
Choroby spowodowane defektem jednego genu
Choroby spowodowane uszkodzeniem chromosomu
tak duże że można obserwować je
pod mikroskopem
ABERACJE CHROMOSOMALNE
CYTOGENETYKA

w wielu aberacjach chromosomalnych znalezione
geny, które są odpowiedzialne za opisywane zespoły
cytogenetyczne
różnica pomiędzy aberacjami chromosomalnymi
a chorobami spowodowanymi defektem
pojedynczego geny nie jest ostra
dzięki badaniom molekularnym jesteśmy w stanie
określić od którego z rodziców pochodzi aberacja
chromosomalna

W latach 50-tych rozwinięto szereg technik, które
pozwalały na obserwowanie chromosomów.
1) użycie kolchicyny, która zatrzymuje podział
komórki somatycznej w metafazie, kiedy to
chromosomy są najbardziej skondensowane
i łatwe do obserwacji
2) użycie hipotonicznego roztworu soli, który
powodował zwiększenie objętości komórki, pęknięcie
jądra komórkowego i lepszą separację pojedynczych
chromosomów
3) technika barwienia chromosomów - prążkowanie
pozwalające na identyfikację chromosomów

Diagnostyka chromosomalna:
uzyskanie żywych komórek
hodowla komórek
(48-72 h przy limfocytach krwi obwodowej)
zatrzymanie podziału komórkowego w metafazie
(kolchicyna)
płyn hipotoniczny
barwienie, fotografia

Najbardziej powszechna technika barwienia to
prążkowanie G (Giemsa).
Prążki G są ciemne, późno ulegają replikacji
i są bogate w AT - zawierają mało genów
Jasne regiony wcześnie ulegają replikacji
i są bogate w GC - zawierają dużo genów

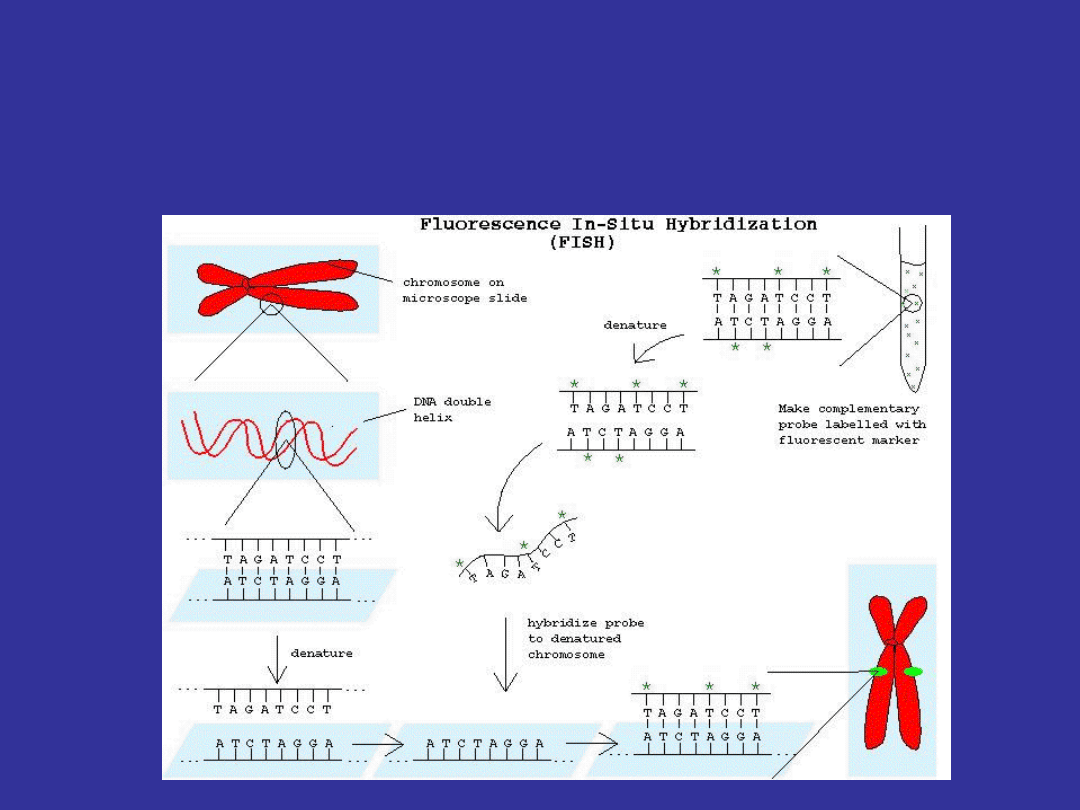
FISH
fluorescence in situ hybridization
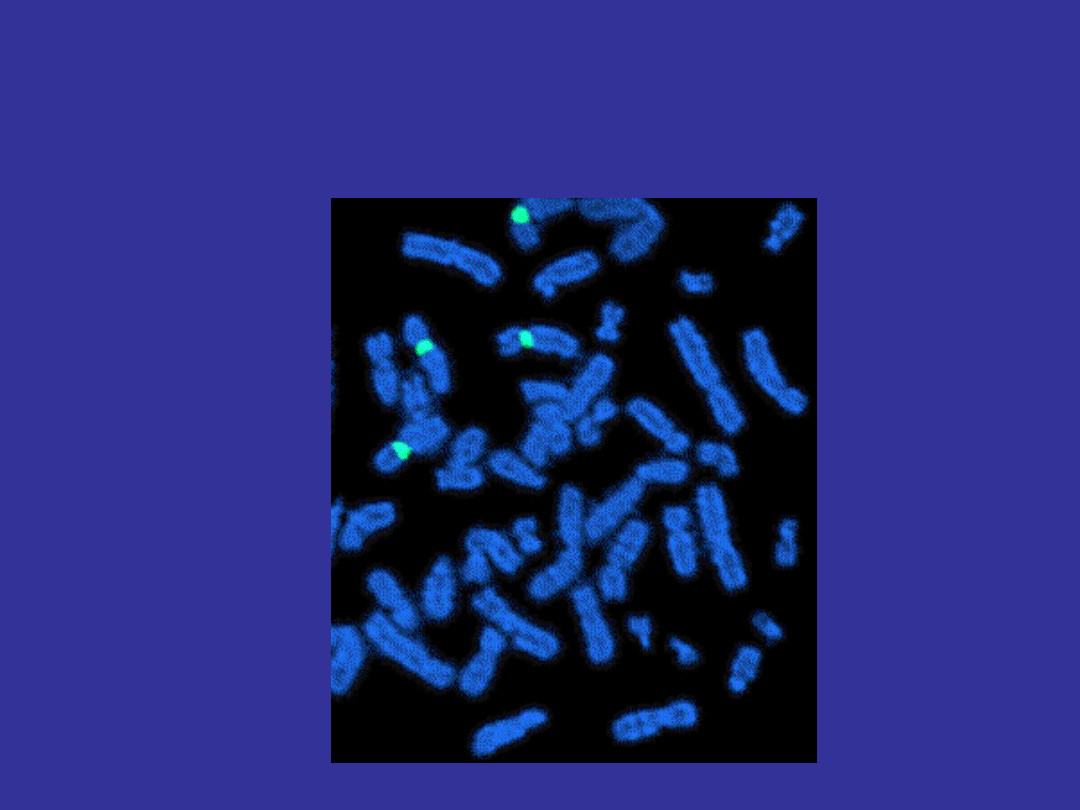
FISH
fluorescence in situ hybridization
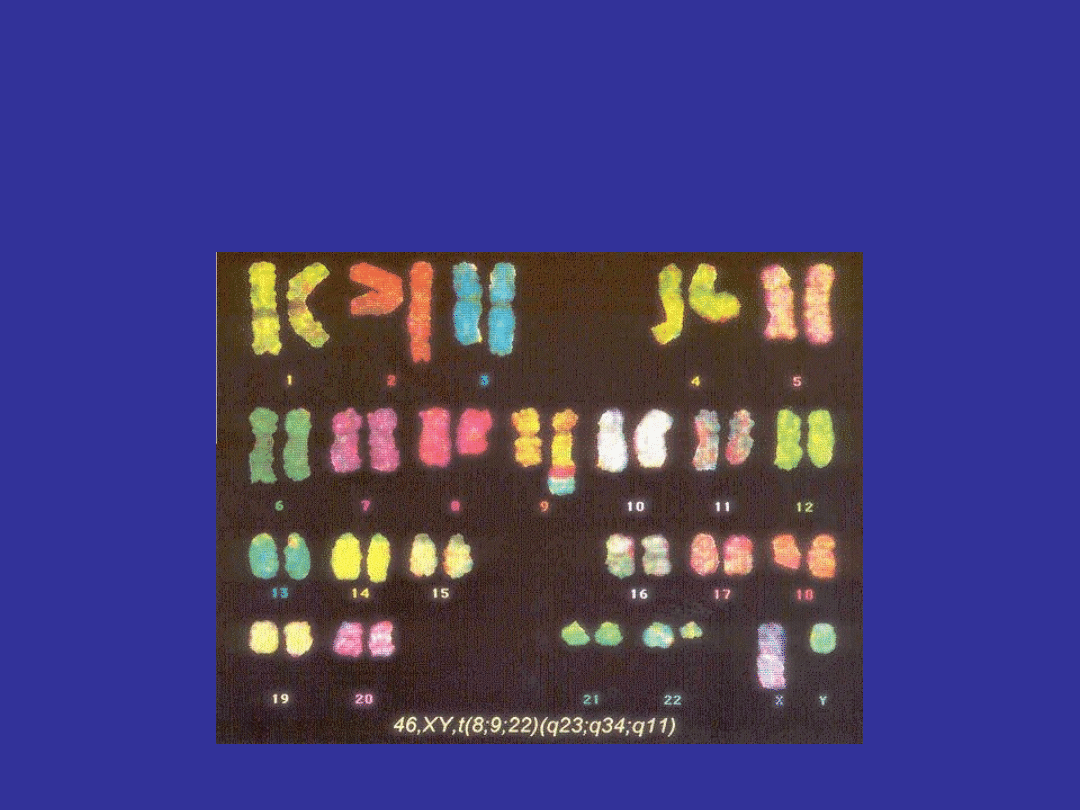
FISH
Kariotyp spektralny

ABERACJE CHROMOSOMALNE
1/150 żywych urodzeń
wiodąca znana przyczyna upośledzenia
umysłowego i poronień
50% wszystkich poronień w 1 trymestrze ciąży
20% wszystkich poronień w 2 trymestrze ciąży

ABERACJE CHROMOSOMALNE
1. ABERRACJE LICZBOWE
ANEUPLOIDIA
-dodanie lub utrata jednego
chromosomu
POLIPLOIDFIA
-dodanie całego
haploidalnego zestawu chromosomów
2. ABERACJE STRUKTURALNE
ZRÓWNOWAŻONE
-nie nastąpiła zmiana
ilości niezbędnego materiału genetycznego
NIEZRÓWNOWAŻONE
-uzyskanie lub
utrata niezbędnego fragmentu chromosomu

CZĘSTOŚĆ ABERRACJI CHROMOSOMALNYCH
1/150 narodzonych dzieci ma
nieprawidłowość chromosomalną
-aneuploidia chromosomów płci
33%
- aneuploidia autosomalna
25%
- zrównoważone aberracje strukturalne
33%
- niezrównoważone aberracje strukturalne
8%
Aberracje strukturalne występują u bezpłodnych
małżeństw w 2-4% !!!!

ABERRACJE LICZBOWE
POLIPLOIDIA
-dodanie całego haploidalnego
zestawu chromosomów
TRIPLOIDIA
69,XXX; 69,XXY; 69,XYY
DISPERMIA
- zapłodnienie jednej komórki jajowej
dwoma plemnikami

ABERRACJE LICZBOWE
Zaburzenia podziału mejotycznego komórek
DIANDRIA
- zapłodnienie haploidalnego jaja
przez diploidalny plemnik
DIGYNIA
- zapłodnienie diploidalnego jaja
przez haploidalny plemnik
TETRAPLOIDIA
92, XXXX; 92,XXYY
brak pierwszego podziału zygoty
-poronienie samoistne
-ryzyko powtórzeń nie jest zwiększone !!!

ABERRACJE LICZBOWE
ANEUPLOIDIA
-dodanie lub utrata jednego chromosomu
TRISOMIA
Obecność trzech zamiast dwóch prawidłowych kopii
chromosomu 47,XX,+21
MONOSOMIA
Obecność tylko jednego z dwóch chromosomów
danej pary w kariotypie 45,XX,-13

ABERRACJE LICZBOWE
ANEUPLOIDIA MOZAIKOWA
47,XX,+21/46,XX
-nierozejście lub opóźnienia chromosomu na etapie
bruzdkowania lub we wczesnym okresie rozwoju
embrionalnego
-objawy kliniczne zależą od proporcji komórek linii
prawidłowej i aneuploidalnej

TRISOMIA AUTOSOMALNA
-poronienie we wczesnym okresie ciąży
-wśród noworodków żywo urodzonych tylko trzy
trisomie chromosomów autosomalnych
47,XX,+21 (Zespół Downa)
47,XX,+18 (Zespół Edwardsa)
47,XX,+13 (zespół Pataua)

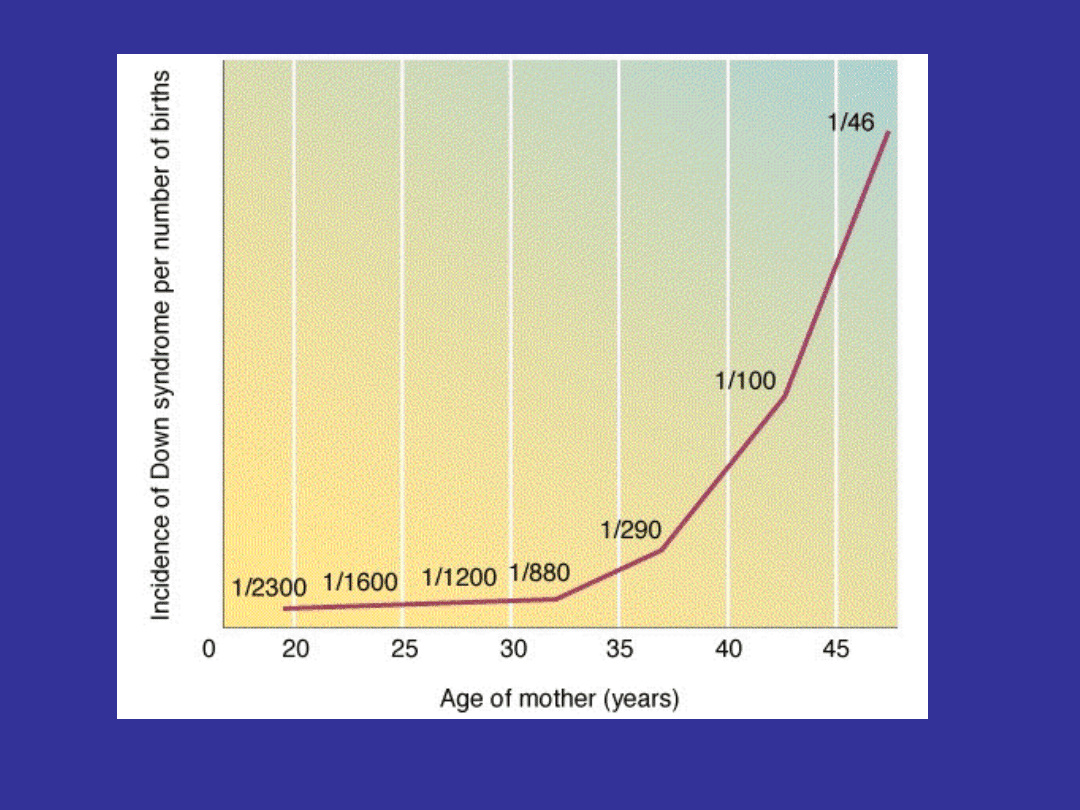
TRISOMIA AUTOSOMALNA
Częstość trisomii na 100,000 zapłodnień
Zapłodnienia
SamoistneŻywe
Poronienia
Porody
Trisomia 21
450 (~1/200)
345 (77%) 105(~1/800)
Trisomia 18
200 (~1/500)
190 (95%) 10 (~1/8500)
Trisomia 13
195 (~1/500)
190 (97,5%)
5 (~1/17000)
Łącznie
845
725 (85%) 120 (~1/700)

TRISOMIA 21 – ZESPÓŁ DOWNA
Cechy zespołu Downa u noworodków
Niskie ciśnienie tętnicze;
Twarz z płaską nasadą nosa;
Zmarszczki nakątne;
Duży język;
Małe uszy;
Płaska potylica
Wady wrodzone serca !!!
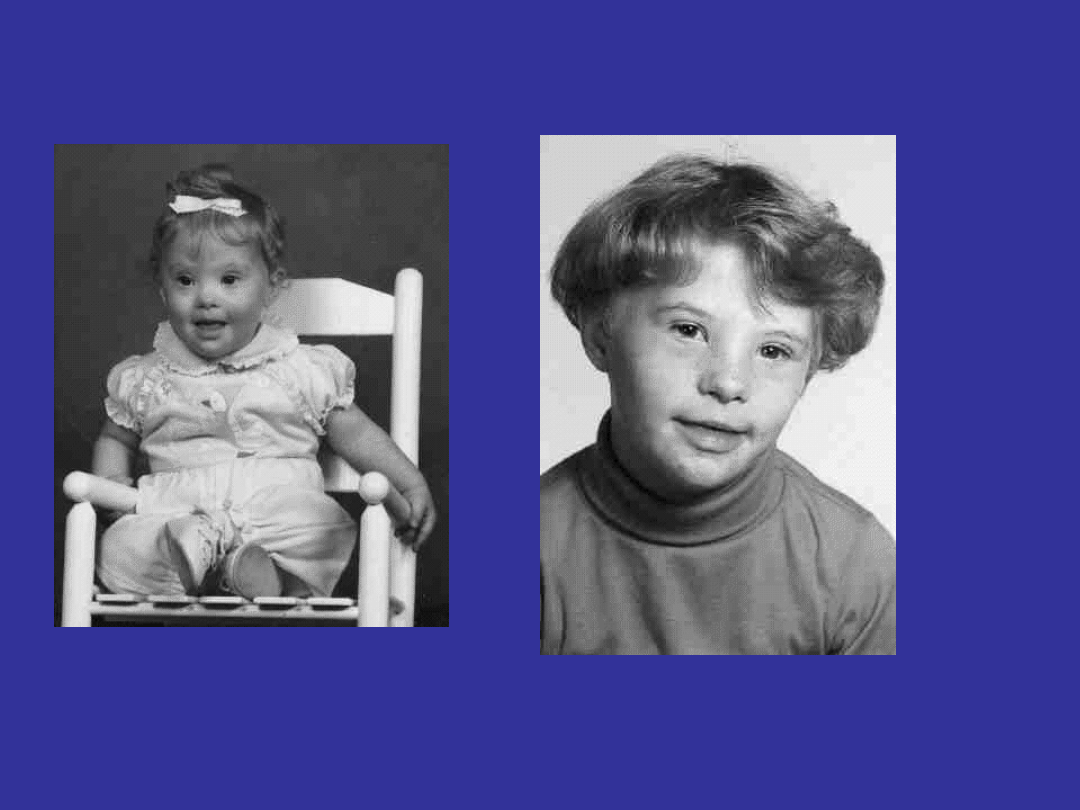
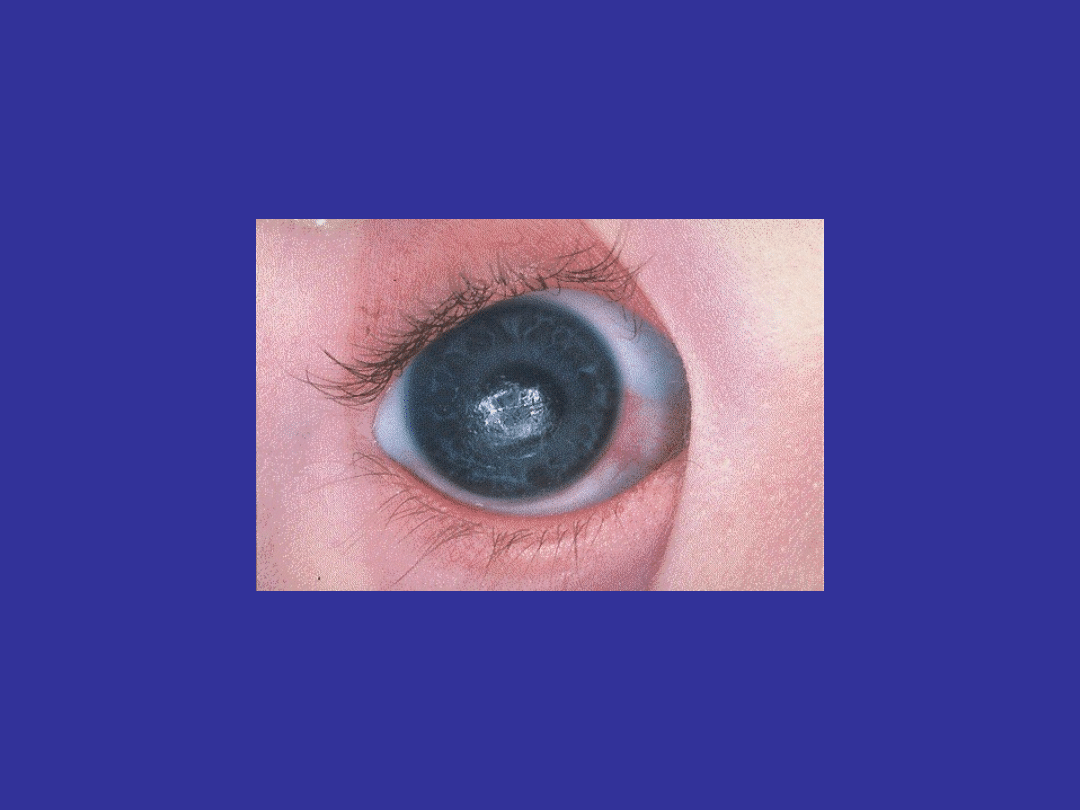
Zmarszczki nakątne;

TRISOMIA 21 – ZESPÓŁ DOWNA
Rozwój
85% noworodków umiera w pierwszym roku życia
z powodu wrodzonych wad serca;
Opóźnienie rozwoju umysłowego
(25% wszystkich przypadków upośledzenia
umysłowego w stopniu umiarkowanym
i znacznym u dzieci w wieku szkolnym);

TRISOMIA 21 – ZESPÓŁ DOWNA
Rozwój
Niski wzrost;
Otyłość w wieku dziecięcym;
Niedosłuch;
Choroby oczu;
Nieprawidłowości układu odpornościowego;
Często białaczki;
Po 40 roku życia dochodzi do wczesnego otępienia
starczego;
W wieku 60 lat 75% ma objawy choroby Alzheimera;

TRISOMIA 21 – ZESPÓŁ DOWNA
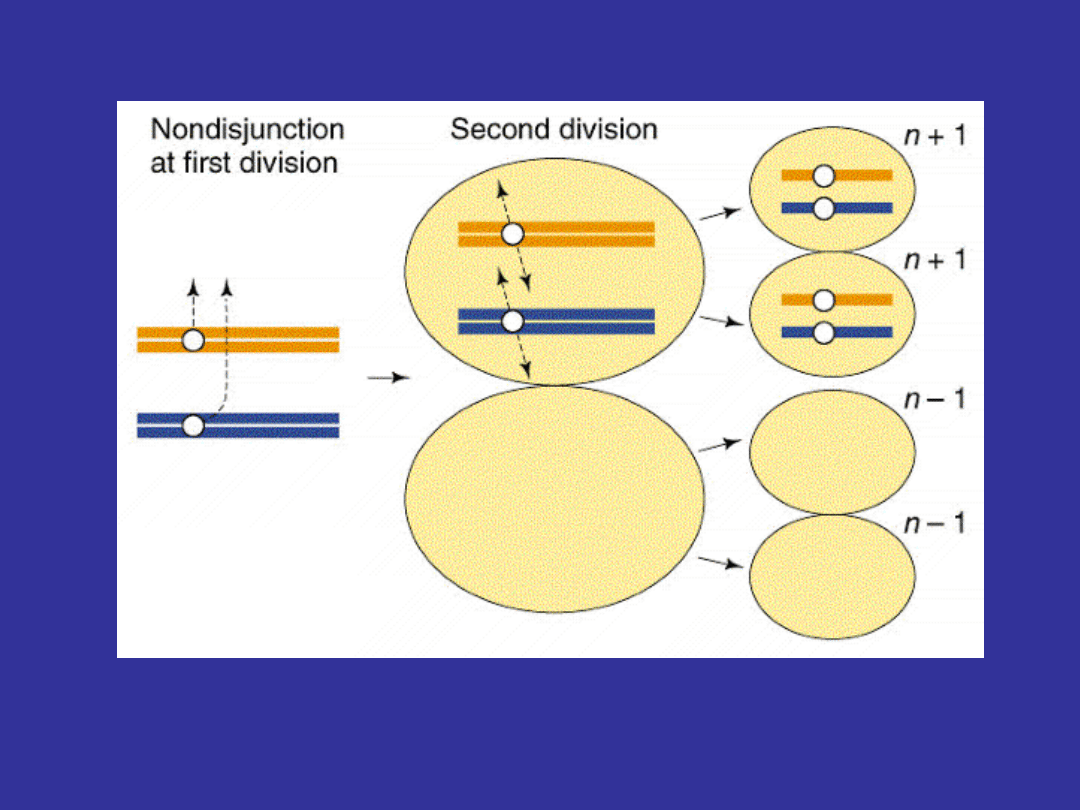
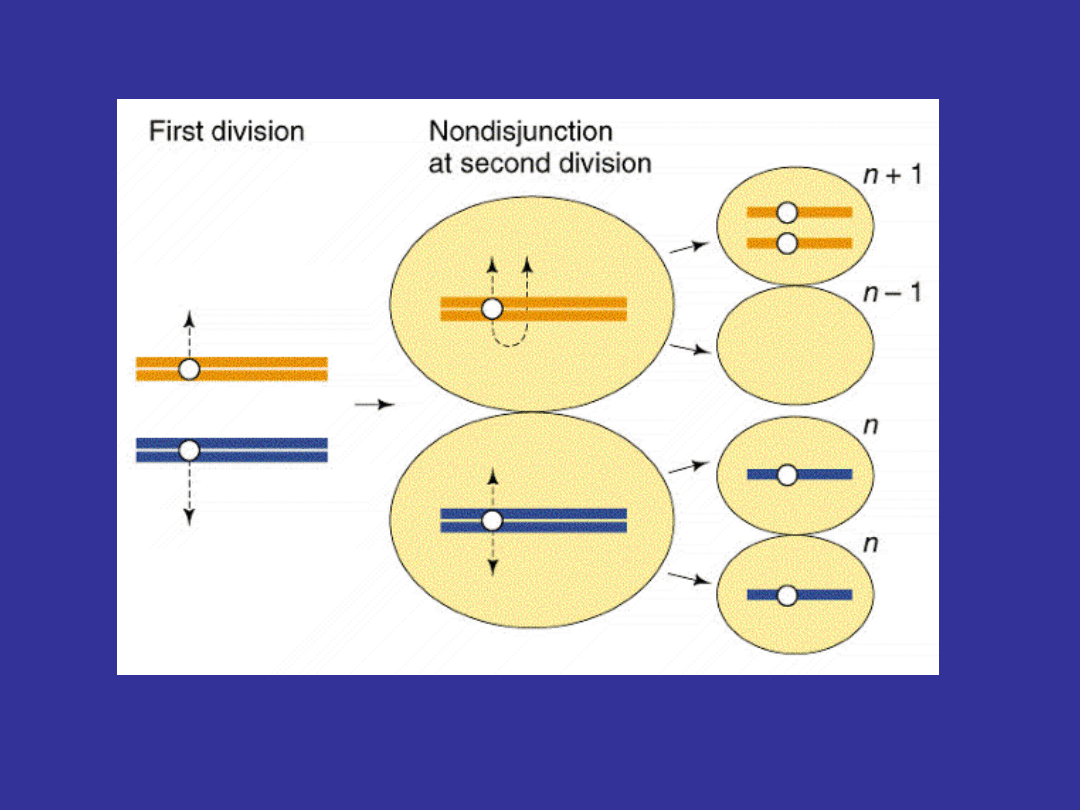
Etiologia
90%
- brak rozdziału chromatyd (nondisjunction)
zazwyczaj podczas pierwszego podziału
mejotycznego oogenezy
5%
- brak rozdziału chromatyd podczas
spermatogenezy
4%
-
jest wynikiem translokacji robertsonowskiej
(częsta przyczyna występującego rodzinnie
zespołu Downa)

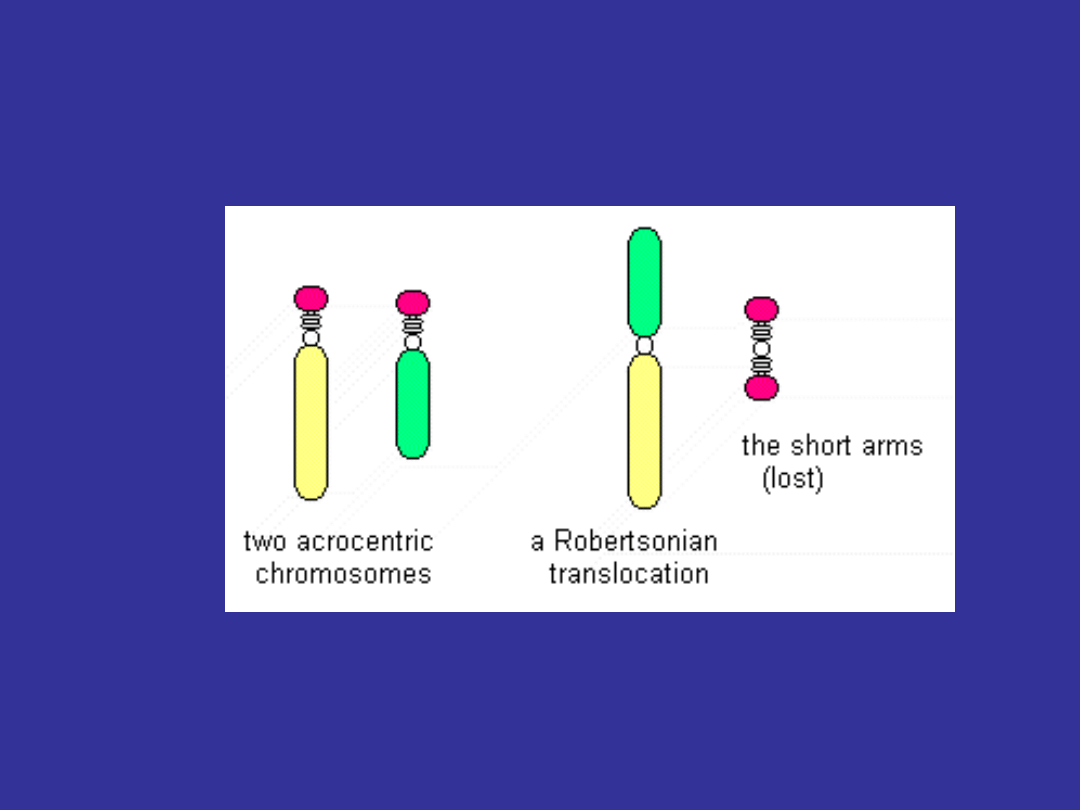
TRANSLOKACJA ROBERTSONOWSKA
Dwa akrocentryczne chromosomy
(13, 14, 15, 21, 22) łączą się ze sobą blisko
centromeru i tracą dwa krótkie ramiona
Nosiciele zrównoważonej translokacji
robertsonowskiej są fenotypowo normalni
z 45 chromosomami lecz charakteryzują się oni
zwiększonym ryzykiem posiadania dziecka
z niezrównoważonym chromosomem
45,XX, der14;21(q10;q10) – prawidłowy fenotyp
46,XX, der14;21(q10;q10), +21 (zespół Downa)
46,XY, der14;21(q10;q10), +21 (zespól Downa)

TRISOMIA 18 – ZESPÓŁ EDWARDSA
~1 /8500 żywych urodzeń
95% samoistne poronienia
częstość ma związek z wiekiem matki
Etiologia
- dodatkowy chromosom 18
- 90% pochodzenia matczynego jako wynik braku
rozdziału chromatyd (nondisjunction) podczas
drugiego podziału mejotycznego oogenezy
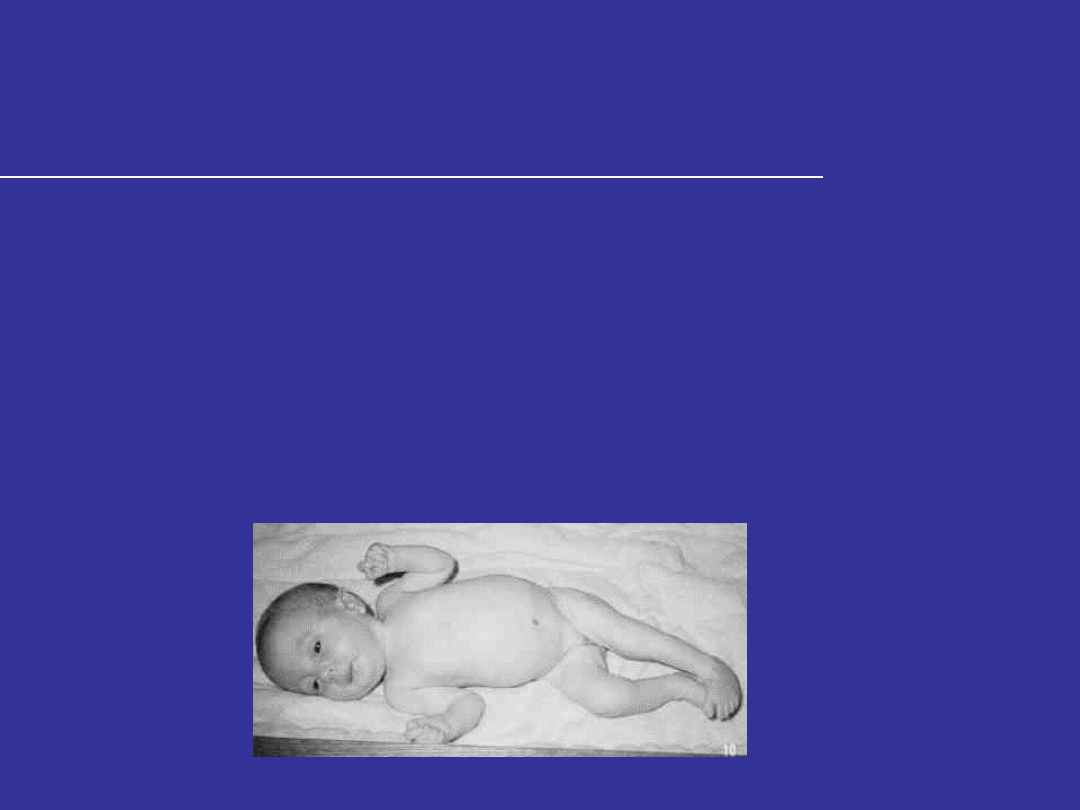
TRISOMIA 18 – ZESPÓŁ EDWARDSA
Cechy zespołu Edwardsa u noworodków
Niska masa ciała; liczne cechy dysmorfii;
charakterystyczny kształt czaszki- mała bródka,
wypukła potylica, nisko osadzone, zniekształcone
małżowiny uszne;
Krótki mostek;
Stopy o łukowato wygiętych podeszwach;
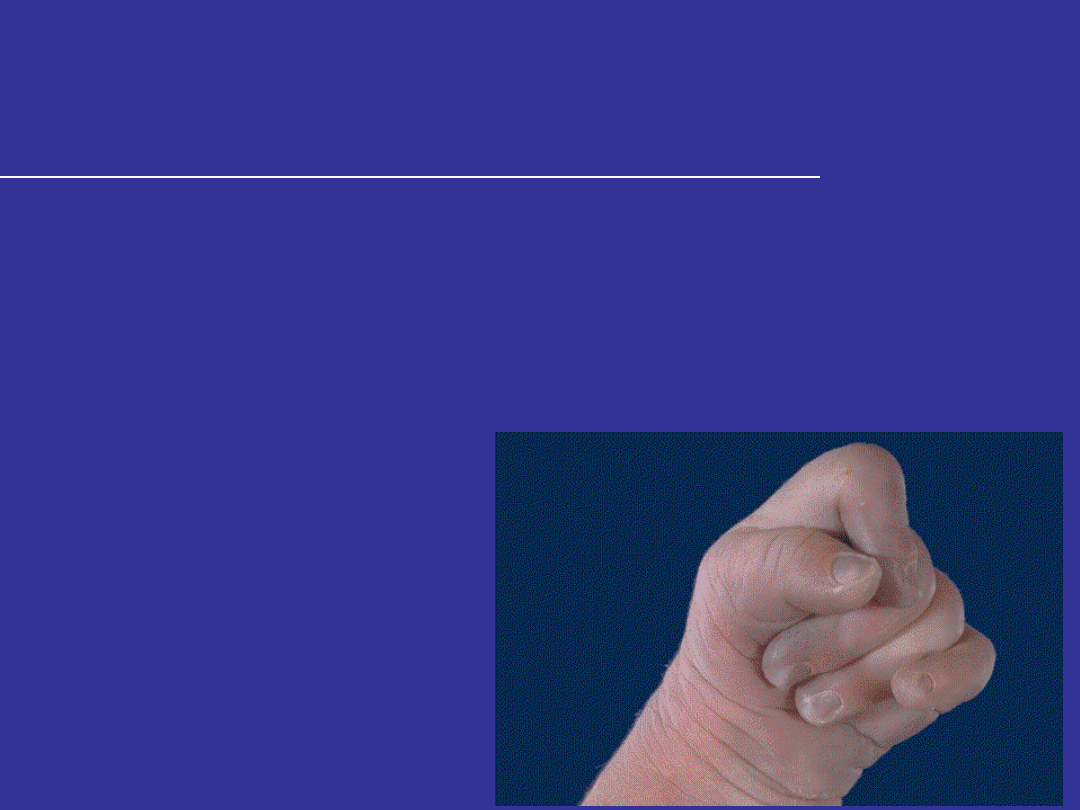
TRISOMIA 18 – ZESPÓŁ EDWARDSA
Cechy zespołu Edwardsa u noworodków
Zaciśnięte w pięści dłonie z zachodzącymi na siebie
palcami wskazującym i piątym;
Pojedyncze bruzdy zgięcia na dłoniach;
Częste wady rozwojowe serca i nerek;
Tylko 5% przeżywa 1 rok życia – głębokie
upośledzenie rozwoju;


TRISOMIA 13 – ZESPÓŁ PATAUA
~1/17000 żywych urodzeń
97,5% samoistne poronienia
częstość ma związek z wiekiem matki
Etiologia
- dodatkowy chromosom 18
- 65% pochodzenia matczynego jako wynik braku
rozdziału chromatyd (nondisjunction) podczas
pierwszego podziału mejotycznego oogenezy
- 20% jedno z rodziców jest nosicielem translokacji
zrównoważonej
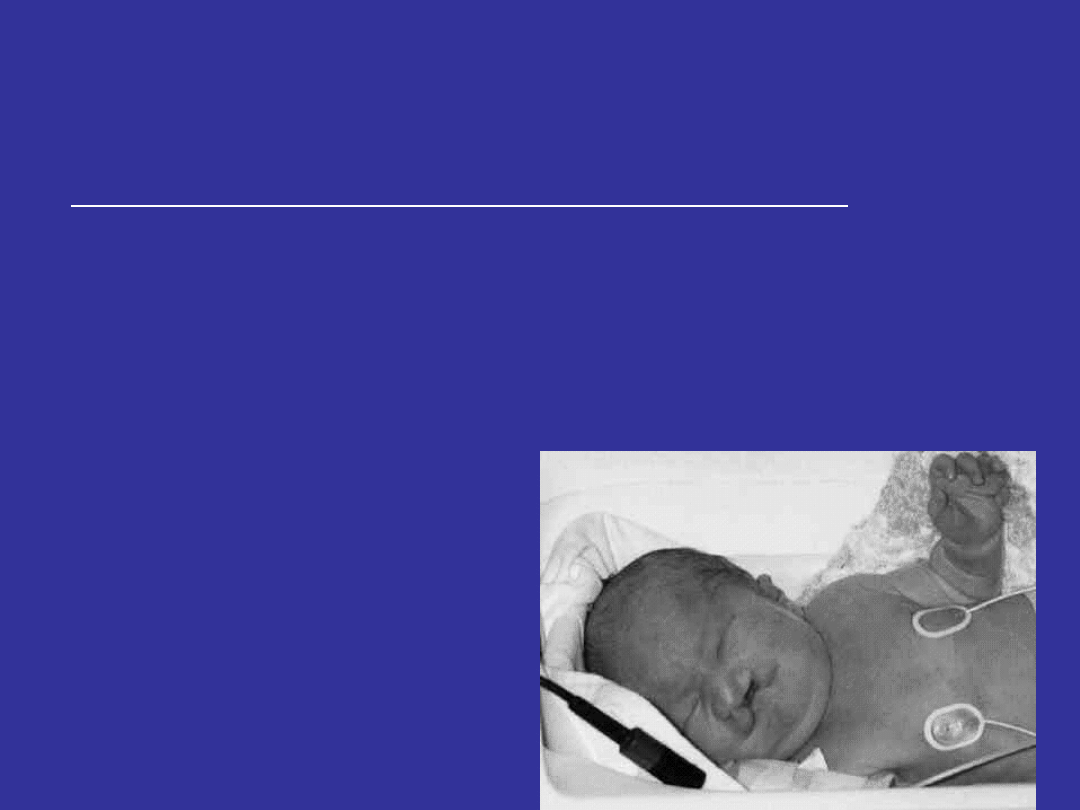
TRISOMIA 13 – ZESPÓŁ PATAUA
Cechy zespołu Pataua u noworodków
Liczne cechy dysmorfii;
Małe oczy; rozszczep wargi i podniebienia;
Nieprawidłowe małżowiny uszne;
Polidaktylia (od strony małego palca);
Wystające pięty;
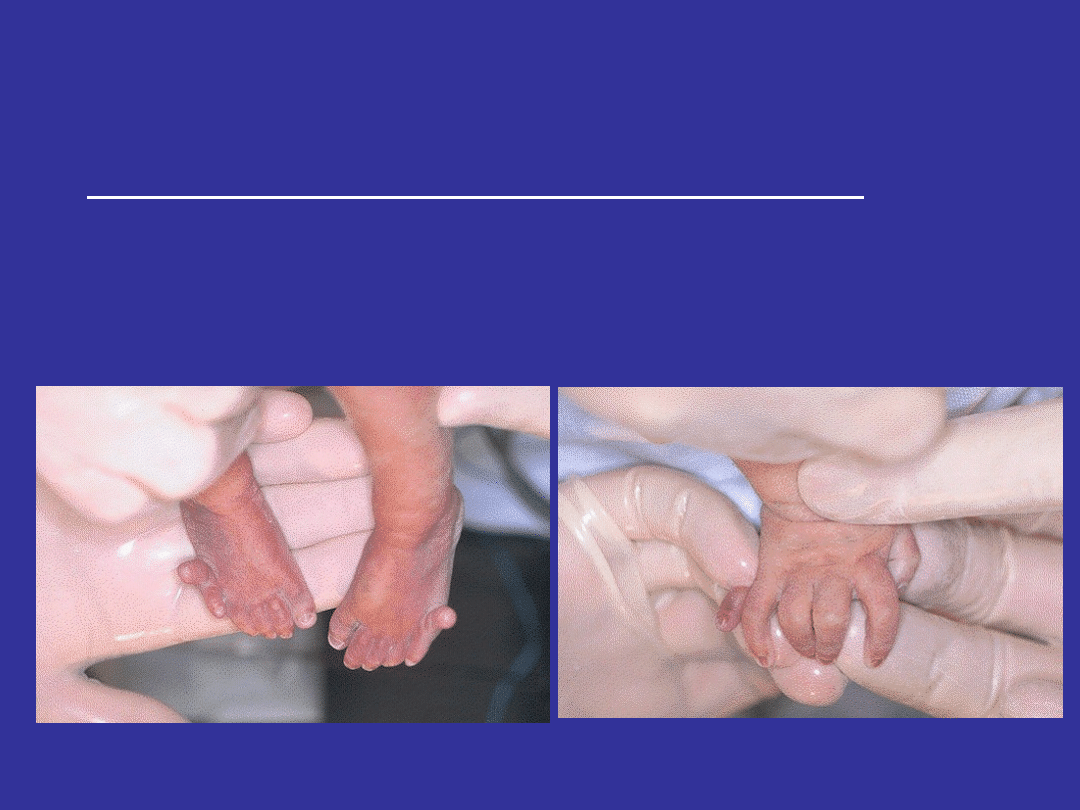
TRISOMIA 13 – ZESPÓŁ PATAUA
Cechy zespołu Pataua u noworodków
Polidaktylia (od strony małego palca);
Wystające pięty;


MONOSOMIA AUTOSOMALNE
- monosomie chromosomów autosomalnych są
letalne i nie są obserwowane wśród żywo
urodzonych dzieci !!!

DISOMIA JEDNORODZICIELSKA
(uniparental disomy)
- w diploidalnej linii komórkowej oba chromosomy
danej pary pochodzą tylko od jednego z rodziców.
- piętnowanie rodzicielskie (imprinting)
- homozygotyczność zmutowanego allelu
Jeżeli brakuje chromosomu 15 od ojca dziecko
rozwija
ZESPÓŁ PRADERA-WILLEGO
Jeżeli brakuje chromosomu 15 od matki dziecko
rozwija
ZESPÓŁ ANGELMANA
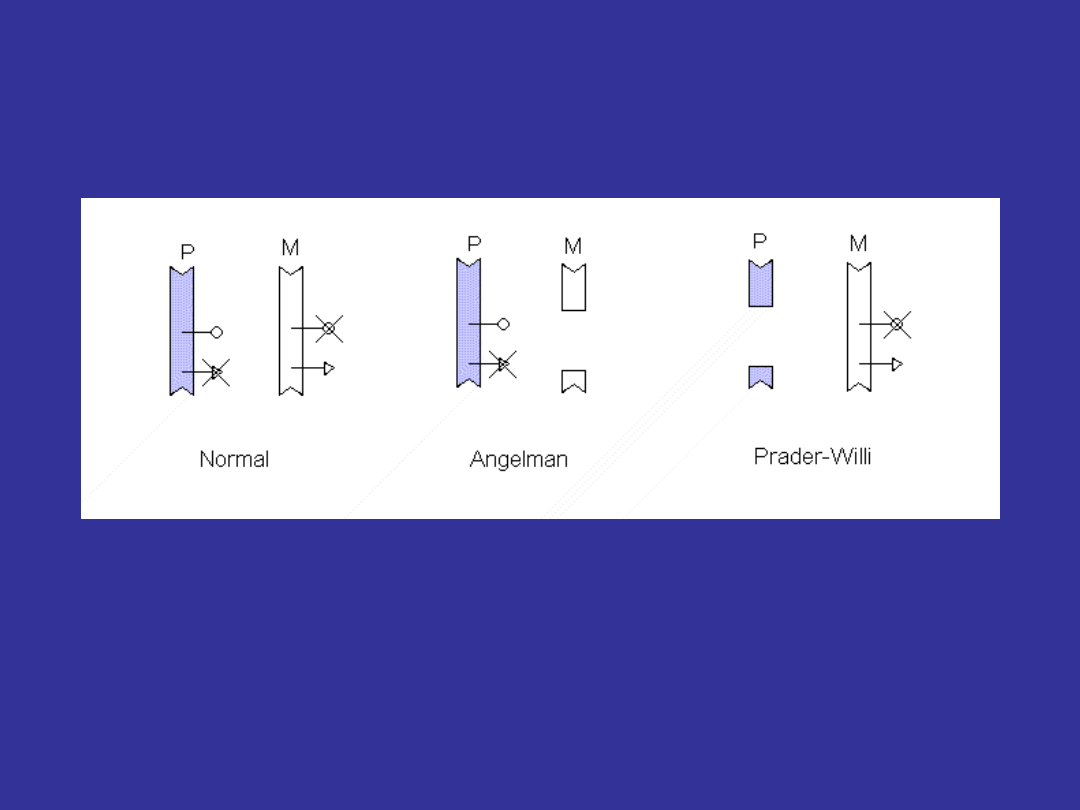

DISOMIA JEDNORODZICIELSKA
(uniparental disomy)
Często PWS lub AS są wynikiem małej delecji
chromosomu 15(q11-q13)
ZESPÓŁ PRADERA-WILLIEGO
Nadmierne łaknienie; otyłość po 2 roku życia;
upośledzenie rozwoju umysłowego;
zaburzenia zachowania;

DISOMIA JEDNORODZICIELSKA
(uniparental disomy)
Często PWS lub AS są wynikiem małej delecji
chromosomu 15(q11-q13)
ZESPÓŁ ANGELMANA (happy puppet syndrome)
Opóźnienie rozwoju; bardzo słaby rozwój mowy;
Gwałtowne ruchy; napady śmiechu; dysmorfia
twarzy; padaczka;

ABERACJE STRUKTURALNE
DELECJE
-terminalne 46,XY,del(5)(p15)
-interstycjalne 46,XX,del(2)(q31q33)
-chromosomy pierścieniowe 46,XX,r(7)(p21q34)
IZOCHROMOSOMY
45,XX,der(21;21)(q10;q10)
46,X,i(X)(q10)

Pierwsza opisana
delecja autosomalna
zespół „kociego krzyku”

ABERACJE STRUKTURALNE
INWERSJE
-pericentryczne 46,XX,inv(6)(p12q23)
-paracentryczne 46,XY,inv(8)(q22q24)
TRANSLOKACJE
-wzajemna 46,XX,t(1;12)(p35;p12)
-robertsonowska 45,XX,der(14;21)(q10;q10)
ZESPOŁY MIKRODELECJI

CHROMOSOMY PŁCIOWE
chromosom X
tylko jeden z chromosomów X jest aktywny
drugi jest nieaktywny i jest rozpoznawany jako
ciałko Barra
chromosom Y
może różnić się wielkością
zawiera niewielką ilość genów
posiada region pseudoautosomalny

ABERACJE STRUKTURALNE
I LICZBOWE CHROMOSOMÓW PŁCI
PŁĘĆKARIOTYP
ZESPÓŁ
ŻEŃSKA
45,X
Zespół Turnera
46,X,i(X)(q10) Zespół Turnera
46,XX,p-
Zespół Turnera
47,XXX
Trisomia X
prawidłowy fenotyp, wysokie kobiety
46,XY
Odwrócenie płci
mutacje w genie SRY
mutacja w genie dla receptora androgenu

ABERACJE STRUKTURALNE
I LICZBOWE CHROMOSOMÓW PŁCI
PŁĘĆKARIOTYP
ZESPÓŁ
MĘSKA
47,XXY
Zespół Klinefeltera
47,XYY
Stan Prawidłowy
46,XX
Odwrócenie płci
translokacja genu SRY na chromosomie X)

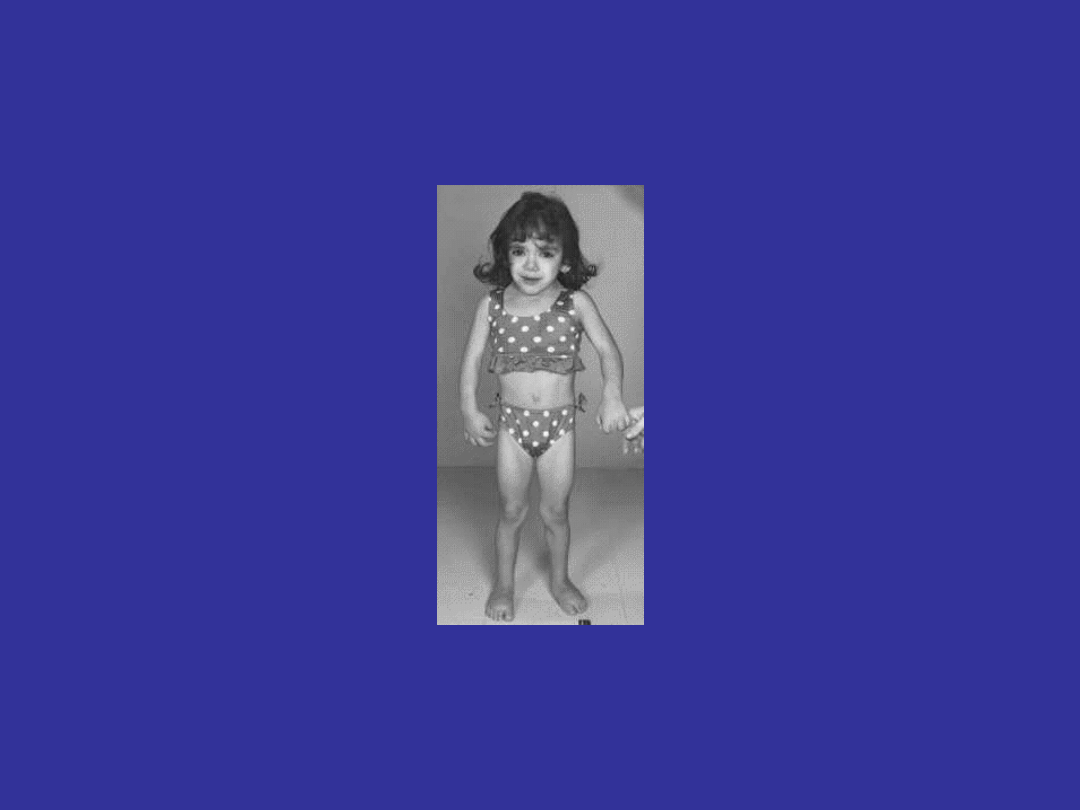
ZESPÓŁ TURNERA
95% samoistne poronienie
~1/2500 żywo urodzonych kobiet
50%
45,X (65% chromosom X od matki)
17%
46,X,i(X)(q10)
24%
46,XX/45,X
7%
chromosom pierścieniowy
2%
46,XX,p-
niski wzrost; opóźnione dojrzewanie płciowe;
bezpłodność;


ZESPÓŁ KINEFELTERA
47,XXY
15% 46,XY/47,XXY
~1/1000 żywo urodzonych chłopców
dodatkowy chromosom X pochodzi od matki (56%)
lub ojca (44%);
wysoki wzrost, małe jądra; bezpłodność; słaby
rozwój wtórnych cech płciowych, ginekomastia;


ABERACJE CHROMOSOMALNE
W KOMÓRKACH SOMATYCZNYCH
odpowiedzialne za niektóre nowotwory
pierwszą rozpoznaną było aberacja odpowiedzialna
za przewlekłą białaczkę szpikową (CML)
wzajemna translokacja pomiędzy chromosomem 9
a chromosomem 22
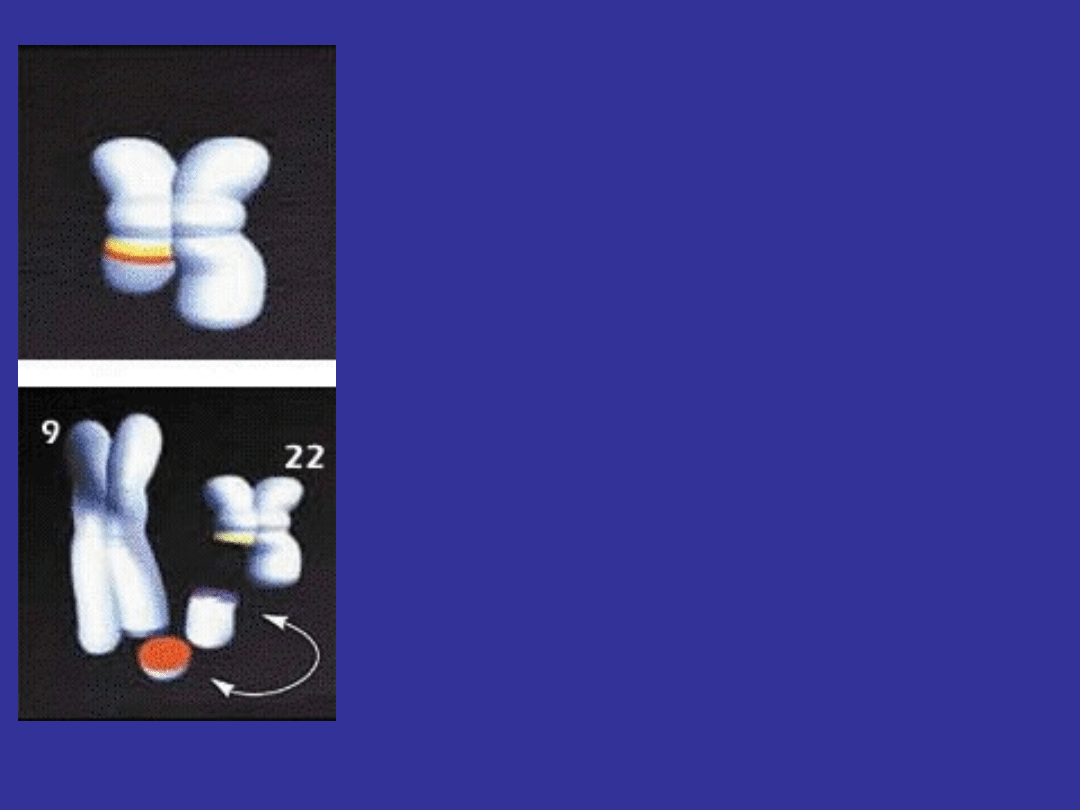
CHROMOSOM FILADELFIA
przewlekła białaczka szpikowa (CML)
protoonkogen abl zostaje
przemieszczony z normalnego
położenia na chromosomie 9q
na chromosome 22
powoduje to że powstałe białko
wykazuje zwiększoną aktywność
kinazy tyrozynowej co powoduje
rozrost nowotworowy

WSKAZANIA DO PRZEPROWADZENIA
ANALIZY CHROMOSOMALNEJ
Osoby u których podejrzewa się znany zespół
chromosomalny (np. Zespół Downa)
Osoby z kilkoma pierwotnymi wadami rozwojowymi
Osoby z nieprawidłowym rozwojem narządów
płciowych
Osoby z nieprawidłowym rozwojem umysłowym wraz
z nieprawidłowościami fizykalnymi

WSKAZANIA DO PRZEPROWADZENIA
ANALIZY CHROMOSOMALNEJ
Rodzice i dzieci osób z:
translokacją
delecją
powtórzeniem

WSKAZANIA DO PRZEPROWADZENIA
ANALIZY CHROMOSOMALNEJ
Płód martwo urodzony
z wadami rozwojowymi
z nierozpoznanym przyczyną zgonu
Osoby płci żeńskiej
z niskim wzrostem i brakiem miesiączek
Osoby płci męskiej
niedorozwojem jąder lub ginekomastią
podejrzenie łamliwego chromosomu X

OPIS KARIOTYPU
46,XY
prawidłowy kariotyp męski
47,XX,+21
kobieta z trisomią 21, Zespół Downa
47/XY,+21/46,XY
mężczyzna który jest mozaiką składającą się
z komórek z trisomią 21 i normalnych komórek
46,XY,del(4)(p14)
mężczyzna z dystalną delecją krótkiego
ramienia chromosomu 4 od prążka p14
46,XX,dup(5p)
kobieta z duplikacją krótkiego ramienia
chromosomu 5

OPIS KARIOTYPU
45,XY,-13,-14,t(13q;14q)
mężczyzna ze zrównoważoną tranlokacją
robertsonowską chromosomów 13 i 14; kariotyp
pokazuje, że brakuje jednego normalnego
chromosomu 13 i jednego normalnego
chromosomu 14
46,XY,t(11,22)(q23;q22)
mężczyzna ze zrównoważoną wzajemną
translokacją pomiędzy chromosomem 11 a 22;
punkty pęknięcia chromosomów są w pozycji
11q23 oraz 22q22

OPIS KARIOTYPU
46,XX,inv(3)(p21;q13)
kobieta z inwersją chromosomu 3, która
rozciąga się od p21 do q13, ponieważ zawiera
ona centromer jest to inwersja perycentryczna
46,X,i(Xq)
kobieta z normalnym chromosomem X
oraz izochromosomem X składającym się
z długich ramion (zespół Turnera)

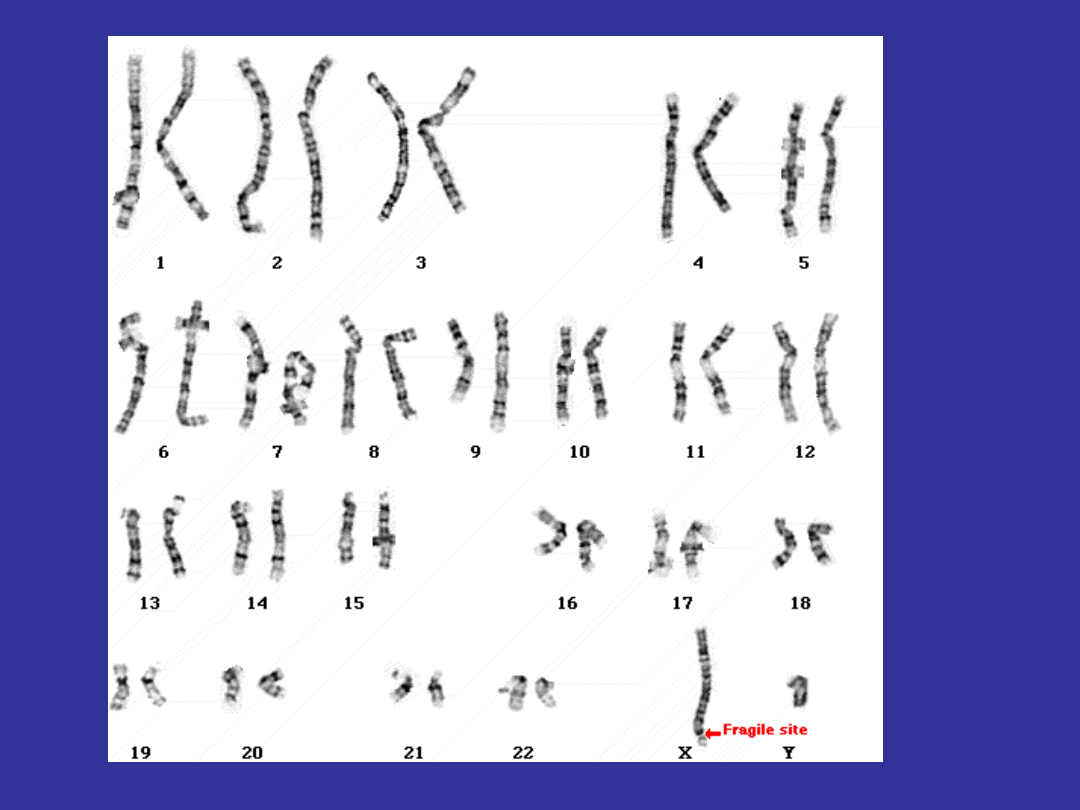
ZESPÓŁ ŁAMLIWEGO CHROMOSOMU X
Od 19 wieku obserwowano około 25% częściej
mężczyzn w zakładach dla umysłowo chorych
przyczyną tego jest zespół łamliwego chromosomu X
obserwowany u 1/4.000 mężczyzn i 1/8.000 kobiet
najczęstsza przyczyna wrodzonego upośledzenia
umysłowego
specyficzny wygląd twarzy z dużymi uszami i
podłużną twarzą
nadmierną elastyczność stawów,
zwiększona objętość jąder
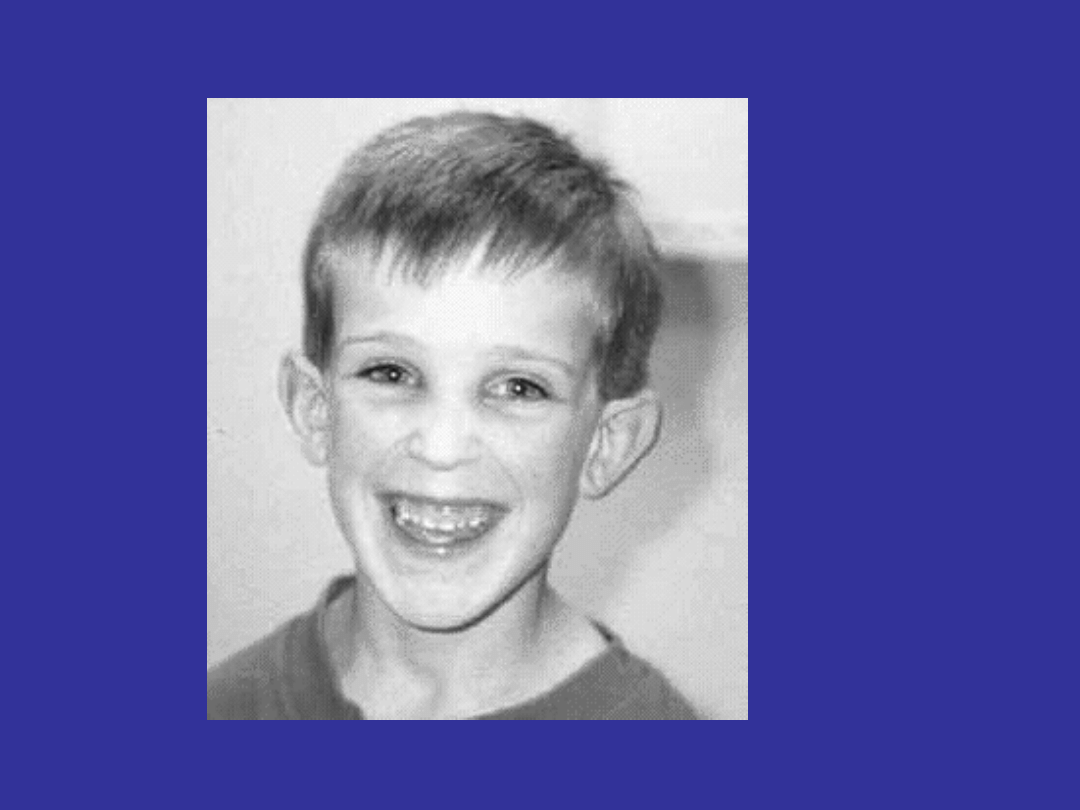

ZESPÓŁ ŁAMLIWEGO CHROMOSOMU X
u kobiet objawy są słabsze
nazwa łamliwy chromosom pochodzi z obserwacji,
że komórki od osoby z tym zespołem kiedy
hodowane są na pożywce bez kwasu foliowego
wykazują złamania i uszkodzenia na końcu długiego
ramienia chromosomu X
zespół ten dziedziczy się w sposób autosomalnie
dominujący sprzężony z chromosomem X
penetracja wynosi 80% u osób płci męskiej
i 30% u osób płci żeńskiej

ZESPÓŁ ŁAMLIWEGO CHROMOSOMU X
obserwuje się tutaj antycypację genetyczną
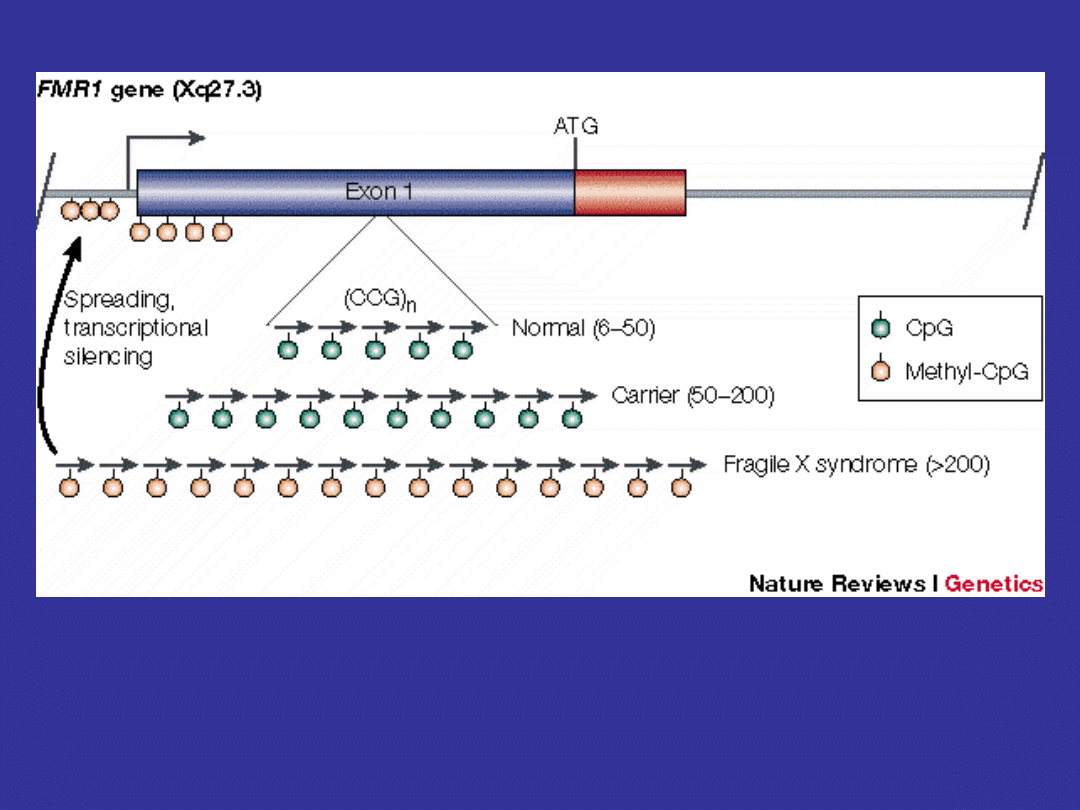
gen odpowiedzialny za tą chorobę to FMR1
38 kb, 17 egzonów, 4,8 kb mRNA
5’UTR zawiera powtórzenia CGG 6-50 kopii
u osoby zdrowej
osoby z zespołem łamliwego chromosomu mają 230
do 1000 i więcej takich powtórzeń

ZESPÓŁ ŁAMLIWEGO CHROMOSOMU X
osoby których dzieci mają ten zespół mają od 50
do 230 powtórzeń
jeżeli kobiety przekazują ten gen do swojego dziecka
następuje powiększenie liczby powtórzeń z 50-230
do ponad 230
to powiększenie nie ma miejsca jeżeli mężczyzna
przekazuje ten gen
przy dużej liczbie powtórzeń nie ma ekspresji
genuFMR1
powtórzenia CGG są silnie metylowane u chorych
z pełną mutacją
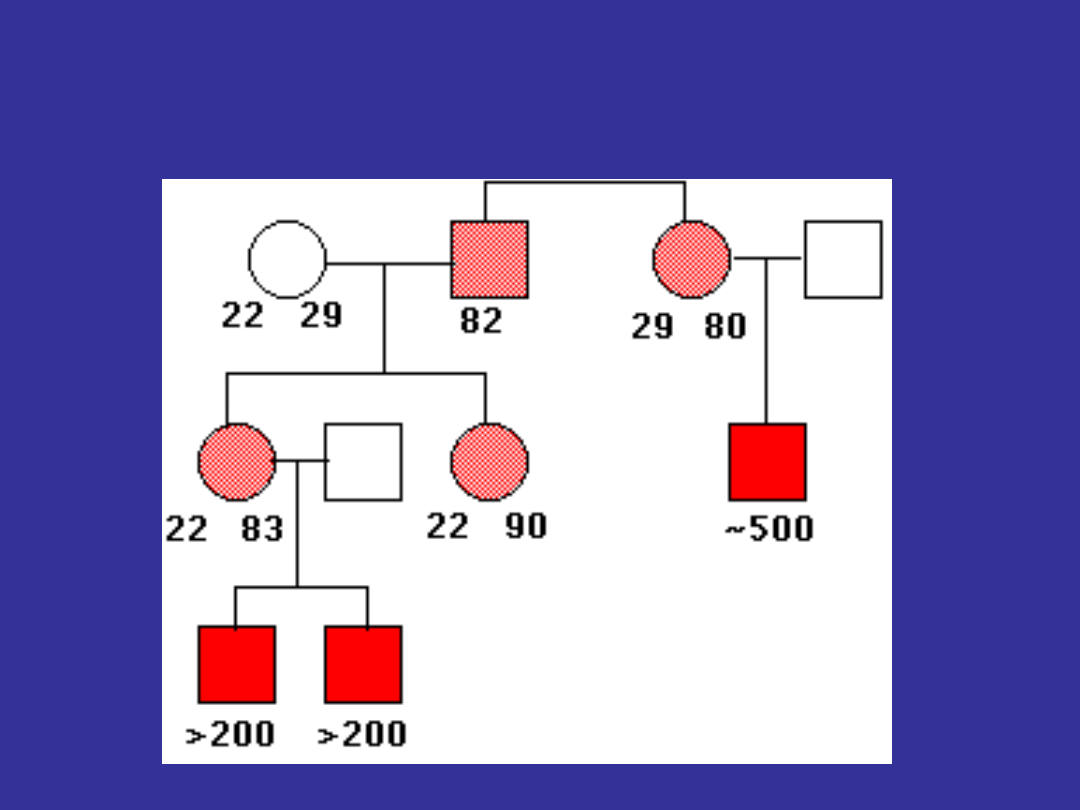
Sherman paradox
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
Wyszukiwarka
Podobne podstrony:
Choroby spowodowane sposobem wykonywania pracy, Ergonomia
Choroby spowodowane niewłaściwą dietą
Choroby spowodowane piciem alkoholu
Choroby spowodowane piciem alkoholu, PSYCHOLOGIA DOKUMENTY
CHOROBY SPOWODOWANE MUTACJAMI GENOWYMI
Rozwiazanie stosunku pracy z powodu czasowej niezdolnosci do pracy spowodowanej choroba, kadry-i-awa
DEFEKTY I ZMIANY CHOROBOWE KOŃCZYNY GÓRNEJ, Kosmetologia
Odkryto mechanizm jednego z najdziwniejszych defektów mózgu
Paznokcie choroby i defekty, Kosmetologia
16 Stany nagłe spowodowane chorobami wewnętrznymiid 16818 ppt
8 Badanie wpływu genu na występowanie choroby
DEFEKTY I ZMIANY CHOROBOWE KOŃCZYNY GÓRNEJ, kosmetologia
Choroba psychiczna jednego z rodziców negatywnie wpływa na rozwój psychiczny dziecka, rozwój dziecka
DEFEKTY I CHOROBY PŁYTKI PAZNOKCIOWEJ
Choroby genetyczne spowodowane aberacjami chromosomowymi 2
więcej podobnych podstron