
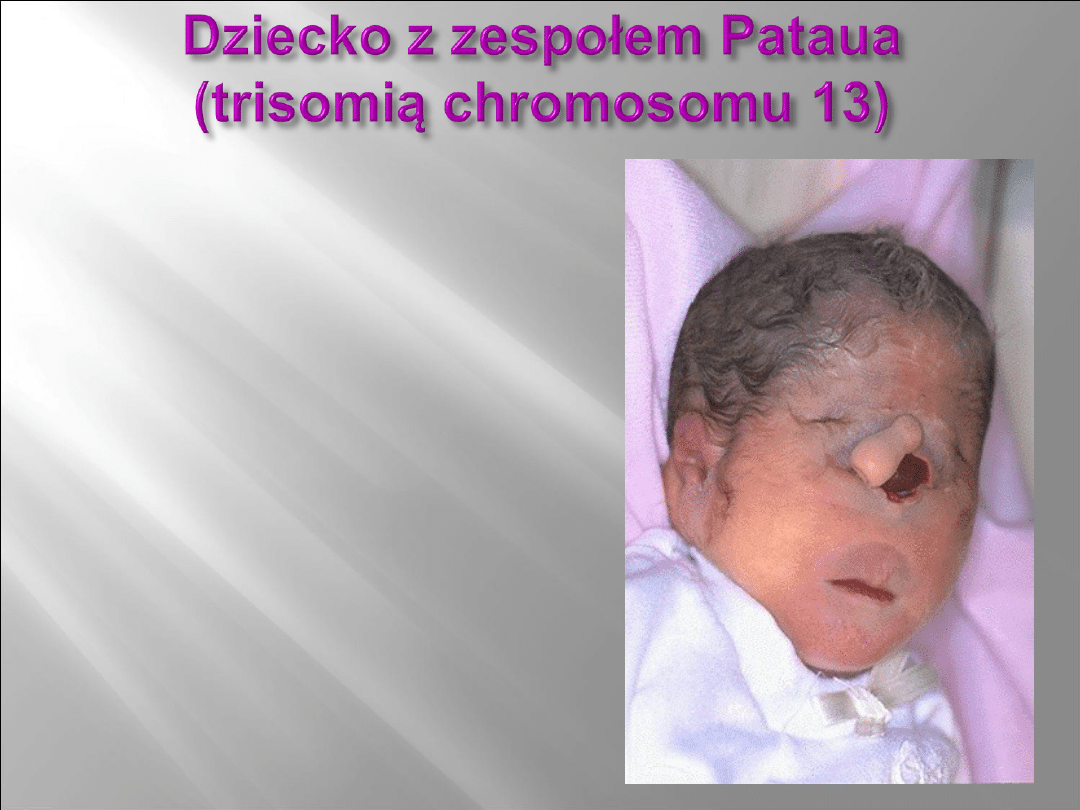
Zespół Pataua jest trzecim co do
częstości, po zespole Downa i
zespole Edwardsa, zespołem
wywołanym trisomią chromosomu
autosomalnego. Szacuje się, że
występuje on średnio raz na 10 000
– 20 000 żywo urodzonych dzieci.
Z badań wynika, że około jedna
czwarta noworodków, które
przychodzą na świat z zespołem
Pataua, umiera podczas pierwszej
doby od urodzenia, a około
połowa w pierwszym tygodniu życia.
Szansa na przeżycie pierwszego
miesiąca przez dzieci z zespołem
Pataua sięga 30%, natomiast
odsetek dzieci, które przeżywają
pierwszy rok, waha się od 5 do 10%.
Czynnikiem, od którego zależy częstość występowania aneuploidii
(określenie oznaczające zaburzenie ilości materiału genetycznego
znajdującego się w jądrze komórkowym) u potomstwa, jest wiek
matki. Im późniejszy wiek zajścia w ciążę, tym większe ryzyko
wystąpienia trisomii chromosomu 13 u płodu.
Rozpoznanie kliniczne powinno być zawsze potwierdzone badaniem
cytogenetycznym. W tym celu od noworodka pobierana jest próbka
krwi, z której następnie są hodowane i izolowane limfocyty.
Badaniami, które mogą wskazywać na istnienie trisomii
chromosomu 13 u płodu, jest m.in. test PAPP-A – biochemiczne
badanie surowicy krwi matki oraz badanie ultrasonograficzne płodu
(amniopunkcja, czyli nakłucie worka owodniowego i pobranie
kilkunastu mililitrów płynu owodniowego).

Dzieci z zespołem Pataua już przy urodzeniu prezentują wiele
małych i dużych anomalii, które dla lekarza
neonatologa stanowią podstawę do klinicznego rozpoznania tego
zespołu. Do najbardziej charakterystycznych należą:
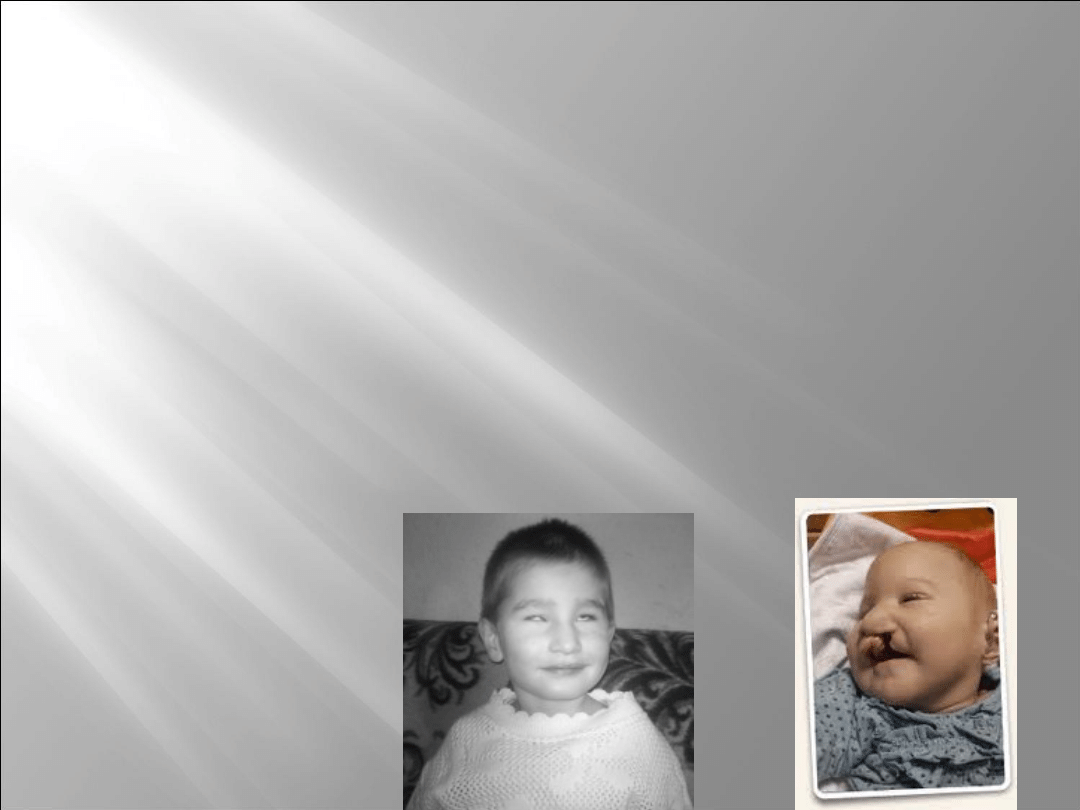
rozszczep wargi i/lub podniebienia
małoocze lub bezocze
dodatkowe palce u rąk i stóp
naczyniaki włośniczkowe
ubytki w skórze głowy okolicy potylicznej
nieprawidłowo wykształcone i/lub osadzone małżowiny uszne.
Ponadto u dzieci z trisomią 13 często współistnieją wady
narządów wewnętrznych, m.in.:
serca (jak: ubytek przegrody międzykomorowej, przetrwały
przewód Botalla, ubytek przegrody międzyprzedsionkowej oraz
dekstrokardia, czyli prawostronne położenie serca)
układu moczowo-płciowego (jak: wielotorbielowatość nerek, ale
zdarza się również nerka podkowiasta czy podwójny moczowód.
U chłopców może występować nieprawidłowa budowa
moszny, u dziewczynek– dwurożna macica)
układu kostnego i innych.

Niemowlęta z trisomią chromosomu 13 słabo przybierają na
wadze, często zarówno obwód głowy, masa ciała, jak i długość
są bardzo małe. Większość dzieci wymaga już od urodzenia
żywienia parenteralnego lub sondą dożołądkową ze względu na
zaburzone odruchy ssania i przełykania. Dodatkowym
problemem jest refluks przełykowo-żołądkowy.
Wady anatomiczne mózgu występujące u dzieci z zespołem
Pataua pociągają za sobą zaburzenia funkcjonalne układu
nerwowego. Początkowo napięcie mięśniowe jest obniżone, z
czasem staje się nadmierne. U około połowy dzieci występują
drgawki. Istotnym problemem jest występowanie bezdechów
pochodzenia centralnego.
W zespole Pataua opisuje się różnego rodzaju wady oczu.
Powikłaniami tych wad mogą być zarówno zaćma, jak i jaskra.
Opisywane przypadki dzieci z trisomią 13, które przeżyły dłużej
niż rok, świadczą o głębokim opóźnieniu rozwoju
psychoruchowego występującym w tym zespole. Takie dzieci
nie chodzą samodzielnie, nie mówią, cierpią na głęboki
niedosłuch.

Rodzice dziecka z trisomią 13, dowiadując się o wysokim
prawdopodobieństwie jego zgonu, wraz z personelem medycznym biorą
udział w podejmowaniu decyzji dotyczących resuscytacji, ewentualnych
operacji, interwencji chirurgicznych, intensywnej terapii i przedłużania
życia. Personel medyczny, oprócz udzielania wyczerpujących informacji
o stanie zdrowia dziecka, powinien unikać raniących słów i
określeń, a także wykazać się taktem i zrozumieniem dla
reakcji rodziców.
Wraz z upływem czasu rodzice muszą liczyć się z jednej strony z nadal
utrzymującym się wysokim ryzykiem śmierci dziecka, a z drugiej –
poważnych zaburzeń rozwojowych.
Doktorzy John Carey i Scott Showalter zwracają uwagę na fakt, że dzieci
te, mimo wielu ograniczeń rozwojowych, osiągają do pewnego stopnia
„kamienie milowe": nawiązują kontakt emocjonalny z najbliższymi
osobami w rodzinie, reagują na ich obecność, potrafią się uśmiechać i
czasem zdobywają takie umiejętności, jak samodzielne spożywanie
posiłków.
Należy pamiętać, że każde dziecko – niezależnie od tego, czy przyszło na
świat z prawidłowym czy nieprawidłowym zestawem chromosomów –
zasługuje na naszą troskę i szacunek. Decyzje odnośnie do
postępowania medycznego i terapeutycznego powinny być
rozpatrywane indywidualnie, w zależności od istniejących warunków i
możliwości, oraz podejmowane w porozumieniu z rodzicami.

SMA jest skutkiem utraty neuronów
ruchowych rdzenia kręgowego, co
prowadzi do postępującego
niedowładu i zaniku mięśni.
Za komunikację pomiędzy rdzeniem
kręgowym a poszczególnymi
grupami mięśni odpowiedzialne jest
białko SMN. Brak tego białka
powoduje zwyrodnienie rogów
przednich rdzenia kręgowego,
wskutek czego występują problemy
z przesyłaniem impulsów z rdzenia
kręgowego do mięśni, które ulegają
powolnemu zanikowi.
Rozpoczyna się w życiu płodowym
i postępuje w okresie
niemowlęcym oraz wczesnego
dzieciństwa.

SMA jest chorobą genetycznie uwarunkowaną, dziedziczoną autosomalnie recesywnie,
spowodowaną mutacjami w genie SMN1 położonym na ramieniu długim chromosomu 5.
Częstość występowania choroby szacuje się na 1:7000–1:10 000 żywych urodzeń,
jednak mutacje w genie SMN1 występują u co 45–50 zdrowej osoby.
Objawy kliniczne SMA typ 1 (choroba Werdniga–Hoffmanna)- ciężka postać dziecięca:
ciężka hipotonia, uogólnione osłabienie, mała masa mięśni, brak odruchów ścięgnistych
oraz zajęcie mięśni języka, twarzy, żwaczy, a zaoszczędzenie mięśni zewnętrznych gałki
ocznej i zwieraczy. Już w momencie urodzenia się dziecka mogą wystąpić zespół
zaburzeń oddechowych i niemożność przyjmowania pokarmu.
Wrodzone przykurcze stawów stwierdza się u około 10% ciężko uszkodzonych
noworodków. Ponad 2/3 dzieci umiera do 2. roku życia. Chore dziecko nie unosi głowy,
nigdy nie siada samodzielnie, krzyk dziecka jest słaby, ssie z trudnością. Zanik mięśni
maskowany jest tkanką tłuszczową. Typowym objawem choroby są rytmiczne drżenia
palców. Rozwój psychiczny dzieci jest przeważnie bardzo dobry. Nie stwierdza się
zaburzeń czucia. Z wiekiem dochodzi u nich do ogromnych deformacji kręgosłupa i
klatki piersiowej, pojawiają się dalsze przykurcze stawowe. Dzieci te są zdumiewająco
zdolne, znacznie przewyższają inteligencją swoich rówieśników.
Najpoważniejszą konsekwencją SMA jest powolne porażenie przepony prowadzące do
utraty możliwości samodzielnego oddychania. Najczęściej dochodzi do tego w
pierwszych sześciu miesiącach życia. Uwagę zwraca również brak grasicy lub jej małe
rozmiary z obecnością tylko warstwy rdzeniowej, a także niedorozwój układu
siateczkowo-środbłonkowego migdałków, śledziony i jelit .

Objawy kliniczne SMA typ 2- postać późnodziecięca, wolniej postępująca: Dzieci rodzą
się pozornie zdrowe. U niemowląt ssanie i połykanie są zwykle zachowane, nie
obserwuje się zaburzeń oddychania. Rozwija się osłabienie mięśni, niemniej wiele dzieci
przeżywa do wieku szkolnego. Mowa nosowa i zaburzenia połykania pojawiają się w
późniejszym okresie. W pierwszych latach rozwija się obraz choroby
podobny do typu 1 SMA, ale zamiast wiotkości na pierwszy plan wysuwają się
zniekształcenia klatki piersiowej i kręgosłupa oraz przykurcze stawowe, a zanik mięśni
jest łatwo zauważalny z powodu szczupłości dzieci; wyraźne widoczne są fascykulacje
(błyskawiczne, drobne skurcze grup włókienek mięśniowych).
Objawy kliniczne SMA typ 3 (choroba Kugelberga–Welandera)- bardziej przewlekła,
młodzieńcza postać. Jest to najłagodniejsza postać SMA. Rozwój dziecka w okresie
niemowlęcym przebiega prawidłowo. Osłabienie dotyczy przede wszystkim mięśni pasa
miednicznego i wykazuje postępujący przebieg. Rzadko występują objawy ze strony
mięśni opuszkowych zagrażające uduszeniem. Objawy choroby rozpoczynają się zwykle
pomiędzy 3. a 18. r.ż. Należą do nich opóźnienie rozwoju ruchowego, niezręczny
chód, trudności we wstawaniu z pozycji leżącej i we wchodzeniu na schody. Zaniki
mięśniowe dotyczą przede wszystkim mięśni obręczy biodrowej z przerostem
rzekomym lub prawdziwym łydek i pośladków. Rzadko powstają
deformacje kręgosłupa. W tym typie drżenia pęczkowe są częstsze niż w typie 1 i 2.
Opisywano również przypadki kardiomiopatii (grupa chorób mięśnia sercowego).
Odmianą szczególną SMA jest choroba Fazio–Londego. Dotyczy ona bardziej pnia
mózgu niż rdzenia kręgowego i wyraża się postępującym porażeniem opuszkowym.
Objawami zespołu opuszkowego są zaburzenia połykania (dysfagia) oraz dyzartria
wiotka i niedowład podniebienia. Mowa jest niewyraźna i zamazana, zniekształcone są
głoski wargowe i językowe. Dochodzi do zaników mięśni języka,
mogą być obecne fascykulacje.

Istnieje możliwość diagnostyki prenatalnej SMA. Dziś najbardziej nowoczesnym
badaniem jest metoda ilościowa MLPA stosowana w Szwecji.
Obecnie w diagnostyce SMA stosuje się trzy testy, SSCP, PCR oraz DHPLC
(metoda szybkiej diagnostyki SMA).
Po urodzeniu dziecka możliwe jest również wykonanie innych badań, np. pobranie
bioptatu z mięśnia.
Badanie elektromiograficzne ma bardzo duże znaczenie w diagnostyce –
potwierdza neurogenny charakter uszkodzenia, a także pozwala wykrywać tzw.
cechy rdzeniowe. Do najważniejszych cech zapisu EMG należą wysoka amplituda
ubogiego zapisu wysiłkowego i pojedynczego potencjału, wydłużenie potencjałów
i wzrost obszaru jednostki ruchowej.
Od kilku lat trwają poszukiwania takiego leku, który podwyższałby stężenie białka
SMN u chorych dzieci. Najbardziej obiecującą grupą wydają się inhibitory
deacetylazy histonowej. Leki te rozluźniają wiązania DNA z białkami, na które DNA
jest nawinięte. Geny są odsłaniane i ułatwiony jest dostęp do transkrypcji DNA.
Spośród szeregu substancji należących do inhibitorów deacetylazy histonowej
najbardziej znanymi są kwas walproinowy i fenylomaślan sodowy. Leki te były już
wcześniej wykorzystywane w medycynie, stąd możliwe było ich szybkie wejście w
fazę prób klinicznych.
Leki podnoszące stężenie białka SMN nie wyleczą wprawdzie choroby SMA, ale
mogą opóźnić lub zahamować jej postęp. Dochodzi tu bowiem do obumierania
neuronów ruchowych w okresie życia płodowego i komórek tych nie można już
odtworzyć ani zregenerować. Toczy się więc walka o obronę tych neuronów, które
przeżyły początkowy okres choroby do momentu rozpoznania i włączenia leczenia
.

Cel i zadania rehabilitacji pokrywają się z celami leczenia usprawniającego w dystrofiach
mięśniowych. Trzeba jednak pamiętać, że w przypadkach rdzeniowych zaników mięśnie lepiej
poddają się bezpośredniej stymulacji biomechanicznej (jak masaż czy ćwiczenia z oporem), gdyż
istota patologii leży poza włóknami mięśniowymi. Czasami jedyną pomocą w typie 1 SMA pozostaje
wentylacja mechaniczna. Konieczne jest zwalczanie wszelkich, nawet najbardziej błahych infekcji
układu oddechowego i systematyczna toaleta drzewa oskrzelowego.
Ważne jest również żywienie dożołądkowe (dojelitowe), odpowiednio zbilansowane jakościowo i
kalorycznie.
Po uzyskaniu wyniku badania genetycznego potwierdzającego rozpoznanie rdzeniowego zaniku
mięśni rodzice niekiedy załamują się, nie godzą się z rzeczywistością, nie chcą myśleć o przyszłości.
Uzyskują informację od lekarzy o braku skutecznego leczenia farmakologicznego. Poszukują więc
pomocy poza medycyną konwencjonalną. Tym samym opóźniają właściwe i skuteczne działanie!
Najtrudniejsze są pierwsze 2 lata życia dziecka z powodu naturalnej niedojrzałości układu
odpornościowego i oddechowego. Mimo znacznych ograniczeń w
rozwoju ruchowym dzieci z SMA cechują się ponadprzeciętnym poziomem rozwoju intelektualnego.
Są bardzo radosne i szczęśliwe. W szkole osiągają bardzo dobre wyniki w nauce. Jest tylko jeden
warunek, muszą mieć zabezpieczony układ oddechowy!
Rodzice zamartwiają się w pierwszym rzędzie ograniczeniem rozwoju fizycznego swoich dzieci.
Zapominają o tym, że brak ruchów kończyn, niemożność siedzenia i chodzenia nie wpływają
bezpośrednio na czas przeżycia. Życie dziecka zależy od jakości układu oddechowego.
Jak najszybciej zakupić lub wypożyczyć należy ssak (pozwala usuwać wydzielinę z gardła i nosa),
pulsoksymetr (urządzeniem służącym do monitorowania saturacji oraz akcji serca), butlę z tlenem
(tlen powinien być stosowany wyłącznie w czasie ciężkiej infekcji układu oddechowego i w trakcie
akcji reanimacyjnej) i ambu (samorozprężalny worek służący do prowadzenia wentylacji ręcznej
powietrzem lub lepiej mieszaniną powietrza i tlenu podczas reanimacji). Są to wszystko urządzenia
niezbędne, służące bezpośrednio do ratowania życia.

Jednostka chorobowa znana
także pod nazwą zespołu
Seemanowej. Jest to rzadka
choroba monogenowa,
dziedziczona w
sposób autosomalny recesywny,
której przyczyną jest wrodzony
defekt jednego
z podstawowych mechanizmów
naprawy dwuniciowych pęknięć
DNA, prowadzący do
niestabilności genomu.
Najczęściej dotyka osoby
pochodzenia
zachodniosłowiańskiego, przy
czym najwięcej przypadków
zdiagnozowano w Polsce.

Na obraz choroby składają się przede wszystkim: pierwotne małogłowie,
zaburzenia wzrastania prowadzące do niedoboru wzrostu, dysfunkcja gonad,
nawracające infekcje spowodowane złożonymi niedoborami odporności,
nadwrażliwość na promieniowanie jonizujące o wysokich energiach oraz
wybitna predyspozycja do rozwoju w młodym wieku nowotworów złośliwych,
zwłaszcza chłoniaków. Zespołowi może towarzyszyć upośledzenie umysłowe,
najczęściej w stopniu lekkim.
Rozwój psychoruchowy i umysłowy w pierwszych czterech latach życia dziecka
z NBS przebiega prawidłowo lub na pograniczu tzw. szerokiej normy. Z
wiekiem jednak dochodzi do stopniowego obniżania się funkcji intelektualnych
i ilorazu inteligencji do uzyskania poziomu lekkiego lub umiarkowanego
upośledzenia umysłowego u wszystkich pacjentów z NBS po ukończeniu 14 r.ż.
Niedostateczny rozwój mózgoczaszki determinuje dysmorfię twarzy, która
narasta z wiekiem i polega przede wszystkim na pochyleniu wąskiego czoła ku
tyłowi i cofnięciu bródki, co znacząco uwydatnia środkową część
twarzy. Ponadto mogą występować dysplastyczne małżowiny uszne,
zmarszczki nakątne szpar powiekowych, względnie wydłużony i zadarty w górę
nos, uwypuklenie rynienki wargowej.
U co drugiego pacjenta występują na skórze plamy w kolorze kawy z mlekiem
oraz postępujące z wiekiem bielactwo.

Występować mogą także niewielkie anomalie rozwojowe
kończyn- palców stóp i rąk, rzadziej występuje zdwojenie kciuka,
zwężenie lub zarośnięcie odbytu i dysplazja stawów biodrowych.
Charakterystyczne są nieprawidłowości w budowie mózgu, które
w badaniu magnetycznym rezonansem są widoczne
pod postacią hipoplazji płatów czołowych.
U 55% dzieci z NBS stwierdza się zachorowania na nowotwory
złośliwe, przy czym nowotwory te pojawiają się zwykle w
pierwszej dekadzie życia dziecka.
Ryzyko pojawienia się NBS u potomstwa jest w każdej ciąży
jednakowo wysokie i wynosi 25%.
Dzieci młodsze wymagają zazwyczaj kompleksowej pomocy
psychologicznej i logopedycznej ze względu na wzmożoną
aktywność psychoruchową z deficytem uwagi i opóźnionym
rozwojem mowy. U dzieci starszych z
niepełnosprawnością umysłową powinien być wdrażany
specjalistyczny program opieki specjalnej, uwzględniający ich
możliwości intelektualne.
Dzieci z NBS cechuje zazwyczaj łagodna osobowość i nieśmiałość,
mimo to są zdolne do nawiązywania prawidłowych interakcji
społecznych.

Francuski neurolog i neuropsychiatra Georges Albert E. B. Gilles de la
Tourette jako pierwszy w 1885 roku opisał zespół zróżnicowanych tików,
nazywany później jego nazwiskiem. Wśród objawów stwierdzono
występowanie tików motorycznych
i wokalnych oraz echolalii i koprolalii, które obecnie nie są wymieniane
wśród kryteriów diagnostycznych wymaganych do rozpoznania
zaburzenia.
Przyjmuje się, że zespół Tourette`a występuje u około 5 przypadków na
10 000 osób, 3-4 razy częściej u chłopców niż u dziewcząt.

Grupa zaburzeń F95 Tiki obejmuje (na podstawie ICD-10, 1997r.):
- F95.0 – Tiki przemijające, utrzymujące się nie dłużej niż przez okres 12
miesięcy, często występujące u dzieci w wieku 4-5 lat.
- F95.1 – Przewlekłe tiki ruchowe i głosowe (wokalne). Specyficzne dla te
grupy jest występowanie tików motorycznych lub wokalnych osobno – nie
jednocześnie. Czas trwania objawów jest dłuższy niż rok. Tiki mogą być
proste i złożone (mnogie).
- F95.2 – Zespół tików głosowych i ruchowych (zespół Gilles de la Tourette`a)
-F95.8 – Inne tiki.
Tiki są to niezależne od woli, nagłe, szybkie, powtarzające się ruchy i/lub
wokalizacje. Tiki ruchowe to mimowolne, szybkie, gwałtowne, nagłe
czynności ruchowe występujące pojedynczo i/lub seryjnie, obejmujące grupy
mięśni twarzy, kończyn ramion.
Są to niecelowe, powtarzające się skurcze pojedynczych mięśni lub grup
mięśni, najczęściej w obrębie głowy, twarzy, ramion, rzadziej kończyn. Tiki
wokalne to mimowolne, szybkie, gwałtowne, nagle wydawane dźwięki i/lub
wypowiadane słowa.

W zespole Tourette`a wyróżnia się trzy grupy przyczyn: genetyczne,
neurochemiczne
i spowodowane zaburzeniami centralnego układu nerwowego. Istotną rolę
w utrwalaniu się objawów zaburzenia odgrywają także czynniki
psychologiczne.
W uwarunkowaniach genetycznych najczęściej wskazuje się na
dziedziczenie autosomalne dominujące. Ryzyko wystąpienia zespołu lub
innych patologii, na przykład zachowań obsesyjno-kompulsyjnych u
potomstwa obojga płci osoby chorej na zespół Tourette`a, wynosi 50%.
Genetyczne uwarunkowanie zaburzeń sugerował już sam G. Gilles de la
Tourette. Badania dotyczące roli dziedziczenia są obecnie szeroko
prowadzone w Anglii i Stanach Zjednoczonych.
W neurochemicznych uwarunkowaniach zakłada się zaburzenia w zakresie
neuroprzekaźników – szczególnie dopaminy, serotoniny i noradrenaliny
(hiperaktywność lub niedobór substancji neuroprzekaźnikowych w mózgu).
U osób z syndromem Tourette`a zaburzenia w zakresie neuroprzekaźników
to nadwrażliwość receptorów dopaminowych oraz obniżone stężenie
serotoniny i noradrenaliny.

Do grupy przyczyn występowania zaburzeń centralnego układu
nerwowego zaliczono nieprawidłowości w obszarach mózgu
odpowiedzialnych za motorykę. Tiki są spowodowane dysfunkcją w
okolicach korowych i podkorowych, w zwojach podstawy i korze
przedniej. Sytuacje stresowe, wydarzenia traumatyczne pogarszają stan
pacjentów. Spokój i wyrozumiały stosunek do dziecka oraz stabilne
środowisko jego stan poprawiają, pomimo słabej odporności
obserwowanej u niego w trudnych sytuacjach.
Pierwsze symptomy zaburzenia można dostrzec około 7. roku życia
dziecka. U większości osób w 90% ujawniają się między 2. a 15. rokiem
życia (Goodman, Scott, 2000, s.119-120). W przypadku tików rokowanie
jest pomyślne. jednak w przypadku zespołu Tourette`a, ze względu na
genetyczny i neuropsychiatryczny charakter symptomów, występuje
tendencja do nawracania i utrwalania się zaburzeń.

U dzieci z zespołem Gilles da la Tourette`a występują złożone tiki
motoryczne oraz jeden lub kilka tików wokalnych, które mogą się
ujawniać od wieku przedszkolnego do około 15. roku życia. Do prostych
tików motorycznych zalicza się między innymi: uporczywe mruganie,
potrząsanie głową, napięciowe ruchy ramion, dłoni, języka, grymasy
twarzy, zaciskanie pieści, otwieranie ust, ruchy przypominające
potakiwanie, uderzanie, dotykanie, obwąchiwanie. Proste tiki wokalne to
na przykład: pomruki, chrząkanie, jęki, czkawka.
Wyróżnia się też złożone tiki motoryczne i wokalne określane jako:
echopraksja (przymusowe naśladowanie ruchów, gestów wykonywanych
przez inne osoby), echolalia (mimowolne powtarzanie zasłyszanych
głosek, sylab, słów, zdań), palilalia (automatyczne powtarzanie własnych,
wypowiadanych przez siebie głosek, sylab, słów, zdań, nawet fraz).

Do tików złożonych zaliczane są także
kopropraksja (mimowolne wykonywanie
obscenicznych gestów i czynności) oraz
koprolalia (przymusowe wypowiadanie
wulgarnych słów), które występują u około
10-30% chorych.
Obraz kliniczny zespołu Tourette`a
charakteryzuje się zmiennością, która
obejmuje: intensywność tików, formę,
lokalizację, częstość występowanie oraz siłę.
Zespół Tourette`a przebiega w sposób
niejednorodny u poszczególnych pacjentów.
Obraz ten zmienia się wraz z rozwojem
dziecka. Tiki raczej wykazują tendencję do
stabilizacji na pewnym poziomie, zdarza się,
że wygasają albo słabną w okresie
adolescencji.

Według Klasyfikacji Amerykańskiego Towarzystwa
Psychiatrycznego, kryteria diagnostyczne będące podstawą
rozpoznania u dziecka zespołu Tourette`a są następujące:
A. Występowanie złożonych tików ruchowych i pojedynczych lub złożonych
współwystępujących razem.
B. Tiki występują wiele razy wciągu dnia (zazwyczaj w seriach) prawie
codziennie lub
z przerwami przez okres powyżej roku; w tym okresie nie występował ani
razu okres wolny
od tików, trwający dłużej niż 3 kolejne miesiące.
C. Początek przed 8. rokiem życia.
D. Zaburzenie nie jest spowodowane bezpośrednim fizjologicznym
wpływem substancji
(np. pobudzających) lub znaną przyczyną chorobową, np. pląsawicą
Huntingtona lub wirusowym zapaleniem mózgu (DSM-IV TR, 2000, s.114).

Dzieci z zespołem Tourette`a najczęściej są poddawane leczeniu
farmakologicznemu, rzadziej terapii psychologicznej. Leczenie
farmakologiczne, stosowane przede wszystkim w stanach przewlekłych i o
dużym nasileniu symptomów, przynosi pozytywne efekty. Leczenie
powinno być wspomagane terapią psychologiczną, zwłaszcza w zakresie
redukcji tików i natręctw (albo ich osłabienia) oraz kontroli zachowań
agresywnych.
Dzieciom z zespołem Tourette`a potrzebna jest także pomoc w nauce
szkolnej. Trudności w koncentracji uwagi – szczególnie podczas
samodzielnej pracy – znacznie obniżają ich wyniki w szkole.
Zorganizowanie im warunków sprzyjających skupianiu się na zadaniu
zwiększa zakres i trwałość przyswajanej wiedzy.

Rodzinom dzieci z tym zespołem
lekarze powinni przekazać rzetelną
wiedzę na temat istoty zaburzenia i
rokowania. Niezbędna jest także
pomoc pedagogiczna, zwłaszcza w
zakresie współdziałania z dzieckiem
opartego na więzi emocjonalnej i
konsekwencji, a nie na nieefektywnym
wychowaniu opartym na
imperatywnych działaniach.
Pomoc psychologiczna dla dziecka
powinna natomiast obejmować trening
behawioralny, a dla rodzin trening
terapeutyczny w zakresie radzenia
sobie ze stresem oraz z problemami,
jakie mogą wystąpić na dalszych
etapach życia dziecka.

Opis trudności związanych z funkcjonowaniem społecznym, dziś
określanych jako fobia społeczna, jest jedną z jednostek podawanych w
oficjalnych klasyfikacjach zaburzeń, która pojawiła się już w starożytności.
W fobii społecznej odczuwane lęki i zachowania unikające są wywoływane
przez różne sytuacje społeczne. To unikanie ludzi w mniejszych grupach,
unikanie bliższego kontaktu dotyczy jakiegokolwiek stykania się albo też
określonych rodzajów kontaktu […]”. Fobia społeczna jest nazywana także
zaburzeniem lęku społecznego.

Współczesne badania wskazują głównie na dwa mechanizmy
związane
z występowaniem fobii społecznej: neurobiologiczne mechanizmy oraz
uwarunkowania genetyczne. Te pierwsze wskazują na rolę
neurotransmiterów w reakcji wycofania się
z kontaktów społecznych. Głównym podejrzanym jest serotonina.
Lękliwość jest z nią łączona. Serotonina wzmaga lęk, depresję i inne
składniki unikania urazów.
Badania etiologii fobii społecznej wskazują też na jej genetyczne
uwarunkowania. Badania rodzin osób z fobią społeczną sugerują
dziedziczne uwarunkowania lęku społecznego. W rodzinach osób z
fobią społeczną częściej występują zaburzenia związane z lękiem, jak
również inne problemy zdrowotne (np.: problemy z żołądkiem, katar
sienny, problemy skórne, oddechowe, alergie).

W klasyfikacji zaburzeń ICD-10 fobia społeczna występuje w grupie
zaburzeń nazywanych zaburzeniami lękowymi pod postacią fobii (F40). To
lęk społeczny w dzieciństwie (F93.2), który jest rozpoznawany na podstawie
następujących kryteriów:
A. Utrwalony lęk przejawiający się zachowaniem o cechach społecznego unikania
w sytuacjach, w których dziecko jest narażone na obecność nieznanych sobie ludzi, w
tym także rówieśników.
B. Dziecko wykazuje koncentrację na sobie, zakłopotanie lub nadmierne skupienie na
sprawie stosowności swego zachowania w czasie kontaktów z nieznanymi sobie
ludźmi.
C. Zachodzi istotne zakłócenie relacji społecznych (w tym relacji z rówieśnikami),
które ulegają konsekwentnemu ograniczaniu. Gdy doświadczane są nowe lub
wymuszone sytuacje społeczne, powodują znaczne cierpienie u dyskomfort
przejawiane w postaci płaczu, braku spontanicznych wypowiedzi lub wycofania się.
D. Dziecko ma zadowalające relacje społeczne z członkami rodziny lub dobrze
znanymi sobie rówieśnikami

E. Początek zaburzenia współistnieje z
fazą rozwoju, w której takie reakcje
lękowe
są adekwatne. nieprawidłowy stopień,
utrwalanie w czasie i towarzyszące
upośledzenie
funkcjonowania
społecznego ujawnia się przed 6. rokiem
życia.
F. Nie są spełnione kryteria uogólnionego
zaburzenia lękowego w dzieciństwie.
G. Zaburzenie nie stanowi części
szerszego
zaburzenia
emocji,
zachowania
lub
osobowości,
ani
całościowego zaburzenia rozwojowego,
zaburzenia
psychotycznego
czy
związanego z używaniem substancji
psychoaktywnych.
H. Czas trwania zaburzenia wynosi co
najmniej 4 tygodnie.

Pierwsze objawy fobii społecznej (zaburzeń lęku społecznego), a właściwie
zwiastuny późniejszych zaburzeń pojawiają się u dziecka już około 1. roku
życia. Małe dziecko reaguje strachem, niepokojem w kontaktach z ludźmi.
W okresie przedszkolnym i szkolnym symptomy trudności stają się coraz
wyraźniejsze. W większości zaburzeń związanych
z lękiem społecznym symptomy ujawniają się przed adolescencją.
Dziecko z fobią społeczną ma trudności z komunikowaniem się z innymi i
ekspresją siebie (postaw, emocji, zachowań). Problemy w zachowaniu są
też widoczne w grupie rówieśniczej oraz w szkole, w sytuacjach
zadaniowych. Dzieci z fobią społeczną nie są samodzielne. Mają bardzo
dużą świadomość doświadczanych problemów, czego konsekwencją może
być pojawienie się objawów depresji.

W przypadku fobii społecznej wczesna interwencja może być profilaktyką
wobec wymienionych wcześniej zjawisk. Mówiąc o wczesnej interwencji
należy mieć na myśli podejmowanie profilaktycznych działań wobec dzieci
do 5. roku życia i ich rodzin. Zaburzenia związane są z lękiem społecznym
rzadko są diagnozowane tak wcześnie. Objawem, który jest bardzo
widoczny w obrazie klinicznym dziecka z lękiem społecznym, jest
wycofanie się. Wycofanie i tego typu problemy występują też u takich
dzieci, u których zachowanie takie nie jest jednak związane z lękiem, ale z
brakiem kompetencji potrzebnych w prawidłowym funkcjonowaniu
społecznym i komunikowaniu się z otoczeniem (np. całościowe zaburzenia
rozwojowe).
Diagnozy są formułowane bowiem wobec dzieci w wieku od 10 do 13 lat.
Szkoła podstawowa i gimnazjum to takie etapy w życiu dziecka, w których
symptomy fobii stają się bardzo wyraźne. Elementarnym błędem może być
bagatelizowanie trudności dziecka w sferze społecznej i tłumaczenie ich
nieśmiałością. Fobia społeczna powinna być odróżniana
od takich zaburzeń, jak: zaburzenia uwagi, napady paniki czy agorafobia
(Jefferson, 2001).

W terapii fobii społecznej można wyodrębnić cztery formy
oddziaływań:
1. Terapie poznawcze – ich cel to zmiana obrazu własnej osoby oraz
nastawienia do relacji interpersonalnych (tamże, s.161).
2. Trening umiejętności społecznych – ważnym elementem pracy z
osobą z fobią społeczną jest umożliwienie jej nauczenia się, jak
najlepiej radzić sobie w sytuacjach społecznych.
3. Relaksacja.
4. Farmakoterapia.

Choroba sieroca jest spowodowana brakiem więzi z osobami bliskimi,
jest choroba braku miłości – bycia kochanym; stanowi zespół
poważnych zaburzeń lub zahamowani rozwoju psychicznego i
fizycznego u dzieci.

Przyczyn nabycia choroby sierocej należy poszukiwać w bardzo wczesnym
dzieciństwie. Dziecko przychodzi na świat jako jednostka zupełnie zależna od opieki
dorosłego, bezbronna i niesamodzielna. Proces kształtowania się indywidualnego
przywiązania dziecka do osoby opiekującej się nim, przeważnie matki, rozpoczyna się
zaraz po urodzeniu.
Jego nasilenie i przebieg zależy od stopnia, w jakim matka odpowiada na wezwania
dziecka i zaspokaja jego potrzeby. Osoba matki ma więc znaczenie kluczowe i
wyjątkowe w poznawaniu przez nie świata i wzbogacaniu doświadczeń. Powstawanie
interakcji między matką a dzieckiem opartej na przywiązaniu i bliskości może być
zaburzone, a nawet w sposób gwałtowny zerwane: czasami z powodu odrzucenia
dziecka przez najbliższe mu osoby, innym razem – odebrania go rodzicom. W takich
sytuacjach dziecko dotyka stan określany w literaturze przedmiotu sieroctwem
społecznym. Przyczyn sieroctwa społecznego można się doszukać w rodzinach z
problemem alkoholowym lub/i narkotykowym, kiedy rodzice dopuszczają się
poważnych zaniedbań i nadużyć wobec swoich dzieci. Czynnikami powodującymi tę
chorobę mogą być także ubóstwo, wielodzietność, bezrobocie bądź choroba
psychiczna opiekunów. Innym zjawiskiem patologicznym, wręcz wymuszającym
separację dziecka od rodziny jest stosowanie przemocy, a szczególnie krzywdzenie
fizyczne i molestowanie seksualne.

W literaturze przedmiotu wyróżnia się zwykle trzy etapy choroby sierocej. W
pierwszej fazie, zwanej protestem dziecko domaga się płaczem kontaktu z matką.
W ten sposób protestuje przeciwko oddzieleniu go od osoby bliskiej, bez której jest
mu źle i za którą tęskni. Faza ta ma różny przebieg, zależny od wieku. Dzieci
starsze zwykle reagują agresją,
a czasem oporem. Agresja może być skierowana przeciwko sobie, przedmiotom lub
innym osobom. U młodszych pojawiają się zaburzenia snu, nadpobudliwość, utrata
łaknienia, krzyk, płacz, a także zaburzenia żołądkowe i wymioty.
Gdy rozłąka trwa przez pewien czas, pojawia się druga faza choroby sierocej –
rozpacz, objawiająca się częstym płaczem, smutkiem, małym ożywieniem dziecka.
Przeciągająca się w czasie separacja od matki powoduje, że na zbliżenie się innej
osoby dorosłej dziecko reaguje krzykiem lub ucieczką. Równocześnie pojawiają się
u niego objawy związane
z układem nerwowym, takie jak ślinienie się, drżenie ciała, pocenie się, zaburzenia
snu, moczenie nocne, utrata wagi.

Ostatnia, trzecia faza choroby sierocej – wyobcowanie lub zobojętnienie –
nie manifestuje się burzliwymi objawami. Dziecko po ostrym buncie
pozornie się uspokaja: już nie płacze, nie protestuje, wydaje się jakby
pogodzone z losem, pozornie przyzwyczajone do swojej sytuacji, staje się
bierne, posłuszne, apatyczne, ma „pusty”, matowy wzrok. U niemowląt
często obserwuje się „sufitowanie” – ciągłe błądzenie pustym wzrokiem po
białej pozbawionej bodźców powierzchni.

Długotrwałe pozbawienie dziecka bliskości z matką, a przede wszystkim
brak jej obecności emocjonalnej oraz fizycznej, ma teratogenny wpływ na
jego dalszy rozwój, szczególnie gdy dotyczy pierwszych dwóch, trzech lat
życia. Skutki choroby sierocej mogą być nieodwracalne lub trudno
odwracalne.
Znana badaczka zjawiska sieroctwa – M. Łopatkowa (1989) wymieni
następujące skutki choroby sierocej obserwowane u wychowanków
placówek opiekuńczo – wychowawczych:
1. Niedojrzałość uczuciowa, zahamowanie rozwoju emocjonalnego.
2. Problemy z nawiązywaniem i podtrzymywaniem kontaktów
uczuciowych.
3. Poczucie niższości, samotności, lęki i depresja.
4. Niska aktywność życiowa i ubóstwo inicjatyw.
5. Niskie zainteresowanie światem i otoczeniem.
6. Huśtawka nastrojów, nieprzewidywalne zachowania.

7. Nieumiejętność radzenia sobie w nowych sytuacjach (bezradność
lub agresja).
8. Brak zainteresowania opiekunami (pracownikami domu dziecka).
9. Duże zainteresowanie obcymi ludźmi.
10. Brak koncentracji uwagi.
11. Uboga wyobraźnia.
12. Słabe myślenie logiczne i abstrakcyjne.

1. Oswajanie dziecka z nową sytuacją życiową. Opiekun dziecka nie może
mu się narzucać (pozorna bierność), a równocześnie musi okazywać mu
życzliwość i zrozumienie. Powinien być zawsze w pobliżu, towarzyszyć mu
i akceptować je takim, jakie jest.
2. Po początkowym, spokojnym okresie dziecko zaczyna się zachowywać
agresywnie, jest niespokojne i zaczyna rozpamiętywać przeszłość
(frustracja). Wtedy można z nim nawiązać nić porozumienia. W tym
okresie reakcje dziecka są nienormalne – dotychczas grzeczne zaczyna
objawiać niepokój, a często nawet agresję w stosunku do matki. Jest to
zatem najtrudniejszy moment dla matki adopcyjnej czy zastępczej, która
zwraca ku dziecku swoją miłość, a w odpowiedzi spotyka się z wrogością.
Osobą akceptowaną przez dziecko staje się ojciec. Być może dlatego, że
w dotychczasowym swoim życiu w placówce opiekuńczej dziecko było
wychowywane przez kobiety, więc traktuje „nową” matkę jako jeszcze
jedną opiekunkę z domu dziecka.

3. Po „oczyszczających” i „wzmacniających” przeżyciach dziecko
zaczyna
wracać
do równowagi. Przywiązuje się zarówno do matki, jak i ojca, staje się
wrażliwsze i „bardziej normalne”.
Specjaliści zajmujący się problemem samotności twierdzą, że jako
społeczeństwo
coraz
częściej
nie
doceniamy
wartości
więzi
międzyludzkich. Rozwiązaniem tego dylematu bywa spychanie napięcia
psychicznego w obręb patologii osamotnionego człowieka. Tymczasem
otaczające go społeczeństwo tę samotność napędza, potęguje i samo
staje się przyczyną „zapętlonej” samotności. Popycha wielu ludzi w
jeszcze
większą
izolację
i nieprzystosowanie. Mylnie przyjmuje się założenie, że to nie w
społeczeństwie, ale w tych osobach dzieje się coś niedobrego. Dziecko
samotnie izoluje się od tłumu, a tłum izoluje go od siebie. Zatem
możemy powiedzieć, że nasze osamotnione i odtrącone dzieci są
skazane na samotność w tłumie. Mimo że żyją w zbiorowości, w licznych
grupach, tak naprawdę są nikomu nie potrzebne. To jest syndrom
spadłego liścia.

Bibliografia:
„Dzieci chore, niepełnosprawne
i z utrudnieniami w
rozwoju”
pod red. B. Cytowskiej, B. Winczury i A. Stawarskiego,
Wyd. „Impuls”, Kraków 2008
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
Wyszukiwarka
Podobne podstrony:
Dzieci chore, niepełnosprawne i z utrudnieniami w rozwoju ebook
Dzieci chore, niepełnosprawne i z utrudnieniami w rozwoju ebook
dzieci chore, niepełnosprawne i z utrudnieniami w rozwoju
Wczesne wspomaganie rozwoju dzieci, Oligofrenopedagogika, NIEPEŁNOSPRAWNOŚĆ, oligofrenopedagogika ku
Nadpobudliwość psychoruchowa u dzieci, Studia rok I, Psychologia rozwoju człowieka
WARTOŚĆ RUCHU W TERAPII DZIECI, Dokumenty o niepełnosprawnościach
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 7, PSYCHOLOGIA ROZWOJOWA DZIECI I MŁODZIE
Funkcjonowanie społeczne dzieci w rodzinach niepełnych
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 3(2), Psychologia rozwojowa dzieci i młod
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 3(2), Psychologia rozwojowa dzieci i młod
4. Rozwój lateralizacji (M.Bogdanowicz - 'Leworęczność u dzieci' s.29-43), PSYCHOLOGIA ROZWOJOWA
Metodyka nauczania zagadnienia 2011 dzienne, Oligofrenopedagogika, Różnice programowe, Metodyka nauc
Program stymulacji funkcji komunikacyjnych u dzieci z głęboką niepełnosprawnością intelektualną
Istota i?le katechizacji dzieci i młodzieży niepełnosprawnej intelektualnie
więcej podobnych podstron