
ćwiczenie 5
Poradnictwo
genetyczne w
chorobach
dziedziczonych
autosomalnie
dominująco

Do poradni genetycznej zgłosili się rodzice z 7-letnim chłopcem, u
którego lekarz okulista stwierdził obecność charakterystycznych
guzków Lischa oraz - pojawiających się od kilku lat na skórze całego
ciała - plam w kolorze kawy z mlekiem. Badanie przedmiotowe
dziecka wykazało 6 plam o średnicy powyżej 15 mm oraz kilka
mniejszych, pojedynczego naczyniaka, piegi w okolicy pachowej,
guzki Lischa oraz skrzywienie kręgosłupa. W wywiadzie rodzinnym
podobne zmiany skórne występują także u matki (badanie fizykalne
wykazało 5 plam "cafe au lait" o średnicy powyżej 15 mm oraz
kilkanaście mniejszych na skórze całego ciała, sinawe guzki
podskórne uwypuklające się ponad powierzchnię skóry), u ojca
stwierdzono kilka plam w kolorze kawy z mlekiem, ale żadna nie
miała średnicy większej od 15 mm. Siostra matki zmarła w wieku
dziecięcym z powodu guza mózgu, brat matki jest niewidomy,
ojciec matki pacjentki poza zmianami skórnymi cierpi z powodu
głuchoty.
Opis przypadku:
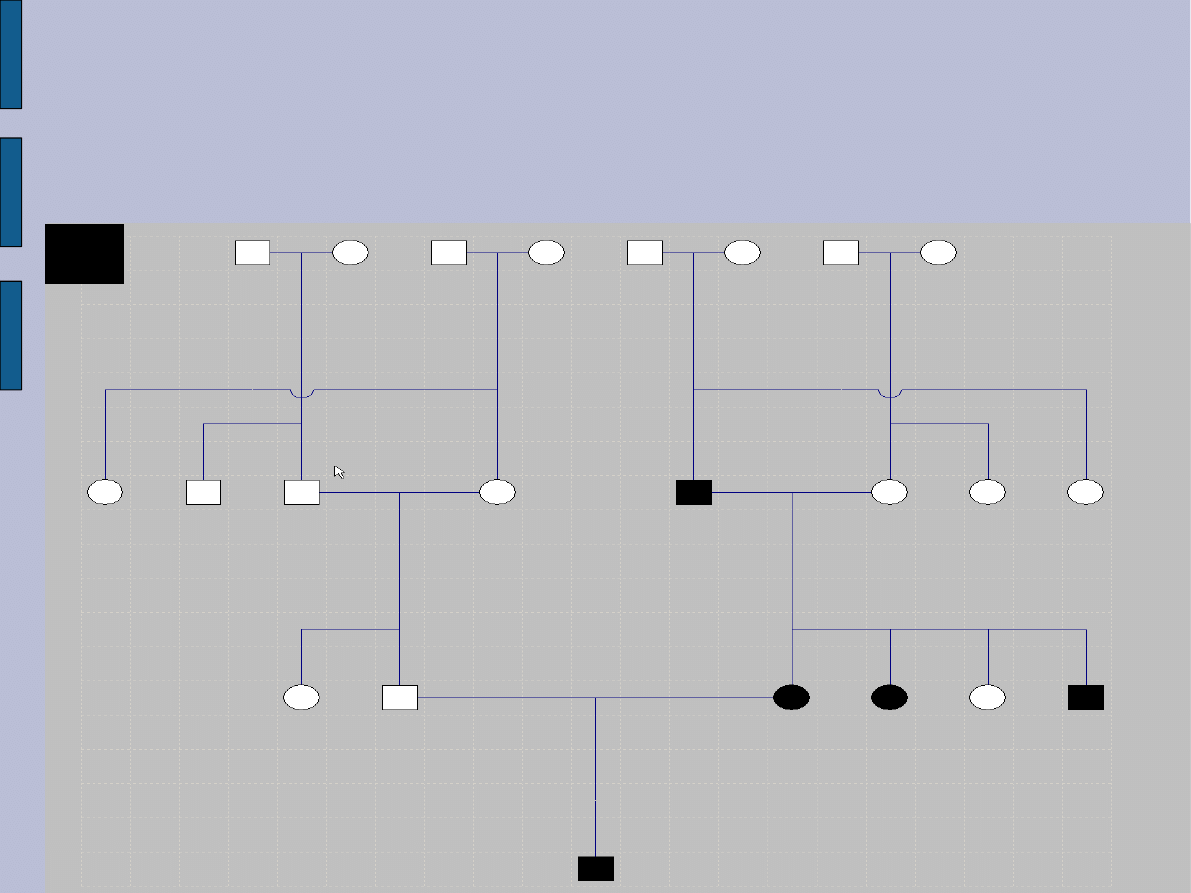
Rodowód 1:
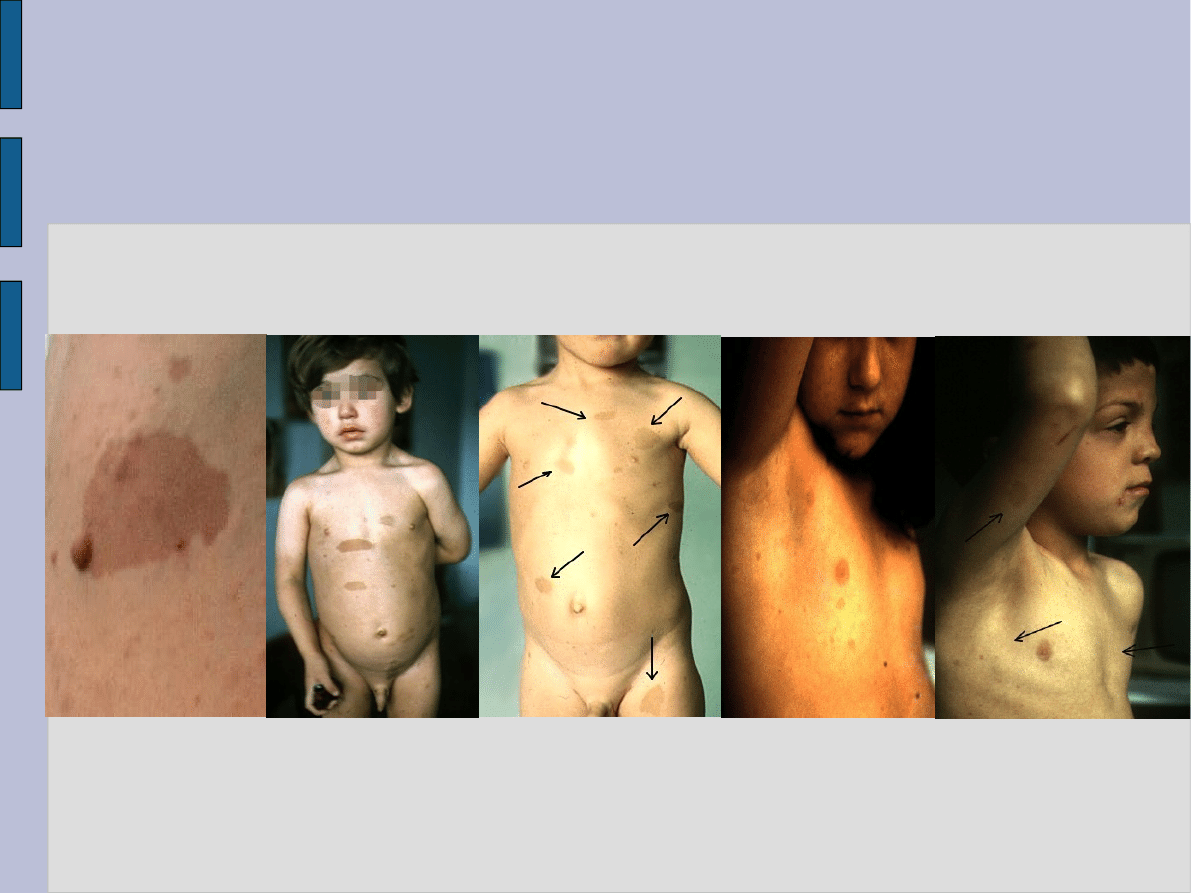
Zmiany skórne w NF-1
Przyjmuje się, że w typie NF-1 liczba plam jest większa niż sześć, a średnica ich nie jest mniejsza niż 15 mm. Trzeba
także pamiętać, że w miarę upływu lat liczba tych plam i ich rozmiary mogą nadal się zwiększać.
Objawy kliniczne nerwiakowłókniakowatości typu 1 (NF-1) manifestują się zmianami skórnymi o typie różnej wielkości plam
"kawy z mlekiem", nierzadko licznymi, o średnicy od kilku do kilkunastu milimetrów. Liczba i wielkość plam zależna jest w
dużej mierze od wieku pacjenta. Mogą one występować już u noworodków, jednak częściej obserwuje się je w dalszych
miesiącach i latach życia. U zdrowych dzieci stwierdzić można także pojedyncze plamy na skórze o zabarwieniu kawy z
mlekiem, jednak nie musi to świadczyć o istnieniu nerwiakowłókniakowatości typu 1.
Plamy café-au-lait są często jedynym objawem choroby u dzieci do 10. roku życia. Ze względu na ich częste
występowanie u około 10-15% osób w ogólnej populacji, jedynie stwierdzenie co najmniej 5 plam o średnicy >=5 mm
przed okresem dojrzewania lub 6 plam o średnicy >=15 mm w późniejszym okresie stanowi kryterium diagnostyczne NF-
1.
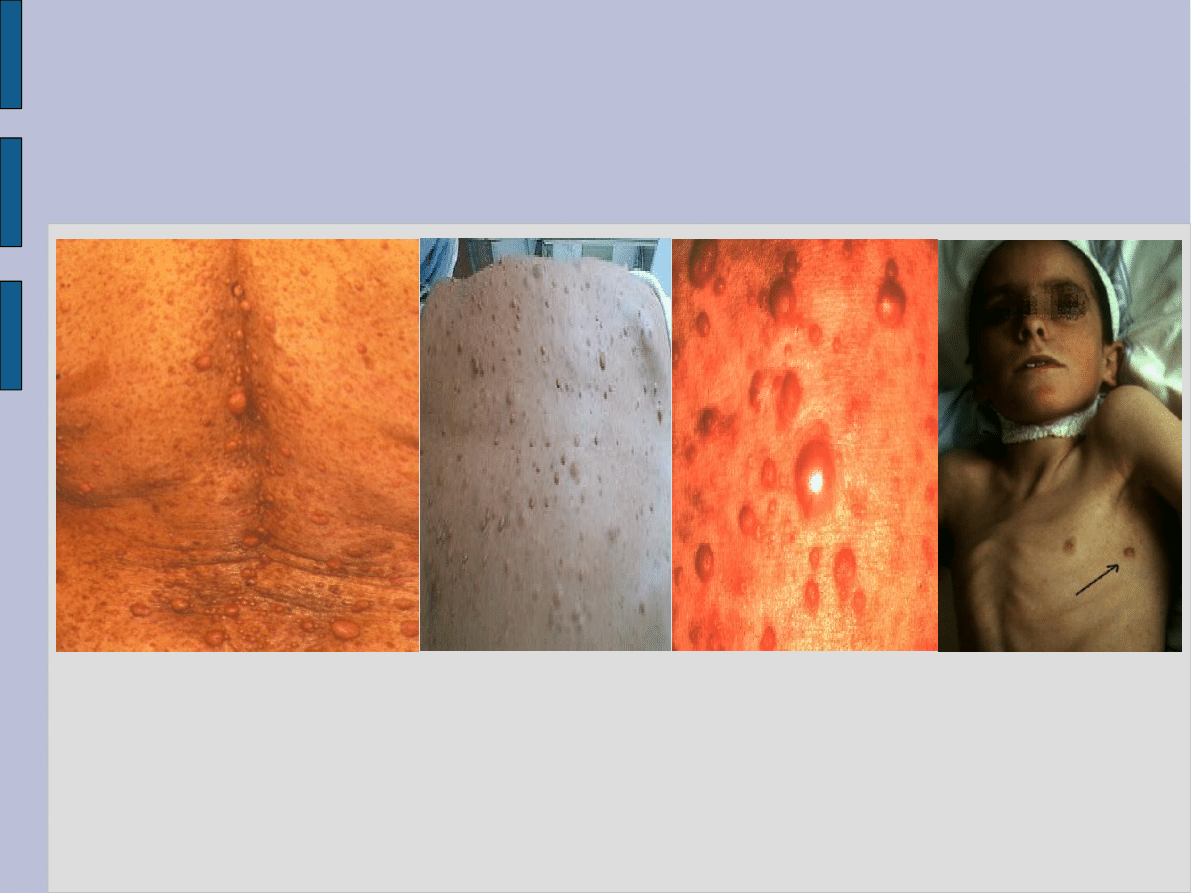
Zmiany skórne w NF-1
sinawe guzki podskórne uwypuklające się ponad powierzchnię skóry.
Szczegółowe badanie histopatologiczne wykazuje zmiany o typie nerwiaków lub nerwiakowłókniaków. Guzki mogą
wywodzić się głównie z nerwów obwodowych kończyn, a także umiejscawiać się wzdłuż naczyń krwionośnych. W miarę
upływu czasu zmiany mogą przeobrazić się w nowotwory z różnym stopniem złośliwości.
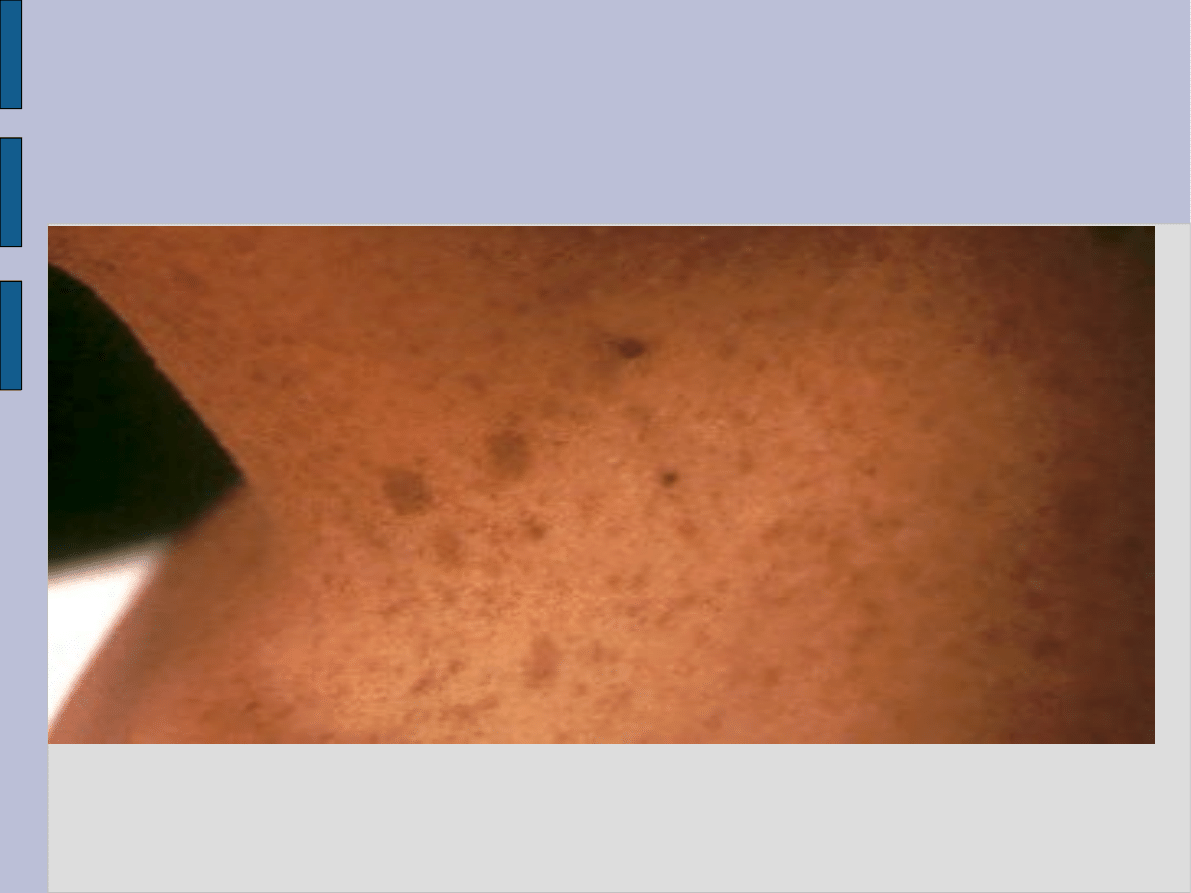
Zmiany skórne w NF-1
liczne zmiany w postaci obfitych piegowatych nakrapiań występujące zwłaszcza w okresie dojrzewania. Umiejscowione są
one głównie w okolicach pach i pachwin, przy czym przybierają wyraźnie większe rozmiary niż powszechnie znane zmiany
piegowate.
Nerwiakowłókniaki
splotowate
nerwiakowłókniaki splotowate powodujące rozlane zgrubienia pni naczyniowych. Nierzadko wrastają one do oczodołów
powodując wytrzeszcz gałek ocznych, a jeśli wrastają do kanału kręgowego, mogą uciskać rdzeń kręgowy i być przyczyną
niedowładu kończyn. Niekiedy nerwiakowłókniaki splotowate doprowadzają do znacznych zniekształceń kości, przerostów
kończyn oraz ich złamań.
Szczególnym rodzajem guzów obwodowych pni nerwowych są nerwiakowłókniaki splotowate (neurofibroma plexiforme).
Mogą one występować u chorych na NF-1 w każdym wieku, a ze względu na swoją wieloogniskową oraz różnorodną
lokalizację sprawiają istotne problemy terapeutyczne. Niekiedy wrastają do kanału kręgowego, narządów wewnętrznych lub
ucha środkowego i powodują znaczne zniekształcenia kości i deformacje kończyn.
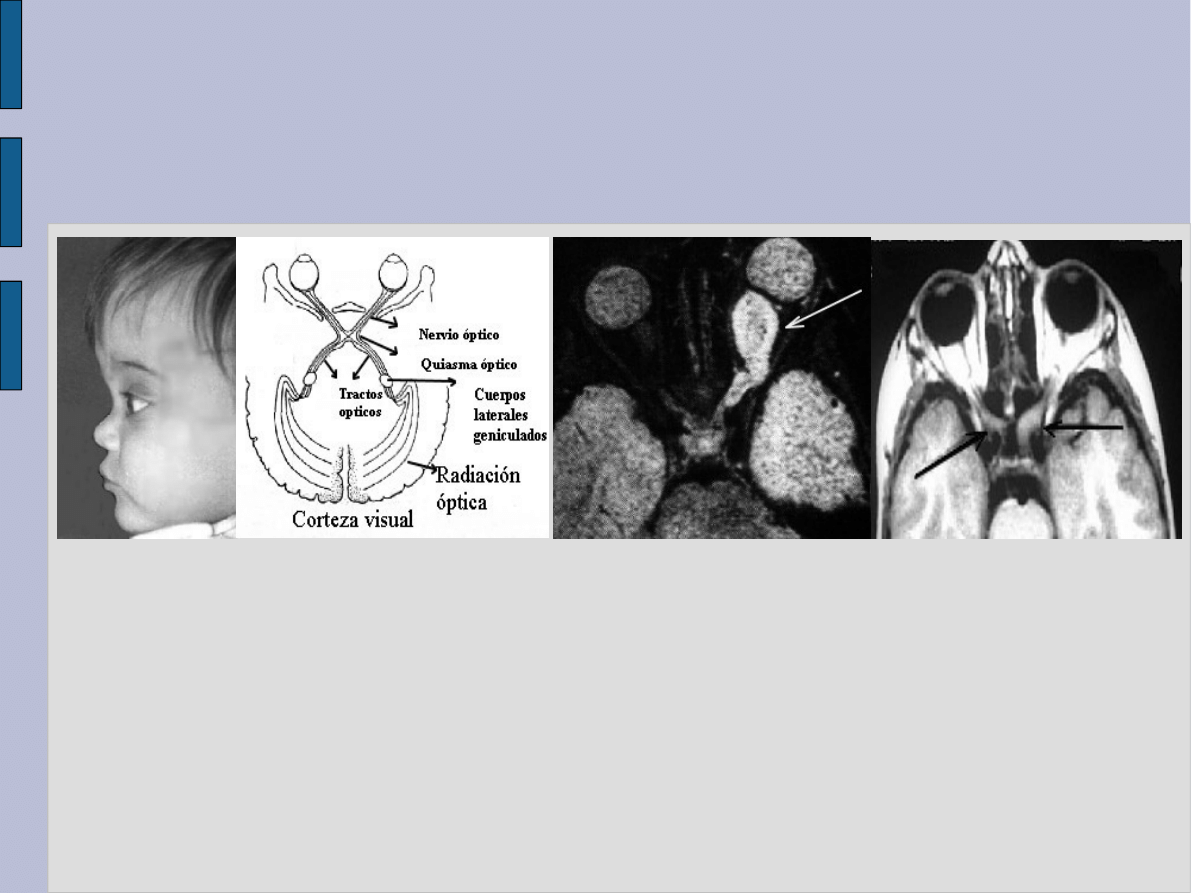
Zmiany w układzie
wzroku
W nerwiakowłókniakowatości typu
1 występują
glejaki nerwów
wzrokowych
, które wywołują
upośledzenie wzroku z zanikiem
nerwów wzrokowych włącznie.
Badając dno oka, można często
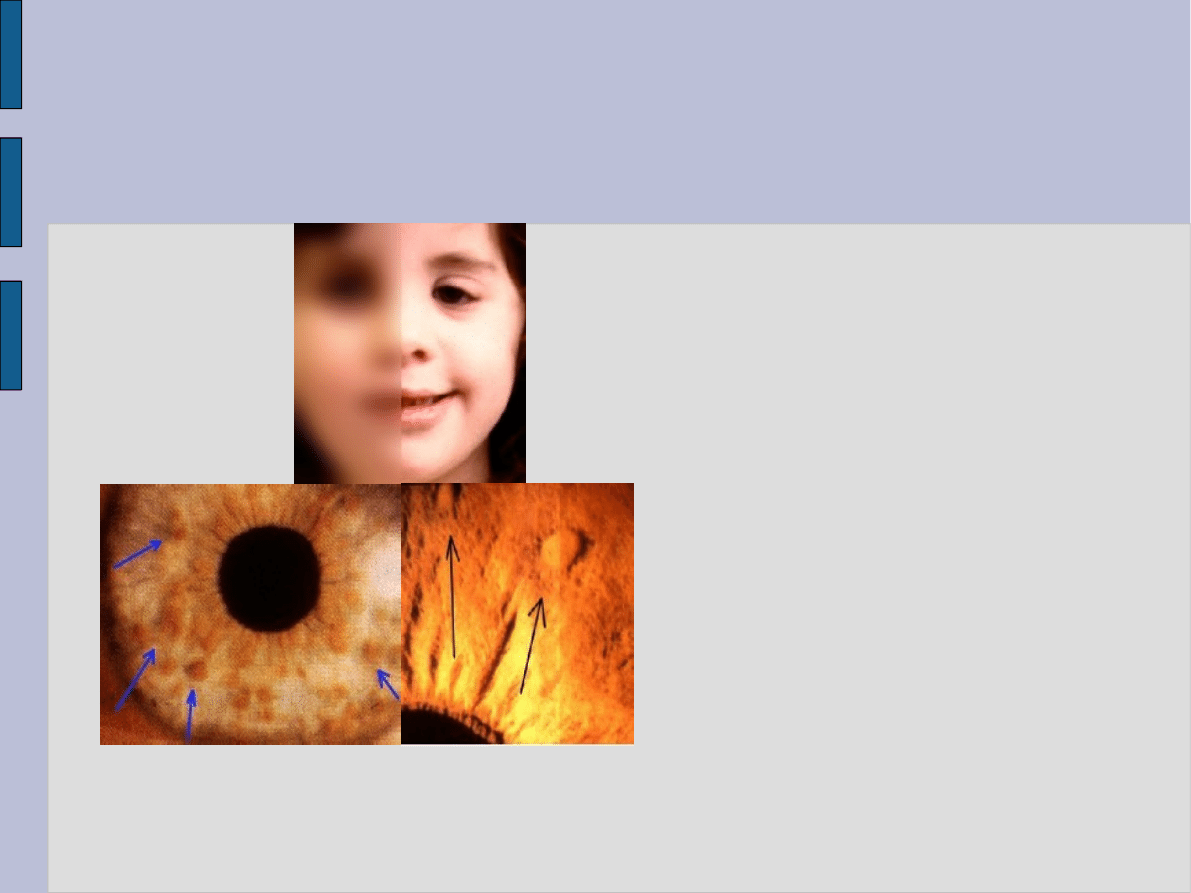
stwierdzić tzw.
guzki Lischa
, które
obejmują tęczówkę oka.
glejaki nerwów wzrokowych,
rosnąc mogą uciskać
skrzyżowanie nerwów
wzrokowych.
Inne nowotwory, występujące u
chorych na NF-1 to np.: oponiaki
mózgu i rdzenia kręgowego,
gwiaździaki, rhabdomyosarcoma,
pheochromocytoma i guz
Wilmsa.
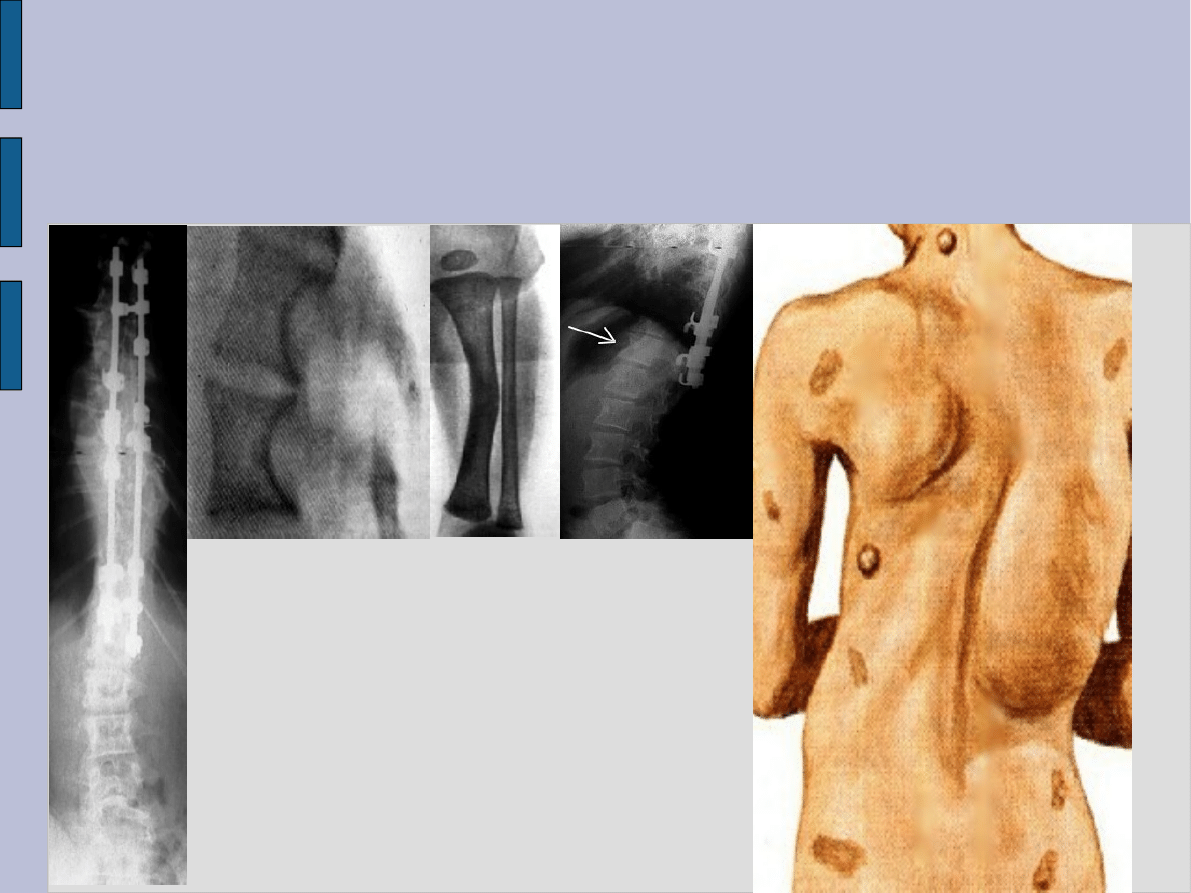
Zmiany w układzie kostnym
Pseudoartroza kości długich
skolioza
zmiany dysplastyczne w
zakresie kręgów
wygięcie kości długich
Objawy kliniczne w różnych grupach
wiekowych:
Od okresu noworodkowego do końca 2
r.ż.:
-plamy cafe au lait
-nerwiakowłókniaki splotowate
-guzy nerwu wzrokowego
-opóźnienie rozwoju
-deformacje kości długich
Od 2-10 r.ż:
-guzy nerwu wzrokowego
-nerwiakowłókniaki splotowate
-skolioza
-nadciśnienie tętnicze
-problemy z nauką
Od 10 r.ż. - wiek dorosły:
-nerwiakowłókniaki
-problemy z uczeniem się
-skolioza
-nerwiakowłókniaki
splotowate
-nadciśnienie tętnicze
U dorosłych:
-chore potomstwo
-progresja
nerwiakowłókniaków
skórnych
-złośliwe guzy nerwów
obwodowych
-nadciśnienie
-nerwiakowłókniaki
splotowate
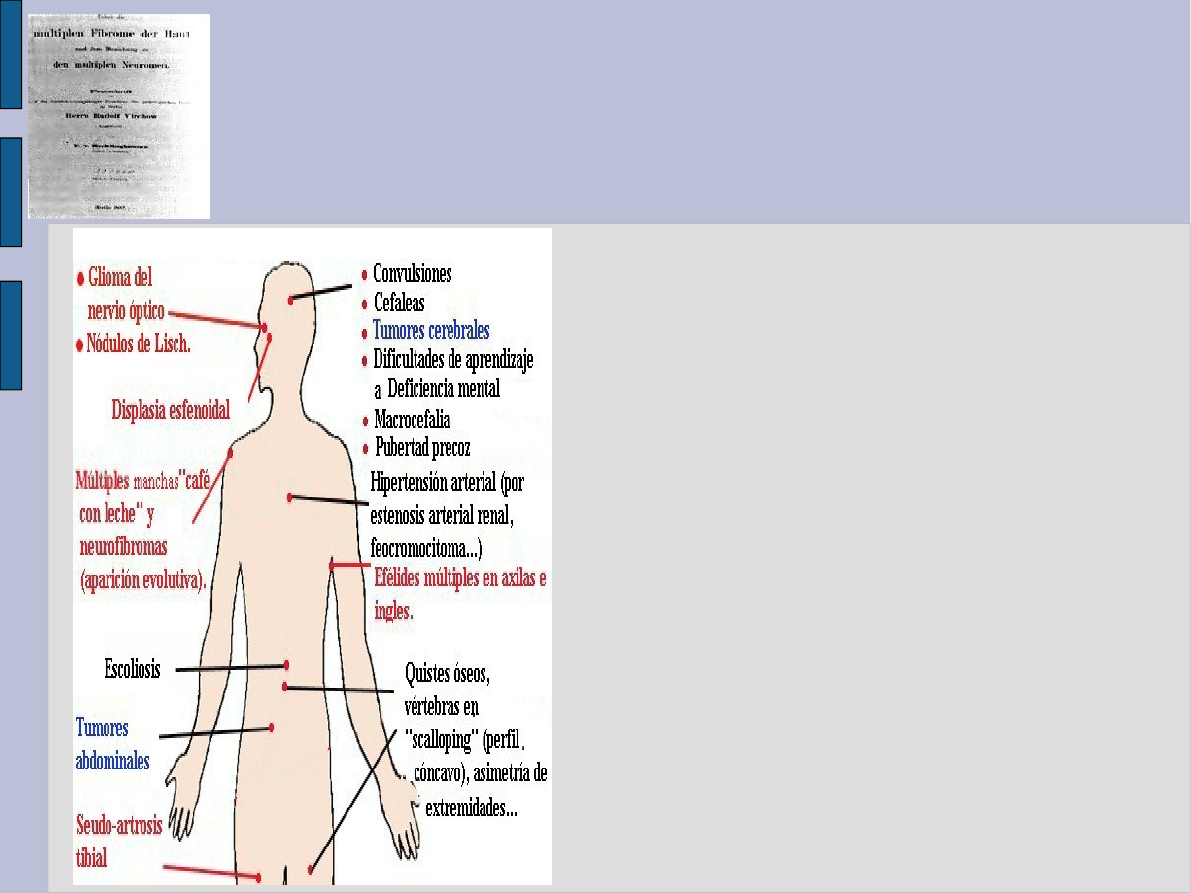
Kryteria rozpoznania
NF-1
●
>=6 plam >5 mm lub > 15
mm w zależności od wieku
●
2 lub> nerwiakowłókniaków
lub 1 nw splotowaty
●
objaw Crewe- gł. w dołach
pachowych lub pachwinowych
●
guz nerwu wzrokowego
●
2 lub> guzów Lischa
●
charakterystyczne zmiany
kostne kręgów lub kości
długich
●
chory krewny I stopnia
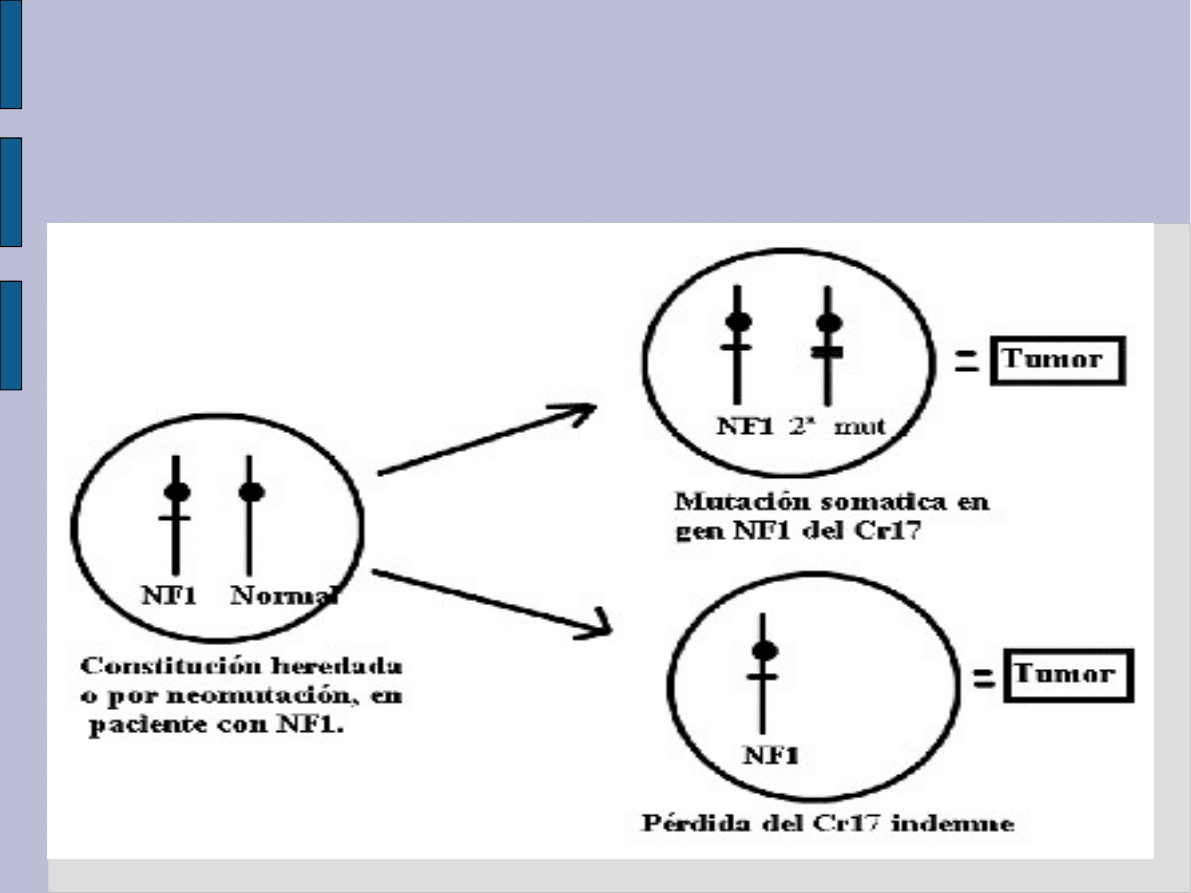
Teoria rozwoju
nowotworowego w NF1

homozygota /
heterozygota
Wyjaśnij pojęcia
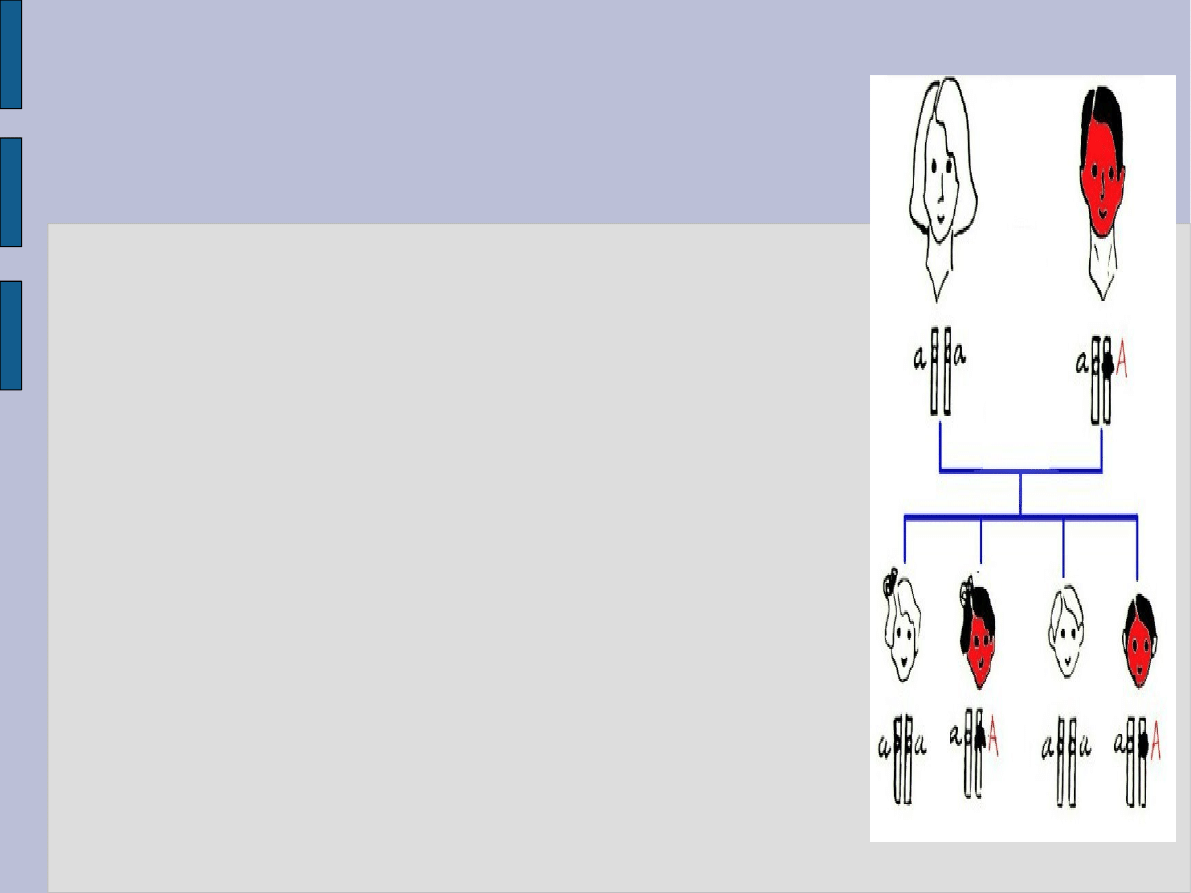
Dziedziczenie autosomalne dominujące
i. Choroba lub cecha autosomalna dominująca
to taka, która w dużym stopniu lub w pełni
ulega ekspresji u heterozygot.
ii. Fenotyp pojawia się w każdym pokoleniu, a
każda choroba osoba ma chorego rodzica (za
wyjątkiem sytuacji, gdy: 1) przypadki choroby
są wynikiem nowej mutacji (mutacji de novo),
2) gen zmutowany nie uległ ekspresji lub tylko
nieznaczna ekspresja genu była przyczyną
niezbyt nasilonego fenotypu);
iii. Każde dziecko chorego rodzica ma 50%
szans na odziedziczenie cechy (choroby)
iv. Fenotypowo prawidłowi członkowie rodziny
nie przekazują choroby swojemu potomstwu
(poza sytuacjami braku penetracji genu lub tak
nieznacznej, że niezauważalnej ekspresji genu)
v. Kobiety i mężczyźni mają jednakowe szanse
na przekazanie choroby dzieciom każdej płci.
W szczególności: występuje przekazywanie z
ojca na syna a mężczyźni mogą mieć chore
córki

Zmienna ekspresja
Ustalenie właściwego sposobu dziedziczenia pozwala na dokładną
ocenę ryzyka powtórzenia się choroby, lecz nie pozwala
przewidzieć nasilenia ciężkości choroby. Różne osoby tą samą
chorobą genetyczną mogą nią być dotknięte w bardzo różnym
stopniu. Zjawisko to określa się mianem zmiennej ekspresji.

„Od zdiagnozowania Iwonki minęły już trzy lata. Dziecko miało na
początku tylko skoliozę i guzki podskórne. Już wtedy jednak
zmuszona byłam rozpocząć samotną walkę z naszymi lekarzami, o
wszystko: gorset ortopedyczny, badania, rehabilitację...
Nasi lekarze zwykle nie są chętni do rozmów z rodzicami,
dokształcania się, szukania nowatorskich rozwiązań. Potrzebują
dużo czasu do oceny sytuacji, które gdzie indziej są już prawie
rutyną. Nazywam to "wyważaniem otwartych drzwi". W
przypadku Iwonki zdarzyło się tak dwukrotnie. Zastanawiam się,
dlaczego ja, matka, potrafię zajrzeć do Internetu, a lekarz nie?”
Opinia znaleziona na jednej ze stron internetowych poświęconych NF-1
Mimo pozornej łatwości rozpoznania, choroba wymaga
dokładnej znajomości przebiegu naturalnego i schorzeń
współistniejących aby we właściwy sposób prowadzić proces
leczenia i profilaktyki.

Do Poradni Genetycznej zgłosił się 52-letni pacjent, u którego od 2 lat występują ruchy
mimowolne kończyn dolnych, trudności w koncentracji, problemy z pamięcią i mową.
Lekarz podejrzewa, że pacjent choruje na chorobę Huntingtona. Z wywiadu zebranego
od pacjenta i jego rodziny wynika, że podobne objawy kliniczne, ale bardziej
zaawansowane obserwowano u ojca pacjenta, który zmarł w 70 r.ż. (brak dokumentacji
lekarskiej potwierdzającej dane z wywiadu). Ojciec pacjenta miał 3 rodzeństwa - u
jednej siostry występowały objawy kliniczne choroby Huntingtona (brak jest jednak
dokumentacji lekarskiej). Druga siostra zmarła w 70 r.ż. bez objawów choroby, ale za to
u jej córki w 50 r.ż. rozpoznano chorobę Huntingtona (chora zmarła). Pacjent ma 5
dzieci wieku od 22 do 28 lat, obecnie wszyscy zdrowi.
Lekarz genetyk stwierdza konieczność przeprowadzenia badań genetycznych
potwierdzających diagnozę u 52-pacjenta. Wyniki badań są jednoznaczne - pacjent
choruje na chorobę Huntingtona. Badania genetyczne zostają następnie wykonane u
innych członków rodziny pacjenta wytypowanych na podstawie rodowodu.
Jakie objawy kliniczne występują w chorobie Huntingtona? Na podstawie, jakich badań
genetycznych potwierdza się rozpoznanie tej choroby u pacjentów? Jakie jest rokowanie
w tej chorobie? W przypadku potwierdzenia rozpoznania, kogo z rodziny typuje się do
wykonania badań genetycznych? Jak wygląda porada genetyczna w chorobie
Huntingtona?
Opis przypadku 2:

Trudno jest wychwycić początek tej choroby, co razem z
rzadkim jej występowaniem, przyczynia się do opóźnień w
rozpoznaniu. Zasadą jest to, że im wcześniej choroba się
pojawia, tym szybciej postępuje.
Bliscy mogą na początku zauważyć niewytłumaczalne
zmiany nastroju. Okresy rozdrażnienia przeplatają się z
apatią, po złości następuje depresja. Ponieważ zmiany
zwyrodnieniowe obejmują obszary odpowiedzialne za
intelekt, to wczesne symptomy mogą dotyczyć też
trudności w uczeniu się nowych rzeczy, przypominaniu
starych, w odpowiedziach na pytania czy w podejmowaniu
decyzji. Mogą pojawić się problemy w pisaniu czy w
prowadzeniu samochodu.
Choroba Huntingtona

U innych osób choroba zaczyna się niekontrolowanymi ruchami
palców, stóp, twarzy czy tułowia, nasilającymi się przy napięciach
emocjonalnych. Zaburzenia ruchowe pojawiające się w większości
przypadków choroby mogą również przybrać formę problemów z
równowagą czy pozorów niezdarności. Chorzy mają trudności z
chodzeniem, potykają się i upadają.
Mowa staje się niewyraźna, pogłębiają się zaburzenia połykania, żucia,
mowy i chodzenia. Występuje również upośledzenie czynności
intelektualnych, choć wielu chorych potrafi do późnego okresu
wyrażać swoje emocje. Choroba trwa zwykle 10-30 lat.
Choroba Huntingtona

Zwiększenie powtórzeń trinukleotydowych CAG
(kodujących kwas glutaminowych) powyżej 36 powoduje
wystąpienie choroby.
Wiek wystąpienia choroby jest tym wcześniejszy im
więcej trójek, lecz zakres wieku wystąpienia choroby dla
danej liczby powtórzeń jest zbyt duży, aby można było
wykorzystać to poradnictwie.
Dziedziczenie jest autosomalne dominujące z pełną
penetracją. Dla liczby powtórzeń wynoszącej 36-39
penetracja jest obniżona.
Choroba Huntingtona

Postacie choroby:
Klasyczna postać choroby Huntingtona występuje u dorosłych w średnim
wieku. Zwykle charakteryzuje ją pląsawica, choć wyróżnia się odmiany bez
nieskoordynowanych ruchów.
Jeśli choroba rozwija się przed 20. r. ż., to mówimy o tzw. chorobie
Huntingtona o wczesnym początku bądź młodzieńczej. Objawia się ona
początkowo trudnościami w nauce, czasami drobnymi zmianami pisma czy
pewną ociężałością ruchów. Niektóre z zaburzeń przypominają symptomy,
które obserwujemy w chorobie Parkinsona, co łączy te dwie z pozoru
odmienne, czy wręcz przeciwstawne choroby (w chorobie Parkinsona
występuje zwykle sztywność mięśniowa, spowolnienie i drżenie). Ta postać
rokuje znacznie gorzej niż klasyczna. W ciągu 10 lat doprowadza zwykle do
zgonu.
Istnieje także grupa osób, u których choroba rozwinęła się po 55. r. ż. Tak
późny początek znacznie utrudnia diagnozę.
Choroba Huntingtona
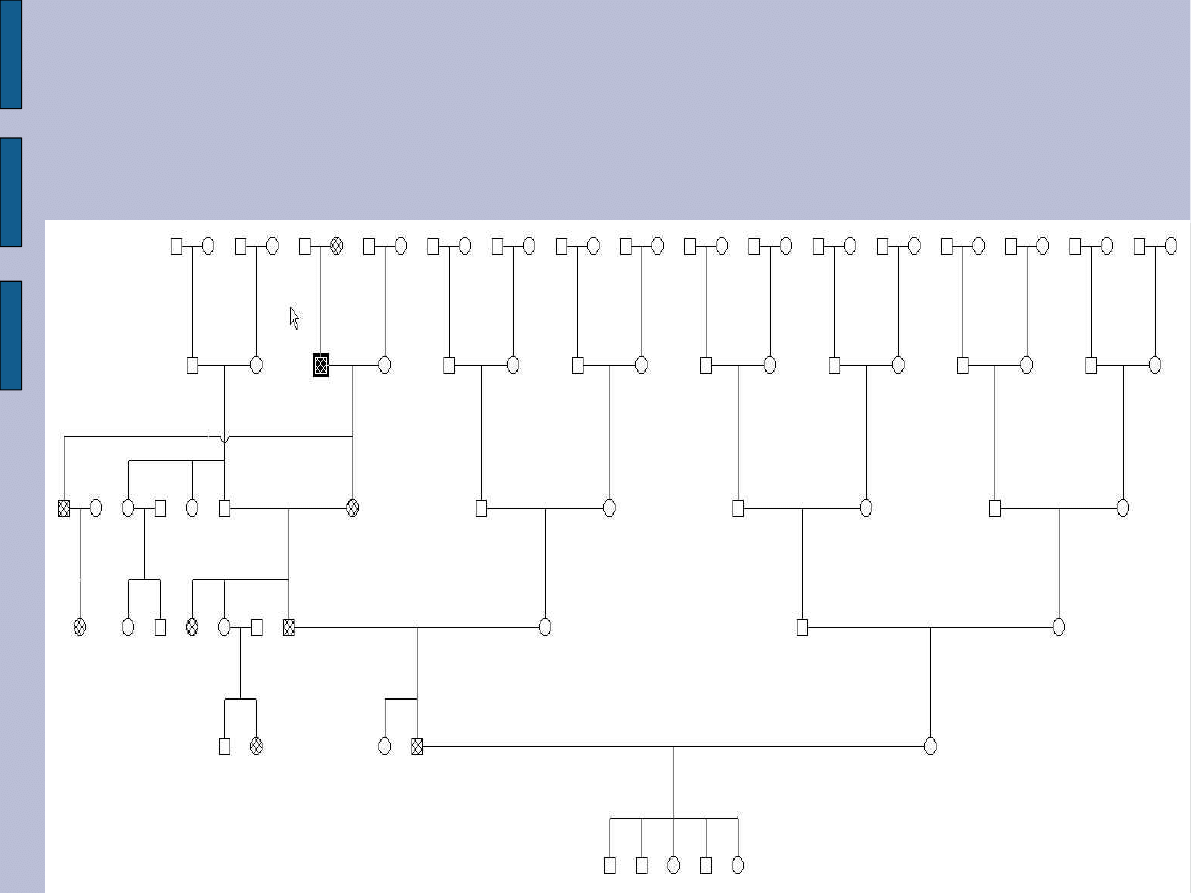
rodowód
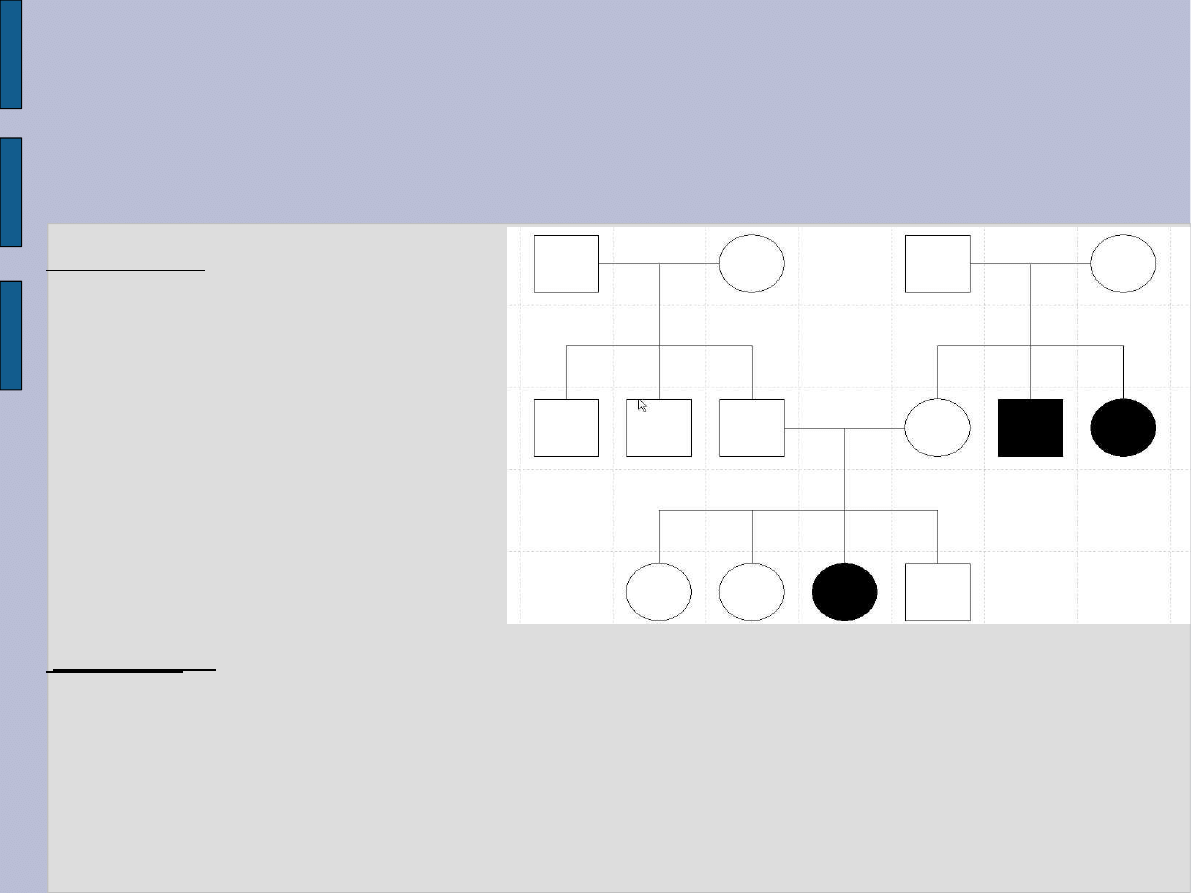
Ekspresja. Ustalenie
właściwego sposobu
dziedziczenia pozwala na
dokładną ocenę ryzyka
powtórzenia się choroby, lecz
nie pozwala przewidzieć
nasilenia ciężkości choroby.
Różne osoby tą samą chorobą
genetyczną mogą nią być
dotknięte w bardzo różnym
stopniu. Zjawisko to określa
się mianem zmiennej
ekspresji.
Ekspresja i penetracja
Penetracja. Jeśli nie u wszystkich osobników posiadający zmutowany
gen (gen choroby), choroba ta rozwija, mówimy o obniżonej
penetracji danego genu (innymi słowy: pomimo obecności
określonego genotypu nie rozwija fenotyp). Penetracja opiera się na
zasadzie wszystko albo nic. Stopień penetracji może być związany z
wiekiem.

Diagnostyka choroby
Huntingtona
1. Wywiad rodzinny.
2. Badanie neurologiczne i psychologiczne.
3. Diagnostyka CT/MRI- powiększenie komór bocznych, atrofia jądra
ogoniastego.
4. Diagnostyka molekularna:
Gen IT15 zlokalizowany jest na chromosomie 4 (4p16.3) i
zbudowany jest z 67 egzonów zawierających 10,366 bp sekwencji
kodujących i niestabilne sekwencje trójnukleotydowe CAG, które
kodują glutaminy na 5'-końcu.
Najczęściej stosowaną metodą jest PCR. Osoby zdrowe posiadają
około 20 (9-29) powtórzeń tripletu CAG w genie IT15.Rozpoznanie
potwierdza obecność =>40 powtórzeń trójnukleotydowych. Osoby
posiadające od 30 do 39 powtórzeń zwykle mają przebieg
bezobjawowy ale mają zwiększone ryzyko że w kolejnym pokoleniu (
u dzieci) może pojawić się pełnopbjawowy zespoł Huntingtona. Im
większa liczba powtórzeń tym większe ryzyko wczesnego początku
choroby (>80 – postać młodzieńcza). Dla 42 i więcej powtórzeń
penetracja choroby wynosi 100%. Niestabilność tripletów CAG jest
wyższa, jesli gen został przekazany przez ojca i w takich
przypadkach początek choroby u dzieci jest wcześniejszy niż u ojca.

Mutacje dynamiczne i
antycypacja
Jednym z najbardziej intrygujących odkryć ostatnich lat było wykazanie,
że kilka genów zawiera serię powtarzających się trzynukleotydowych
sekwencji, które mogą stawać się niestabilne i powiększać się.
6
Kiedy
rozprzestrzenianie (ekspansja) osiągnie punkt krytyczny, gen przestaje
podlegać ekspresji albo produkuje nieprawidłowe białko. Bez względu na
mechanizm, który to powoduje, skutki są poważne, chociaż niektóre
ekspansje po prostu kończą się niemal stycznie do łamliwych miejsc na
chromosomie X
Zaburzenia genetyczne spowodowane przez mutacje dynamiczne mają
niezwykłe właściwości.
Antycypacja" dotyczy tendencji do występowania choroby o cięższym
przebiegu i/albo wcześniejszego wieku wystąpienia choroby w
następnych pokoleniach. Jako ogólną zasadę można przyjąć, że większa
ekspansja skutkuje gorszym rokowaniem. Godne uwagi są obserwacje,
że na gwałtowność przebiegu choroby ma wpływ płeć rodzica-
przekaziciela. Na przykład ekspansja łamliwego chromosomu (fragile) X i
mutacje dystrofii miotonicznej są dużo większe, jeśli są przekazywane
przez matkę, w przeciwieństwie do choroby Huntingtona, która ujawnia
się w młodszym wieku u potomków zaatakowanych tą chorobą
mężczyzn.

Testy przedobjawowe
Badanie DNA powinno być wykonane tylko po uzyskaniu
świadomej zgody. Należy pacjentom uświadomić wszystkie
możliwe konsekwencje (dla badanego i jego bliskich)
wykrycia lub wykluczenia mutacji. Istotne mogą byc także
konsekwencje socjoekonomiczne (praca, ubezpieczenie,
opieka społeczna, ochrona danych).
Nie należy wykonywać testów u dzieci, z wyjatkiem stanów
klinicznych mogących sugerować rozpoznanie choroby
Huntingtona.
Etyczne problemy diagnostyki prenatalnej.

Plejotropizm
Mendel zakładał, że każdy gen warunkuje
jedną cechę organizmu. Gdyby tak było, to
musiałoby istnieć tyle genów, ile jest
niezależnie dziedziczących się cech. Późniejsze
badania wykazały, że jeden gen może wpływać
jednocześnie na bardzo różne, istotnie lub
pozornie nie związane ze sobą własności
organizmu. Określa się to mianem
plejotropowego działania genu.
zjawisko warunkowania przez jeden gen kilku,pozornie nie związanych ze sobą właściwości fenotypowych.

Mozaikowatość
somatyczna i germinalna
Wyjaśnij pojęcia i podaj znaczenie dla okreslenia ryzyka
powtórzenia się choroby u innych członków rodziny.
Krótki sprawdzian:
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
Wyszukiwarka
Podobne podstrony:
3 ćwiczenia BADANIE asfaltów
Ćwiczenie7
Cwiczenia 2
Ćwiczenia V
metody redukcji odpadów miejskich ćwiczenia
Ćwiczenia1 Elektroforeza
cwiczenia 9 kryzys
Ćwiczenia 1, cz 1
Ćwiczenie 8
9 ćwiczenie 2014
Cwiczenie 1
Ćwiczenie 2 Polska w europejskim systemie bezpieczeństwa
11 CWICZENIE 1 SEMESTR LETNIid 12747 ppt
więcej podobnych podstron