Z W I Ą Z K I K A R B O N Y L O W E
2007. 08.
Aleksander Kołodziejczyk
Grupa karbonylowa -C=O występuje w wielu związkach organicznych, ale tylko aldehydy i
ketony nazywane są związkami karbonylowymi. W jedynie nich karbonylowy atom węgla jest
związany wyłącznie z atomami wodoru lub/i węgla. W pozostałych związkach zawierających
grupę -C=O, tj. w kwasach karboksylowych i ich pochodnych do karbonylowego atomu węgla
dołączona jeszcze inna grupa funkcyjna, np. -OH, -OR, -COR czy -X.
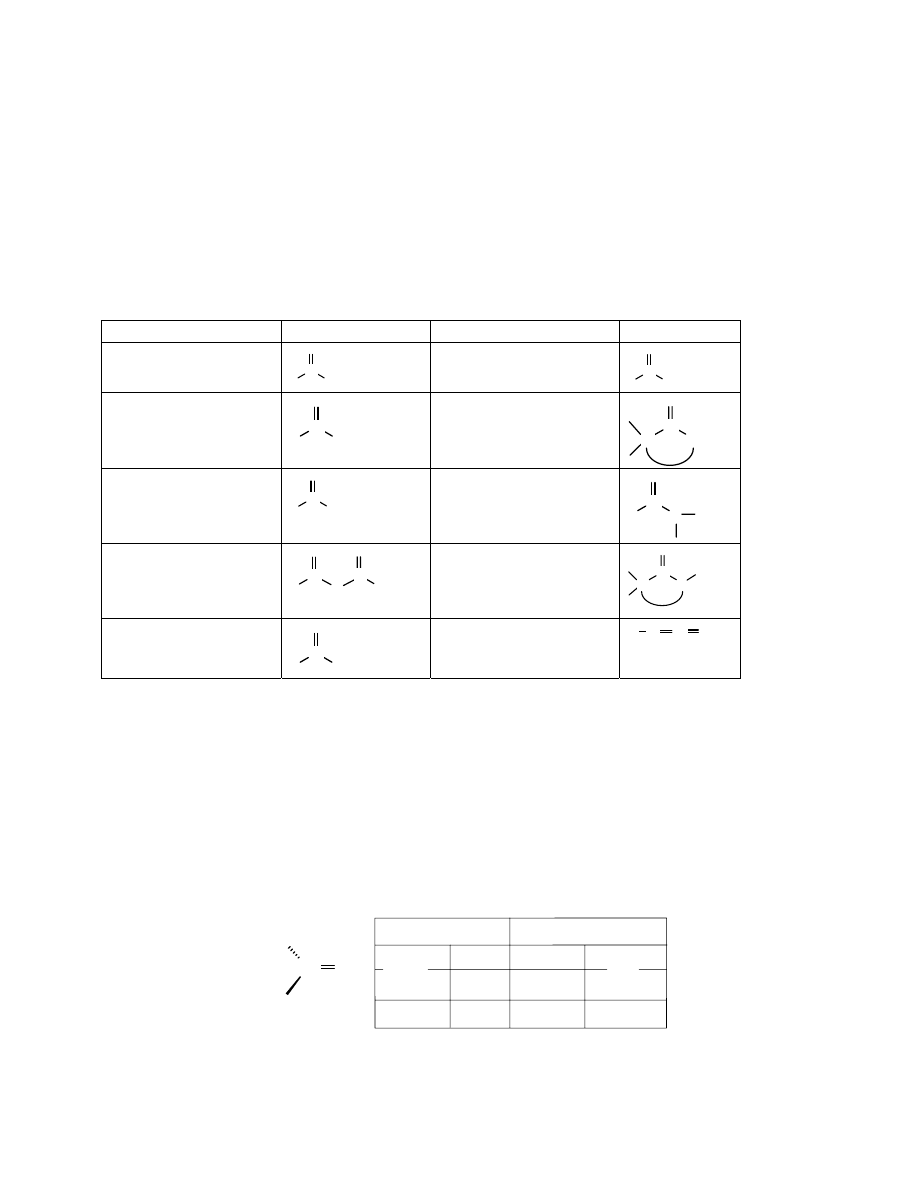
Związki organiczne zawierające grupę karbonylową
Tabela 11.1
Nazwa Wzór
ogólny
Nazwa
Wzór ogólny
aldehyd
O
R
H
C
ester
O
R
OR'
C
keton
O
R
R'
C
lakton (ester cykliczny)
O
O
C
C
kwas karboksylowy
O
R
OH
C
amid
O
R
N
C
bezwodnik kwasowy
O
R
O
O
R'
C
C
laktam (amid cykliczny)
O
N
C
C
chlorek kwasowy
(halogenek)
O
R
Cl
C
keten
R C C O
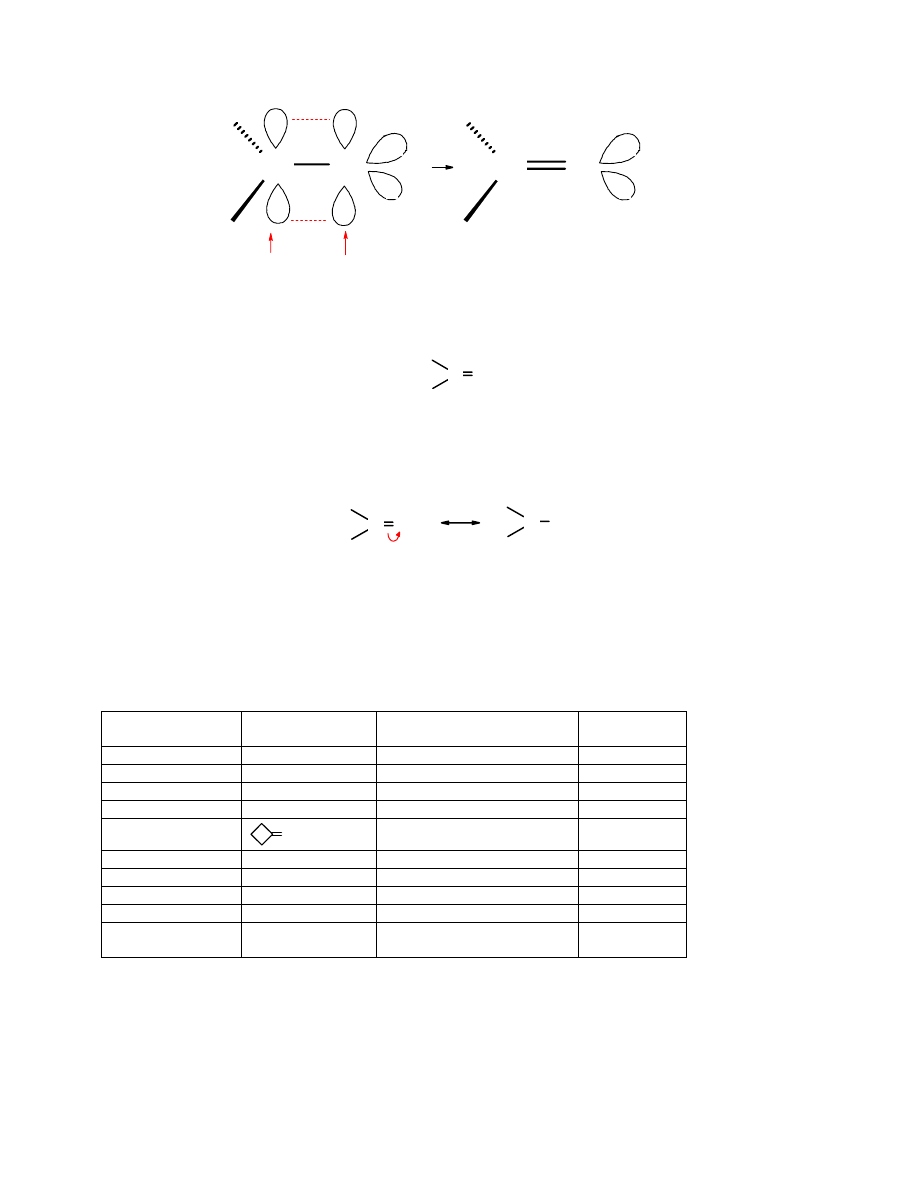
Budowa grupy karbonylowej
Grupa karbonylowa C=O jest pod względem geometrycznym podobna do podwójnego wiązania
C=C. Wokół atomu C o hybrydyzacji sp
2
rozmieszczone są 3 podstawniki. Ułożone są one w
jednej płaszczyźnie pod kątem zbliżonym do 120
o
. Wiązanie C=O jest krótsze niż C=C (1,22 Å i
1,43Å odpowiednio) i znacznie silniejsze – 175 kcal/mol (732 kJ/mol) wobec 92 kcal/mol (385
kJ/mol). Silniejsze nie oznacza mniej reaktywne. Wiązanie karbonylowe jest mocniej
spolaryzowane, co rzutuje na jego właściwości chemiczne i wysoką reaktywność.
Tabela 11.2
H
C
H
3
C O
parametry fizyczne cząsteczki etanalu
Kąty walencyjne [
o
]
Długości wiązań [A]
H-C-C
118
C=O
1,22
C-C-O
121
H-C-O
121
C-C
1,5
OC-H
1,09
Karbonylowy atom węgla o hybrydyzacji sp
2
tworzy 3 wiązania typu
σ i jedno π.
1
C
O
.
.
..
..
C
O
..
..
elektrony na niezhybrydyzowa-
nych orbitalach p po nałożeniu
tworzą wiązanie typu
π
orbitale p
Związki zawierające grupę karbonylową są polarne, ponieważ wiązanie C=O jest
spolaryzowane. Łatwo się domyślić, że częściowy ładunek dodatni jest zlokalizowany na atomie
węgla, zaś ujemny na atomie tlenu.
O
C
+δ -δ
..
..
Grupę karbonylową można przedstawić w postaci wzorów mezomerycznych, przy czym na
jednym z nich są całkowicie rozdzielone ładunki. Prawdopodobieństwo występowania grupy
karbonylowej z rozdzielonymi ładunkami jest niskie, ale wyjaśnia ono podatność tego typu
związków na atak odczynników nukleofilowych i kwasów:
O
O
C
..
..
C ..
..
:
+ -
+δ -δ
Dodatkowe podstawniki i grupy funkcyjne mogą zwiększać lub zmniejszać polarność tego
wiązania, a tym samym wpływać na wielkość momentu dipolowego cząsteczki i reaktywność
grupy. W tabeli przedstawiono wartości momentów dipolowych wybranych związków
zawierających grupę karbonylową.
Wartości momentu dipolowego wybranych związków
Tabela 11. 3
zawierających grupę karbonylową
Nazwa Wzór Grupa
związku Moment
dipolowy [D]
metanal
H
2
C=O
aldehyd
2,33
etanal
CH
3
HC=O
aldehyd
2,72
aceton
(CH
3
)
2
C=O
keton
2,88
acetofenon
PhCOCH
3
keton
3,02
cyklobutanon
O
keton
2,99
kwas octowy
CH
3
COOH
kwas karboksylowy
1,74
chlorek acetylu
CH
3
COCl
chlorek kwasowy
2,72
octan metylu
CH
3
COOCH
3
ester
1,72
acetamid
CH
3
CONH
2
amid
3,72
N,N-dimetylo-
acetamid
CH
3
CON(CH
3
)
2
amid
3,81
2
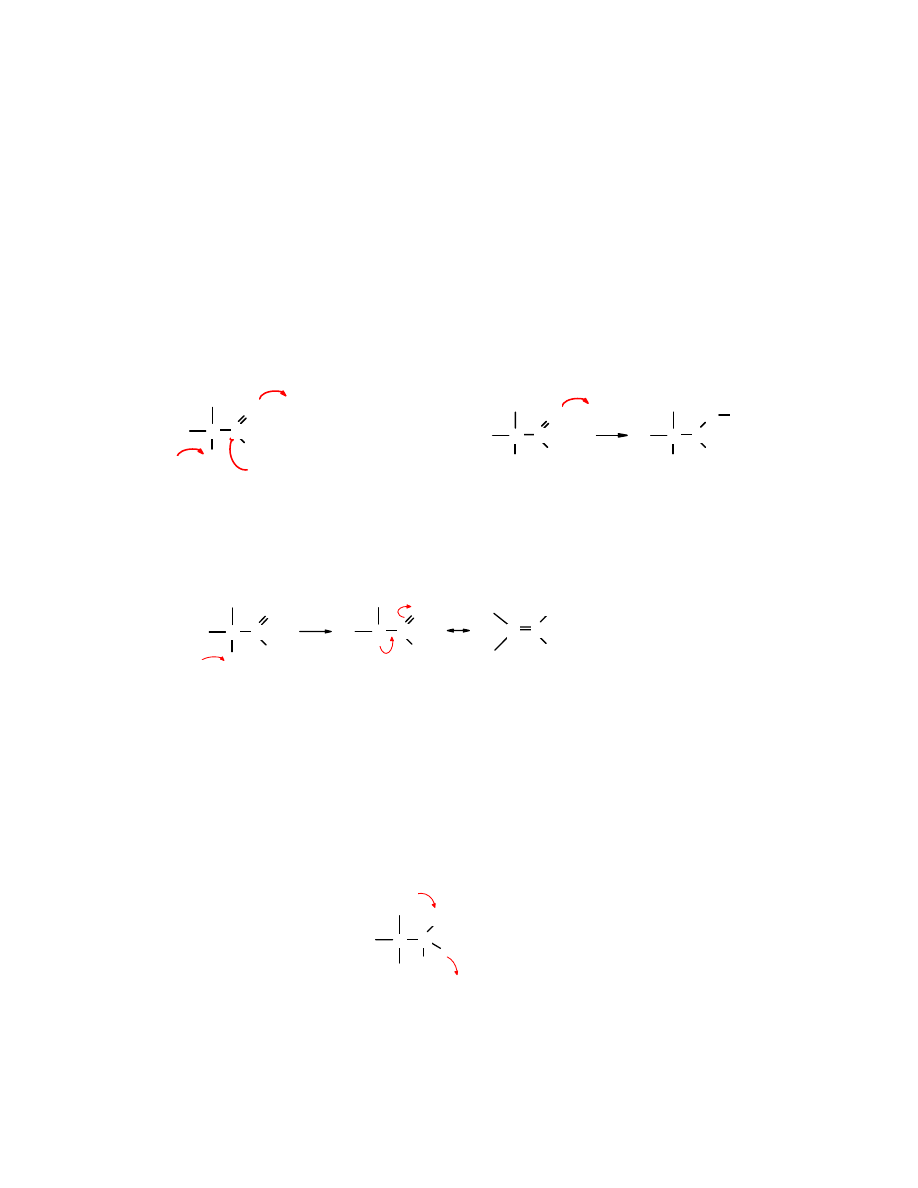
Reaktywność związków karbonylowych
Reaktywność związków zawierających grupę karbonylowa jest związana z właściwościami
elektronowymi atomów wchodzących w jej skład, przede wszystkim z dodatnio naładowanym
karbonylowym atomem węgla C
+δ
i ujemnie naładowanym atomem tlenu O
-
δ
.
Karbonylowy atom węgla C
+δ
jest podatny na atak odczynników nukleofilowych (
:Nu
), a atom
tlenu O
-
δ
na działanie kwasów (
A
). Powinowactwo karbonylowego atomu węgla do nukleofila
:Nu
będzie modyfikowane właściwościami elektronowymi związanej z nim grupy funkcyjnej Y.
Grupy elektronoakceptorowe będą zwiększały powinowactwo, a eletronodonorowe –
zmniejszały. Reaktywne są również atomy wodoru przy C
α
. Mają one podwyższoną ruchliwość.
Sąsiedztwo grupy karbonylowej zwiększa ich kwasowość w porównaniu z typowym
alifatycznym atomem wodoru, a przez to łatwiej ulegają oderwaniu pod wpływem zasad (
B:
).
H
Y
O
H
O
Y
H
O
Y
C
:Nu
:
..
A
:B
+
δ
-
δ
C
:B
:Nu
nukleofil
zasada
A
kwas
C C
:
..
+
A
+
δ
-
δ
C C
:
..
A
+
A
– kwasy Lewisa (np.:
+
H,
+
NH
2
, BF
3
, AlCl
3
, itp.); wykazują duże powinowactwo do
karbonylowego atomu tlenu O; tworzą z nim kompleksy.
:B
– zasady Lewisa (np.:
-
H,
-
NH
2
,
-
OH,
-
OR, NR
3
, itp.) mają powinowactwo do ruchliwych
protonów – odrywają H przy C
α
w stosunku do grupy karbonylowej:
H
O
Y
O
Y
O
Y
:B
C C
:
..
+
δ
-
δ
C C
:
..
-
-
+
BH
C C
:
..
-
:
↑
Powstający po oderwaniu protonu karboanion jest podatny na atak odczynnika elektrofilowego –
E
+
; ten etap zapoczątkowuje reakcje kondensacji karbonylowej.
:Nu
– wykazuje powinowactwo do dodatnio naładowanego węgla grupy karbonylowej i reakcja z
nim zaczyna się od addycji do karbonylowego atomu węgla. Dalszy bieg reakcji zależy od
właściwości podstawnika
Y
:
– może nastąpić protonowanie atomu O lub
– eliminacja podstawnika
-
Y
, jeżeli jest dobrą grupą odchodzącą.
O
Y
Nu
+
H
C C
:
..
-
:
protonowanie
atomu O
eliminacja
grupy Y
albo
Do najpopularniejszych reakcji, jakim ulegają związki zawierające grupę karbonylową należą:
addycja nukleofilowa substytucja
α
i
acylowa substytucja nukleofilowa
kondensacja karbonylowa
3
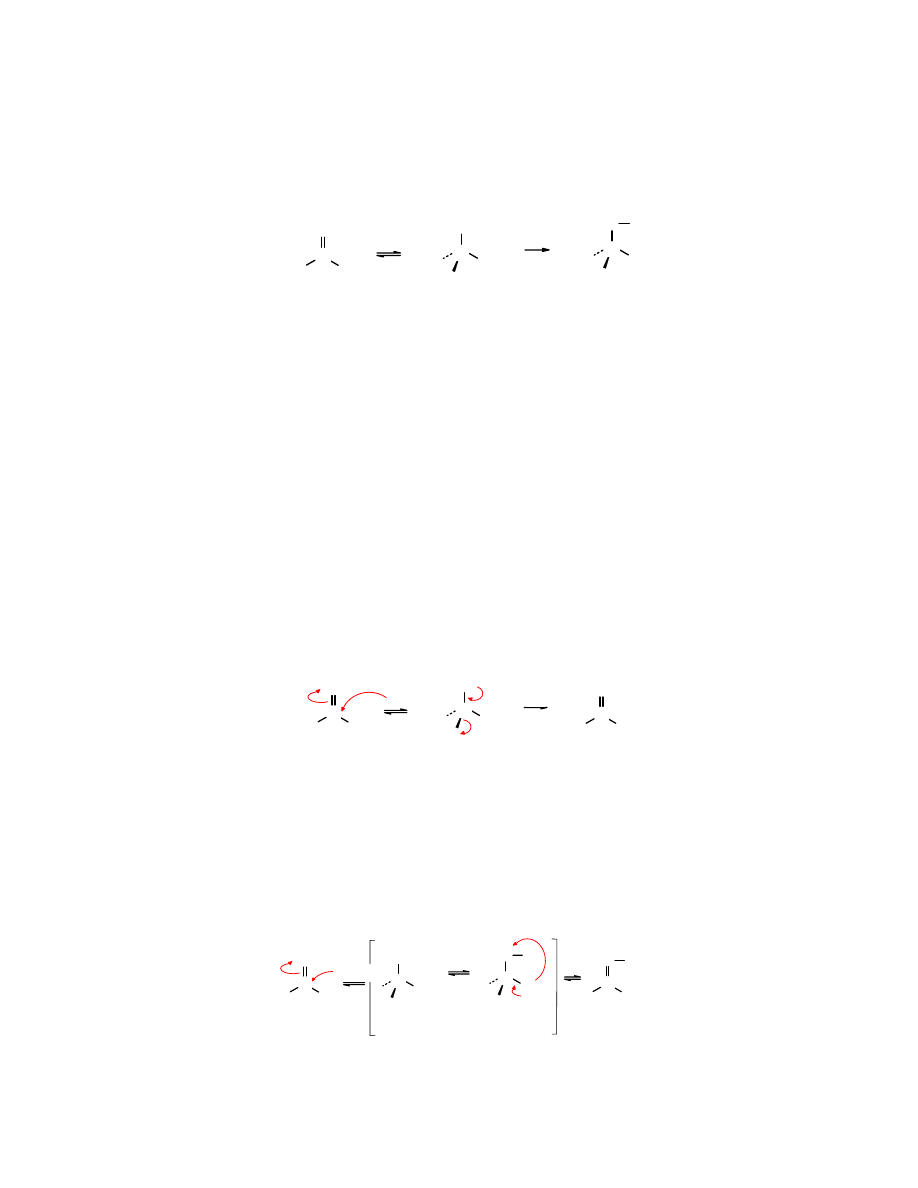
Addycja nukleofilowa do grupy karbonylowej
Jeżeli podstawnik grupy karbonylowej -Y jest złą grupą odchodzącą (-H lub -R) to w reakcji z
nukleofilem dochodzi do
addycji nukleofilowej
, przy czym zmienia się hybrydyzacja obu
atomów tworzących grupę karbonylową (z sp
2
na sp
3
), a po sprotonowaniu produktu addycji
(produktu pośredniego) powstaje alkohol. Tak z nukleofilami reagują aldehydy i ketony.
..
O
R
Y
O
R
Y
Nu
O
R
Y
Nu
H
C
:Nu
C
-
: :
: :
..
Csp
2
Csp
3
+
H
C
:
Csp
3
aldehyd lub keton produkt addycji alkohol
R i Y: H, alkil lub aryl Nu:
-
H (jon wodorkowy) lub związki Grignarda (R
’
MgX)
Substytucja na karbonylowym atomie węgla
Reakcja substytucji
na grupie C=O może biec dwoma sposobami (
A
i
B
). Wg
A
w końcowym
produkcie zostaje zachowana grupa C=O, a wg
B
tworzy się ugrupowanie C=Nu.
Sposób
A
Związki karbonylowe zawierające łatwo odchodząca grupę Y, np. -OH, -OR, -SR, -C(O)OR,
lub -halogen po przyłączeniu nukleofila
:Nu
tworzą produkt pośredni (addukt), z którego
odszczepia się grupa Y i powstaje inny związek zawierający grupę karbonylową, np. kwas
karboksylowy zostaje przekształcony w ester, chlorek kwasy w amid itp. Są to reakcje
charakterystyczne dla kwasów karboksylowych i ich pochodnych. Nie wszystkie przedstawione
powyżej podstawniki Y należą do grup łatwo odchodzących, ale w odpowiednich warunkach
mogą zostać w takie grupy przekształcone, np. -OH czy -OR po protonowaniu do H
-
O
+
H
2
lub -
O
+
RH stają się grupami łatwo odchodzącymi.
O
R
Y
O
R
Y
Nu
O
R
Nu
C
:Nu
C
-
: :
: :
..
Csp
2
Csp
3
-
Y
-
Csp
2
+
δ
-
δ
C
: :
+
δ
-
δ
R: H, alkil lub aryl Y: halogen; -OR, -OH, -SR, -C(O)OR
Kwasy karboksylowe i ich pochodne reagują z nukleofilami według sposobu
A
.
Sposób
B
Aldehydy i ketony po addycji do karbonylowego atomu niektórych związków zawierających
grupę aminową (-NH
2
) eliminują atomu tlenu w postaci cząsteczki wody (łatwo odchodząca
grupa) i powstaje imina.
O
R
Y
O
R
Y
NH
2
R'
O
R
Y
NHR'
H
N
R
Y
R'
C
:NH
2
R'
C
-
: :
: :
..
Csp
2
Csp
3
+
δ
-
δ
+
C
:
..
Csp
3
..
C
:
+
δ
-
δ
Csp
2
- HOH
aldehydy lub ketony iminy
Y: H, alkil lub aryl : NH
2
R’: aminy 1
o
, hydrazyna i inne związki zawierające grupę -NH
2
4
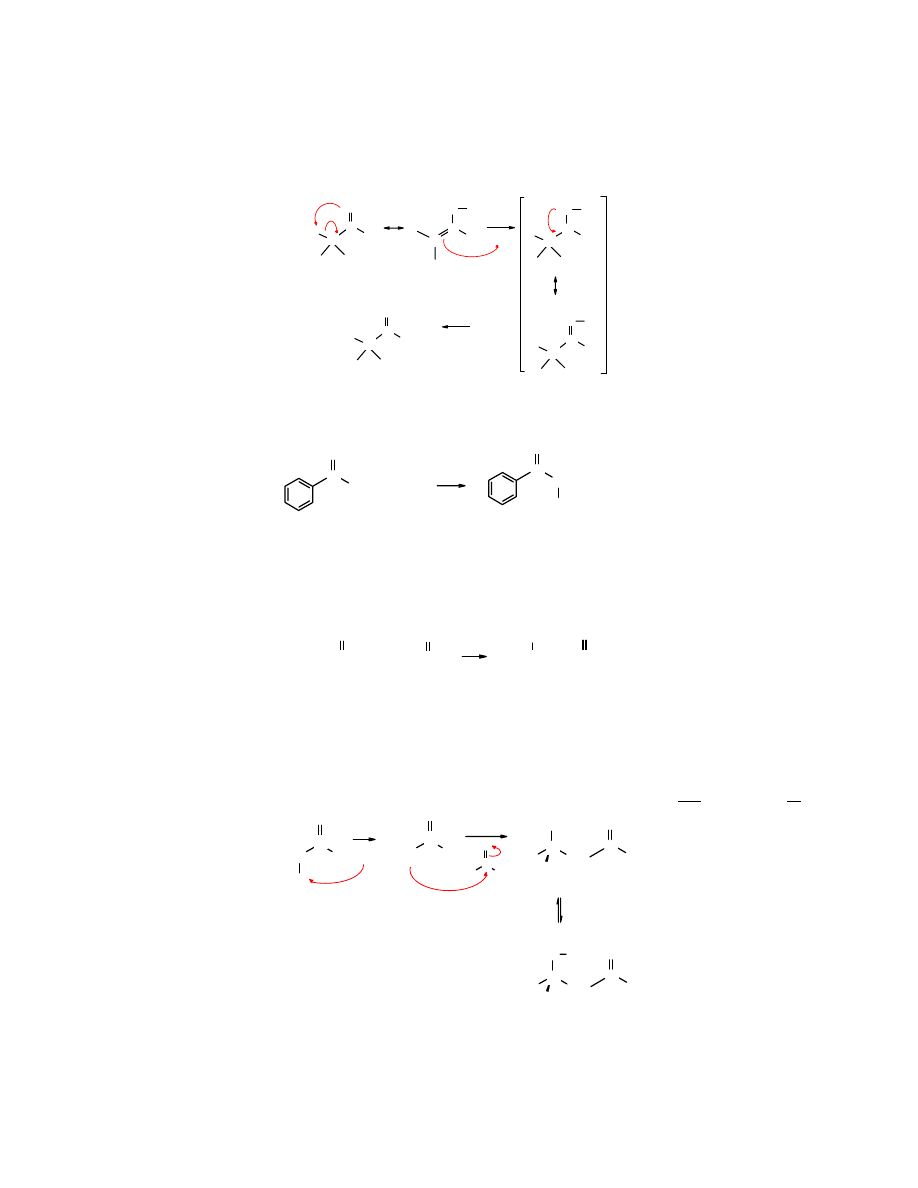
Substytucja
α
W reakcji substytucji
α
dochodzi do podstawienia uaktywnionego atomu wodoru przy C
α w
stosunku do grupy karbonylowej. Miejsce H
α
może zająć elektrofil, np. halogen lub alkil.
Reakcja biegnie poprzez formę enolanową.
H
O
Y
O
Y
E
O
Y
E
O
Y
H
H
H
E
O
Y
C
C
- H
+
C
C
..
:
+
E
..
C
C
:
:
C
C
: :
+
+
:
C
C
:
związek karbonylowy
α-podstawiony związek karbonylowy
Przykładem takiej reakcji może być otrzymywanie
α-bromoacetofenonu
.
Związek ten jest silnym
lakrymatorem (drażni oczy).
O
CH
3
O
CH
2
Br
C
+ Br
2
C
+ HBr
AcOH
acetofenon
α-bromoacetofenon
(72%)
Reakcje kondensacji związków karbonylowych
W środowisku zasadowym aldehydy i ketony zawierające H
α
ulegają dimeryzują.
O
O
OH
O
CH
3
CH + CH
3
CH
CH
3
CHCH
2
CH
-
OH
etanal etanal 3-hydroksybutanal
Mechanizm
Reakcja zaczyna się od oderwania protonu H
α
i utworzenia karboanionu, który jako nukleofil
przyłącza się do karbonylowego atomu węgla wg uprzednio poznanych reguł. Po zakwaszeniu
powstałego enolanu tworzy się
aldol
. Ta nazwa to skrót sporządzony ze słowa aldehydoalkohol.
..
..
O
H
2
C
H
H
O
H
2
C
H
O
C
H
3
CH
2
O
C
H
3
H
H
H
O
O
C
H
3
CH
2
H
H
O
H
:
C
:B
-
- HB
C
..
-
C
C
:
..
-
C
:
C
..
C
+
H/HOH
etanal
aldol
(3-hydroksybutanal)
:
:
Do tego typu reakcji należy kondensacja aldolowa aldehydów i ketonów, kondensacja Claisena
estrów i szereg innych podobnych reakcji.
5
A L D E H Y D Y I K E T O N Y
Do aldehydów zalicza się związki zawierające przy karbonylowym atomie węgla resztę
organiczną i atom wodoru. Szczególnym przypadkiem jest
metanal
(
aldehyd mrówkowy
inaczej
formaldehyd
), ponieważ z karbonylowym atomem węgla związane są dwa atomy wodoru.
W ketonach do grupy karbonylowej przyłączone są dwie reszty organiczne.
O
R
H
O
R
R'
C
C
aldehydy, R: alkil, aryl lub H ketony, R, R’: alkil lub aryl
Występowanie
Aldehydy są związkami nietrwałymi, ponieważ łatwo ulegają utlenieniu. Dlatego chociaż
występują często w naturze, rzadko spotyka się je w dużych stężeniach. Są składnikami
niektórych olejków eterycznych, np.:
cytronellal
,
neral
(
cytral b
),
geranial
(
cytral a
),
aldehyd
benzoesowy
(
benzaldehyd
) czy
aldehyd anyżowy
. Znane są też pochodne
aldehydu 3,4-
dihydroksybenzoesowego
. Aldehydy należą do ważnych substratów w biosyntezie.
CHO
CHO
CHO
H
H
CHO
CHO
aldehyd
benzoesowy
α
β
cytranellal neral geranial
Aldehyden jest
aldosteron
, przedstawiciel hormonów kortykosterydowych (hormonów
nadnercza). O obecności grupy aldehydowej świadczy nazwa tego związku –
aldosteron
.
O
O
H
COCH
2
OH
H
H
H
OHC
aldosteron
Popularnymi aldehydami są cukry – aldozy, np.
D-glukoza
. W odróżnieniu od innych aldehydów
charakteryzują się dużą trwałością, ponieważ jako polihydroksyaldehydy tworzą hemiacetale –
związki odporne na utleniające działanie tlenu z powietrza.
OH
H
H
O
H
OH
H
OH
H
CH
2
OH
O
O
H
O
H
OH
OH
H
O
H
CHO
C
C
C
C
D
-glukoza
hemiacetal
Niezwykle interesującymi (ze względu na możliwość praktycznego zastosowania do zwalczania
owadów) są substancje utrudniające żerowanie owadów - IAF (ang. insect antifeedants). Są
6
pośród nich aldehydy należące do terpenoidów, np.
poligodial
(
tadeonal
), ajugaryny (I-V) lub
ajugomaryna. IAF wydzielają niektóre rośliny, żeby chronić się przed żerującymi na nich
owadami, np. przed szarańczą. Jak dotychczas naturalne IAF nie znalazły praktycznego
zastosowania (są zbyt skomplikowane, żeby je syntezować, a w naturze występują w małym
stężeniu). Jednak na ich podstawie zaprojektowano prostsze związki o zbliżonej aktywności.
OCOCH
3
O
O
CH
2
OCOCH
3
CH
3
H
C
H
3
O
H
CHO
CHO
H
3
C
poligolial
(
tadeonal
)
ajugaryna I
Naturalne ketony są znacznie popularniejsze od aldehydów. Nietrudno zauważyć, że
aldosteron
jest także ketonem. Pozostałe kortykosterydy też zawierają grupę lub grupy ketonowe. Do
ketonów należy większość hormonów płciowych:
estron
,
progesteron
,
testosteron
i
androsteron
.
O
H
H
H
O
O
H
H
H
OH
progesteron testosteron
Pośród terpenoidów spotyka się wiele związków zawierających grupę ketonową; jako przykłady
można podać
menton
,
karwon
,
tujon
czy
kamforę
:
O
O
O
O
(-)-menton (+)-karwon (-)-
α-tujon kamfora
Ketonami są substancje zapachowe pochodzenia zwierzęcego, takie jak
cybeton
i
muskon
.
O
O
cybeton muskon
Grupę ketonową zawierają cukry – ketozy, czyli polihydroksyketony, a najpopularniejszą pośród
nich jest
D-fruktoza
. Ketozy, tak jak i aldozy występują w formie hemiacetalowej.
Nomenklatura aldehydów
1.Systematyczna
Nazwy aldehydów tworzy się przez dodanie końcówki
-al
do nazwy węglowodoru
macierzystego.
7
O
H
H
O
C
H
3
H
O
CH
3
CH
2
H
CH
2
CH
3
CH
3
C
C
C
CH
3
CHCH
2
CHCHO
etanal
metanal propanal 2,4-dimetyloheksanal
2. Karboaldehydowa
Inny systematyczny sposób nazywania aldehydów polega na dodaniu do nazwy węglowodoru
związanego z grupą -CHO przyrostku
karboaldehyd
wraz z łącznikiem „
o
”. Stosowany
najczęściej w nazywaniu związków o skomplikowanej budowie.
CHO
H
Et
H
H
CHO
cykloheksanokarboaldehyd (1R,2R)-2-etylocyklo-heksano-1-karboaldehyd
3. Przerostkowa – podobną rolę pełni przedrostek
formylo
:
CH
2
CHO
CH
3
OHC
COOH
OCHCH
2
CH
2
CHCHCHO
3-(formylometylo)-2-metyloheksano-1,6-dial kwas 4-formylobenzoesowy
4. Odkwasowa
Popularny, półsystematyczny sposób nadawania nazw aldehydom wywodzi się od nazw kwasów
karboksylowych. Te dwie grupy związków są z sobą spokrewnione, ponieważ utlenienie
aldehydów prowadzi do kwasów zawierających tę samą resztę organiczną. Mamy więc
aldehyd
mrówkowy
(
metanal
, zwany także
formaldehydem
) wywodzący się od
kwasu mrówkowego
,
podobnie
aldehyd
octowy
(
etanal
),
propionowy
(
propanal
),
masłowy
(
butanal
),
izomasłowy
(
2-
metylopropanal
),
benzoesowy
czy
salicylowy
.
5. Nazwy zwyczajowe
Niektóre aldehydy znane są prawie wyłącznie pod nazwami zwyczajowymi, np.
aldehyd
salicylowy
czy
anyżowy
.
OH
CHO
CHO
(CH
3
)
2
CHCHO
H
3
CO
aldehyd izomasłowy
aldehyd anyżowy
(
2-metylopropanal
)
aldehyd salicylowy
(
p-metoksybenzoesowy
)
(
o-hydroksybenzoesowy
)
Nomenklatura ketonów
1. Systematyczna
Zgodnie z zasadami IUPAC nazwy ketonów tworzy się przez dodanie końcówki „
on
” do nazwy
węglowodoru macierzystego:
O
C
H
3
CH
3
O
O
O
CH
3
CH
3
CH=CCH
2
CCH
3
C
CH
3
CCH
2
CH
2
CH
3
pentan-2-on
3-metyloheks-2-en-5-on
propanon
(
aceton
)
cykloheksanon
8
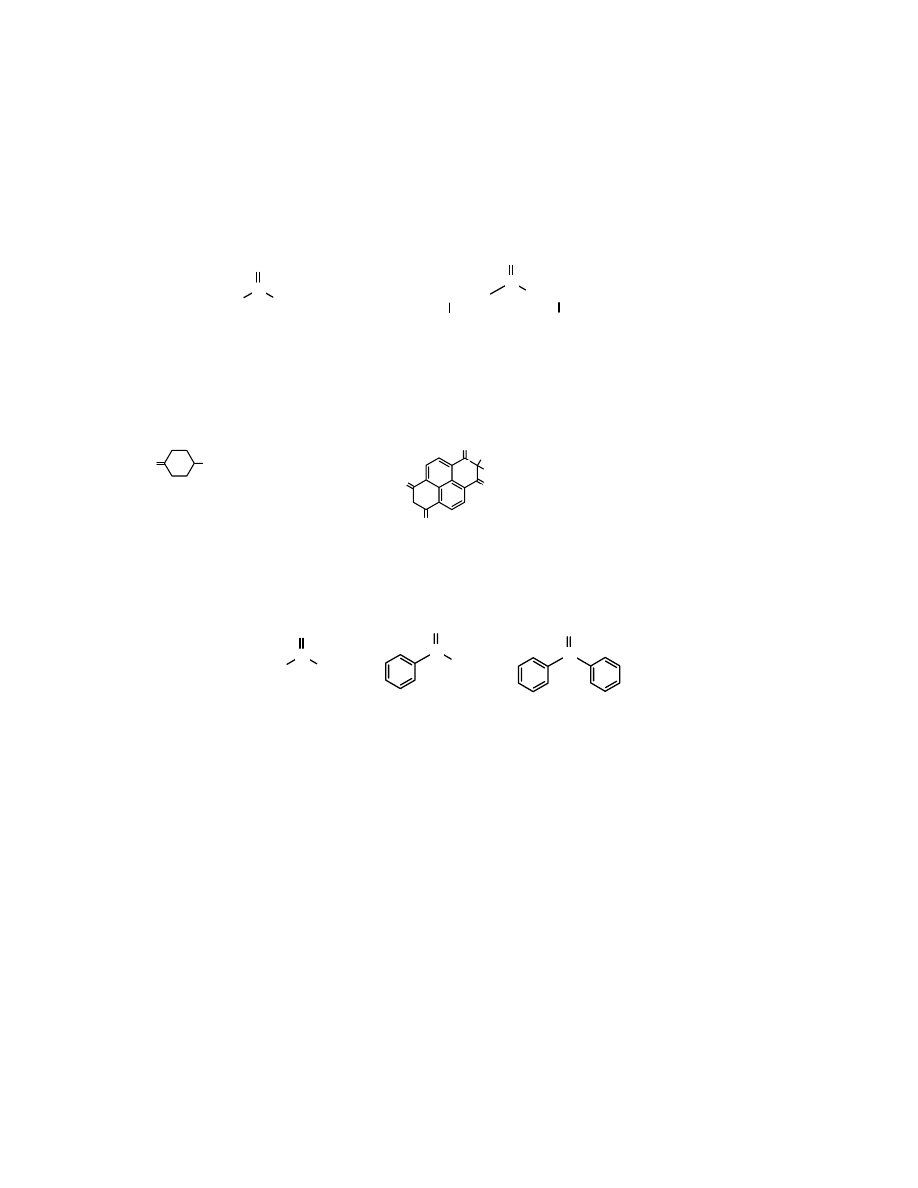
2. Grupowo-funkcjonalna
Powszechnie stosowany sposób nazywania ketonów, tzw. grupowo-funkcyjny polega na dodaniu
po słowie keton (w porządku alfabetycznym i rozdzielone myślnikiem) nazwy reszt alkilowych
(arylowych) związanych z grupą karbonylową. Należy pamiętać o tym, żeby używać nazw reszt
organicznych w formie przymiotnikowej, tj. np.
keton metylowo-propylowy
, a nie
metylo-
propylowy
, gdyż w wymowie ta druga forma jest równoznaczna z
ketonem metylopropylowym
,
a to oznacza inny związek.
O
C
H
3
CH
2
CH
2
CH
3
O
CH
3
CHCH
2
CH
2
CHCH
3
CH
3
CH
3
C
C
keton metylowo-propylowy
(
keton metylopropylowy
)
keton bis(2-metylopropylowy)
3. Podstawnikowa
Tlen grupy ketonowej =O, jako podstawnik nosi nazwę
okso
i można tego przedrostka używać w
tworzeniu nazw podstawnikowych. Zastępuje on dawniej używany przedrostek „
keto
”.
O
CH
2
CH
2
CH
2
COOH
O
O
O
O
H
COOH
1
3
4
5
6
8
9
10
kwas 4(4-oksocykloheksylo)butanowy
kwas 1,3,6,8-tetraokso-1,2,3,6,7,8,-
-heksahydropireno-2-karboksylowy
4. Zwyczajowa
Wiele ketonów ma nazwy zwyczajowe. Oprócz wymienionych uprzednio ketonów naturalnych
należy znać takie związki, jak
aceton
,
acetofenon
czy
benzofenon
:
O
CH
3
O
O
C
H
3
CH
3
C
C
acetofenon
benzofenon
C
aceton
Właściwości fizykochemiczne
Metanal
jest gazem (tw. -19
o
C),
etanal
zaś bardzo lotna cieczą, lotniejszą niż
eter etylowy
(tw.
20,8
o
C), a
propanal
wrze w temp. 48
o
C.
Metanal
i
etanal
mieszają się z wodą w każdym
stosunku; rozpuszczalność w wodzie wyższych aldehydów maleje wraz ze wzrostem ich masy
cząsteczkowej; przykładowo
propanal
,
n-butanal
i
aldehyd benzoesowy
rozpuszczają się w
wodzie odpowiednio: 28, 4 i 3 g/100 ml w 20
o
C. Aldehydy i ketony rozpuszczają się dobrze w
większości rozpuszczalników organicznych. Wodny roztwór
metanalu
(25-37%) znany pod
nazwą
formaliny
zawiera często
metanol
jako stabilizator.
Formalina
ma silne działanie
antyseptyczne i służy do przechowywania preparatów organicznych.
Metanal
ma bardzo silny, drażniący i duszący zapach,
etanal
nieco słabiej, ale też bardzo
intensywnie oddziałuje na zmysł powonienia. Zapach wyższych aldehydów alifatycznych jest
bardzo nieprzyjemny („smrodliwy”).
Benzaldehyd
i wiele innych aldehydów aromatycznych
charakteryzują się specyficznym zapachem gorzkich migdałów.
Aceton
wrze w temperaturze 56
o
C i miesza się z wodą w każdym stosunku. Ma
charakterystyczny rozpuszczalnikowy zapach.
Aceton
jest popularnym rozpuszczalnikiem
stosowanym do rozpuszczania związków organicznych (w tym farb i lakierów).
9
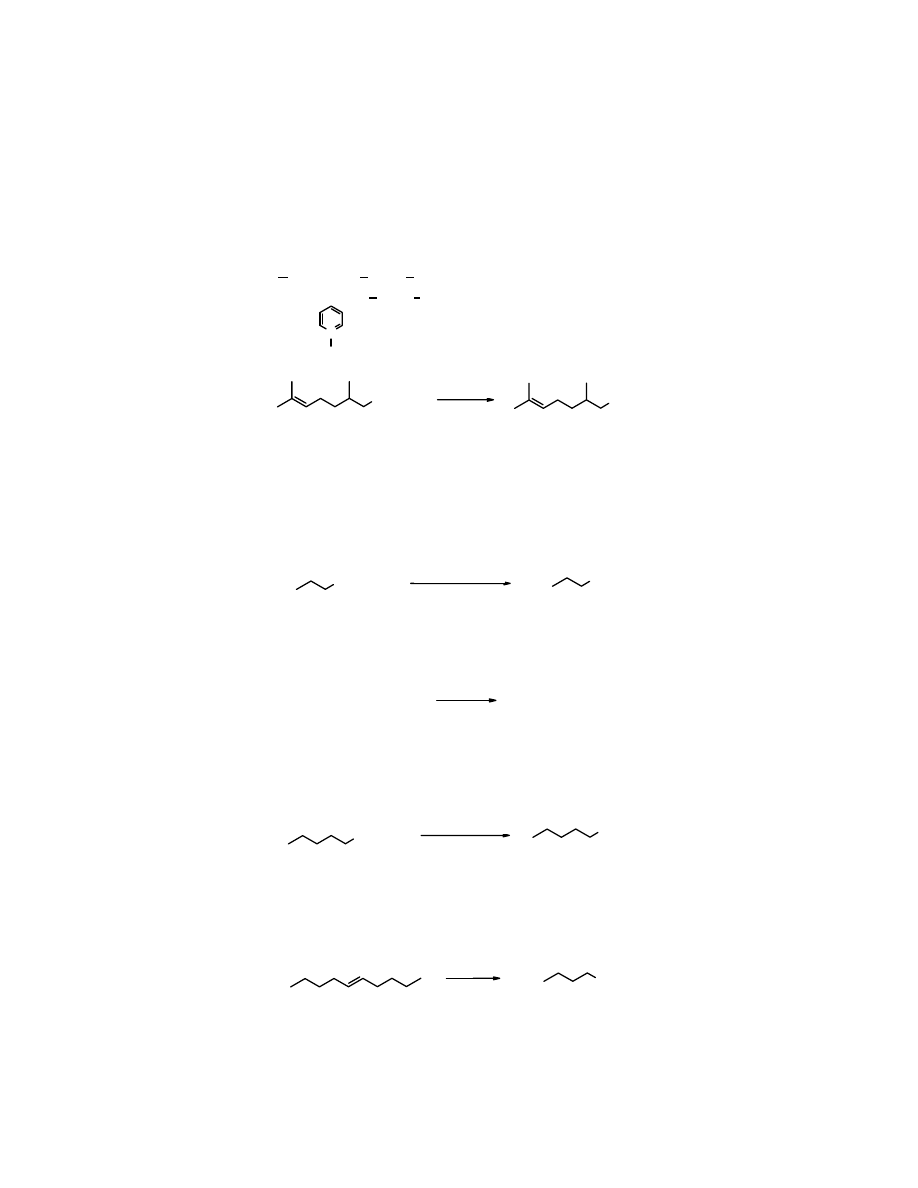
Otrzymywanie aldehydów
1. Utlenianie
Alkohole 1
o
można przeprowadzić w aldehydy pod wpływem odpowiednio dobranych
utleniaczy, należy jednak zapewnić warunki zabezpieczające przed dalszym utlenieniem ich do
kwasów, ponieważ aldehydy łatwiej
utleniają się
do kwasów niż alkohole do aldehydów.
Rekomendowanym utleniaczem 1
o
alkoholi do aldehydów jest
chlorochromian pirydyny
– w
skrócie PCC (ang. pyridium chlorochromate). Reakcja biegnie w
dichlorometanie
w
temperaturze pokojowej (RT – ang. room temperature).
N
H
+
CrO
3
Cl
-
PCC, chlorochromian pirydyny
CH
2
OH
CHO
PCC, RT
CH
2
Cl
2
cytronellol cytronellal
(82%)
Aldehydy lotniejsze od wody otrzymuje się z odpowiednich alkoholi poprzez ich
utlenianie
za
pomocą dichromianu sodu we wrzącej wodzie. W tych warunkach powstający aldehyd usuwany
jest z utleniającego środowiska przez oddestylowanie. Przykładem może być przekształcanie
n-
butanolu
(tw. 118
o
C) w
n-butanal
(tw. 75
o
C). Wydajności są jednak dużo niższe niż poprzez
utlenianie
za pomocą PCC.
CH
2
OH
CHO
Na
2
Cr
2
O
7
/H
2
SO
4
HOH/tw
n
-butanolu
n-butanal
(32%)
Jedna z przemysłowych metod otrzymywania
aldehydu octowego
polega na
utlenianiu
etanolu
w
fazie gazowej w obecności katalizatora srebrowego.
CH
3
CH
2
OH + 0,5 O
2
500
o
C
Ag
CH
3
CHO + HOH
etanol
etanal
(90%)
2. Odwodornianie
Alkohole 1
o
można
odwodornić
katalitycznie w obecności, np. katalizatora złożonego z miedzi i
tlenku chromu osadzonego na pumeksie. Jest to metoda preferowana w przemyśle.
CH
2
OH
CHO
Cu-CrO
3
/pumeks
- H
2
, 330
o
C
n-heksanol n-hesanal
(30%)
3.Ozonoliza
Z alkenów zawierających winylowy atom wodoru powstają aldehydy w reakcji
ozonolizy
.
Korzystnymi substratami są alkeny symetryczne.
CHO
1.
O
3
2.
Zn, AcOH
2
dec-5-en n-pentanal
10
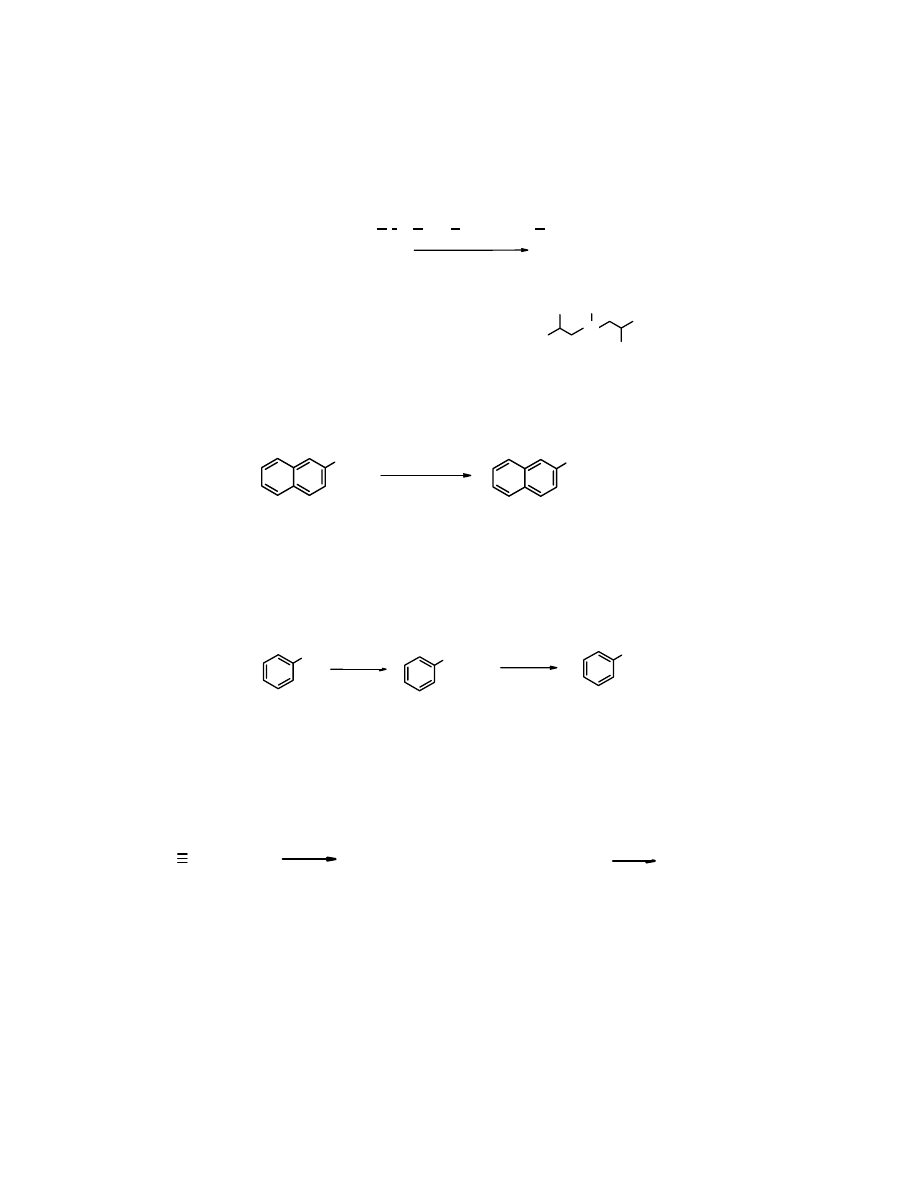
4. Redukcja pochodnych kwasów karboksylowych
W syntezie aldehydów wykorzystywane są pochodne kwasów karboksylowych, głównie estry i
chlorki kwasowe. Większość reduktorów przeprowadza kwasy karboksylowe i ich pochodne w
alkohole 1
o
. Do otrzymania aldehydów potrzebne są reduktory specjalne, umożliwiające
selektywną redukcję
. Należy do nich
hydroglinian diizobutylowy
(
wodorek
didizobutyloglinowy
– DIBAH, ang. diisobutylaluminium hydride).
1.
DIBAH, toluen,-78
o
C
2.
H
+
/HOH
CH
3
(CH
2
)
10
COOCH
3
CH
3
(CH
2
)
10
CHO
dodekanian metylu dodekanal
(88%)
H
DIBAH: (CH
3
)
2
CHCH
2
-Al(H)-CH
2
CH(CH
3
)
2
Al
5. Redukcja Rosenmunda
Aldehydy można otrzymać z chlorków kwasowych poprzez uwodornienie wobec
zdezaktywowanego katalizatora palladowego. Metoda ta nazywana redukcją Rosenmunda ma
obecnie znaczenie historyczne.
COCl
COCl
H
2
Pd/BaSO
4
/chinolina
(81%)
chlorek 2-naftoesowy aldehyd 2-naftoesowy
6. Hydroliza geminalnych dihalogenopochodnych
Zarówno aldehydy, jak i ketony można otrzymać poprzez hydrolizę geminalnych
dihalogenopochodnych. Jest to sposób przydatny wówczas, kiedy takie pochodne są łatwo
dostępne. Do popularnych należy
chlorek benzylidenu
, otrzymywany w reakcji chlorowania
toluenu
. W trakcie jego hydrolizy łatwo powstaje
benzaldehyd
:
CH
3
CHCl
2
CHO
Cl
2
, h
ν
-
OH/HOH
toluen chlorek benzylidenu aldehyd benzoesowy
7. Hydratacja alkinów
Aldehydy i ketony tworzą się w reakcji
addycji
wody do alkinów. Reakcja jest katalizowana
przez sole rtęci. Z
etynu
powstaje
etanal
, a z pozostałych alkinów tworzą się ketony. W ten
sposób otrzymywano
aldehyd octowy
przemysłowo, obecnie w przemyśle dominuje
utlenianie
etenu
tlenem gazowym.
C
H
CH + HOH
HgSO
4
H
2
SO
4
/HOH
CH
3
CHO
etyn
etanal
CH
2
=CH
2
+ 0,5 O
2
120
o
C
Pd/CuCl
2
CH
3
CHO
(94%)
eten
etanal
metoda przemysłowa
Otrzymywanie ketonów
1. Utlenianie
Ketony najłatwiej otrzymać z alkoholi 2
o
, nie ma w tym przypadku obawy o ich dalsze
utlenienie. W ten sposób powstaje
cykloheksanon
z
cykloheksanolu
.
11
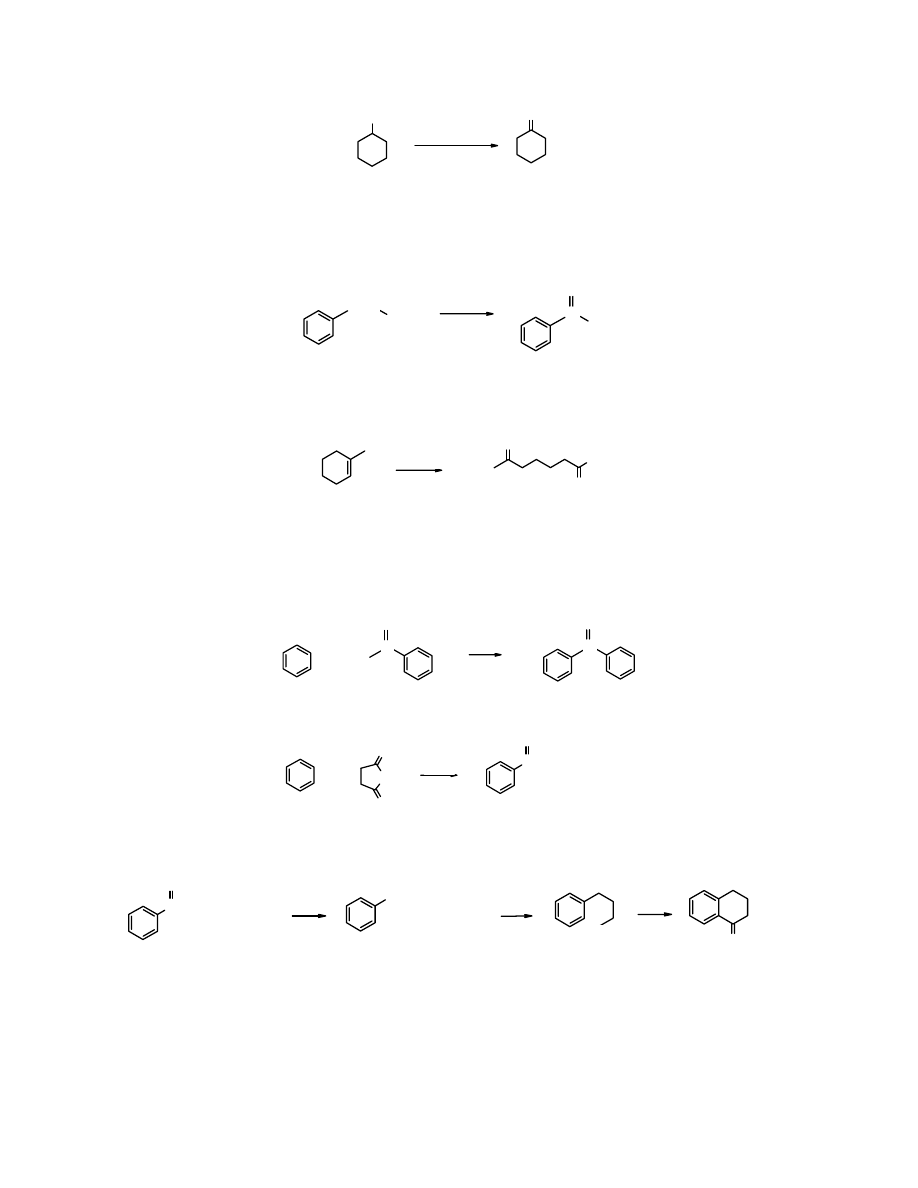
OH
O
Na
2
Cr
2
O
7
/H
2
SO
4
HOH
cykloheksanol
cykloheksanon
(79%)
Utleniaczem alkoholi 2
o
, tak jak alkoholi 1
o
może być PCC.
Cykloheksanon
przemysłowo
produkuje się przez
utlenienie
powietrzem
cykloheksanu
.
Utlenianie
powietrzem arenów zawierających odpowiednie łańcuchy boczne jest szczególnie
przydatne w otrzymywaniu ketonów alifatyczno-aromatycznych.
CH
3
CH
3
O
CH
2
powietrze
100
o
C
C
etylobenzen benzofenon
2. Ozonoliza
Ozonoliza
odpowiednich alkenów prowadzi do ketonów:
O
O
H
1.
O
3
2.
Zn, AcOH
1-metylocykloheksen 6-oksoheptanal
(86%)
3. Acylowanie
Niezwykle wygodnym sposobem otrzymywania ketonów alifatyczno-aromatycznych lub
aromatycznych jest reakcja acylowania arenów Friedla-Craftsa. Polega ona na działaniu
chlorkami lub bezwodnikami kwasowymi na związki zawierające pierścień aromatyczny.
O
Cl
O
+
C
AlCl
3
∆
C
benzen chlorek benzoilu benzofenon
(85%)
O
O
O
C-CH
2
CH
2
COOH
O
+
AlCl
3
benzen
kwas 3-benzoilo-
propanowy
bezwodnik
bursztynowy
(94%)
Za pomocą reakcji Friedla-Craftsa można otrzymywać związki cykliczne; w ten sposób z
kwasu
fenylobutanowego
powstaje
tetralon
:
O
C-CH
2
CH
2
COOH
O
(CH
2
)
3
COOH
ClC=O
AlCl
3
kwas 3-benzoilo-
propanowy
NaBH
4
kwas 4-fenylo-
butanowy
SOCl
2
(89%)
tetralon
(79%)
chlorek
kwasowy
12
Właściwości chemiczne
1. Utlenianie
Aldehydy są podatne na
utlenienie
bardziej niż ketony; nawet pod wpływem słabych utleniaczy
zostają przekształcane w kwasy karboksylowe.
R
O
H
R
O
OH
C
C
[O]
aldehyd
kwas
karboksylowy
Utleniacze:
HNO
3
,
KMnO
4
,
O
2
,
Na
2
CrO
7
, odczynniki Jonesa, Tollensa, Fehlinga i inne.
Odczynnik Jonesa –
CrO
3
/H
2
SO
4
/HOH – należy do najłagodniejszch utleniaczy przekształcających aldehydy w
kwasy.
Odczynniki Tollensa – (
AgNO
3
/NH
3
/HOH) i Fehlinga – (CuSO
4
/H
2
SO
4
/
HOH-NaOH/KNaC
4
H
4
O
6
/
HOH) są
stosowane w testach do wykrywania aldehydów.
Odczynnik Tolensa w obecności reduktora typu aldehydu ulega
redukcji
do srebra
metalicznego, które tworzy lustro, ponieważ w postaci cienkiej warstwy osadza się na dnie
naczynia szklanego. Powstawanie takiego lustra jest testem na obecność aldehydu. Reakcja ta
wykorzystywana jest do produkcji luster.
R
O
H
R
O
OH
C
+ 2 Ag
+
C
+ 2 Ag
lustro srebrowe
aldehyd
kwas karboksylowy
W próbie Fehlinga obserwuje się pomarańczowy osad wytrącającego się tlenku miedzi (I) pod
wpływem aldehydu.
R
O
H
R
O
OH
C
+ 2 Cu
2+
C
+ Cu
2
O
-
OH
pomarańczowy
osad
aldehyd
kwas karboksylowy
Ketony też ulegają utlenieniu, ale pod wpływem silniejszych utleniaczy, następuje przy tym
rozerwanie wiązania pomiędzy atomem C
α
a grupą karbonylową. Z ketonów alifatycznych
powstaje mieszanina kwasów karboksylowych.
O
R-CH
2
-C-CH
2
-R'
KMnO
4
-
OH/HOH,
∆
RCOOH + RCH
2
COOH +
HOOCR' + HOOCCH
2
R'
keton alifatyczny mieszanina kwasów karboksylowych
Z ketonów cyklicznych otrzymuje się jednorodne kwasy dikarboksylowe.
O
O
H
O
O
OH
1.
KMnO
4
/NaOH/HOH,
∆
2.
H
+
/HOH
cykloheksanon kwas adypinowy
(
kwas heksano-1,6-dionowy
) (65%)
2. Addycja nukleofilowa
Addycja nukleofilowa
do grupy karbonylowej jest charakterystyczną reakcją aldehydów i
ketonów. Na atomie węgla grupy C=O zlokalizowany jest częściowy ładunek dodatni, a przez to
karbonylowy atom węgla jest podatny na atak nukleofilowy.
13
O
R
R(H)
C
δ
+
δ
-
:Nu
Nukleofilami (
Nu:
-
)
mogą być atomy lub grupy obdarzone ładunkiem ujemnym, np.:
..
..
RO:
-
..
HO:
-
..
N
C:
-
R
3
C:
-
H:
-
aniony: cyjankowy karboanion wodorkowy alkoksylowy hydroksylowy
lub bez formalnego ładunku (cząsteczki obojętne –
:Nu-H
), ale zawierające elektroujemne atomy
z wolną para elektronową, np.:
RNH
2
R
2
NH
ROH
..
..
..
..
HOH
..
..
H
3
N:
amoniak amina 1
o
amina 2
o
alkohol woda
Addycja nukleofilowa do grupy karbonylowej biegnie jednym z dwóch wariantów:
A
jedynie
addycja
lub
B
addycja
i następcza
eliminacja
. Kierunek reakcji zależny jest głównie od
charakteru odczynnika nukleofilowego. W obu przypadkach pierwszym etapem jest atak
nukleofila na karbonylowy atom węgla i utworzenie adduktu, przy czym atom ten zmienia
hybrydyzację na sp
3
. W wariancie
A
addukt stabilizuje się przez
protonowanie
atomu tlenu, a w
drugim przypadku –
B
, następuje
odszczepione
cząsteczki wody (
eliminacja
) i atom węgla
powraca do poprzedniej hybrydyzacji sp
2
.
Odszczepienie
cząsteczki wody jest możliwe dzięki
temu, że nukleofil zawiera 2 ruchliwe atomy wodoru.
O
R
R'
O
R
Nu
R'
O
R
NuH
2
R'
O
R
Nu
R'
H
Nu
R
R'
C
aldehyd
lub keton
:Nu-
:NuH
2
C
H
+
A
B
: :
..
: : -
C
..
: : -
+
C
..
:
C
- HOH
Rys. 11.1 Mechanizm addycji nukleofilowej do karbonylowego atomu węgla
Addycja nukleofilowa
do grupy karbonylowej aldehydów i ketonów jest szeroko
wykorzystywana w syntezie chemicznej, można za jej pomocą otrzymywać wiele cennych
produktów. Poniższy schemat obrazuje te możliwości:
14
O
OH
R
OH
CN
N
Y
R
2
N
H
H
OR
OR
R
R
OH
H
C
aldehydy lub ketony
RMgX
alkohole
HCN
C
cyjanohydryny
H
2
N-Y
C
C
pochodne azotowe
iminy
oksymy
hydrazony
itp
R
2
NH
C
C
1.
HSCH
2
CH
2
SH
2.
Ni
Ra
C
alkany
ROH
C
acetale
(Ph)
3
P=C(R)
2
C
C
alkeny
NaBH
4
C
:H
-
alkohole
enaminy
ylidy
2.1 Addycja wody – tworzenie geminalnych dioli
W wyniku
addycji
cząsteczki wody do grupy karbonylowej powstają geminalne diole, zwane w
skrócie gemdiolami lub gemglikolami (grec. geminalny czyli bliźniaczy; w tym przypadku
odnosi się do dwóch grup -OH na tym samym atomie węgla).
Hydratacja
grupy karbonylowej
jest reakcją odwracalną i najczęściej jej równowaga jest przesunięta na korzyść formy
karbonylowej. Zdarzają się jednak trwałe gemglikole (tzw. hydraty).
O
R
R'
OH
R
OH
R'
C
+ HOH
C
forma karbonylowa forma uwodniona (hydrat)
Trwałymi gemdiolami są
hydrat metanalu
i
chloralu
. W większości wodnych roztworów
aldehydów i ketonów udział formy uwodnionej nie przekracza 0,1%, podczas gdy
metanal
i
chloral
występują głównie w formie uwodnionej (99,9%). Hydrat chloralu jest silnym lekiem
usypiającym, o działaniu zbliżonym do barbituranów. Jego działanie uboczne polega na
drażniącym działaniu na skórę i błony śluzowe. Stosowany także jako surowiec do produkcji
DDT.
O
H
H
2
C(OH)
2
Cl
3
CC(OH)
2
Cl
3
CC
ciecz wrząca
w temp. 97
o
C
t.t. = 57
o
C
hydrat metanalu chloral hydrat chloralu
Hydrat metanalu
występuje jedynie w roztworze wodnym, podczas gdy
hydrat
chloralu
jest
trwałym, krystalicznym związkiem.
Występowanie hydratów związków karbonylowych udowodniono na podstawie widm
spektroskopowych, a także za pomocą reakcji izotopowej z wodą zawierającą
18
O. Po pewnym
15
czasie od rozpuszczenia aldehydu w takiej wodzie izolowano aldehyd zawierający w grupie
C=O tlen
18
O.
RCHO + H
18
OH
RCH(OH)(
18
OH)
RCH
18
O + HOH
Reakcja hydratacji aldehydów i ketonów jest katalizowana zarówno zasadowo, jak i kwasowo. W
środowisku obojętnym biegnie wolno.
Mechanizm hydratacji grupy karbonylowej
O
O
OH
H
O
OH
O
H
O H
O H
O
O
H
H
H
O
O
H
H
H
C
:OH
..
..
C
:
..
..
:
:
..
..
HO
..
..
C
..
:
..
..
+ :OH
..
..
C
:
..
H
2
O
..
..
C
:
..
+
C
..
+
H
2
O
..
..
C
:
..
..
+
H
2
O
..
..
C
:
..
+
H
3
O
+
+
..
-
-
kataliza zasadowa kataliza kwasowa
-
2.2 Addycja cząsteczki alkoholu - tworzenie hemiacetali, acetali i tioacetali
Cząsteczka alkoholu, podobnie jak cząsteczka wody przyłącza się do grupy karbonylowej, a
produkt takiej addycji nosi nazwę hemiacetalu (grec. hemi = w połowie), dla odróżnienia od
acetali (pełnych acetali). Dawniej tego typu związki nazywano
półacetalami
. Reakcja jest
katalizowana kwasami.
O
O H
O H
O
O
R
H
H
OH
OR
C
:
..
C
:
..
+
C
..
+
ROH
..
..
C
:
..
..
+
C
:
..
..
H
+
- H
+
..
aldehyd
lub keton
hemiacetal
Zwykle hemiacetale są nietrwałe i występują jedynie w roztworze alkoholowym w stanie
równowagi z formą karbonylową. W środowisku kwaśnym reagują dalej z alkoholem w kierunku
acetali. Acetale pochodzące od ketonów nazywa się często ketalami.
16
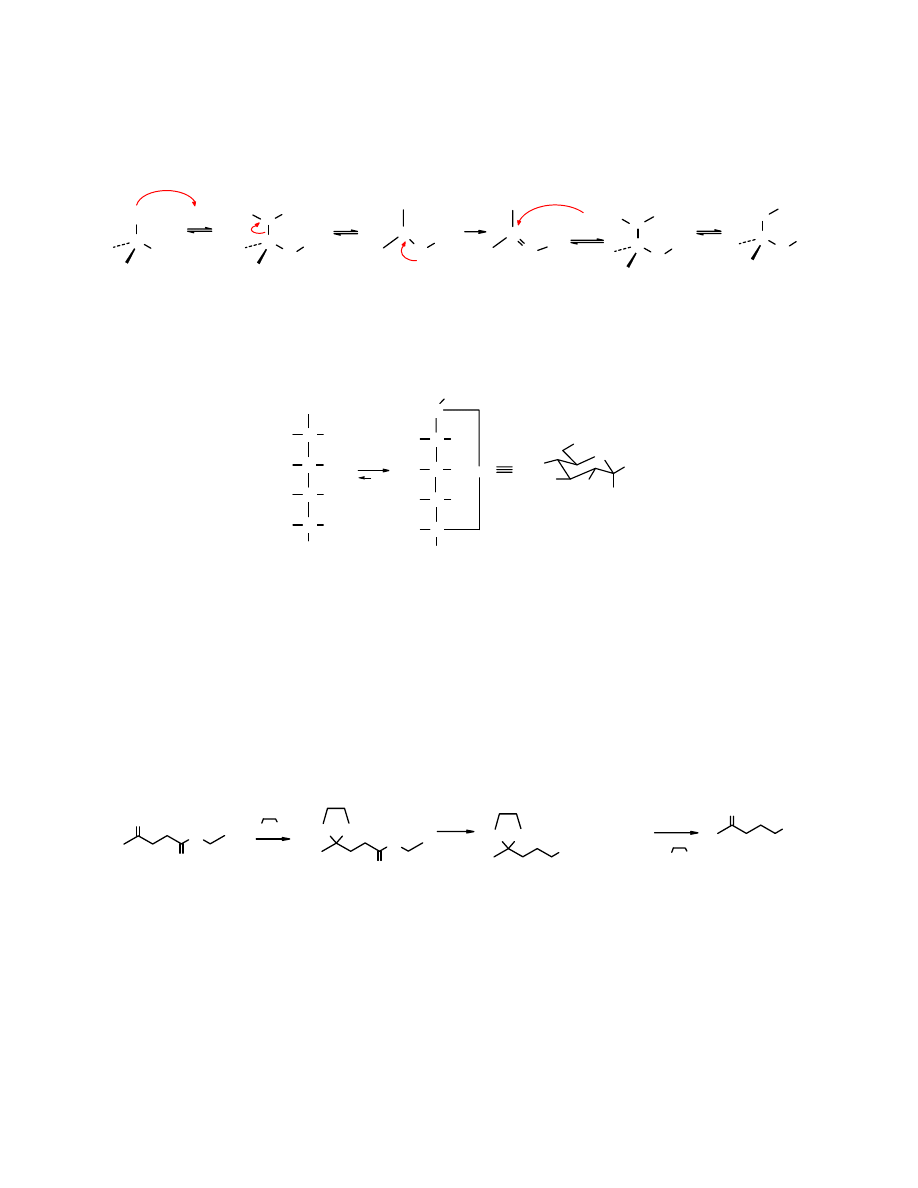
Mechanizm
Reakcja tworzenia acetali zaczyna się od protonowania grupy -OH, eliminacji cząsteczki wody,
przyłączenia cząsteczki alkoholu do atomu węgla i kończy się stabilizacją przez odszczepienie
protonu:
OH
OR
O
O
R
R
O
O
H
H
R
O
R
O R
H
O
O
R
R
..
C
:
..
..
C
:
..
+
..
ROH
H
+
- H
+
C
..
+
..
-HOH
C
+
..
..
C
+
..
..
..
C
..
..
hemiacetal
acetal
Chociaż większość hemiacetali występuje jedynie w roztworach alkoholowych w równowadze
ze związkami karbonylowymi, to hemiacetale wewnątrzcząsteczkowe, do których należą cukry
są trwałe. W cukrach równowaga przesunięta jest na korzyść hemiacetali.
OH
H
H
O
H
OH
H
OH
H
CH
2
OH
O
O
H
O
H
OH
OH
H
O
H
OH
H
H
O
H
OH
H
O
H
CH
2
OH
OH
CHO
C
C
C
C
D
-glukoza
HC
C
C
C
C
Wiązanie hemiacetalowe w
D-glukozie
tworzy się pomiędzy grupą -OH przy C5 i grupą
aldehydową. Cząsteczka
glukozy
przyjmuje konformację krzesłową. Acetale utworzone z
cukrów nazywają się glikozydami. Występują powszechnie w przyrodzie.
Synteza acetali polega na ogrzewaniu aldehydów lub ketonów z bezwodnym alkoholem w
kwaśnym środowisku. Ulegają one łatwo
hydrolizie
w środowisku kwaśnym, a są odporne na
hydrolizę zasadową
. Stosuje się je do czasowej osłony grupy karbonylowej, tak żeby można
było przeprowadzić reakcję w innej części cząsteczki, chroniąc grupę karbonylową, np. przed
utlenieniem
lub
redukcją
. Po zaplanowanych przekształceniach grupę karbonylową łatwo
odzyskuje się w reakcji
hydrolizy kwaśnej
. Często dla ochrony funkcji karbonylowej używa się
glikolu etylowego
, ponieważ z nim tworzy się wyjątkowo łatwo acetal cykliczny.
O
O
O
O
H
OH
O
O
O
O
OH
O
O
OH
O
O
H
OH
H
+
1.
LiAlH
4
2.
+
H/HOH
+
H/HOH
4-oksopentanian etylu
-
+ EtOH
4-oksopen-1-ol
Selektywna redukcja
przedstawiona na powyższym schemacie byłaby niemożliwa do
przeprowadzenia bez czasowej ochrony grupy ketonowej.
Podobną rolę pełnią tioacetale, które tworzą się jeszcze łatwiej, a ulegają usunięciu, np. podczas
katalitycznej redukcji. Warto wiedzieć, że związki siarki dezaktywują katalizatory typu Pt czy
Pd, ale nie nikiel Renaya (Ni
Ra
). W ten sposób można zredukować grupę karbonylową
całkowicie, czyli do węglowodoru.
17

O
RS
SR
C
2 RSH
+
H
C
Ni
Ra
EtOH
CH
2
+ NiS
aldehyd lub keton tioacetal węglowodór
Do osłony grupy karbonylowej można też użyć
tioglikolu etylenowego
.
S
H
SH
O
S
S
BF
3
Ni
Ra
4-metylocykloheksanon
tioacetal
4-metylocykloheksan
2.3 Reakcje aldehydów i ketonów ze związkami Grignarda
Związki Grignarda reagują z grupą karbonylową w ten sposób, że ujemnie naładowana reszta
organiczna R przyłącza się do karbonylowego atomu węgla, a -MgX tworzy wiązanie
alkoholanowe z atomem tlenu. Po hydrolizie alkoholanu otrzymuje się alkohole.
O
MgX
O
R
OH
R
:
:
..
C
-
δ
+
δ
+ R
C
MgX
+
H/HOH
-
+
:
..
C
-
δ +δ
: :
związek Grignarda
aldehyd lub keton alkoholan alkohol
W reakcji związków Grignarda z
metanalem
powstają alkohole 1
o
, z innymi aldehydami –
alkohole 2
o
, a z ketonami – alkohole 3
o
.
Br
MgBr
H
H
O
H
H
CH
2
OH
Mg
eter
C=O
C
-
MgBr
+
+
H/HOH
bromek
cyklopentylu
bromek cyklopentylowo-
magnezowy
cyklopentylometanol
(48%)
(alkohol 1
o
)
MgCl
CH
3
CH
3
C
H
3
O
H
CH
3
CH
3
C
H
3
OH
H
C
+
C
C
C
bromek-t-butylo - benzaldehyd 1-fenylo-2,2-dimetylo-
magnezowy propanol
(alkohol
2
o
)
MgI
O
IMgO
C
H
3
O
H
C
H
3
CH
3
+
+H/HOH
jodek metylo- cyklopentanon 1-metylocyklopentanol
magnezowy
(alkohol
3
o
)
2.4 Addycja nukleofilowa HCN; otrzymywanie cyjanohydryn
Cyjanowodór przyłącza się do grupy karbonylowej tych aldehydów i ketonów, które nie mają
zawady przestrzennej wokół tej grupy. Produktem reakcji są cyjanohydryny (inaczej nitryle
α
-
hydroksykwasów), czyli związki zawierające funkcję -CN i -OH przy tym samym atomie węgla.
W wyniku addycji cyjanowodoru do
benzaldehydu
powstaje
nitryl kwasu migdałowego
.
18
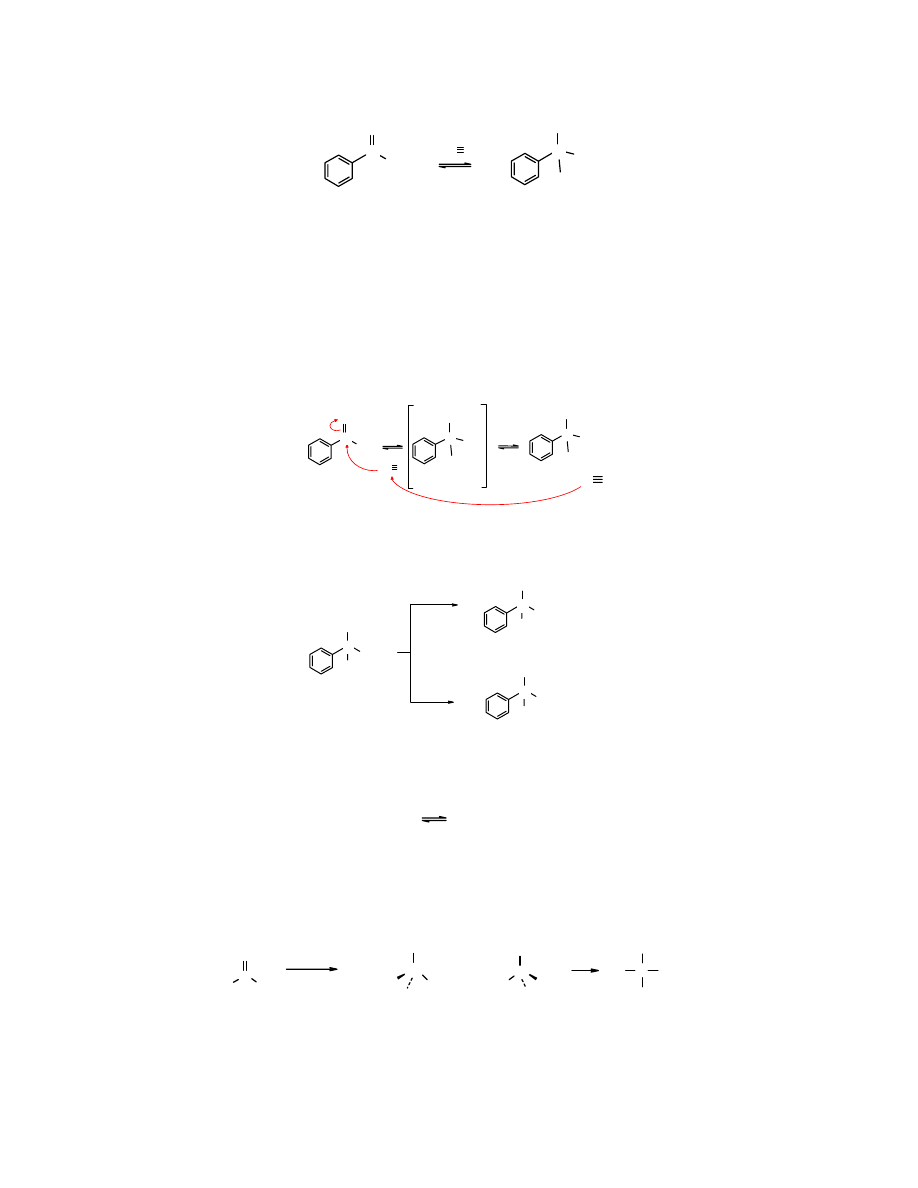
O
H
N
OH
H
CN
C
HC
C
benzaldehyd nitryl kwasu migdałowego
(cyjanohydryna) (88%)
Addycja cyjanowodoru do aldehydów lub ketonów jest reakcją odwracalną, zwykle z równowagą
przesuniętą na korzyść produktu. Reakcję ułatwia kataliza zasadowa, gdyż jon
-
CN przyłącza się
łatwiej niż HCN. Jon cyjankowy jest silnym nukleofilem i dlatego równowaga jego addycji do
grupy karbonylowej jest przesunięta w stronę produktów.
Inne kwasy, np. halogenowodory,
kwas siarkowy czy kwasy karboksylowe są słabymi nukleofilami i nie tworzą adduktów ze
związkami karbonylowymi
.
W reakcji addycji cyjanowodoru do grupy karbonylowej wystarczają śladowe ilości zasady, jako
katalizator, ponieważ w warunkach równowagi jon cyjankowy odtwarza się:
O
H
OH
H
CN
O
H
CN
N
N
:
C
C
-
C
C
:
: :
..
..
:C
-
HCN
+
-
aldehyd produkt przejściowy cyjanohydryna
Cyjanohydryny stanowią dogodny półprodukt w syntezie organicznej. Poprzez ich redukcję
można otrzymać
β
-aminoalkohole, a w wyniku hydrolizy
α
-hydroksykwasy.
OH
H
CN
OH
H
CH
2
NH
2
OH
H
COOH
C
1.
LiAlH
4
,THF
2.
HOH
+
H/HOH
lub
-
OH/HOH
C
C
cyjanohydryna
benzaldehydu
2-amino-1-fenylo-
etanol
kwas migdałowy
∆
2.5 Addycja jonu wodorkowego – redukcja wodorkami
Wodorki typu NaBH
4
lub LiAlH
4
redukują aldehydy i ketony do odpowiednich alkoholi, przy
czym czynnikiem redukującym jest
anion wodorkowy
– H
-
.
Na
+
BH
4
-
Na
+
BH
3
+ :H
-
tetrahydroboran sodu anion wodorkowy
Szczególnie przydatnym okazał się tetrahydroboran sodu, ponieważ jest trwalszy (odporniejszy
na hydrolizę) od LiAlH
4
, działa selektywnie (nie redukuje kwasów karboksylowych i ich
pochodnych), a reakcję można prowadzić w
metanolu
, nawet uwodnionym.
O
R
R'
O
R
R'
H
O
H
R'
R
OH
H
R
R'
C
: :
:H
-
NaBH
4
lub
LiAlH
4
C
: :
.. -
+
C
: :
.. -
+
H/HOH
C
R, R': H, alkil lub aryl
alkohol
aldehyd lub keton
19
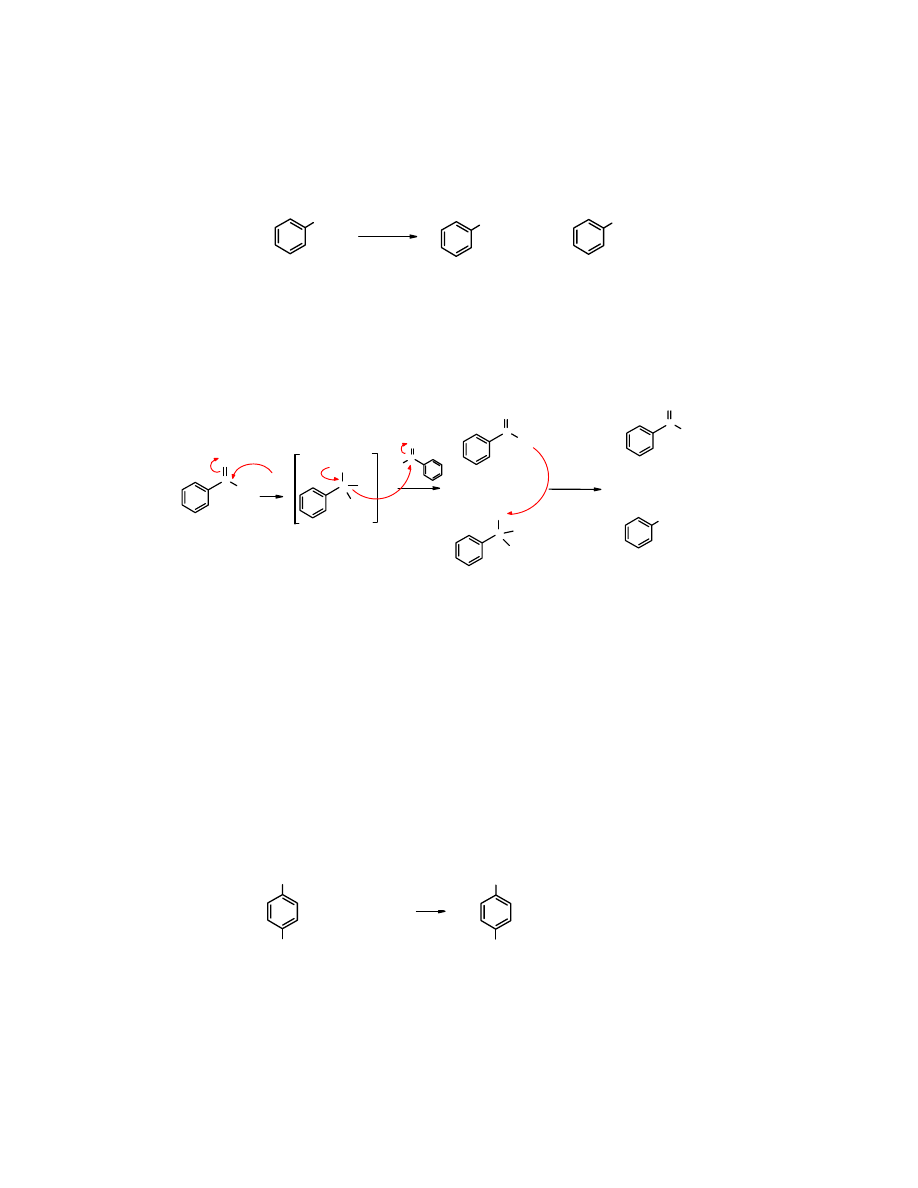
3. Reakcja Cannizzaro
Jest to
reakcja dysproporcjonowania
, w wyniku której z aldehydu tworzy się alkohol
(
redukcja
) i kwas karboksylowy (
utlenienie
). Zachodzi ona w środowisku alkalicznym, a
ulegają jej aldehydy
nie zawierające atomów wodoru przy C
α
, np.
metanal
i aldehydy
aromatyczne.
CHO
CH
2
OH
COOH
2
+
1. -
OH/HOH
2.
H
+
/HOH
(86%)
(79%)
benzaldehyd alkohol benzylowy kwas benzoesowy
Charakterystyczną cechą tej reakcji jest to, że bierze w niej udział
anion wodorkowy
. W
pierwszym etapie reakcji grupa hydroksylowa ulega addycji do karbonylowego atomu węgla, po
czym
anion wodorkowy
, jako nukleofil atakuje grupę karbonylową drugiej cząsteczki aldehydu.
Następnie proton z kwasu karboksylowego (silniejszy kwas) przechodzi do alkoholanowego
atomu tlenu:
H
O
OH
O
H
O
H
H
O
H
OH
O
O
O
CH
2
OH
..
..
-
..
C
OH
..
:..
-
:
C
:
:
C
:
..
:
C
..
:
-
+
C
C
:
..
-
+
benzaldehyd
anion benzoesanowy
alkohol benzoesowy
Chociaż produkt reakcji Cannizzaro jest złożony (kwas i alkohol), to łatwo udaje się go
rozdzielić, ponieważ oba składniki różnią się właściwościami chemicznymi i fizycznymi. Z
zasadowego, wodnego środowiska alkohol wyekstrahuje się nie mieszającym się z wodą
rozpuszczalnikiem organicznym, a kwas wytrąci z wodnego roztworu jego soli przez
zakwaszenie. Kwas można izolować też za pomocą jonitów.
Pomimo tych ułatwień nie jest to szeroko stosowana metoda otrzymywania ani
kwasu
benzoesowego
, ani
alkoholu benzylowego
, ponieważ znane są lepsze sposoby syntezy tych
związków. Większe znaczenie preparatywne ma tzw. krzyżowa reakcja Cannizzaro, w której
wykorzystuje się fakt, że w obecności aldehydów aromatycznych
metanal
ulega głównie
utlenieniu
do mrówczanu, natomiast większa część aldehydu aromatycznego ulega
redukcji
alkoholu aromatycznego. Ta różnica w reaktywności spowodowana jest większą
elektrofilowością karbonylowego atomu węgla
metanalu
– jest on bardziej podatny na atak reszty
hydroksylowej niż karbonylowy atom aldehydu aromatycznego.
CH
3
CHO
CH
3
CH
2
OH
HCHO
+
KOH
+ HCOO
-
K
+
metanal
mrówczan potasu
aldehyd
p
-toluilowy alkohol p-metybenzylowy
(72%)
20
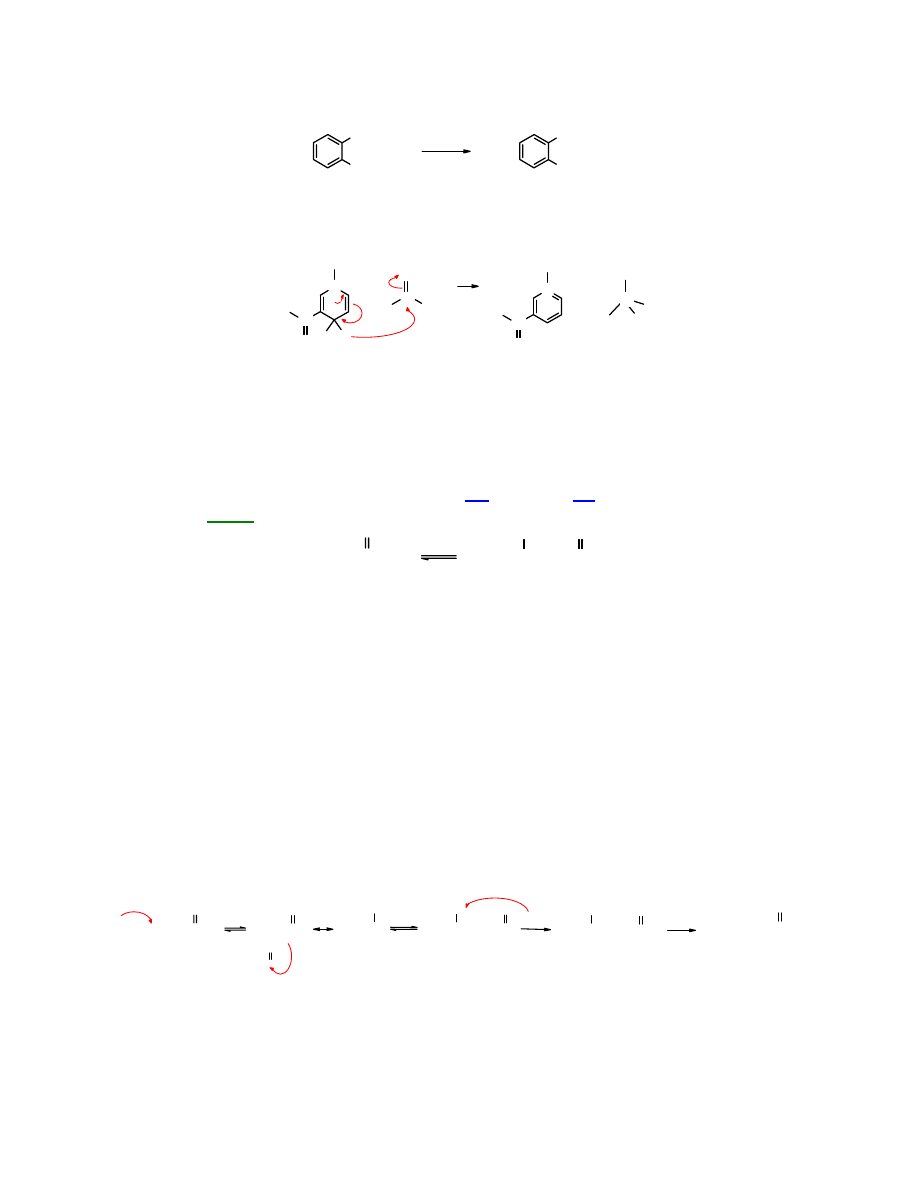
Z
aldehydu ftalowego
w reakcji Cannizzaro powstaje
kwas (hydroksymetylo)benzoesowy
.
CHO
CHO
CH
2
OH
COOH
1.
KOH
2.
+
H/HOH
aldehyd o-ftalowy kwas o-(hydroksymetylo)benzoesowy
W najważniejszym procesie
redukcji biochemicznej
, z udziałem zredukowanego dinukleotydu
nikotynoamidoadeninowego (NADH), widoczny jest mechanizm reakcji Cannizzaro:
O
R
R'
N
H H
R''
O
N
H
2
N
R''
O
N
H
2
R
R'
H
OH
+
C
..
:
..
C
+
+
C
C
zw. karbonylowy
hydroksy-
związek
NADH NAD
+
R’’: reszta cukrowo-fosforanowa
4. Reakcje kondensacji
4.1 Kondensacja aldolowa
Aldehydy i ketony zawierające w atom wodoru przy C
α
ulegają
kondensacji
katalizowanej przez
zasady i kwasy. W reakcji tej powstają dimery –
ald
ehydoalkoh
ole
, dlatego została ona nazwana
kondensacją aldolową
a jej najprostszy produkt dimeryzacji
etanalu
zwany jest
aldolem
.
O
O
OH
2 CH
3
CH
NaOEt
EtOH
CH
3
CH-CH
2
CH
acetaldehyd aldol
(
3-hydroksybutanal
)
W odwracalnej reakcji kondensacji aldolowej dla monopodstawionych aldehydów (RCH
2
CHO)
równowaga jest przesunięta na prawo, zaś dla rozgałęzionych aldehydów i większości ketonów
na lewo. Z aldehydami biegnie ona z dobrą szybkością. Zwykle w warunkach reakcji tworzą się
nienasycone aldehydy lub ketony, ponieważ w produktach dochodzi do
eliminacji
cząsteczki
wody.
β
-podstawione aldehydy i ketony są bardzo podatne na
eliminację
.
Dehydratacja
powoduje przesunięcie równowagi reakcji w kierunku dimerów
.
Z podatności produktów do
dehydratacji
reakcja ta powszechnie nazywana jest
kondensacją
, a
nie
dimeryzacją
.
Kondensacja
bowiem to reakcja
dimeryzacji
(oligomeryzacji lub
polimeryzacji), w której obok głównego produktu wydzielają się małe cząsteczki – w tym
wypadku wody.
Kondensacja aldolowa
polega na addycji nukleofilowej do karbonylowego atomu węgla anionu
powstałego w wyniku oderwania protonu z C
α
.
..
..
O
O
O
O
O
O
O
OH
O
HO:
..
-
+ H-CH
2
-CH
H
2
C-CH
..
-
H
2
C=CH
:
: :
-
-
:
CH
3
CH
CH
3
-CH-CH
2
-CH
: :
HOH
..
..
CH
3
-CH-CH
2
-CH
- HO
-
CH
3
CH=CH-CH
aldol
acetaldehyd
aldehyd krotonowy
- HOH
- HOH
(but-2-enal)
21
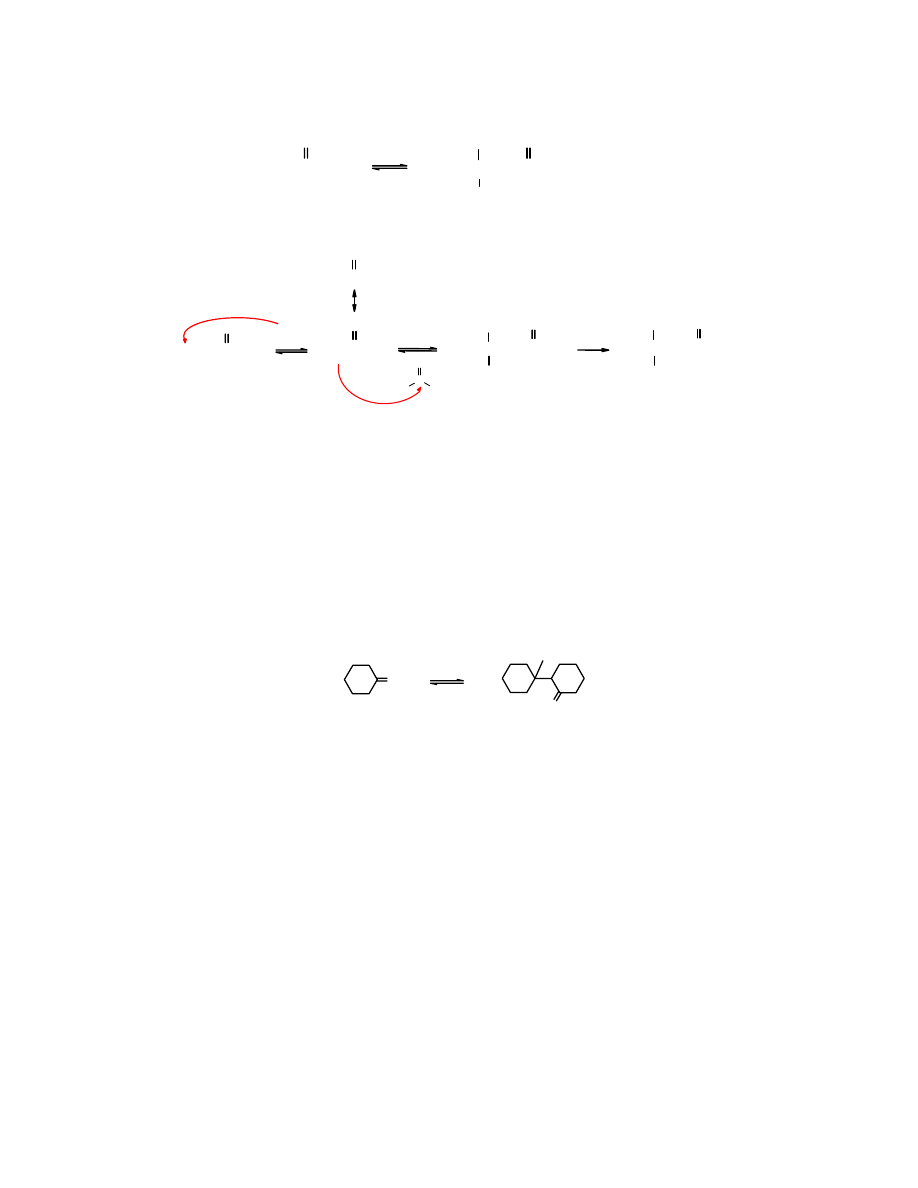
Podobnie wygląda kondensacja aldolowa
acetonu
, jednak wydajność tej reakcji jest niewielka:
O
OH
CH
3
O
H
3
C-C-CH
3
-
OH
H
3
C-C-CH
2
-C-CH
3
aceton
4-hydroksy-
- 4-metylo-
pent-2-on
(5%)
Mechanizm:
O
O
O
O
C
H
3
CH
3
OH
CH
3
O
O
CH
3
O
..
: :
H-H
2
C-C-CH
3
aceton
OH
: ..
..
-
H
2
C-C-CH
3
-
..
H
2
C=C-CH
3
-
C
H
3
C-C-CH
2
-C-CH
3
H
3
C-C-CH
2
-C-CH
3
:
:
..
-
HOH
-
-
OH
4-hydroksy-4-metylopent-2-on
Wydajność
kondensacji aldolowej
acetonu
można zwiększyć przez usuwanie produktu ze
środowiska reakcji, tzn. oddzielając go od alkalicznego katalizatora. Tę reakcję można
prowadzić, np. w kolumnie wypełnionej kawałkami Ba(OH)
2
.
Aceton
skrapla się w chłodnicy
zwrotnej do kolumny, gdzie w zetknięciu z Ba(OH)
2
ulega częściowej
dimeryzacji
i spływa do
kolby zawierając kilka procent
4-hydroksy-4-metylopentan-2-onu
.
Aceton
w kolbie wrze, a jego
pary skraplając się w chłodnicy zwrotnej ponownie przepływają przez kolumnę z Ba(OH)
2
,
podczas czego kolejna porcja
acetonu
ulegnie dimeryzacji. Ten proces trwa aż do wyczerpania
acetonu
, a w kolbie pozostaje prawie czysty produkt.
W
cykloheksanonie
zawada przestrzenna jest mniejsza niż w
acetonie
(sztywny układ) i dzięki
temu wydajność jego
dimeryzacji
jest kilka razy większa (22%):
O
OH
O
2
NaOH
cykloheksanon 1-hydroksy-2’-oksodicykloheksyl
Kondensację aldolową
stosuje się często w syntezie różnorodnych związków organicznych,
zarówno w laboratorium, jak i w skali technicznej. Powstające w reakcji aldole można poddawać
dehydratacji
, dalszej
kondensacji aldolowej
lub
selektywnej redukcji
.
22
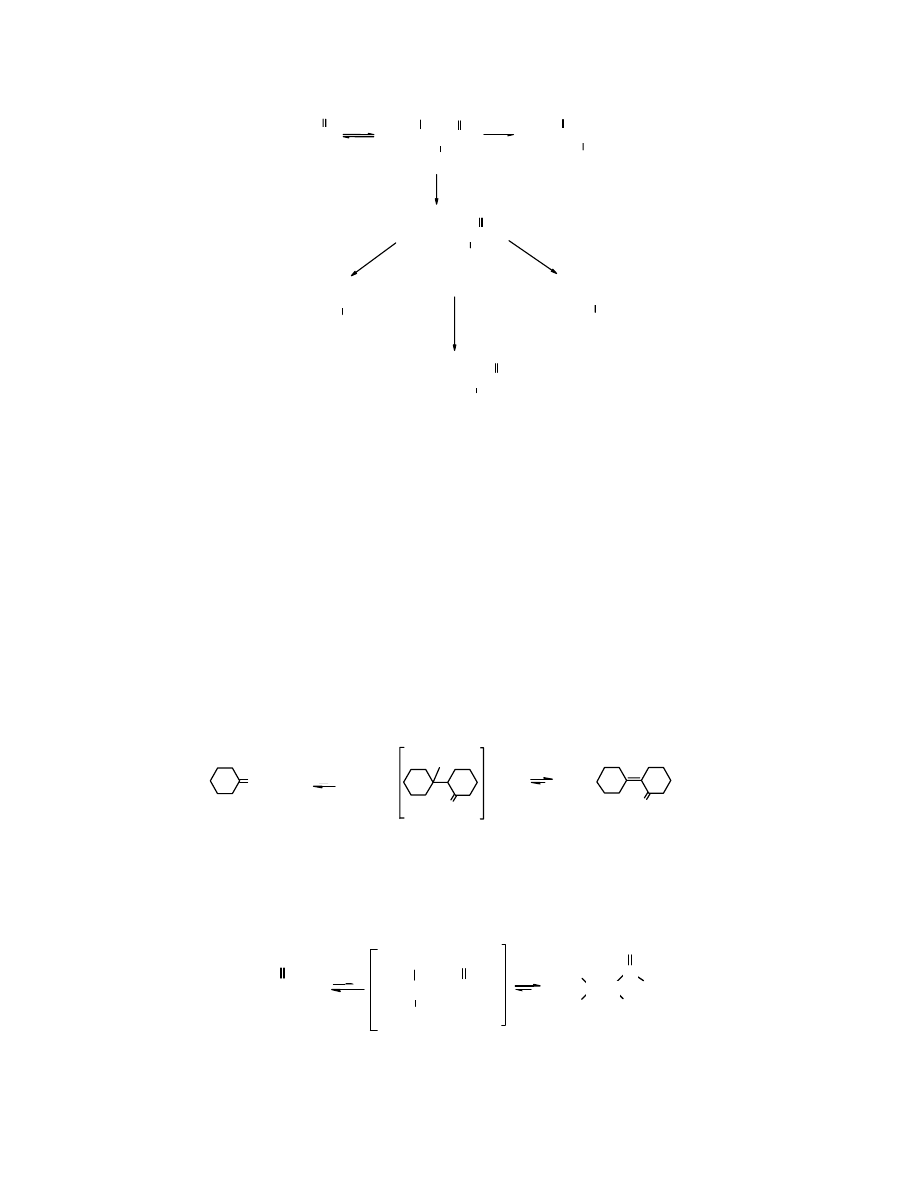
O
OH
R
O
OH
R
O
R
R
R
R
O
RCH
2
CH
NaOH
RCH
2
CHCHCH
NaBH
4
RCH
2
CHCHCH
2
OH
1,3-diol
2
aldehyd
aldol
- HOH
RCH
2
CH=CCH
RCH
2
CH
2
CHCH
2
OH
RCH
2
CH=CCH
2
OH
H
2
/Pd/C
RCH
2
CH
2
CHCH
H
2
/Ni
NaBH
4
alkohol allilowy
aldehyd
alkohol
aldehyd
α,β-nienasycony
Kondensacja aldolowa jest szeroko stosowana w przemyśle, szczególnie do otrzymywania
alkoholi. Tą drogą produkuje się
butan-1-ol
.
Zadanie: zaproponuj schemat reakcji prowadzących do
butan-1-olu
.
Dehydratacja
β
-hydroksyaldehydów i
β
-hydroksyketonów jest charakterystyczną reakcją tych
związków. Często już w warunkach
kondensacji aldolowej
dochodzi do
dehydratacji
, jeżeli nie
zachowuje się specjalnego reżimu (niskiej temperatury). Powstają przy tym
α
,
β
-nienasycone
aldehydy lub ketony, zwane inaczej sprzężonymi enonami. Reakcja
dehydratacji
β
-
hydroksyaldehydów i ketonów jest katalizowana zarówno przez kwasy, jak i zasady. Usunięcie
cząsteczki wody z produktu kondensacji aldolowej przesuwa równowagę reakcji na prawo i w ten
sposób otrzymuje się sprzężone enony z wysoką wydajnością pomimo, tego że równowaga samej
reakcji nie sprzyja
dimeryzacji
. W warunkach równowagi z
cykloheksanonu
powstaje jedynie
22% produktu dimeryzacji, ale w wyniku
dehydratacji
tworzy się ponad 90% sprzężonego
enonu, co świadczy o prawie całkowitym zużyciu substratu (ilościowej dimeryzacji wyjściowego
ketonu).
O
OH
O
O
2
NaOH
+ HOH
cykloheksanon
1-hydroksy-2’oksodicykloheksyl cykloheksylideno-cykloheksan-2-on
(90%)
Jak uprzednio wspomniano produkt
kondensacji aldolowej
acetonu
–
4-hydroksy-4-
metylopenta-2-on
powstaje w reakcji równowagowej jedynie z wydajnością 5%. Jeżeli jednak
reakcja będzie prowadzona w temperaturze powyżej 50
o
C, to z dobrą wydajnością tworzy się
sprzężony enon, zwany
tlenkiem mezytylu
.
O
OH
CH
3
O
C
H
3
C
H
3
H
O
CH
3
Ba(OH)
2
H
3
C-C-CH
3
H
3
C-C-CH
2
-C-CH
3
aceton
C=C
C
50
o
C
4-hydroksy-4-metylopenta-2-on
(5%)
tlenek mezytylu
(95%)
23

Reakcja podobna do
kondensacji aldolowej
zachodzi również w środowisku kwaśnym.
Aceton
po nasyceniu chlorowodorem ulega przekształceniu w
tlenek mezytylu
; równocześnie tworzy się
produkt
trimeryzacji
i
dehydratacji
, nazywany
foronem
.
C
H
3
C
H
3
H
O
CH
3
CH
3
H
C=C
C
C=C
foron
Zadanie: napisz etapy powstawania
foronu
i jego
cyklizacji
do
mezytylenu
, który tworzy się z
acetonu
pod wpływem stężonego kwasu siarkowego.
4.2 Mieszane (krzyżowe) reakcje kondensacji aldolowej
W reakcji
kondensacji aldolowej
dwóch różnych aldehydów, np.
etanalu
i
propanalu
powstaną
cztery produkty
dimeryzacji
: dwa w wyniku połączenia się wzajemnego 2 takich samych
cząsteczek (
etanal
+
etanal
) i (
propanal
+
propanal
) oraz dwa jako efekt
dimeryzacji mieszanej
:
(
etanal
+
propanal
) i (
propanal
+
etanal
).
OH
CH
3
CHCH
2
CHO
OH
CH
3
CH
2
CHCH
2
CHO
OH
CH
3
OH
CH
3
CH
3
CHO
:B
-
+
CH
3
CH
2
CHO
etanal
propanal
3-hydroksybutanal
3-hydroksy-2-metylopentanal
3-hydroksypentanal
3-hydroksy-2-metylobutanal
CH
3
CH
2
CHCHCHO
CH
3
CHCHCHO
+
+
+
+
Reakcja, w której powstają 4 różne produkty o podobnych właściwościach fizycznych i
chemicznych jest mało przydatna, nie tylko dlatego, że wydajności poszczególnych związków są
niskie, ale również z tego powodu, że trudno je rozdzielić. Syntezę chemiczną należy tak
prowadzić, żeby z
maksymalnie wysoką wydajnością otrzymywać jeden czysty produkt
.
Wobec tego wydawać się może, że
mieszane kondensacje aldolowe
są praktycznie
nieprzydatne. Jeżeli jednak w reakcji będzie brał udział jeden z substratów, który nie zawiera
atomu wodoru przy C
α
, to teoretycznie powstaną dwa produkty: autokondensat i mieszany. Jest
to sytuacja znacznie korzystniejsza. Ponadto tę reakcję można tak prowadzić, żeby zmniejszyć
wydajność autokondensatu, który w takiej reakcji jest produktem ubocznym (niepożądanym).
Założony cel osiągną się przez wkraplanie substratu zawierającego H
α
(
A
) do alkalicznego
środowiska reakcji, w którym znajduje się składnik bez H
α
(
B
). W ten sposób substrat
A
dopiero
w środowisku zasadowym może autokondesować, spotyka jest jednak ze znacznym nadmiarem
składnika
B
i wobec tego powstaje głównie produkt mieszany (
A
+
B
, a nie
A
+
A
). Dodatkowo
należy tak dobierać reagenty, żeby grupa karbonylowa składnika
B
była silniejszym elektrofilem
niż reagenta
A
, np. grupy karbonylowe
benzaldehydu
czy
metanalu
są silniejszymi elektrofilami
niż u większości innych aldehydów, nie wspominając o ketonach. Substrat
A
nazywany jest
często donorem (eletronów), a
B
akceptorem.
Z
benzaldehydu
i
etanalu
otrzymuje się w ten sposób
aldehyd cynamonowy
(główny składnik
aromatu cynamonu), a z
benzaldehydu
i
propanalu
powstaje
aldehyd
α-metylocynamonowy
.
24
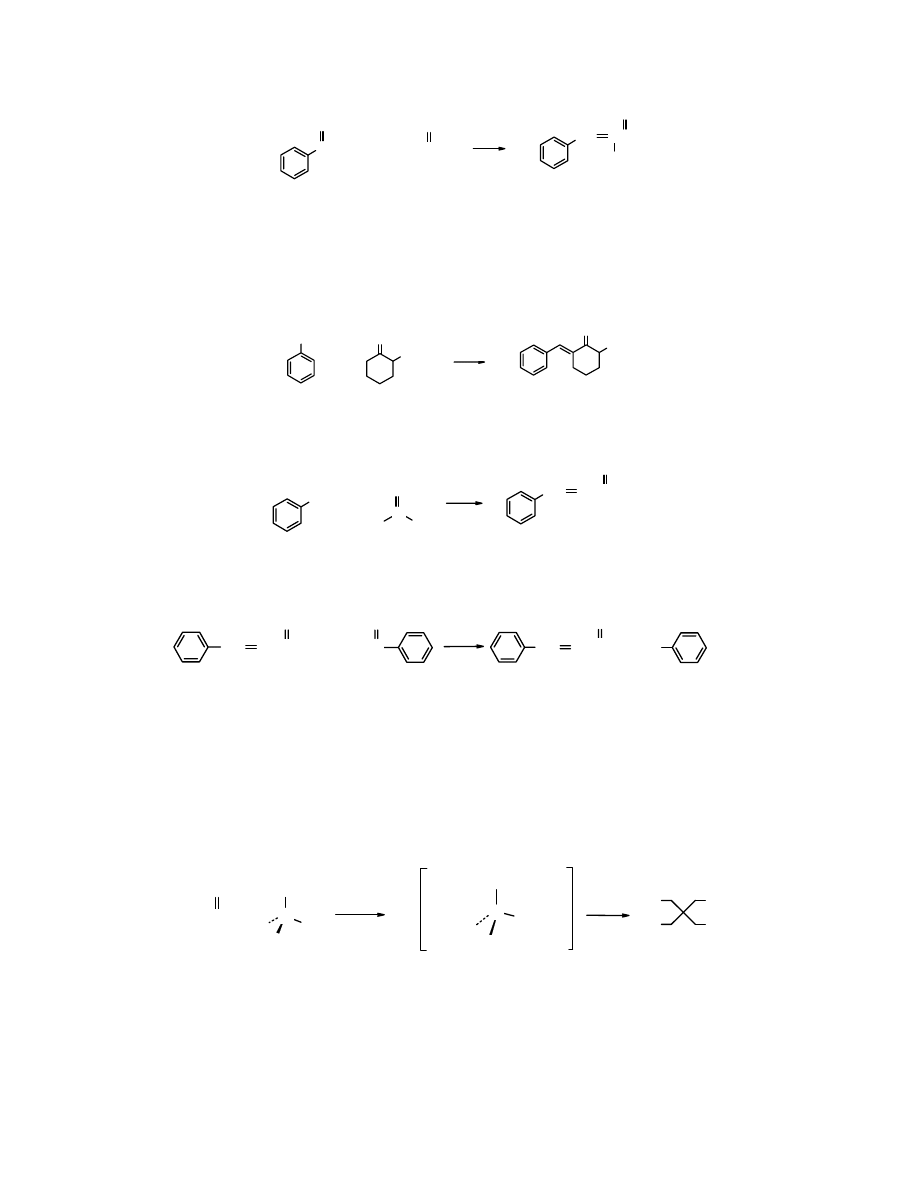
CH
O
O
CH
O
CH
3
+ CH
3
CH
2
CH
CCH
NaOH
10
o
C
benzaldehyd
B
propanal
A
aldehyd
α-metylocynamonowy
(80%)
(akceptor) (donor)
Zadanie: wyjaśnij dlaczego należy roztwór
propanalu
wkraplać do intensywnie mieszanego roztworu
benzaldehydu
w środowisku zasadowym, a nie odwrotnie.
Jako inny przykład krzyżowej kondensacji aldolowej służy reakcja
benzaldehydu
z ketonem, np.
metylocykloheksanonem
.
CHO
O
CH
3
CH
3
O
+
NaOEt
- HOH
benzaldehyd 2-metylocyloheksanon 2-benzylideno-6-metylocykloheksanon
(78%)
W reakcji
benzaldehydu
z
acetonem
powstaje
benzylidenoaceton
. Reakcja biegnie szybko z
dobra wydajnością.
CHO
O
C
H
3
CH
3
CH CH
O
+
C
NaOH
CCH
3
benzylidenoaceton
(77%)
- HOH
beznaldehyd aceton
Benzylidenoaceton
można przekształcić w
dibenzylidenoaceton
przez traktowanie go kolejną
porcją
benzaldehydu
.
CH CH
O
CH
CH CH=CH
O
O
CCH
3
benzylidenoaceton
(77%)
+
C
benzaldehyd
dibenzylidenoaceton
NaOH
HC
- HOH
Akceptorem często
wykorzystywanym
w
krzyżowej kondensacji aldolowej
jest
metanal
. W
reakcji
metanalu
z
etanalem
powstaje produkt addycji trzech cząsteczek
metanalu
do
etanalu
,
który ulega spontanicznej
redukcji
nadmiarem
metanalu
do polialkoholu zwanego
pentaerytrytolem
. Jest on używany jako preparat w leczeniu zaparć, a jego ester z kwasem
azotowym –
tetraazotan pentaerytrytolu
– podobnie jak
nitrogliceryna
rozszerza naczynia
wieńcowe i stosuje się go leczniczo oraz zapobiegawczo w przypadku dusznicy bolesnej. Działa
dłużej niż
nitrogliceryna
.
Tetraazotan pentaerytrytolu
również jak
nitrogliceryna
jest silnym
materiałem wybuchowym.
O
CHO
HOCH
2
CH
2
OH
O
H
O
H
OH
OH
CHO
H
H
H
3 HCH +
Ca(OH)
2
C
HCHO
krzyżowa reakcja
Cannizzaro
- HCOOH
metanal etanal
pentaerytrytol
CH
2
OH
(trihydroksymetylo)etanal
C
(55%)
Wysoką wydajność jednego tylko produktu w krzyżowych kondensacjach aldolowych zapewnia
użycie jako donora związku posiadającego bardziej kwaśne atomy wodoru niż H
α
aldehydów i ketonów
. Do takich związków należy, np.
acetylooctan etylu
. Dwie grupy
25
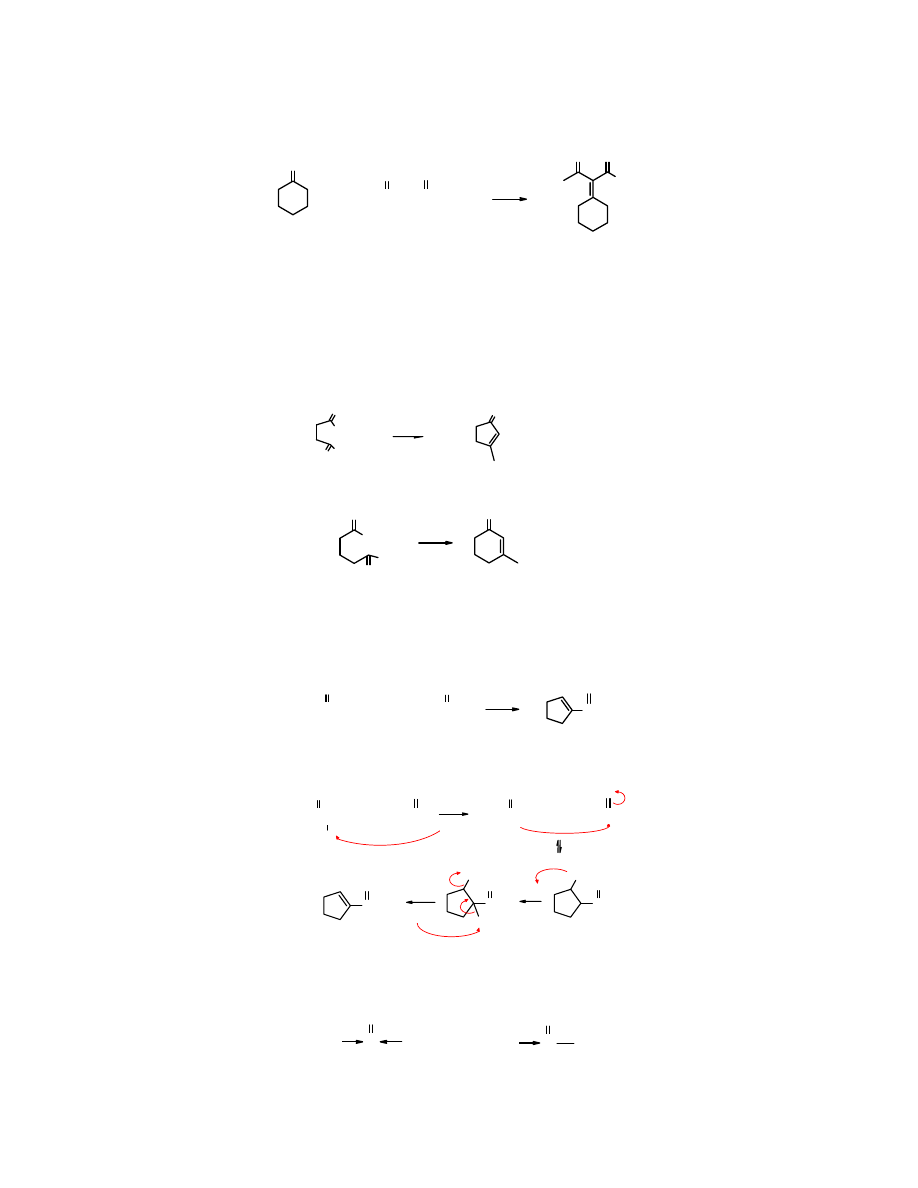
karbonylowe mocniej uaktywniają atomy wodoru, które przez to łatwiej ulegają oderwaniu
tworząc karboanion przyłączający się do grupy karbonylowej akceptora.
O
O
O
O
O
OEt
+ CH
3
CCH
2
COEt
NaOEt
acetylooctan etylu
- HOH
cykloheksan
(akceptor) (donor)
cykloheksylidenoacetylooctan etylu
(80%)
4.3 Wewnątrzcząsteczkowa kondensacja aldolowa – reakcje cyklizacji
Cząsteczki zawierające grupę akceptorową i donorową mogą ulegać
wewnątrzcząsteczkowej
kondensacji aldolowej
w wyniku, czego dochodzi do cyklizacji. W tego typu reakcjach biorą
udział dialdehydy i diketony (także oksoaldehydy); najłatwiej kondensuję te, w których grupy
karbonylowe oddalone są od siebie tak, żeby powstawały pięcio- lub sześcioczłonowe
pierścienie. Z 1,4-diketonu tworzy się pochodna
cyklopentanonu
.
CH
3
O
O CH
3
O
NaOH
- HOH
heksano-
-2,5-dion
3-metylocyklopent-2-enon
(40%)
Z 1,5-ketonu powstanie pochodna
cykloheksanonu
.
CH
3
O
CH
3
O
O
NaOH
- HOH
heptano-
2,6-dion
3-metylocyklo-
heks-2-enon
(44%)
Natomiast z
6-oksoheptanalu
otrzymuje się głównie pochodną
cyklopetanonu
pomimo tego, że
odległość pomiędzy grupami karbonylowymi jest taka sama jak w
hepta-2,6-dionie
. Jest to
spowodowane tym, że lepszym akceptorem jest grupa aldehydowa, zaś atomy wodoru przy C5 są
bardziej kwaśne niż przy C7.
O
O
CCH
3
O
CH
3
CCH
2
CH
2
CH
2
CH
2
CH
NaOH
- HOH
6-oksoheptanal keton 1-cyklopentenylowometylowy
(73%)
Mechanizm reakcji:
O
O
H
O
O
CCH
3
O
CCH
3
OH
H
CCH
3
O
O
O
CH
3
CCHCH
2
CH
2
CH
2
CH
-
OH
CH
3
CCHCH
2
CH
2
CH
2
CH
..
-
..
:
..
:
:
-
HOH
..
:
-
OH
6-oksoheptanal
keton 1-cyklopentenylowo-metylowy
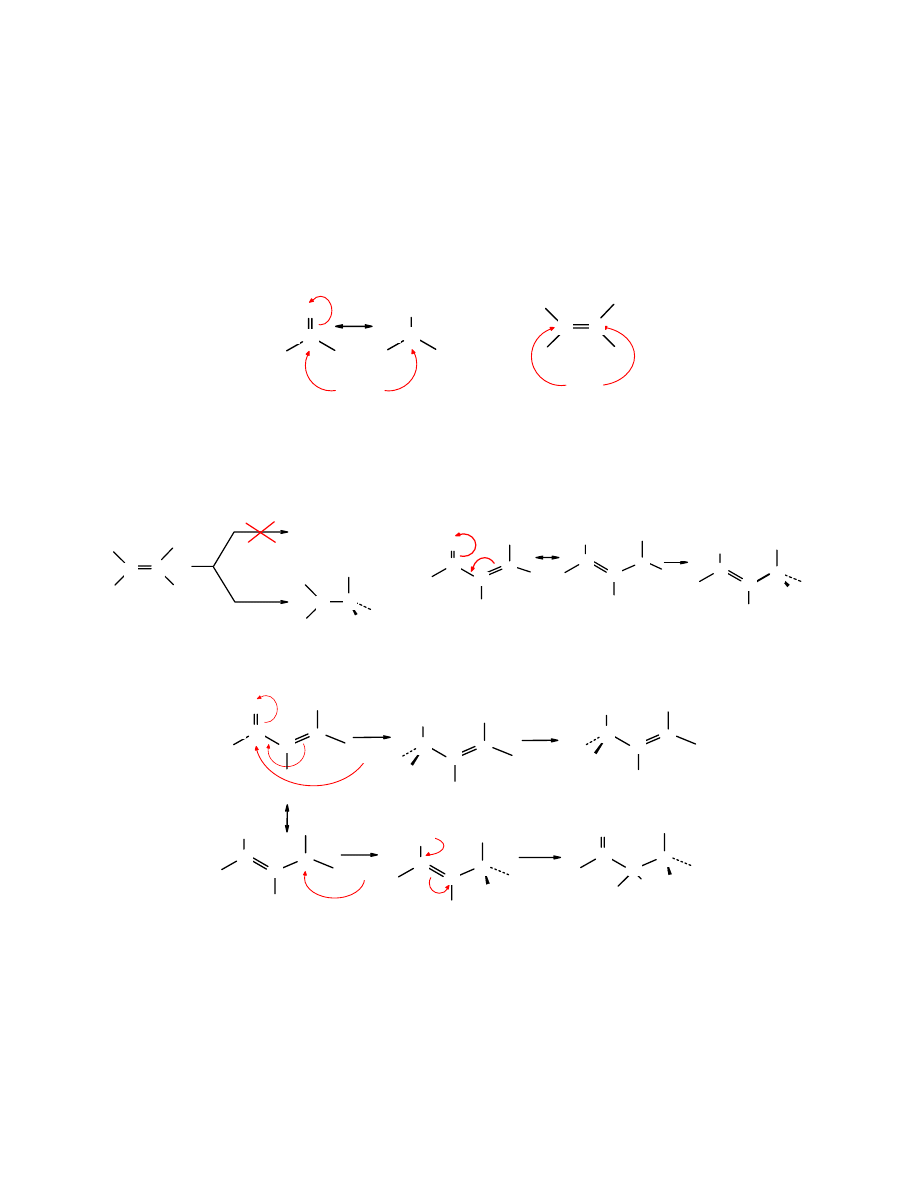
Aktywność elektronoakceptorowa ketonowej grupy karbonylowej w stosunku do aldehydowej
jest zmniejszona w wyniku elektrodonorowego oddziaływania dwóch reszt alkilowych:
O
O
C
R
R'
C
R
H
w ketonach w aldehydach
26
4.4 Addycja nukleofilowa do
α,β-nienasyconych aldehydów i ketonów
Przyłączenie nukleofila do karbonylowego atomu węgla sprzężonych enonów nie zawsze
prowadzi do
addycji 1,2
.
Może nastąpić addycja 1,4
. Kierunek reakcji w dużej mierze zależy
od właściwości nukleofilowo-zasadowych odczynnika nukleofilowego.
Grupa karbonylowa jest spolaryzowana, przy czym częściowy ładunek dodatni jest
zlokalizowany przy atomie C i w to miejsce skierowany jest atak nukleofila –
:Nu
-
. W alkenach
i enonach izolowanych znajduje się podwójne wiązanie C=C, które ma powinowactwo do
elektrofila
E
+
.
O
O
C
: :
δ+
δ-
C
: :
+
C
C
:Nu
-
E
+
..
-
Natomiast enonach sprzężonych na skutek oddziaływania elektronów
π obu podwójnych wiązań
(C=C i C=O) dochodzi do polaryzacji cząsteczki i utworzenia częściowego ładunku dodatniego
na atomie węgla C4, który tym samym staje się podatny na oddziaływanie odczynnika
nukleofilowego
:Nu
-
.
O
E
O
Nu
O
:Nu
-
C
: :
C
C
+
C
C
:Nu
-
..
E
+
brak reakcji
C
C
+
-
C
: :
C
C
..
-
C
: :
C
C
alken
sprzężony enon
Mechanizm addycji 1,2 i 1,4
:
O
O
O
Nu
O
Nu
OH
Nu
O
Nu
H
C
: :
δ+
δ-
C
C
2 3
4
C
: :
C
C
2
3
4
+
:Nu
-
addycja
1,2
addycja
1,4
C
: :
C
C
1
2
4
..
3
C
: :
C
C
2
3
4
..
-
-
+
H/HOH
+
H/HOH
C
:
C
C
1
2
4
..
3
C
: :
C
C
2
3
4
nienasycony alkohol
aldehyd lub keton
1
1
..
1
:Nu
-
-
Silnie zasadowe odczynniki nukleofilowe, np.
związki Grignarda
przyłączają się głównie w
sposób 1,2.
27
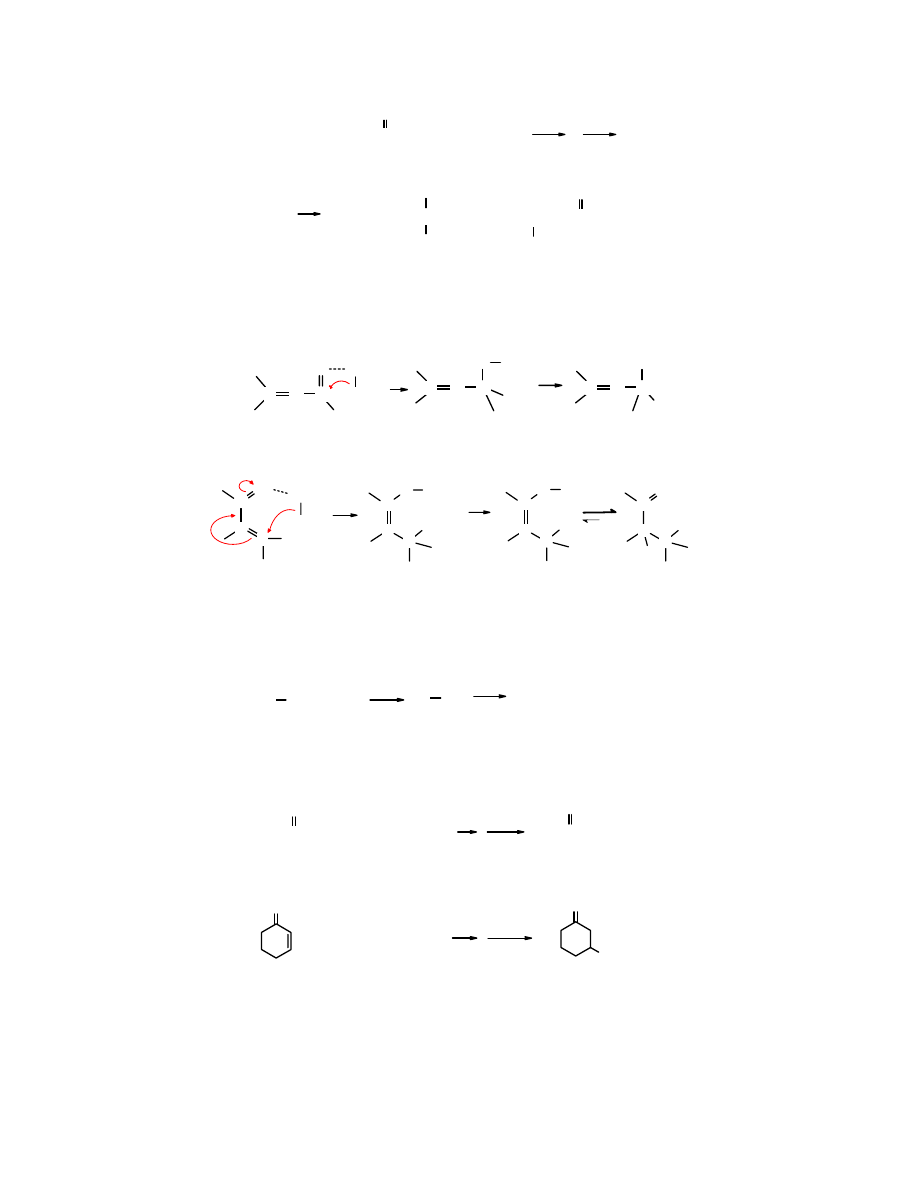
O
OH
CH
3
CH
3
O
CH
3
CH=CHCCH
3
+ CH
3
MgBr
THF
+
H/HOH
CH
3
CH=CHCCH
3
+
CH
3
CHCH
2
CCH
3
pent-3-en-2-on bromek metylomagnezowy
3-metylopent-2-en-3-ol
(72%)
4-metylopent-2-on
(20%)
addycja 1,2 addycja 1,4
Mechanizm reakcji
addycji 1,2
związków Grignarda:
O
O
R
MgX
OH
R
MgX
C C C
δ+
δ-
C C C
:
..
..
:
C C C
..
:
HX
- MgX
2
R
δ-
δ+
addycja 1,2
Mechanizm reakcji
addycji 1,4
związków Grignarda:
..
..
O
MgX
O
R
MgX
O
R
H
O
R
H
C
C
C
:
R
δ-
δ+
C
C
C
:
HX
- MgX
2
C
C
C
:
..
C
C
C
:
..
enol
aldehyd lub keton
sprzężony enon
addycja 1,4
Mniej zasadowe nukleofile, np.
odczynniki Gilmana
przyłączają się prawie wyłącznie w sposób
1,4.
Otrzymywanie odczynników Gilmana:
X
Li
R
pentan
+ 2 Li
R
- LiX
CuI
eter
Li
+
(
-
R-Cu-R) + LiI
halogenek alkilu alkilolit dialkilomiedzian litu
(odczynnik Gilmana)
Za pomocą
dimetylomiedzianu litu
można wprowadzić grupę metylową do łańcucha węglowego
sprzężonego enonu:
O
O
CH
3
CCH=CH
2
+ Li(CH
3
)
2
Cu
+H/HOH
CH
3
CCH
2
CH
2
CH
3
but-1-en-3-on dimetylomiedzian litu pentan-2-on
(97%)
Tym sposobem przyłącza się również reszty nienasycone lub aromatyczne:
O
O
CH=CH
2
+H/HOH
Li(H
2
C=CH)
2
Cu
(65%)
+
cykloheks-2-enon diwinylomiedzian litu 3-winylocykloheksanon
28
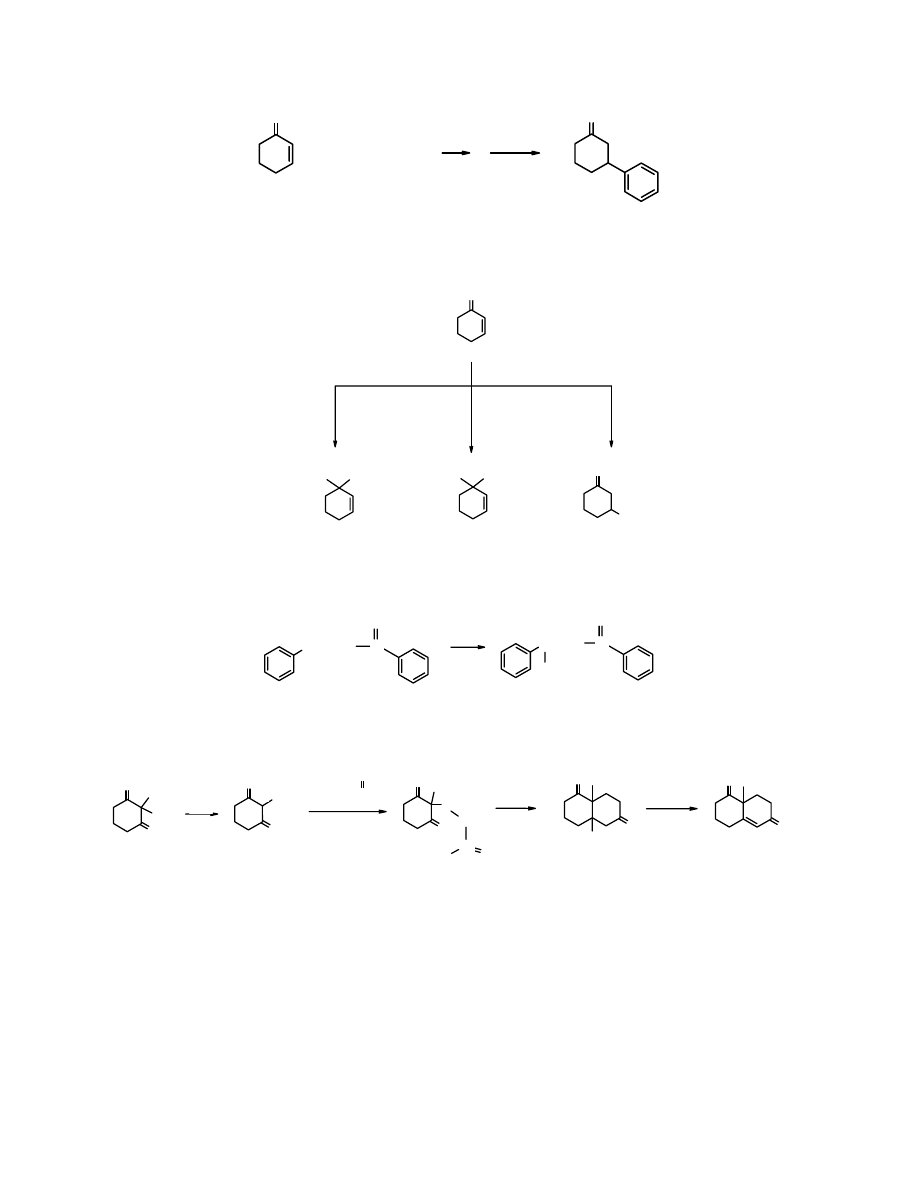
O
O
+H/HOH
Li(C
6
H
5
)
2
Cu
+
(70%)
cykloheks-2-enon difenylomiedzian litu 3-fenylocykloheksanon
Odczynniki Gilmana stanowią wyjątek pośród związków metaloorganicznych, np.
butylolit
i
inne alkilolity jako silnie zasadowe ulegają
addycji 1,2
.
O
O
CH
3
CH
3
O
H
CH
3
O
H
2.
+
H/HOH
1. Li(CH
3
)
2
Cu
1.
CH
3
MgBr
1. CH
3
Li
2.
+
H/HOH
2.
+
H/HOH
cykloheks-2-enon
1-metylocykloheks-2-enon 3-metylocykloheksanon
Do grupy nukleofili, które do sprzężonych enonów przyłączają się w sposób 1,4 należy jon
cyjankowy. Jest on silnym nukleofilem o umiarkowanej zasadowości.
CH=CH
O
CHCH
2
O
CN
C
1.
CN
-
1.
H
+
C
keton fenylowo-(2-fenylowinylowy) 3-cyjano-1,3-difenylo-3-oksopropan
(95%)
Także karboaniony powstałe z C-kwasów są silnymi nukleofilami i przyłączają się zwykle w
sposób 1,4. Wykorzystując je można przeprowadzać skomplikowane syntezy.
O CH
3
H
O
O
CH
3
O
O
O CH
3
CH
2
O CH
2
C
H
3
O
O
O
OH
O
O
-
OH
MeOH
-
..
CH
2
=CHCCH
3
C
-
OH/MeOH
-
OH/MeOH
- HOH
2-metylocyklo-
heksa-1,3-dion
karboanion
5,6-didehydro-1,7-diokso-
-10-metylodekalina
(65%)
5. Azotowe pochodne aldehydów i ketonów
Aldehydy i ketony reagują z aminami i ich analogami tworząc pochodne, których budowa zależy
od rzędowości amin. Z aminami 1
o
powstają iminy (aldiminy lub ketiminy) zwane zasadami
Schiffa, podczas gdy z aminami 2
o
tworzą się enaminy (grupa aminowa przyłączona do C=C).
Reakcja jest katalizowana kwasami, jednak środowisko silnie kwaśnie nie sprzyja reakcji;
optymalne pH wynosi ~ 4.
29
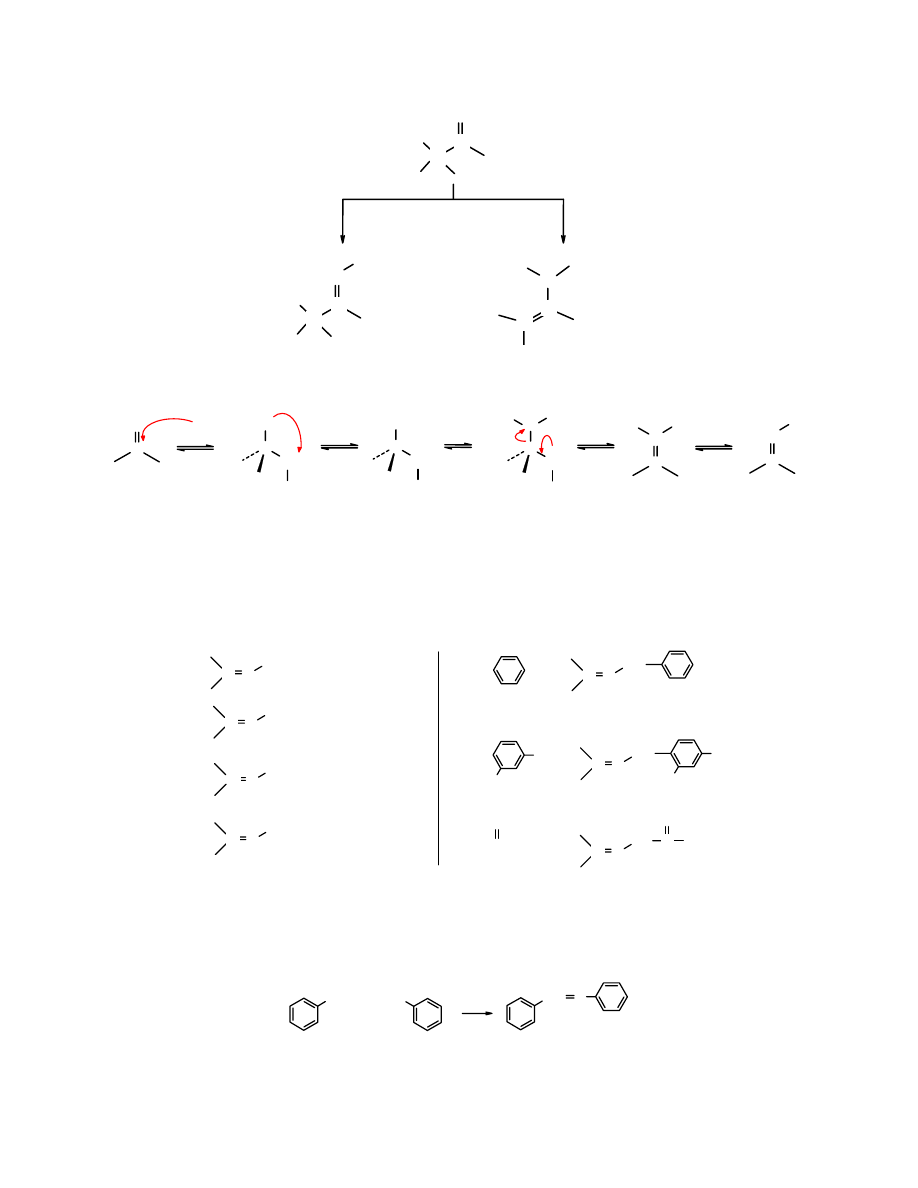
O
H
R
2
NH
..
R-NH
2
..
N
H
R
N
R
R
C
C
C
C
:
+ HOH
C
C
+ HOH
aldehyd
lub keton
imina (aldimina lub ketimina) enamina
..
Mechanizm addycji amin
1
o
O
O
NH
2
Y
OH
NH
Y
O
NH
Y
H
H
N
Y
H
N
Y
:
C
aldehyd
lub keton
H
2
N-Y
..
C
..
..
: :
-
+
C
..
:
..
+
H
C
..
:
..
- HOH
+
C
+
- H
+
C
aminoalkohol
(karbinoloamina)
zasad Schiffa
W zależności od reszty Y przyłączonej do grupy aminowe otrzymuje się szereg azotowych
pochodnych aldehydów i ketonów :NH
2
-Y.
Y: wzór nazwa Y: wzór nazwa
N
H
N
R
N
OH
N
NH
2
N
NH
NO
2
O
2
N
N
NH
NO
2
O
2
N
O
N
NH
NH
2
O
semikarbazon
-H
C ..
aldimina lub ketimina
-R (alkil, aryl)
C ..
zasada Schiff
(imina)
-OH
-NH
2
C ..
oksym
hydrazon
-NH-
C ..
fenylohydrazon
-NH-
C ..
2,4-dinitrofenylohydrazon
-NH-C-NH
2
C ..
C
C
Jedną z najbardziej znanych imin jest
benzylidenoanilina
, preparat, który prawie każdy student
wydziału chemicznego zamodzielnie syntezuje w trakcie zajęć laboratoryjnych z chemii
organicznej, ponieważ jest on niezwykle łatwy w wykonaniu – miesza się ze sobą oba substraty i
krystalizuje produkt.
CH N
CHO
N
H
2
+
- HOH
benzaldehyd anilina benzylidenoanilina
(84%)
30
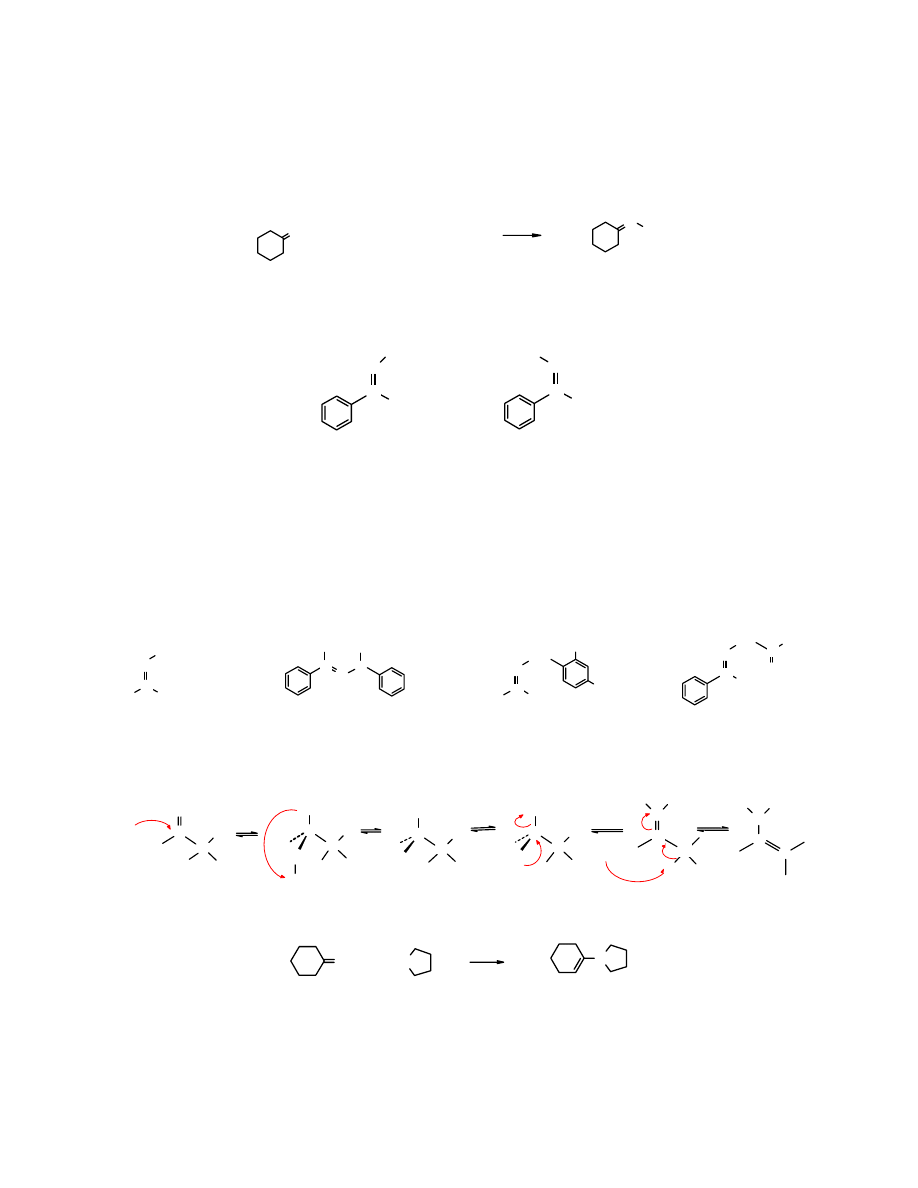
Duże znaczenie pośród azotowych pochodnych ketonów ma o
ksym cykloheksanonu
. Z niego po
przeształceniu w
kaprolaktam
(przegrupowanie Hofmanna) wytwarzana jest
lizyna
, służy także
jako monomer w przemyśle włókien syntetycznych.
Oksym cykloheksanonu
otrzymuje się w
reakcji
cykloheksanonu
z chlorowodorkiem hydroksyloaminy w obecności
octanu sodu
(buforu
obniżającego pH).
O
N
OH
..
+
H
2
NOH
.
HCl
NaAc
cykloheksanon
chlorowodorek hydroksyloaminy
oksym cykloheksanonu
(87%)
Oksymy i innego podobne związki zawierające układ =N- tworzą stereoizomery typu cis-/trans-
zwane syn- i anti-.
H
N
OH
H
N
O
H
.
.
..
C
C
syn
anti
stereoizomery oksymu benzaldehydu
Takie pochodne aldehydów i ketonów, jak oksymy, fenylohydrazony,
2,4-
dinitrofenylohydrazony i semikarbazony wykorzystuje się do charakteryzowania związków
karbonylowych. Są one zwykle krystaliczne i dzięki temu po specyficznych temperaturach
topnienia można zidentyfikować aldehydy i ketony, z których powstały. Ten sposób identyfikacji
stracił obecnie na znaczeniu, z uwagi na powszechne stosowanie metod spektralnych, szczególnie
użytecznego do tego celu NMR.
Przykłady krystalicznych pochodnych ciekłych związków karbonylowych:
N
H
C
H
3
OH
N
N
H
H
N
C
H
3
CH
3
NH
NO
2
NO
2
N
H
NH
O
NH
2
C
C
C
C
C
oksym etanalu
fenylohydrazon benzaldehydu
2,4-dinitrofenylohydrazon semikarbazon
(tt. 47
o
C) (tt. 158
o
C)
acetonu
(tt. 126
o
C)
benzaldehydu
(tt. 222
o
C)
Mechanizm addycji 2
o
amin do związków karbonylowych
O
H
O
R
2
N
H
H
OH
R
2
N
H
OH
2
R
2
N
H
N
R
R
N
H
R
R
C
C
R
2
NH
..
+
C
C
+
+
: :
:
:
..
-
C
C
:
..
..
C
C
:
..
..
+
H
HOH
C
C
..
C
C
..
..
+
-
+
H/HOH
- HOH
enamina
Często do otrzymywania enamin używa się 2
o
amin cyklicznych, np.
pirolidyny
.
O
N
H
N
:
+
- HOH
:
cykloheksanon pirolidyna pirolidynowa enamina cykloheksanonu
6. Redukcja Wolffa-Kiżnera
Grupę karbonylową można zredukować do CH
2
. Służy do tego
redukcja Wolffa-Kiżnera
.
31
Ludwig Wolff (1857-1919); ur. w Neustadt/Hardt (Niemcy), doktorat w Strasburgu, Francja, u Fittiga; prof. Uniw.
w Jenie.
N. M. Kiżner (1867-1935); ur. w Moskwie; doktorat w Moskwie u Markownikowa; prof. w Tomsku i w Moskwie.
Reakcja
addycji
hydrazyny do grupy karbonylowej jest wykorzystywana do redukcji tej grupy
do -CH
2
-, czyli przekształcania związku karbonylowego w węglowodór; nosi ona nazwę
redukcji Wolffa-Kiżnera
. Polega ona na ogrzewaniu aldehydu lub ketonu z hydrazyną w
obecności KOH w temperaturze 240
o
C. Pośrednio powstaje fenylohydrazon. Późniejsza
modyfikacja polegająca na zastosowaniu DMSO jako rozpuszczalnika pozwoliła na znaczne
obniżenie temperatury reakcji. Redukcja Wolffa-Kiznera jest alternatywnym redukcji grupy
karbonylowej do metody Clemmensena (za pomocą amalgamatu rtęci).
O
CH
2
CH
3
CH
2
CH
2
CH
3
C
H
2
NNH
2
/KOH
DMSO
+ HOH + N
2
propiofenon propylobenzen
(
keton etylowo-fenylowy
) (82%)
7. Reakcja haloformowa
Ketony metylowe i acetaldehyd rozpadają się w środowisku zasadowym pod wpływem
halogenów; wydziela się przy tym haloform (CHX
3
), a z reszty tworzy się sól kwasu
karboksylowego.
O
CH
3
COO
O
CX
3
-
C
X
2
-
OH
+ CHX
3
C
-
OH
acetofenon
trihalogenoacetofenon
benzoesan
haloform
Ketony metylowe ulegają w środowisku zasadowym
halogenowaniu
pod wpływem jodu, bromu
lub chloru. Reakcja nie zatrzymuje się na
monohalogenowaniu
, ale biegnie dalej aż do
podstawienia
wszystkich atomów wodoru grupy metylowej. Taki trihalogenometylowy keton
jest nietrwały w obecności
zasad i
rozpada
się na haloform oraz jon karboksylanowy:
..
..
O
CH
2
O
CH
2
H
O
CH
2
O
CH
2
Br
O
CBr
3
O
COO
:B
-
R C
R C
R C
-
-
:
:
:
: :
-
jon enolanowy
Br
2
R C
:
:
+ Br
-
Br
2
Br
2
:B
-
:B
-
R C
:
:
:B
-
R C
:
:
+ CHBr
3
jon
karboksylanowy
chloroform
Podobnej reakcji ulegają 2
o
alkohole zawierające grupę CH
3
przy hydroksylowanym atomie
węgla. Tego typu alkohole łatwo utleniają się pod wpływem halogenu do metylowego ketonu:
OH
O
RCHCH
3
X
2
RCCH
3
- HX
2
o
alkohol zawierający grupę CH
3
keton metylowy
przy hydroksylowanym atomie C
Działanie jodem na ketony metylowe, acetaldehyd i 2
o
alkohole zawierające grupę CH
3
przy
hydroksylowanym atomie C, służy jako test do wykrywania tego typu ugrupowań:
O
OH
-CCH
3
i
-CHCH
3
32
Po dodaniu jodu do zasadowego roztworu takich związków wypada
jodoform
w formie żółtych
kłaczków.
Test ten nosi nazwę
reakcji jodoformowej.
O
-CCH
3
I
2
-
OH
-COO
-
+ CHI
3
jodoform
8. Kondensacja pinakolinowa
Pinakolanami nazywane są wicynalne glikole zawierające grupy hydroksylowe przy 3
o
atomach
węgla, przy czym najprostszy z nich, czyli
2,3-dimetylo-2,3-dihydroksybutan
nosi nazwę
pinakolu
. Pinakole powstają w środowisku aprotonowym w wyniku
dimeryzacji
ketonów pod
wpływem reaktywnych metali. W obecności donorów protonów metale te redukują ketony do
alkoholi. Pierwszy etapem reakcji w obu przypadkach jest utworzenie anionorodnika (ketylu) w
procesie przeniesienia pojedynczego elektronu (SET, ang. single electron transfer).
..
O
R'
O
R'
-
R C
+ M
o
SET
:
:
R C
:
:
.
anionorodnik
(ketyl)
Dalsze etapy reakcji zależą od środowiska:
W środowisku
protonowym
anionorodnik przyłącza proton i przekształca się w rodnik,
który ponownie w reakcji SET pobiera elektron od atomu metalu przechodząc w anion.
Z anionu po kolejnym protonowaniu tworzy się alkohol.
A.
B.
Zaś w środowisku
aprotonowym
anionorodnik
dimeryzuje
tworząc dianion
(alkoholan), z którego po zakwaszeniu powstaje pinakol.
..
O
R'
OH
R'
O
R'
O
R'
O
O
R'
OH
R'
OH
R'
H
R
OH
R' R'
R'
R C.
anionorodnik
(ketyl)
środowisko
protonowe
środowisko
aprotonowe
H
+
..
R C
:
.
-
..
R C
:
.
+
..
-
..
R C
:
. ..
-
..
R C
:
..
-
..
R C
:
..
H
+
SET
M
o
..
R C
:
..
H
+
R C
:
..
alkohol
R C C
HO
pinakol
-
A
B
rodnik
anionorodnik
anionorodnik
: :
-
anion
alkoholan
33
Dowodem na to, że w reakcji
kondensacji pinakolinowej
pośrednio powstaje anionorodnik jest
zmiana barwy roztworu, w którym zachodzi reakcja. Zarówno substrat – keton, jak i produkt –
pinakol są bezbarwne, natomiast w trakcie reakcji pojawia się intensywnie niebieskie (lub
fioletowe) zabarwienie, typowe dla anionorodników.
O
O
OH
+
:
.
..
C
K
THF
C
:
: :
-
K
ketyl benzofenonu
intensywnie
niebieski
benzofenon
bezbarwny
+
H
C
C
HO
benzopinakol
bezbarwny
Pinakol
, czyli
2,3-dimetylobutano-2,3-diol
jest najczęściej otrzymywanym pinakolem.
Otrzymuje się z bardzo dobrą wydajnością w reakcji
acetonu
z magnezem; krystalizuje w postaci
hydratu.
O
O
OH
CH
3
C
H
3
CH
3
CH
3
C
H
3
CH
3
C
H
3
CH
3
O
C
H
3
CH
3
O
O
CH
3
C
H
3
CH
3
CH
3
2+
:
.
..
C
Mg
benzen
C
:
: :
-
aceton
+
H
C
C
HO
2
.
..
C
: :
-
Mg
C
C
-
-
..
..
: : : :
HOH
6 HOH
.
hydrat pinakonu
95%
9. Addycja wodorosiarczanu (IV) – połączenia bisulfitowe
Wodorosiarczany (IV) przyłączają się do grupy karbonylowej aldehydów i niektórych ketonów
(nie zawierających objętościowo dużych podstawników) tworząc krystaliczne, trudno
rozpuszczalne w wodzie sole kwasów
α
-hydroksysulfonowych, zwane połączeniami
bisulfitowymi.
O
OH
SO
3
C
+ Na
HSO
3
C
Na
-
+
-
+
aldehyd lub
keton
połączenie bisulfitowe
(
α
-hydroksysulfonian sodu)
Z połączeń bisulfitowych łatwo odtwarza się wyjściowe związki karbonylowe. Ulegają one
rozkładowi pod wpływem kwasów lub zasad. Połączenia bisulfitowe służą często do
wyodrębniania związków karbonylowych z mieszanin (ze środowiska reakcji) lub do usuwania
ich resztek, kiedy były użyte jako substraty.
OH
SO
3
O
O
C
-
+
H
-
OH
C
+ SO
2
+ HOH
C
+ SO
3
-2
+ HOH
10. Reakcja Wittiga
Georg Wittig (1897-1987), ur. w Berlinie; doktorat w Marburgu u von Auwersa; prof. uniwersytetów we Freiburgu,
Tybindze i Heiderbergu; laureat nagrody Nobla w 1979 r.
Reakcja Wittiga służy do otrzymywania alkenów z aldehydów lub ketonów. Kluczowym
reagentem w tej reakcji są ylidy – specyficzne pochodne
trifenylofosfiny
.
34
P
..
trifenylofosfina
Trifenylofosfina
ulega łatwo
alkilowaniu
do 4
o
soli fosfoniowej.
Br
CH
3
Ph
3
P: + CH
3
S
N
2
Ph
3
P
Br
-
+
bromek trifenylometylofosfoniowy
trifenylo-
fosfina
bromek
metylu
4
o
sole fosfoniowe, np.
bromek trifenylometylofosfoniowy
pod wpływem silnych zasad
przekształca się w betainę, będącą izomeryczną formą ylidu. Zasada odrywa z halogeneku
trifenyloalkilofosfoniowego kwaśny atom wodoru znajdujący się przy C
α
w stosunku do atomu
fosforu.
CH
2
H
CH
2
CH
2
..
BuLi
Ph
3
P
Br
-
+
bromek
trifenylometylofosfoniowy
Ph
3
P
-
+
Ph
3
P
ylid
betaina
Betaina tym różni się od ylidu, że w betainie rozdzielone są ładunki.
Obie te formy są
odmianami mezomerycznymi tego samego związku
.
Ylid reaguje ze związkami karbonylowymi tworząc nietrwały addukt, który się rozpada na alken i
tlenek trifenylofosfiny
.
CH
2
O
CH
2
CH
2
O
C
H
2
..
Ph
3
P
ylid
C
:
Ph
3
P
-
+
Ph
3
P
-
+
C
..
..
:
C
- Ph
3
P=O
alken
tlenek trifenylofosfiny
Reakcja Wittiga, pośród wielu metod otrzymywania alkenów, należy do najbardziej
selektywnych, szczególnie gdy produktem ma być związek o rozbudowanym szkielecie
węglowym.
Z aldehydów lub ketonów można trzymać alkeny również innym sposobem: poprzez ich reakcję
ze związkami Grignarda, a następnie dehydratację otrzymanych alkoholi pod wpływem POCl
3
,
jednak produktem jest zwykle mieszanina izomerów.
O
CH
3
CH
2
CH
2
1.
CH
3
MgBr
2.
POCl
3
Ph
3
P=CH
2
+
1-metylocykloheksen
metylenocykloheksen
metyleno-
cykloheksen
(84%)
35
O efektywności reakcji Wittiga świadczy to, że znalazła ona zastosowanie przemysłowe, np. do
wytwarzania
β-karotenu
(prowitaminy A).
CH
PPh
3
O
HC
+
retinal
ylid retinylideno-
trifenylofosfoniowy
β-karoten
reakcja
Wittiga
11. Tautomeria keto-enolowa
Związki karbonylowe zawierające atom wodoru w położeniu
α ulegają
tautomerii
, przemianie
polegającej na przemieszczaniu się H
α
pomiędzy atomami O
↔C. W większości przypadków
równowaga jest przesunięta w kierunku grupy karbonylowej (na lewo):
..
..
H
R'
O
R''
R
R'
O
R''
H
R C
C
C
C
:
:
Zjawisko przejścia atomu wodoru (przegrupowanie H) w obrębie tej samej cząsteczki nazywane
jest
tautomerią
(grec. tauto – takie samo, meros – część). Poszczególne izomery tautomeryczne
nazywane są tautomerami.
Tautomeria
jest związana z
mezomerią
. Etapem pośrednim
tautomeryzacji jest jon enolanowy, a powstający tautomer jest enolem.
Nazwa enol pochodzi od połączenia końcówek dwóch grup funkcyjnych „en” (od alkenu) i „ol”
(od alkoholu) i oznacza, że grupa -OH jest przyłączona do atomu węgla tworzącego podwójne
wiązanie (tzn. o hybrydyzacji sp
2
).
R
H
O
R''
R'
R
R'
O
R''
H
R
R'
O
R''
C C
C C
C
C
- H
+
+ H
+
- H
+
+ H
+
jon enolanowy
:
: :
..
..
:
-
..
enol
forma
karbonylowa
W środowisku obojętnym tautomeria jest reakcją wolną, natomiast w obecności zarówno
kwasów jak i zasad jej szybkość znacznie wzrasta.
Kataliza kwasowa
:
H
O
H
O
H
O
H
H
O H
+
C
C
C
..
:
+
H
C
..
C
C
..
:
+
-
+
H
C
C
..
:
forma ketonowa
forma enolowa
Kataliza zasadowa:
H
O
O H
O
O
C
C
..
:
-
+
H
C
C
..
:
forma ketonowa
forma enolowa
:
C
-
OH
C
..
-
:
C
C
..
-
:
- HOH
36
Aldehydy i ketony, chociaż występuję w obu formach (ketonowej i enolowej) to najczęściej
równowaga jest zdecydowanie przesunięta w kierunku grupy karbonylowej. Znane są jednak
związki, w których forma enolowa przeważa, należy do nich
fenol
, a także 1,3-diony.
Równowaga tautomeryczna przykładowych związków
CH
OH
O
O
C
H
2
OH
O
OH
OH
O
O
O
OH
O
forma ketonowa forma enolowa
aldehyd
octowy
CH
3
CH
CH
2
aceton
CH
3
CCH
3
99,99%
<0,001%
CCH
3
99,999%
<0,0001%
cykloheksanon
98,8%
1,2%
fenol
<0,001%
99,999%
penta-2,4-dion
CH
3
CCHCCH
3
CH
3
C=CHCCH
3
24%
76%
Aromatyczność cząsteczki
fenolu
jest przyczyną jej stabilności; przeważa forma enolowa,
ponieważ
fenol
ma niższą energię potencjalną niż tautomerczny keton.
W przypadku 1,3-dionów wiązanie wodorowe H
…
O
…
H w formie enolowej obniża energię
cząsteczki.
O
O
H
H
CH
3
C
H
3
O
O
H
H
CH
3
C
H
3
O
O
H
CH
3
C
H
3
H
:
:
:
:
:
:
:
..
:
:
:
:
..
forma ketonowa forma enolowa
penta-2,4-dion
Z powodu tautomerii dochodzi czasami do nieoczekiwanych, a nawet zaskakujących przemian.
Przykładem może być racemizacja (przemiana enancjomeru w racemat) optycznie czynnych
związków karbonylowych.
Ph
Et
Me
H
O
Ph
Et
Me
H
O
Ph
Et
H Me
O
C
C
-
OH
lub
+
H
C
C
C
C
+
keton (+)-sec-butylowo-fenylowy keton (
±)-sec-butylowo-fenylowy
37
Przyczyną tej racemizacji jest tworząca się przejściowo, pozbawiona chiralnego atomu węgla
forma enolowa. W trakcie odtworzania grupy karbonylowej -C=O dochodzi do powstanie takiej
samej ilości izomeru (+) jak i (-), czyli racematu.
Zadanie: napisz wzory mezomeryczne jonu enolanowego powstałego z
ketonu sec-butylowo-fenylowego
.
Dzięki tautomerii można wprowadzać izotopy wodoru w położenie
α do grupy karbonylowej.
O
H
O
D
CH
3
CH
2
CC
H
3
C
+
D/DOD
lub
-
OD/DOD
CH
3
CH
2
CC
H
3
C
1-fenylo-2-metylobutan-1-on 2-deutero-1-fenylo-2-metylobutan-1-on
Zadanie: przedstaw mechanizm
α deuterowania związków karbonylowych.
Stosunkowo łatwa wymiana protonu przy C
α
na deuteron (lub tryton) jest możliwa dzięki
stosunkowo dużej wartości pK
a
(kwasowości) atomu wodoru w tej pozycji – pK
a
przy C
α
wynosi 19-20, podczas gdy przy C
β
jedynie 40-50.
R
O
H H
C C C
α
β
pK
a
19-20
40-50
↑ ↑
kwasowy atom H niekwasowy alifatyczny atom H
1
o
i 2
o
nitrozwiązki też ulegają
tautomerii
, tworząc przy tym stosunkowo silne kwasy.
Nitrozwiązki w formie tautomerycznej noszą nazwę aci-nitropochodnych. Stała kwasowa H
α
dla
nitrometanu
wynosi 6,1.10
-11
, podczas gdy jego formy aci – 5,6.10
-4
. Roztwór
nitrometanu
nie
daje odczynu kwaśnego wobec papierka uniwersalnego, natomiast
nitrometan
w formie aci
zabarwia ten papierek podobnie jak
kwas octowy
. W nitrozwiązkach w środowisku obojętnym
równowaga przesunięta jest w kierunku formy nitrowej; w
nitrometanie
formy aci jest jedynie
około 0,00001%, w
nitroetanie
0,009%, a w
2-nitropropanie
0,3%.
N
R
O
O
H
N
R
O
O
H
C
:
:
..
..
..
+
-
C
:
:
..
..
..
+
-
forma nitro forma aci
W obecności zasad nitrozwiązki wolno tworzą rozpuszczalne w wodzie sole formy aci. Sole te
pod wpływem mocnych kwasów szybko zostają przekształcane w nietrwałe kwasy – aci-
nitrozwiązki. Po ponownym zalkalizowaniu świeżo otrzymanych aci-nitrozwiązków powstają z
nich sole tak szybko, jak z kwasów mineralnych. Aci-nitrozwiązki z ciągu kilku godzin
samorzutnie przechodzą w formę nitro.
38
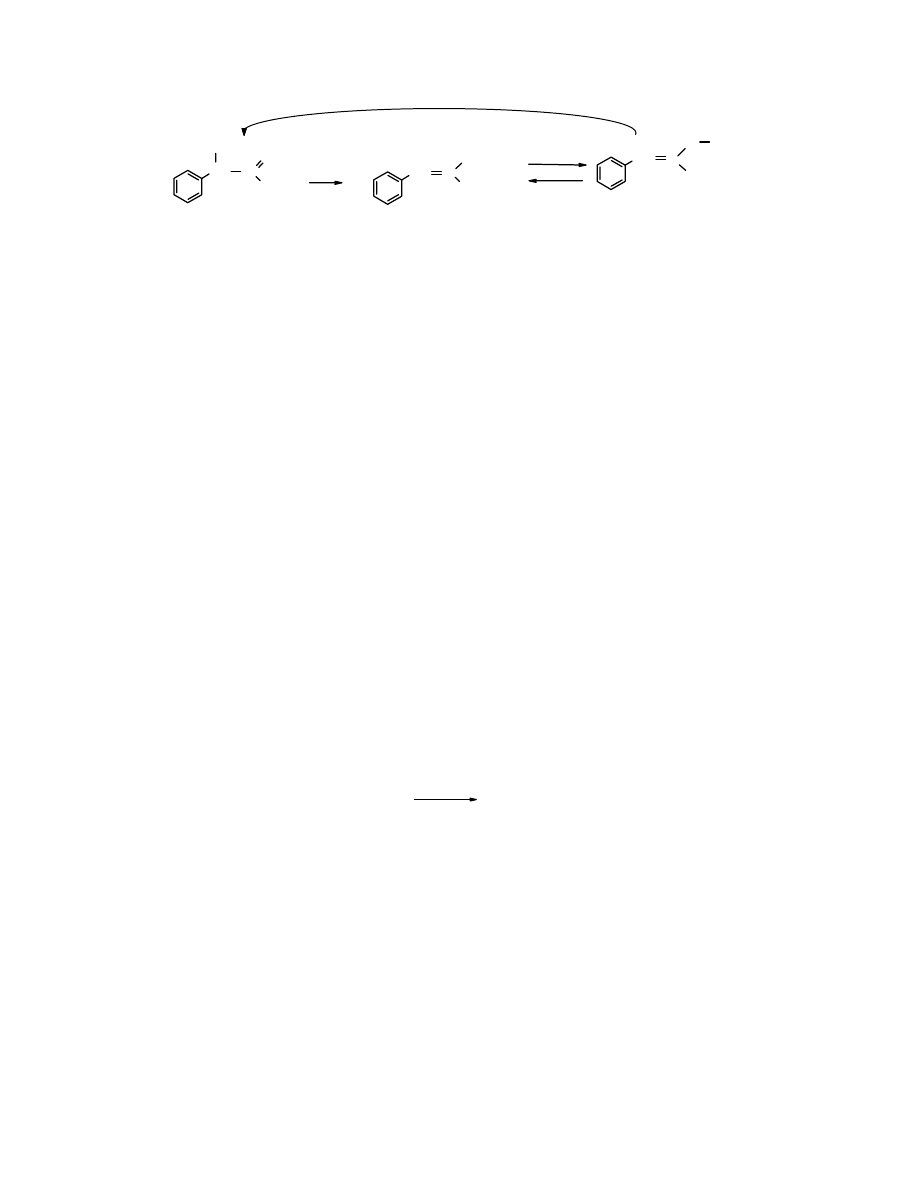
CH N
O
O
CH N
O
O
H
CH N
O
O
H
:
:
..
..
+
-
NaOH
wolno
..
:
:
..
..
+
-
..
fenylonitrometan
ciecz, tw. 226
o
C
Na
+
-
:
:
..
..
+
-
..
H
+
szybko
NaOH szybko
kilka godzin
forma nitro
fenylonitrometan
ciało stałe, tt. 84
o
C
forma aci
Większość nitrozwiązków w formie aci jest tak nietrwała, że trudno je wyizolować w postaci
indywiduów chemicznych.
Fenylonitrometan
należy do tych nitrozwiązków, które są trwałe przez
kilka godzin, co wystarcza, żeby je wyodrębnić i scharakteryzować.
Aci-nitropochodne są silniejszymi kwasami niż kwas węglowy i rozpuszczają się w roztworach
węglanu sodu czy potasu, natomiast nitrozwiązki w formie nitro nie rozpuszczają się w
roztworach węglanów. Aci-nitrozwiązków pod wpływem roztworu chlorku żelaza (III) przyjmują
czerwone zabarwienie, podobnie jak enole i fenole.
Fenole są przykładem trwałych enoli. Do trwałych enoli należą również 1,3-diony,
β
-
oksyaldehydy,
β
-oksynitryle czy
β
-oksyestry.
12. Polimeryzacja aldehydów
Aldehydy niskocząsteczowe (
metanal
i
etanal
) ulegają
cyklicznej oligomeryzacji
lub
liniowej
polimeryzacji
. Tworzą się przy tym produkty zawierające wiązania acetalowe. Aldehydy
aromatyczne zawierające grupę karbonylową przy pierścieniu nie ulegają
polimeryzacji
.
Tendencja do
polimeryzacji
aldehydów alifatycznych zmniejsza się wraz ze wzrostem ich masy
cząsteczkowej.
Metanal
jest trwały w stanie gazowym. Ślady wody i wiele innych zanieczyszczeń powodują jego
szybką
polimeryzację
. W roztworach wodnych
polimeryzacja
jest wolniejsza, ale widoczny
osad (polimer
metanalu
) na dnie formaliny (wodnego roztworu
metanalu
) świadczy, że
polimeryzacja
ma miejsce. Można ją przyspieszyć. Katalitycznie działają zarówno kwasy, jak i
zasady. Z wodnego roztworu
metanalu
pod wpływem wodorotlenków alkalicznych powstaje
α-
polioksymetylen
.
(n + 2) HCHO + HOH
HOCH
2
(OCH
2
)
n
OCH
2
OH
+
H lub
-
OH
metanal
polioksymetylen
Pod wpływem stężonego kwasu siarkowego wytrąca się
β-polioksymetylen
, a
paraformaldehyd
tworzy się w trakcie odparowywania pod obniżonym ciśnieniem wodnych roztworów
metanalu
.
Wszystkie te trzy formy są liniowo spolimeryzowanym
metanalem
. Różnica polega na tym, że
stopień polimeryzacji n
paraformaldehydu
mieści się w granicach 10-50, zaś stopień
polimeryzacji polioksymetylenów jest większy.
β-Polioksymetylen
zawiera małe ilości kwasu
siarkowego związanego estrowo. Obecnie
paraformaldehyd
jest również zaliczany do
polioksymetylenów.
Paraformaldehyd
jest produkowany na skalę przemysłową, stanowi bowiem dogodną formę
przechowywania i transportowania
metanalu
.
Paraformaldehyd
łatwo przekształcić w wolny
39
aldehyd, ponieważ
depolimeryzuje
on w temperaturze 140-160
o
C bezpośrednio do gazowego
produktu. Również w niektórych reakcjach zachowuje się jak wolny aldehyd.
Trioksan
(
trioksymetylen
), cykliczny trimer
metanalu
powstaje w trakcie ogrzewania
paraformaldehydu
(polioksymetylenu) z katalityczną ilością kwasu siarkowego w zamkniętym
naczyniu. Jest to w odróżnieniu od polimerów liniowych związek jednorodny i w odróżnieniu od
metanalu
ma przyjemny zapach.
O
O
O
HOCH
2
(OCH
2
)
n
OCH
2
OH
H
2
SO
4
trioksan
tt. 64
o
C
polioksymetylen
Etanal
i wyższe aldehydy przechowywane w stanie czystym, w szczelnie zamkniętym naczyniu są
trwałe – nie ulegają
samorzutnej polimeryzacji
. Jednak pod wpływem katalitycznych ilości
silnych kwasów
etanal
szybko
trimeryzuje
. Ten trimer nazywany jest
paraacetaldehydem
lub
krócej
paraaldehydem
(
paraldehydem
). Równocześnie powstaje tetramer zwany
metaaldehydem
(
metaldehydem
).
O
O
O
CH
3
C
H
3
CH
3
O
O
O
O
CH
3
CH
3
C
H
3
C
H
3
3 CH
3
CHO
+
H
+
etanal paraaldehyd metaaldehyd
(tw. 21
o
C, tt. –125
o
C) (tw. 124
o
C, tt. 10
o
C) (tt. 190
o
C, subl.)
Reakcja
oligomeryzacji
etanalu
jest reakcją odwracalną, przy czym stan równowagi zależy od
temperatury; w temp. 50
o
C 40% aldehydu ulega
oligomeryzacji
. W niższych temperaturach
równowaga przesuwa się w kierunku oligomerów. Czysty
paraaldehyd
otrzymuje się przez
oddestylowanie go po zobojętnieniu mieszaniny równowagowej. Z pozostałości wydziela się
metaaldehyd
. Dawniej
metaaldehyd
w formie kostek służył jako bezpieczne paliwo turystyczne.
Nie stwarzało ono tyle problemów, co np. benzyna w szklanych butelkach. Wadą
metaaldehydu
jako paliwa jest jego niska wartość energetyczna, ponieważ jest w znacznym stopniu utleniony.
Obecnie metalowe butle z ciekłym gazem (propan/butan) są najpopularniejszym paliwem
turystycznym.
Z
paraaldehydu
po zakwaszeniu wydziela się
etanal
. Jest to jedna z metod wprowadzania
etanalu
jako reagenta do wielu syntez.
Wielocząsteczkowy, liniowy polimer etanalu powstaje poprzez
polimeryzację
etanalu
wobec
tlenku glinu. Można go również otrzymać z zamrożonego do -120
o
C monomeru. Po jego
podgrzaniu do temperatury pokojowej otrzymuje się gęstą ciecz, która samorzutnie przekształca
się w stały produkt.
40
Document Outline
- Z W I Ą Z K I K A R B O N Y L O W E 20
- chlorek kwasowy
- 2. Karboaldehydowa
- Inny systematyczny sposób nazywania aldehydów polega na dodani
- Mechanizm
- Reakcja tworzenia acetali zaczyna się od protonowania grupy -O
- Y: wzór nazwa Y: wzór nazwa
- stereoizomery oksymu benzaldehydu
- 6. Redukcja Wolffa-Kiżnera
Wyszukiwarka
Podobne podstrony:
Enzymatyczna redukcja związków karbonylowych i zawierających wiązania C=C
pochodne związków karbonylowych zadania
Związki karbonylowe(13)gohar
zwiazkikarbonylowe[1], Związki karbonylowe
instrukcja enzymatyczna redukcja zwiazkow karbonylowych i wiazan CC, Biotechnologia Enzymatyczna
W -- Zwiazki karbonylowe cz. II, podstawy chemii organicznej
W -- Zwiazki karbonylowe cz. I, podstawy chemii organicznej
wyk 7 etery,związki karbonylowe
ZWIĄZKI KARBONYLOWE
Związki karbonylowe
Enzymatyczna redukcja związków karbonylowych i zawierających wiązania C=C
pochodne związków karbonylowych zadania
Rodzaj alkoholu otrzymywanego w syntezie metodą Grignarda zależy od typu użytego związku karbonylowe
związki karbonylowe
Rodzaj alkoholu otrzymywanego metoda a zalezyuzytego zwiazku karbonylowego(1)
Rodzaj alkoholu otrzymywanego metoda a zalezyuzytego zwiazku karbonylowego
Związki karbonylowe(13)gohar
więcej podobnych podstron