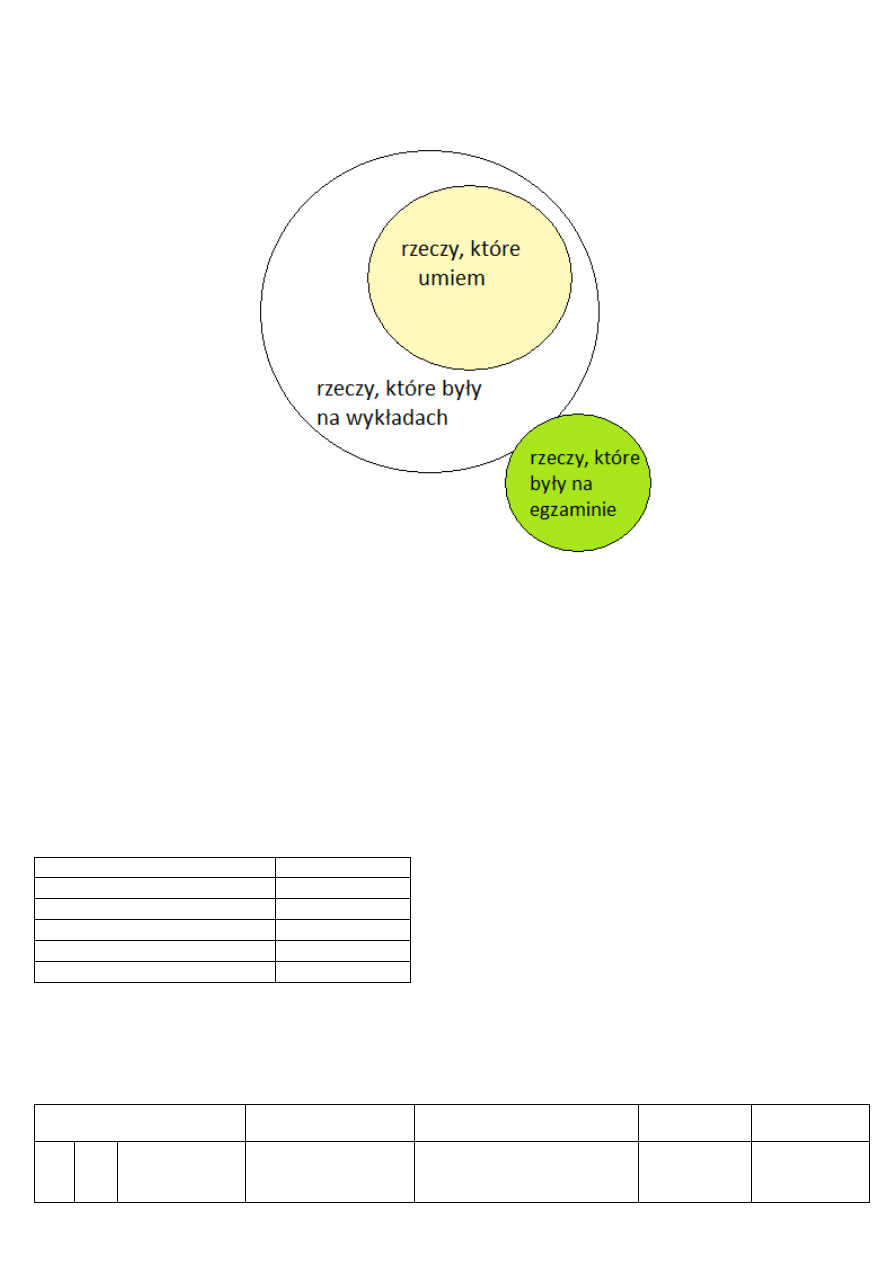
Wersja opracowania – 1.3
dodane biocząsteczki
Międzywydziałowa Szkoła Inżynierii Biomedycznej 2008/2009
Podczas tworzenia niniejszego opracowania wykorzystane zostały materiały zamieszczone przez Sebastiana (Ziza) na forum MSIB:
http://student.agh.edu.pl/~forsby/forum/index.php
Dodatkowo korzystałem z informacji od Dr Inż. Hasik oraz notatek z wykładów Kasi L. Pozdrawiam. ;)
Egzamin z chemii organicznej składać się ma z około 19tu pytań zamkniętych (ABCD) po 0,5 punktu za każde, kilku pytań
otwartych za 1 punkt każde oraz 3 pytań, które będą wymagały większej wiedzy na temat mechanizmów reakcji
chemicznych. Nie widziałem byśmy na wykładach zajmowali się otrzymywaniem wszystkich związków (podobnie w
wymaganiach na egzamin nie zostało to zawarte), dlatego część oznaczona (*) jest podana jako najprawdopodobniej
nieobowiązkowa.
Zagadnienia do egzaminu z chemii organicznej 2008/2009
1. Klasyfikacja związków organicznych na podstawie charakterystycznych grup funkcyjnych.
Klasy związków :
Węglowodory
C, H
Halogenopochodne
C, H, X (F, Cl, Br, I)
Estry, alkohole, aldehydy, ketony
C, H, O
Aminy, iminy, nitryle, zw.azotowe C, H, N
Amidy, związki nitrowe
C, H, O, N
Chlorki kwasowe
C, H, O, Cl
2. Hybrydyzacja orbitali elektronowych atomów węgla w cząsteczkach związków organicznych.
Rodzaje wiązań i geometria cząsteczek wynikające z hybrydyzacji orbitali.
Nazwa
Budowa cząsteczki
Rodzaje
wiązań C-C
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
A
li
fa
t
y
cz
n
e
N
a
sy
c
o
n
e
Alkany
łańcuchowa
(prosta lub rozgałęziona)
tylko pojedyncze (σ)
tylko sp
3
C
n
H
2n+2
Cykloalkany
cykliczna
(z podstawnikami)
tylko pojedyncze (σ)
tylko sp
3
C
n
H
2n
N
ie
n
a
sy
co
n
e
Alkeny
łańcuchowa
(prosta lub rozgałęziona)
jedno podwójne
(składające się z 1 σ i 1 π),
pozostałe pojedyncze
sp
2
(wiązanie
C=C) i sp
3
C
n
H
2n
Cykloalkeny
cykliczna
(z podstawnikami)
jedno podwójne
(składające się z 1 σ i 1 π),
pozostałe pojedyncze
sp
2
(wiązanie
C=C) i sp
3
C
n
H
2n-2
Alkadieny
łańcuchowa
(prosta lub rozgałęziona)
dwa podwójne (składające się z 1
σ i 1 π każde),
pozostałe pojedyncze
sp
2
(wiązania
C=C) i sp
3
C
n
H
2n-2
Alkiny
łańcuchowa
(prosta lub rozgałęziona)
jedno potrójne
(składające się z 1 σ i 2 π),
pozostałe pojedyncze
sp (wiązanie
CΞC) i sp
3
C
n
H
2n-2
A
ro
m
a
ty
cz
n
e
Areny
cykliczna
(z podstawnikami)
w pierścieniu – pośrednie
(składające się z układu wiązań σ i
zdelokalizowanych wiązań π),
w podstawnikach - różne
w pierścieniu –
tylko sp
2
C
n
H
2n-6
(dla szeregu
benzenu)
Węglowodory o
skondensowanych
pierścieniach
aromatycznych
cykliczna
(z podstawnikami)
w pierścieniu – pośrednie
(składające się z układu wiązań σ i
zdelokalizowanych wiązań π),
w podstawnikach - różne
w pierścieniu –
tylko sp
2
-
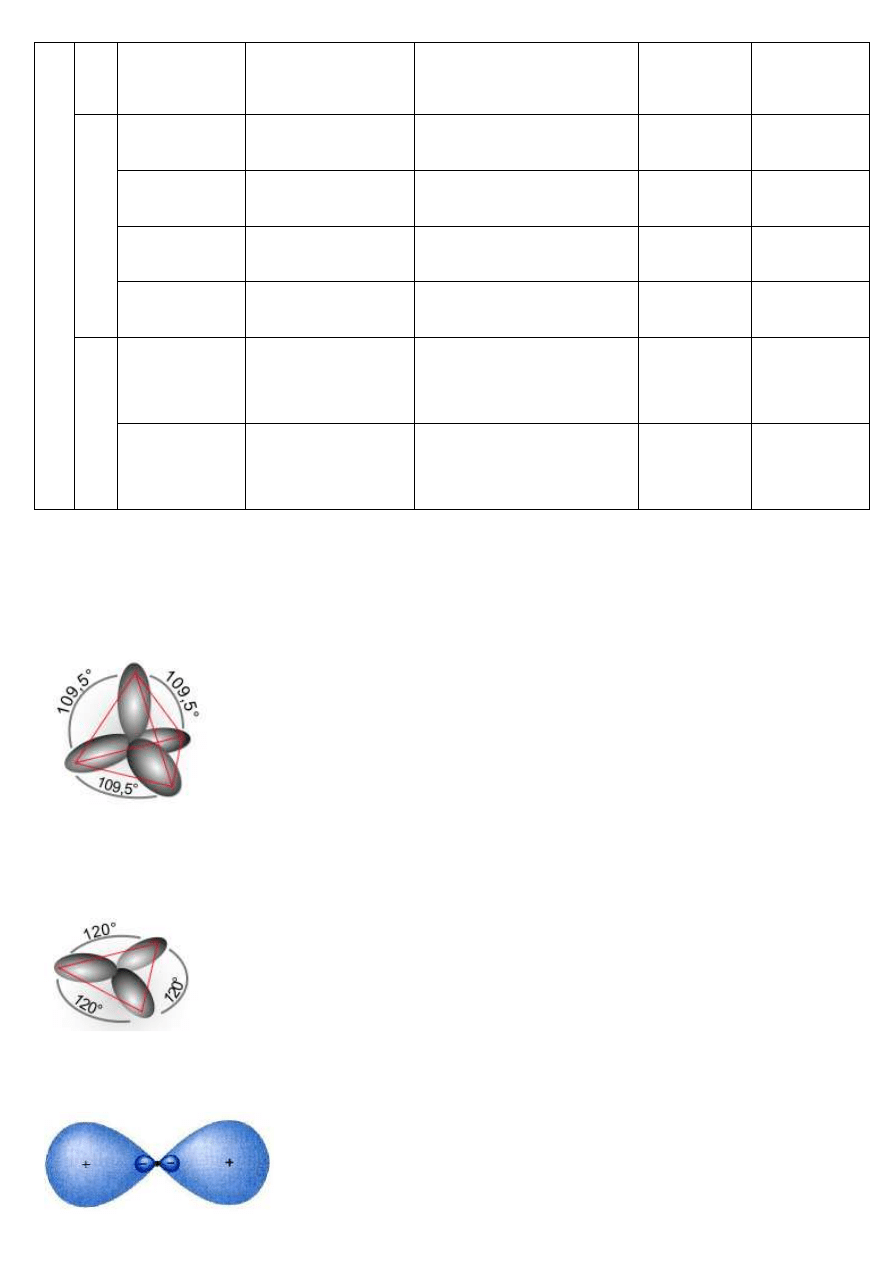
Możliwe hybrydyzacje:
1) 2s
1
2p
x
1
2p
y
1
2p
z
1
– sp
3
węgiel tetraedryczny (4 hybrydy; 109,5
o
)
wszystkie związki organiczne gdzie węgiel ma wiązania pojedyncze (alkany, cykloalkany)
wiązania pojedyncze C-C i C-H to wiązania σ powstające przez czołowe nałożenie się orbitali
2) 2s
1
2p
x
1
2p
y
1
2p
z
1
– sp
2
węgiel trygonalny (3 hybrydy; 120
o
)
wszystkie związki organiczne gdzie C ma wiązania podwójne (alkeny, cykloalkeny, związki aromatyczne)
wiązanie podwójne C=C to jedno wiązanie σ (sp
2
-sp
2
) i jedno π (p-p; boczne nakładanie się orbitali)
3) 2s
1
2p
x
1
2p
y
1
2p
z
1
– sp
węgiel liniowy(2 hybrydy; 180
o
)
wszystkie związki organiczne, gdzie C ma wiązania potrójne (alkiny, cykloalkiny)
wiązanie potrójne to jedno wiązanie σ i dwa π
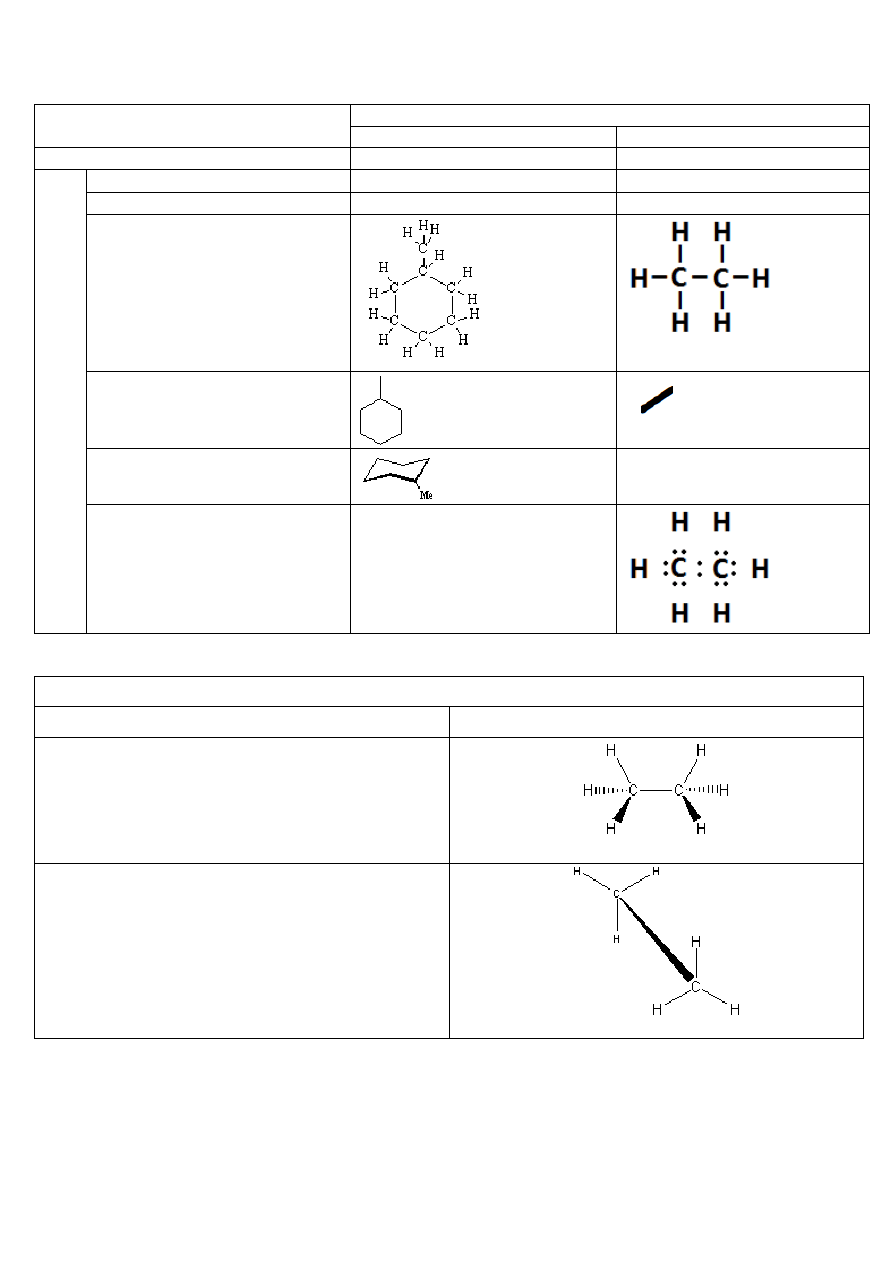
3. Różne sposoby zapisu wzorów cząsteczek związków organicznych: wzory sumaryczne, strukturalne (Lewisa,
kreskowe, półstrukturalne, grupowe, szkieletowe) i rzutowe (projekcyjne) - projekcje konikowe, Newmana i Fischera.
rodzaj zapisu
przykład zapisu
metylocykloheksanu
etanu
prosty wzór sumaryczny
C
7
H
13
C
2
H
6
w
zo
ry
s
tr
u
k
tu
ra
ln
e
wzór półstrukturalny
C
6
H
10
–CH
3
CH
3
–CH
3
wzór grupowy
C
6
H
10
CH
3
CH
3
CH
3
wzór kreskowy
(pełny wzór strukturalny)
wzór szkieletowy
(płaski)
wzór szkieletowy
(przestrzenny, łódkowy)
wzór Lewisa
wzory projekcyjne
konwencja zapisu wzoru
przykładowy zapis
wzór stereostrukturalny
grube kreski to wiązania wysunięte przed płaszczyznę
rysunku, zwykłe są na płaszczyźnie rysunku, a przerywane
wchodzą pod nią
(etan)
wzór konikowy (kozłowy)
(etan)
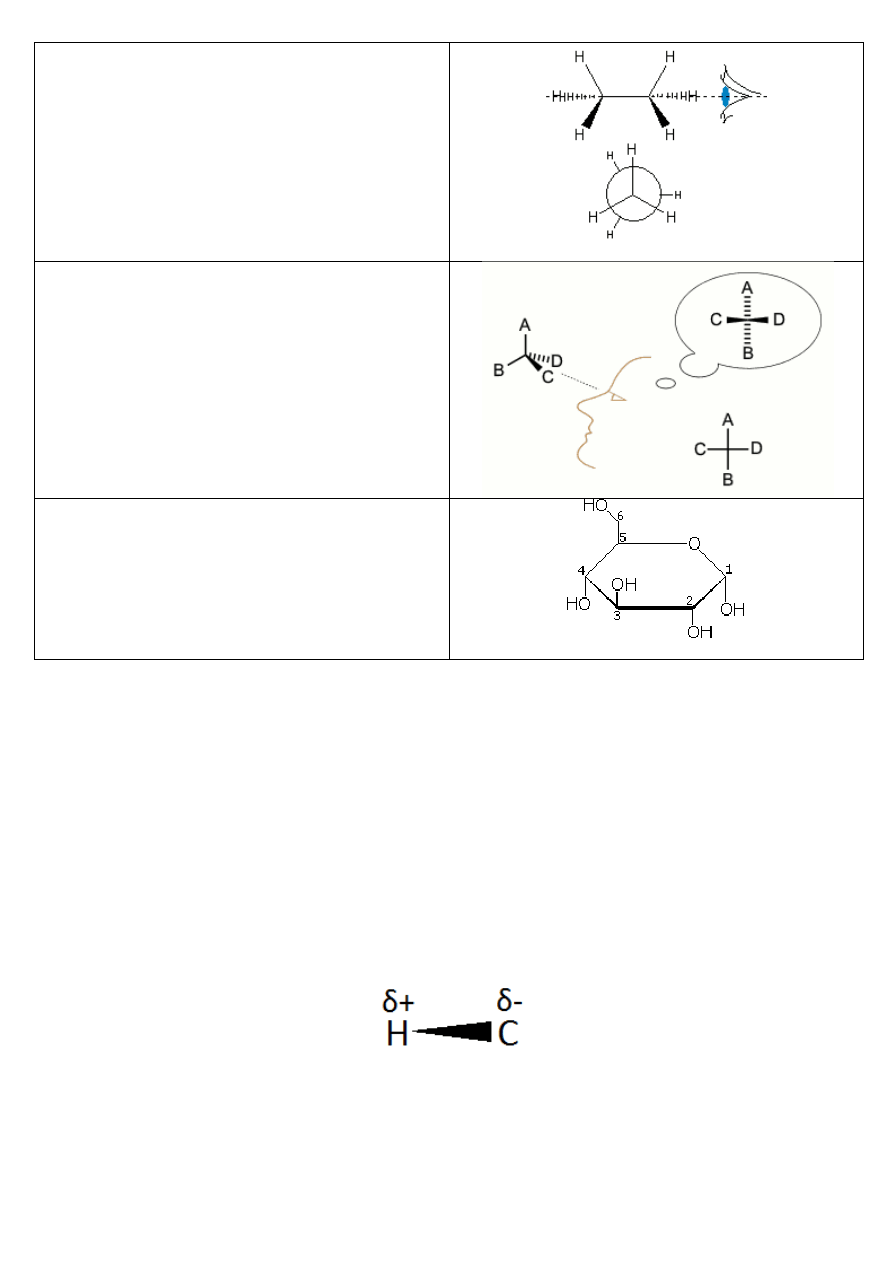
projekcja Newmana
duże koło oznacza tylny atom węgla; przedni atom węgla
to punkt, w którym zbiegają się trzy odcinki z przodu; ze
względu na poprawienie wyrazistości na rysunku i na
podobnych projekcjach Newmana tylną grupę obraca się
lekko, aby nie pokrywała się z przednią, jednak należy
pamiętać, że ma się tu namyśli taką sytuację, w której
właśnie te grupy na siebie zachodzą
(etan)
projekcja Fishera
ten sposób przedstawiania cząsteczek umożliwia
prezentację konfiguracji absolutnej enancjomerów i
diastereoizomerów(izomeria optyczna)
projekcja Hawrotha (*)
stosowana w przypadku prezentacji półacetalowych
(cyklicznych) odmian cukrów
(glukoza)
(źródło - http://www.chemmix.artnet.pl/index.php?s1=02&s2=004&s3=006)
4. Polaryzacja wiązań w związkach organicznych – efekty indukcyjne i mezomeryczne. Podstawniki elektronodonorowe
i elektronoakceptorowe.
Polaryzacja wiązań chemicznych jest związana z różnicą elektroujemności pierwiastków. Spontaniczna polaryzacja wiązań
chemicznych wynika w pierwszym rzędzie z różnej elektroujemności związanych z sobą atomów. W uproszczeniu atom o
większej elektroujemności "ściąga" w swoim kierunku chmurę elektronową tworzącą wiązanie na skutek czego uzyskuje
cząstkowy ładunek ujemny, zaś atom mniej elektroujemny jest "odsłaniany" i uzyskuje cząstkowy ładunek dodatni. W
rezultacie między atomami powstaje elektryczny moment dipolowy, którego wektor jest skierowany wzdłuż wiązania w
kierunku atomu bardziej elektroujemnego. W układzie okresowym, pierwiastki po prawej stronie węgla (w tym samym
okresie) są bardziej elektroujemne. Umownie przyjmuje się, że wiązania pojedyncze pomiędzy atomami, dla których
różnica elektroujemności nie przekracza wartości 1,7, należą jeszcze do kategorii wiązań atomowych spolaryzowanych
(powyżej są to już wiązania jonowe – czasami też oznacza się dolną granicę polaryzacji wiązania, co daje przedział różnicy
elektroujemności od 0,4 do 1,7). W przypadku węgla i wodoru, węgiel ma elektroujemność 2,5, zaś wodór 2,1. Daje to
różnicę elektroujemności 0,4, co oznacza, iż jest to wiązanie kowalencyjne słabo spolaryzowane.
Ładunek dodatni na atomie oznacza się δ+, zaś ujemny δ-.
Polaryzacja często decyduje o kierunku przebiegu reakcji chemicznej (np. reakcja z odczynnikiem Grignarda RMgX
tworzenia ze związku karbonylowego (keton, aldehyd) alkohol):
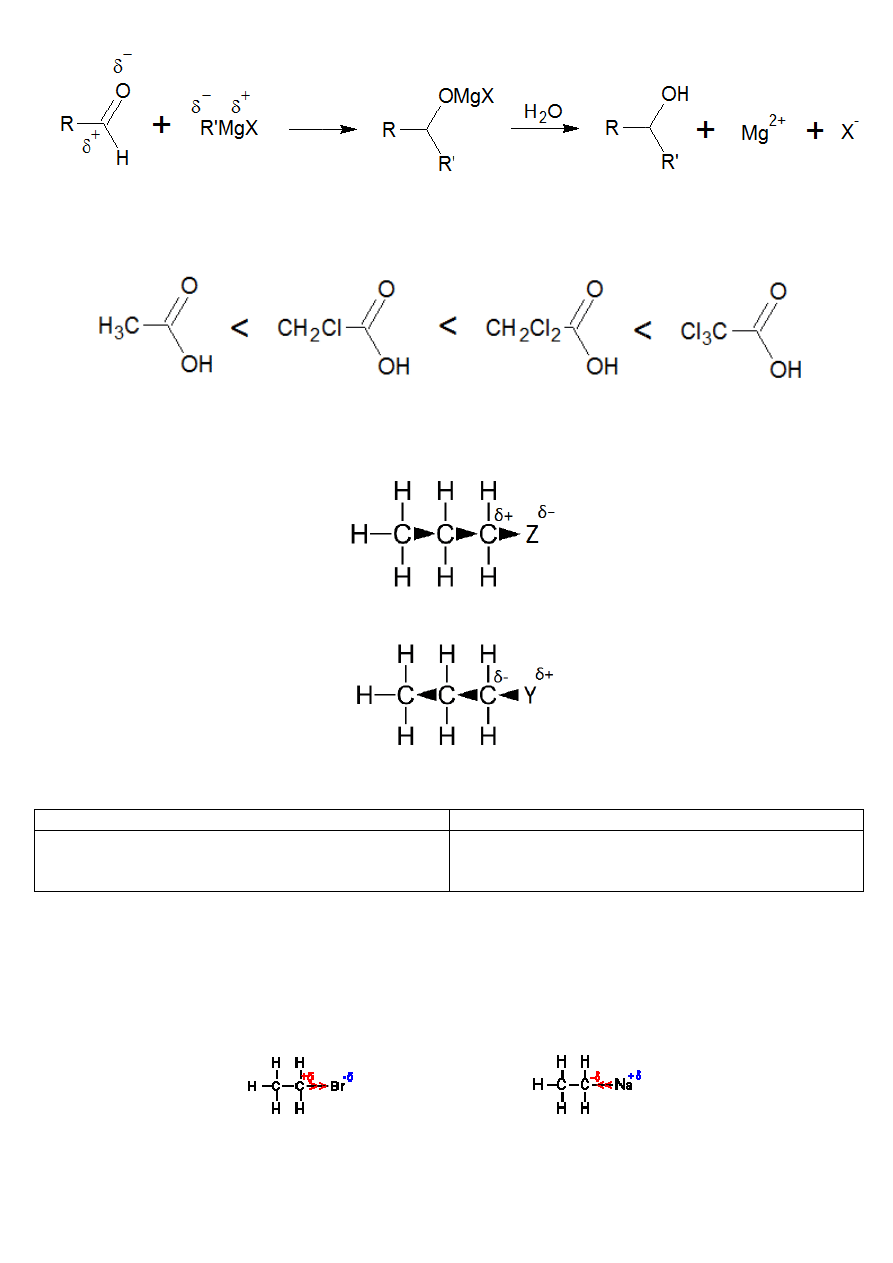
Efekt indukcyjny.
Polaryzacja także wpływa na kwasowość pods
słabiej jest związany wodór w kwasie):
Należy pamiętać, iż wiązanie węgiel-heteroatom wpływa na rozkład
zawierający atom bardziej elektroujemny od węgla będzie powodował następujący efekt (Z
akceptorowy –I):
Natomiast podstawnik mniej elektroujemny od węgla powoduje
Przesunięcie elektronów w kierunku atomu bardziej elektroujemnego to
podstawniki elektrono akceptorowe
fluorowce (F, Cl, Br, I)
grupy -CO, -CHO, -COOH, -COOR, -C(O)Cl, -
-OH, -OR, -CN, -NO
2
, -NH
2
, -NHR, NR
2
Efekt mezomeryczny (zwany także efektem rezonansowym) jest zazwyczaj silniejszy od efektu indukcyjnego. Ma spory
wpływ na reakcje związków organicznych, szczególnie aromatycznych. Ponieważ efekt mezomeryczny sprawia trochę
kłopotów z interpretacją, więc poświęcimy mu nieco w
W każdym przypadku, by uzupełnić równanie reakcji chemicznej należy rozpatrzyć efekty indukcyjne
M podstawników obecnych w substracie, a następnie znaleźć centra aktywne, czyli potencjalne miejsca, które mogą być
atakowane przez odczynnik. Efekt indukcyjny i mezomeryczny rozpatruje się z punktu widzenia grupy, która go wywołuje:
Grupa wywołująca efekt indukcyjny
wyciąga elektrony ze związku, zubaża
go w elektrony, ale sama przez to
uzyskuje cząstkowy ładunek ujemny.
Polaryzacja także wpływa na kwasowość podstawionych kwasów karboksylowych (im więcej przyłączonych chlorów, tym
heteroatom wpływa na rozkład elektronów w całej cząsteczce. Podstawnik
zawierający atom bardziej elektroujemny od węgla będzie powodował następujący efekt (Z
Natomiast podstawnik mniej elektroujemny od węgla powoduje efekt odwrotny (Y – podstawnik elektrono donorowy +I):
Przesunięcie elektronów w kierunku atomu bardziej elektroujemnego to efekt indukcyjny
podstawniki elektrono akceptorowe -I
podstawniki elektrono donorowe +I
-C(O)NH
2
grupy alkilowe C
n
H
2n+1
atomy metali – Mg, Li
(zwany także efektem rezonansowym) jest zazwyczaj silniejszy od efektu indukcyjnego. Ma spory
wpływ na reakcje związków organicznych, szczególnie aromatycznych. Ponieważ efekt mezomeryczny sprawia trochę
kłopotów z interpretacją, więc poświęcimy mu nieco więcej miejsca.
W każdym przypadku, by uzupełnić równanie reakcji chemicznej należy rozpatrzyć efekty indukcyjne
podstawników obecnych w substracie, a następnie znaleźć centra aktywne, czyli potencjalne miejsca, które mogą być
Efekt indukcyjny i mezomeryczny rozpatruje się z punktu widzenia grupy, która go wywołuje:
Grupa wywołująca efekt indukcyjny
wyciąga elektrony ze związku, zubaża
go w elektrony, ale sama przez to
uzyskuje cząstkowy ładunek ujemny.
W tym przypadku grupa wzbogaca związek
w elektrony, sama zaś uzyskuje
ładunek dodatni. Mówimy tu, że atom sodu
wykazuje efekt +I
tawionych kwasów karboksylowych (im więcej przyłączonych chlorów, tym
elektronów w całej cząsteczce. Podstawnik
zawierający atom bardziej elektroujemny od węgla będzie powodował następujący efekt (Z – podstawnik elektrono
podstawnik elektrono donorowy +I):
(I).
podstawniki elektrono donorowe +I
(zwany także efektem rezonansowym) jest zazwyczaj silniejszy od efektu indukcyjnego. Ma spory
wpływ na reakcje związków organicznych, szczególnie aromatycznych. Ponieważ efekt mezomeryczny sprawia trochę
W każdym przypadku, by uzupełnić równanie reakcji chemicznej należy rozpatrzyć efekty indukcyjne I oraz mezomeryczne
podstawników obecnych w substracie, a następnie znaleźć centra aktywne, czyli potencjalne miejsca, które mogą być
Efekt indukcyjny i mezomeryczny rozpatruje się z punktu widzenia grupy, która go wywołuje:
W tym przypadku grupa wzbogaca związek
w elektrony, sama zaś uzyskuje cząstkowy
ładunek dodatni. Mówimy tu, że atom sodu

Mówimy wtedy, że taka grupa
wykazuje efekt -I
Grupa aminowa wzbogaca pierścień
w elektrony, sama uzyskuje ładunek
formalny dodatni. Wykazuje więc
efekt +M. O takiej grupie mówi się, że
jest elektrodonorowa.
Ze względu na powyższe oddziaływania podstawniki dzielimy na:
mezomeryczne dodatnie +M i należą tutaj
obecnością wolnych par elektronowych na centralnym atomie węgla podstawnika. Na przykład tlen w podstawniku
ma dwie wolne pary elektronowe.
podstawniki drugiego rodzaju wykazujące efekt
Wszystkie te podstawniki charakteryzują się obecnością wiązań wielokrotnych pomiędzy centralnym atomem
podstawnika a pozostałymi atomami. Uwzględniając efekty I i M podstawników należy stwierdzić, że odgrywają one
znaczną rolę w przebiegu reakcji jonowych.
Nukleofil – to cząsteczka lub grupa, która, jak sama nazwa wskazuje, "lubi" dodatnio naładowane
Sama posiada nadmiar elektronów i w odpowiednich warunkach jest skłonna się nimi podzielić, czyli być ich donorem
(benzen jest tzw. miękkim nukleofilem - cząsteczka duża, z rozmytym "centrum nukleofilowości"
konkretnego miejsca, które ma nadmiar elektronów).
Elektrofil – to cząsteczka lub grupa która, jak sama nazwa wskazuje, "lubi"
odpowiednich warunkach jest w stanie je przyjąć, czyli być ich akceptorem. Elektrofilami są wszystkie
zgodne z definicją Brønsteda, jak i te zgodne z definicją Lewisa. Oprócz tego mogą to być jednak cząsteczki, które nie
wykazują żadnych kwasowych własności, lecz tylko mają "zwykły" deficyt elektronów
szersze od pojęcia kwasu (elektrofilem twardym jest np. AlCl
"centrum elektrofilowości" – czyli jednym konkretnym miejscu w cząsteczce, które jest szczególnie skłonne przyjmować
elektrony).
Reasumując, dodatni efekt mezomeryczny, wytwa
nukleofilem (chętnie odda elektrony dodatnio naładowanej cząstce
zachodzić łatwiej.
5. Izomeria konstytucyjna (szkieletu, położenia, budowy
związków organicznych. Asymetryczne atomy w
asymetrycznym atomie węgla.
Izomeria konstytucyjna (izomeria strukturalna):
występowanie związków różniących się kolejnością i sposobem połączenia atomów w cząsteczkach
Izomeria szkieletowa:
cząsteczki różnią się budową
szkieletu węglowego
podstawnika, grupy
Mówimy wtedy, że taka grupa
Grupa aminowa wzbogaca pierścień
w elektrony, sama uzyskuje ładunek
formalny dodatni. Wykazuje więc
. O takiej grupie mówi się, że
Grupa karbonylowa zubaża pierścień w
elektrony, dlatego nazywamy ją grupą
elektroakceptorową i mówimy o niej, że
wykazuje efekt -M
Ze względu na powyższe oddziaływania podstawniki dzielimy na:
+M i należą tutaj -R, -OH, -NH
2
, -X. Wszystkie podstawniki
obecnością wolnych par elektronowych na centralnym atomie węgla podstawnika. Na przykład tlen w podstawniku
podstawniki drugiego rodzaju wykazujące efekt mezomeryczny ujemny -M i należą tutaj
Wszystkie te podstawniki charakteryzują się obecnością wiązań wielokrotnych pomiędzy centralnym atomem
podstawnika a pozostałymi atomami. Uwzględniając efekty I i M podstawników należy stwierdzić, że odgrywają one
gu reakcji jonowych.
, która, jak sama nazwa wskazuje, "lubi" dodatnio naładowane
i w odpowiednich warunkach jest skłonna się nimi podzielić, czyli być ich donorem
cząsteczka duża, z rozmytym "centrum nukleofilowości"
konkretnego miejsca, które ma nadmiar elektronów).
która, jak sama nazwa wskazuje, "lubi" elektrony, czyli sama posiada ich niedomiar i w
odpowiednich warunkach jest w stanie je przyjąć, czyli być ich akceptorem. Elektrofilami są wszystkie
cją Brønsteda, jak i te zgodne z definicją Lewisa. Oprócz tego mogą to być jednak cząsteczki, które nie
wykazują żadnych kwasowych własności, lecz tylko mają "zwykły" deficyt elektronów
twardym jest np. AlCl
3
, H
+
- cząsteczka mała, zwarta i o bardzo skoncentrowanym
czyli jednym konkretnym miejscu w cząsteczce, które jest szczególnie skłonne przyjmować
odatni efekt mezomeryczny, wytwarza na pierścieniu benzenowym ujemny ładunek, który
nukleofilem (chętnie odda elektrony dodatnio naładowanej cząstce np. H
+
), a więc reakcja addycji elektrofilowej będzie
5. Izomeria konstytucyjna (szkieletu, położenia, budowy) i przestrzenna (geometryczna, konformacyjna, enancjomeria)
zków organicznych. Asymetryczne atomy węgla: konfiguracja względna (D, L) i absolutna (R, S) podstawników przy
Izomeria konstytucyjna (izomeria strukturalna):
występowanie związków różniących się kolejnością i sposobem połączenia atomów w cząsteczkach
Izomeria podstawienia (położenia):
cząsteczki różnią się położeniem
podstawnika, grupy funkcyjnej lub wiązania
wielokrotnego
cząsteczki posiadają różne grupy
baża pierścień w
elektrony, dlatego nazywamy ją grupą
elektroakceptorową i mówimy o niej, że
Wszystkie podstawniki tej grupy charakteryzują się
obecnością wolnych par elektronowych na centralnym atomie węgla podstawnika. Na przykład tlen w podstawniku -OH
M i należą tutaj -NO
2
, -SO
3
H, COOH.
Wszystkie te podstawniki charakteryzują się obecnością wiązań wielokrotnych pomiędzy centralnym atomem
podstawnika a pozostałymi atomami. Uwzględniając efekty I i M podstawników należy stwierdzić, że odgrywają one
, która, jak sama nazwa wskazuje, "lubi" dodatnio naładowane jądra innych atomów.
i w odpowiednich warunkach jest skłonna się nimi podzielić, czyli być ich donorem
cząsteczka duża, z rozmytym "centrum nukleofilowości" – czyli brakiem jednego
, czyli sama posiada ich niedomiar i w
odpowiednich warunkach jest w stanie je przyjąć, czyli być ich akceptorem. Elektrofilami są wszystkie kwasy, zarówno te
cją Brønsteda, jak i te zgodne z definicją Lewisa. Oprócz tego mogą to być jednak cząsteczki, które nie
wykazują żadnych kwasowych własności, lecz tylko mają "zwykły" deficyt elektronów – pojęcie elektrofila jest więc
cząsteczka mała, zwarta i o bardzo skoncentrowanym
czyli jednym konkretnym miejscu w cząsteczce, które jest szczególnie skłonne przyjmować
iu benzenowym ujemny ładunek, który czyni go
), a więc reakcja addycji elektrofilowej będzie
) i przestrzenna (geometryczna, konformacyjna, enancjomeria)
dna (D, L) i absolutna (R, S) podstawników przy
występowanie związków różniących się kolejnością i sposobem połączenia atomów w cząsteczkach
Izomeria funkcyjna:
cząsteczki posiadają różne grupy
funkcyjne
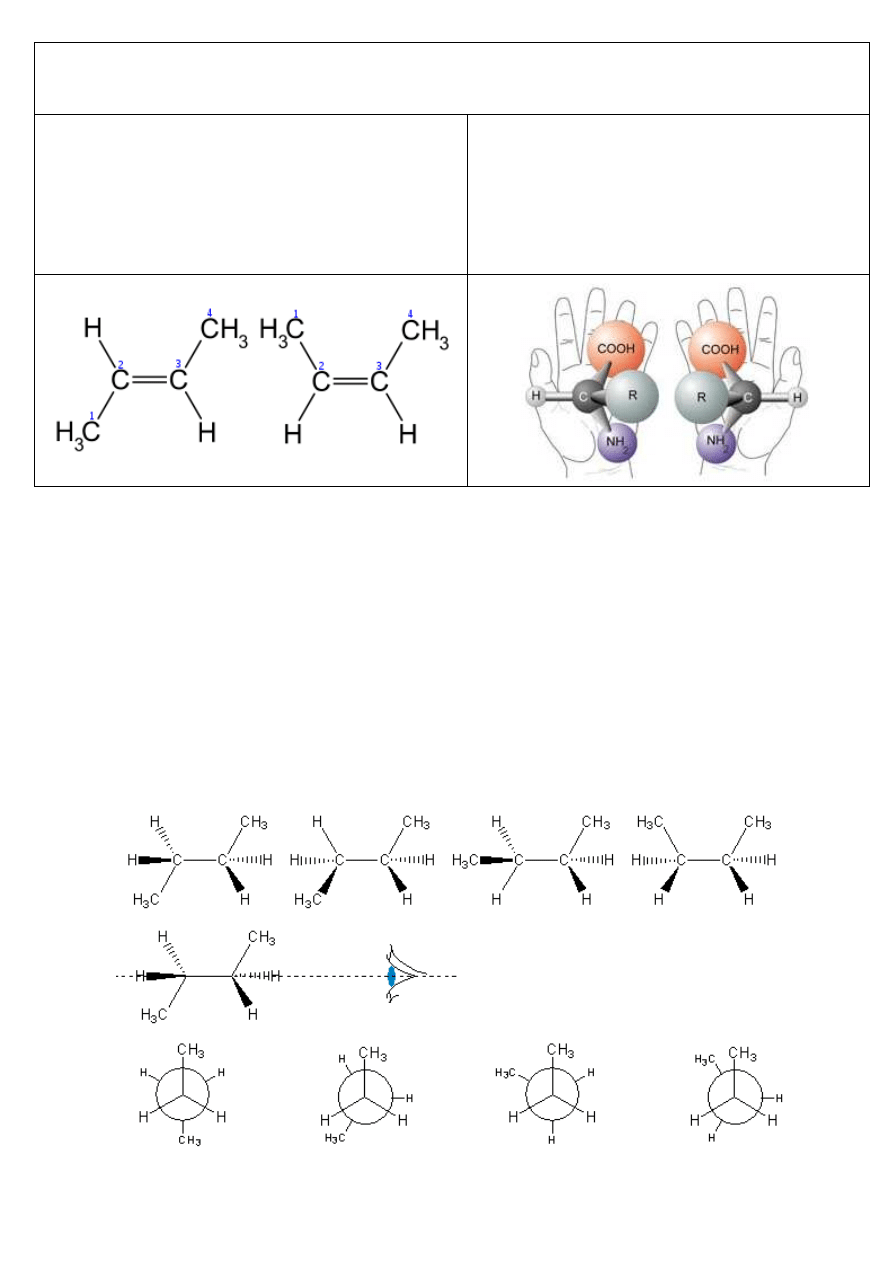
Stereoizomeria (izomeria konfiguracyjna):
występowanie związków o identycznej strukturze(konstytucji),
a różnym rozmieszczeniu atomów w przestrzeni (konfiguracji)
Izomeria geometryczna (cis/trans):
cząsteczki różnią się położeniem podstawników względem
wiązania podwójnego lub płaszczyzny pierścienia węglowego
Izomeria optyczna:
cząsteczki są chiralne, czyli nieidentyczne ze swymi
lustrzanymi odbiciami; warunkiem jest obecność atomu
węgla połączonego z czterema różnymi podstawnikami i
brak płaszczyzny symetrii w cząsteczce
(np. kwas mezowinowy nie spełnia 2 warunku – ma
element symetrii)
Substancja jest czynna optycznie jeżeli skręca płaszczyznę polaryzacji światła.
enancjomery – izomery będące wzajemnymi odbiciami lustrzanymi
chiralność – zachodzi, kiedy przedmioty nie pokrywają się ze swoimi odbiciami lustrzanymi,
nie wykazują żadnych elementów symetrii.
centrum chiralności – atom węgla połączony z 4 różnymi podstawnikami
(ilość chiralnych atomów węgla decyduje o ilości enancjomerów)
diastereoizomery – różnią się konfiguracją przy co najmniej jednym centrum chiralności
epimery – różnią się konfiguracją przy tylko jednym centrum chiralności
forma mezo – dwa enancjomery mające wspólną płaszczyznę symetrii, uznaje się je za jedną odmianę
Izomeria konformacyjna - to rodzaj izomerii cząsteczek chemicznych, polegająca na częściowym zablokowaniu swobodnej
rotacji podstawników lub pojedynczych atomów, występujących przy dwóch atomach połączonych pojedynczym
wiązaniem chemicznym. Izomery konformacyjne często przedstawia się za pomocą tzw. projekcji Newmana.
Izomeria konformacyjna na przykładzie butanu (rozpatrując konformery n-butanu przy tworzeniu projekcji Newmana
spoglądaliśmy wzdłuż środkowego wiązania C−C; przykład dla konformacji antyperiplanarnej (180°)):
Wzory stereostrukturalne i projekcje Newmana konformerów butanu (kolejno: 180°, 120°, 60°, 0°).
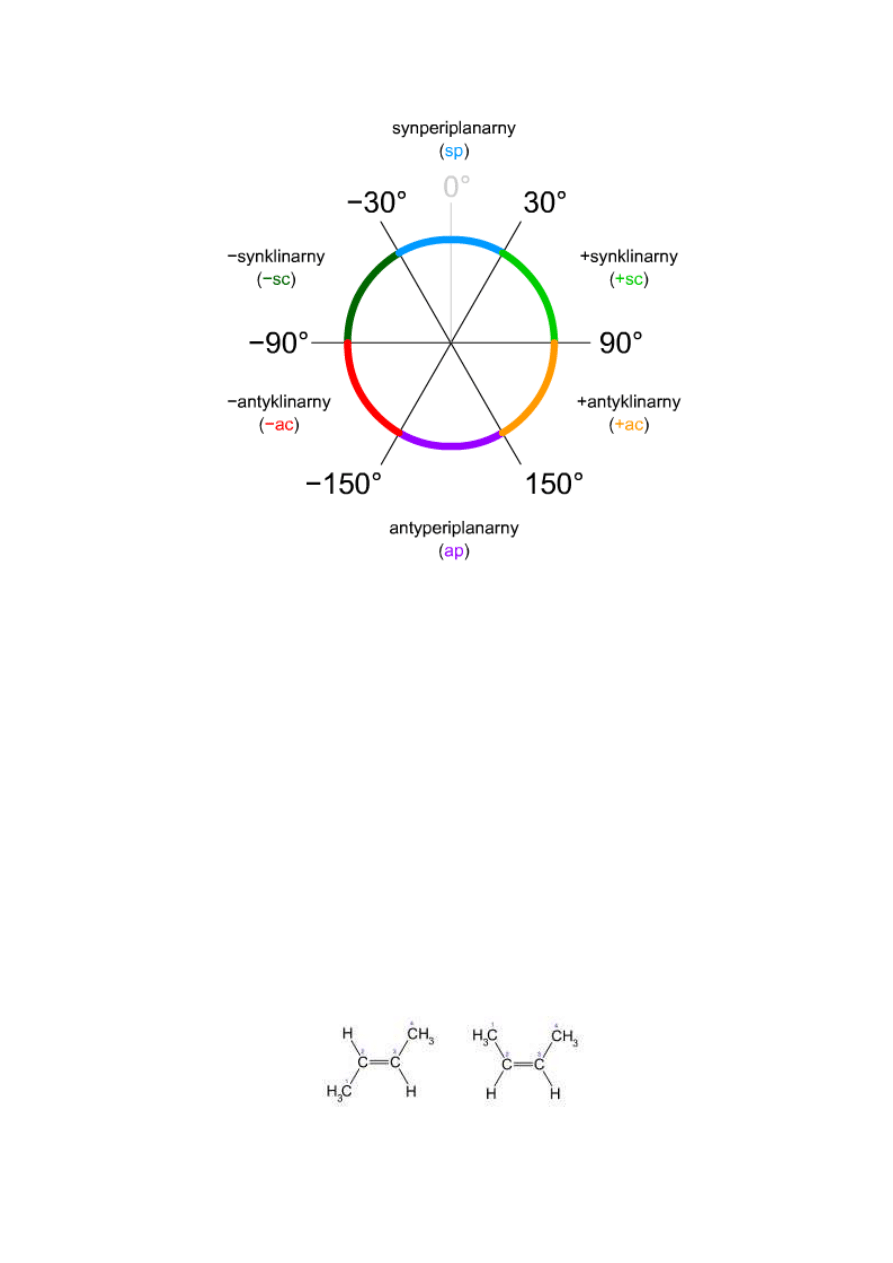
Konformery n-butanu mają swoje nazwy w zależności od kąta jaki zawarty jest pomiędzy grupami metylowymi. 180° to
konformacja antyperiplanarna, 60° synklinarna, 0° synperiplanarna. Dla uproszczenia posługiwać się będziemy jednak
wartościami tych kątów.
Wiemy już że konformacje naprzeciwległe (120° i 0°) są mniej trwałe od naprzemianległych (180° i 60°). Ale czy nawet
mniej trwałe 120° i 0° są równocenne? Okazuje się, że nie. Oprócz naprężenia torsyjnego występuje też inne - naprężenie
steryczne. Powstaje ono kiedy grupy funkcyjne fizycznie sobie przeszkadzają zachodząc na siebie.
Naprężenie steryczne pojawia się w konformacji 60° i 0° oraz pomiędzy nimi a także w dalszej rotacji aż do 60° po drugiej
stronie. Im bliżej konformacji 0° tym naprężenie steryczne jest silniejsze i tym większa energia jest potrzebna do
osiągnięcia takiego kształtu.
Konwencja Z/E – zusammen/entgegen (razem/naprzeciw).
Istnieją dwie, równoległe konwencje nazewnicze tego rodzaju izomerii. Tzw. trans-cis oraz E-Z:
- forma trans ma miejsce gdy dwa identyczne podstawniki znajdują się po przeciwnych stronach względem
płaszczyzny przechodzącej wzdłuż wiązania wielokrotnego lub pierścienia.
- forma cis ma miejsce gdy wcześniej wspomniane podstawniki znajdują się po tej samej stronie płaszczyzny
wiązania podwójnego lub pierścienia.
Jak widać, jeżeli nie ma dwóch identycznych podstawników przy podwójnym wiązaniu (lub pierścieniu), system cis-trans
nie daje możliwości jednoznacznego nazwania konfiguracji, dlatego stworzono system E-Z, opierający się na regułach wagi
podstawników Cahna-Ingolda-Preloga.
- o formie E (niem. entgegen 'naprzeciw') mówi się, gdy dwa podstawniki o większej wadze znajdują się po
przeciwnych stronach względem płaszczyzny przechodzącej wzdłuż wiązania wielokrotnego lub pierścienia.
- o formie Z (niem. zusammen 'razem') mówi się, gdy wcześniej wspomniane podstawniki znajdują się po tej samej
stronie płaszczyzny wiązania podwójnego lub pierścienia.
Po lewej mamy (E)-But-2-en, a po prawej (Z)-But-2-en.
Tutaj można znaleźć trochę informacji na temat izomerii E/Z:
http://www.acdlabs.com/iupac/nomenclature/93/r93_626.htm
6. Rodzaje reakcji organicznych i reagenty bior
kowalencyjnego.
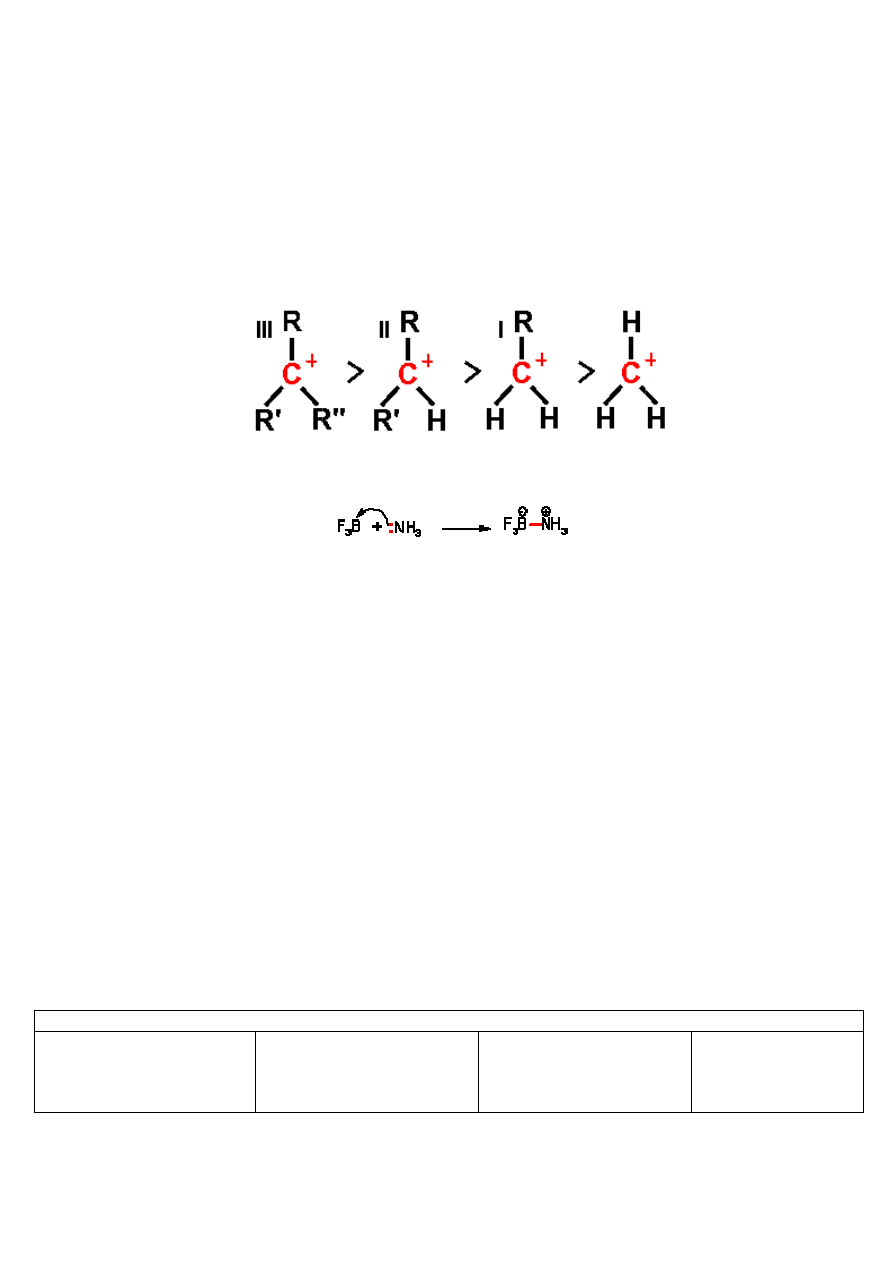
Na początek zaczniemy od wprowadzenia pojęcia karbokationu:
Karbokation to jon, a dokładnie kation, w którym elektryczny ładunek dodatni jest zlokalizowany na jednym lub więcej
atomach węgla. Dodatnio naładowany atom węgla posiada 6.
reaguje z cząsteczkami, które mogą mu dostarczyć dwóch elektr
posiadający znaczny deficyt elektronów, jest silnym
i łatwo reaguje z wszelkimi zasadami, np. jonem
istnieje tylko jako produkt przejściowy wielu
mechanizmu SN1. Szczególnie nietrwałe i reaktywne są karbokationy
ich rzędowości, tzn. od tego, czy ładunek dodatni znajduje się na atomie węgla, do którego jest przyłączony jeden inny
atom węgla (pierwszorzędowy), dwa takie atomy (drugorzędowy), czy trzy takie atomy (trzeciorzędowy):
A teraz wracamy do tematu:
Pomijając nieliczne przypadki reakcji kwasu Lewisa z zasadą Lewisa, w których tworzy się nowe wiązanie koordynacyjne,
to w większości przemian chemicznych musi nastąpić rozpad jednych wiązań na rzecz powstawania drugich. W przypadku
gdy dwa atomy związane wiązaniem σ nie r
wiążąca para elektronów ulega podziałow
pojedynczy, niesparowany elektron. Takie rozerwanie wiązania nazywane jest rozpadem homolitycznym lub rodnikowym.
Jeżeli jednak atomy różnią się między sobą elektroujemnością, to p
cząsteczka rozpada się na dwa jony: dodatni i ujemny. Mamy tu do czynienia z rozerwaniem wiązania, które nazywane
jest rozpadem heterolitycznym lub jonowym. Heterolityczny rozpad wiązań niekoniecznie musi powod
się karbokationów lub karboanionów istniejących jako niezależne elementy. Najczęściej nowe wiązanie tworzy się
równocześnie z rozpadem wiązania starego i w żadnym momencie reakcji, na atomie węgla nie wytwarza się deficyt
elektronów. Tym niemniej jednak, jon odchodzący zabiera ze sobą parę elektronów wiążących, lub zostawia je przy
atomie węgla.
Istnienie znacznych różnic między reakcjami jonowymi i rodnikowymi powodują różne właściwości jonów organicznych i
wolnych rodników, oraz odmienne warunki homolitycznego i heterolitycznego rozpadu wiązań.
sprzyja wysoka temperatura, zachodzą one łatwo w fazie gazowej oraz w rozpuszczalnikach niepolarnych. Nie
są
wrażliwe na katalityczny wpływ kwasów lub zasad, ulegają natomiast
reagujących z wolnymi rodnikami. Natomiast reakcjom jonowym sprzyjają rozpuszczalniki polarne, zachodzą zwykle w
fazie ciekłej, lub na powierzchni ciał stałych, czyli polarnych katalizatorów. Reakcje jonowe są wrażliwe n
wpływ kwasów i zasad, natomiast niewrażliwe na działanie inhibitorów reagującymi z wolnymi rodnikami. Należy jednak
zaznaczyć, że nie istnieją reakcje pośrednie pomiędzy reakcjami rodnikowymi a jonowymi. W zależności od sposobu
rozerwania wiązań uzyskujemy cząsteczki o charakterze rodnika lub elektrofila i nukleofila.
substytucja
W zależności od charakteru odczynnika atakującego (elektrofil, nukleofil, rodnik), każdy z tych trzech rodzajów reakcji
może należeć do jednej z trzech podstawowych kategorii czyli, homolitycznej (rodnikowej) i dwóch heterolitycznych
(elektrofilowej i nukleofilowej). W sumie otrzymujemy dziewięć typów możliwych reakcji, które zostały
usystematyzowane przez Ingolda. Typy reakcji organicznych ze względu na rodzaj odczynnika:
6. Rodzaje reakcji organicznych i reagenty biorące w nich udział. Homolityczny i heterolityczny rozpad wi
zaczniemy od wprowadzenia pojęcia karbokationu:
, w którym elektryczny ładunek dodatni jest zlokalizowany na jednym lub więcej
. Dodatnio naładowany atom węgla posiada 6. elektronów walencyjnych
, które mogą mu dostarczyć dwóch elektronów, zgodnie z tzw. regułą oktetu
posiadający znaczny deficyt elektronów, jest silnym elektrofilem i kwasem Lewisa. Jest więc podatny na atak
, np. jonem OH- czy jonem Cl-. Większość karbokationów jest bardzo nietrwała i
istnieje tylko jako produkt przejściowy wielu reakcji chemicznych, np. podstawienia nukleofilowego
Szczególnie nietrwałe i reaktywne są karbokationy alkilowe. Ich trwałość zależna jest jednak silnie od
ich rzędowości, tzn. od tego, czy ładunek dodatni znajduje się na atomie węgla, do którego jest przyłączony jeden inny
ęgla (pierwszorzędowy), dwa takie atomy (drugorzędowy), czy trzy takie atomy (trzeciorzędowy):
Pomijając nieliczne przypadki reakcji kwasu Lewisa z zasadą Lewisa, w których tworzy się nowe wiązanie koordynacyjne,
ości przemian chemicznych musi nastąpić rozpad jednych wiązań na rzecz powstawania drugich. W przypadku
σ nie różnią się zbytnio elektroujemnością to podczas rozerwania tego wiązania,
wiążąca para elektronów ulega podziałowi a więc powstają wolne rodniki, czyli atomy lub grupy atomów, posiadające
jedynczy, niesparowany elektron. Takie rozerwanie wiązania nazywane jest rozpadem homolitycznym lub rodnikowym.
Jeżeli jednak atomy różnią się między sobą elektroujemnością, to para elektronów zostaje przy jednym z atomów a
cząsteczka rozpada się na dwa jony: dodatni i ujemny. Mamy tu do czynienia z rozerwaniem wiązania, które nazywane
jest rozpadem heterolitycznym lub jonowym. Heterolityczny rozpad wiązań niekoniecznie musi powod
się karbokationów lub karboanionów istniejących jako niezależne elementy. Najczęściej nowe wiązanie tworzy się
równocześnie z rozpadem wiązania starego i w żadnym momencie reakcji, na atomie węgla nie wytwarza się deficyt
emniej jednak, jon odchodzący zabiera ze sobą parę elektronów wiążących, lub zostawia je przy
Istnienie znacznych różnic między reakcjami jonowymi i rodnikowymi powodują różne właściwości jonów organicznych i
warunki homolitycznego i heterolitycznego rozpadu wiązań.
sprzyja wysoka temperatura, zachodzą one łatwo w fazie gazowej oraz w rozpuszczalnikach niepolarnych. Nie
wrażliwe na katalityczny wpływ kwasów lub zasad, ulegają natomiast zahamowaniu pod wpływem inhibitorów
reagujących z wolnymi rodnikami. Natomiast reakcjom jonowym sprzyjają rozpuszczalniki polarne, zachodzą zwykle w
fazie ciekłej, lub na powierzchni ciał stałych, czyli polarnych katalizatorów. Reakcje jonowe są wrażliwe n
wpływ kwasów i zasad, natomiast niewrażliwe na działanie inhibitorów reagującymi z wolnymi rodnikami. Należy jednak
zaznaczyć, że nie istnieją reakcje pośrednie pomiędzy reakcjami rodnikowymi a jonowymi. W zależności od sposobu
ązań uzyskujemy cząsteczki o charakterze rodnika lub elektrofila i nukleofila.
REAKCJE ORGANICZNE
addycja
eliminacja
W zależności od charakteru odczynnika atakującego (elektrofil, nukleofil, rodnik), każdy z tych trzech rodzajów reakcji
może należeć do jednej z trzech podstawowych kategorii czyli, homolitycznej (rodnikowej) i dwóch heterolitycznych
eofilowej). W sumie otrzymujemy dziewięć typów możliwych reakcji, które zostały
Typy reakcji organicznych ze względu na rodzaj odczynnika:
ce w nich udział. Homolityczny i heterolityczny rozpad wiązania
, w którym elektryczny ładunek dodatni jest zlokalizowany na jednym lub więcej
elektronów walencyjnych i dlatego nadzwyczaj chętnie
regułą oktetu. Karbokation, jako
. Jest więc podatny na atak nukleofilowy
arbokationów jest bardzo nietrwała i
podstawienia nukleofilowego zachodzącego wg
. Ich trwałość zależna jest jednak silnie od
ich rzędowości, tzn. od tego, czy ładunek dodatni znajduje się na atomie węgla, do którego jest przyłączony jeden inny
ęgla (pierwszorzędowy), dwa takie atomy (drugorzędowy), czy trzy takie atomy (trzeciorzędowy):
Pomijając nieliczne przypadki reakcji kwasu Lewisa z zasadą Lewisa, w których tworzy się nowe wiązanie koordynacyjne,
ości przemian chemicznych musi nastąpić rozpad jednych wiązań na rzecz powstawania drugich. W przypadku
óżnią się zbytnio elektroujemnością to podczas rozerwania tego wiązania,
i a więc powstają wolne rodniki, czyli atomy lub grupy atomów, posiadające
jedynczy, niesparowany elektron. Takie rozerwanie wiązania nazywane jest rozpadem homolitycznym lub rodnikowym.
ara elektronów zostaje przy jednym z atomów a
cząsteczka rozpada się na dwa jony: dodatni i ujemny. Mamy tu do czynienia z rozerwaniem wiązania, które nazywane
jest rozpadem heterolitycznym lub jonowym. Heterolityczny rozpad wiązań niekoniecznie musi powodować utworzenie
się karbokationów lub karboanionów istniejących jako niezależne elementy. Najczęściej nowe wiązanie tworzy się
równocześnie z rozpadem wiązania starego i w żadnym momencie reakcji, na atomie węgla nie wytwarza się deficyt
emniej jednak, jon odchodzący zabiera ze sobą parę elektronów wiążących, lub zostawia je przy
Istnienie znacznych różnic między reakcjami jonowymi i rodnikowymi powodują różne właściwości jonów organicznych i
warunki homolitycznego i heterolitycznego rozpadu wiązań.
Reakcjom rodnikowym
sprzyja wysoka temperatura, zachodzą one łatwo w fazie gazowej oraz w rozpuszczalnikach niepolarnych. Nie
zahamowaniu pod wpływem inhibitorów
reagujących z wolnymi rodnikami. Natomiast reakcjom jonowym sprzyjają rozpuszczalniki polarne, zachodzą zwykle w
fazie ciekłej, lub na powierzchni ciał stałych, czyli polarnych katalizatorów. Reakcje jonowe są wrażliwe na katalityczny
wpływ kwasów i zasad, natomiast niewrażliwe na działanie inhibitorów reagującymi z wolnymi rodnikami. Należy jednak
zaznaczyć, że nie istnieją reakcje pośrednie pomiędzy reakcjami rodnikowymi a jonowymi. W zależności od sposobu
ązań uzyskujemy cząsteczki o charakterze rodnika lub elektrofila i nukleofila.
przegrupowanie
(reorganizacja wiązań
w cząsteczce pod
wpływem kwasu )
W zależności od charakteru odczynnika atakującego (elektrofil, nukleofil, rodnik), każdy z tych trzech rodzajów reakcji
może należeć do jednej z trzech podstawowych kategorii czyli, homolitycznej (rodnikowej) i dwóch heterolitycznych
eofilowej). W sumie otrzymujemy dziewięć typów możliwych reakcji, które zostały
Typy reakcji organicznych ze względu na rodzaj odczynnika:
Rodzaj reakcji
Addycja
Substytucja
Eliminacja
Substytucja nukleofilowa – S
N
:
Zachodzi w wyniku ataku czynnika nukleofilowego na elektronowe centrum związku chemicznego. Czynnikami
nukleofilowymi są cząsteczki lub jony odznaczające
elektronów (NH
3
), cząsteczki z niepolarnym wiązaniem
pierwszego lub drugiego rzędu. Odpowiednio wtedy dla reakcji pierwszego rzędu (szybkość reakcji zależy tylko od stężenia
jednego substratu), mechanizm reakcji określa się symbolem
reakcji zależy od stężenia dwóch substratów) mechan
Typ reakcji
S
N
1
Ilość
etapów
Reakcja 2-etapowa.
1 etapem jest wytworzenie karbokationu,
który w drugim etapie jest atakowany przez
nukleofil
Rząd
reakcji
Reakcja I rzędu, jej szybkość zależy wyłącznie
od stężenia R-X w środowisku reakcji.
Czynność
optyczna
Z czynnego optycznie substratu powstaje
nieczynna optycznie mieszanina racemiczna
Co jej
ulega?
Najłatwiej ulegają jej III rzędowe halogenki
alkilowe
Substytucja elektrofilowa – S
E
:
Cząsteczka elektrofilowa atakuje związek nukleofilowy. Takimi substancjami elektrofilowymi są cząsteczki albo jony
odznaczające się niedoborem elektronów
reakcji substytucji elektrofilowej są halogenowanie, nitrowanie, sulfonowanie, alkilowanie.
Substytucja wolnorodnikowa – S
R
:
Reakcja przebiegająca z udziałem wolnych rodników, które powstają stale w wyniku reakcji łańcuchowej.
Addycja nukleofilowa - A
N
:
Reakcja przyłączania, która przebiega w wyniku ataku czynnika nuklefilowego na elektronowe centrum związku
chemicznego.
Charakter odczynnika
elektrofil
nukleofil
rodnik
A
E
A
N
A
R
S
E
S
N
S
R
E
E
E
N
E
R
achodzi w wyniku ataku czynnika nukleofilowego na elektronowe centrum związku chemicznego. Czynnikami
odznaczające się nadmiarem elektronów (OH
-
, Cl
), cząsteczki z niepolarnym wiązaniem
π
- eten, benzen). Sama reakcja może przebiegać według kinetyki
Odpowiednio wtedy dla reakcji pierwszego rzędu (szybkość reakcji zależy tylko od stężenia
jednego substratu), mechanizm reakcji określa się symbolem S
N
1 i odpowiednio dla reakcji drugiego rzędu (szybkość
reakcji zależy od stężenia dwóch substratów) mechanizm reakcji określa się symbolem S
N
2.
etapowa.
1 etapem jest wytworzenie karbokationu,
który w drugim etapie jest atakowany przez
Reakcja 1-etapowa. W jednym etapie do cząsteczki
następuje zbliżenie się nukleofila, który się podstawia i
odejście nukleofila odchodzącego. Tworzy się pośredni etap,
w którym powstaje częściowe wiązanie z nukleofilem
atakującym a wiązanie z nukleofilem odchodzącym zanika.
Nukleofil atakujący zawsze atakuje ze strony przec
nukleofila odchodzącego. Warunkuje to najczęściej zmianę
konfiguracji formalnej atomu, przy którym następuje
podstawienie (substytucja).
Reakcja I rzędu, jej szybkość zależy wyłącznie
X w środowisku reakcji.
Reakcja II rzędu, jej szybkość zależy od stężenia R
środowisku reakcji
Z czynnego optycznie substratu powstaje
nieczynna optycznie mieszanina racemiczna
Z czynnego optycznie substratu powstaje czynny optycznie
produkt
Najłatwiej ulegają jej III rzędowe halogenki
Najłatwiej ulegają jej I rzędowe halogenki alkilowe
Cząsteczka elektrofilowa atakuje związek nukleofilowy. Takimi substancjami elektrofilowymi są cząsteczki albo jony
niedoborem elektronów (H
+
, AlCl
3
, biegun dodatni wiązań atomowych spolaryzowanych). Przykładami
elektrofilowej są halogenowanie, nitrowanie, sulfonowanie, alkilowanie.
Reakcja przebiegająca z udziałem wolnych rodników, które powstają stale w wyniku reakcji łańcuchowej.
zania, która przebiega w wyniku ataku czynnika nuklefilowego na elektronowe centrum związku
rodnik
achodzi w wyniku ataku czynnika nukleofilowego na elektronowe centrum związku chemicznego. Czynnikami
, Cl
-
, cząsteczki z wolnymi parami
eten, benzen). Sama reakcja może przebiegać według kinetyki
Odpowiednio wtedy dla reakcji pierwszego rzędu (szybkość reakcji zależy tylko od stężenia
i odpowiednio dla reakcji drugiego rzędu (szybkość
.
S
N
2
etapowa. W jednym etapie do cząsteczki
się nukleofila, który się podstawia i
odejście nukleofila odchodzącego. Tworzy się pośredni etap,
w którym powstaje częściowe wiązanie z nukleofilem
atakującym a wiązanie z nukleofilem odchodzącym zanika.
Nukleofil atakujący zawsze atakuje ze strony przeciwnej do
nukleofila odchodzącego. Warunkuje to najczęściej zmianę
konfiguracji formalnej atomu, przy którym następuje
podstawienie (substytucja).
rzędu, jej szybkość zależy od stężenia R-X i Nu w
środowisku reakcji
Z czynnego optycznie substratu powstaje czynny optycznie
produkt
Najłatwiej ulegają jej I rzędowe halogenki alkilowe
Cząsteczka elektrofilowa atakuje związek nukleofilowy. Takimi substancjami elektrofilowymi są cząsteczki albo jony
, biegun dodatni wiązań atomowych spolaryzowanych). Przykładami
elektrofilowej są halogenowanie, nitrowanie, sulfonowanie, alkilowanie.
Reakcja przebiegająca z udziałem wolnych rodników, które powstają stale w wyniku reakcji łańcuchowej.
zania, która przebiega w wyniku ataku czynnika nuklefilowego na elektronowe centrum związku
Addycja elektrofilowa - A
E
:
Reakcja przyłączenia, która przebiega w wyniku ataku czynnika elektrofilowego na cząsteczkę nukleofilową.
Addycja rodnikowa - A
R
:
Reakcja przyłączenia przebiegająca w postaci łańcuchowej z udziałem wolnych rodników.
Eliminacja:
Jest to reakcja chemiczna, w której od jednej cząsteczki substratu oddzielają się dwa atomy albo grupy atomów i nie są
zastępowane innymi. Otrzymane produkty zwierają wiązania wielokrotne. Przykładami reakcji eliminacji są reakcje
odwodornienia i odwodnienia. Reakcje eliminacji mogą przebiegać zgodnie z kinetyką pierwszego rzędu
drugiego rzędu E2.
7. Alkany: budowa (w tym: rzędowo
(podstawienie wolnorodnikowe, spalanie). Łatwo
selektywność halogenowania alkanów.
Budowa:
Nazwa
Budowa cząsteczki
A
li
fa
t
y
cz
n
e
N
a
sy
c
o
n
e
Alkany
łańcuchowa
(prosta lub rozgałęziona)
Rzędowość atomów węgla:
Rzędowość to po prostu ilość wiązań węgla z innymi węglami. Maksymalna rzędowość to IV, minimalna to 0. Np.
cząsteczka butanu składa się z dwóch pierwszorzędowych węgli i dwóch drugorzędowych.
rzędowości jest - liczba określająca ile atomów
organicznego. Rzędowość tradycyjnie oznacza się l
Reakcja przyłączenia, która przebiega w wyniku ataku czynnika elektrofilowego na cząsteczkę nukleofilową.
Reakcja przyłączenia przebiegająca w postaci łańcuchowej z udziałem wolnych rodników.
Jest to reakcja chemiczna, w której od jednej cząsteczki substratu oddzielają się dwa atomy albo grupy atomów i nie są
innymi. Otrzymane produkty zwierają wiązania wielokrotne. Przykładami reakcji eliminacji są reakcje
odwodornienia i odwodnienia. Reakcje eliminacji mogą przebiegać zgodnie z kinetyką pierwszego rzędu
Eliminacja elektrofilowa E
E
Eliminacja nukeofilowa E
N
Eliminacja rodnikowa E
R
dowość atomów węgla), nazewnictwo, izomeria, charakterystyczne reakcje
(podstawienie wolnorodnikowe, spalanie). Łatwość tworzenia wolnych rodników alkilowych.
Budowa cząsteczki
Rodzaje
wiązań C-C
łańcuchowa
(prosta lub rozgałęziona)
tylko pojedyncze (σ)
Rzędowość to po prostu ilość wiązań węgla z innymi węglami. Maksymalna rzędowość to IV, minimalna to 0. Np.
cząsteczka butanu składa się z dwóch pierwszorzędowych węgli i dwóch drugorzędowych.
atomów węgla o hybrydyzacji sp³ jest przyłączona do określonego atomu
. Rzędowość tradycyjnie oznacza się liczbami rzymskimi.
Reakcja przyłączenia, która przebiega w wyniku ataku czynnika elektrofilowego na cząsteczkę nukleofilową.
Jest to reakcja chemiczna, w której od jednej cząsteczki substratu oddzielają się dwa atomy albo grupy atomów i nie są
innymi. Otrzymane produkty zwierają wiązania wielokrotne. Przykładami reakcji eliminacji są reakcje
odwodornienia i odwodnienia. Reakcje eliminacji mogą przebiegać zgodnie z kinetyką pierwszego rzędu E1 lub z kinetyką
gla), nazewnictwo, izomeria, charakterystyczne reakcje
tworzenia wolnych rodników alkilowych. Mechanizm i
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
tylko sp
3
C
n
H
2n+2
Rzędowość to po prostu ilość wiązań węgla z innymi węglami. Maksymalna rzędowość to IV, minimalna to 0. Np.
cząsteczka butanu składa się z dwóch pierwszorzędowych węgli i dwóch drugorzędowych. Bardziej precyzyjną definicją
sp³ jest przyłączona do określonego atomu związku
Nazewnictwo – metan, etan, propan, butan, pentan, heksan, heptan, oktan, nonan, dekan, undekan, dodekan, tridekan,
teradekan, pentadekan
Izomeria - szkieletowa, położenia, podstawnika oraz dla niektórych związków tej grupy możliwe jest występowanie
izomerii optycznej
Charakterystyczne reakcje:
SUBSTYTUCJA RODNIKOWA – S
R
:
INICJACJA
w pierwszym etapie chlor, najczęściej pod wpływem
promieniowania UV, ulega rozpadowi na wolne rodniki,
inicjujące łańcuch reakcji.
PROPAGACJA
wolne rodniki chloru reagują z metanem, tworząc
rodnik metylowy, który następnie w reakcji z wolnym
chlorem tworzy produkt (jeden z wielu) chlorowania i
kolejny wolny rodnik zdolny do dalszych reakcji;
chlorowanie przebiega do mono- i wielopodstawionych
chloropochodnych metanu
CH
3
Cl, CH
2
Cl
2
, CHCl
3
oraz CCl
4
.
ZAKOŃCZENIE ŁAŃCUCHA REAKCJI
może zajść na wiele sposobów. Najczęściej jest to
spowodowane rekombinacją się dwóch wolnych
rodników, która prowadzi do powstania nie
reaktywnych cząsteczek
Selektywność halogenowania:
Reaktywność wiązań C-H zależy od rzędowości atomu węgla, z którym związany jest atom wodoru. Najłatwiej substytucji
wolnorodnikowej ulegają alkany posiadające atomy wodoru przy III rzędowych atomach węgla, potem przy II, a na końcu
przy I rzędowych. Fluorowce charakteryzują się różną reaktywnością, która w reakcji substytucji rodnikowej alkanów
maleje w kolejności F > Cl > Br > I. Fluor reaguje z alkanami wybuchowo, a jod jest bierny chemicznie, co można
wytłumaczyć dużą energią wiązań I-I. W reakcji chlorowania wyższych alkanów powstaje mieszanina izomerycznych
monochloropodstawionych. Biorąc pod uwagę względy statystyczne (częstość zderzeń) jak i względne szybkości reakcji
substytucji atomu wodoru do chlorowania propanu wydajność 1-chloropropanu wynosi ok. 45%, a dla 2-chloropropanu ok.
55%. Jeżeli o wyniku reakcji decyduje względna reaktywność poszczególnych pozycji w cząstecze substratu, to mówi się o
selektywności danej reakcji. Oznacza to, że reakcja chlorowania nie jest reakcją regioselektywną. Wolne rodniki alkilowe
oraz rodniki cząsteczki chloru powstają w wyniku rozpadu termicznego (ok. 400
o
C) lub podczas fotolizy – reakcja
zachodząca pod wpływem dal o długości 250 nm. Reakcja monobromowania na przykładzie propanu jest natomiast
selektywna (1-bromopropan – 1%; 2-bromopropan – 99%). Brom jest odczynnikiem o dużej selektywności.
SPALANIE:
spalanie całkowite – produkty spalania to dwutlenek węgla i para wodna (woda); na przykładzie butanu:
2 C
4
H
10
+ 13 O
2
→ 8 CO
2
+ 10 H
2
O
półspalanie – produkty spalania to tlenek węgla i para wodna (woda); na przykładzie etanu:
2 C
2
H
6
+ 5 O
2
→ 4 CO + 6 H
2
O
spalanie niecałkowite – produkty spalania to para wodna (woda), sadza (węgiel); na przykładzie metanu:
CH
4
+ O
2
→ C + 2 H
2
O
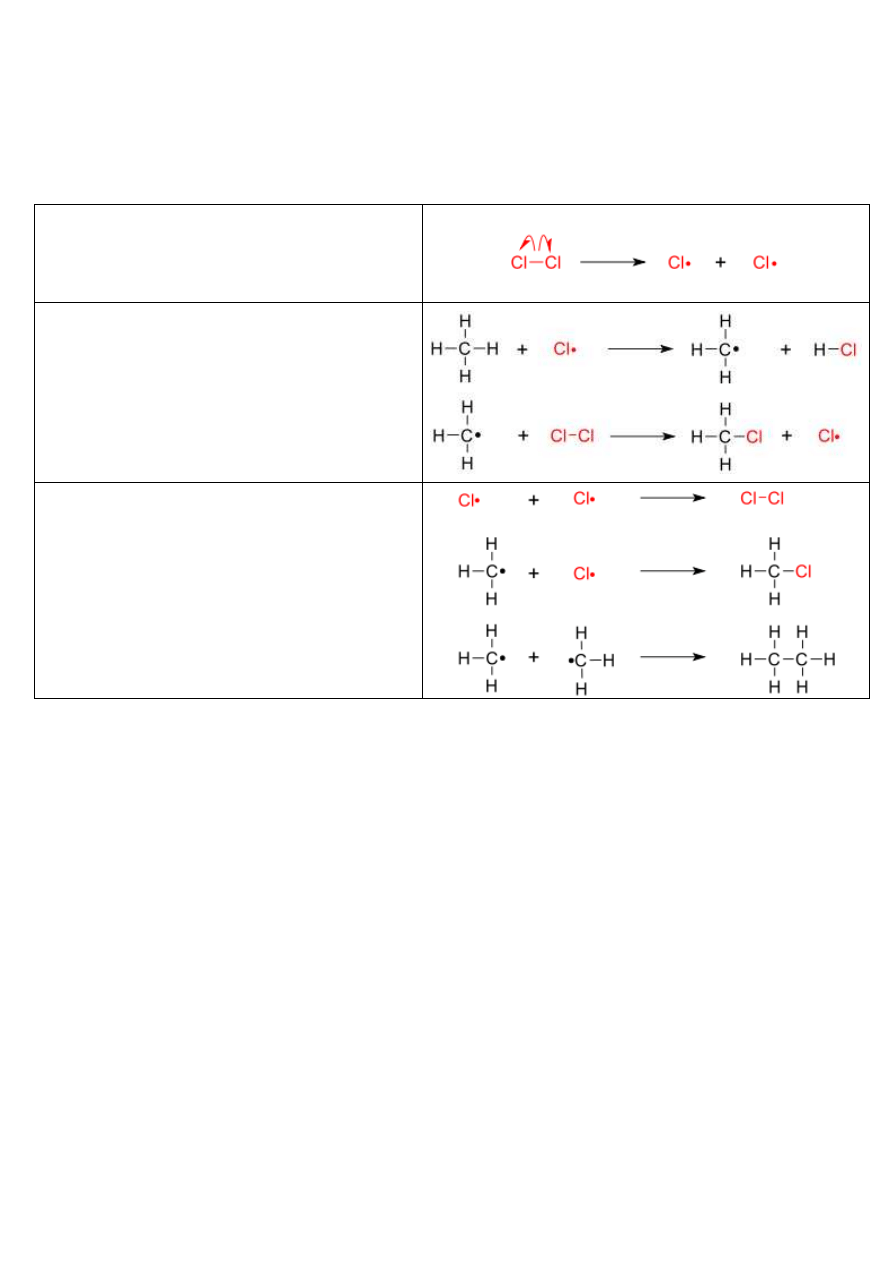
8. Cykloalkany: budowa (w tym: konformacje cykloheksanu), trwałość, nazewnictwo, charakterystyczne reakcje
(podstawienie wolnorodnikowe, reakcje połączone z rozpadem pierścieni 3-, 4-członowych).
Budowa:
Nazwa
Budowa cząsteczki
Rodzaje
wiązań C-C
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
A
li
fa
t
y
cz
n
e
N
a
sy
c
o
n
e
Cykloalkany
cykliczna
(z podstawnikami)
tylko pojedyncze (σ)
tylko sp
3
C
n
H
2n
Trwałość – cykloalkany są stosunkowo trwałe, z wyjątkiem cyklopropanu i cyklobutanu, które ze względu na
silne naprężenia kątowe wiązań chemicznych węgiel-węgiel łatwo ulegają rozkładowi z wytworzeniem
odpowiednich rodników.
konformery cykloheksanu
1 – krzesłowa
2 – pół-krzesło
3 – skręcona łódka
4 – łódka
5 – skręcona łódka
W pierścieniu łódkowym występują większe naprężenia, dlatego forma krzesłowa jest trwalsza i właśnie taką praktycznie
zawsze się rysuje (np. w cukrach).
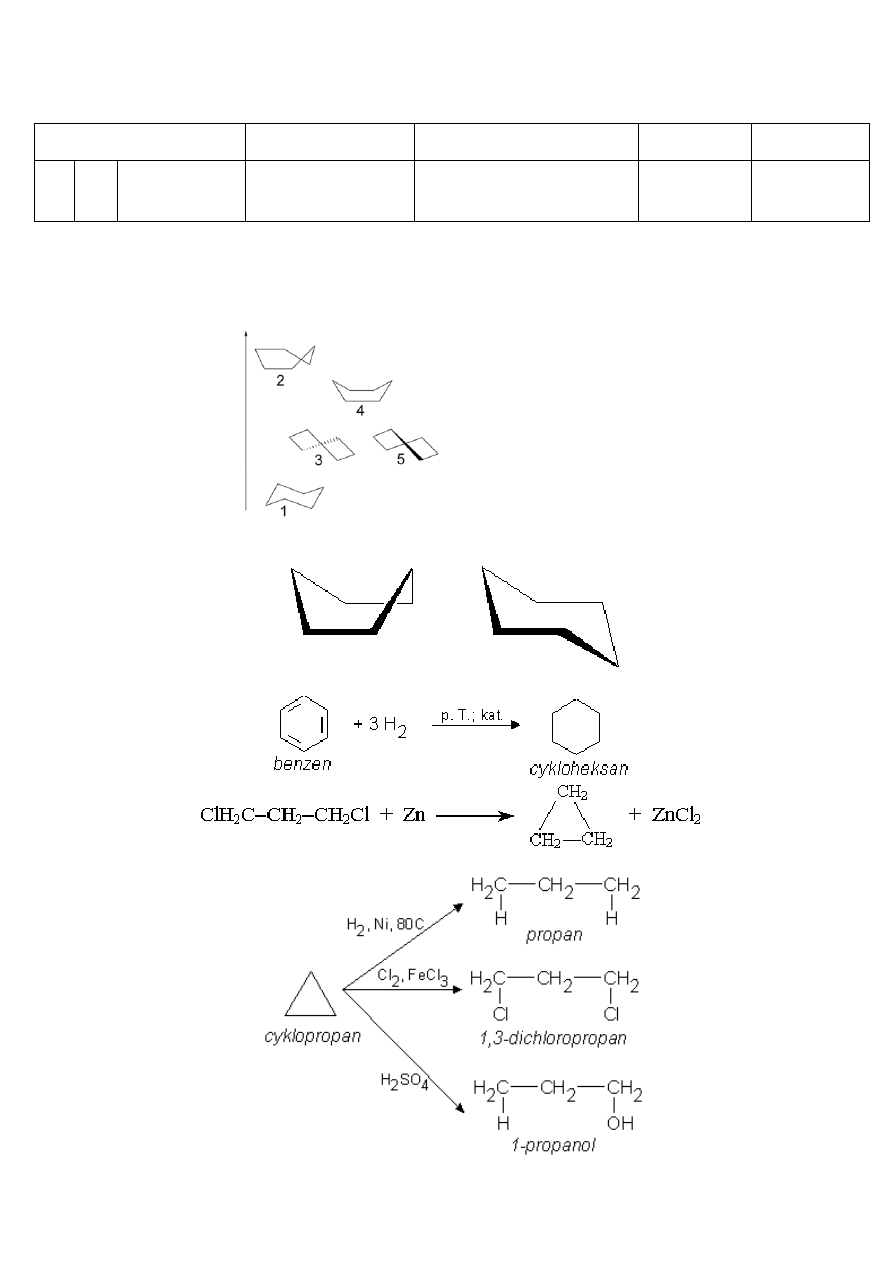
Otrzymywanie (*):
Charakterystyczne reakcje:
(na rysunku przedstawiona jest decyklizacja cyklopropanu)
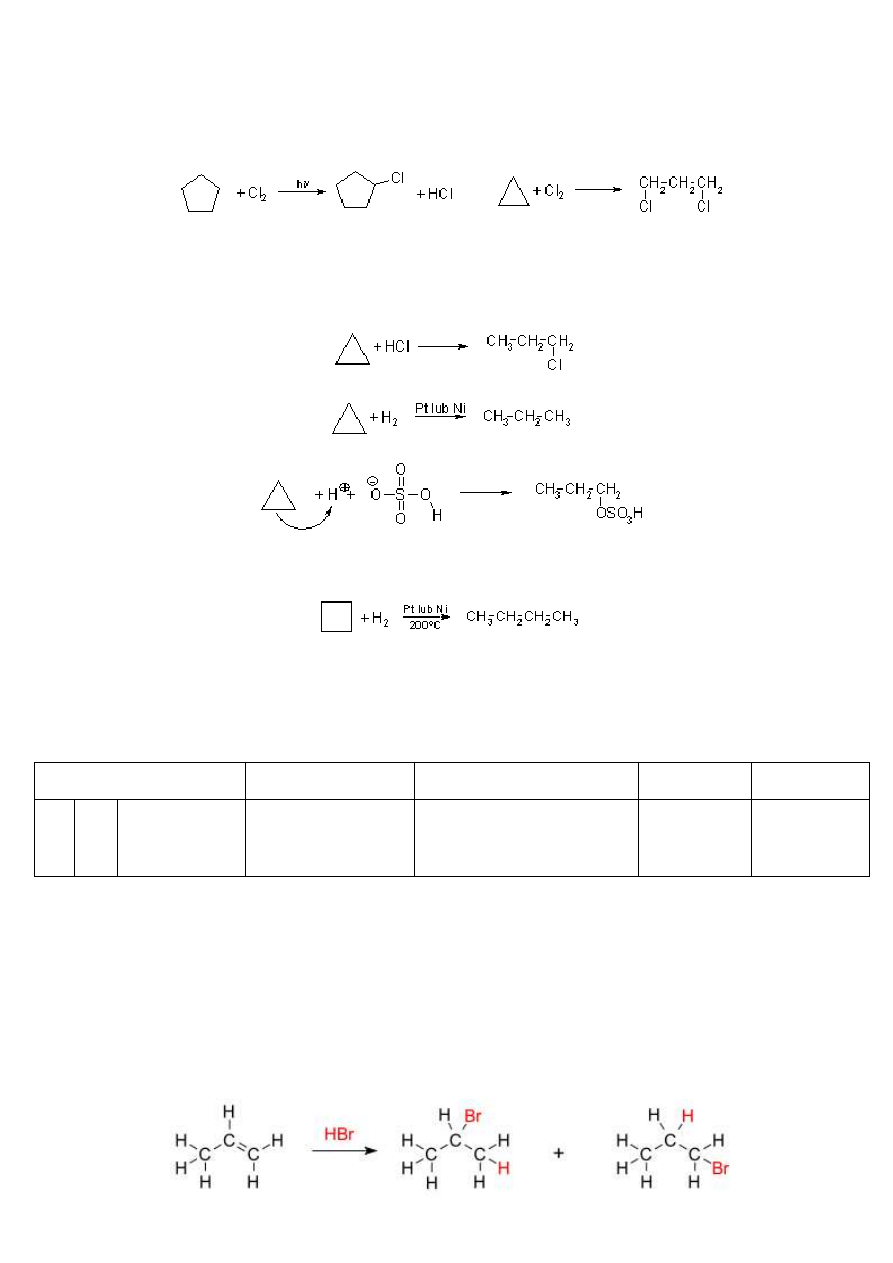
Cykloalkany o pierścieniach pięcioczłonowych i większych mają identyczne właściwości chemiczne jak alkany, a co za tym
idzie, ulegają reakcjom substytucji rodnikowej: chlorowaniu i nitrowaniu. Mechanizm tej reakcji jest identyczny jak
mechanizm chlorowania alkanów. Natomiast chyklopropan ze względu na specyficzny rodzaj wiązań C-C (wiązania
bananowe), który raczej przypomina wiązanie π niż σ, łatwiej ulega reakcjom z rozerwaniem pierścienia, czyli addycji niż
reakcjom substytucji:
Addycja ta zachodzi bardzo łatwo w obecności chlorku glinu, AlCl
3
.
Nukleofilowy charakter wiązania C-C w cyklopropanie powoduje, że reaguje on również z chlorowodorem lub z
bromowodorem. Reakcje tę zaliczamy również do reakcji addycji. Z uwagi na charakter odczynnika, addycja ta
klasyfikowana jest jako addycja elektrofilowa:
W obecności katalizatora, nawet w łagodnych warunkach przyłącza wodór i redukuje się do propanu:
Cyklopropan jest jedynym węglowodorem, który dzięki temu rozpuszcza się w stężonym kwasie siarkowym:
Cyklobutan ulega podobnym reakcjom jak inne, wyższe cykloalkany. Jedynym wyjątkiem jest reakcja uwodornienia na
katalizatorze. Wyższe cykloalkany nie ulegają tej reakcji, natomiast cyklobutan, dość łatwo się uwodornia w wyższej
temperaturze:
9. Alkeny: budowa, nazewnictwo, charakterystyczne reakcje (addycja elektrofilowa, addycja wolnorodnikowa HBr,
ozonoliza, utlenianie, polimeryzacja związków winylowych). Reguła Markownikowa. Mechanizm reakcji A
E
, A
R
.
Trwałość i przegrupowania karbokationów.
Budowa:
Nazwa
Budowa cząsteczki
Rodzaje
wiązań C-C
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
A
li
fa
ty
cz
n
e
N
ie
n
a
sy
co
n
e
Alkeny
łańcuchowa
(prosta lub rozgałęziona)
jedno podwójne
(składające się z 1 σ i 1 π),
pozostałe pojedyncze
sp
2
(wiązanie
C=C) i sp
3
C
n
H
2n
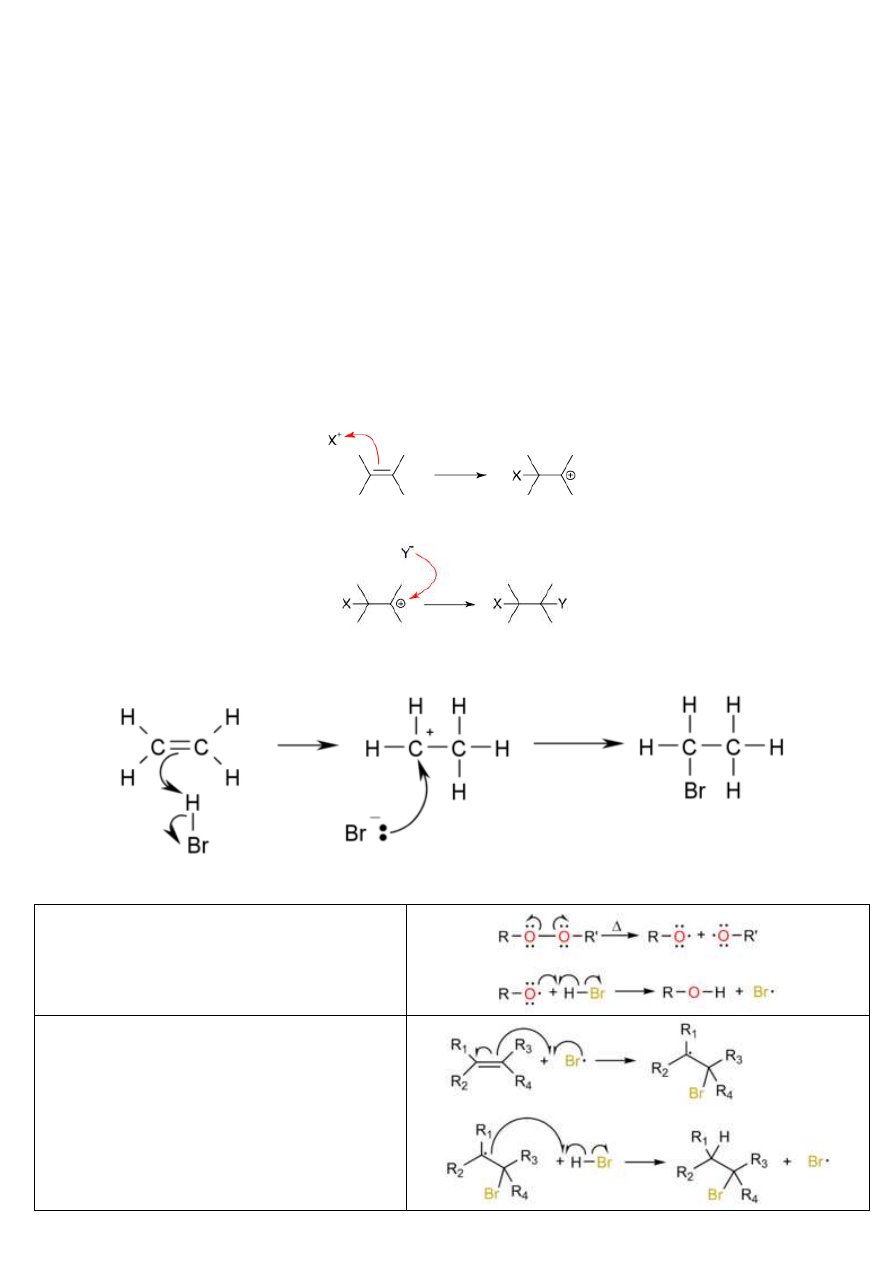
Reguła Markownikowa:
Zasada określająca kierunek reakcji addycji do podwójnego wiązania węgiel-węgiel (-C=C-). Nazwa tej reguły pochodzi od
jej twórcy, rosyjskiego chemika Władymira Markownikowa, który zaproponował ją w 1869 r. Zasada ta głosi, że na ogół w
reakcjach addycji do wiązania -C=C- występujących w wielu związkach organicznych (np. alkenach) atomy lub grupy o
mniejszej elektroujemności przyłączają się do tego z dwóch atomów węgla, do którego już wcześniej było przyłączone
więcej atomów lub grup o własnościach elektrododatnich. Mnemotechnicznie najłatwiej jest zapamiętać tę regułę jako "ci,
którzy już mają, dostaną jeszcze więcej" - albo "każdy idzie do swojego" (addycja HX do wiązania podwójnego przebiega
tak, że atom wodoru przyłącza się przede wszystkim do tego atomu węgla, przy którym jest już więcej atomów wodoru).
Dlatego też, na ilustracji produkt po lewej stronie (2-bromopropan) będzie wytwarzany w większości, natomiast ten po
prawej (1-bromopropan) w znacznie mniejszej ilości:
Charakterystyczne reakcje:
W tym miejscu zamieszczam ważną notatkę – addycja rodnikowa (wolnorodnikowa, free radical addition) nie zachodzi z
każdym halogenowodorem (HX), albo zachodzi w sposób wybuchowy, przez co reakcja nie jest preferowana w chemii
laboratoryjnej. Informacja ta pochodzi z Wiki, ale ponieważ nie jestem w stanie poprzeć tych stwierdzeń w konkretniejszy
sposób, więc tylko zamieszczam krótką adnotację. Na pewno najbezpieczniej mechanizmy przedstawiać za pomocą reakcji
z bromem i bromowodorem. Nie należy także zapominać, iż bromowodór kwas i bromowodór gaz to dwie różne rzeczy –
reakcje zachodzące z bromowodorem niekoniecznie zajdą z jego wodnym roztworem (odbarwianie wody bromowej).
ADDYCJA ELEKTROFILOWA (na przykładzie HBr) – A
R
:
W chemii organicznej addycja elektrofilowa zachodzi najczęściej do związków posiadających wielokrotne wiązania węgiel-
węgiel (np: alkeny). W wyniku tej reakcji jedno lub więcej wiązań π między atomami węgla rozpada się z utworzeniem
wiązań σ między atomami węgla a elektrofilem:
X-Y + C=C → X-C-C-Y
Wyjściowy związek X-Y nie jest często sam z siebie elektrofilowy, lecz staje się nim w wyniku dysocjacji elektrolitycznej, w
wyniku której powstaje kation X
+
.
X-Y → X
+
+ Y
-
To właśnie jon X
+
pełni rolę faktycznego czynnika z silnym deficytem elektronów, który atakując wiązanie C=C "pobiera"
elektron z wiązania π aby utworzyć między sobą i jednym z atomów węgla wiązanie σ:
W wyniku tego procesu powstaje nowy kation X-C-C
+
- - który z kolei atakuje anion Y
-
, będący silnym nukleofilem, czyli
posiadający nadmiar elektronów:
Reakcja addycji elektrofilowej do wiązań wielokrotnych węgiel-węgiel jest zwykle regioselektywna i zazwyczaj zachodzi
zgodnie z regułą Markownikowa. W chemii metaloorganicznej, wiele reakcji addycji elektrofilowych zachodzi jednak
często przeciwnie z regułą Markownikowa.
ADDYCJA WOLNORODNIKOWA – A
R
:
INICJACJA
PROPAGACJA
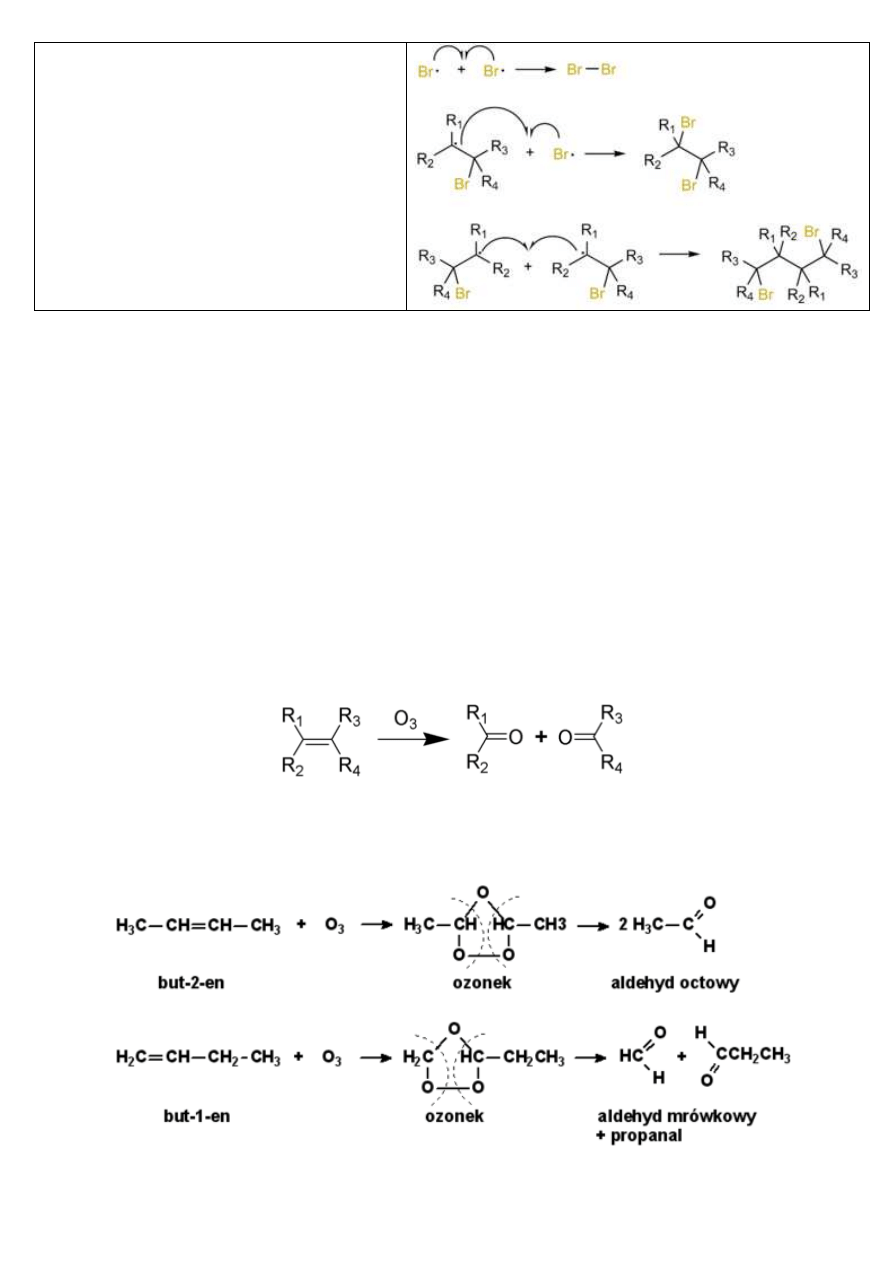
TERMINACJA
UTLENIANIE:
Standardowo, do utleniania używa się nadmanganianu potasu (manganian(VII) potasu – KMnO
4
), albo chromianu(VI)
potasu – K
2
CrO
4
.Dodatkowo utleniaczami są – kwas siarkowy(VI) H
2
SO
4
, kwas azotowy(V) HNO
3
, tlenek miedzi(II) CuO
(próba Trommera), tlenek srebra(I) Ag
2
O (pod wpływem zw. organicznych redukuje się do wolnego srebra – próba
Tollensa), nadtlenek wodoru H
2
O
2
. Na podstawie reakcji redoks jesteśmy w stanie stwierdzić jakie mniej-więcej produkty
powstaną. W chemii organicznej szereg utleniania wygląda następująco – alkan, alkohol, aldehyd, kwas karboksylowy (i
np. w przypadku kwasu octowego, dalsze utlenianie powoduje rozpad na dwutlenek węgla i wodę). Cały szereg działa
także w drugą stronę w przypadku dodania związków redukujących w postaci np. H
2
z katalizatorem, albo w podwyższonej
temperaturze. Nie należy również zapominać o tym, że ketony oraz alkohole trzeciorzędowe nie ulegają już dalszemu
utlenianiu lub dalsze utlenianie powoduje degeneracje cząsteczki. A teraz wracając do reakcji utleniania alkenów:
2 KMnO4 + 3 C2H4 + 4 H2O = 3 C2H4(OH)2 + 2 MnO2 + 2 KOH
Przy redoksie z nadmanganianem należy pamiętać o stopniach utlenienia w poszczególnych środowiskach –
nadmanganian w środowisku zasadowym z VII przechodzi na VI, w środowisku obojętnym (tak jak w przykładzie powyżej)
z VII na IV oraz w środowisku kwasowym z VII na II.
OZONOLIZA:
Podczas ozonolizy stosunkowo nietrwałe ozonki pod wpływem wody rozkładają się do odpowiednich aldehydów. Rodzaj
aldehydów dowodzi miejsca występowania wiązania podwójnego. Gdy wiązanie podwójne występuje w pozycji 1 -
powstaje aldehyd mrówkowy (metanal), gdy wiązanie występuje w pozycji 2 powstaje aldehyd octowy (etanal), gdy w
pozycji 3 aldehyd propionowy (propanal) itd. W toku rozpadu ozonku pod wpływem wody powstaje też nadtlenek wodoru.
Aby nie utleniał on tworzących się aldehydów do środowiska reakcji dodaje się cynk.
BROMOWANIE:
Bardzo ważną reakcją, pozwalającą na wykrywanie wiązań podwójnych jest bromowanie – odbarwianie wody bromowej
(nie mylić z reakcją z gazowym bromem!):
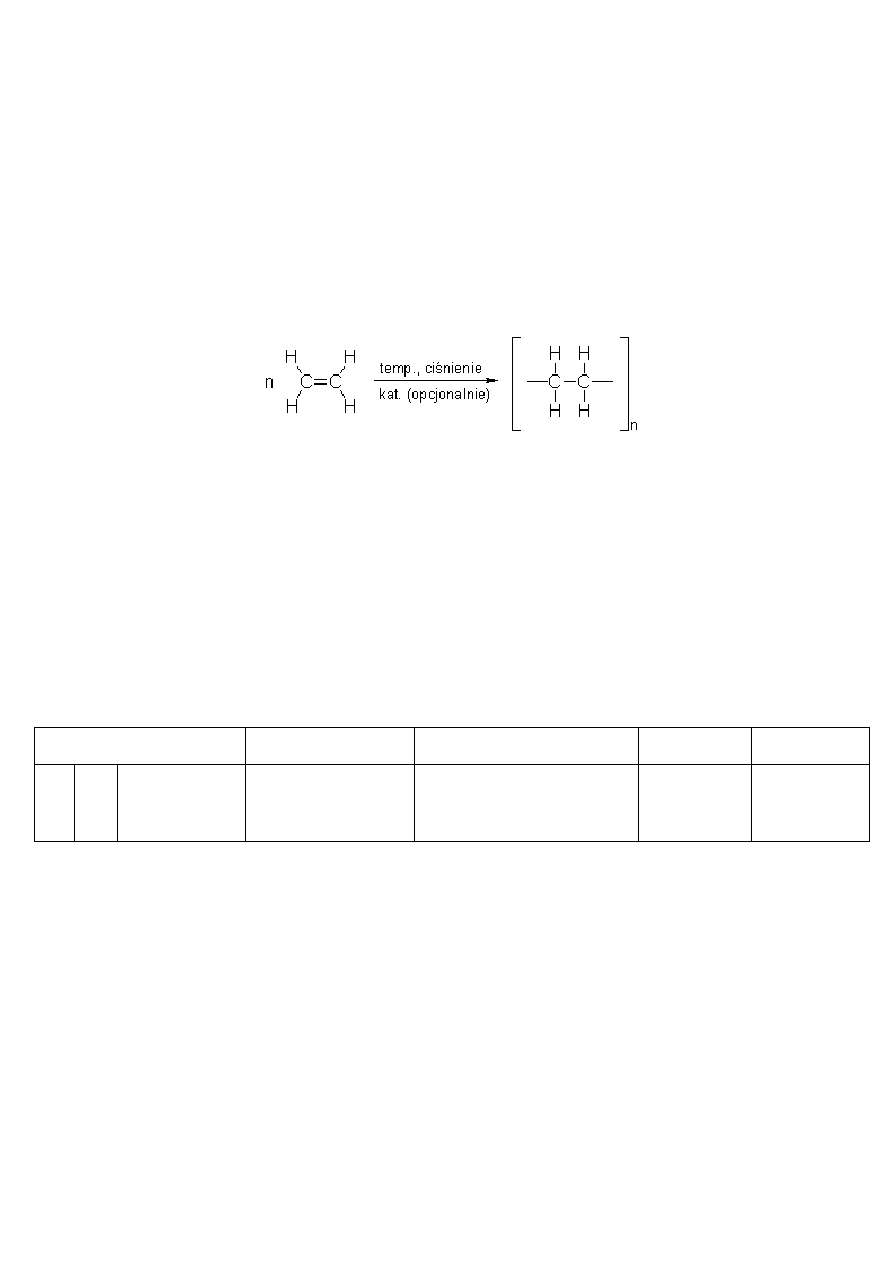
C
2
H
4
+ Br
2
→ CH
2
BrCH
2
Br
Woda bromowa jest odbarwiana przez nienasycone związki organiczne, ale nie jest odbarwiana przez związki
aromatyczne. Brom Br
2
może być stosowany w mieszaninie z czterochlorkiem węgla CCl
4
(odbarwienie z czerwonego
zabarwienia roztworu). Zwykle reakcje te wykonuje sie działając na ciekły alken wodą bromową (Br
2
+ H
2
O). Gdy mamy
mało alkenu Br
2
musi być rozpuszczony w medium, w którym alken choć trochę rozpuszcza się. Ponieważ aceton i alkohole
odpadają, pozostaje CCl
4
i etery. Tutaj nadobowiązkowa rozpiska procesu reakcyjnego(*):
reakcja biegnie na zimno, HBr nie wydziela się - alkeny;
reakcja po ogrzaniu, HBr nie wydziela się - styreny Ph-CH=CH-R, Ph-CH=CH-COOH, kwas cynamonowy itp.;
reakcja na zimno, HBr wydziela się - czasami fenole - raczej mało takich związków!
reakcja po ogrzaniu HBr wydziela się - aldehydy, metyloketony i inne, terpeny,
fenole, bardziej reaktywne aromaty (piren).
POLIMERYZACJA ZWIĄZKÓW WINYLOWYCH:
Grupa winylowa to CH
2
=CH-
Polimery winylowe to produkty polimeryzacji monomerów winylowych:
eten – polietylen PE (torebki foliowe)
propen – polipropylen PP (opakowania)
chlorek winylu – Poli(chlorek winylu) PCW = PCV (okna, panele podłogowe)
styren (winylobenzen) – polistyren PS (materiały izolacyjne)
ADDYCJA WODORU:
Wiązanie podwójne w alkenach można łatwo uwodornić na drodze addycji (często stosuje się jako katalizatory Ni i Al):
CH
2
=CH
2
+ H
2
→ CH
3
-CH
3
10. Alkadieny: podział ze względu na położenie wiązań podwójnych w cząsteczce. Reakcje addycji do dienów
sprzężonych. Kauczuki – polimery dienów sprzężonych.
Nazwa
Budowa cząsteczki
Rodzaje
wiązań C-C
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
A
li
fa
ty
cz
n
e
N
ie
n
a
sy
co
n
e
Alkadieny
łańcuchowa
(prosta lub rozgałęziona)
dwa podwójne (składające się z 1
σ i 1 π każde),
pozostałe pojedyncze
sp
2
(wiązania
C=C) i sp
3
C
n
H
2n-2
W alkadienach rozróżniamy 3 układy położenia wiązań podwójnych względem siebie:
układ skumulowany C=C=C
układ sprzężony C=C-C=C
układ izolowany C=C-C-C-C=C
Alkadieny skumulowane są bardzo nietrwałe, przechodzą w alkiny.
Charakterystyczne reakcje:
ADDYCJA:
W alkadienach izolowanych każde wiązanie reaguje niezależnie, natomiast w alkadienach sprzężonych dochodzi do
addycji 1,2 i 1,4, przy czym addycja 1,4 stanowi główny produkt reakcji (reakcja na przykładzie buta-1,3-dienu):
CH
2
=CH–CH=CH
2
+ YZ → CH
2
Y–CHZ–CH=CH
2
+ CH
2
Y–CH=CH–CH
2
Z
11. Alkiny: budowa, nazewnictwo, charakterystyczne reakcje (przyłączanie do wiązania potrójnego, reakcje
terminalnych alkinów jako kwasów), polimeryzacja acetylenu.
Nazwa
Budowa cząsteczki
Rodzaje
wiązań C-C
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
A
li
fa
ty
cz
n
e
N
ie
n
a
sy
co
n
e
Alkiny
łańcuchowa
(prosta lub rozgałęziona)
jedno potrójne
(składające się z 1 σ i 2 π),
pozostałe pojedyncze
sp (wiązanie
CΞC) i sp
3
C
n
H
2n-2
enyn – zawiera w cząsteczce wiązania podwójne i potrójne
Charakterystyczne reakcje:
ADDYCJA:
CH≡CH + Cl
2
→ CHCl=CHCl + Cl
2
→ CHCl
2
–CHCl
2
W tym miejscu należy zwrócić uwagę, iż d
obierając odpowiednie proporcje możemy rozróżnić alkeny od alkilów w
reakcji z wodą bromową, np. 5g Br
2
odbarwi się po dodaniu 0.5dm
3
acetylenu, ale nie odbarwi się po dodaniu
takiej samej ilości etylenu.
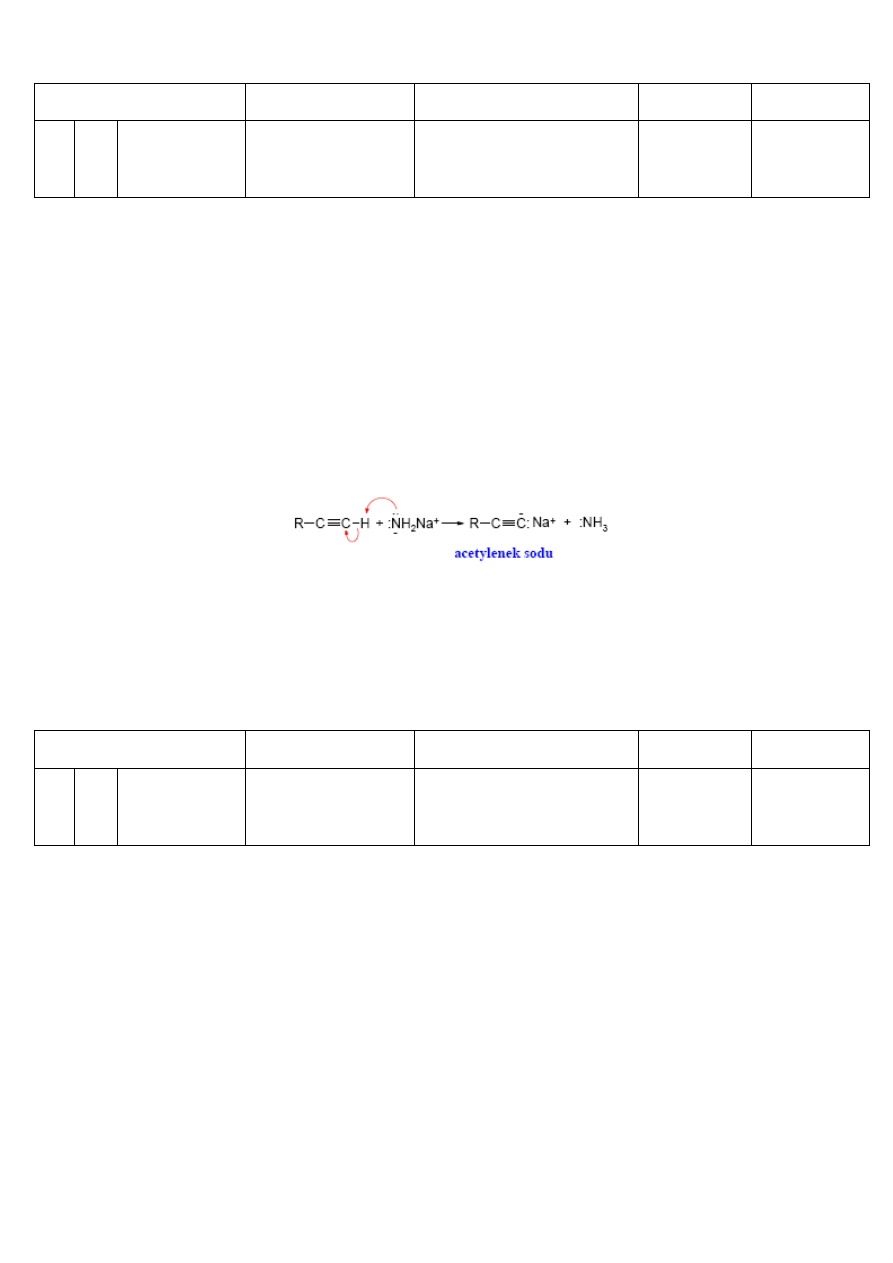
ALKINY TERMINALNE:
Spośród wszystkich atomów wodoru występujących w węglowodorach atom H związany z atomem C o hybrydyzacji sp jest
najbardziej kwaśny.
Kwas, w którym proton odrywany jest od atomu węgla nazywa się C-kwasem. Z silnymi zasadami, np.
z amidkiem sodu z terminalnych alkinów powstają
acetylenki:
12. Węglowodory aromatyczne: budowa, trwałość pierścieni aromatycznych, struktury rezonansowe, reguła Hückla.
Najważniejsi przedstawiciele związków aromatycznych. Nazewnictwo. Podstawienie elektrofilowe jako reakcja
charakterystyczna węglowodorów aromatycznych. Mechanizmy reakcji nitrowania i sulfonowania benzenu (w tym:
struktury rezonansowe przejściowych karbokationów). Podstawniki w pierścieniach aromatycznych – efekty
elektronowe i wpływ na reakcje S
E
(podstawniki aktywujące i dezaktywujące pierścień w reakcjach S
E
, kierujące
działanie podstawników w pochodnych benzenu na reakcje S
E
– podstawniki I i II rodzaju).
Nazwa
Budowa cząsteczki
Rodzaje
wiązań C-C
Hybrydyzacja
atomów węgla
Wzór szeregu
homologicznego
A
li
fa
ty
cz
n
e
A
ro
m
a
t
y
cz
n
e
Areny
cykliczna
(z podstawnikami)
w pierścieniu – pośrednie
(składające się z układu wiązań σ i
zdelokalizowanych wiązań π),
w podstawnikach - różne
w pierścieniu –
tylko sp
2
C
n
H
2n-6
(dla szeregu
benzenu)
Z nazw, które pojawiły się na wykładzie, i której najprawdopodobniej trzeba znać to:
benzen, toluen (metylobenzen), ksyleny (orto, meta, para), styren (winylek benzenu), kumen, anilina (fenyloamina), fenol
(hydroksybenzen), aldehyd i kwas benzoesowy, fenyl, nitrobenzen, difenylometan, difenyloeten, tri fenyloetan, naftalen,
antracen, fenantren, piren, benzopiren, koronen
Właściwości chemiczne węglowodorów aromatycznych są ściśle związane z budową pierścieni. Występuje w nich
delokalizacja elektronów π, dzięki czemu areny to związki bogate w elektrony, czyli nukleofile (czyli są podatne na atak
reagentów elektrofilowych). Pierścień aromatyczny jest trwały, trudno ulega rozpadowi i aby ten problem „ominąć”,
przebiegają tzw. reakcje z zachowaniem pierścienia (substytucja elektrofilowa).
Reguła Hückla:
Reguła zwana też regułą 4n+2 - prosta metoda pozwalająca na sprawdzenie, czy określony, organiczny, cykliczny związek
chemiczny jest aromatyczny. Reguła ta wynika z analiz kwantowo-mechanicznych zjawiska aromatyczności wykonanych
przez Ericha Hückla w 1931 r. Sama reguła 4n+2 została jednak sformułowana przez von Doeringa w 1951 r. Reguła ta
głosi, iż związek jest prawdopodobnie aromatyczny gdy w układzie wiązań wielokrotnych tworzących układ cykliczny tego
związku występuje 4n+2 elektronów zlokalizowanych na wiązaniach π, gdzie n = dowolna liczba naturalna. Reguła ta w
praktyce jest sprawdzona dla n = od 1 do 6, przy czym zawodzi ona dla związków w których występują więcej niż 3
sprzężone pierścienie. Np.: piren i koronen są związkami aromatycznymi, mimo że nie spełniają reguły Hückla. Podobnie
jest też z fullerenami.
Reaktywność, a przyłączanie podstawników do pierścienia benzenowego:
W celu lepszego zrozumienia charakteru zjawiska polecam powrót do części o efekcie mezomerycznym i indukcyjnym.
Podstawnik już przyłączony do pierścienia wykazuje dwa efekty:
1.Wpływa na reaktywność pierścienia aromatycznego w reakcji substytucji elektrofilowej. Niektóre podstawniki aktywują
pierścień, czyniąc go bardziej reaktywnym niż benzen, a niektóre dezaktywują pierścień czyniąc go mniej reaktywnym niż
benzen.
2.Ma wpływ na orientację (kierunek) reakcji.
Trzy możliwe produkty dwupodstawione: orto-, meta-i para-, nie tworzą się w równych ilościach.
Charakter podstawnika już obecnego w pierścieniu benzenowym określa położenie drugiego podstawnika.
Niektóre grupy kierują następny podstawnik przede wszystkim w położenie orto- i para-, podczas gdy inne grupy kierują w
położenie meta-.
grupy kierujące w położenie –orto i –para
grupy kierujące w położenie –meta
silnie aktywujące: -NH
2
, -NHR, -NR
2
, -OH
średnio aktywujące: -NHCOR, -OR
słabo aktywujące: -R, -C
6
H
5
słabo dezaktywujące: -X (-F, -Cl, -Br, -I)
silnie dezaktywujące: -NO
2
,-NR
3
średnio dezaktywujące: -CN, -SO
3
H, -COOH,
-COOR, -COR, -CHO
(R oznacza łańcuch węglowodorowy; X to symbol halogenków, od fluoru do jodu – 17 grupa)
Charakterystyczne reakcje:
SUBSTYTUCJA ELEKTROFILOWA – S
E
:
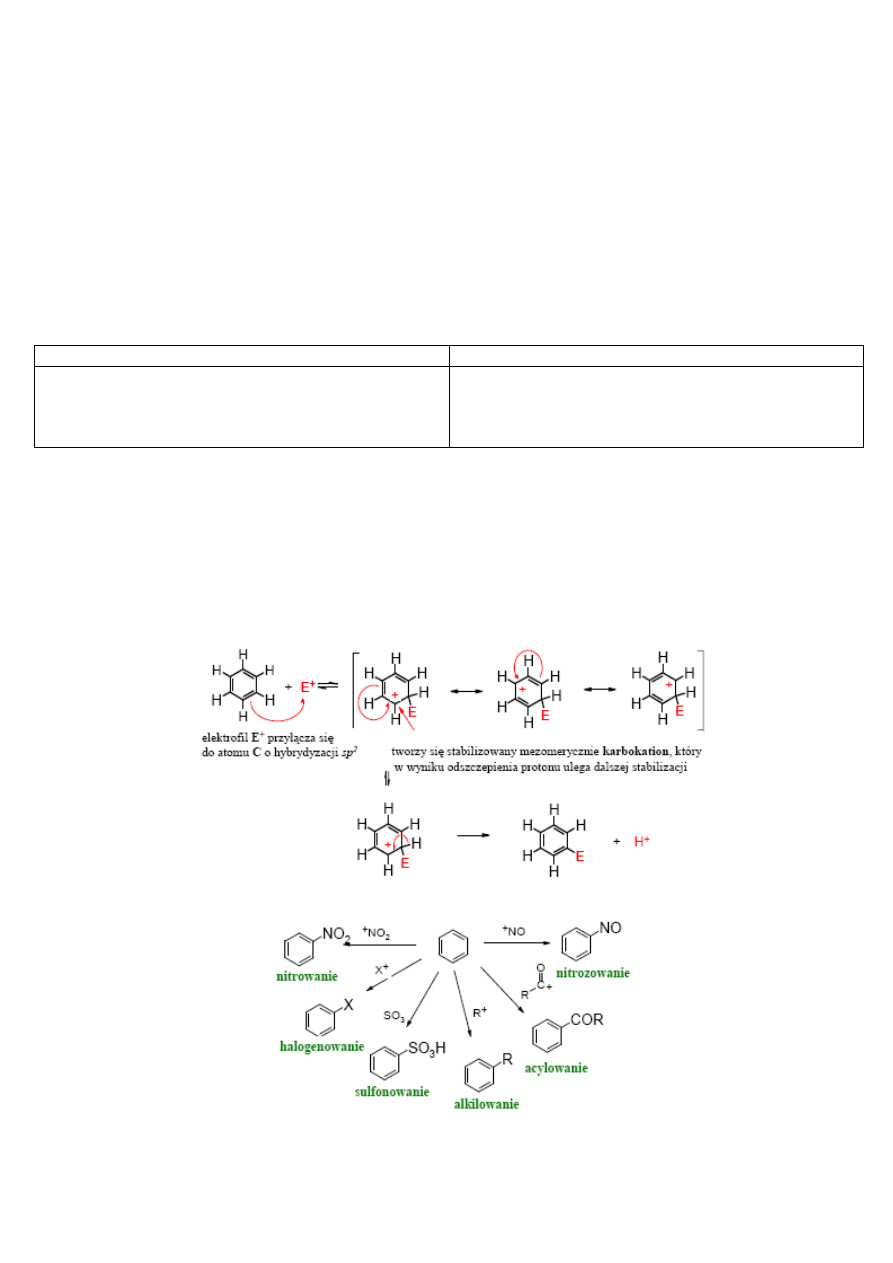
Charakterystyczną reakcją związków aromatycznych jest substytucja elektrofilowa (SE), zwana również reakcją
aromatycznej substytucji elektrofilowej. Polega ona najczęściej na tym, że elektrofil (E+) podstawia proton związany z
pierścieniem aromatycznym. W pierwszym etapie dochodzi do addycji elektrofila E+ do pierścienia aromatycznego, w
wyniku czego powstaje stabilizowany mezomerycznie karbokation, po czym po odszczepieniu protonu odtwarza się układ
aromatyczny (na podstawie poniższego schematu można tworzyć struktury rezonansowe karbokationów w procesie
sulfonowania i nitrowania benzenu):
W ten sposób zachodzą reakcje typu halogenowania, alkilowania, acylowania, nitrowania, sulfonowania i inne.
Nitrowanie:
Grupę nitrową do pierścienia aromatycznego najczęściej wprowadza się za pomocą mieszaniny nitrującej, czyli kwasu
azotowego i siarkowego o różnych stężeniach, zależnych od reaktywności arenu. W mieszaninie tych kwasów wytwarza
się kation nitroniowy
+
NO
2
, który jako silny elektrofil wchodzi na miejsce jednego z aromatycznych atomów wodoru.
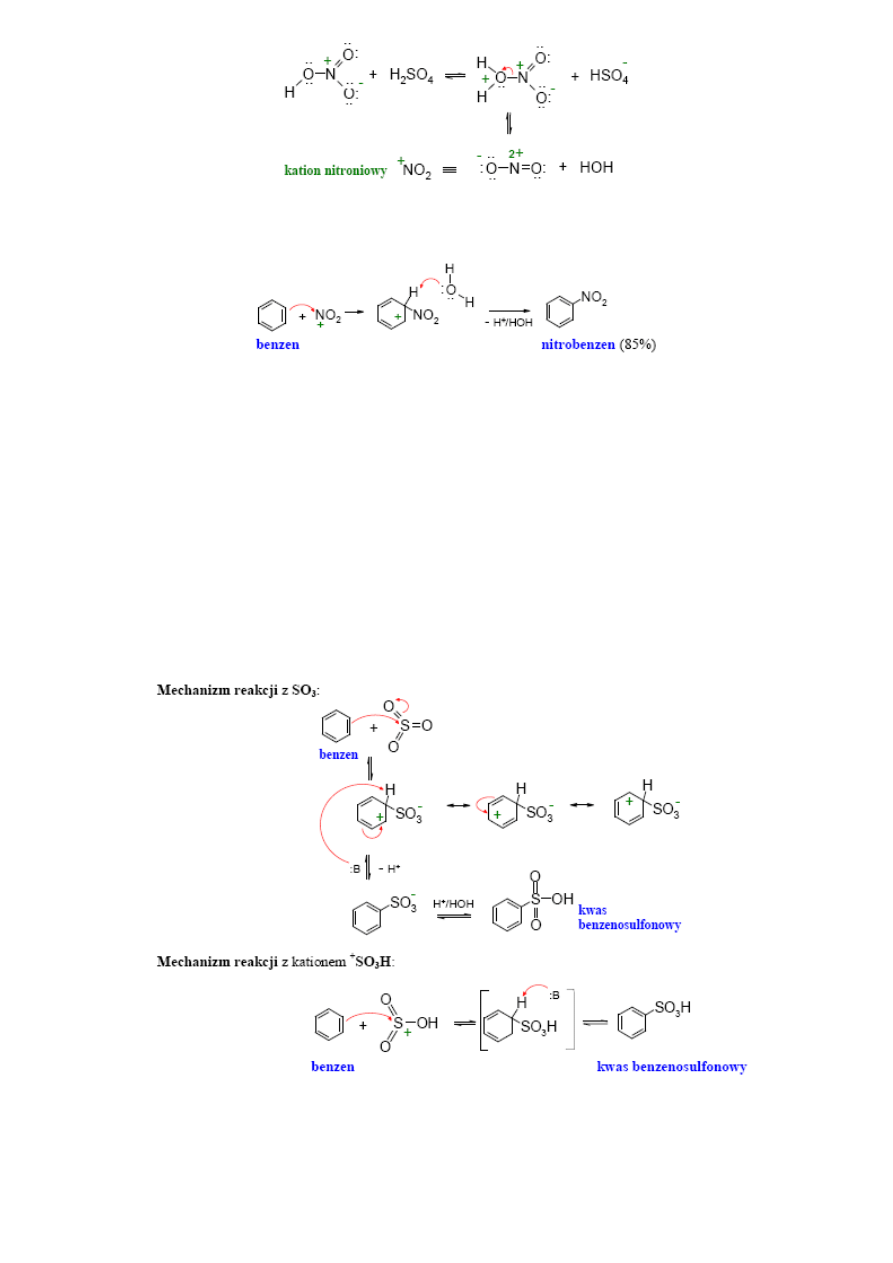
Kwas siarkowy pełni rolę dawcy protonu, czynnika wiążącego wodę oraz ułatwia rozpuszczanie arenów. Dwie pierwsze
funkcje kwasu siarkowego sprzyjają powstawaniu kationu nitroniowego. Kation nitroniowy jako elektrofil reaguje z
arenem tworząc addukt, który stabilizuje się przez odszczepienie protonu. Jest to typowy mechanizm reakcji S
E
.
Nitrowanie arenów jest reakcją wysoko egzotermiczną.
Sulfonowanie:
Utworzenie wiązania pomiędzy atomem węgla związku organicznego a atomem siarki grupy sulfonowej –
-
SO
3
H lub jej
odpowiednikiem chlorosulfonowym –
-
SO
2
Cl nazywa się sulfonowaniem. Sulfonowanie arenów biegnie wg mechanizmu
S
E
. Prowadzi się je za pomocą SO
3
, jego kompleksów, np. z pirydyną oraz stężonym kwasem siarkowym lub dymiącym
kwasem siarkowym (oleum). Podczas sulfonowania kwasem siarkowym powstają duże ilości trudnych do
zagospodarowania kwaśnych odpadów. W zasadzie sulfonowanie tritlenkiem siarki jest bezodpadowe. Trójtlenek siarki
jest jednak substancją stałą, co utrudnia prowadzenie reakcji. Często stosuje się procedurę mieszaną – sulfonowanie
rozpoczyna się w niewielkiej ilości kwasu siarkowego i w miarę postępu reakcji dodaje się SO
3
. Ten sposób postępowania
zapewnia nie tylko utrzymywanie stałego stężenia środka sulfonującego, ale i wiązanie wody powstającej w reakcji,
ponadto kwas siarkowy dodatkowo pełni rolę rozpuszczalnika. Podczas sulfonowania benzenu lub toluenu wodę można
usuwać azeotropowo. Wiązanie lub usuwanie wody ze środowiska reakcji jest koniecznie, gdyż reakcja sulfonowania jest
odwracalna i tylko odpowiednie stężenie kwasu (zależne od aktywności arenu) zapewnia przesunięcie równowagi na
korzyść produktów. Wodę powstającą w reakcji sulfonowania można usuwać azeotropowo. Czynnikiem sulfonującym
jest SO
3
lub tworzący się z kwasu siarkowego i SO
3
kation
+
SO
3
H.
(pozostałe procesy, jako nie zawarte w wymaganiach pomijam w omawianiu)

13. Halogenki alkilów: budowa (w tym: rzędowość), nazewnictwo. Podstawienie nukleofilowe jako reakcja
charakterystyczna halogenków alkilów. Mechanizm, kinetyka i stereochemia reakcji S
N
1 i S
N
2. Reakcje eliminacji (E1 i
E2) jako reakcje konkurencyjne w stosunku do S
N
. Reguła Zajcewa.
Rzędowość w przypadku halogenków alkilów określa się tak samo jak w przypadku omówionym w punkcie 7.
Halogenki mają wyższe temperatury wrzenia niż alkany o tej samej liczbie atomów węgla. Są one nierozpuszczalne w
wodzie, ale rozpuszczają się w typowych rozpuszczalnikach organicznych. Są bardzo reaktywne, używane jako substraty
do otrzymywania innych związków.
Reguła Zajcewa:
Reguła dotycząca chemicznej reakcji eliminacji, w której powstają nowe wiązania podwójne węgiel-węgiel. Regułę tę
sformułował jako pierwszy rosyjski chemik Aleksander Zajcew w 1875 r. W reakcjach eliminacji, w których powstaje
wiązanie C=C, powstają zawsze w przewadze bardziej rozgałęzione izomery.
W przypadku reakcji eliminacji halogenowodorów (HX) z halogenków alkilowych z reguły tej wynika, że jako produkt
główny powstaje alken zawierający maksymalną liczbę grup alkilowych przy atomach węgla posiadających wiązanie
podwójne. Reguła ta obowiązuje dla reakcji zachodzących według mechanizmu dwucząsteczkowego (E
2
) o ile w
substratach nie ma blisko powstającego wiązania podwójnego podstawników posiadających charakter silnie nukleofilowy
oraz nie wchodzą w grę zjawiska zawady sterycznej.
Mechanizm E
1
:
Reakcja eliminacji E1 jest reakcją dwuetapową i normalnie zachodzi bez obecności zasady. W pierwszym etapie powstaje
karbokation. Etap ten zachodzi powoli a więc decyduje on o szybkości reakcji. W etapie drugim rozpuszczalnik odrywa
proton od węgla β karbokationu, następuje przejście pary elektronowej i utworzenie podwójnego wiązania. Odłączenie
wodoru jest etapem szybkim a więc nie wpływa na szybkość reakcji. Na szybkość reakcji ma wpływ również budowa
łańcucha węglowego. Mianowicie wraz ze wzrostem rozgałęzienia łańcucha reaktywność rośnie, jest to spowodowane
tworzeniem się możliwie najtrwalszego karbokationu. Pierwszy etap jest analogiczny do mechanizmu Sn1.
Skłonność do eliminacji E1 wykazują głównie trzeciorzędowe halogenopochodne i to wtedy gdy w mieszaninie reakcyjnej
nie znajdują się silne zasady. Dowody istnienia eliminacji jednocząsteczkowej:
- Reakcja jest pierwszego rzędu w stosunku do substratów
- Jeśli reakcji ulegają dwie cząsteczki różniące się jedynie grupą odchodzącą to szybkości ich reagowania powinny być
rożne, gdyż zależy to od zdolności cząstek do jonizacji. Jeżeli karbokation już się utworzy to w tym samym rozpuszczalniku
i w tych samych warunkach powinny zachodzić identycznego jego przemiany. Jest to spowodowane brakiem wpływu
grupy odchodzącej na drugi etap reakcji.
- Wiele reakcji prowadzonych w łagodnych warunkach przebiega łatwiej, gdy oderwaniu musi ulec wodór z pozycji cis niż z
pozycji trans
- Jeśli powstają przejściowo karbokationy, to przy użyciu odpowiednich substratów można oczekiwać przegrupowań
Mechanizm E
2
:
Jest to eliminacja dwucząsteczkowa i obydwie grupy odszczepiają się równocześnie, z tym, że proton jest odciągany przez
zasadę. Reakcja ta zachodzi w jednym etapie i jest drugiego rzędu.
Stwierdzono, że reakcje eliminacji E2 tak jak w przypadku Sn2 przebiegają najczęściej stereospecyficznie, co oznacza ze
wszystkie pięć zaangażowanych atomów leży w jednej płaszczyźnie, natomiast wodór z grupą odchodzącą muszą być w
pozycji trans.
Typ halogenku
Sn1
Sn2
E1
E2
RCH
2
X
Nie zachodzi
Uprzywilejowana
Nie zachodzi
Zachodzi przy
użyciu silnych zasad
R
2
CHX
Może zachodzić w
przypadku
halogenków
benzylowych i
allilowych
Konkuruje z reakcją
E2
Może zachodzić w
przypadku
halogenków
benzylowych i
allilowych
Uprzywilejowana
przy użyciu silnych
zasad
R
3
CX
Uprzywilejowana w
rozpuszczalnikach
hydroksylowych
Nie zachodzi
Konkuruje z Sn1
Uprzywilejowana
przy użyciu zasad
Pierwszorzędowe halogenki alkilowe
Drugorzędowe halogenki alkilowe
Trzeciorzędowe halogenki alkilowe
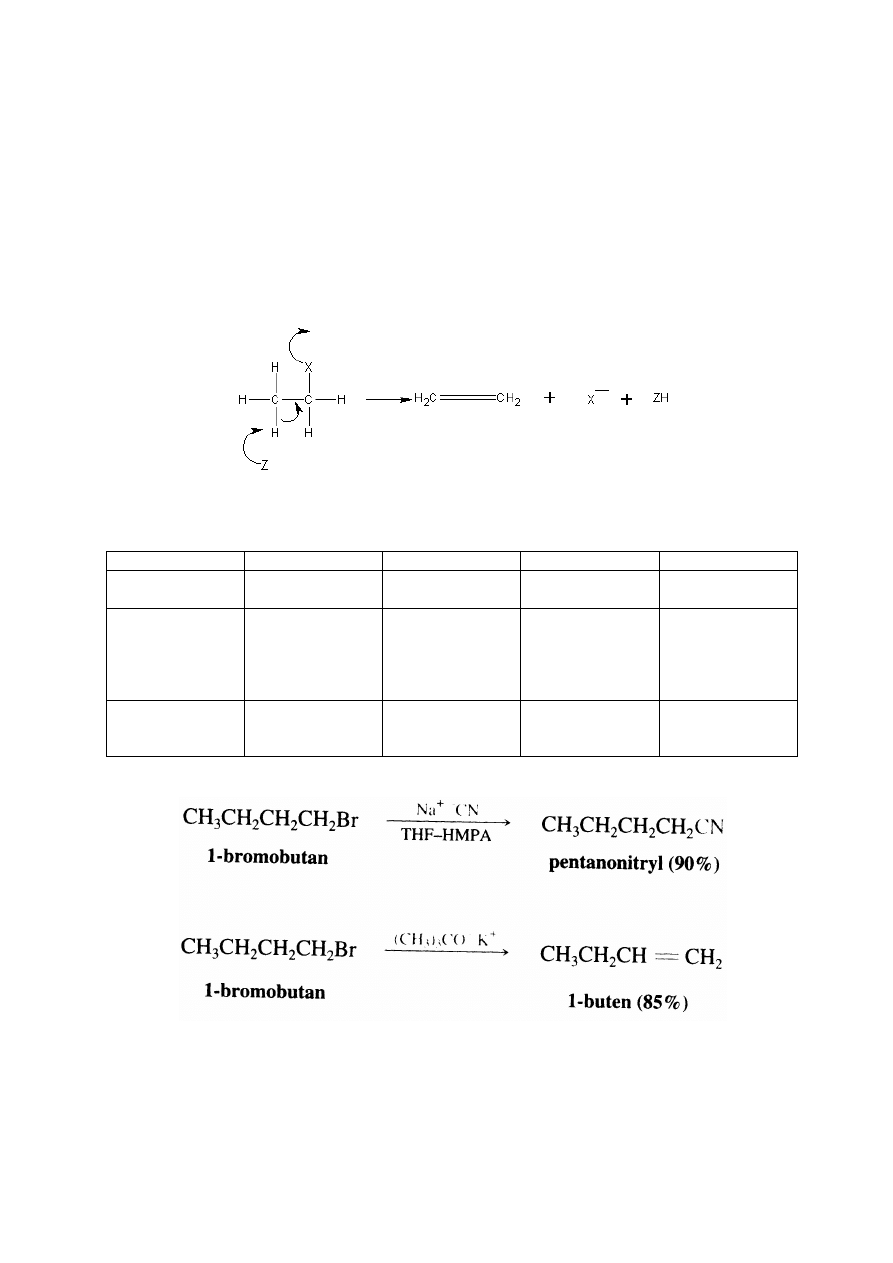
Reakcje S
N
:
R-X
OH
-
H
2
O
OR
-
CN
-
NH
3
R’NH
2
R’NHR”
SH
-
SR’
-
R-OH+X
-
R-OH+HX
R-OR+X
-
R-CN+X
-
R-NH
2
+X
-
R-NHR’+X
-
R-NR’R”+HX
R-SH+X
-
R-SR
-
+X
-
alkohol
alkohol
eter
nitryl
amina
I-rzędowa
amina
II-rzędowa
amina
III-rzędowa
tiol
(tioalkohol)
sylfid
(tioeter)
14. Halogenki arylów – budowa i reakcje.
15. Halogenopochodne alkenów: halogenki allilowe i winylowe – budowa i reaktywność.
Halogenki Alkilów
Halogenki Arylów
Atom halogenu związany z tetraedrycznym
atomem węgla
Atom halogenu związany bezpośrednia z atomem węgla w pierścieniu
aromatycznym
Łatwo ulegają reakcjom Sn i eliminacji (po
wpływem np. KOH)
Nie ulegają reakcjom Sn ani eliminacji. W szczególnych przypadkach
możliwa reakcja SnAr
Tworzą związki metaloorganiczne

16. Alkohole – budowa (rzędowość), nazewnictwo, wiązania wodorowe i ich wpływ na właściwości fizyczne alkoholi.
Reakcje alkoholi jako kwasów (tworzenie alkoholanów). Reakcje alkoholi jako zasad (reakcje z halogenowodorami:
podstawienie nukleofilowe SN1, SN2 - mechanizmy). Dehydratacja (tj. eliminacja wody) i utlenianie alkoholi.
Właściwości fizyczne alkoholi uwarunkowane są wpływem obu grup funkcyjnych – zarówno części hydrofilowej, polarnej
w postaci –OH, jak i części hydrofobowej (lipofilowa), która stanowi łańcuch węglowodorowy –R. Wraz ze wzrostem
łańcucha alkilowego, rosną właściwości lipofilowe i spadają hydrofilowe. Wraz ze zwiększoną ilością grup –OH, wzrasta
rozpuszczalność w wodzie (decydują o tym także wiązania wodorowe).
Wiązania wodorowe w alkoholach:
Ze względu na obecność silnie elektroujemnego atomu tlenu i związanego z nim atomu wodoru alkohole tworzą wiązania
wodorowe, ulegając asocjacji w większe struktury. Niższe alkohole takie jak metanol, etanol i propanol mają niskie
temperatury wrzenia i dużą lotność. Wiązania wodorowe zwiększają temperatury topnienia i wrzenia, obniżają lotność.
Charakterystyczne reakcje:
CHARAKTER CHEMICZNY ALKOHOLI:
charakter zasadowy alkoholi
charakter kwasowy alkoholi
alkohole ulegają reakcjom z rozerwaniem
wiązania C-O (wskazujące na charakter
zasadowy alkoholi)
C
2
H
5
OH + HBr → C
2
H
5
Br + H
2
O
alkohole pod wpływem metali o właściwościach
silnie redukujących tworzą alkoholany
2CH
3
OH + 2Na → 2CH
3
ONa + H
2
w procesie estryfikacji alkohole ulegają reakcji rozerwania wiązania O-H
DEHYDRATACJA:
Charakterystyczna dla alkoholi reakcja eliminacji (nazywana również reakcją dehydratacji) przebiega w środowisku
kwasowym, np. pod wpływem stężonego H
2
SO
4
lub w obecności tlenku glinu Al
2
O
3
w podwyższonej temperaturze.
Produktem reakcji eliminacji cząsteczki wody z etanolu jest eten – węglowodór nienasycony (mechanizm E
1
):
CH
3
CH
2
OH → CH
2
=CH
2
+H
2
O
UTLENIANIE ALKOHOLI:
Tak jak odnotowałem już wcześniej, pierwszorzędowe alkohole utleniają się do aldehydów (a później do kwasów
karboksylowych), natomiast alkohole drugorzędowe utleniają się do ketonów. Alkohole trzeciorzędowe nie ulegają
procesowi utleniania (w bardzo drastycznych warunkach dochodzi do degeneracji cząsteczki). Do przeprowadzenia tych
reakcji używa się K
2
Cr
2
O
7
(dichromian(VI) potasu) lub KMnO
4
(manganian(VI) potasu), a w przypadku alkoholi
drugorzędowych, dodatkowo stosuje się do dichromianu jego tlenek CrO
3
.
CH
3
CH
2
OH + CuO -(pod wpływem T)→ CH
3
COH + Cu + H
2
O
CH
3
C(OH)CH
3
+ CuO -(pod wpływem T)→ CH
3
C(O)CH
3
+ Cu + H
2
O
REAKCJE S
N
W ALKOHOLACH:
S
N
1
protonowanie alkoholu
CH
3
CH
2
OH + H(+) → CH
3
CH
2
O(+)[H]H
wytwarzanie karbokationu
CH
3
CH
2
O(+)[H]H → CH
3
CH
2
(+) + H
2
O
reakcja karbokationu z nukleofilem
CH
3
CH
2
(+) + Cl(-) → CH
3
CH
2
Cl
S
N
2
protonowanie alkoholu
CH
3
OH + H(+) → CH
3
O(+)[H]H
reakcja kationu etylooksoniowego z nukleofilem
CH
3
O(+)[H]H + Br(-) → (stan przejściowy)
Br- CH
3
O(+)[H]H → BrCH
3
+ H
2
O
17. Fenole – budowa, przykładowe związki, wiązania wodorowe i ich wpływ na właściwości fizyczne. Właściwości
kwasowe fenoli. Podstawienie elektrofilowe w fenolach.
Monohydroksylowe
Polihydroksylowe
Fenole
Budowa
grupa OH połączona z at. C o hybrydyzacji sp
3
Grupa OH przy at. C o
hybrydyzacji sp
2
Najprostszy
przedstawiciel
metanol
CH
3
OH
glikol etylenowy
CH
2
OHCH
2
OH
fenol
Dysocjacja
Nie
Nie
Tak (słabo)
Reakcja z NaOH
Nie
Nie
Tak
Reakcja z Na
Tak
Tak
Tak
Charakter
kwasowy
Tylko w reakcji z metalami aktywnymi
(bardzo słaby)
Bardzo
słaby,
choć
trochę
mocniejszy
od
monohydroksylowych
Słaby, ale wyraźny
Reakcja z HX
Tak, choć zależy od rzędowości i rodzaju
HX – najlepiej 3 rzędowe z HI
Tak,
choć
słabiej
niż
monohydroksylowe
Nie
Reakcje
charakterystyczne
Reakcja z Cu(OH)
2
– daje
szafirowy roztwór
Reakcja
z
FeCl
3
–
powstają
barwne
kompleksy
Reakcja z X2
Tylko gdy jest wiązanie podwójne
tak
Oprócz kwasowości, fenole charakteryzują się dużą reaktywnością pierścienia w reakcjach substytucji elektrofilowej.
Fenole ulegają nie tylko reakcjom podstawienia elektrofilowego, typowym dla większości związków aromatycznych, lecz
także wielu innym reakcjom, które możliwe są tylko dzięki niezwykłej reaktywności pierścienia. Do tych reakcji zaliczamy
m. in. estryfikację, sulfonowanie, halogenowanie, nitrowanie, etc.
18. Aldehydy i ketony - budowa, przykładowe związki. Addycja nukleofilowa jako charakterystyczna reakcja grupy
karbonylowej. Reakcje utleniania aldehydów (w tym: próba Tollens’a).
Charakterystyczne reakcje:
ADDYCJA NUKLEOFILOWA – A
N
:
Addycję nukleofilową do cząsteczki aldehydu, bądź ketonu można przeprowadzać za pomocą następujących związków,
które stanowią Nu na schemacie - HCN, ROH, RMgX (odczynnik Grigranda).
Drugą możliwością jest zastosowanie związków aminowych - NH
3
, RNH
2
(aminy), NH
2
OH (hydroksyloamina).
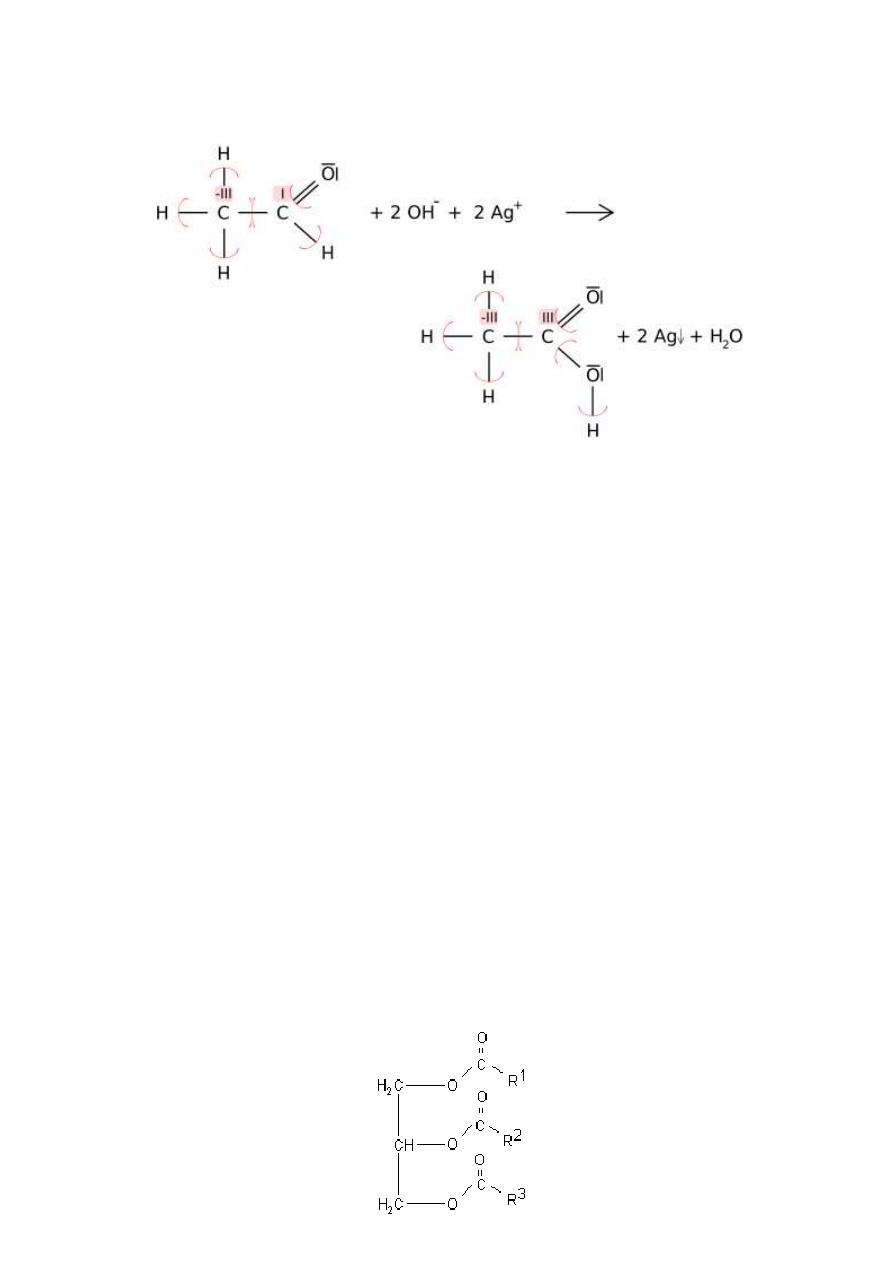
UTLENIANIE ALDEHYDÓW I KETONÓW:
Próba Tollens’a
Próba lustra srebrnego to reakcja chemiczna służąca do wykrywania aldehydów. Ketony dają negatywny wynik próby, z
wyjątkiem ketoz, takich jak fruktoza. Podczas pozytywnej próby Tollensa powstaje srebro metaliczne, osadzające się w
postaci lustrzanej powłoki na szklanej powierzchni naczynia reakcyjnego.
Próba Trommer’a
Reakcja chemiczna, która służy do wykrywania aldehydów. Ketony dają negatywny wyniki próby. Służy również do
określania właściwości redukujących cukrów. Monosacharydy oraz większość disacharydów dają pozytywny wynik próby.
Przykładem disacharydu niewykazującym właściwości redukujących jest sacharoza. Polisacharydy w większości wykazują
wynik negatywny. Odczynnik Trommera przygotowuje się, dodając roztwór wodorotlenku sodu do roztworu siarczanu(VI)
miedzi(II). Otrzymuje się niebieski, koloidalny osad wodorotlenku miedzi(II). Osad ten ogrzewa się z próbką. W przypadku
obecności aldehydów powstaje ceglastoczerwony osad tlenku miedzi(I).
2 Cu(OH)
2
+ CH
3
CHO + OH
-
→ Cu
2
O + CH
3
COO
-
+ 3 H
2
O
19. Kwasy karboksylowe – budowa, nazewnictwo, przykładowe związki. Wiązania wodorowe i ich wpływ na
właściwości fizyczne kwasów. Właściwości chemiczne kwasów (reakcje grupy karboksylowej, reakcje zachodzące na
atomie węgla a). Moc kwasów karboksylowych zawierających podstawniki przy atomach węgla a. Podstawowe
pochodne kwasów karboksylowych (estry, chlorki kwasowe, amidy).
Kwasy karboksylowe otrzymuje się przez utlenianie aldehydów. Mają właściwości kwasowe, które maleją ze wzrostem
długości łańcucha wodorowęglowego. Są to związki, które reagują z metalami (pierwiastki, które w szeregu napięciowym
posiadają niższy potencjał od wodoru) oraz tlenkami metali, a także wodorotlenkami i amoniakiem (tworząc sole). W
reakcji z alkoholami, kwasy ulegają estryfikacji.
Charakterystyczne reakcje:
POWSTAWANIE SOLI:
CH3COOH + NaOH → CH3COONa + H2O
POCHODNE - ESTRY
ESTRYFIKACJA:
Estry gliceryny i wyższych kwasów tłuszczowych to tłuszcze – nasycone (twarde, zwierzęce) i nienasycone – (płynne ,
roślinne).
HCOOH + CH3(stęż. H2SO4)→ HCOOCH3 + H2O
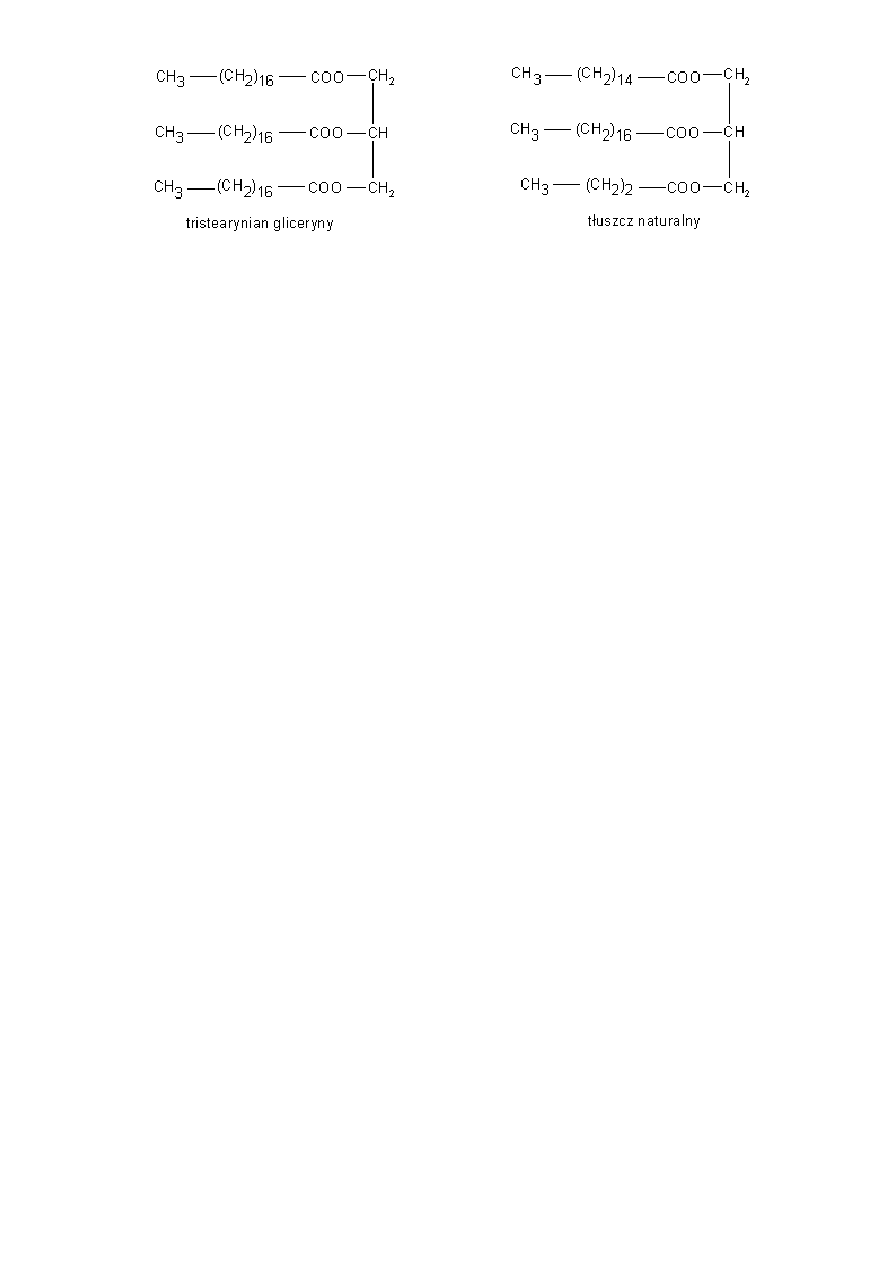
POCHODNE - AMIDY
OTRZYMYWANIE AMIDÓW:
W amidach występuje wiązanie peptydowe.
CH
3
COOH + NH
3
→ CH
3
COONH
4
-[temperatura]→ CH
3
CONH
2
POCHODNE – CHLORKI KWASOWE:
OTRZYMYWANIE CHLORKÓW ACYLOWYCH:
Chlorki kwasów karboksylowych czyli chlorki acylowe otrzymuje się w wyniku substytucji nukleofilowej w grupie acylowej
kwasu karboksylowego lub bezwodnika kwasu karboksylowego, przy użyciu związku z reaktywnymi atomami chloru,
najczęściej: SOCl
2
, PCl
3
, PCl
5
, POCl
3
, czyli w wyniku reakcji z chlorkami kwasów nieorganicznych.
ܴ − ܥܱܱܪ + ܱܵܥ݈
ଶ
→ ܴ − ܥܱܥ݈ + ܱܵ
ଶ
+ ܪܥ݈
20. Aminy – budowa (rzędowość), nazewnictwo, przykładowe związki. Wiązania wodorowe i ich wpływ na właściwości
fizyczne amin. Zasadowość amin.
Aminy to związki, które można potraktować jako pochodne amoniaku (NH
3
), w którym 1 atom wodoru (aminu 1 rzędowe),
lub więcej atomów wodoru (2 i 3 rzędowe) zostały zastąpione grupą alkilową, lub arylową. Aminy
alifatyczne mają
silniejszy charakter zasadowy niż amoniak, aromatyczne zaś – słabszy.
I i II rzędowe aminy z grupami alkilowymi mają charakterystyczny rybi zapach. Wszystkie trzy aminy z grupami
metylowymi są dobrze rozpuszczalnymi w wodzie gazami, podobnie jak amoniak. Aminy z wyższymi grupami alifatycznymi
są ciekłe lub stałe i ze wzrostem długości łańcuchów węglowych coraz gorzej rozpuszczają się w wodzie. Aminy
aromatyczne są wysokowrzącymi cieczami lub ciałami stałymi o ostrym, charakterystycznym ale nie rybim zapachu.
Własności chemiczne amin są zbliżone do amoniaku. Są to więc związki o silnych własnościach zasadowych, łatwo reagują
z kwasami nieorganicznymi i organicznymi oraz posiadają odczyn zasadowy w roztworach wodnych, gdyż w wodzie
ulegają one reakcji wg schematu:
R-NH
2
+ H
2
O → RNH
3
OH ↔ RNH
3
(+) + OH(-)
Zasadowość amin zależy od podstawników przy atomie azotu. Aminy alifatyczne są bardziej zasadowe od amoniaku i ich
zasadowość wzrasta wraz z rzędowością, zaś w przypadku amin aromatycznych jest dokładnie na odwrót – mają one
mniej zasadowe własności od amoniaku i spadają one ze wzrostem rzędowości. Z punktu widzenia biochemii
najważniejszą reakcją amin jest reakcja z kwasami karboksylowymi prowadząca do powstania wiązań peptydowych.

21. Biocząsteczki:
a) Lipidy – podział ze względu na budowę chemiczną. Tłuszcze - budowa i podstawowe reakcje-zmydlanie,
uwodornienie tłuszczów nienasyconych; działanie detergentów na przykładzie mydeł. Woski – budowa chemiczna,
funkcje biologiczne. Fosfolipidy – glicerofosfolipidy i sfingofosfolipidy – budowa chemiczna, funkcje biologiczne.
Steroidy – budowa chemiczna, funkcje biologiczne.
UTWARDZANIE TŁUSZCZY:
ZMYDLANIE TŁUSZCZY:
Hydroliza tłuszczy w środowisku zasadowym (nieodwracalna).
Woski - mieszaniny estrów kwasów tłuszczowych i alkoholi o długich łańcuchach węglowych
Fosfolipidy – zawierają ugrupowania estrowe kwasu fosforowego i alkoholi
Nie-estry :
Steroidy - organiczne związki chemiczne, lipidy, których wspólną cechą jest występowanie w ich cząsteczkach szkieletu
węglowego w formie czterech sprzężonych pierścieni, czyli steranu (cyklopentanoperhydrofenantrenu).
W tkankach roślin i zwierząt, jak dotąd wykryto istnienie kilkuset różnych steroidów, które pełnią w ich organizmach
rozmaite funkcje. W fizjologii i medycynie najważniejszymi steroidami są cholesterol i jego pochodne oraz hormony
steroidowe.
b) Sacharydy. Monosacharydy – definicja, pojęcia aldozy, ketozy; aldehyd glicerynowy jako najważniejsza trioza,
heksozy występujące w przyrodzie, wzory projekcyjne Fischera i Hawortha monosacharydów. Pojęcia anomerycznego
atomu węgla i anomerów, zjawisko mutarotacji. Disacharydy – definicja, najważniejsi przedstawiciele. Polisacharydy –
najważniejsi przedstawiciele;
Gwoli wstępu – cukry (węglowodany, sacharydy) składają się z wielu grup –OH (hydroksylowych) oraz jednej grupy, która
decyduje o przynależności do ketoz lub aldoz – grupa karbonylowa. Jeśli cząsteczka monocukru (mono, ponieważ składa
się tylko z jednej cząsteczki cukru) jest zakończona grupą aldehydową –CHO, to mamy doczynienia z aldozą. W przypadku
występowania grupy >C=O, jest to ketoza.
Na początek musimy poznać kilka podstawowych związków należących do szeregu węglowodanów. Najprostszą aldozą
jest aldehyd glicerynowy, który jest triozą (nazwy cukrów pochodzą od ilości atomów węgla w głównym łańcuchu – triozy,
tetrozy, pentozy, heksozy, etc. - większość biologicznie ważnych monosacharydów ma 5 lub 6 atomów węgla, choć w
fizjologii komórek (fotosynteza, cykl Krebsa) znaczenie mają też monosacharydy 3- i 4-węglowe, a spotyka się też
monosacharydy i ich pochodne o większej niż 6 liczbie atomów węgla):
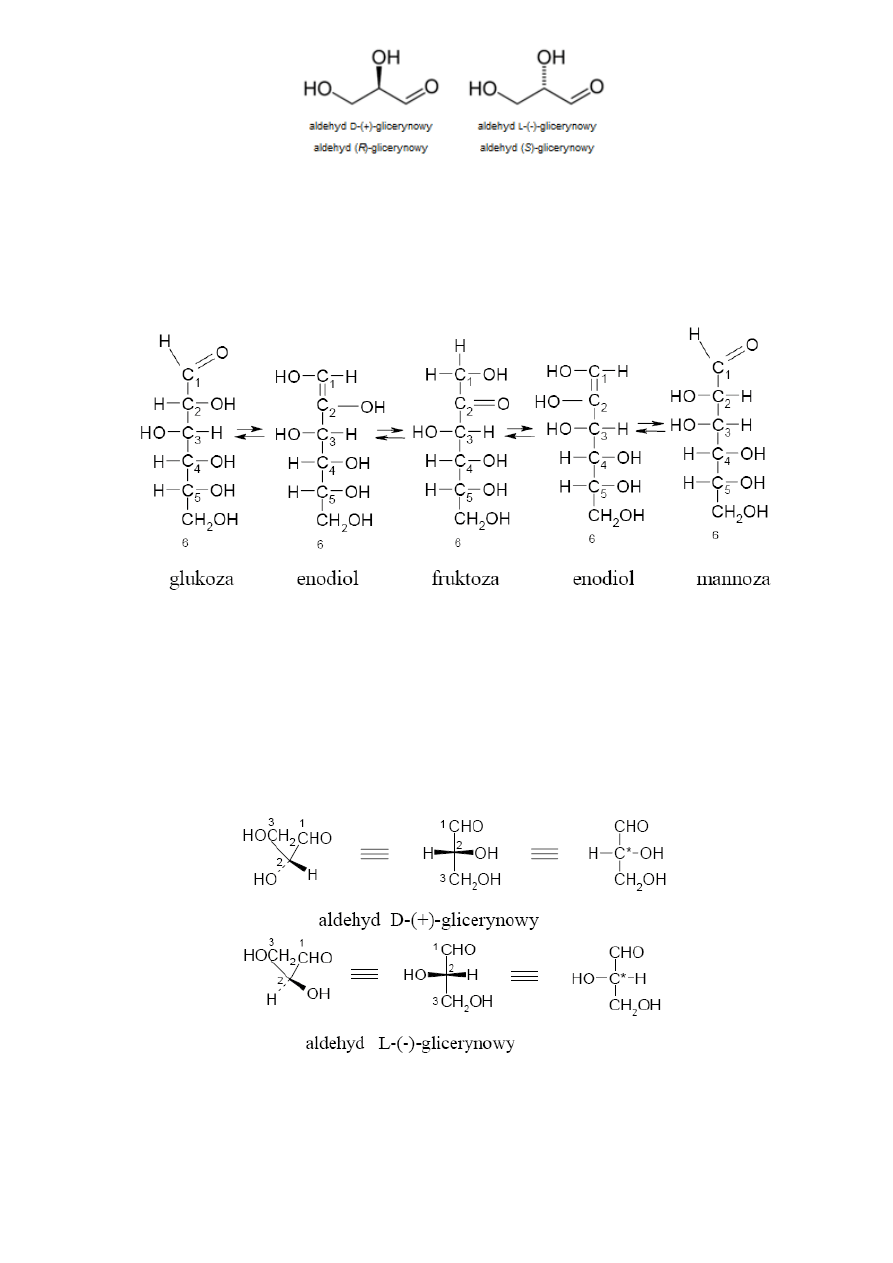
Należy pamiętać o izomerii optycznej, która odgrywa ważną rolę w przypadku cukrów. Np. D-glukoza jest wchłaniana
przez ludzki organizm, ale już L-glukoza nie. Swego czasu (o ile dobrze pamiętam) był taki lek przeciwbólowy, który w
przypadku zażywania przez kobiety w ciąży dawał dwojakie skutki – jedna z konformacji powodowała poronienia. Niby
malutka różnica, a jednak zupełnie inny związek. Podobnie jest z aminokwasami – znanych jest ponad 300 aminokwasów
występujących naturalnie. W skład białek wszystkich organizmów żywych wchodzi głównie 20 podstawowych
aminokwasów (aminokwasy biogenne lub białkowe), które są α-aminokwasami szeregu L (przeciwnie do sacharydów).
Następnymi związkami są heksozy występujące w przyrodzie – glukoza, fruktoza i mannoza.
Granica podziału na ketozy i aldozy jest dość umowna ze względu na duże możliwości przegrupowania grup funkcyjnych.
Dla węglowodanów, w środowisku zasadowym zachodzi reakcja epimeryzacji – reakcja w wyniku której następuje zmiana
konfiguracji podstawników przy węglu asymetrycznym połączonym z grupą aldehydową (powstaje epimer). Proces ten
zachodzi poprzez stadium endioli (powyżej schemat przedstawiający epimeryzację glukozy).
Teraz coś nie coś o samej budowie cukrów, czyli o co chodzi z D i L, wzory projekcyjne Fishera i Hawortha monosacharydów
oraz jak się tworzą pierścienie i czym jest mutarotacja.
Asymetryczny atom węgla (centrum asymetrii), oznacza taki atom węgla, który ma cztery różne podstawniki. Aldehyd
glicerynowy, czyli aldotrioza jest wzorcem do ustalania konfiguracji cukrów. Ma jeden asymetryczny atom węgla.
Jeśli grupa OH znajduje się po prawej stronie centrum asymetrii mówi się o odmianie D, a jeśli grupa OH znajduje się po
lewej stronie centrum asymetrii mówi się o odmianie L. Cukry naturalne należą w większości do szeregu D. Otrzymane
syntetycznie enancjomery tych cukrów, należące do szeregu L, nie są przyswajalne przez organizmy żywe. W przypadku
cukrów o większej ilości atomów węgla, o przynależności do odpowiedniego szeregu decyduje położenie podstawników
przy ostatnim asymetrycznym atomie węgla.
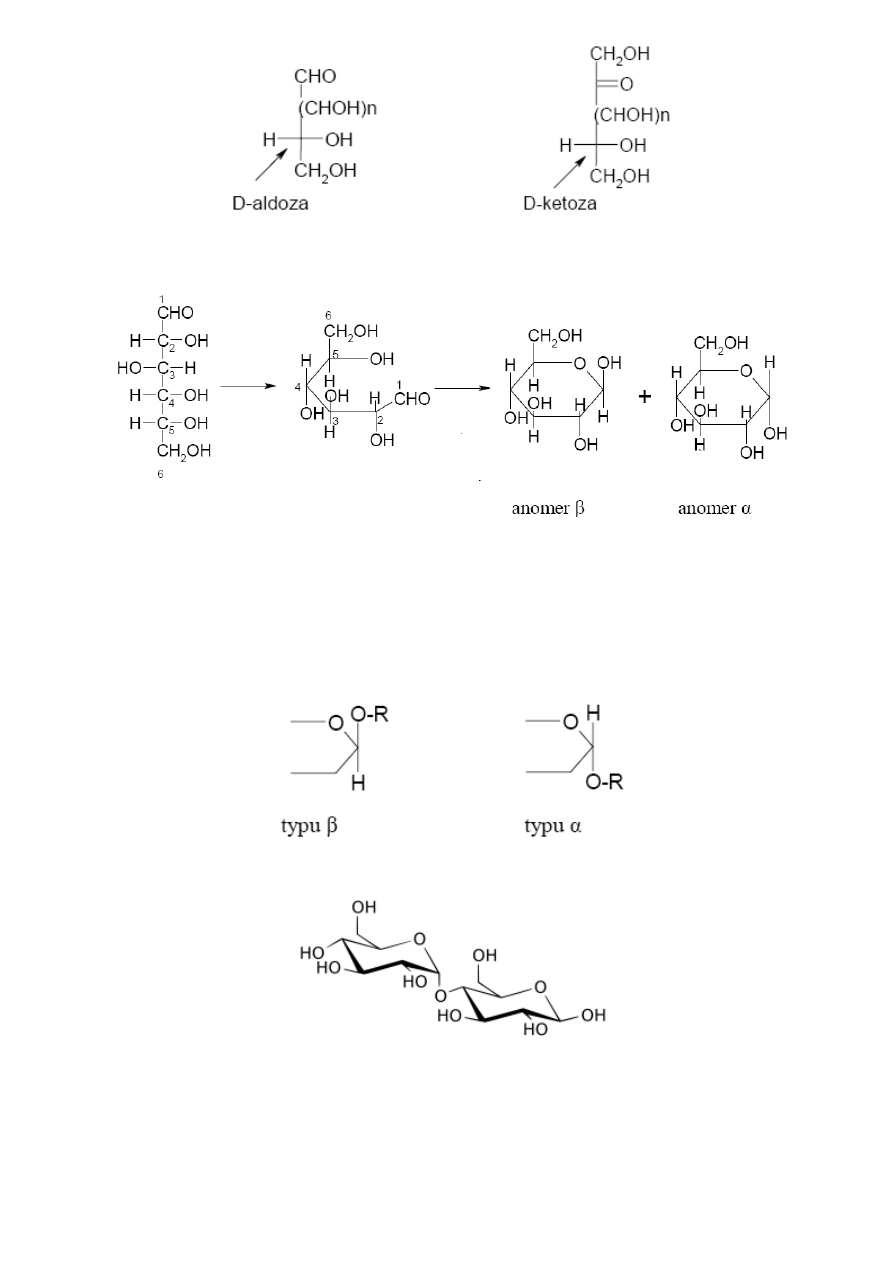
Na skutek utworzenia pierścienia półacetalowego pierwszy atom węgla staje się węglem asymetrycznym, w związku z
czym powstają dwie odmiany D-glukozy: α-D-glukoza, β-D-glukoza. Gdy grupa OH przy tym atomie węgla znajduje się pod
płaszczyzną pierścienia wówczas jest to anomer α, a jeśli nad płaszczyzną pierścienia, to anomer β.
Mutarotacja to zjawisko jakie zachodzi dość swobodnie przy rozpuszczeniu cukru w roztworze (np. wodnym). Jest to
zmiana kąta skręcalności optycznej roztworu substancji czynnej, aż do pewnej stałej wartości. Przyczyną mutarotacji jest
przejście w roztworze jednego z izomerów w drugi o innej skręcalności światła spolaryzowanego. Między izomerami ustala
się równowaga dynamiczna. α-D-glukoza po krystalizacji z metanolu, rozpuszczona w wodzie posiada początkową
skręcalność roztworu [α]D = +112º lecz w miarę upływu czasu skręcalność ta stopniowo spada i osiąga stałą wartość [α]D
= +52,5º. W stanie równowagi jest różna ilość odmian α i β. α–D-glukozy jest 36%, formy łańcuchowej 0,02%, a β-D-
glukozy 64%. Mutarotację katalizują zarówno kwasy jak i zasady. Reakcja przebiega poprzez odmianę łańcuchową
(aldehydową).
Cukry mogą tworzyć wiązania glikozydowe, których rodzaje są przedstawione powyżej. Dzięki takim połączeniom między
cząsteczkami węglowodanów powstają cukry złożone (disacharydy, trisacharydy, oligosacharydy, polisacharydy). Na
przykład maltoza to disacharyd składający się z dwóch cząsteczek glukozy, połączonych wiązaniem α-1,4 glikozydowym:
Ważnymi disacharydami są również laktoza(4-O-β-D-galaktopiranozylo-D-glukopiranoza – D-glukoza i D-galaktoza
połączone wiązaniem β) i sacharoza(β-D-fruktofuranozylo-α-D-glukopiranozyd - fruktoza (β-D-fruktofuranoza) oraz
glukoza (α-D-glukopiranoza) - grupy te są połączone za pomocą wiązania α,β-1,2-glikozydowego).
Na koniec zajmiemy się jeszcze wzorami projekcyjnymi Fishera i Hawortha, ale tylko przedstawiając konkretny przykład w
postaci glukozy. Projekcja Hawortha dotyczy przedstawiania cukrów w ich formie cyklicznej, z wiązaniem półacetalowym
(według wielkości pierścienia, monosacharydy nazywamy furanozami lub piranozami; furanoza zawiera 5 członowy
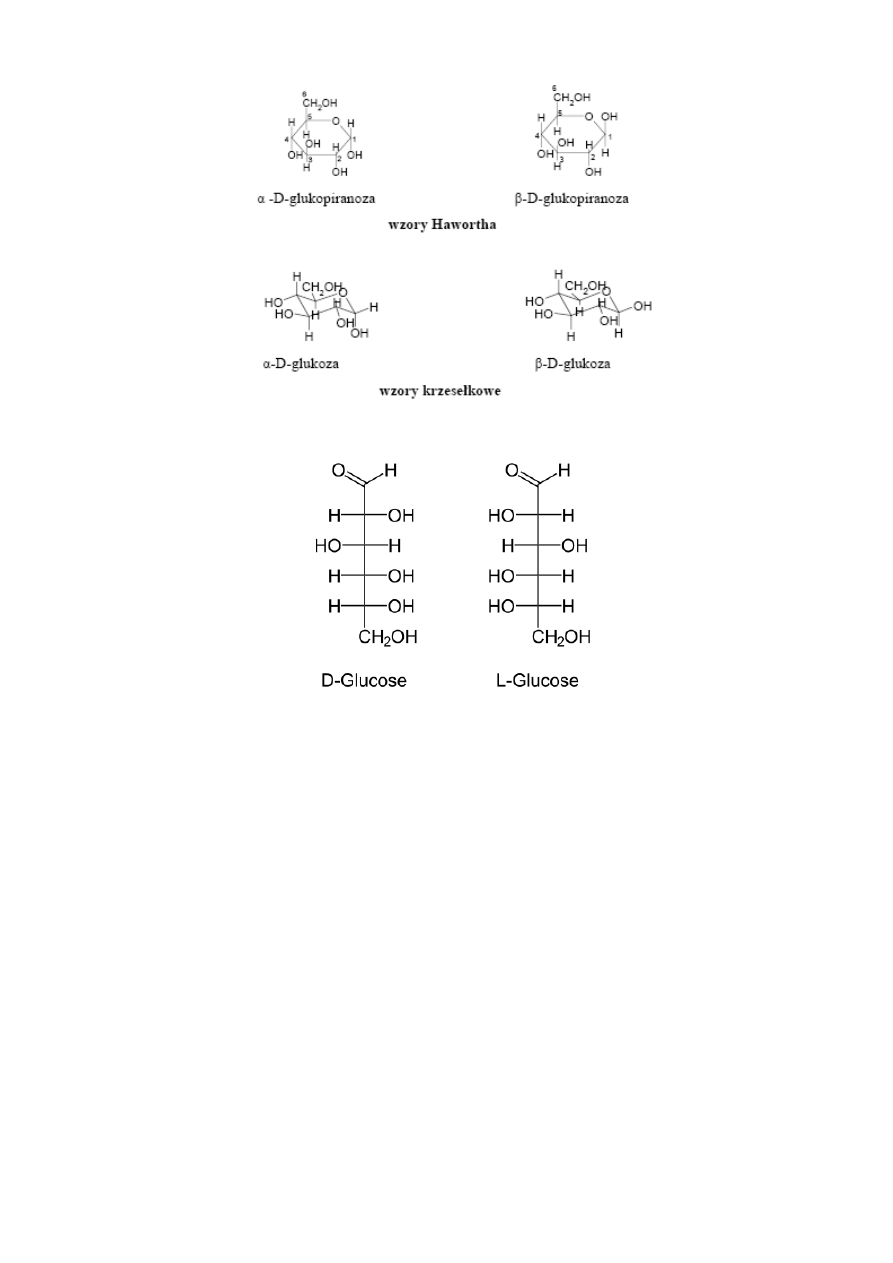
pierścień z 4 węgli i jednego atomu tlenu, zaś piranoza ma 5 atomów węgla w pierścieniu połączonym atomem tlenu; w
nazwie, przed rdzeniem -furanoza lub -piranoza umieszcza się przedrostek określający budowę cukru).
Projekcja Fishera dotyczy przedstawiania związków z pominięciem atomów węgla w łańcuchu – oczywiście krańcowe
atomy węgla, zawierające grupy funkcyjne przedstawia się już normalnie (np. COOH, CHO, CH
2
OH – zależy od inwencji
autora grafiki :P ).
Ponieważ w zaleceniach nic nie pisze o reakcjach cukrów więc nasze słodkie rozważania zakończymy przedstawicielami
polisacharydów w postaci skrobi, która składa się z bardzo wielu glukoz ((C
6
H
10
O
5
)n gdzie n>300). :P
c) Aminokwasy – budowa, tworzenie soli wewnętrznych, właściwości amfoteryczne, pojęcie punktu izoelektrycznego.
Tworzenie peptydów. Białka jako polipeptydy.

Pytania sesja A.D. 2008
1. Podane były dwa związki: trzeba było podać ich nazwę i do jakich grup związków organicznych należą (na zerówce był
akurat alkohol i keton, wzorów nie pamiętam niestety).
2. Podane były 2 związki (znowu keton i aldehyd) trzeba było zaznaczyć, który związek ulega próbie Tollensa i uzasadnić
wybór.
3. Było podane kilka związków, spośród nich trzeba było wybrać taki, który zawiera asymetryczny atom węgla i wyjaśnić
jakie konsekwencje niesie on dla związku.
4. Były podane 3 reakcje chemiczne, spośród nich trzeba było wybrać reakcje eliminacji i nazwać pozostałe dwie (na
zerówce była Sn1 i Sn2).
5. Były podane 3 związki i trzeba było wybrać malejącą kolejność rozpuszczania ich w wodzie (chodzi o ilość grup -OH, im
jest ich więcej tym lepiej związek się rozpuszcza itd.).
6. Były podane związki trzeba było wybrać parę izomerów i ogólnie nazwać rodzaje izomerii.
7. 3 związki podane, trzeba było wybrać parę takich które z wodorotlenkiem sodu reagowały i uzasadnić odpowiedź.
8. Była opisana przemiana czegoś w coś (nie pamiętam związków) i trzeba było nazwać reakcje składowe: pełna nazwa +
symbol.
9. Był podany jakiś związek aromatyczny i reakcja przyłączania, trzeba było nazwać produkt przyłączania.
10. Wybrać spośród związków taki który posiada anomeryczny atom węgla i wyjaśnić co to jest anomeryczny atom węgla.
11. Wybrać reakcję którą ulegają tłuszcze (spośród wymienionych) i napisać równanie reakcji .
12. Duże otwarte zadanie o Nitrowaniu (mechanizm, wpływ kierujący podstawników, produkty reakcji otd./, związki były
podane).
13. Duże otwarte zadanie o Sn1 (albo Sn2, nie pamiętam) - napisać równanie reakcji, stereochemia, nazwa pełna +
symbol, związki ofc tez podane były
14. Duże zadanie o substytucji wolnorodnikowej (etapy, mechanizm, itd.)
W przypadku błędów w notatce lub pytań i sugestii, proszę kontaktować się z autorem.
Grupa II laboratorium IV.
Międzywydziałowa Szkoła Inżynierii Biomedycznej.
mail – michalgasior89@gmail.com
www - http://student.agh.edu.pl/~bonesaaa/
Pozdrawiam,
Mike.
Wyszukiwarka
Podobne podstrony:
Attribution of Hand Bones to Sex and Population Groups
ANIMAL BONES
Your Bones in Space
bones&fracture
Egil`s Bones
Swanwick Bones of the?rth
BONES METASTASES pol dla studentów
Asterisk A Bare Bones VoIP Example
Attribution of Hand Bones to Sex and Population Groups
Debt of Bones
BONES Nowe życie ale inne niż wyśnione
BONES Ja też umarłam
BONES Dlaczego oddalasz się odemnie
BONES 16
BONES Terapia
BONES Wypadek
BONES Czemu NIE WOLNO dawać broni Zezulcom
więcej podobnych podstron