylenowego
(wykorzystanie błęktu metylenowego jako końcowego akceptora elektronów)
a) Ponumerować pięć probówek.
b) Do każdej probówki dodać 0,1 ml wodnego roztworu błękitu metylenowego.
c) Przygotować rozcieńczenia młodej hodowli bulionowej E. coli wg schematu:
nr probówki
bulion (ml)
hodowla (ml)
1
9
1
2
7
3
3
5
5
4
-
10
5 (kontrola)
10
-
d) Zawartość probówek dokładnie wymieszać i wstawić je do termostatu o temp. 37
o
C.
BAKTERIA
Bulion
odżywczy
Bulion
odżywczy
+ KNO
3
Mleko
z lakmusem
Stosunek bakterii
do tlenu
niski
słup
wysoki słup
+ glukoza
+ ogrzanie
+ parafina
Escherichia
coli
Micrococcus
luteus
Serratia
marcescens
Pseudomonas
stutzeri
Pseudomonas
fluorescens
Staphylococcus
epidermidis
Bacillus
subtilis
Enterococcus
faecalis
Paracoccus
versutus

31
e) W odstępach 10-minutowych obserwować zabarwienie zawartości probówek. Zanotować
czas odbarwienia błękitu w poszczególnych probówkach.
e) Po zakończeniu obserwacji zawartość probówek ponownie wymieszać. Wyjaśnić
obserwowane zmiany zabarwienia.
III. ZAGADNIENIA DO OPRACOWANIA
1. Stosunek bakterii do tlenu: tlenowce bezwzględne i względne, mikroaerofile, beztlenowce
bezwzględne i aerotolerancyjne.
2. Metody hodowli tlenowców, mikroaerofilów i beztlenowców.
3. Porównanie procesów oddechowych i fermentacji:
a) końcowe akceptory elektronów
b) końcowe produkty
c) zysk energetyczny w różnych typach oddychania i w fermentacjach.
32
Tabela 5. Podsumowanie właściwości metabolicznych badanych szczepów
BAKTERIA
Źródło węgla
Sposób uzyskiwania
energii
Typ pokarmowy
Źródło azotu
Wymagania
pokarmowe
Końcowy akceptor
elektronów
Stosunek do tlenu
Bakterie
nitryfikacyjne
Halothiobacillus
neapolitanus
Paracoccus
versutus
Escherichia
coli
Serratia
marcescens
Pseudomonas
stutzeri
Azotobacter sp.
Clostridium
pasteurianum
Bacillus
subtilis
Enterococcus
faecalis
Micrococcus
luteus
Staphylococcus
epidermidis
33
Ćwiczenie 8
Temat: Koniugacja u bakterii
I. WPROWADZENIE
1. Plazmidy i koniugacja
Koniugacja jest jednym ze sposobów przekazywania materiału genetycznego u bakterii,
warunkujących horyzontalny transfer genów. Proces ten wymaga bezpośredniego kontaktu
dwu komórek, z których jedna pełni rolę dawcy, a druga biorcy DNA. Zdolność do
przekazywania materiału genetycznego (bycia dawcą) jest uwarunkowana obecnością w
komórce plazmidu koniugacyjnego. Plazmidy to dwuniciowe, autonomiczne cząsteczki DNA
(najczęściej koliście zamknięte), zdolne do replikacji w komórce odpowiedniego gospodarza.
Niektóre z nich zawierają geny nadające komórce określony fenotyp. Plazmidy
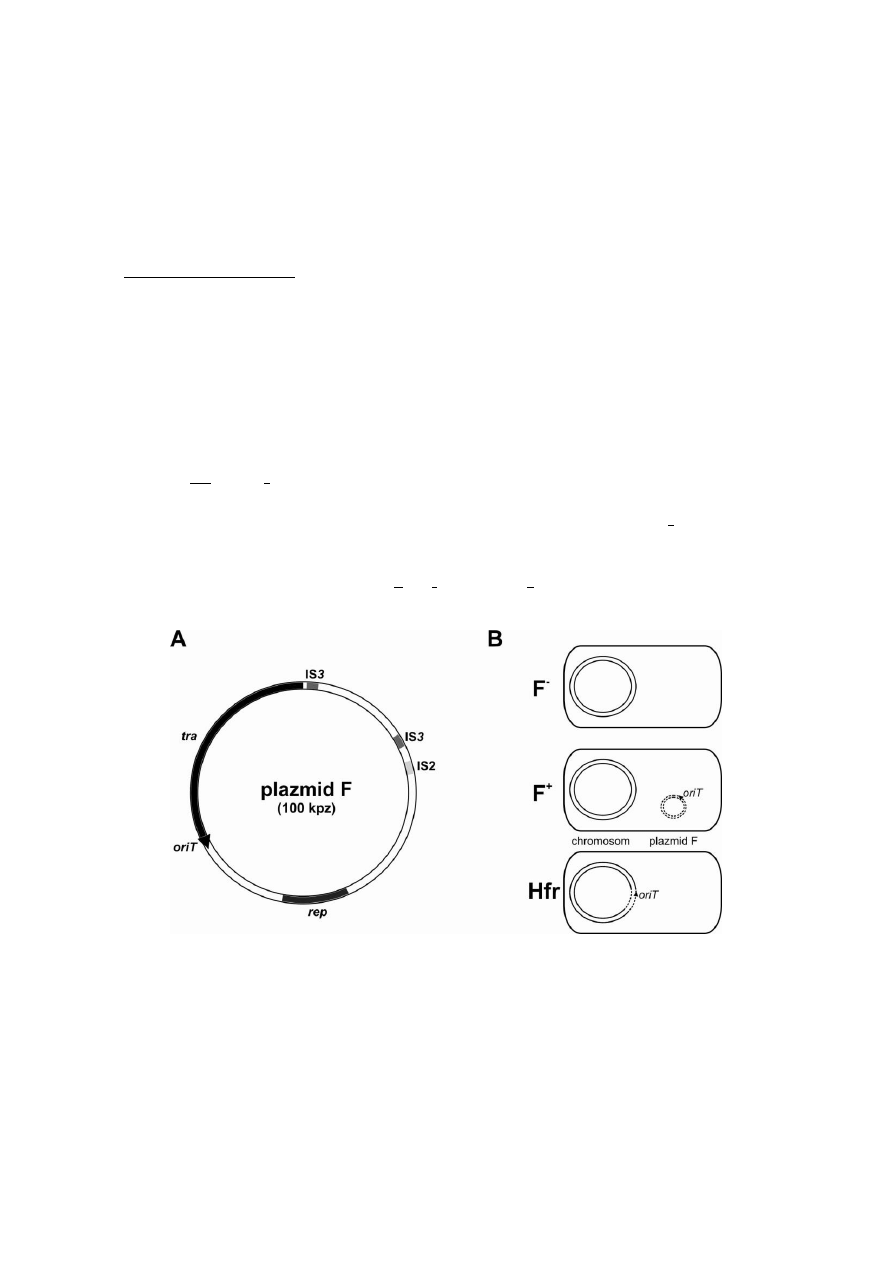
koniugacyjne (np. plazmid F; rys. 4A) mają zestaw genów tra, warunkujących między
innymi (i) syntezę pilusów, które umożliwiają wytworzenie pary koniugacyjnej i (ii) miejsce
oriT (ang. origin of transfer), od którego rozpoczyna się transfer plazmidu od dawcy do
biorcy.
Jednym z badanych na ćwiczeniach plazmidów jest plazmid F (ang. fertility) E. coli.
Może on występować albo w stanie autonomicznym (w szczepach nazwanych F
+
), w którym
replikuje się niezależnie od chromosomu gospodarza, albo w stanie zintegrowanym z
chromosomem (w szczepach Hfr; ang. high frequency of recombination), w którym replikuje
się jako jego część. Szczepy E. coli bez plazmidu F określa się jako F
-
(rys. 4B).
Rys. 4. A. Organizacja genetyczna plazmidu F (uproszczony schemat).
B. Typy komórek E. coli w zależności od obecności plazmidu F.
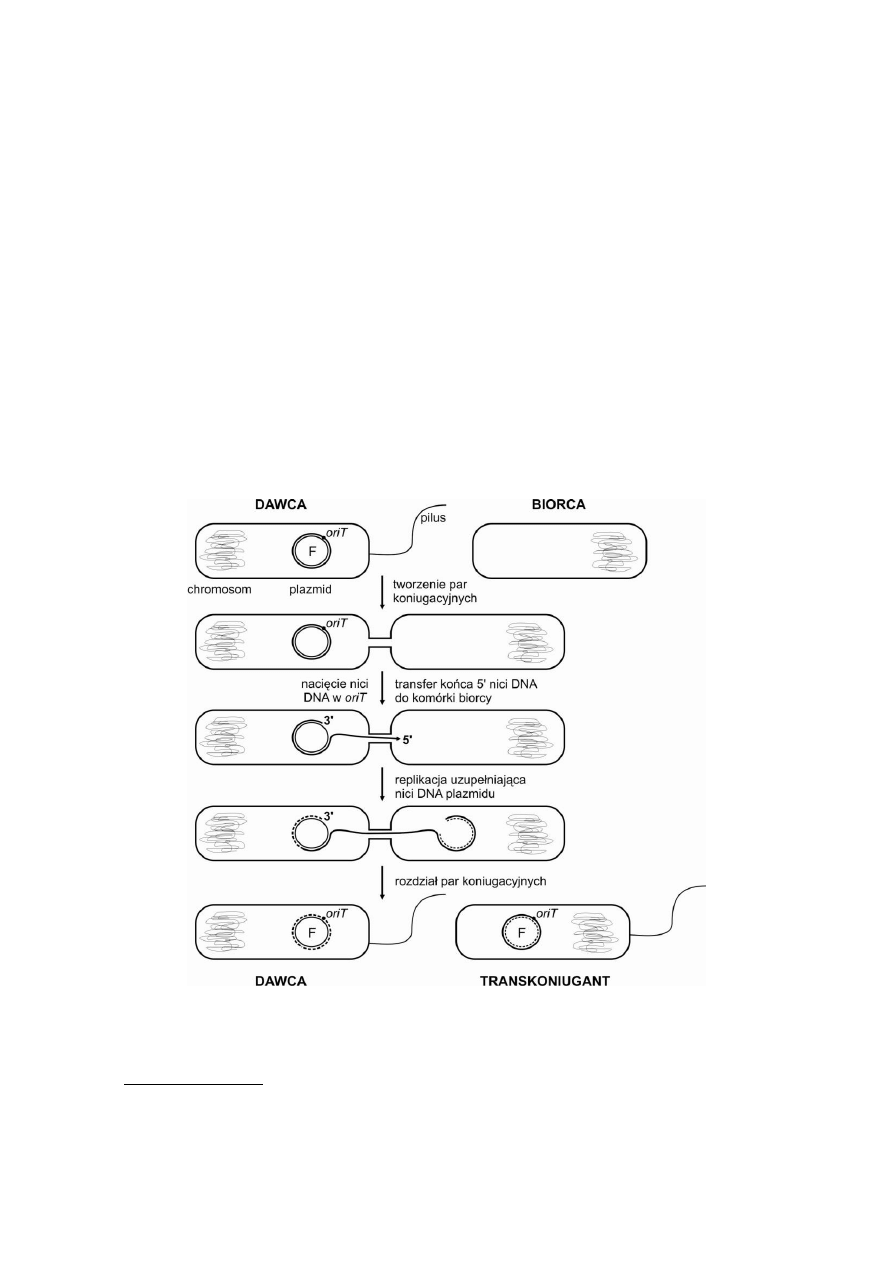
W wyniku koniugacji biorcy F
-
z dawcą F
+
powstają transkoniuganty, które uzyskują
plazmid F (rys. 5). Koniugacja biorcy F
-
z dawcą typu Hfr prowadzi do ukierunkowanego
przekazywania chromosomu dawcy, w wyniku czego mogą powstawać z dużą częstością
rekombinanty chromosomowe (nie zawierające plazmidu F). Częstość pojawiania się
różnego typu rekombinantów jest zależna od odległości danego genu od punktu
początkowego transferu – oriT i jest tym większa, im bliżej tego punktu leży dany gen.
W zależności od miejsca integracji plazmidu F z chromosomem bakteryjnym,
34
powstają różnego typu szczepy Hfr. Każdy z nich ma plazmid włączony w inne, ściśle
określone miejsce. Ponieważ transfer koniugacyjny DNA zawsze rozpoczyna się od miejsca
oriT, każdy typ Hfr przekazuje inne geny chromosomowe jako pierwsze. Zastosowanie
różnych typów Hfr pozwoliło, wiele lat temu, na określenie położenia różnych genów w
chromosomie E. coli.
Podobnie jak plazmid F mogą być przekazywane inne plazmidy koniugacyjne. Niosą
one często, poza genami warunkującymi replikację i transfer koniugacyjny, także geny
nadające komórce gospodarza określone cechy fenotypowe, np. oporność na antybiotyki,
metale ciężkie, czy też zdolność do metabolizowania określonych źródeł węgla (np. toluenu).
Cechy te w pewnych warunkach środowiska mogą stać się selektywnie korzystne dla bakterii,
która uzyskała plazmid. Na ćwiczeniach badany jest transfer koniugacyjny plazmidu pMAO–
oriT, warunkującego oporność na ampicylinę i kanamycynę.
Stosując odpowiednie podłoża selekcyjne, można uzyskać kolonie transkoniugantów,
a więc komórek biorcy, które uzyskały plazmid dzięki transferowi koniugacyjnemu. Podłoże
selekcyjne musi mieć taki skład, aby możliwy był wzrost kolonii transkoniugantów, natomiast
zahamowany wzrost komórek biorcy i dawcy. Stosując tę samą zasadę, można
wyselekcjonować kolonie rekombinantów chromosomowych po koniugacji szczepu F
-
i Hfr.
Rys. 5. Koniugacja szczepów F
-
(biorca) i F
+
(dawca) E. coli.
1. Fenotyp i genotyp
Geny i operony prokariotów oznacza się, stosując symbole trzyliterowe, pisane małą literą i
kursywą (np. operon lac), które pochodzą od nazw angielskich (np. lac od ang. lactose).
Wielka litera po tej nazwie trzyliterowej oznacza konkretny gen (np. lacZ – gen β-galakto-
35
zydazy), którego produkty uczestniczą w danym szlaku biosyntezy, degradacji, itd.
Fenotyp (np. rekombinantów) zapisuje się wielką literą, zamieszczając w górnej
frakcji „+” lub „-”, np. Lac
+
– oznacza bakterię zdolną, a Lac
-
– niezdolną, do wykorzystania
laktozy jako źródła węgla i energii. Oporność lub wrażliwość na antybiotyki zapisuje się
symbolami dwuliterowymi, np. Ap
r
, jeśli geny oporności znajdują się w plazmidzie lub
trzyliterowymi, np. Amp
s
, jeśli geny mają lokalizację chromosomową (Ap i Amp oznaczają
ampicylinę). Występujące w górnej frakcji symbole „s” i „r”, oznaczają, że szczep jest
odpowiednio – wrażliwy (ang. sensitive) lub oporny (ang. resistant) na dany antybiotyk.
Zapis: E. coli lacZ met Str
r
oznacza, że ten szczep E. coli (i) ma mutację w genie lacZ,
kodującym β-galaktozydazę (a więc jest niezdolny do wykorzystania laktozy jako źródła
węgla), (ii) ma mutację w jakimś genie związanym z biosyntezą metioniny (więc wymaga
metioniny do wzrostu) i (iii) jest oporny na streptomycynę, ale przyczyna tej oporności nie
jest znana. Pozostałe geny są takie jak w szczepie dzikim.
II. CZĘŚĆ PRAKTYCZNA
Szczepy bakteryjne:
Escherichia coli
a) F
-
108
pro met thy Str
r
- biorca
Hfr C
prototrof Str
s
- dawca
b) DH5αR
Rif
r
- biorca
TG1 (pMAO1-oriT)
Ap
r
Km
r
- dawca
Podłoża i roztwory:
- agar odżywczy
- podłoże Davisa płynne (150 ml wody dest., 40 ml soli Davisa, 4 ml 20 % glukozy)
- podłoże Davisa stałe (150 ml agaroidu, 40 ml soli Davisa, 4 ml 20 % glukozy)
- roztwór fizjologiczny
- roztwór hydrolizatu kazeiny (20 %)
- roztwory aminokwasów: metioniny i proliny (1 mg/ml)
- roztwór tyminy (3 mg/ml)
- roztwór streptomycyny (5 mg/ml)
- roztwór ampicyliny (5 mg/ml)
- roztwór kanamycyny (5 mg/ml)
- roztwór ryfampicyny (5 mg/ml).
Uwaga: Studenci otrzymują gotowe, jałowe roztwory metioniny, proliny, tyminy i
antybiotyków. Należy dodawać je w ilości 1 ml na 100 ml podłoża.
Każdy zespół wykonuje jedną z koniugacji:
1) F
-
108 x HfrC
2) DH5αR x TG1(pMAO-oriT)
Koniugacja 1
1. Szczepy rodzicielskie hodować przez 18 godz., w temp. 37
o
C, w podłożu Davisa
uzupełnionym wymaganymi przez szczep czynnikami oraz hydrolizatem kazeiny.

36
2. Nocną hodowlę każdego szczepu rozcieńczyć 1:20 świeżym podłożem i inkubować w
37
o
C do osiągnięcia wykładniczej fazy wzrostu (2,5-3 godz.).
3. Hodowle dawcy i biorcy zmieszać w stosunku 1:10 (4,5 ml hodowli F
-
i 0,5 ml hodowli
Hfr).
4. Mieszaninę koniugacyjną wstawić do termostatu o temp. 37
o
C na 60 min.
5. Rozcieńczyć hodowlę szczepu dawcy i wysiać (rozc. 10
-6
i 10
-7
) na płytki z agarem
odżywczym w celu określenia liczby komórek użytych do koniugacji (jtk/ml). Płytki
inkubować przez 24 godz. w 37
o
C.
6. Po zakończeniu koniugacji mieszaninę koniugacyjną rozcieńczyć i wysiać na odpowiednie
podłoża selekcyjne wg schematu:
a) selekcja rekombinantów Pro
+
Str
r
– na podłożu Davisa z dodatkiem metioniny, tyminy
i streptomycyny – wysiać rozcieńczenie 10
-2
i 10
-3
(po dwa powtórzenia);
b) selekcja rekombinantów Met
+
Str
r
– na podłożu Davisa z dodatkiem proliny, tyminy i
streptomycyny – wysiać rozcieńczenie 10
-1
i 10
-2
(po dwa powtórzenia).
Inkubować w 37
o
C przez 48 godzin.
7. Policzyć kolonie rekombinantów i szczepu dawcy, a następnie przeliczyć na jtk/ml;
obliczyć częstość rekombinacji (X) wg wzoru:
liczba rekomb. w 1 ml miesz. koniug. [jtk/ml] x 100 %
X = ----------------------------------------------------------------------
liczba dawcy w 1 ml miesz. koniug. [jtk/ml]
8. Otrzymane wyniki wstawić do tabeli 6 i zinterpretować.
Koniugacja 2
1. Szczepy rodzicielskie hodować przez 18 godz., w temp. 37
o
C, w bulionie odżywczym:
szczep DH5αR – bez dodatków, TG1(pMAO1-oriT) – z ampicyliną (stęż. 50 g/ml) lub z
kanamycyną (50 g/ml).
2. Nocne hodowle rozcieńczyć 10-krotnie świeżą pożywką bez antybiotyku i inkubować w
37
o
C do uzyskania wykładniczej fazy wzrostu (2-3 godziny).
3. Zmieszać w probówce hodowle biorcy i dawcy (w wykładniczej fazie wzrostu) w stosunku
1:1 i inkubować 60 min w 37
o
C.
4. Rozcieńczyć mieszaniny koniugacyjne i wysiać rozcieńczenia 10
-1
– 10
-3
na podłoża
selekcyjne:
a) agar odżywczy z ryfampicyną i ampicyliną (po 50 g/ml) – dla transkoniugantów
Rif
r
Ap
r
;
b) agar odżywczy z ryfampicyną (50
g/ml) i kanamycyną (50
g/ml) – dla
transkoniugantów Rif
r
Km
r
.
Płytki inkubować w 37
o
C przez 24 godziny.
5. Rozcieńczyć hodowlę szczepu dawcy i wysiać (rozc. 10
-5
i 10
-6
lub 10
-6
i 10
-7
, według
wskazówek prowadzącego zajęcia) na płytki z agarem odżywczym w celu określenia liczby
komórek użytych do koniugacji. Płytki inkubować przez 24 godz. w 37
o
C.
6. Policzyć kolonie transkoniugantów i szczepów dawców, a następnie przeliczyć na jtk/ml;
obliczyć częstość przekazywania plazmidu (X) wg wzoru:
liczba transkoniug. w miesz. koniug. [jtk/ml] x 100 %
X = -------------------------------------------------------------------
liczba dawcy w miesz. koniug. [jtk/ml]
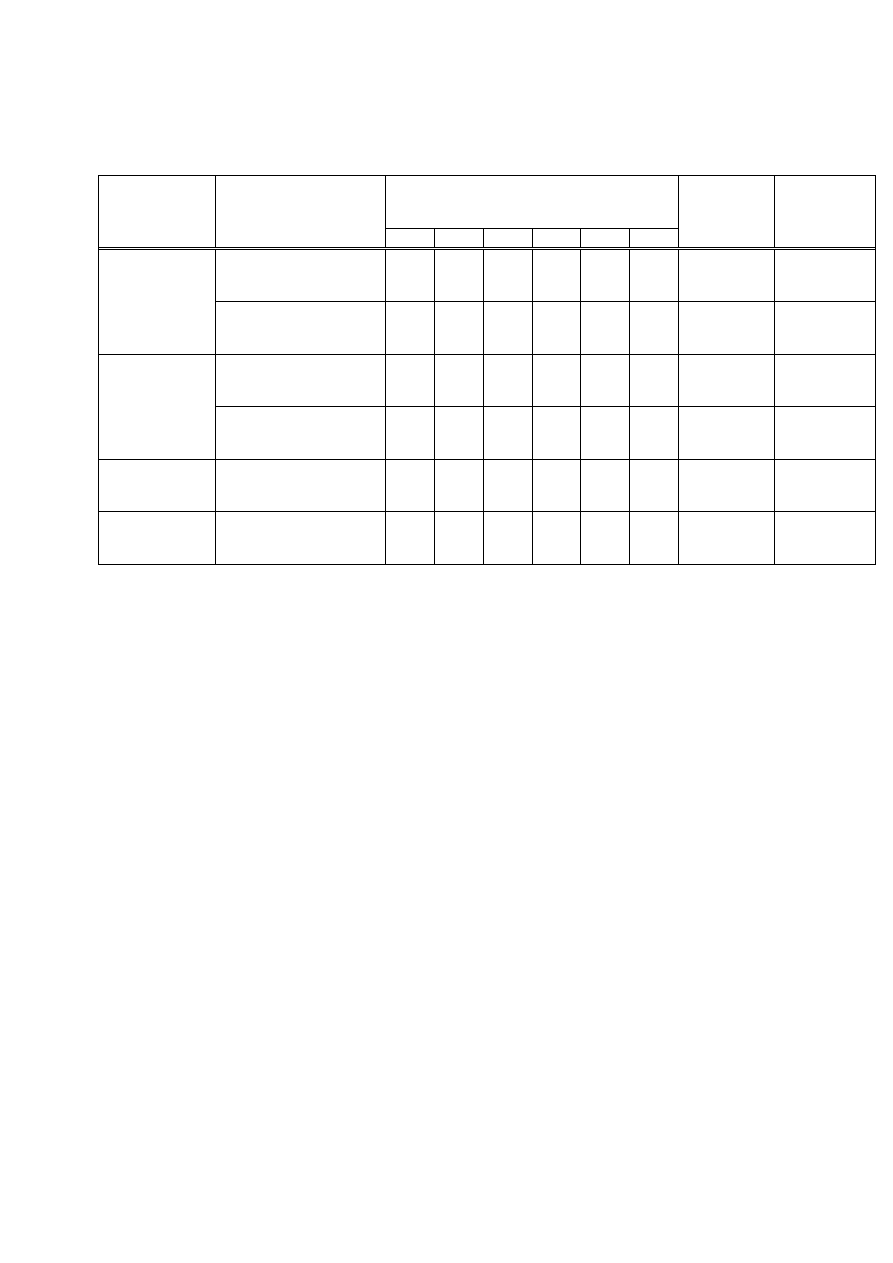
37
7. Otrzymane wyniki wstawić do tabeli 6 i zinterpretować.
Tabela 6. Koniugacja u bakterii
Rodzaj
wysiewu
Fenotyp
selekcjonowanego
rekomb. lub transkon.
Średnia liczba kolonii rekomb.,
transkoniug. lub dawców uzyskanych z
rozcieńczenia:
jtk/ml
[%]
rekomb./
transkon.
10
-1
10
-2
10
-3
10
-5
10
-6
10
-7
Koniugacja I
Fenotyp I
XXX XXX XXX
Fenotyp II
XXX XXX XXX
Koniugacja II
Fenotyp I
XXX XXX XXX
Fenotyp II
XXX XXX XXX
Dawca I
XXXXXXXXXX
XXX XXX XXX
XXXXXXX
Dawca II
XXXXXXXXXX
XXX XXX XXX
XXXXXXX
III. ZAGADNIENIA DO OPRACOWANIA
1. Plazmidy i koniugacja.
2. Różne typy dawców w koniugacji warunkowanej plazmidem F.
3. Przekazywanie cech chromosomowych i plazmidowych.
4. Selekcjonowanie różnych typów rekombinantów i transkoniugantów.

38
Ćwiczenie 9
Temat: Analiza mikrobiologiczna wody
Oznaczanie bakterii grupy coli
I. WPROWADZENIE
W wodach powierzchniowych, oprócz typowej mikroflory wodnej, mogą się też znajdować
mikroorganizmy, które zostały wypłukane z gleby lub przedostały się do niej wraz ze
ściekami. W skład mikroorganizmów ściekowych wchodzą mikroorganizmy stanowiące
normalną mikroflorę przewodu pokarmowego ludzi i zwierząt oraz mikroorganizmy
chorobotwórcze. Woda jest konsumowana w znacznych ilościach, więc jeśli nawet zawiera
niewielką liczbę mikroorganizmów patogennych (takich jak Salmonella spp., Shigella spp.,
Vibrio cholerae, enterowirusy, itd.) może stanowić źródło zakażenia. Zmusza nas to do
prowadzenia stałej kontroli sanitarnej wody pitnej, wód zbiorników powierzchniowych i
wody w basenach. O możliwości występowania w badanej wodzie mikroorganizmów
patogennych wnioskuje się pośrednio, badając obecność tzw. bakterii wskaźnikowych, które
stale żyją jako saprofity w przewodzie pokarmowym człowieka i zwierząt wyższych.
Najważniejszą bakterią wskaźnikową jest Escherichia coli, która wchodzi w skład
tzw. grupy coli. Bakterie grupy coli to gramujemne pałeczki, nieprzetrwalnikujące,
względnie tlenowe, fermentujące laktozę po 48 godz. z wytworzeniem kwasu oraz gazu i
tworzące na podłożu różnicującym z laktozą charakterystyczne kolonie (np. na podłożu Endo
i EMB – kolonie bordowe z metalicznym połyskiem). Bakterie grupy coli dzielą się na dwa
typy:
(i)
kałowy (fekalny), w skład którego wchodzą bakterie fermentujące laktozę z
wytworzeniem kwasu i gazu w 37
o
i 44
o
C (E. coli);
(ii)
ziemny, w skład którego wchodzą bakterie ziemne, zdolne do fermentacji
laktozy w temperaturze 37
o
C, ale nie w 44
o
C (Citrobacter sp. i
Enterobacter sp.).
Obecność w badanej wodzie bakterii grupy coli świadczy o jej skażeniu ściekami bytowymi
(jeśli wykrywa się bakterie grupy coli typu kałowego) lub glebą (jeśli wykrywa się bakterie
grupy coli typu ziemnego).
Analiza sanitarna wody obejmuje następujące badania mikrobiologiczne:
(i) badanie obecności bakterii grupy coli, z zastosowaniem (w zależności od spodziewanego
stopnia skażenia badanej wody):
- metody fermentacyjno-probówkowej
- metody filtrów membranowych.
Miano coli jest to najmniejsza objętość badanej wody (wyrażona w cm
3
), w której znajdują
się jeszcze bakterie grupy coli.
(ii) oznaczanie liczby:
- bakterii psychrofilnych (typowych autochtonicznych bakterii wodnych)
- bakterii mezofilnych (bakterii allochtonicznych, pochodzących z gleby lub ścieków).
W tym celu badaną wodę (lub jej rozcieńczenia) sieje się na agar odżywczy, metodą
posiewu powierzchniowego lub wgłębnego, w zależności od spodziewanej liczby bakterii.
Płytki inkubuje się w dwóch temperaturach: (i) 20
o
C – w celu określenia jtk/ml bakterii
psychrofilnych i (ii) 37
o
C – w celu określenia jtk/ml bakterii mezofilnych.

39
II. CZĘŚĆ PRAKTYCZNA
Materiał:
Podłoża:
woda wodociągowa
1) agar odżywczy
woda ze zbiornika naturalnego
2) podłoże Eijkmana o składzie:
ścieki komunalne
- pepton
- laktoza
- NaCl
- purpura bromokrezolowa
1. Badanie wody wodociągowej
a) Pobrać próbkę wody wodociągowej.
- W tym celu wylot kranu należy wymyć, wytrzeć do sucha i opalić palnikiem.
- Następnie spuszczać wodę z kranu przez 10 minut.
- Po tym czasie, nie zakręcając dopływu, pobrać około 200 ml wody do jałowej kolby à 300
ml. Kolbę zamknąć jałowym korkiem.
b) Oznaczyć liczbę bakterii psychrofilnych i mezofilnych metodą posiewu wgłębnego.
- W tym celu na 4 płytki Petriego wlać po 1 ml pobranej wody.
- Następnie na każdą z nich wlać 20 ml upłynnionego agaru odżywczego ostudzonego do
temperatury około 46
o
C.
- Całość rozprowadzić równomiernie po powierzchni płytki.
- Po zakrzepnięciu dwie płytki inkubować w 20
o
C (przez 72 godz.) i dwie w 37
o
C (przez 24
godz).
- Po inkubacji policzyć liczbę kolonii na płytkach i uśrednić. Porównać liczbę psychrofili i
mezofili.
c) Oznaczyć bakterie grupy coli.
- Do 10 probówek zawierających po 10 ml podłoża Eijkmana wlać po 1 ml wody
wodociągowej
- 5 probówek inkubować w 37
o
C, a 5 w 44
o
C.
- Po 24 i 48 godzinach inkubacji obserwować zmiany pożywki. Zakwaszenie podłoża
(zmiana barwy podłoża z fioletowej na żółtą) i obecność gazu w rurce Durhama świadczą
o występowaniu bakterii grupy coli. Takie zmiany podłoża obserwowane w probówkach
inkubowanych w obu temperaturach wskazują na obecność bakterii grupy coli typu
fekalnego. Brak gazu i zakwaszenia po 48 godzinach uznaje się za wynik ujemny.
2. Badanie wody ze zbiornika naturalnego
a) Pobrać próbkę wody ze zbiornika.
b) Oznaczyć liczbę bakterii psychrofilnych i mezofilnych.
- Rozcieńczyć badaną wodę 10
-1
do 10
-4
.
- Po 0,1 ml każdego z rozcieńczeń wysiać na 4 płytki z agarem odżywczym.
-
Dwie z nich inkubować w 37
o
C, a dwie w temperaturze pokojowej.
- Policzyć wyrosłe kolonie. Na podstawie wzoru z punktu 3d ze str. 12 obliczyć liczbę
bakterii psychrofilnych i mezofilnych w 1 ml badanej wody. Wyniki zamieścić w tabeli 7 i
krytycznie je porównać.
c) Oznaczyć miano coli metodą fermentacyjno-probówkową.
- Do probówek zawierających po 10 ml podłoża Eijkmana i rurki Durhama,
ponumerowanych od 1 do 10 dodajemy kolejno wg schematu:
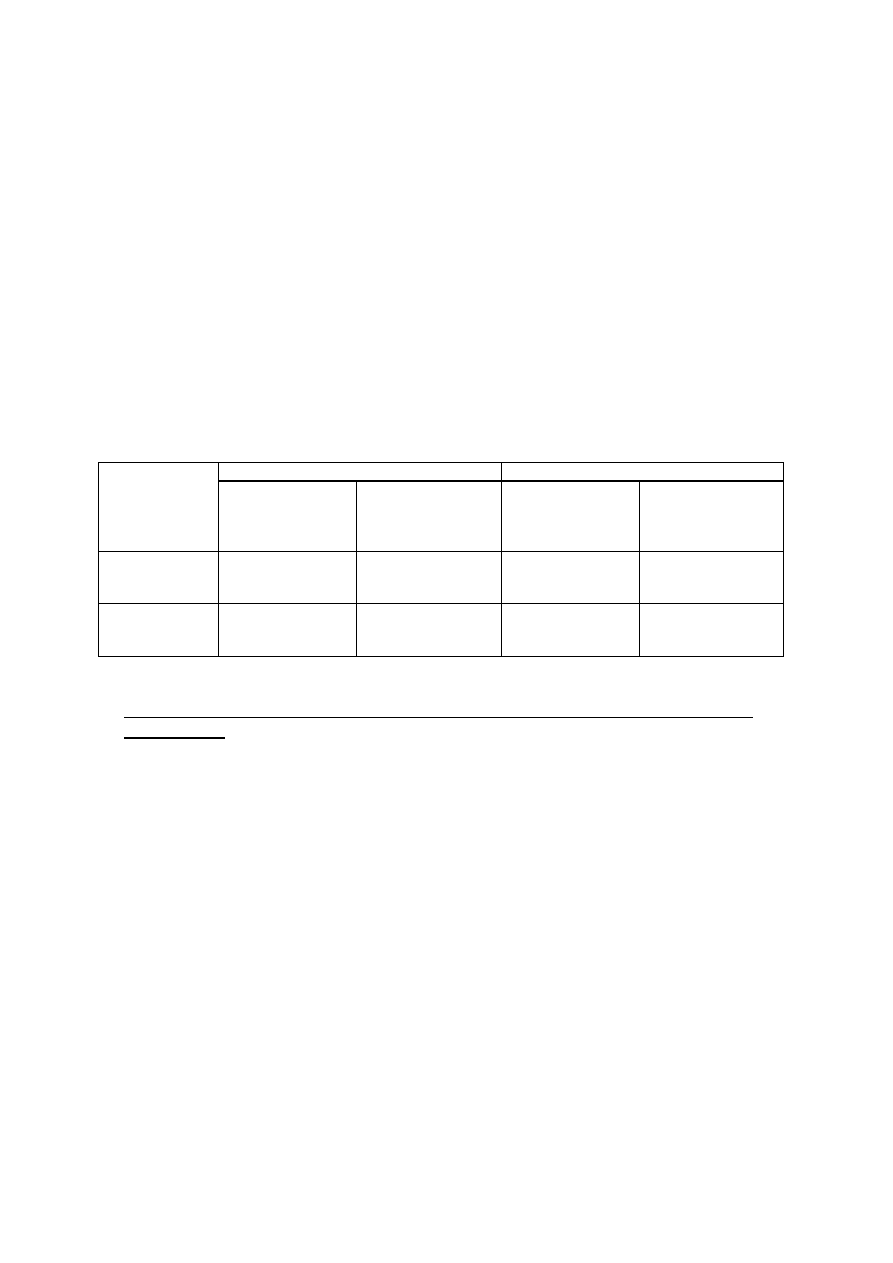
40
1 ml wody nierozcieńczonej
probówka nr 1 i 2
1 ml wody rozcieńczonej 10
-1
probówka nr 3 i 4
1 ml wody rozcieńczonej 10
-2
probówka nr 5 i 6
1 ml wody rozcieńczonej 10
-3
probówka nr 7 i 8
1 ml wody rozcieńczonej 10
-4
probówka nr 9 i 10
- Szereg probówek o numerach parzystych inkubować w temperaturze 37
o
C, a o numerach
nieparzystych w 44
o
C. Obserwacje należy przeprowadzić po 48 godz. inkubacji.
Zakwaszenie podłoża (zmiana barwy podłoża z fioletowej na żółtą) i obecność gazu w rurce
Durhama świadczą o występowaniu bakterii z grupy coli, przy czym jeśli takie zmiany
podłoża obserwuje się w probówkach inkubowanych w 44
o
C – bakterii z grupy coli typu
fekalnego. Ostatnia probówka w szeregu z wynikiem pozytywnym stanowi podstawę do
oznaczenia miana coli i miana coli typu fekalnego. Brak gazu i zakwaszenia po 48 godz.
przyjmuje się jako wynik ujemny.
Tabela 7. Liczba bakterii z grupy coli w próbach wody pobranych z wód powierzchniowych
3. Oznaczenie bakterii z grupy coli w ściekach komunalnych metodą fermentacyjno-
probówkową
a) Przygotować rozcieńczenia badanych ścieków 10
-1
do 10
-6
.
b) Oznaczyć miano coli metodą fermentacyjno-probówkową tak jak w punkcie 2c).
III. ZAGADNIENIA DO OPRACOWANIA
1. Właściwe bakterie wodne.
2. Mikroorganizmy chorobotwórcze, które mogą się dostać do wody wraz ze ściekami.
3. Bakterie wskaźnikowe świadczące o skażeniu wody.
4. Sanitarna analiza bakteriologiczna wody.
5. Wykrywanie bakterii grupy coli metodą fermentacyjno-probówkową i metodą filtrów
membranowych.
6. Miano coli.
Woda
powierzchniowa
Temperatura inkubacji 22
o
C
Temperatura inkubacji 37
o
C
Wysiane
rozcieńczenie i
otrzymana liczba
kolonii bakterii
jtk/ml
Wysiane
rozcieńczenie i
otrzymana liczba
kolonii bakterii
jtk/ml
Zbiornik I
Zbiornik II

41
Ćwiczenie 10
Temat: Podstawowe techniki pracy z bakteriofagami
I. WPROWADZENIE
1. Etapy infekcji fagowej
Bakteriofagi,
zwane w skrócie fagami, to wirusy będące bezwzględnymi,
wewnątrzkomórkowymi pasożytami bakterii i archeonów. Jak wszystkie wirusy, mają one
budowę niekomórkową. Cząstka wirusa (wirion) zawiera tylko jeden rodzaj kwasu
nukleinowego, otoczony przez białkowy kapsyd. Fagi są zdolne do namnażania się w
komórkach właściwego gospodarza, ale nie mają własnego metabolizmu. Wykorzystują do
tego celu aparat metaboliczny tych prokariotów, które są w stanie zainfekować.
Zwykle dany bakteriofag infekuje tylko określony gatunek, bądź nawet szczep czy
grupę szczepów bakterii. Kolejne etapy infekcji fagowej: to (i) adsorpcja do wrażliwej
komórki, posiadającej określony receptor; (ii) przedostanie się kwasu nukleinowego do
komórki; (iii) replikacja kwasu nukleinowego wirusa; (iv) synteza podjednostek białkowych
kapsydu; (v) łączenie się podjednostek białkowych kapsydu i pakowanie kwasów
nukleinowych do kapsydu (czyli tworzenie potomnych cząstek wirusa); (vi) uwolnienie
namnożonych cząstek wirusa z komórki, najczęściej w wyniku jej lizy (np. fag λ). Niektóre
fagi nitkowate po namnożeniu opuszczają komórki gospodarza, nie doprowadzając do ich lizy
(np. fag M13).
Fagi łagodne (umiarkowane, np. fag λ) mają zdolność do wejścia w stan stabilnej
równowagi (zwanej lizogenią) z komórkami gospodarza. W czasie infekcji populacji bakterii
fagiem łagodnym większość komórek ulega lizie, a tylko niewielka część populacji –
lizogenizacji. W szczepach E. coli zlizogenizowanych fagiem λ [E. coli (λ)], genom faga jest
zintegrowany z chromosomem gospodarza (w postaci tzw. profaga) i replikuje się wraz z
nim. Bakterie te są odporne na superinfekcję tym samym wirusem. Z niewielką częstością w
populacji E. coli (λ) genom profaga ulega wycięciu z chromosomu i dochodzi do cyklu
litycznego, dzięki któremu powstają cząstki wirusów potomnych.
2. Techniki pracy z fagami
Fagi hoduje się i bada na (i) podłożu stałym porośniętym gęsto bakteriami (tzw. murawa
bakteryjna), gdzie w wyniku ich działalności można obserwować tzw. łysinki, lub (ii) w
płynnej hodowli bakterii wrażliwych, gdzie aktywność fagów przejawia się stopniowym
spadkiem mętności hodowli, w miarę lizy komórek gospodarza.
Łysinka jest to widoczna gołym okiem strefa przejaśnienia w gęstym wzroście
bakterii na podłożu półpłynnym (tzw. metoda płytek dwuwarstwowych), powstająca
najczęściej wskutek lizy części lub wszystkich komórek przez fagi. Zwykle łysinka powstaje
w wyniku pierwotnej infekcji jednej bakterii przez jeden wirion. Morfologia łysinki, tzn.
wielkość, kształt brzegów, stopień przejrzystości, może być pomocna w identyfikacji faga,
np. fagi zjadliwe tworzą łysinki przejrzyste, zaś fagi łagodne – mętne, gdyż nie lizują
wszystkich komórek. Dzięki możliwości uzyskiwania łysinek nie tylko (i) identyfikujemy
niektóre fagi, ale także (ii) mianujemy lizaty fagowe i (iii) uzyskujemy czyste klony fagów.
II. CZĘŚĆ PRAKTYCZNA
Szczepy bakteryjne:
Podłoża:
E. coli JM109 F’ lub TG1
agar odżywczy

42
E. coli DH1 lub DH5
agar odżywczy półpłynny (0,7 %)
E. coli( )
bulion odżywczy
Fagi:
vir
i M13
1. Oznaczenie miana faga techniką agaru dwuwarstwowego
a) Rozcieńczyć odpowiednio zawiesinę faga w bulionie odżywczym (wg wskazówek osoby
prowadzącej zajęcia).
b) Do 4 probówek wlać po 0,1 ml hodowli E. coli DH1, a następnie dodać po 0,1 ml
sąsiednich rozcieńczeń zawiesiny faga w dwóch powtórzeniach (np. do dwóch 10
-4
i do
dwóch 10
-5
). Uwaga! Zawiesinę bakterii i faga wlewać bezpośrednio na dno probówki,
nie po ściankach! Inkubować 20 min w 37
o
C.
c) Do każdej probówki dodać po 3 ml upłynnionego i schłodzonego do około 46
o
C agaru
półpłynnego. Wymieszać, obracając probówkę między dłońmi.
d) Natychmiast wylewać zawartość probówki na szalkę z agarem odżywczym (uprzednio
odpowiednio podpisaną) i rozprowadzić szybko i równomiernie po całej jej powierzchni.
Szalkę położyć poziomo, aby agar zastygł.
e) Po zastygnięciu agaru, inkubować płytki w 37
o
C przez 24 godziny.
f) Obliczyć liczbę łysinek fagowych. Uśrednić wyniki z dwóch płytek i uwzględniając
rozcieńczenie obliczyć miano faga (X) wg wzoru:
X = a x b x 10 [pfu/ml]
a - średnia liczba łysinek
b - odwrotność rozcieńczenia
10 - przeliczenie na 1 ml wyjściowej zawiesiny
(pfu – ang. plaque forming unit, jednostka tworząca łysinkę)
2. Rodzaje łysinek fagowych
a) Do probówki dodać 0,1 ml hodowli E. coli TG1 (wlewać bezpośrednio na dno probówki,
nie po ściankach!) i 3 ml upłynnionego, schłodzonego agaru półpłynnego. Wymieszać i
wylać na szalkę z agarem odżywczym, rozprowadzając szybko i równomiernie po całej
powierzchni.
b) Po zastygnięciu podzielić płytkę na 3 sektory i na każdy z nich nałożyć kroplę (nie więcej
niż 10 l!) zawiesiny:
- faga
vir
- faga M13
- hodowli E. coli( )
c) Po wsiąknięciu zawiesin, odwrócić płytki do góry dnem i inkubować w 37
o
C.
d) Opisać rodzaje stref lizy na murawie bakteryjnej.
3. Test wrażliwości na infekcję fagiem (cross-streaking)
a) Na szalkę z agarem odżywczym nanieść w postaci rysy, za pomocą ezy, zawiesinę faga
vir
i równomiernie ją rozprowadzić wzdłuż linii posiewu (Rys. 6).
b) Równolegle w podobny sposób nanieść faga M13.
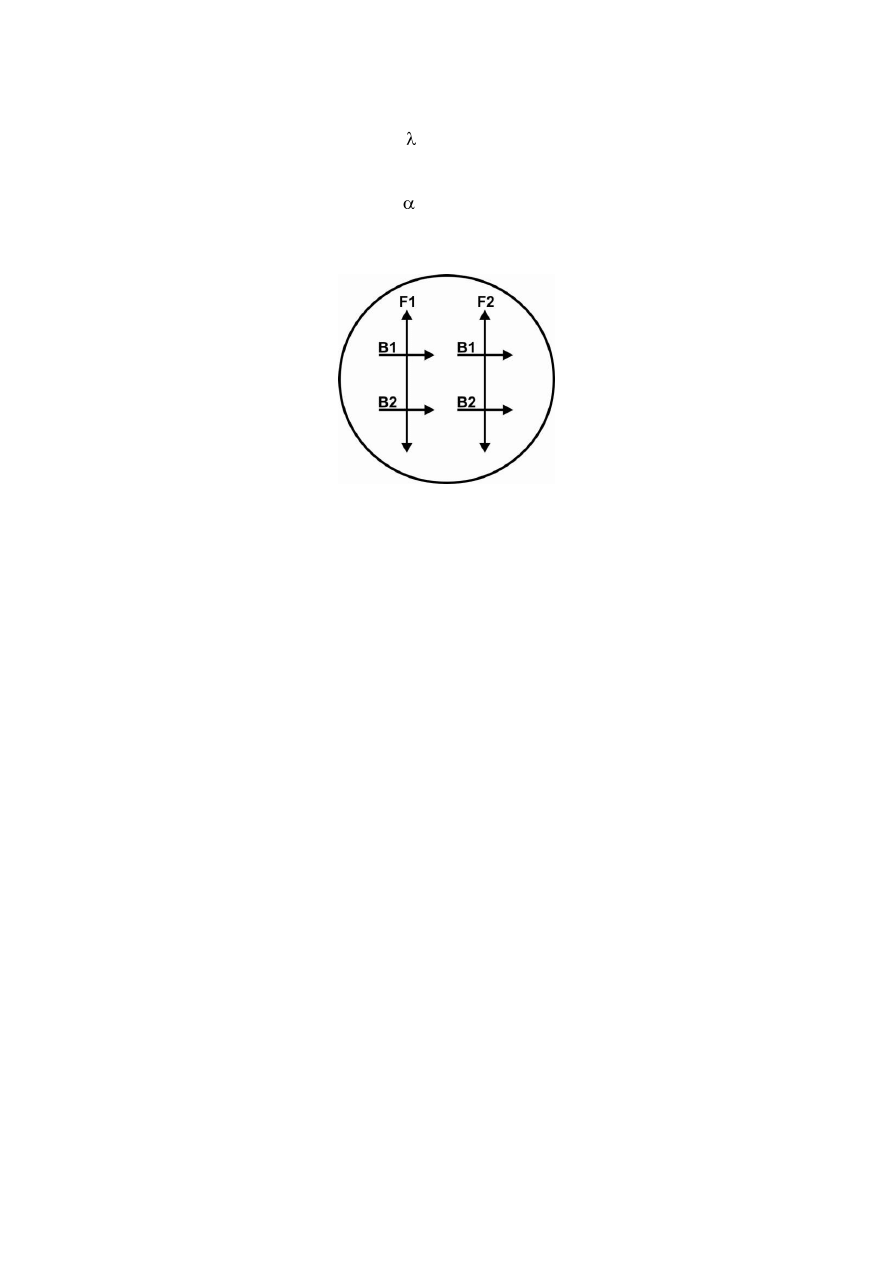
43
c) Po wsiąknięciu zawiesin fagowych nabrać na ezę hodowlę E. coli TG1 i jednym ruchem
posiać ją rysą w poprzek posiewu faga
vir
.
d) Ezę opalić, ponownie nabrać hodowli tego samego szczepu i posiać ją rysą w poprzek faga
M13.
d) W taki sam sposób posiać E. coli DH5 .
e) Po wsiąknięciu hodowli w pożywkę inkubować płytki w 37
o
C.
f) Ocenić wrażliwość obu szczepów na badane fagi.
Rys. 6. Określanie wrażliwości bakterii na fagi (test cross-streaking).
F1 i F2 – linie posiewu fagów; B1 i B2 – linie posiewu bakterii
III. ZAGADNIENIA DO OPRACOWANIA
1. Charakterystyka bakteriofagów.
2. Etapy infekcji fagowej w cyklu litycznym i lizogennym.
3. Wrażliwość szczepów bakterii na infekcję fagami.
4. Charakterystyka faga λ i faga M13.
5. Podstawowe techniki pracy z bakteriofagami i ich zastosowanie.
6. Łysinka fagowa.
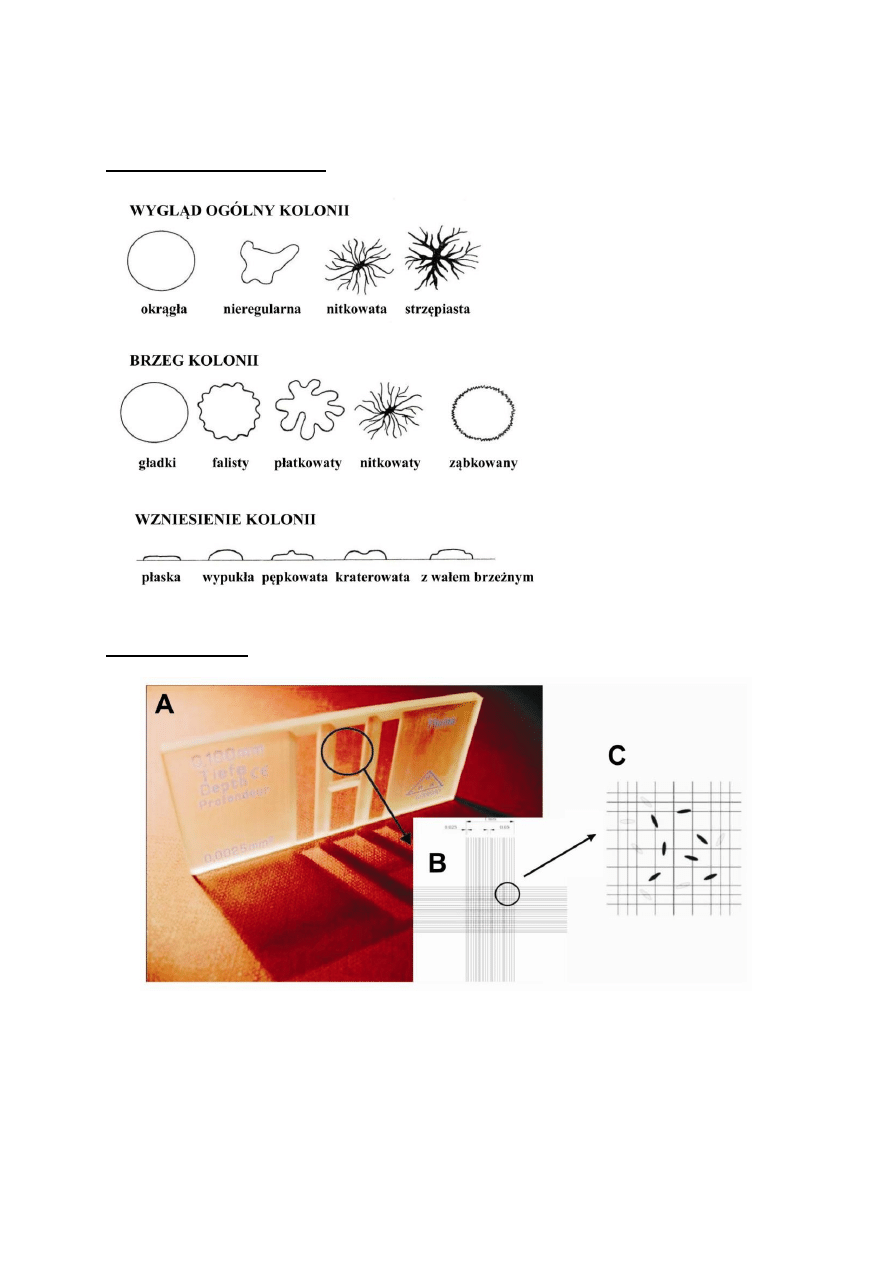
44
III. ADDENDUM
1. Opis kolonii bakteryjnych
2. Komora Thomy
Rys. 7. Komora Thomy. (A) Widok ogólny. (B) Duży „kwadrat”. (C) Mały „kwadrat”. Komora Thomy zawiera
kwadratową siatkę, którą tworzą pionowe i poziome linie, wydzielające 16 dużych „kwadratów”. Każdy z nich
składa się z 16 małych „kwadratów” o boku długości 1/20 mm. W rzeczywistości każdy mały „kwadrat” jest
prostopadłościanem o powierzchni podstawy równej 1/400 mm
2
i wysokości 0,1 mm, zatem jego objętość
wynosi 1/4000 mm
3
. Znając średnią liczbę komórek badanego mikroorganizmu, określoną na podstawie ich
liczby w 50 kolejnych prostopadłościanach, można określić zagęszczenie tego drobnoustroju w 1 ml zawiesiny.
45
IV. KRÓTKA CHARAKTERYSTYKA BAKTERII STOSOWANYCH NA
ĆWICZENIACH
(na podstawie „Bergey’s Manual of Systematic Bacteriology”, wydanie II, tomy 2-5, 2001-2012,
Springer)
TYP FIRMICUTES
Rodzaj Bacillus
(łac. bacillum – laseczka/pałeczka)
Laseczki gramdodatnie, poruszające się za pomocą rzęsek. Występują pojedynczo lub tworzą
łańcuszki. Tlenowce lub warunkowe tlenowce. Chemoorganoheterotrofy – są wśród nich
prototrofy i auksotrofy. Tworzą endospory. Ich pierwotnym siedliskiem jest gleba, ale dzięki
endosporom można je izolować z najróżniejszych środowisk. Niektóre gatunki są patogenami,
np. B. anthracis (laseczka wąglika).
B. megaterium (laseczka olbrzymia)
(gr. mega – wielki; gr. teras – potwór, bestia)
Tworzy łańcuszki. Centralnie położone endospory nie powodują rozdęcia komórki
macierzystej. Prototrof. Temp. optymalna 30
o
C.
B. subtilis (laseczka sienna)
(łac. subtilis – wysmukły, cienki)
Tworzy łańcuszki. Centralnie położone endospory nie powodują rozdęcia komórki
macierzystej. Prototrof. Temp. optymalna 37
o
C. Łatwo można wyizolować tę bakterię z siana.
Rodzaj Clostridium
(gr. kloster – wrzeciono; clostridium – małe wrzeciono)
Laseczki gramdodatnie. Beztlenowce. Chemoorganoheterotrofy. Tworzą endospory owalne
bądź okrągłe, często o średnicy większej od średnicy komórki macierzystej. Niektóre gatunki
są chorobotwórcze, np. C. tetani (laseczka tężca), C. botulinum (laseczka jadu kiełbasianego).
C. pasteurianum
(łac. pasteurianum – należący do Pasteura)
Prototrof zdolny do wiązania azotu cząsteczkowego. Gromadzi granulozę. Owalne endospory
położone są subterminalnie i powodują rozdęcie komórki macierzystej. Temp. optymalna 30-
37
o
C. Występuje w glebie na całym świecie.
Rodzaj Enterococcus
(paciorkowce)
(gr. enteron – jelito; gr. kokkos – pestka, ziarno, jagoda; Enterococcus – ziarniak jelitowy)
E. faecalis (paciorkowiec kałowy, dawniej Streptococcus faecalis)
(łac. faex – odchody; faecalis – kałowy)
Gramdodatni, nieruchliwy ziarniak występujący w parach i krótkich łańcuszkach.
Beztlenowiec aerotolerancyjny. Metabolizm fermentacyjny. Fermentuje węglowodany z
wytworzeniem głównie kwasu mlekowego (bez gazu). Chemoorganotrof, auksotrof.
Występuje w przewodzie pokarmowym ludzi i zwierząt. Można go też znaleźć w glebie, na
owadach i roślinach, a także w żywności (np. produkty mleczne, mięso). Zdolny do wzrostu
w temp. 10
o
i 45
o
C, w obecności 6,5 % NaCl i pH 9,6. Patogen oportunistyczny
46
odpowiedzialny za wiele zakażeń szpitalnych, np. powoduje infekcje dróg moczowych,
zakażenia ran pooperacyjnych.
Rodzaj Staphylococcus
(gronkowce)
(gr. staphyle – winne grono; gr. kokkos – pestka, ziarno, jagoda)
S. epidermidis (gronkowiec skórny, dawniej gronkowiec biały)
(łac. epidermidis – skórny)
Gramdodatni ziarniak występujący głównie w postaci dwoinek i tetrad (tworzy też grona).
Nieruchliwy. Wytwarza biały barwnik. Warunkowy tlenowiec. Chemoorganoheterotrof,
auksotrof. Wzrost w temp. 15
o
-45
o
C (temp. optymalna 30-37
o
C). Rośnie w podłożach
zawierających do 7,5 % NaCl. Powszechnie występuje na skórze i błonach śluzowych
zwierząt i ludzi. Może stać się patogenem oportunistycznym.
TYP ACTINOBACTERIA
Rodzaj Micrococcus
(gr. micros – mały; gr. kokkos – pestka, ziarno, jagoda)
M. luteus (pakietowiec żółty)
(łac. luteus – złoto-żółty)
Gramdodatni, nieruchliwy ziarniak, tworzący układy złożone z 4 komórek (pakiety), a także
nieregularne
skupiska
pakietów.
Tlenowiec,
metabolizm
ściśle
oddechowy.
Chemoorganoheterotrof, auksotrof. Temp. optymalna 25-37
o
C. Występuje na skórze ludzi i
zwierząt, w glebie, wodzie, kurzu, mleku i jego przetworach.
TYP PROTEOBACTERIA
I Klasa Alphaproteobacteria
Rodzaj Paracoccus
(gr. para – podobny do; gr. kokkos – pestka, ziarno, jagoda)
P. versutus
(łac. versutus – zmienny)
Gramujemna pałeczka. Warunkowy chemolitoautotrof zdolny do wzrostu autotroficznego na
podłożu mineralnym z tiosiarczanem. Prototrof. Rośnie chemoorganotroficznie na pożywkach
zawierających różne związki organiczne jako jedyne źródło węgla i energii. Warunkowy
tlenowiec. Zdolny do denitryfikacji. Temp. optymalna 30
o
C. Występuje w glebie.
II. Klasa Gammaproteobacteria
Rodzina Enterobacteriaceae (pałeczki jelitowe)
Gramujemne pałeczki. Chemoorganoheterotrofy. Warunkowe tlenowce.
Rodzaj Escherichia
(Teodor Escherich – mikrobiolog niemiecki, który wyizolował tę bakterię)
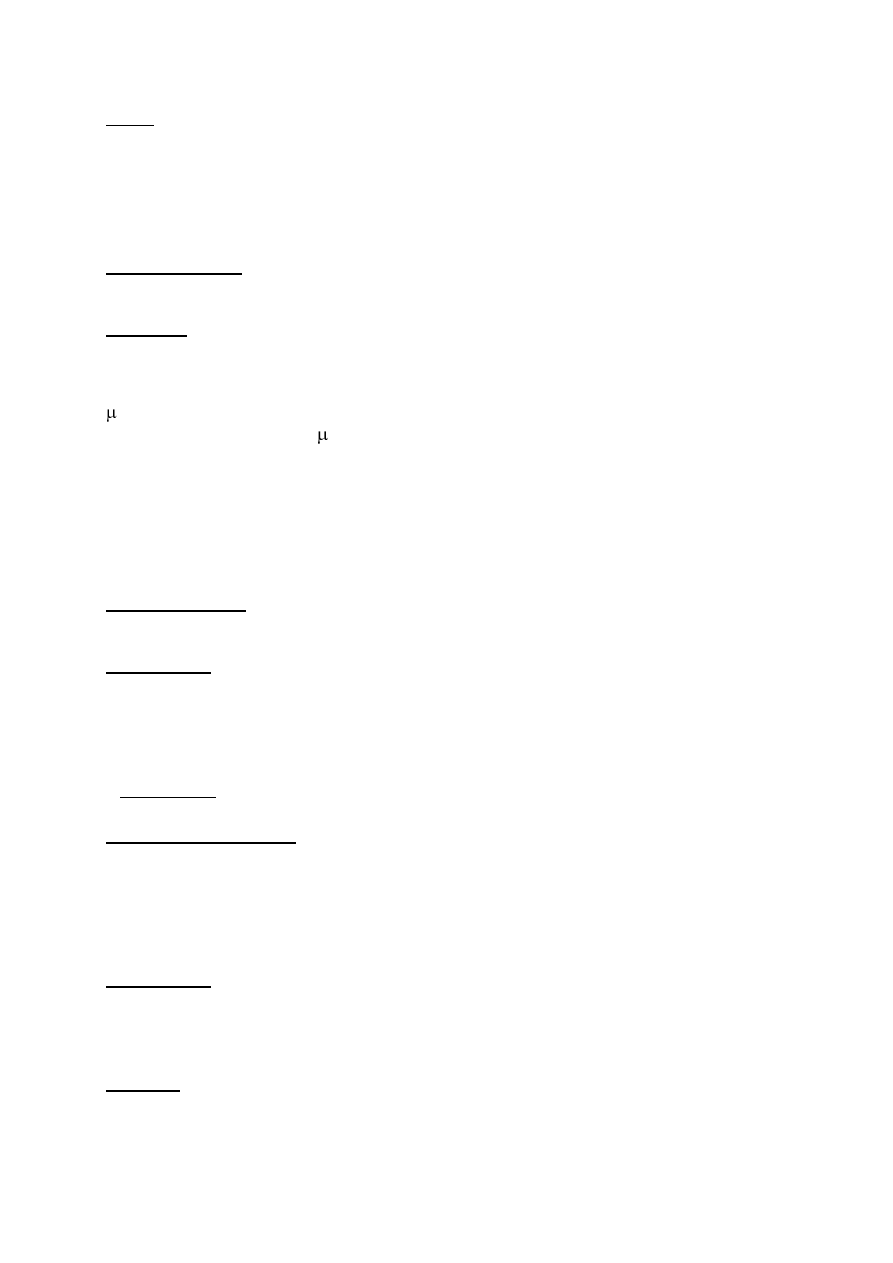
47
E. coli (pałeczka okrężnicy)
(gr. colon – jelito grube/okrężnica; miejsce występowania tej bakterii)
Pałeczka jelitowa. Warunkowy tlenowiec. Chemoorganoheterotrof. Prototrof. Temp.
optymalna 37
o
C. Występuje w jelicie grubym człowieka i licznych zwierząt. Szeroko
rozpowszechniona w środowisku życia człowieka. Najlepiej poznana bakteria pod względem
fizjologicznym i genetycznym.
Rodzaj Proteus
(pałeczka odmieńca)
(gr. Proteus – grecki bożek morski zdolny do przybierania różnorodnych kształtów)
P. vulgaris
(łac. vulgaris – pospolity)
Urzęsiona pałeczka, bardzo ruchliwa. W czasie wzrostu na wilgotnych podłożach stałych
komórki wielu szczepów podlegają cyklicznym przemianom. Formy osiadłe są krótkie (1-3
m długości) i mają nieliczne rzęski. Po osiągnięciu pewnego zagęszczenia przekształcają się
one w formy długie (20-80 m długości), posiadające liczne rzęski. Formy długie migrują
(ang. swarming), a następnie osiadają i tworzą formy krótkie. Takie powtarzające się cykle
dają w rezultacie wzrost w postaci koncentrycznych pierścieni wokół pierwotnego punktu
posiewu (wzrost „rozpełzliwy”). Bakteria odgrywa ważną rolę w rozkładzie materii
organicznej. W organizmie człowieka jest na ogół komensalem, ale niekiedy staje się
patogenem. Może powodować wtórne infekcje u ludzi dorosłych, zakażenia układu
moczowego oraz biegunki i zatrucia u niemowląt.
Rodzaj Serratia
(Serafino Serrati – włoski fizyk)
S. marcescens (pałeczka cudowna, pałeczka krwawa)
(łac. marcescens – zwiędły, przekwitły)
Prototrof. W temp. poniżej 30
o
C, w warunkach tlenowych, wiele szczepów wytwarza
czerwony barwnik prodigiozynę. Występuje na roślinach, w glebie i wodzie, niekiedy jest
komensalem człowieka. Może by patogenem oportunistycznym u ludzi hospitalizowanych.
Inne rodziny:
Rodzaj Pseudomonas
(gr. pseudes – fałszywy, rzekomy; gr. monas – jednostka, monada)
Gramujemne pałeczki poruszające się dzięki rzęskom. Tlenowce, niektóre wykorzystują
azotany jako końcowe akceptory elektronów. Większość to chemoorganoheterotrofy,
prototrofy. Niektóre gatunki są warunkowymi chemolitoautotrofami, zdolnymi do
wykorzystania H
2
lub CO jako źródła energii.
P. fluorescens (pałeczka fluoryzująca)
(łac. fluorescens – fluoryzująca)
Chemoorganoheterotrof, prototrof. Zdolny do denitryfikacji. Temp. optymalna 25-30
o
C.
Występuje w wodach i glebie. Może spowodować zanieczyszczenie żywności.
P. stutzeri
(łac. stutzeri – należący do Stutzera; Albert Stutzer – mikrobiolog niemiecki, który
wyizolował tę bakterię)

48
Chemoorganoheterotrof, prototrof. Zdolny do denitryfikacji. Temp. optymalna 30-35
o
C.
Występuje w wodach i glebie.
Rodzaj Halothiobacillus
(gr. hals – morze, sól; gr. thios – siarka; bacillum – pałeczka/laseczka)
Gramujemne pałeczki. Tlenowce. Bezwzględne chemolitoautotrofy. Uzyskują energię z
utleniania siarki i jej związków nieorganicznych. Niektóre szczepy są umiarkowanymi
halofilami, wymagającymi NaCl (opt. stęż. 0,4-1,0 M).
H. neapolitanus
(łac. neapolitanus – neapolitański)
Wyizolowany w 1902 r. z Zatoki Neapolitańskiej. Występuje w wodzie morskiej i na
korodującym betonie.

49
V. SŁOWNICZEK POLSKO-ANGIELSKI
(terminy mikrobiologiczne)
l.m. – liczba mnoga
l.p. – liczba pojedyncza
agar odżywczy – nutrient agar
barwienie – staining
barwienie Grama – Gram staining
barwienie proste –simple staining
bulion odżywczy – nutrient broth
czynnik wzrostowy – growth factor
czysta kultura – pure culture
endospora – endospore
eza – loop
faza zastoju (adaptacyjna) – lag phase
gęstość optyczna – optical density
głaszczka – spreader
grupa coli – coliform
hodowla – culture
hodowla ciągła – continuous culture
hodowla okresowa – batch culture
hodowla synchroniczna – synchronized culture
hodowla wzbogacająca – enrichment culture
jednostka tworzaca kolonię (jtk) – colony forming unit (cfu)
kolonia – colony
komora Thomy – Thoma chamber
kropla wisząca – hanging-drop
metoda rozcieńczeń – spread plate metod
metoda suchych rozcieńczeń (posiew redukcyjny) – streak plate
laseczka – rod
mureina – murein
odporność – immunity
oporność – resistance
otoczka – capsule
pałeczka – rod
peptydoglikan – peptidoglycan
pipeta – pipette
płytka Petriego – Petri plate, Petri dish
podłoże mikrobiologiczne – l.p. medium, l.m. media
podłoże mineralne – mineral medium
podłoże minimalne – minimal medium
podłoże płynne – liquid medium
podłoże półpłynne – semi-solid medium
podłoże różnicujące – differential medium lub indicator medium
podłoże selekcyjne – selective medium
podłoże stałe – solid medium
podłoże syntetyczne – synthetic medium lub defined medium
podłoże wzbogacone – enriched medium

50
podłoże złożone – complex medium lub undefined medium
posiew powierzchniowy – spread plate
posiew redukcyjny (metoda suchych rozcieńczeń) – streak plate
posiew wgłębny – pour plate method
rozmaz – smear
rzęska – l.p. flagellum, l.m. flagella
skos – slant
słupek – stab
szczep – strain
szczepić – inoculate
szkiełko nakrywkowe – cover-slip (cover-glass)
szkiełko podstawowe – slide (microscope slide)
szkiełko z łezką – depression slide
ściana komórkowa – cell wall
utrwalanie – fixation
ziarniak – l.p. coccus, l.m. cocci

51
VI. ZALECANA LITERATURA
1) Kunicki-Goldfinger W.J.H. „Życie bakterii”. PWN, 2001.
2) Madigan M.T., Martinko J.J., Parker J. „Brock biology of microorganisms”.
Pearson Education, Inc. Upper Saddle River, 2003.
(oraz wszystkie następne wydania tej książki)
3) Schlegel H.G. „Mikrobiologia ogólna”. PWN, 1996.
4) Salyers A.A, Whitt D.D. „Mikrobiologia – różnorodność, chorobotwórczość i
środowisko”. PWN, 2003.
5) Baj J., Markiewicz Z. (red.) „Biologia molekularna bakterii”. PWN, 2006.
6) Różalski A. „Ćwiczenia z mikrobiologii ogólnej – skrypt dla studentów biologii”.
Wyd. Uniwersytetu Łódzkiego, Łódź, 1996.
7) Duszkiewicz-Reinhard W., Grzybowski R., Sobczak E. „Teoria i ćwiczenia z
mikrobiologii ogólnej i technicznej”. Wyd. SGGW, Warszawa, 1996.
8) Grabińska-Łoniewska (red). „Ćwiczenia laboratoryjne z mikrobiologii ogólnej”.
Oficyna Wyd. Politechniki Warszawskiej, Warszawa, 1996.
Wyszukiwarka
Podobne podstrony:
Cwiczenie 01 id 98935 Nieznany
HIGIENA, CWICZENIE 7, 13 11 201 Nieznany
Antropologia Cwiczenia 01 id 65 Nieznany (2)
CwiczenieArcGIS 01 id 125936 Nieznany
Hstoria?ministracji 10 201 Nieznany
cwiczenie 01 id 125027 Nieznany
HIGIENA, CWICZENIE 8, 27 11 201 Nieznany
Pediatria Cwiczenia 01 id 35420 Nieznany
Fizjologia Cwiczenia 01 id 1743 Nieznany
Cwiczenie 01 id 98935 Nieznany
Cwiczenia nr 10 (z 14) id 98678 Nieznany
FINANSE PRZEDSIĘBIORSTW ĆWICZENIA 2 (21 10 2012)
HIGIENA, ĆWICZENIE 3, 16 10 2012
pieniadz PL 2012 slajdy 1 10 id Nieznany
algebra Skrypt Algebra KSzW 201 Nieznany
Wykład 2012-01-10, psychologia drugi rok, psychologia ról
01 10 2012
więcej podobnych podstron