niedokrwistość Blackfana-Diamonda
choroba Charcota-Mariego-Tootha
wrodzona niedoczynność tarczycy
Chromosom 19
{kind=link}
Chromosom 20
• Choroby
•
•
•
•
•
•
•
zespół Gerstmanna-Sträusslera-Sc
•
rzekoma niedoczynność przytarczy
•
•
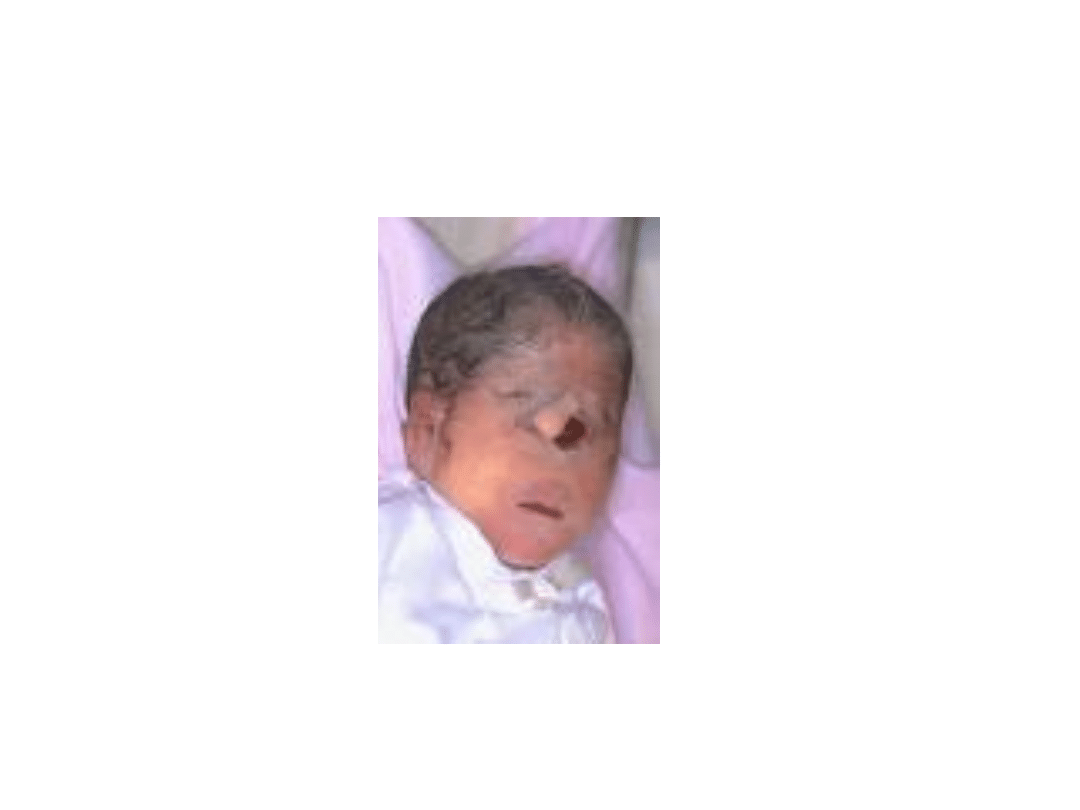
Zespół Pateau
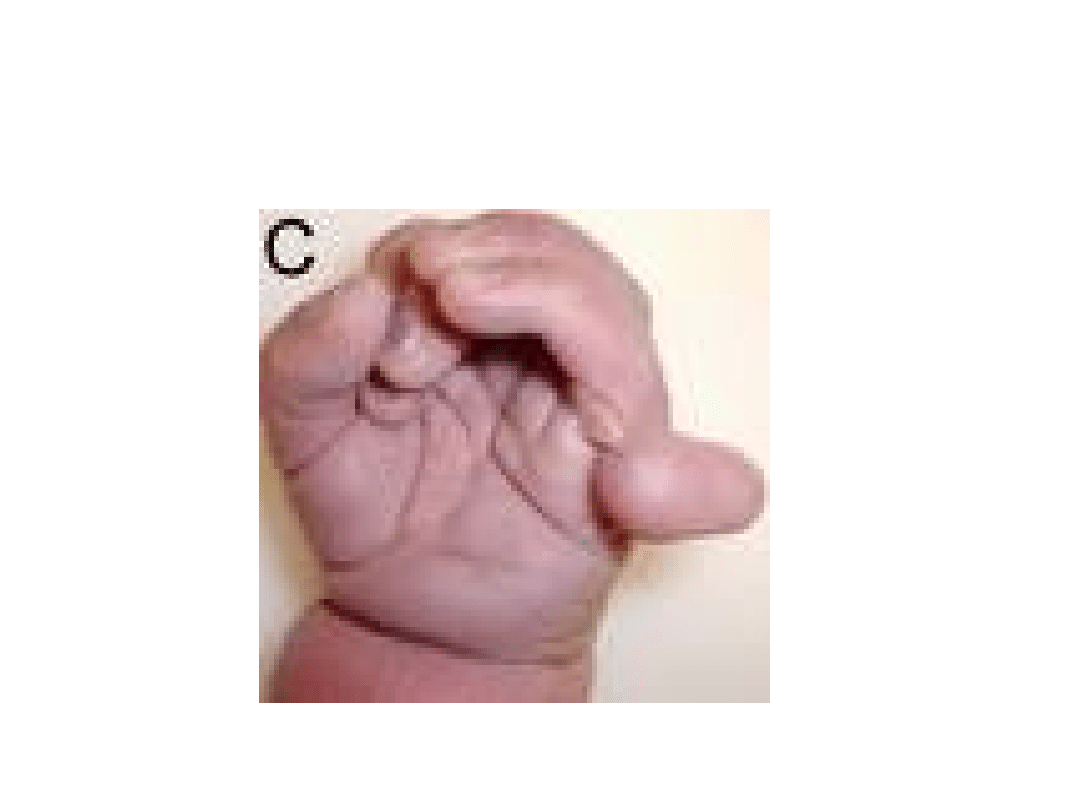
polidaktylia

• Zespół Pataua (
Patau syndrome) –
spowodowany
(
47,XX,+13 albo 47,XY,+13).
• Trisomia 13 spowodowana jest
się chromosomów podczas
I lub II podziału
u któregoś
z rodziców (opóźnione rozchodzenie się
chromosomów w
).
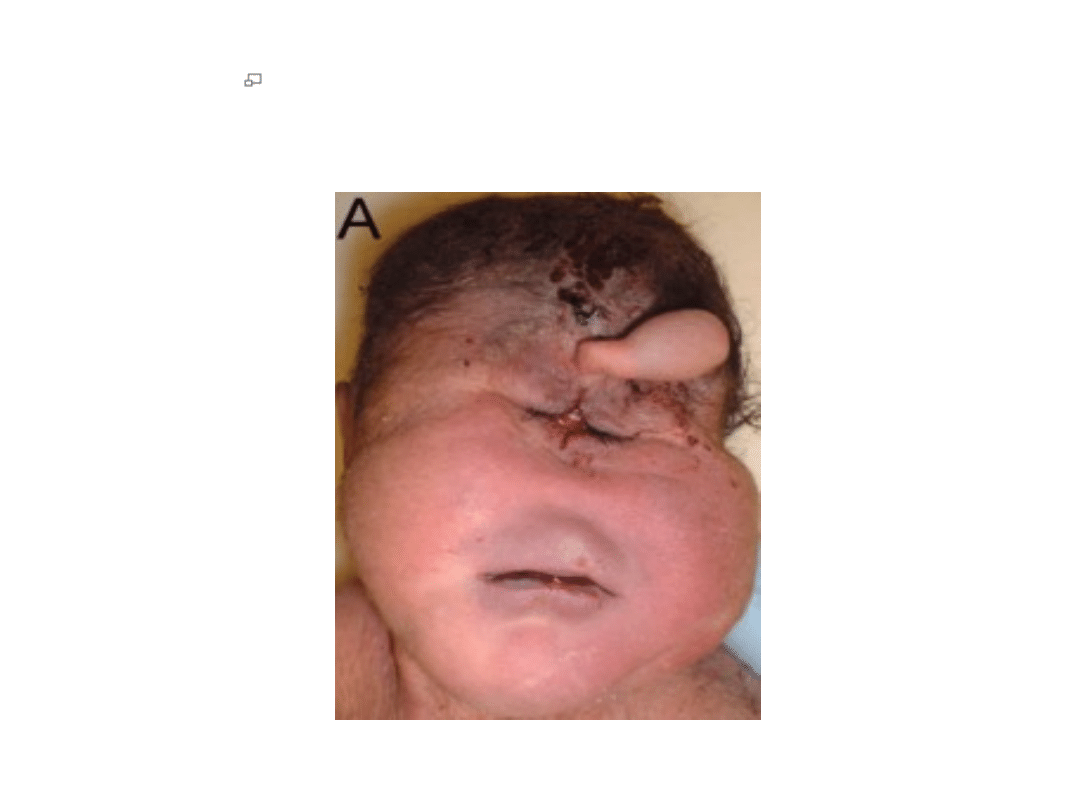
Twarz niemowlęcia płci męskiej z zespołem Pataua
urodzonym w 37 i 1/2 tygodniu ciąży, z
holoprozencefalią i trąbką (proboscis) nad
pojedynczą szparą powiekową
.

niska masa urodzeniowa
ubytek skóry skalpu (aplazja skóry głowy)
holoprozencefalia przebiegająca z wadami narządu wzroku (anoftalmią,
mikroftalmią, hipoteloryzmem lub cyklopią) nieprawidłowo wykształconym
nosem, i tzw. trąbką (proboscis), rozszczepem wargi i (lub) podniebienia
wady małżowin usznych (niskie osadzenie, zniekształcenie małżowin)
anomalie kończyn (polidaktylia pozaosiowa, ustawienie palców w kształcie
"kurka od strzelby", pojedyncza bruzda zgięciowa, wydatna pięta)
wady sercowo-naczyniowe (przetrwały przewód tętniczy, dekstrokardia,
ubytek przegrody międzykomorowej, ubytek przegrody
międzyprzedsionkowej)
wady nerek (torbielowatość nerek, wodonercze)
wady rozwojowe mózgowia i cewy nerwowej
napady drgawek
hipotonia mięśniowa
przepukliny, przepuklina pępkowa
naczyniaki włośniczkowe w okolicy czoła
wnętrostwo
wady rozwojowe macicy.

• Zespół Edwardsa (
Edwards'
syndrome, trisomy 18) jest to
spowodowany
(47,XX,+18 albo
47,XY,+18). Częstość występowania
zespołu szacowana jest na 1:8.000
urodzeń
. Przyczyną jest
w I lub II podziale mejotycznym;
nierzadko spotyka się
.
Występuje przyrost masy
o około
2,8%

niska masa urodzeniowa noworodka (dystrofia
wewnątrzmaciczna)
deformacje czaszki:
(microcephalia)
(scaphocephalia)
wydatna
(91%
)
wąskie szpary powiekowe (80%
)
wady
(86%
)
poszerzenie szwów i ciemiączek
nisko osadzone,
małżowiny uszne
pojedyncza bruzda zgięciowa dłoni
nadmiar skóry na szyi (86%
3
]szeroko rozstawione brodawki sutkowe (hiperteloryzm
brodawek sutkowych - 90%
)

anomalie szkieletu:
krótki
)
zwichnięcie stawów biodrowych (82%
)
deformacje zgięciowe palców, nakładające się na siebie palce
)
wydatne pięty
zgięcie podeszwowe paluchów ("młotkowaty paluch" - 89%
)
:
(PDA)
ubytek przegrody międzykomorowej
(VSD)
wady rozwojowe nerek
paznokci (blisko 100%
)
zmiany dermatoglifów
wady narządów płciowych
hipoplazja
(blisko 100% [3])
przerost łechtaczki (89% [3])
drgawki (62% [3])
głębokie upośledzenie umysłowe.

kariotyp pacjenta z Zespołem Edwardsa,
dodatkowy chromosom pary 18 zaznaczono
strzałką; 46,XX,+18

Zespół Edwardsa - spowodowany jest występowaniem u dziecka
dodatkowego chromosomu z 18 pary. U dzieci z tym zespołem występują
mnogie wady. Tylko 10% z nich przeżywa pierwszy rok życia i wszystkie z nich
wykazują głębokie opóźnienie rozwoju umysłowego i fizycznego. Częstość
występowania tego zespołu wśród noworodków wynosi około 1 na 3500.
Ryzyko urodzenia dziecka z zespołem Edwardsa wzrasta wraz z wiekiem
matki.
Otwarta wada ośrodkowego układu nerwowego - powstaje w wyniku
defektu w procesie formowania się cewy nerwowej w czasie rozwoju
zarodkowego. Istnieją dwie grupy otwartych wad OUN:
rozszczep kręgosłupa - wada polegająca na braku spojenia łuków kręgowych
co doprowadza do uwypuklenia się opon i/lub rdzenia kręgowego i powstania
przepukliny. Stan taki może doprowadzić do umysłowego i fizycznego
kalectwa.
bezmózgowie - polega na braku mózgu lub większości jego struktur. Wada ta
wyklucza możliwość utrzymania noworodka przy życiu.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
Wyszukiwarka
Podobne podstrony:
śr.mieszkaniowe, 16 pomieszczenia-choroby, Wyodrębniono zespoły chorobowe związane z ujemnym wpływem
Zespoły chorobowe, ۩ NAUKA, Genetyka
Zespół chorób metabolicznych, PWSZ, Żywienie kliniczne i kliniczny zarys chorób, semestr V, Kliniczn
Podstawowe zespoly chorobowe w neurologii, FIZJOTERAPIA, neurologia
Zespół?S, choroby a ciąża
Zespoły chorobowe 2
Zespoły chorobowe w neurologii
Podstawowe zespoly chorobowe w neurologii
Dziedziczne zespoły chorobowe, którym towarzyszy krótkowzroczność
Choroba niedokrwienna, ostre zespoły wieńcowe
choroby genetyczne zespoly meta Nieznany (2)
choroba alzheimera i inne zespoły otępienne wieku podeszłego
Choroby z?erracji chromosomowych Zespół Downa
choroba zespol downa, Psychologia
Pytania-Książka-opracowane, Choroba Huntingtona a zespół atetotyczny
masaz w jednostkach chorobowych, ZESPOŁY BÓLOWE KRĘGOSŁUPA, ZESPOŁY BÓLOWE KRĘGOSŁUPA - RWA KULSZOWA
Biologia część V, Zespół Turnera choroba genetyczna
Zespół Sudeck'a, CHOROBY CHARAKTERYSTYKA
Zespół Sudecka, Patologia i choroby
więcej podobnych podstron