
ĆWICZENIE 29.
BIOKATALIZATORY I ICH ZASTOSOWANIE W PRZEMYŚLE
ĆWICZENIE 30.
BIOKATALITYCZNA INWERSJA SACHAROZY W PRZEPŁYWOWYCH
REAKTORACH KOLUMNOWYCH
OSOBY PROWADZĄCE:
mgr inż. Katarzyna Jodko (pok.134, konsultacje: środy 10
30
-11
30
, czwartki 13
00
-14
00
)
dr hab. Grzegorz Litwinienko (pok. 132, konsultacje: środy 10
30
-11
30
, czwartki 13
00
-14
00
)
LITERATURA:
1. Atkins, P. W. Równowaga chemiczna. W: Chemia fizyczna. PWN 2007, 201-227.
2. Atkins, P. W. Szybkość reakcji chemicznych. W: Chemia fizyczna. PWN 2007, 735-763.
3. Berg, J. M., Tymoczko, J. L. i L. Stryer. Enzymy. Podstawowe pojęcia i kinetyka.
W: Biochemia. PWN 2007, 189-228.
4. Ishikawa, T., Karnkowska, A., Lilpop, J. i J. Urbański. Słodki świat enzymów. Szkoła
Festiwalu Nauki (materiały dostępne na stronie: www.sfn.edu.pl).
5. Walory, J., Pilarek, M., Kalinowska, M. i H. Jaworowska-Deptuch. Kinetyka reakcji
enzymatycznej. W: Biochemia. Ćwiczenia laboratoryjne. Oficyna Wydawnicza PW 2003,
44-60.
6. Adamczak, M. Biokataliza i jej zastosowanie. W: Podstawy biotechnologii przemysłowej.
Red. W. Bednarski i J. Fiedurek. WNT 2007, 317-378.
7. Murray R. K., Granner D. K., Mayes P. A., i V. Rodwell. rozdz. I.8-I.11 w: Biochemia
Harpera, Wydawnictwo Lekarskie PZWL Warszawa 1995, str. 82-118
8. Malepszy, S. Zabiegi technologiczne zwiększające produkcję metabolitów wtórnych.
W: Biotechnologia roślin. PWN 2001, 331-334.

2
Wymagania do ćwiczeń:
1. Definicje: szybkość / rzędowość / cząsteczkowość / stała równowagi reakcji chemicznej,
równanie Arrheniusa, reguła van’t Hoffa, energia aktywacji, kataliza homogeniczna
i heterogeniczna (przykłady), opis oddziaływań katalizatora z substratem, biokataliza,
biokatalizator, enzym, rybozym, kofaktor, enzymy allosteryczne, teoria kompleksu
aktywnego.
2. Właściwości enzymów / biokatalizatorów: aktywność, wydajność syntezy produktu, liczba
obrotów enzymu, selektywność substratowa i typu reakcji.
3. Mechanizm działania enzymów: modele opisujące działanie enzymów, klasyfikacja
enzymów.
4. Kinetyka Michaelisa-Menten – założenia i wyprowadzenie równania, sens fizyczny stałych
obecnych w równaniu, możliwe uproszczenia, sposób wyznaczania parametrów
charakteryzujących enzym.
5. Regulacja reakcji enzymatycznych: inhibicja kompetytywna i niekompetytywna, wpływ
parametrów intensywnych na szybkość reakcji enzymatycznej.
6. Techniki instrumentalnej analizy ilościowej (spektroskopia UV-Vis, prawo Lamberta-
Beera, sporządzanie krzywej wzorcowej, przeliczanie stężeń).
7. Dodatkowo studenci są proszeni o wybranie dowolnego enzymu i samodzielne (pisemne!)
opracowanie jego charakterystyki w oparciu o dostępne bazy literaturowe. Opracowanie to
powinno obejmować – numer wg klasyfikacji E.C., typ katalizowanej reakcji, znaczenie
katalizowanego procesu w organizmie, struktura centrum aktywnego (wraz z kofaktorem,
jeżeli występuje), ewentualne inhibitory i mechanizm ich działania. Postuluje się, żeby
studenci z jednej grupy wybierali różne enzymy do scharakteryzowania.

3
Poniższy wstęp zakłada znajomość podstawowych definicji i pojęć kinetyki chemicznej,
z którymi student zapoznał się podczas kursu chemii fizycznej. Ponadto, studenci są proszeni
o zapoznanie się z materiałem dotyczącym enzymów prezentowanym na wykładzie Elementy
Biotechnologii (Wykłady nr 3-5).
I. BIOKATALIZA
Procesy biochemiczne, przebiegające w organizmach żywych, są kontrolowane przez
biokatalizatory – naturalnie występujące cząsteczki przyspieszające bądź hamujące przebieg
reakcji, do których zaliczamy enzymy, hormony i witaminy (często pełniące funkcję
koenzymów). W analogii do procesów przebiegających in vivo, podejmowane są próby
przemysłowego wykorzystania materiału biologicznego do wydajnego prowadzenia reakcji
chemicznych. Zabieg taki określa się mianem biokatalizy, a zachodzącą pod wpływem
materiału biologicznego reakcję – biotransformacją lub biokonwersją.
Rolę biokatalizatora w procesach przemysłowych mogą pełnić całe komórki (w formie
zawiesiny lub immobilizowane), ekstrakt komórkowy lub enzymy o różnym stopniu
oczyszczenia, wolne lub immobilizowane. Zastosowanie hormonów (zaliczanych do
naturalnych biokatalizatorów) do przemysłowego prowadzenia reakcji chemicznych nie jest
możliwe, gdyż regulują one procesy zachodzące w komórce tylko za pośrednictwem
specyficznych receptorów, obecnych we wnętrzu komórek lub umieszczonych śródbłonowo.
Biokatalizatory stosowane są w przemyśle na coraz większa skalę - obecnie już około
130 procesów z użyciem enzymów lub komórek mikroorganizmów zostało opracowanych dla
skali produkcji przewyższającej 100 kg. Enzymy znajdują zastosowanie głównie w przemyśle
spożywczym (przemysł piekarniczy, browarnictwo, gorzelnictwo, mleczarstwo, przemysł
mięsny), przy produkcji detergentów (biodetergentów), w przemyśle papierniczym oraz
w medycynie (w tym w diagnostyce genetycznej).
II. ENZYMY
Enzymy to związki wielkocząsteczkowe wykazujące właściwości katalityczne,
umożliwiające prawidłowe tempo przemian metabolicznych w organizmach żywych
i wirusach. Wykazano wpływ enzymów na przebieg wszystkich szlaków anabolicznych
i
katabolicznych w komórce. Przykładowo, fundamentalna w procesach oddychania
komórkowego i oddychania płucnego reakcja:
CO
2
+ H
2
O ' H
2
CO
3
(1)

4
przebiega 10
7
– razy szybciej w obecności enzymu anhydrazy węglanowej. Działanie enzymu
umożliwia więc wydajne usunięcie dwutlenku węgla, który powstaje w procesach
metabolicznych poprzez rozpuszczenie go we krwi, a następnie uwolnienie go z krwi do
wnętrza pęcherzyków płucnych. Śmiało możemy więc powiedzieć, że aktywność katalityczna
anhydrazy węglanowej warunkuje efektywną wymianę gazową.
II.1. Budowa enzymów
II.1.1. Rybozymy jako pozostałość „świata RNA”
Niemal wszystkie enzymy są białkami. Jednak w „świecie RNA” (hipotetycznym,
wczesnym etapie rozwoju życia na Ziemi) rolę katalizatorów prawdopodobnie pełniły
cząsteczki kwasu rybonukleinowego (RNA). W latach 80., Thomas Cech i Sidney Altman
niezależnie odkryli cząsteczki RNA wykazujące aktywność katalityczną - rybozymy (za co
w 1989 roku otrzymali Nagrodę Nobla w dziedzinie chemii). Cech udowodnił, że proces
splicingu RNA u
jednokomórkowego orzęska Tetrahymena thermophila polega na
autokatalitycznym wycinaniu intronów. Katalityczne właściwości RNA wynikają ze
zdolności przyjmowania przez te cząsteczki skomplikowanych struktur, umożliwiających
idealne przestrzenne dopasowanie do siebie enzymu i substratu katalizowanej reakcji.
Obecnie wiemy, że rybozymy występują w komórkach wszystkich organizmów i uczestniczą
przede wszystkim w mechanizmach syntezy białek (na przykład rRNA, wchodzący w skład
rybosomów, katalizuje reakcję tworzenia wiązań peptydowych miedzy aminokwasami).
Odkrycie cząsteczek RNA o właściwościach katalitycznych potwierdza przedstawioną wyżej
hipotezę „świata RNA”. Należy w tym miejscu podkreślić, że w „świecie RNA” kwas
rybonukleinowy obok funkcji katalitycznej, pełnił też rolę nośnika informacji genetycznej,
czego pozostałością we współczesnym świecie są retrowirusy (w tym wirus HIV). Postuluje
się, że cząsteczkę, która jest jednocześnie nośnikiem informacji i katalizuje swoje własne
przemiany, można uznać za pierwszą, niekomórkową formę życia na Ziemi.
II.1.2. Enzymy białkowe
Jak już wspomniano, podstawową rolą enzymów w organizmie jest katalizowanie
przebiegu ściśle określonych reakcji metabolicznych. Wobec tego, najbardziej istotnymi
właściwościami enzymów, warunkującymi prawidłowe pełnienie przez te cząsteczki swojej
funkcji, będą siła katalityczna (aktywność) i selektywność (specyficzność). Porównywanie
aktywności różnych enzymów stało się możliwe po wprowadzeniu przez Międzynarodową
Unię Biochemiczną międzynarodowej jednostki standardowej U. Jest to ilość enzymu,
która katalizuje przemianę 1µmola substratu w czasie 1 minuty w temperaturze 30ºC,

5
w odpowiednim dla danego enzymu pH i przy optymalnym stężeniu substratu. Wyrażana jest
w µmol/min. Inne jednostki aktywności, które wciąż używane są w analizach biochemicznych
to: aktywność właściwa, aktywność molekularna (inaczej liczba obrotów enzymu) i katal.
Selektywność enzymu dotyczy zarówno katalizowanej reakcji (selektywność typu reakcji)
jak i substratów biorących w niej udział (selektywność substratowa). Selektywność
substratowa enzymu polega na wybraniu spośród wielu podobnych substancji tylko tej
właściwej, która będzie podlegała procesowi enzymatycznemu. Ten typ selektywności jest
efektem precyzyjnego dopasowania trójwymiarowej struktury enzymu do cząsteczki
substratu. Łańcuch polipeptydowy zbudowany z dwudziestu różnych aminokwasów zapewnia
większą selektywność niż cząsteczka kwasu nukleinowego, która jest kombinacją tylko
czterech typów nukleotydów. Z powodu większej różnorodności białek, to właśnie te
biocząsteczki stanowią główny materiał z którego zbudowana jest większość znanych
enzymów. Selektywność typu reakcji oznacza, że spośród wielu możliwych reakcji, jakim
ulegać może substrat, enzym katalizuje tylko jeden, ściśle określony proces. Efektem
selektywności enzymu jest właściwie brak zachodzenia reakcji ubocznych.
Wiele enzymów jest aktywnych (albo wykazuje podwyższoną aktywność) tylko
w obecności niebiałkowych cząsteczek – kofaktorów. W przypadku tych enzymów, ich
białkową część określamy mianem apoenzymu, a tworzony aktywny kompleks (apoenzym +
kofaktor) – holoenzymem. W roli kofaktorów mogą występować cząsteczki nieorganiczne,
jony metali, lub małocząsteczkowe związki organiczne. W oparciu o chemiczny charakter
kofaktora i moc wiązania, które tworzy z białkową częścią enzymu, w niektórych
opracowaniach kofaktory podzielono na dwie grupy:
- koenzymy, małe cząsteczki organiczne, które w sposób specyficzny łączą się
z apoenzymem; często są wiązane tylko na czas reakcji i uwalniane wraz z jej
produktami.
- grupy prostetyczne są silnie (często kowalencyjnie) związane z apoenzymem; w tej roli
mogą występować jony metali, małe cząsteczki nieorganiczne bądź organiczne.
Reakcja katalityczna zachodzi w miejscu aktywnym enzymu, czyli w obszarze
wiązania substratu i ewentualnych kofaktorów. W enzymach białkowych miejsce aktywne
składa się z reszt aminokwasowych, zwanych grupami katalitycznymi enzymu, które biorą
bezpośredni udział w zachodzącej reakcji. Miejsce aktywne enzymu jest często opisywane
jako „szczelina” w strukturze enzymu, w którą wpasowują się substraty biorące udział
w reakcji. Pełni ono dwojaką funkcję: selekcjonuje cząsteczki, które mogą brać udział w
reakcji, a ponadto umożliwia właściwe ułożenie reagenta / reagentów względem siebie.

6
Miejsce aktywne często jest tworzone przez reszty aminokwasowe oddalone od siebie w
sekwencji aminokwasowej – dopiero przybranie przez białko odpowiednio „zwiniętej”
trzeciorzędowej struktury powoduje zbliżenie tych reszt do siebie i utworzenie miejsca
aktywnego. Ponadto, miejsce aktywne przeważnie zajmuje jedynie małą część w strukturze
cząsteczki enzymu – gdy jednak weźmiemy pod uwagę, jak odległe aminokwasy biorą udział
w jego tworzeniu, jasne stanie się, że reszta cząsteczki jest niezbędna do przyjęcia przez
enzym właściwej struktury, pełniąc rolę rusztowania dla miejsca aktywnego. Za wiązanie
substratu w miejscu aktywnym, w zależności od chemicznego charakteru substratu i reszt
aminokwasowych tworzących miejsce aktywne, odpowiedzialne mogą być: oddziaływania
elektrostatyczne i hydrofobowe, wiązania wodorowe oraz siły van der Waalsa.
II.2. Modele ilustrujące działanie enzymów
Enzymy w sposób wysoce specyficzny wiążą substraty reakcji i ustawiają je
w odpowiedniej konformacji przestrzennej, sprzyjającej zajściu katalizowanego procesu.
W 1890 roku Hermann Emil Fischer podjął próbę opisania zjawiska specyficzności
substratowej enzymów przy użyciu modelu „zamka i klucza”. Zgodnie z założeniami
modelu, enzym (a dokładniej jego miejsce aktywne) i substrat doskonale odpowiadają sobie
przestrzennie. To dopasowanie jest warunkiem, po pierwsze, wzajemnego rozpoznania
substratu i enzymu, a następnie - zajścia katalizowanej reakcji. Według modelu „zamka i
klucza” (ang. lock and key model – Rysunek 1a), specyficzność substratowa enzymu zależy
od precyzyjnego ułożenia atomów w miejscu aktywnym, które oddziałują z przylegającą do
nich cząsteczką substratu. Obecnie wiemy, że postulowany przez Fischera
1
model nie
tłumaczy w sposób właściwy zachodzącego procesu. Dopasowanie przestrzenne, zgodne z
modelem „zamka i klucza” uniemożliwiałoby bowiem efektywne obniżenie energii aktywacji
zachodzącej reakcji (czyli w zasadzie blokowałoby działanie enzymu, patrz rozdział II.3).
Zgodnie z kolejnym modelem, zaproponowanym w 1958 przez Daniela E. Koshlanda
Juniora
2
, struktury przestrzenne miejsca aktywnego i substratu wykazują względem siebie
powinowactwo (umożliwiające ich wzajemne rozpoznanie), ale dopiero podczas wiązania
substratu, kształt miejsca aktywnego nieznacznie zmienia się i idealnie dopasowuje
przestrzennie do substratu. Nieznaczne zmiany konformacyjne w strukturze miejsca
aktywnego są źródłem naprężeń wiązań w strukturze enzymu, co obniża energię aktywacji
katalizowanej reakcji chemicznej. W modelu zaproponowanym przez Koshlanda, określanym
1
H. E. Fisher ostatecznie został laureatem Nagrody Nobla w dziedzinie chemii, ale za prace dotyczące syntezy
cukrów i puryn, a nie za prace nad mechanizmem działania enzymów.
2
Daniel E. Koshland Jr. był dziedzicem fortuny Levi Straussa
7
mianem „indukowanego dopasowania” (ang. induced fit model – Rysunek 1b), enzymy
traktowane są jako dynamiczne struktury przestrzenne, które mogą zmieniać swoją
konformację w obecności substratu.
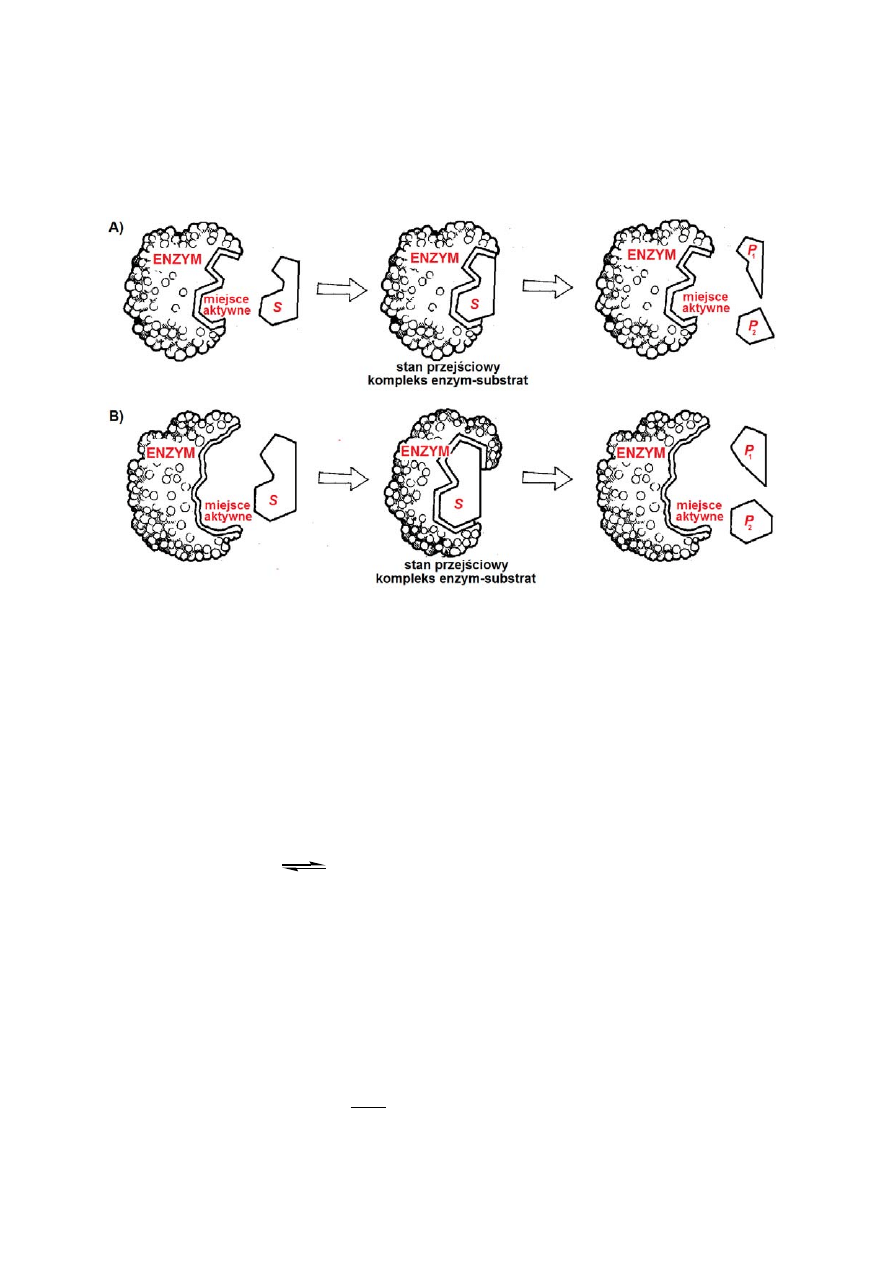
Rysunek 1. Mechanizm wiązania substratu (substratów) przez enzym. Na rysunku przedstawiono
schematycznie reakcję rozpadu substratu S do produktów P
1
i P
2
., zachodzącą w obecności enzymu. Proces
przedstawiono za pomocą modelu „zamka i klucza” (A), zgodnie z którym miejsce aktywne enzymu jest
komplementarne do kształtu substratu i modelu „indukowanego dopasowania” (B), według którego wiązanie
substratu pociąga za sobą zmiany konformacyjne w obrębie enzymu.
II.3. Mechanizm działania enzymów
Żeby zrozumieć mechanizm działania enzymów, rozważmy hipotetyczną reakcję
przemiany substratu S w produkt P, katalizowaną przez enzym E:
S P
E
(2)
Enzym E w tym samym stopniu obniża barierę aktywacyjną reakcji tworzenia produktu P, co
reakcji ponownej przemiany produktu P w substrat S, a zatem równocześnie przyspiesza
reakcje zachodzące w obydwu kierunkach. Wobec tego obecność enzymu nie zmienia stanu
równowagi zachodzącej reakcji chemicznej, opisanego przez stałą równowagi K, a jedynie
przyspiesza jego osiągnięcie. Stała równowagi K zależy od standardowej entalpii swobodnej
reakcji zgodnie z zależnością:
∆
−
=
RT
G
K
0
exp
(3)
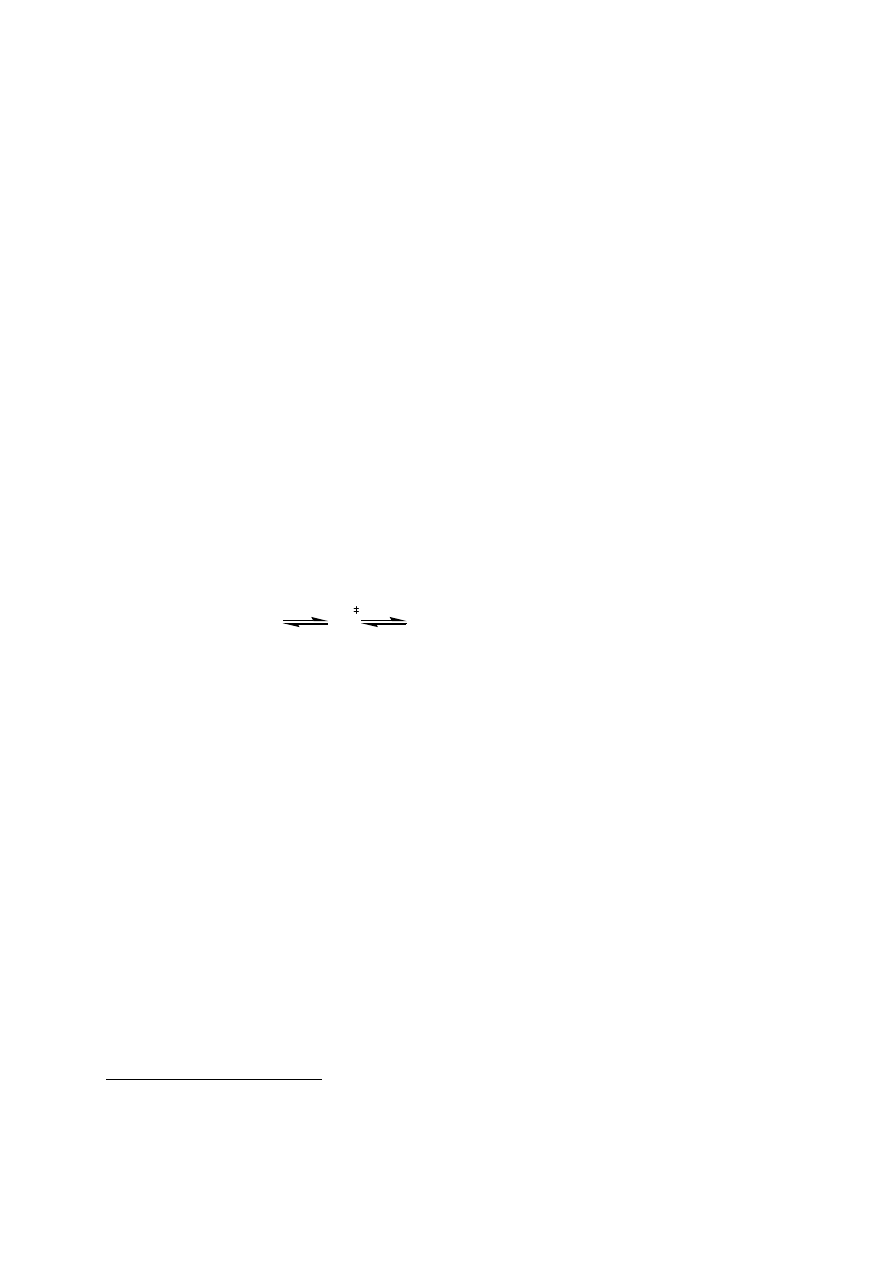
8
Standardowa entalpia swobodna reakcji
0
G
∆
jest z kolei różnicą pomiędzy standardową
entalpią swobodną produktu
0
P
G i standardową entalpią swobodną substratu
0
S
G . Ponieważ
obecność enzymu nie wpływa na wartość standardowych entalpii swobodnych związków
biorących udział w reakcji (
0
P
G i
0
S
G ), nie może też zmienić wartości
0
G
∆
, a co za tym idzie
– nie może wpłynąć na położenie stanu równowagi określonego przez stałą równowagi
K.
Przebieg zachodzącej reakcji można jednak opisać za pomocą innego parametru –
entalpii swobodnej aktywacji
‡
G
∆
(w literaturze częściej określanej mianem energii aktywacji
Gibbsa, lub po prostu energii aktywacji). Parametr ten to różnica pomiędzy entalpią
swobodną stanu przejściowego reakcji
‡
TS
G
a standardową entalpią swobodną substratu
0
S
G .
Stanem przejściowym reakcji
TS
‡
(ang. transition state) będzie najrzadziej występujący stan,
w którym mogą się znaleźć związki uczestniczące w reakcji, charakteryzujący się najwyższą
wartością entalpii swobodnej. Stan ten należy sobie wyobrazić jako jeden z etapów na drodze
przemian prowadzących od substratu
S do powstania produktu P, zatem równanie (2)
przyjmuje postać:
S TS
P
(4)
Osiągnięcie stanu przejściowego
TS
‡
, czyli najmniej korzystnego etapu przemiany, wymaga
dostarczenia do układu energii niezbędnej do pokonania bariery energetycznej pomiędzy
substratem a stanem przejściowym. Po osiągnięciu stanu przejściowego następuje jego
samorzutne przekształcenie w produkt reakcji
3
. Działanie enzymów polega na obniżaniu
energii aktywacji dla katalizowanej reakcji poprzez stabilizację stanu przejściowego (czyli
obniżenie entalpii swobodnej i zwiększenie prawdopodobieństwa jego wystąpienia – Rysunek
2). Stabilizacja stanu przejściowego reakcji jest możliwa np. poprzez zmianę środowiska
reakcji (substrat reakcji zostaje ukryty w „szczelinie” miejsca aktywnego, która stabilizuje
stan przejściowy reakcji). Warto tu podkreślić, że centrum aktywne często utworzone jest
przez hydrofobowe reszty aminokwasowe. Wobec tego, w przypadku reakcji zachodzących w
cytoplazmie (będącej złożonym koloidem wodnym), wejście substratu w „szczelinę” miejsca
aktywnego wiąże się ze zmianą środowiska z hydrofilowego na hydrofobowe, co może
sprzyjać tworzeniu stanu przejściowego.
3
Omawiany tu model jest bardzo uproszczony i zakłada istnienie tylko jednego stanu przejściowego.
W rzeczywistości na współrzędnej reakcji można czasem wyodrębnić kilka stanów przejściowych a przejście
jednego w drugi jest możliwe po pokonaniu dodatkowych barier energetycznych. Można wtedy powiedzieć, że
metastabilne struktury (TS
1
)
‡
,
(TS
2
)
‡
,
(TS
3
)
‡
są pewnymi lokalnymi maksimami energetycznymi.
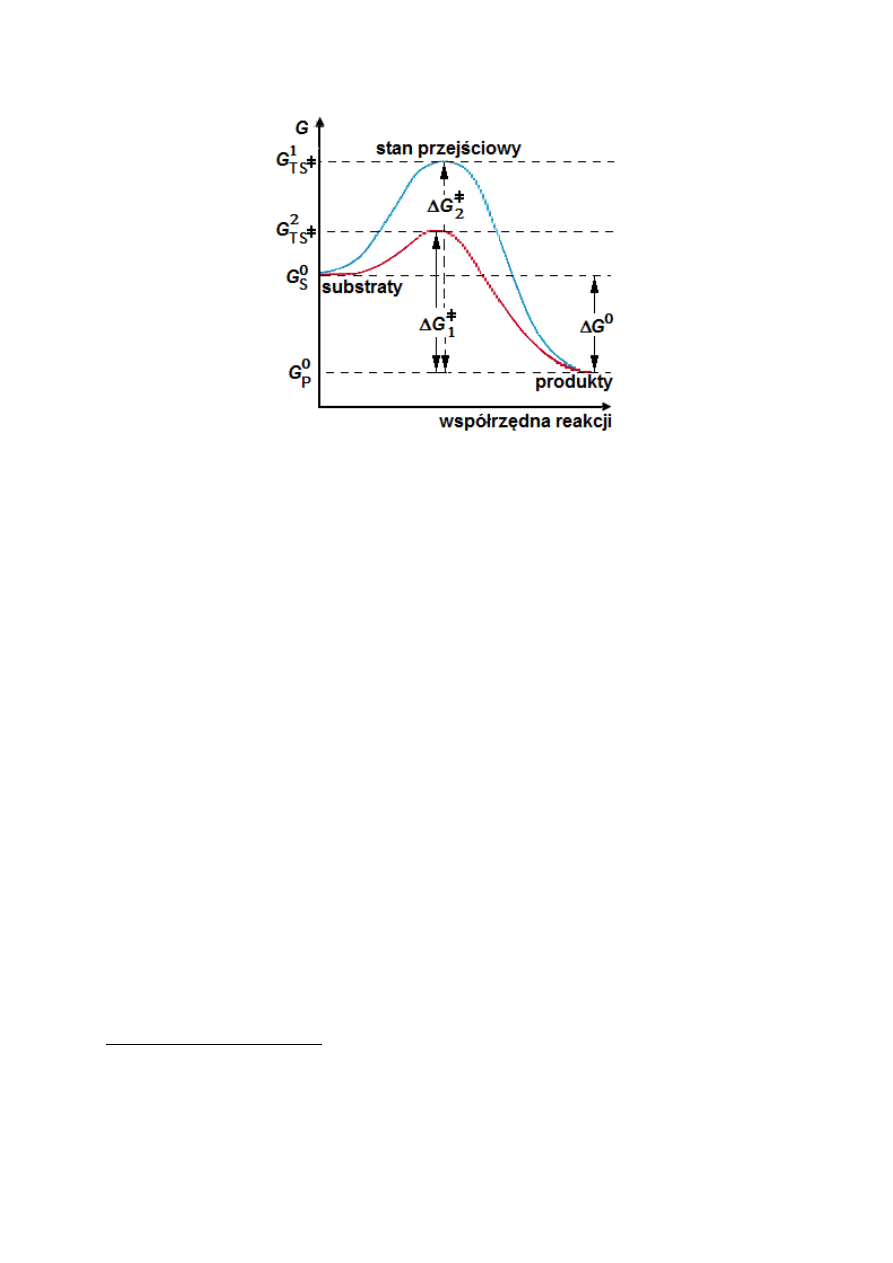
9
Rysunek 2. Mechanizm działania enzymów. Na rysunku przedstawiono jak zmienia się energia swobodna G
podczas przemiany substratów S w produkty P, zachodzącej przez stan przejściowy TS
‡
(2), charakteryzujący się
najwyższą wartością entalpii swobodnej. Enzymy ułatwiają tworzenie stanu przejściowego – w ich obecności
reakcja zachodzi przez stan przejściowy TS
‡
(1).
II.4. Kinetyka reakcji enzymatycznych
Najprostszy model matematyczny opisujący kinetykę reakcji enzymatycznej
zaproponowali w 1913 roku: niemiecki biochemik, Leonor Michaelis i Maud Leonora
Menten
4
. Forma matematyczna zaproponowanego przez nich równania kinetycznego,
znanego jako równanie Michaelisa-Menten, okazała się na tyle uniwersalna, że została
potem wykorzystana do opisu kinetyki wzrostu mikroorganizmów
5
.
Model Michaelisa-Menten zakłada, że etapem pośrednim w procesie przekształcenia
substratu
S w produkt P jest utworzenie kompleksu aktywnego enzym-substrat E-S. Na
istnienie stanu przejściowego w postaci kompleksu
E-S wskazuje fakt „wysycania” enzymu
w obecności wysokiego stężenia substratu. Oznacza to, ze jeżeli prowadzimy eksperyment
przy stałym stężeniu enzymu, to na początku zwiększanie stężenia substratu powoduje wzrost
szybkości reakcji. Jednak po przekroczeniu pewnego stężenia granicznego, dalsze
zwiększanie stężenia substratu nie przyspiesza już zachodzącej reakcji. Jest to efektem
wykorzystania wszystkich miejsc aktywnych w strukturze enzymu. Wówczas, kolejne
kompleksy
E-S mogą być tworzone tylko po rozpadzie już istniejących kompleksów,
4
M. L. Menten (1876-1960) jako jedna z pierwszych kobiet w Kanadzie zdobyła stopień doktora medycyny.
Ponieważ nie mogła pracować naukowo w swoim kraju (tytuł lekarza otrzymała na Uniwersytecie w Toronto
w 1911 roku na podstawie wyników badań prowadzonych w Chicago), zdecydowała się na emigrację do
Niemiec, gdzie w 1916 roku obroniła pracę doktorską właśnie pod kierunkiem Michaelisa. Potem pracowała
naukowo w Stanach Zjednoczonych a dopiero od 1950 roku w Kanadzie.
5
Równanie Monoda (Monod, J. The growth of bacterial cultures. A. Rev. Microbiol. 3, 371-394, 1949).

10
prowadzącym do uwolnienia cząsteczek enzymu. Ponadto, istnienie stanu przejściowego
E-S
zostało wykazane metodami krystalograficznymi i spektroskopowymi.
Enzym po związaniu substratu (substratów) ustawia je w optymalnej orientacji
przestrzennej do zajścia katalizowanej reakcji. Zgodnie z założeniami modelu Michaelisa-
Menten, po odwracalnej reakcji tworzenia kompleksu aktywnego
E-S, następuje jego
nieodwracalny rozpad do produktu
P i enzymu E:
E+S E-S E+P
k
+1
k
-1
k
2
(5)
Szybkości reakcji cząstkowych tego procesu wynoszą:
]
[
]
[
]
[
]
[
2
2
1
1
1
1
S
E
k
v
S
E
k
v
S
E
k
v
−
=
−
=
⋅
=
−
−
+
+
(6)
Szybkość zużywania substratu
S przedstawia równanie:
]
[
]
[
]
[
]
[
1
1
1
1
S
E
k
S
E
k
v
v
dt
S
d
−
−
⋅
=
−
=
−
−
+
−
+
(7)
podczas gdy szybkość tworzenia produktu P jest równa:
]
[
]
[
2
2
S
E
k
v
dt
P
d
−
=
=
(8)
Zmiany stężenia kompleksu E-S w czasie przedstawia zależność:
]
[
]
[
]
[
]
[
]
[
2
1
1
2
1
1
S
E
k
S
E
k
S
E
k
v
v
v
dt
S
E
d
−
−
−
−
⋅
=
−
−
=
−
−
+
−
+
(9)
Gdy szybkość tworzenia kompleksu E-S (równa v
+1
) jest równa szybkości jego rozpadu w
obydwu kierunkach (v
-1
+v
2
), to stężenie kompleksu aktywnego E-S nie zmienia się:
0
]
[
=
−
dt
S
E
d
(10)
W tak zdefiniowanym stanie ustalonym (stacjonarnym) możliwe jest rozwiązanie równań (7)
i (8). Zauważmy, że w rozpatrywanym układzie enzym występuje w formie wolnej E
i w formie kompleksu aktywnego E-S. Wobec tego, całkowite stężenie enzymu w układzie:
]
[
]
[
]
[
0
S
E
E
E
−
+
=
(11)
Z równań (9-11) otrzymujemy wyrażenie na stężenie kompleksu aktywnego E-S:
]
[
]
[
]
[
]
[
0
1
1
2
1
E
S
k
k
k
S
k
S
E
+
−
+
+
+
=
−
(12)

11
Po wstawieniu tej zależności do równania (7) lub (8), otrzymujemy wyrażenie na szybkość
prowadzonej reakcji enzymatycznej (wyrażoną jako szybkość zużywania substratu S lub
szybkość powstawania produktu P). Zgodnie z założeniem o stanie ustalonym, szybkość
zużywania substratu jest równa szybkości powstawania produktu - patrz równania (9) i (10):
]
[
]
[
]
[
0
1
1
2
2
1
E
S
k
k
k
S
k
k
V
+
−
+
+
+
=
(13)
Kolejne założenia, o stałości stężenia E
0
w trakcie procesu i znacznym nadmiarze substratu S
w stosunku do ilości enzymu
6
pozwalają nam uprościć równanie (13) do formy
zaproponowanej przez Michaelisa i Menten:
]
[
]
[
]
[
]
[
]
[
]
[
]
[
]
[
max
1
1
2
1
1
0
2
1
1
2
1
0
2
S
K
S
V
S
k
k
k
k
S
k
E
k
S
k
k
k
S
k
E
k
V
M
+
=
+
+
×
=
+
+
×
=
+
−
+
+
+
−
+
(14)
gdzie
1
1
2
+
−
+
=
k
k
k
K
M
stała Michaelisa
]
[
0
2
max
E
k
V
=
maksymalna szybkość reakcji, kiedy [E-S]=[E
0
], czyli gdy cały
enzym jest zaangażowany w tworzenie kompleksu - proszę porównać z równaniem (8)
Stała Michaelisa ma wymiar stężenia i z równania (14) wynika, że dla [S] = K
M
, szybkość
katalizowanej reakcji osiąga połowę szybkości maksymalnej:
max
2
1
]
[
V
V
K
S
M
=
⇔
=
(15)
Wobec tego, prowadząc serię pomiarów szybkości procesu enzymatycznego dla różnych
stężeń substratu można wyznaczyć stałą Michaelisa, która jest takim stężeniem substratu, przy
którym szybkość katalizowanej reakcji osiąga połowę wartości maksymalnej (Rysunek 3)
7
.
6
[S] >> [E
0
] – proszę się zastanowić, w którym momencie wykorzystano to założenie?
7
Dokładniejsze wyznaczenie parametrów równania Michaelisa-Menten jest możliwe po przekształceniu tego
równania do postaci Lineweavera-Burke’a, Hanesa lub Eadie-Hofstee’a (po dobraniu odpowiedniego układu
współrzędnych otrzymujemy wykresy liniowe).
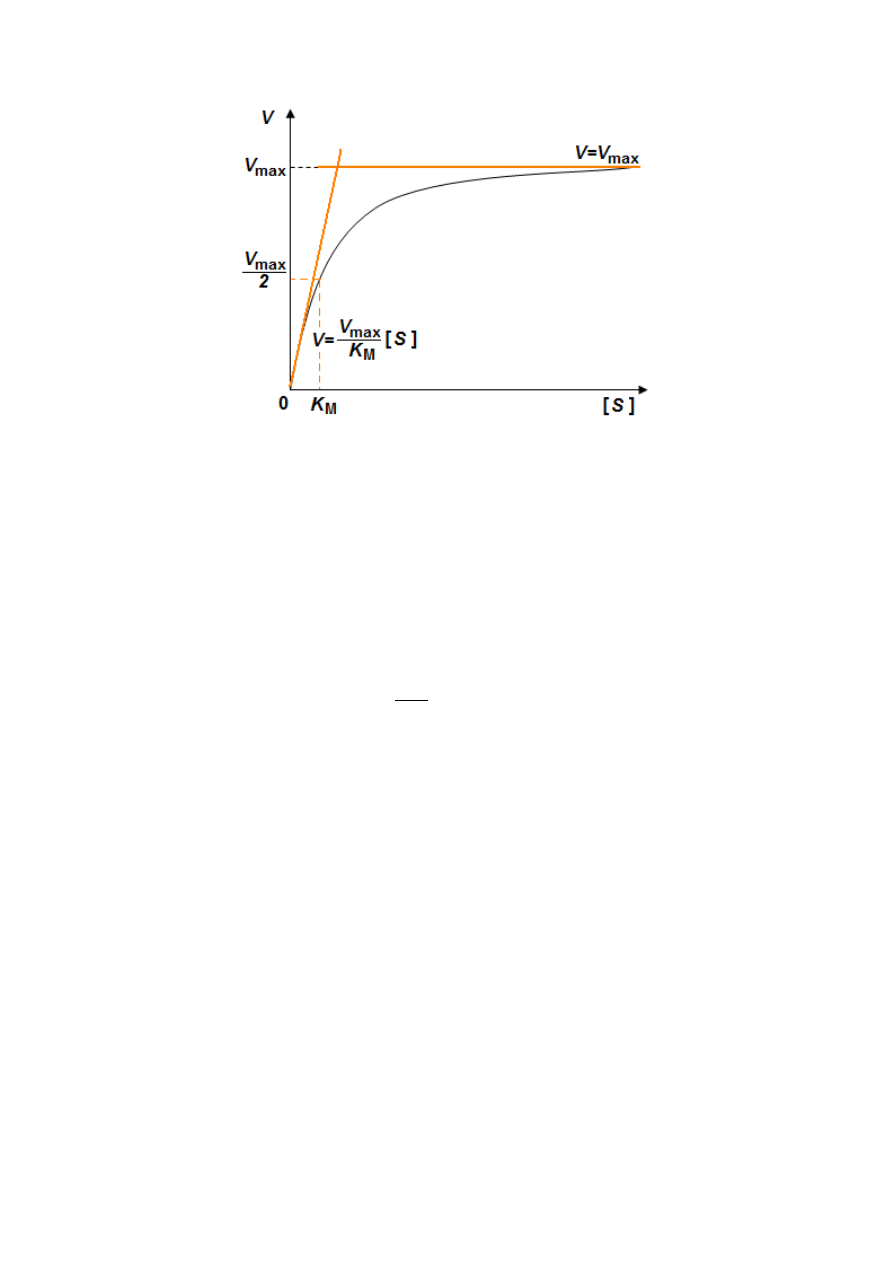
12
Rysunek 3. Szybkość reakcji enzymatycznej V w funkcji stężenia substratu S dla procesu przebiegającego
zgodnie z modelem Michaelisa-Menten. Na wykresie przedstawiono sposób wyznaczenia wartości stałej K
M
oraz uproszczone formy równania Michaelisa-Menten właściwe dla skrajnych stężeń substratu S.
Stała K
M
jest odwrotnie proporcjonalna do powinowactwa enzymu do substratu - jej małe
wartości świadczą o dużym powinowactwie. Na Rysunku 3 przedstawiono jak szybkość
reakcji V zależy od stężenia substratu S. Na krzywej możemy wyróżnić dwa obszary
kinetyczne. Dla bardzo niskich stężeń substratu, równanie (14) można uprościć do postaci:
]
[
]
[
max
S
K
V
V
K
S
M
M
=
⇔
〈〈
(16)
Przy niskich stężeniach substratu proces przebiega zatem zgodnie z mechanizmem reakcji
I-rzędowej. Z kolei, przy wysokich stężeniach substratu, następuje całkowite „wysycenie”
enzymu i reakcja zachodzi z szybkością maksymalną dla danego stężenia enzymu:
max
]
[
V
V
K
S
M
=
⇔
〉〉
(17)
Należy zaznaczyć że równanie Michaelisa-Menten nie jest właściwe do opisu enzymów
allosterycznych, czyli enzymów wielojednostkowych, zawierających kilka miejsc aktywnych
w obrębie jednej cząsteczki. W przypadku tego typu enzymów, cząsteczki substratu wiązane
są kolejno do miejsc aktywnych, przy czym związanie substratu z jednym miejscem może za
sobą pociągać zmiany konformacyjne w obrębie enzymu, które ułatwiają wiązanie substratów
do następnych miejsc aktywnych.
II.5. Regulacja procesów enzymatycznych
Szybkość reakcji enzymatycznej zależy od ilości substratu (zgodnie z równaniem
Michaelisa-Menten, jeżeli stężenie enzymu jest stałe). Przy nadmiarze substratu, enzym ulega

13
„wysyceniu” i szybkość reakcji można zwiększyć przez dodanie nowej porcji enzymu.
W obszarze maksymalnej szybkości reakcji, jej postęp zależy bowiem liniowo od stężenia
enzymu:
]
[
0
2
max
E
k
V
V
=
=
(18)
Zależność ta wykorzystywana jest przy oznaczaniu stężenia enzymów w próbkach
biologicznych, w celach diagnostycznych.
Ponadto, enzymy podlegają regulacji za pomocą wysoce specyficznych mechanizmów
kontroli (z wykorzystaniem tak zwanego centrum allosterycznego) oraz niespecyficznie,
w efekcie zmieniających się parametrów układu takich jak odczyn pH, temperatura, siła
jonowa i innych.
Centrum allosteryczne jest miejscem w strukturze biocząsteczki, do którego wiązane
są inhibitory bądź aktywatory danego enzymu. Istnienie tego miejsca warunkuje szybką
odpowiedź enzymu na zmieniające się warunki środowiska. Jest to szczególnie ważne
w przypadku enzymów regulujących kluczowe dla organizmu procesy, gdyż czyni z enzymu
precyzyjną maszynę, przyspieszającą dany cykl biochemiczny dokładnie wtedy, gdy jest to
potrzebne. Zapobiega to niepohamowanemu wzrostowi stężenia metabolitów (co, w myśl
zasady Dosis facit venenum może być zabójcze dla organizmu) a przede wszystkim zapobiega
marnotrawieniu energii na prowadzenie procesów, które są zbędne. I tak, w przypadku
szlaków metabolicznych, rolę inhibitorów często pełnią ich końcowe produkty, które na
zasadzie ujemnego sprzężenia zwrotnego hamują enzym katalizujący pierwszą reakcję danego
szlaku (w czym znowu przejawia się oszczędność natury).
Inhibicja może być nieodwracalna - polegać na trwałym związaniu cząsteczki
inhibitora (I) z enzymem, bądź może mieć charakter odwracalny, kiedy utworzony kompleks
enzym-inhibitor (E-I) szybko dysocjuje. Zjawisko inhibicji nieodwracalnej jest
wykorzystywane w działaniu niektórych antybiotyków – na przykład penicylina blokuje
transpeptydazę – enzym niezbędny do syntezy bakteryjnych ścian komórkowych.
Podczas inhibicji odwracalnej cząsteczka inhibitora jest czasowo wiązana
z miejscem aktywnym enzymu bądź z innym centrum allosterycznym. W inhibicji
o charakterze kompetytywnym, inhibitor, charakteryzujący się podobną strukturą do
substratu, rywalizuje z nim o miejsce w centrum aktywnym:
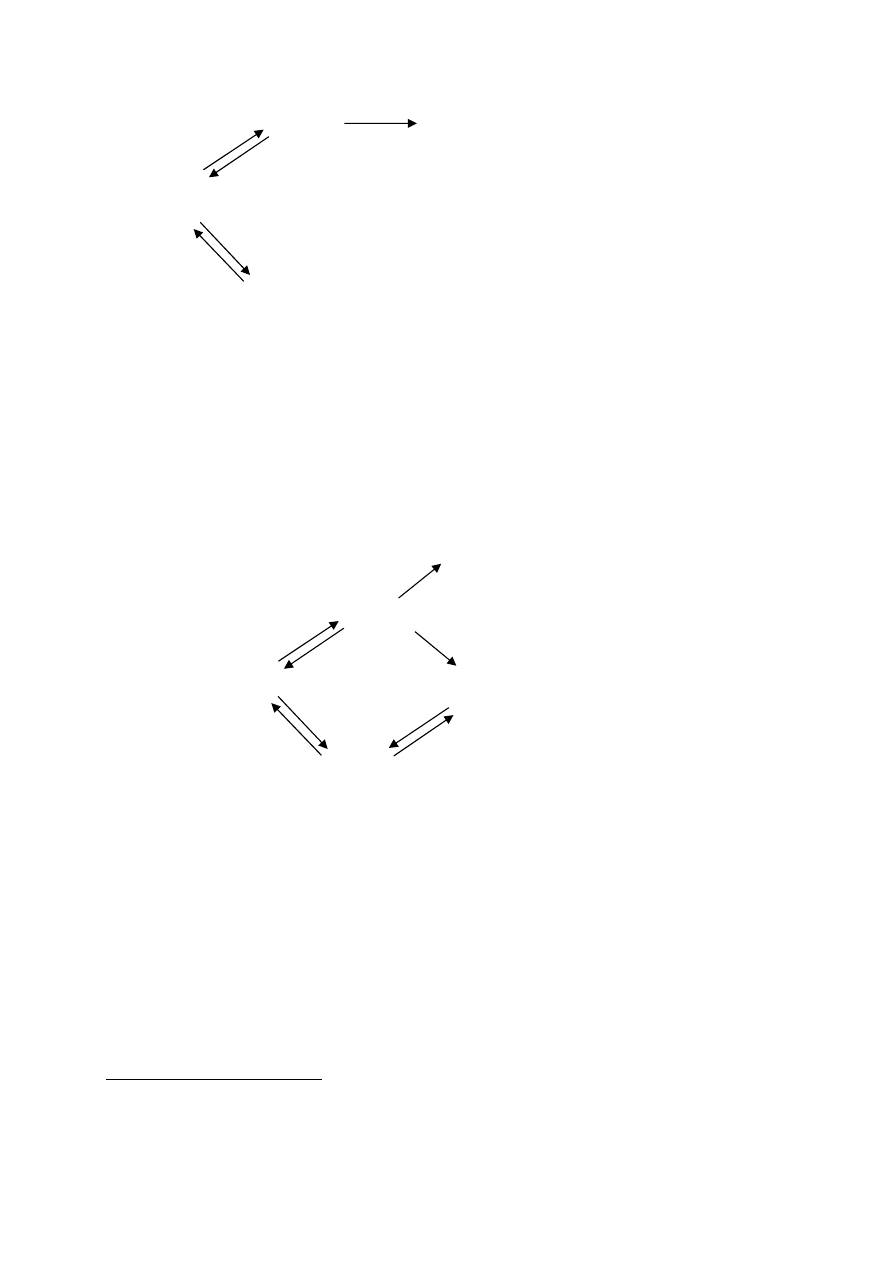
14
W zależności od stosunku stężeń substratu i inhibitora, jeden z tych związków wygrywa
rywalizację o miejsce aktywne. Przy dużym nadmiarze substratu wpływ inhibicji
kompetencyjnej może być całkowicie zniesiony.
W inhibicji niekompetytywnej w strukturze enzymu istnieją oddzielne miejsca
wiązania substratu i inhibitora, jednak cząsteczka ze związanym inhibitorem zmienia swoją
konformację tak, że niemożliwe jest już wiązanie substratu, lub związany substrat nie może
być przekształcony w produkt reakcji:
W inhibicji niekompetytywnej tworzone są dwa typy kompleksów enzym - inhibitor:
kompleks E-I, w którego skład wchodzą cząsteczka enzymu i inhibitor, oraz kompleks E-I-S
powstający, gdy z cząsteczką enzymu jest równocześnie związany inhibitor i substrat.
Aktywność enzymów zależy również od intensywnych parametrów środowiska.
I tak, przebieg reakcji enzymatycznych, podobnie jak wszystkich reakcji chemicznych, silnie
zależy od temperatury, co jest efektem zmiany aktywności biorących w niej udział
cząsteczek. Wpływ temperatury T na szybkość reakcji chemicznej k opisuje równanie
sformułowane przez Svante Arrheniusa, szwedzkiego chemika, w 1889 roku
8
:
8
Oprócz ww. równania, na cześć Arrheniusa zostały też nazwane: laboratorium na Uniwersytecie
Sztokholmskim i … krater na księżycu. Wynika to z wszechstronnych zainteresowań naukowca, który zajmował
się również toksykologią, geologią, astronomią i astrofizyką. Należałoby tu również wspomnieć o czynnym
zaangażowaniu Arrheniusa w tworzenie Narodowego Instytutu Biologii Ras w Uppsali (Statens Institut för
E
+ S
+ I
E-I
E-S P + E
forma zdezaktywowana enzymu
E
+ S
+ I
E-I
E-S
E-I-S
P + E
+ I
+ S
formy
zdezaktywowane
enzymu
15
(
)
RT
E
A
k
a
/
exp
−
⋅
=
(19)
Gdzie
A – stała (czynnik przedwykładniczy)
E
a
– energia aktywacji
R – stała gazowa
Zgodnie z regułą van’t Hoffa, wzrost temperatury o każde 10°C powoduje 2-4-krotny wzrost
szybkości reakcji. Oczywiście, w przypadku reakcji enzymatycznych nie jest możliwe
nieograniczone zwiększanie temperatury, gdyż prowadziłoby to do dezaktywacji enzymu
9
.
Enzymy białkowe są szczególnie wrażliwe na denaturację termiczną, która prowadzi do
zniszczenia ich trzeciorzędowej struktury, kluczowej dla działania miejsca aktywnego.
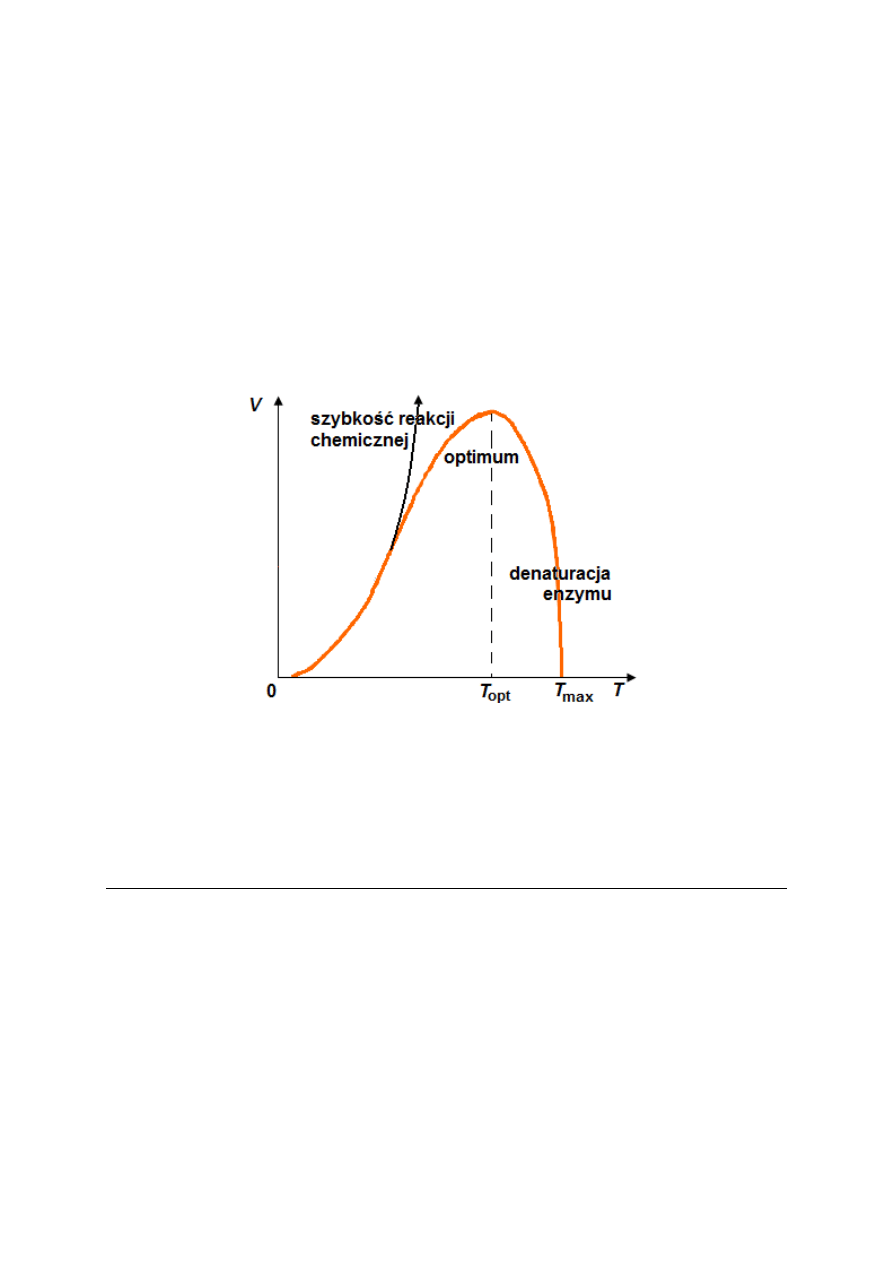
Rysunek 4. Wpływ temperatury na aktywność enzymu. Na wykresie przedstawiono jak szybkość reakcji
enzymatycznej zmienia się wraz ze wzrostem temperatury, zaznaczono temperaturę optymalną T
opt
dla przebiegu
reakcji enzymatycznej oraz temperaturę maksymalną T
max
, po przekroczeniu której następuje denaturacja
enzymu.
W przypadku enzymów wykorzystywanych przemysłowo, kiedy regulacja szybkości reakcji
przy pomocy temperatury wydaje się być kuszącym rozwiązaniem, postuluje się stosowanie
Rasbiologi). Badania prowadzone w Instytucie wywarły duży wpływ na rozwój nazistowskiej eugeniki
i stanowiły „naukowe” uzasadnienie dla programu sterylizacyjnego, realizowanego w Szwecji w latach 1936-
1976 (21 tysięcy przymusowych sterylizacji!)
9
Nawet niewielkie zmiany temperatury mogą powodować dramatyczny wzrost lub spadek aktywności
enzymów, odczuwalny wyraźnie przez organizm – długotrwałe stany gorączkowe lub stany osłabienia
organizmu wynikają ze zmiany temperatury ciała tylko o
±2°C! Już tak niewielkie zmiany w temperaturze
znacząco wpływają na funkcjonowanie organizmu jako całości. Dalsze obniżanie temperatury ciała prowadzi do
hipotermii, czyli przechłodzenia. Ostre objawy hipotermii, w tym sztywność mięśni i utrata świadomości,
pojawiają się, gdy temperatura ciała spadnie do 30°C. Dalsze wychłodzenie, poniżej 28°C prowadzi do śmierci,
której bezpośrednią przyczyną jest zbyt niska temperatura serca i mózgu. Wychłodzenie organizmu zachodzi
znacznie (ok. 20-razy) szybciej w wodzie, ze względu na jej większe przewodnictwo cieplne. Szacuje się, że
czas przeżycia człowieka w wodzie o temperaturze poniżej 10°C wynosi tyle minut ile temperatura wody!
Doskonale tłumaczy to zasięg jednej z największych tragedii morskich – na pokładzie zatopionego
transatlantyku Titanic zginęło ponad 1500 osób. W nocy 14/15.04.1912, kiedy doszło do tragedii, temperatura
wody spadła bowiem do 0°C, co znacząco obniżyło szanse przeżycia pasażerów statku.
16
enzymów wyizolowanych z organizmów termofilnych. Cały metabolizm termofili jest
dostosowany do wysokich temperatur i ich białka są stabilne w temperaturach sięgających
80°C (a nawet 105°C dla hipertermofili!).
Czynnikiem istotnym dla szybkości reakcji enzymatycznej jest też stężenie jonów
wodorowych w mieszaninie reakcyjnej. W zależności od odczynu środowiska różny jest
bowiem stopień dysocjacji reszt aminokwasowych, co bezpośrednio wpływa na
oddziaływanie enzymu z substratem (poprzez zmianę konformacji całego enzymu lub zmianę
właściwości jego miejsca aktywnego). Skrajne wartości pH mogą nawet doprowadzić do
denaturacji enzymu.
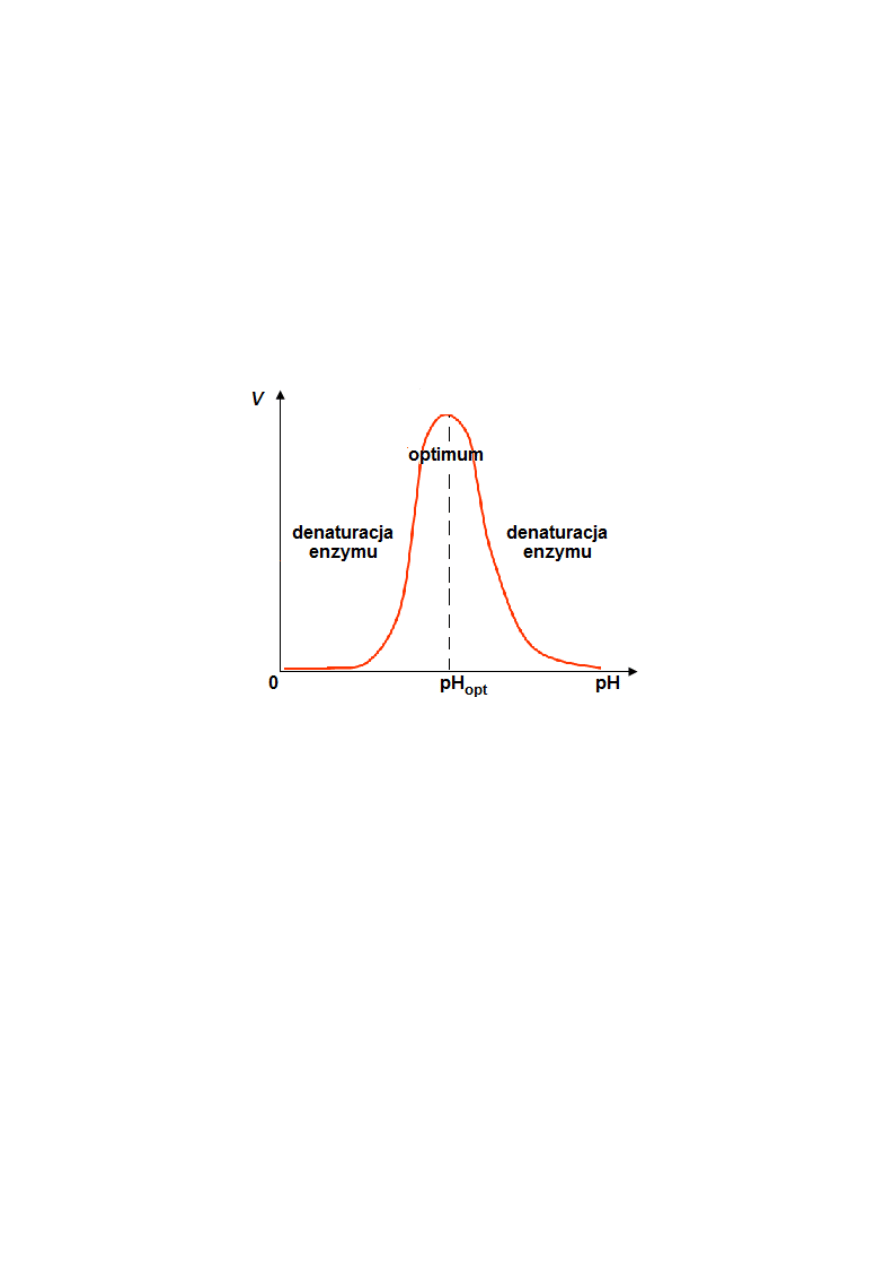
Rysunek 5. Wpływ pH na aktywność enzymu. Na wykresie przedstawiono jak zmiany pH wpływają na
szybkość reakcji enzymatycznej. Zaznaczono optymalną wartość pH – pH
opt
, przy której reakcja przebiega
najszybciej oraz zakresy pH, w których zachodzi denaturacja enzymu.
Enzymy działają więc najefektywniej w ściśle określonym przedziale pH, zależnie od ich
struktury i charakteru oddziaływań enzym-substrat. Umożliwia to na przykład regulację
aktywności enzymów działających w przewodzie pokarmowym (enzymy aktywne w silnie
kwaśnym pH żołądka, jak pepsyna czy lipaza żołądkowa, są dezaktywowane w dwunastnicy,
która zawdzięcza swoje alkaliczne środowisko sokowi trzustkowemu).
III. Immobilizacja enzymów
III.1. Zalety i wady procesu immobilizacji
Jak już wspomniano, enzymy mogą być stosowane w przemyśle w formie zawiesiny
(o różnym stopniu czystości) lub w postaci immobilizowanej. Immobilizacja jest procesem
polegającym na uwięzieniu enzymu w pewnej określonej przestrzeni. Enzym
immobilizowany powinien charakteryzować się aktywnością katalityczna i być przeznaczony
do wielokrotnego stosowania.

17
Termin enzym immobilizowany został precyzyjnie zdefiniowany dopiero w 1971
roku, w wiele lat po pierwszym praktycznym wykorzystaniu procesu immobilizacji. Już w
XVII wieku do produkcji kwasu octowego z etanolu używano bowiem bakterii Acetobacter
umieszczonych na wiórach bukowych. Z kolei enzymu immobilizowanego użyto po raz
pierwszy podczas I wojny światowej – wobec deficytu kwasu siarkowego przeprowadzono
reakcję inwersji sacharozy w obecności inwertazy immobilizowanej na węglu aktywnym
(otrzymanym poprzez zwęglenie kości).
Immobilizacja enzymów umożliwia zwiększenie wydajności procesów
biotechnologicznych
10
. Wydajność procesu możemy zdefiniować poprzez wydajność syntezy
produktu, czyli stosunek masy otrzymanego produktu w stosunku do masy substratu. Wzrost
wydajności procesu jest możliwy, ponieważ immobilizacja zwiększa trwałość enzymów
i ułatwia ich odzyskiwanie z mieszaniny reakcyjnej, co z kolei umożliwia ich wielokrotne
używanie w procesach prowadzonych w układach okresowych lub ciągłych. Enzymy
immobilizowane charakteryzują się zwiększoną stabilnością w zmieniających się warunkach
pH i temperatury oraz w środowisku rozpuszczalników organicznych.
Z drugiej strony, w trakcie procesu immobilizacji może nastąpić częściowa
dezaktywacja enzymu. Enzym immobilizowany często charakteryzuje się niższą aktywnością
niż enzym „wolny” pracujący w tych samych warunkach
11
(aktywności enzymu nie należy
mylić z ostateczną wydajnością prowadzonego procesu). Może to wynikać ze sterycznego
zablokowania centrum aktywnego, niewłaściwych naprężeń w obrębie cząsteczki enzymu a
także mechanicznych uszkodzeń enzymu. Nie do pominięcia są też opory dyfuzyjne, które
zmniejszają efektywne stężenie substratu docierającego do katalizatora, i tym samym
ograniczają szybkość zachodzącej reakcji. Należy tez pamiętać o kosztach samej
immobilizacji, które muszą być uwzględnione w analizie ekonomicznej całego procesu.
10
Immobilizacja może też wpływać na wzrost wydajności procesów prowadzonych w obecności homogenatów
komórkowych i całych komórek. Na przykład wydajność reakcji redukcji kodeinonu do kodeiny pod wpływem
wyciągu z maku lekarskiego (Papaver somniferum) rośnie z 60% (reakcja w zawiesinie) do 70% (biokatalizator
unieruchomiony w żelu alginianowym) a nawet do ponad 80% wskutek immobilizacji biokatalizatora w piance
poliuretanowej. Kodeina (metylomorfina) jest substancją czynną o działaniu przeciwbólowym
i przeciwkaszlowym. Podczas II wojny światowej była używana jako substytut morfiny. Immobilizacja komórek
na nośnikach jest z kolei wskazana w produkcji metabolitów wtórnych. Zagęszczenie komórek wywołane
immobilizacją imituje warunki panujące w tkankach roślinnych (gradient pożywki i metabolitów, zwiększona
odporność na uszkodzenia mechaniczne). I tak, wzrost produkcji alkaloidów purynowych zaobserwowano po
immobilizacji komórek Coffea arabica w alginianie wapnia.
11
Immobilizacja może też wpływać na wzrost aktywności enzymów – taki efekt zaobserwowano w przypadku
enzymów katalizujących reakcje stereoselektywnej estryfikacji i transestryfikacji, po ich unieruchomieniu
metodą sieciowania kryształów enzymu.
18
Alternatywną w stosunku do immobilizacji metodą unieruchomienia enzymów, może
być prowadzenie procesu w reaktorze membranowym, gdzie przestrzeń zajęta przez enzym
jest ograniczona przez półprzepuszczalną membranę.
III.2. Metody immobilizacji enzymów
Metoda immobilizacji i właściwy dobór nośnika determinują właściwości katalityczne
związanego enzymu. Decydując się na określony typ nośnika należy zwrócić uwagę zarówno
na właściwości biochemiczne samego enzymu (w tym jego wielkość, charakter grup
funkcyjnych, aktywność oraz pH- i termostabilność), typ katalizowanej reakcji, jak
i charakterystykę fizykochemiczną nośnika (jego stabilność chemiczną i wytrzymałość
mechaniczną, dostępne grupy funkcyjne, porowatość, stopień rozwinięcia powierzchni).
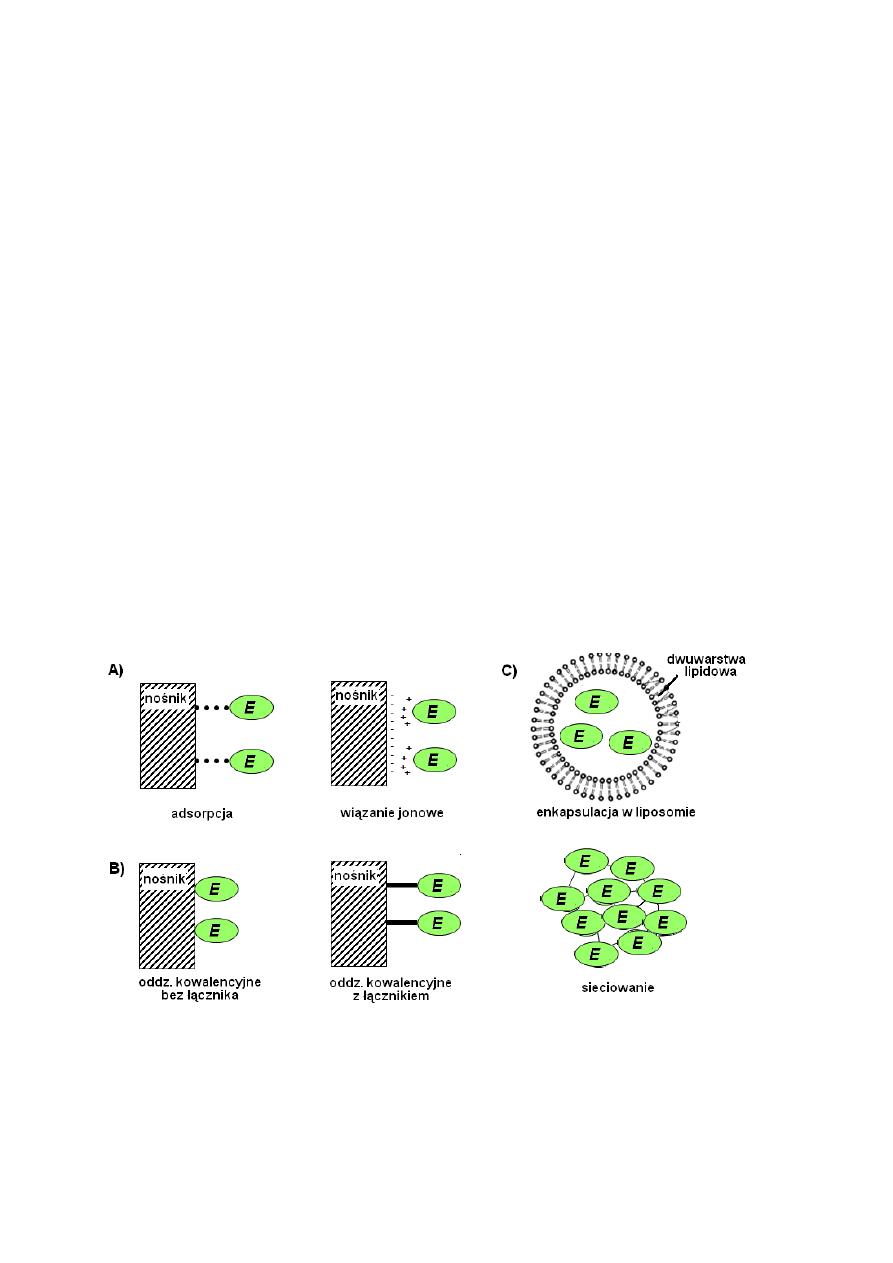
W zależności od wybranej metody immobilizacji, enzym może być zaadsorbowany na
nośniku lub związany z nim wskutek oddziaływań elektrostatycznych. Jest to immobilizacja
fizyczna. Enzym może również być immobilizowany chemicznie: tworzyć z nośnikiem
wiązanie kowalencyjne lub być uwięziony w sieci tworzonej przez czynniki sieciujące.
Trzeci rodzaj immobilizacji, immobilizacja mechaniczna, polega na unieruchomieniu
enzymu w matrycy lub jego okapsułkowaniu. Podstawowe metody immobilizacji zostały
przedstawione na Rysunku 6.
Rysunek 6. Główne metody immobilizacji enzymów: A) immobilizacja fizyczna; B) immobilizacja
chemiczna; C) immobilizacja mechaniczna.
Immobilizacja fizyczna jest efektem niespecyficznych oddziaływań pomiędzy
enzymem a nośnikiem, które są efektem sił van der Waalsa, oddziaływań elektrostatycznych,

19
oddziaływań hydrofobowych i powstawania wiązań wodorowych. Siła tych oddziaływań jest
w ewidentny sposób zależna od środowiska (siły jonowej medium, odczynu pH, stopnia
rozwinięcia powierzchni nośnika). Pociąga to za sobą łatwość desorpcji enzymu i ograniczoną
trwałość wobec zmieniających się warunków środowiska.
Zdecydowaną zaletą immobilizacji chemicznej nad immobilizacją opartą na
oddziaływaniach niespecyficznych jest jej większa trwałość. Wiązanie kowalencyjne może
być tworzone bezpośrednio pomiędzy grupami funkcyjnymi enzymu i nośnikiem albo
powstawać z udziałem łącznika. Cząsteczki łącznika są niezbędne w procesie sieciowania
(określane są wówczas mianem czynników sieciujących). Spośród metod immobilizacji
mechanicznej na szczególną uwagę zasługuje kapsułkowanie enzymów w sztucznych
liposomach, czyli pęcherzykach lipidowych, co umożliwia łatwą inkorporację enzymu
w błony biologiczne.
IV. Podstawowe informacje o inwertazie
Inwertaza (właściwie β-fruktofuranozydaza, E.C. 3.2.1.26) jest enzymem z klasy
hydrolaz (podklasa: glikozydazy), który katalizuje reakcję rozkładu sacharozy do glukozy
i fruktozy - łatwo przyswajalnych cukrów prostych. U człowieka, na wewnętrznej
powierzchni komórek nabłonkowych wyściełających jelito cienkie, występują dwa typy
enzymów umożliwiających przeprowadzenie dwucukrów do łatwo wchłanialnych cukrów
prostych – oprócz inwertazy, stwierdzono obecność laktazy – enzymu, który katalizuje
reakcję hydrolizy laktozy do glukozy i galaktozy. Z kolei inwertaza obecna w ślinie pszczół,
umożliwia im przeprowadzenie cukrów złożonych obecnych w nektarze kwiatowym do
postaci monocukrów, warunkując tym samym proces wytwarzania miodu.
Inwertaza jest wykorzystywana do celów komercyjnych głównie w przemyśle
spożywczym (w cukiernictwie). Przykładem może być produkcja czekoladek z nadzieniem.
Nadzienie w formie stałej, o dużej zawartości sacharozy i z dodatkiem inwertazy, jest
pokrywane warstwą czekolady. W trakcie kilkudniowego (a czasami nawet
kilkutygodniowego) procesu „dojrzewania” następuje hydroliza sacharozy, prowadząca do
uwolnienia cukrów prostych - w efekcie tego procesu nadzienie zostaje upłynnione.
Do celów przemysłowych, inwertaza jest otrzymywana z komórek drożdży.
Najwyższą aktywność tego enzymu stwierdzono w pH 4.5 i w temperaturze 60°C.
20
ĆWICZENIE 29/30
INSTRUKCJA WYKONANIA
Cel ćwiczenia
Celem
ćwiczenia jest poznanie procesu ciągłej produkcji cukru inwertowanego
będącego mieszaniną glukozy i fruktozy. Proces ten prowadzony jest metodą chemiczną (na
złożu jonitowym) lub biokatalityczną (na złożu zawierającym inwertazę drożdżową) w
mikroskali w reaktorach kolumnowych z ciągłym dozowaniem roztworu substratu. Integralną
częścią ćwiczenia jest wykonanie złoża zawierającego inwertazę unieruchomioną na żelu
alginianowym.
Osoby
wykonujące ćwiczenie badają wpływ rodzaju katalizatora, temperatury oraz
objętościowego natężenia przypływu na wydajność procesu inwersji sacharozy. Detekcja
glukozy w mieszaninie poreakcyjnej odbywa się metodą wstrzykowej analizy przepływowej
FIA (ang. Flow Injection Analysis) z użyciem odczynnika zawierającego oksydazę glukozową
oraz peroksydazę (metoda Trindera, nazwa komercyjna zestawu: Glucose GOD-PAP).
W
trakcie
laboratorium
osoby, które zgodnie z planem zajęć wykonują ćwiczenie 30,
są odpowiedzialne za proces przebiegający w reaktorach kolumnowych, podczas gdy osoby,
które wykonują ćwiczenie 29, przygotowują złoże zawierające immobilizowany enzym
i przeprowadzają inwersję sacharozy w układzie okresowym. Uczestnicy laboratorium
zobowiązani są zapoznać się z przebiegiem obydwu prowadzonych doświadczeń (również
z informacjami zawartymi w tym skrypcie) - wyniki eksperymentów omawiane są wspólnie.
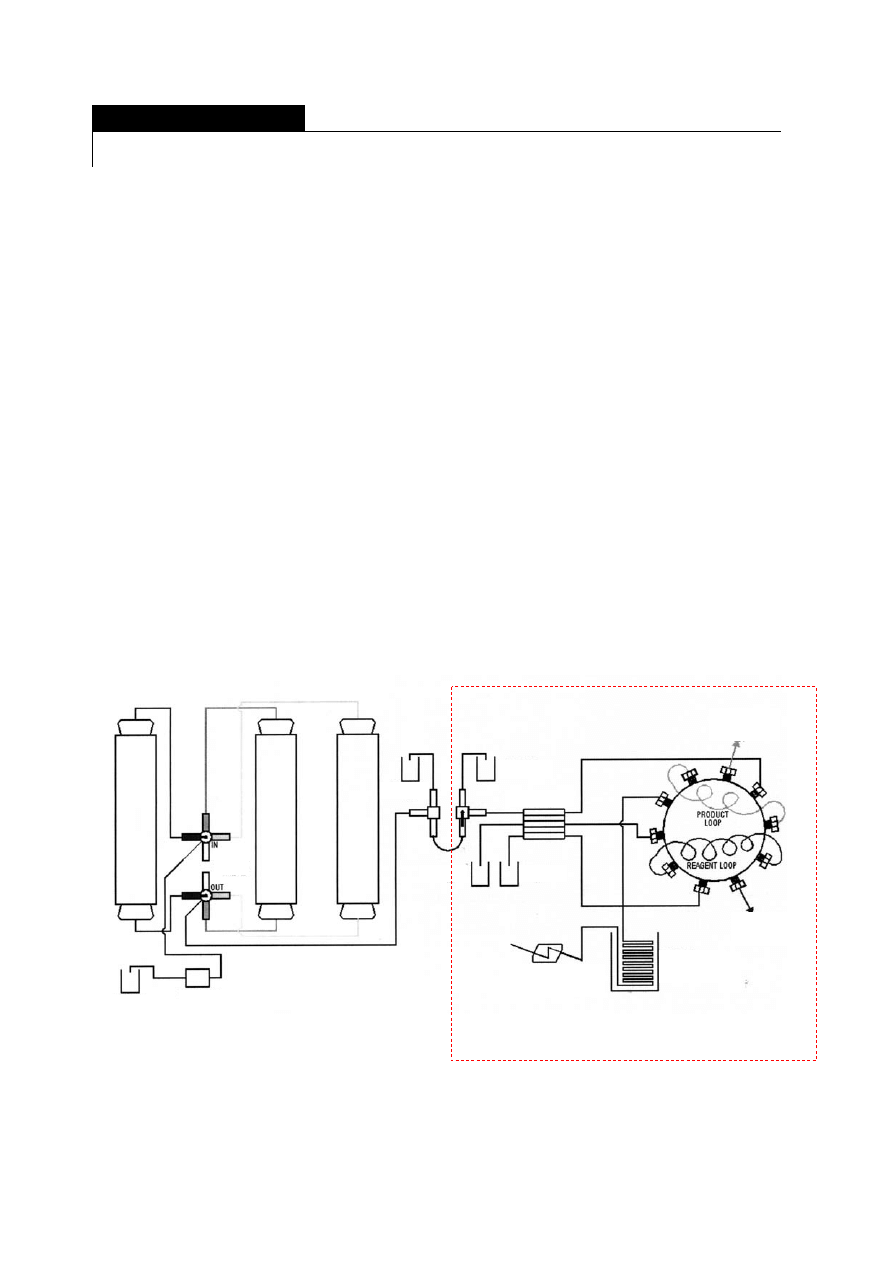
Rysunek 1. Schemat zestawu do prowadzenia reakcji inwersji sacharozy w reaktorach kolumnowych
z unieruchomionym katalizatorem z zaznaczonym modułem analitycznym.
bioreaktor
reaktory chemiczne
krany
produkt
wzorzec
woda
roztwór
zasilający
pompa
zasilająca
reagent
pompa FIA
pętla
reakcyjna
odbiór
reagenta
odbiór
produktu
przepływowa
kuwetka optyczna
MODUŁ FIA
(WSTRZYKOWEJ ANALIZY PRZEPŁYWOWEJ)

21
Sprzęt Odczynniki
Zestaw reaktorów kolumnowych CEU
(zestaw zawiera termostat, reaktory, pompę
perystaltyczną, układ wstrzykowej analizy
przepływowej, komputer
z oprogramowaniem)
pompa perystaltyczna
Butelki 1 L
3 szt.
Butelki 100 mL
5 szt.
Zlewki 100 mL
5 szt.
kolba miarowa 0,5 L
1 szt.
pipeta 20,00 mL
1 szt.
pompka do pipety
1 szt.
cylinder 100 mL
1 szt.
cylinder *
)
500 mL
1 szt.
miniaturowy lejek
1 szt.
zlewka*
)
150 mL
1 szt.
zlewka*
)
1 L
1 szt.
zlewka szklana 1 L
1 szt.
zlewka szklana 600 mL
1 szt.
zestaw do sączenia próżniowego
roztwór HCl (stęż. 2 M)
roztwór sacharozy (stęż. 7,6 g/L)
roztwór CaCl
2
(stęż. 2M)
glukoza
odczynnik Trindera (do analizy glukozy)
jonity DOWEX
alginian sodu
inwertaza drożdżowa lub drożdże
kwas octowy
octan sodu
*sprzęt polipropylenowy, używany tylko do roztworów wodnych
Opis obsługi aparatury oraz kolejność wykonywanych czynności – proces przebiegający
w reaktorach kolumnowych.
1.
Sporządzić następujące roztwory:
roztwór A: sacharoza o stężeniu 7,6 g L
-1
(3
L)
roztwór B: kwas solny 2 M
(1 L)
roztwór C: glukoza o stężeniu 2,0 g L
-1
(500
mL)
Z roztworu C sporządzić w kolbce miarowej po 100 mL roztworów glukozy o następujących
stężeniach 1,0, 0,75, 0,5 i 0,25 g L
-1
i przelać do podpisanych buteleczek o pojemności 100
mL z nakrętkami zawierającymi otwory do wężyków teflonowych. Wszystkie wymienione
powyżej roztwory odgazowywać przez 5 minut na myjce ultradźwiękowej. W ten sam sposób
odgazować 1 L wody destylowanej.
2.
Zapoznać się ze schematem przedstawionym na Rysunku 1 oraz oznaczeniami na Rysunku 2.
Sprawdzić ustawienie kranów i prześledzić drogę cieczy, począwszy od naczynia, z którego
jest pobierana, przez pompę zasilającą (3), reaktory (4), zawory sterujące kierunkiem cieczy
pompowanej do reaktorów i wypływającej z reaktorów (6) aż do naczynia odbierającego
produkt (Rysunek 2).
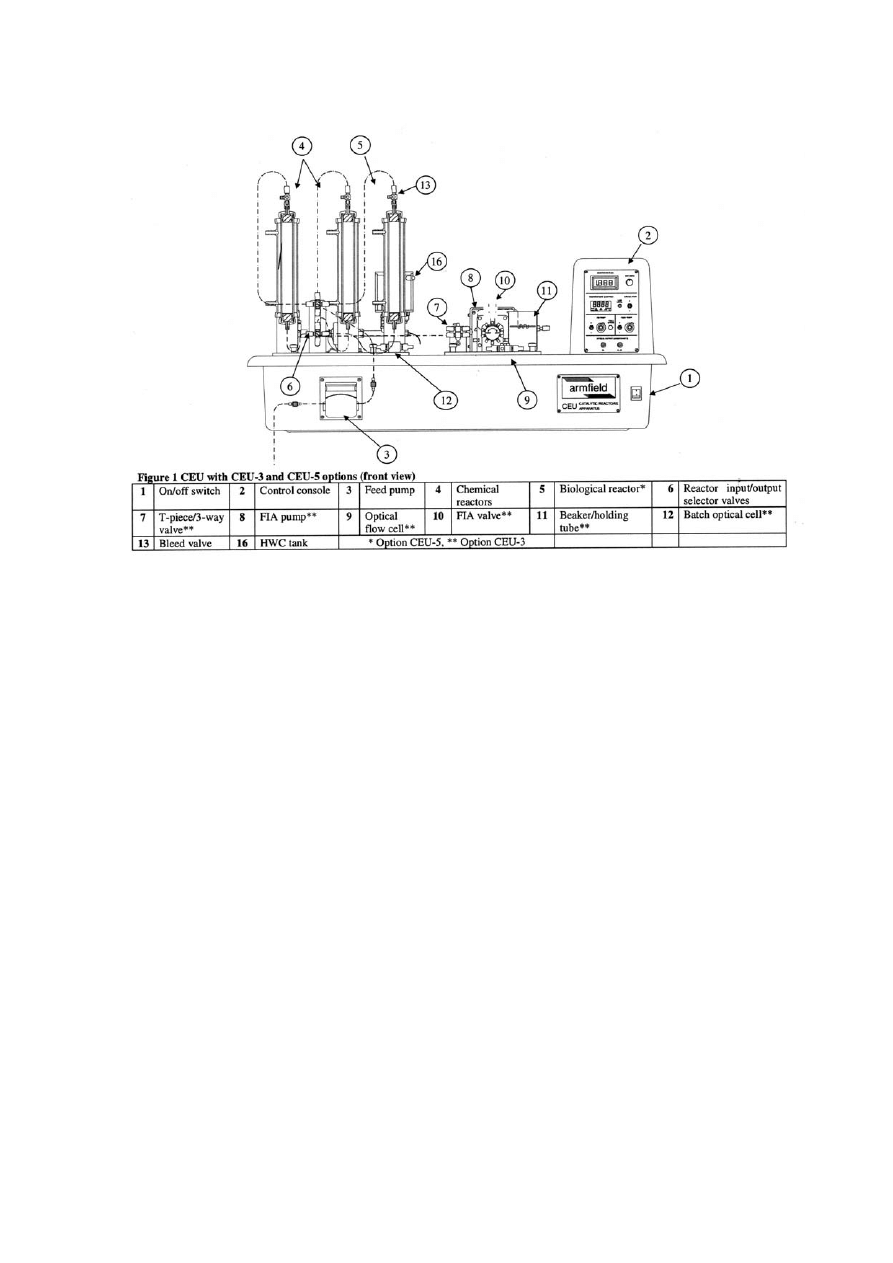
22
Rysunek 2. Schemat zestawu do prowadzenia reakcji w układzie przepływowym - widok z przodu: (1) włącznik
główny; (2) panel sterujący; (3) pompa perystaltyczna zasilająca (dozująca substrat); (4) szklane reaktory
kolumnowe z płaszczami grzejnymi; (5) trzeci, opcjonalny reaktor z biokatalizatorem; (6) zawory dozujące i
odbierające; (7) trójdrożny zawór sterujący wejściem do wstrzykowego analizatora przepływowego (FIA); (8)
pompa dozująca reagenty i ciecz nośną do FIA; (9) przepływowa kuweta optyczna; (10) zawór FIA; (11) pętla
reakcyjna; (12) kuweta optyczna (tylko wtedy, gdy nie ma zestawu FIA); (13) zawory odpowietrzające; (16)
łaźnia termostatu.
3.
Włączyć komputer z oprogramowaniem rejestrującym parametry procesu.
4.
Zapoznać się z obsługą panelu sterującego (oznaczony jako 2 na Rysunku 2), sposobem
regulacji natężenia objętościowego przepływu i innych parametrów procesu. Schemat panelu
sterującego przedstawiono na Rysunku 3. Upewnić się, że termostat jest wyłączony
(TEMPERATURE CONTROL PUMP CIRCULATOR, przełącznik 3 w pozycji „0”).
Podobnie, wyłączona powinna być pompa zasilająca reaktor (FEED PUMP, przełącznik 7a) i
pompa zasilająca analizator przepływowy (FIA PUMP, przełącznik 6a)
23
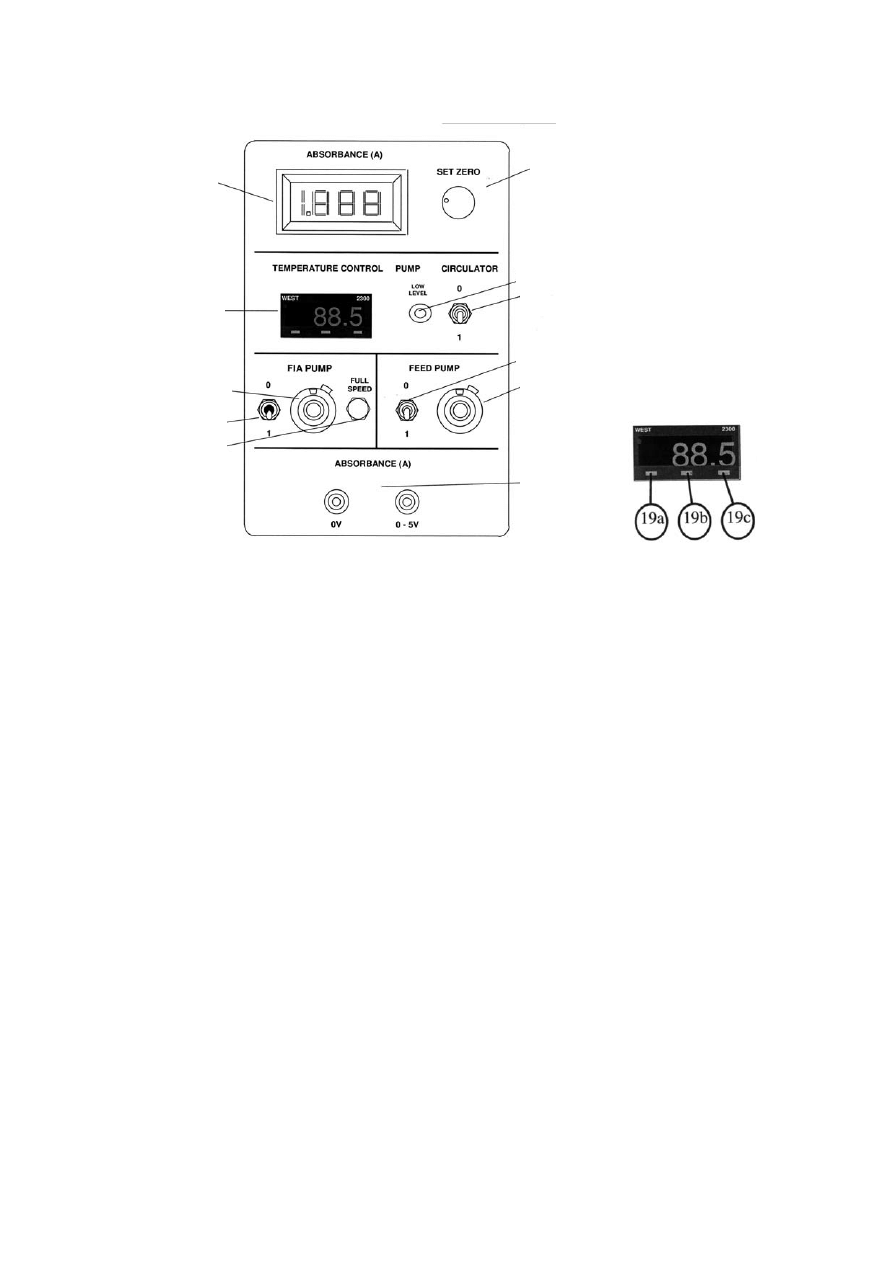
A)
B)
Rysunek 3. (A) Schemat panelu sterującego: ekran wyświetlający wartość absorbancji, rejestrowanej w module
FIA (1); regulator zerowania absorbancji (2); kontroler termostatu (4); włącznik termostatu (3); ekran
wyświetlający temperaturę (5); włącznik pompy FIA (6a); regulacja szybkości pompowania FIA (6b); przycisk
maksymalnej prędkości pompowania (6c); włącznik pompy zasilającej reaktory (7a); regulacja szybkości
pompowania (7b); gniazdko wyjścia sygnału absorbancji (8).
(B) Wygląd ekranu kontrolera termostatu: przyciski umożliwiające wprowadzenie żądanej temperatury (19a-
19c).
5.
Włączyć zasilanie całego urządzenia (włącznik 1 na Rysunku 2).
6.
Ustawić temperaturę termostatu: wcisnąć lewy przycisk (oznaczony jako 19a na Rysunku 3)
aż ukaże się napis SP (set point), wtedy posługując się przyciskami 19b i 19c doprowadzić
wyświetlaną wartość temperatury do wartości pożądanej.
7.
Włączyć termostat (przycisk 3 na Rysunku 3).
8.
URUCHOMIENIE REAKTORÓW
Końcówkę wężyka pompy zasilającej włożyć do butelki z roztworem 2M HCl. Upewnić się,
że wężyk dotyka dna butelki i jest nieruchomy. Sprawdzić drożność układu (zawór zasilający
reaktor oraz zawór odprowadzający roztwór z reaktora powinny być otwarte). Podstawić
naczynie odbierające roztwór produktu. Włączyć pompę zasilającą reaktor. Po
przepompowaniu 400 mL roztworu wykonać czynności opisane w punkcie 15.
9.
URUCHOMIENIE MODUŁU ANALITYCZNEGO – sporządzenie krzywej wzorcowej.
Wężyki pompy zasilającej FIA (Rysunek 1 i 2) podłączyć do trzech buteleczek
zawierających: wodę destylowaną oraz uprzednio przygotowane roztwory: reagenta i
glukozy (wzorca) o najniższym stężeniu (0,25 g L
-1
). Sprawdzić ustawienie kranu (oznaczony
jako 7 na Rysunku 2) i ustawić go w pozycji umożliwiającej pobieranie roztworu wzorca.
Wężyki z odprowadzeniem roztworów (wychodzące z pętli FIA) włożyć do zlewki
zbierającej przereagowane substraty.
1
2
5
6b
4
3
6a
6c
7a
7b
8
24
10.
Skontrolować poziomy zanurzenia wężyków teflonowych pobierających roztwory. Włączyć
pompę FIA, ustawić odpowiednią szybkość pompowania (Rysunek 3, przyciski 6a i 6b).
11.
Przed rozpoczęciem pomiarów absorbancji wzorcowego roztworu glukozy, ustawić
pokrętłem 2 (Rysunek 3) wartość absorbancji równą zero. Włączyć komputerową ciągłą
rejestrację absorbancji (punkty pomiarowe w odstępie czasu 2s).
12.
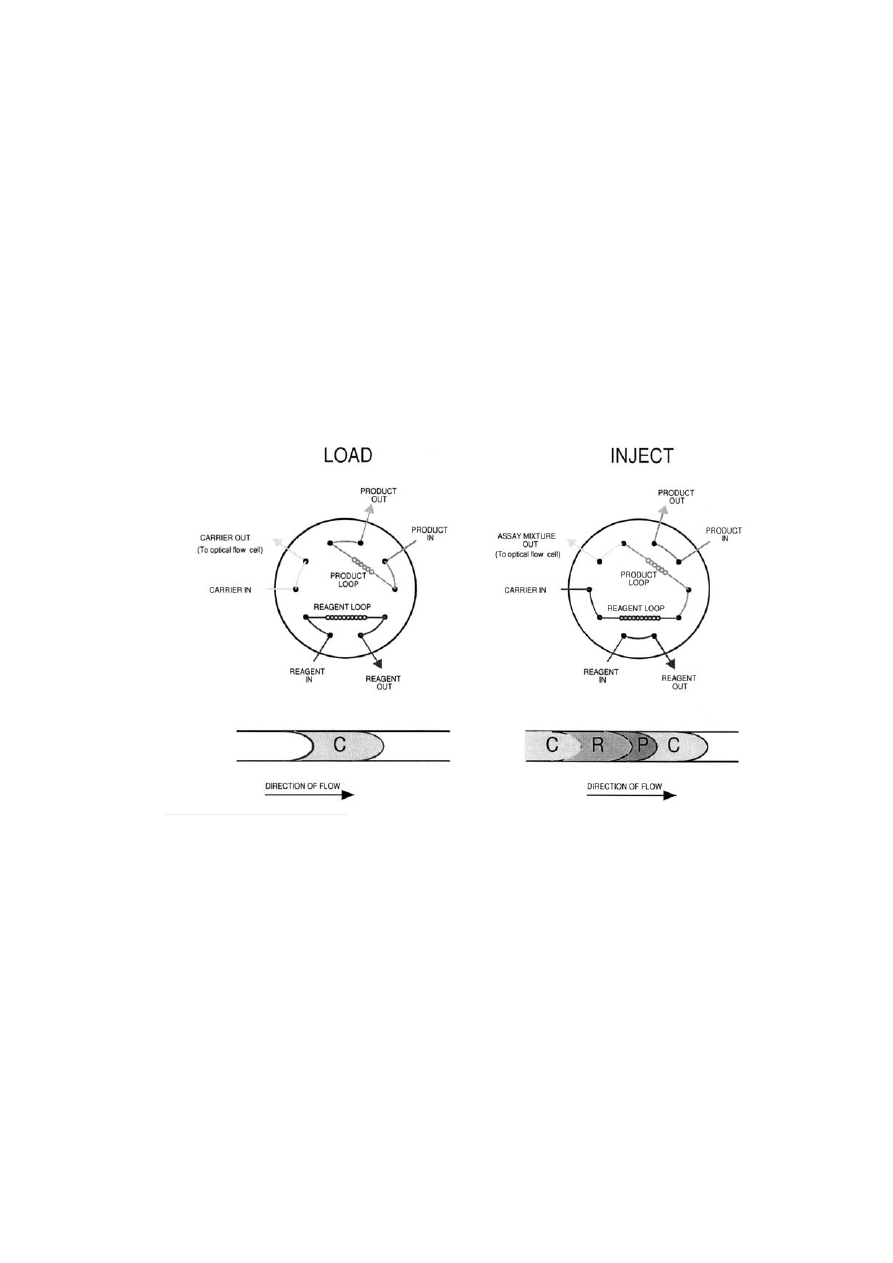
POMIARY ZAWARTOŚCI GLUKOZY.
Przestawić pokrętło pętli FIA z pozycji LOAD do pozycji INJECT. Czynność ta spowoduje
zmieszanie roztworu nośnego z roztworem reagenta i roztworem badanym (lub roztworem
wzorca). Zasadę działania pętli mieszającej przedstawiono na Rysunku 4. Pokrętło powinno
znajdować się w pozycji INJECT przez 60 s, następnie należy je przestawić do pozycji
LOAD. Obserwować zmiany absorbancji na wykresie powstającym na monitorze komputera.
Zanotować maksymalną wartość absorbancji. Pomiar powtórzyć kilkukrotnie aż do uzyskania
powtarzających się wyników.
Rysunek 4. Obieg cieczy w pętli FIA w pozycji LOAD (roztwory nie mieszają się ze sobą) i w pozycji INJECT
(trzy roztwory mieszają się ze sobą zawsze w takich samych proporcjach).
13.
Zatrzymać pompę FIA. Zamienić roztwór wzorca na roztwór o wyższym stężeniu. Włączyć
pompę, odczekać kilka minut i powtórzyć czynności z punktu 12. Wykonać pomiary dla
każdego przygotowanego roztworu glukozy. Wykonać również pomiar dla roztworu
sacharozy (pobrać roztwór zasilający reaktory, stężenie glukozy powinno wynosić zero).
14.
W programie Excel wykreślić krzywą kalibracyjną (wykres absorbancji w funkcji stężenia
glukozy).
15.
ZMIANA POMPOWANEJ CIECZY ZASILAJĄCEJ REAKTORY.
Skontrolować poziom roztworu HCl zasilającego reaktory. Jeśli ubyło 400 mL, zatrzymać
pompę zasilającą (przycisk 7a na Rys. 3). Zamienić butelkę z HCl na butelkę zawierającą
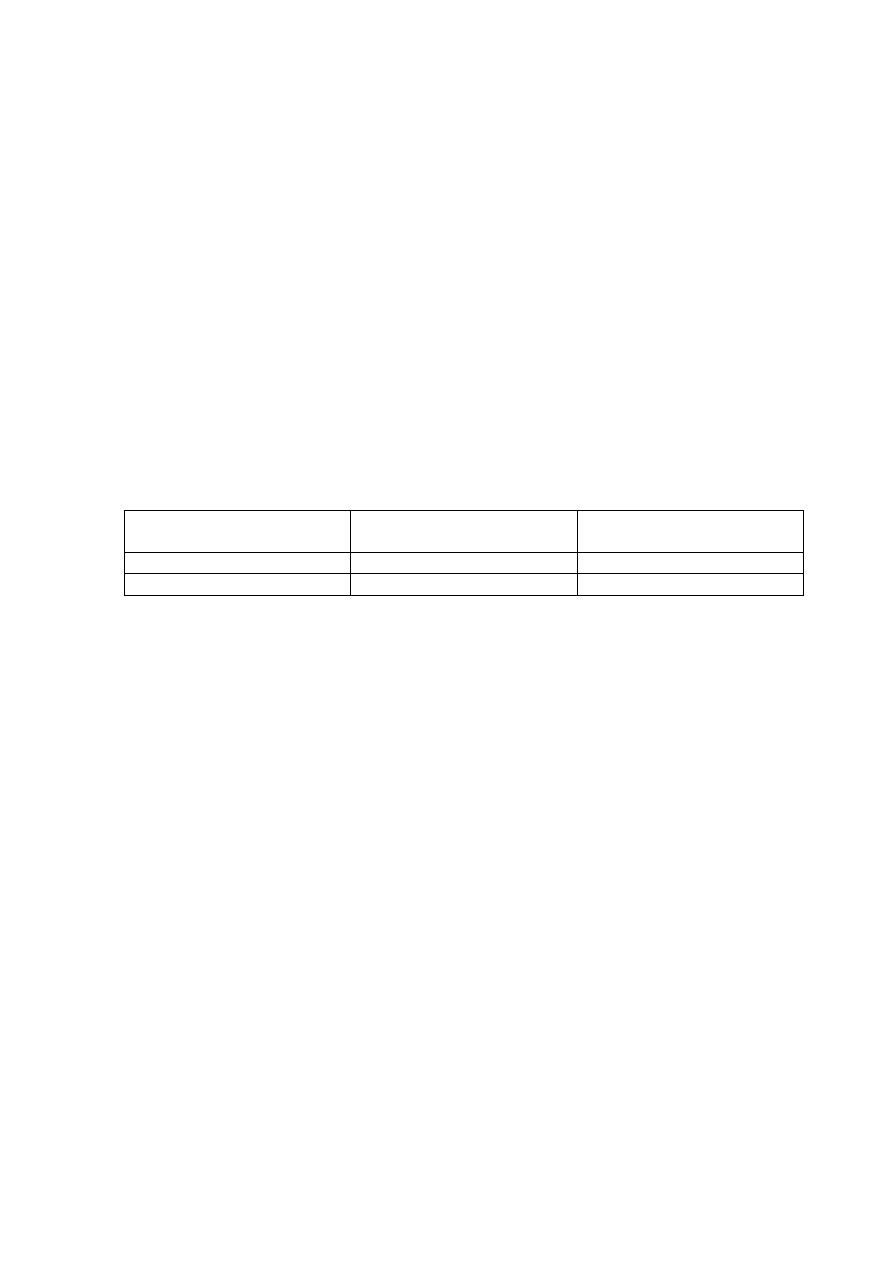
25
odgazowaną wodę destylowaną. Włączyć pompę. Pompować wodę aż roztwór wypływający
z reaktorów nie będzie wykazywał odczynu kwaśnego (sprawdzać papierkiem
wskaźnikowym). Jeśli wyciek z reaktorów jest obojętny, zmienić roztwór zasilający reaktory
– rozpocząć pompowanie roztworu sacharozy. Wyznaczyć natężenie objętościowe przepływu
roztworu sacharozy zbierając przez 10 minut roztwór produktu do cylindra o objętości
100 mL- odczytać objętość cieczy i zanotować natężenie przepływu w mL/min.
16.
Po upływie 15 minut od czasu rozpoczęcia dozowania roztworu sacharozy zmienić ustawienie
kranu (oznaczonego jako 7 na Rysunku 2) w pozycję umożliwiającą pobieranie do analizy
roztworu produktu wypływającego z kolumny.Dokonać analizy zawartości glukozy w
produkcie reakcji wykonując czynności opisane w punkcie 12. Pomiar powtórzyć
kilkukrotnie aż do ustalenia się stałej wartości absorbancji.
17.
Obliczyć stężenie glukozy i wydajność procesu inwersji dla danej temperatury i natężenia
przepływu. Wyniki przedstawić w postaci:
RODZAJ KATALIZATORA:
TEMPERATURA REAKCJI:
NATĘŻENIE OBJĘTOŚCIOWE PRZEPŁYWU:
PRZYCHÓD
[g/godz]
ROZCHÓD
[g/godz]
sacharoza
glukoza
wydajność:
18.
Powtórzyć czynności z punktów 16-17 zmieniając temperaturę procesu, rodzaj katalizatora,
natężenie objętościowe przepływu lub stężenie surowca. UWAGA: Dla procesu
prowadzonego z użyciem immobilizowanej inwertazy, konieczne jest wykonanie czynności z
pkt. 20.
19.
Bioreaktor kolumnowy przepłukiwany jest tylko wodą destylowaną! Immobilizowana
inwertaza wykazuje optymalne działanie w pH 4,8, dlatego roztwór sacharozy powinien być
zakwaszony kwasem octowym do pH 4,5-5,5. UWAGA: Roztwór ten może być skierowany
tylko do bioreaktora, nie do reaktorów z żywicą jonowymienną.
20.
Do opisu ćwiczenia załączyć krzywą kalibracyjną, odczytane wartości absorbancji i obliczone
stężenie glukozy przy określonych warunkach reakcji. Sporządzić bilans materiałowy dla
jednej godziny ruchu ciągłego dla poszczególnych typów reaktorów (chemicznego, z żywicą
jonowymienną oraz biokatalitycznego, z unietuchomioną inwertazą) oraz dla różnych
warunków prowadzenia procesu. Obliczyć wydajności dla jednej godziny ruchu ciągłego.
26
Immobilizacja biokatalizatorów w alginianie wapnia.
Jedną z technik unieruchamiania enzymów lub całych komórek jest ich immobilizacja
w sieci tworzonej przez polimer (czynnik sieciujący). W trakcie tego ćwiczenia inwertaza
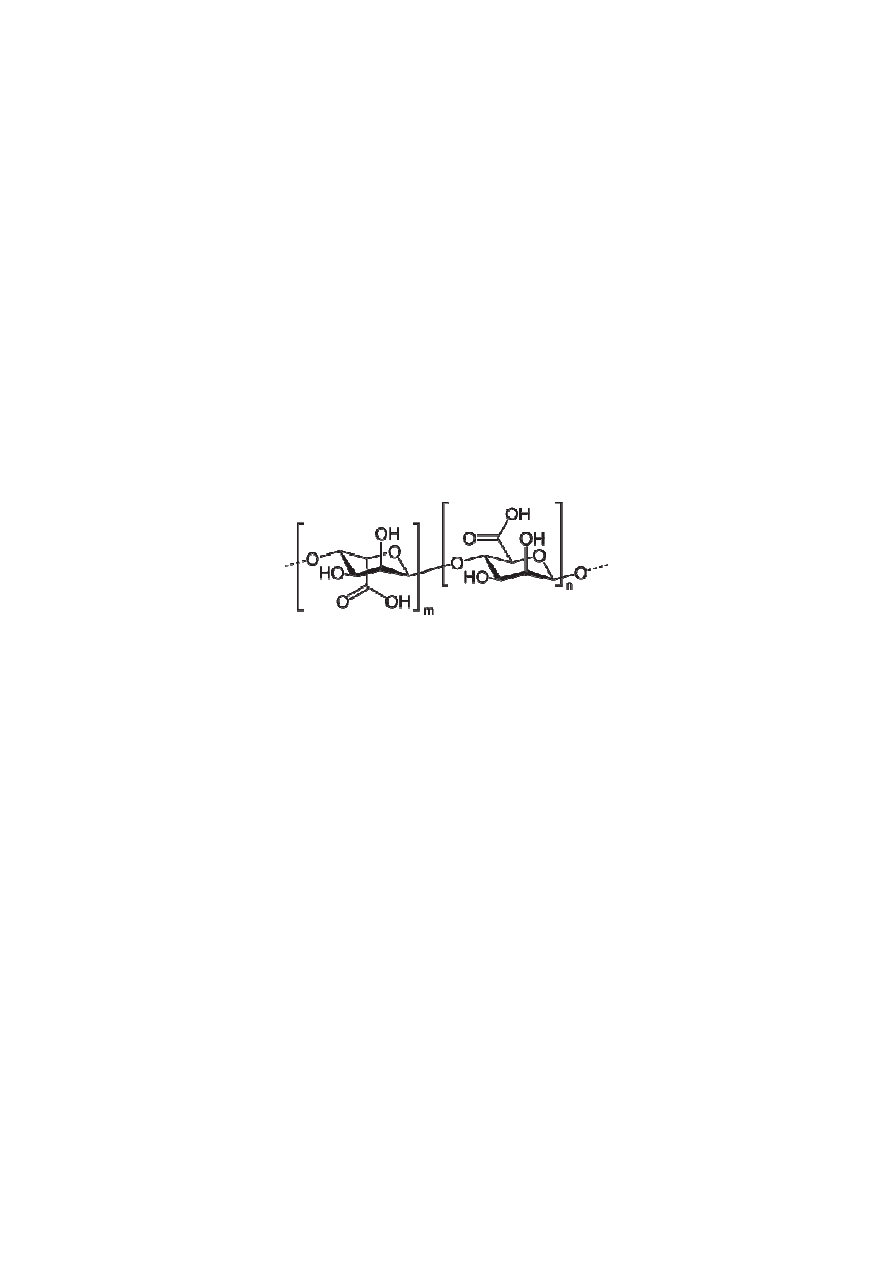
jest u nieruchomiona w alginianie wapnia (soli kwasu alginowego). Kwas alginowy jest
liniowym polimerem, złożonym z monomerów kwasu D-mannuronowego i L-guluronowego,
połączonych wiązaniem β-1-4 glikozydowym (Rysunek 5). W obecności jonów
dwuwartościowych (w tym jonów wapnia) następuje wytrącanie alginianu wapnia z
jednoczesnym sieciowaniem tego polimeru, prowadzącym do utworzenia trójwymiarowego
żelu, w którym mogą zostać uwięzione cząsteczki enzymów bądź całe komórki. Metoda ta
jest szeroko stosowana, ponieważ nie prowadzi do uszkodzenia immobilizowanych
biokatalizatorów. Przy użyciu alginianu wapnia otrzymuje się złoża, którymi wypełnia się
bioreaktory. Ponadto, alginian jest powszechnie używany w produkcji tzw. „sztucznych
nasion”, czyli zarodków somatycznych ukrytych w ochronnej kapsułce zbudowanej z
polimeru.
Proces unieruchamiania biokatalizatorów w alginianie wapnia polega na wymieszaniu
materiału biologicznego z płynnym alginianem sodu, a następnie wkropleniu otrzymanej
zawiesiny do roztworu zawierającego jony wapnia.
Rysunek 5. Kwas alginowy – liniowy polimer będący składnikiem ścian komórkowych alg.
Instrukcja sporządzenia złoża zawierającego immobilizowany enzym.
UWAGA: każda grupa ćwiczeniowa korzysta z preparatu przygotowanego przez poprzednią
grupę studencką (ćwiczenie rozpoczyna się od czynności opisanych punktami 6-7, natomiast
w trakcie ćwiczenia należy przygotować preparat dla następnej grupy, wykonując czynności
z punktów 1-5).
1.
Rozpuścić 0,5 g inwertazy w 300 mL wody destylowanej, delikatnie mieszając na mieszadle
magnetycznym.
2.
Do roztworu inwertazy dodać 15 g alginianu sodowego – mieszać aż do uzyskania
jednorodnego, mętnego roztworu (ok. jedną godzinę ).
3.
Na mieszadle magnetycznym ustawić zlewkę 1 L, nalać 500 mL 0,2 M roztworu CaCl
2
,
zmontować układ do wkraplania alginianu (wężyk z pompy perystaltycznej powinien być
ok. 20 cm ponad lustrem cieczy).
4.
Rozpocząć powolne wkraplanie roztworu alginianu sodu zawierającego inwertazę do
roztworu chlorku wapnia. Obserwować wytrącanie się perełek alginianu wapnia.
5.
Po zakończonym wkraplaniu pozostawić zawiesinę na jeden dzień.

27
6.
Zdekantować roztwór CaCl
2
. Dwukrotnie zalać perełki alginianu (zawierającego
unieruchomioną inwertazę) wodą destylowaną (2 ×500 mL), odsączyć.
7.
Napełnić bioreaktor kolumnowy złożem zawierającym immobilizowaną inwertazę,
uzupełnić wodą destylowaną, a następnie rozpocząć przepłukiwanie złoża wodą
destylowaną. Dalej postępować jak w instrukcji dotyczącej pracy na reaktorach
przepływowych.
Inwersja sacharozy w układzie okresowym.
W alginianie wapnia można immobilizować całe komórki mikroorganizmów i celem
tej części ćwiczenia jest sprawdzenie, czy unieruchomione komórki są nadal aktywne.
Komórki drożdży naturalnie wytwarzają inwertazę, zatem skuteczność procesu immobilizacji
można ocenić, sprawdzając czy drożdże unieruchomione w alginianie wapnia są w stanie
przeprowadzać charakterystyczne dla nich reakcje biochemiczne.
W wyniku hydrolizy sacharozy powstają równomolowe ilości glukozy i fruktozy, tzw.
cukier inwertowany. Sacharoza wykazuje dodatnią skręcalność właściwą (+66,5°), podobnie
jak glukoza (+53,3°), jednak ujemny znak skręcalności właściwej fruktozy (-133,5°)
powoduje, że cały roztwór po hydrolizie ma przeciwny (inwersja) znak skręcalności niż
roztwór wyjściowy. W poniższym eksperymencie postęp reakcji hydrolizy sacharozy jest
śledzony poprzez monitorowanie zmiany skręcalności optycznej przesączu pobranego znad
złoża zawierającego immobilizowane komórki drożdży.
Badanie wpływu immmobilizacji na aktywność drożdży składa się z trzech testów.
I. Komórki drożdży immobilizowane w alginianie wapnia, traktowane roztworem sacharozy
(właściwe doświadczenie).
II. Komórki drożdży immobilizowane w alginianie wapnia, traktowane wodą (kontrola 1).
III. Złoże alginianu wapnia (bez drożdży) traktowane sacharozą (kontrola 2).
Przebieg doświadczenia:
1. Przygotować 50 mL 2% roztworu alginianu sodu w wodzie, pozostawić na mieszadle
magnetycznym do rozpuszczenia.
2. 3 g chlorku wapnia rozpuścić w 200 mL wody destylowanej.
3. Sporządzić zawiesinę 1 g drożdży w 10 mL ciepłej wody destylowanej.
4. W zlewce 25 mL wymieszać 12 mL alginianu sodu i 3 mL zawiesiny drożdży
(w wariancie III mieszamy 12 mL alginianu sodu z 3 mL wody destylowanej bez
dodawania zawiesiny drożdży
).
5. Nabrać przy pomocy strzykawki 15 mL zawiesiny otrzymanej w pkt 4, wkraplać powoli
(dokładnie po kropli) do 75 mL roztworu chlorku wapnia.. Pozostawić kulki w roztworze
chlorku wapnia na około 10 minut, następnie odsączyć i przemyć wodą destylowaną.
6. Przygotować 100 mL 5% roztworu sacharozy. Sprawdzić jego skręcalność optyczną
posługując się polarymetrem.
7. Umieścić otrzymane złoża w probówkach, uzupełnić odpowiednim roztworem (wodą lub
roztworem sacharozy) do objętości 30 mL.
8. Monitorować przebieg reakcji poprzez pomiar skręcalności optycznej przesączu znad
złoża. Pierwszy pomiar przeprowadzić zaraz po uzupełnieniu probówek wodą/sacharozą,
kolejne w odstępach 30 minut. Każdorazowo po pomiarze zlewać pobrany do analizy
przesącz z powrotem do probówki.
28
Wstrzykowa analiza przepływowa
Wstrzykowa analiza przepływowa FIA (ang. Flow Injection Analysis) jest automatyczną
metodą analizy, w której próbka pobierana z przepływowego układu reakcyjnego jest
automatycznie mieszana z pompowanym w sposób ciągły nośnikiem oraz z reagentem
wywołującym reakcję analityczną. W pętli reakcyjnej następuje tworzenie kompleksu lub
derywatyzacja substancji do postaci wykrywanej przez detektor (np. spektrofotometrycznie).
Idea procesu została przedstawiona na Rysunku 6.
czas
DETEKTOR
PETLA
REAKCYJNA
NOSNIK
REAGENT
(odczynnik analityczny)
PRODUKT
SUBSTRAT
Rysunek 6. Zasada działania FIA.
Mała część próbki jest wstrzykiwana do nośnika, który jest mieszany z jednym lub kilkoma
odczynnikami. Schemat układu mieszającego przedstawiono na Rysunku 7.
Rysunek 7. FIA: wygląd i schemat układu pobierającego i mieszającego reagent, badaną próbkę i roztwór
nośnika. Zasada działania została wyjaśniona na Rysunku 4.

29
Ponieważ reakcja analityczna wymaga dokładnego zmieszania reagentów lub może nie
następować od razu, mieszanina przechodzi przez pętlę reakcyjną. Odpowiedni dobór
długości pętli reakcyjnej w stosunku do jej średnicy gwarantuje optymalne wymieszanie
substratów reakcji. Barwa mieszaniny wypływającej z pętli reakcyjnej jest rejestrowana przez
detektor. Odstęp czasu pomiędzy wstrzyknięciem próbki do nośnika a rejestracją zmiany
barwy może wynosić od kilkudziesięciu sekund do kilku minut.
W metodzie FIA nie jest konieczne osiągnięcie stanu równowagi, o wiele ważniejsze
jest, aby mieszanina zawsze docierała do detektora po upływie takiego samego czasu od
momentu zmieszania reagentów, dlatego podczas analizy musi być zachowana stała prędkość
przepływu wszystkich mieszanych cieczy. Spełnienie tego warunku jest konieczne do
zapewnienia powtarzalności i dokładności analizy.
Zaletami FIA są: krótki czas trwania analizy, możliwość analizowania procesów w
trybie on-line, prowadzenie skomplikowanej analizy (wymagającej mieszania kilku
reagentów) metodą ciągłą, prostota czynności wykonywanych przez operatora urządzenia,
możliwość prowadzenia wielu analiz w krótkim czasie, niewielkie zużycie odczynników.
Dzięki tym zaletom, automatyczna metoda wstrzykowej analizy przepływowej jest
wszechstronnie stosowana tam, gdzie wymagana jest duża liczba automatycznych analiz oraz
w miejscach, gdzie istnieje konieczność monitorowania parametrów w sposób cykliczny w
niewielkich odstępach czasu: do kontroli przebiegu procesów przemysłowych, w
laboratoriach medycznych i biologicznych.
Zasada oznaczania glukozy
Metoda polaga na przeprowadzeniu reakcji enzymatycznej z kolorymetryczą detekcją
produktu. W trakcie reakcji glukoza jest utleniana przez oksydazę glukozy (GOD) do
glukonolaktonu w obecności tlenu atmosferycznego:
GOD
glukoza
+
O
2
glukonolakton + H
2
O
2
powstający w tej reakcji nadtlenek wodoru reaguje w obecności peroksydazy (POD) z 4-
aminofenazonem i fenolem tworząc 4-(para-benzochino-monoimino)-fenazon:
POD
2 H
2
O
2
+ 4-aminofenazon + fenol
4-(p-benzochino-monoimino)-fenazon+ 4H
2
O
Produkt tej reakcji ma purpurową barwę i wykazuje maksimum absorbancji w 510 nm. Przy
zachowaniu stałości stężenia wszystkich pozostałych reagentów, intensywność zabarwienia
jest zależna tylko od zawartości glukozy.
Zestaw enzymatyczny używany w ćwiczeniu (GLU, Glukoza GOD-PAP firmy Roche) jest
używany w laboratoriach chemii klinicznej do analiz zawartości glukozy w surowicy i osoczu
krwi. Zakres pomiarowy wynosi 0,11 – 25 mmol glukozy/L (2-450 mg/dL)
Literatura:
Trinder P. Determination of Glucose in Blood using Glucose Oxidase with alternative
oxygen acceptor. Ann. Clin. Biochem. 1969, 6, 24-27.
Tietz N. W. Clinical Guide to Laboratory Tests, 3
rd
ed. Philadelphia, Pa: WB Saunders
Company, 1995, 268-273.
Wyszukiwarka
Podobne podstrony:
Kinetyka reakcji inwersji sacharozy
9 szybkość inwersji sacharozy
kinetyka inwersji sacharozy1
inwersja sacharozy, Farmacja, II rok farmacji, I semstr, fizyczna, Fizyczna, Sprawozdania z fizyczne
Kinetyka reakcji inwersji sacharozy, Studia, Politechnika
Kinetyka inwersji sacharozy tabela pomiariowa
INWERSJA SACHAROZY, NAUKA, WIEDZA
ćw4 - Inwersja sacharozy, studia, chemia fizyczna
Inwersja sacharozy!, Studia, Chemia fizyczna
Szybkość inwersji sacharozy
Instrukcja INWERSJA SACHAROZY
Kinetyka reakcji inwersji sacharozy
Kinetyka inwersji sacharozy
Kinetyka inwersji sacharozyM
Opracowanie wyników inwersja sacharozy
opracowanie wyników inwersja sacharozy
więcej podobnych podstron